

Abstract
Background & Aims:
Liver sinusoidal endothelial cells (LSEC) are ideally situated to sense stiffness and generate angiocrine programs potentially regulating liver fibrosis, portal hypertension. We explored how specific focal adhesion (FA) proteins parlay LSEC mechanotransduction into stiffness-induced angiocrine signaling in vitro and in vivo.
Methods:
Primary human, murine LSEC were placed on gels with incremental stiffness (0.2kP vs. 32kPa). Cell response was studied by FA isolation, actin polymerization assay, RNA sequencing and electron microscopy. Glycolysis was assessed using radioactive tracers. Epigenetic regulation of stiffness-induced genes was analyzed by chromatin immunoprecipitation (ChIP) analysis of histone activation marks, ChIP sequencing and circularized chromosome conformation capture (4C). Mice with LSEC-selective deletion of glycolytic enzymes (HK2fl/fl/VCad-ERT2cre) or treatment with the glycolysis inhibitor 3PO were studied in a portal hypertension (partial ligation of the inferior vena cava, pIVCL) and CCl4 model.
Results:
Glycolytic enzymes, particularly Phosphofructokinase 1 isoform P (PFKP), are enriched in isolated FA from LSEC on gels with incremental stiffness. Stiffness resulted in PFKP recruitment to FA, which paralleled an increase in glycolysis. Glycolysis was associated with expansion of actin dynamics and attenuated by inhibition of Integrin beta 1. Inhibition of glycolysis attenuated a stiffness-induced CXCL1-dominant angiocrine program. Mechanistically, glycolysis promoted CXCL1 expression through nuclear pore changes and increases in NFkB translocation. Biochemically, this CXCL1 expression was mediated through spatial re-organization of nuclear chromatin resulting in formation of super-enhancers, histone acetylation and NFkB interaction with CXCL1 promoter. HK2fl/fl/VCad-ERT2cre mice showed attenuated neutrophil infiltration, portal hypertension after pIVCL. 3PO treatment attenuated liver fibrosis in a CCl4 model.
Conclusion:
Glycolytic enzymes are involved in stiffness-induced angiocrine signaling in liver sinusoidal endothelial cells representing druggable targets in early liver disease.
Keywords: angiocrine signaling, actin polymerization, CXCL1, glycolysis, liver sinusoidal endothelial cells, mechanosensing, nuclear pores, portal hypertension
INTRODUCTION
The hepatic sinusoid with the close proximity of liver sinusoidal endothelial cells (LSEC) and the pericyte-like hepatic stellate cells (HSC) acts as a microvascular niche, which is involved in early liver fibrogenesis.(1) Within this niche, LSEC interact with the portal circulation by expression of surface receptors and release of paracrine factors.(2, 3) These endothelial cell-released paracrine molecules in turn regulate organ function, referred to as angiocrine signaling.(4) Chemokines could be an important part of an angiocrine signaling program given they can attract neutrophils and thrombocytes, thereby promoting inflammation, intrasinusoidal thrombosis and increasing portal venous pressure.(5) Exploration of the mechanisms by which these chemokines are triggered and released from LSEC may lead to the discovery of novel therapeutic targets for the treatment of end-stage liver disease and portal hypertension.
Edema and inflammation result in an increased tissue stiffness.(6) Increased stiffness affects cell behavior, disease progression and promotes fibrogenesis.(7) Cells sense stiffness of their environment through the Integrin family of cell surface receptors.(8) This process called mechanosensing relies on focal adhesions (FA), which consist of numerous integrins and Integrin-associated cytoplasmic proteins. FA proteins mediate force and signal transduction through conformational changes of cytoplasmic proteins resulting in exposure of previously buried sites, which promotes downstream signaling such as stress fiber formation.(9, 10) Downstream signaling then affects gene transcription through various mechanisms including epigenetic modifications.(11) The precise mechanisms by which LSEC mechanosensing parlays into angiocrine signaling are poorly understood.
In this study, we explored novel focal adhesion (FA) proteins involved in LSEC mechanotransduction and stiffness-induced angiocrine signaling in vitro and in vivo, and their possible role for development of early liver fibrosis and portal hypertension.
METHODS
Ethics statement.
Investigation of human samples was approved by the local ethics committee (IRB 15-008251) with written informed consent given by patients where applicable. All animal experiments were approved by IACUC and carried out in accordance with institutional guidelines (Mayo Clinic, Rochester, MN).
Experimental procedures.
Experimental procedures including animal studies are discussed in detail in the supplementary methods (Supplementary Material and CTAT Table).
Statistics.
Experiments were performed at least 3 independent times. Numerical data are expressed as mean ± SEM. Analysis of variance with Bonferroni post-test (ANOVA), paired and unpaired 2-tailed Student’s t test were used to assess the statistical significance between groups as appropriate with GraphPad Prism 5 software (GraphPad Software, Inc, La Jolla, CA). A P value less than 0.05 was considered significant.
RESULTS
Glycolysis and mechanotransduction in liver sinusoidal endothelial cells
Stiffness induces recruitment of glycolytic enzymes, particularly Phoshofructokinase 1, to focal adhesions.
Stiffness results in recruitment of specific Integrin-associated proteins such as Talin and Vinculin to FA.(12) We sought to identify novel proteins recruited to FA in LSEC in response to stiffness. We seeded primary human LSEC on soft (0.2kPa) and hard (32kPa) collagen-I-coated gels mimicking stiffness of healthy liver and liver with advanced fibrosis. Characterization of human LSEC (and comparison with hepatic stellate cells and endothelial cells) can be found in Supplementary Figure 1. Proteomic analysis of isolated FA proteins revealed considerable changes in FA composition with incremental stiffness (Figure 1A). For a detailed list of upregulated proteins, see Supplementary Table 1. Unexpectedly, gene ontology analysis of FA proteins that were more than 1.5 fold up-regulated on hard vs. soft gel identified glycolysis as the most enriched pathway (13.1 fold enrichment, Figure 1B). Fold enrichment was higher than that of the anticipated Integrin-signaling pathway (8.1 fold enrichment). Phosphofructokinase 1 platelet isoform (PFKP), one of the key rate-limiting glycolytic enzymes, was the most abundant glycolytic protein on hard vs. soft substrate. PFKP was up-regulated to a similar extent (13 fold increase) as typical FA proteins (Figure 1C). Proteomic analysis of isolated FA proteins from primary murine LSEC (on hard vs. soft gel) showed a similar involvement of glycolytic enzymes (13.8 fold enrichment, Supplementary Figure 2). For a detailed list of upregulated proteins, see Supplementary Table 2. Glycolysis, measured by radioactive-labeled tracers, was significantly increased with incremental stiffness (+46%, Figure 1D). Congruently, protein levels of PFKFB3, a key activator of glycolysis,(13) were significantly elevated (+59%) in response to stiffness (Figure 1E). These results led us to further explore the involvement of glycolysis and glycolytic enzymes in mechanotransduction through recruitment to FA.
Fig. 1. Increased stiffness results in formation of focal adhesions and recruitment of glycolytic enzymes, particularly Phoshofructokinase 1, to focal adhesions.

A. Heatmap for isolated FA proteins from human LSEC plated on hard (32kPa) vs. soft gel (0.2kPa), log2 fold change compared to integrin beta 1 (relative abundance), based on total spectrum counts obtained from mass spectrometry. Green color indicates relative enrichment of FA proteins compared to Integrin beta 1 (main protein within focal adhesions), red color indicates that the FA protein is less abundant than Integrin beta 1. Heatmap reveals considerable differences in the composition of FA on hard vs. soft gels. B. Gene ontology analysis of FA proteins upregulated ≥1.5 fold on hard vs. soft gels reveals glycolysis as the most enriched pathway, which is more enriched than the Integrin signaling pathway. Insert shows glycolytic enzymes contributing to the enrichment of glycolysis and their log2 fold increase on hard vs. soft gels. All values are normalized to Integrin beta 1. PFK is identified as the most upregulated glycolytic enzyme in FA on hard vs. soft gels. C. Mass spectrometry revealed upregulation of typical FA proteins in FA isolates from human LSEC plated on hard vs. soft gels, with upregulation of PFK being comparable to these typical FA proteins. Values represent log2 fold change of total spectrum counts on hard vs. soft gels, normalized to Integrin beta 1. D. Glycolysis was significantly increased in human LSEC plated on hard gels compared to soft gels using radiolabeled glucose experiments (n=4, **p<0.01). E. Protein levels of PFKFB3, a key activator of glycolysis, are significantly upregulated in human LSEC seeded on hard vs. soft gels (western blots on the left, quantification on the right, n=3, *p<0.05), GAPDH was used as a housekeeping cytosolic gene without changes in the whole cell lysate.
Integrin signaling promotes glycolysis and recruitment of PFKP to FA.
Glycolysis is up-regulated in endothelial cell (EC) migration and angiogenesis.(13, 14) Its role in FA-mediated mechanotransduction has yet to be determined. We sought to analyze the local interaction between Integrin and glycolytic enzymes and to explore changes in glycolysis in response to stiffness with regards to Integrin signaling. PFKP co-localized together with Vinculin (=prototypical FA protein) on hard, but not soft gels (Figure 2A), while no increase in total PFKP expression was detected (Supplementary Figure 3A). For quantification (32kPa vs 0.2kPa) see Figure 2A. For further evaluation of stiffness-induced changes within FA, we used plastic dish as a prototype for hard environment (10MPa). Whole-cell immunostaining of LSEC seeded in collagen-coated plastic dishes showed co-localization of PFKP and Vinculin with a correlation co-efficient of 0.712 (Figure 2B, Supplementary Figure 3B, Supplementary Figure 4). Immunostaining of isolated FA proteins showed a significant and strong overlap of Vinculin and PFKP (Figure 2C and D). Immunoprecipitation studies further demonstrated an association of PFKP with Integrin β1 (Figure 2E). Inhibition of Integrin signaling by treatment with MAb13, an Integrin β1 blocking antibody, significantly decreased mechanotransduction-induced glycolysis (−52%) as did replacement of Integrin activating collagen coating of gels with Poly-D-Lysine coating (−31%, Figure 2F). Congruently, PFKFB3 protein levels were decreased with MAb13 treatment (Figure 2G). To further explore the involvement of glycolysis in FA in vivo, we performed immunofluorescence for Vinculin in LSEC from mice with an LSEC-specific knockdown of glycolysis (HK2 fl/fl Cdh5cre/ERT2). Compared to control mice (HK2 fl/fl), these cells showed absence of FA formation at the cell membrane and disintegration of the cytoskeleton 18h after being seeded on a stiff environment (plastic dish, 10MPa, Supplementary Figures 5 and 6). Taken together, our findings indicate that recruitment of the glycolytic enzyme PFKP to FA and increased glycolysis in response to stiffness is mediated through Integrin signaling.
Fig. 2. Mechanotransduction activates glycolysis through Integrins.

A. Human LSEC plated on hard gels (32kPa) demonstrated increased co-localization of PFK with the focal adhesion marker, vinculin (yellow in high magnitude inserts) compared to soft gels (0.2kPa). Bar indicates 10μm (insert 5μm). Right panel shows quantification of co-localization of Vinculin and PFK on hard (32kPa) vs soft (0.2kPa) gels with a significant increase of co-localization with incremental stiffness. Co-localization was determined by measuring the Pearson’s coefficient calculated by JACoP plug-in in ImageJ. B. On plastic dish (10MPa), PFK and vinculin show strong and significant co-localization (correlation coefficient r=0.712, p<0.001), respective immunofluorescence can be found in Supplementary Figure 4B). C. Note overlap in PFK and Vinculin (immunofluorescence) in isolated FA of human LSEC plated on plastic dishes. Bar indicates 10μm. D. Quantification of co-localization of PFK and Vinculin in isolated FA (r=0.832, p<0.001). E. Immunoprecipitation studies demonstrated an association of PFK with the focal adhesion protein Integrin beta 1, in human LSEC (after crosslinking). F. Stiffness-induced increase in glycolysis was attenuated by Poly-D-lysine coating promoting non-integrin mediated cell attachment on plastic dishes (10MPa), n=3, ***p<0.001. G. Protein levels of PFKFB3 were decreased with the anti-integrin antibody MAb13 (western blots on the left, quantification on the right, n=3, *p<0.05). AU arbitrary units.
Mechanotransduction-induced glycolysis promotes actin polymerization, which results in increased nuclear pore size.
Given the unknown function of PFKP in FA, we first sought to investigate the impact of PFKP on FA and the actin cytoskeleton. Inhibition of glycolysis in LSEC using the small molecule 3-(3-Pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3PO) attenuated the typical formation of lamellipodia/filopodia and stress fiber formation (Figure 3A and C). This effect was not seen, when LSEC were treated with Oligomycin, a compound targeting mitochondrial respiration (Supplementary Figure 7A). Inhibition of glycolysis led to a disassembly of FA (Figure 3B) and fewer number of FA per cell (Supplementary Figure 7B). These findings demonstrate that glycolysis promotes F-actin polymerization and assembly, stabilization of FA.
Fig. 3. Mechanotransduction-induced glycolysis promotes actin polymerization, which results in increased nuclear pore size.

A. Inhibition of glycolysis by 3PO (15μM) in human LSEC on plastic (10MPa) attenuated stress fiber (Phalloidin staining, red) and FA formation (Vinculin staining, green). Bar indicates 10μm. B. Immunostaining of FA isolates reveals decreased FA formation with 3PO treatment (15μM). Bar indicates 10μm. C. Quantification of actin polymerization with a decrease in F/G actin ratio upon inhibition of glycolysis with 15 μM 3PO (western blots on the left, quantification on the right, n=3, *p<0.05). D. Inhibition of glycolysis by 15 μM 3PO in human LSEC plated on glass coverslip (represents stiffness above gigapascals) decreased nuclear pore size visualized by electron microscopy on the left (TEM; example of 50 nm nuclear pore is shown en fasse). Quantification of nuclear pore size for cells treated with vehicle, 3PO and cytochalasin shows significant reduction in nuclear pore size with 3PO and cytochalasin (on the right). For each experiment, >30 pores per condition measured, experiments were run in biological triplicates (*p<0.05).
Mechanosensitive transcriptional regulators can translocate to the nucleus in response to stiffness through changes in nuclear pore size.(15) We sought to investigate whether glycolysis and glycolysis-mediated actin polymerization can affect nuclear pores in LSEC. Disruption of actin polymerization and inhibition of glycolysis both resulted in a significant decrease of nuclear pore size assessed by TEM (Figure 3D). For lower magnification images, see Supplementary Figure 8. These findings indicate that mechanotransduction-induced, glycolysis-mediated actin polymerization increases nuclear pore size.
Glycolysis and gene transcription in liver sinusoidal endothelial cells
RNA sequencing reveals CXCL1-dominant neutrophil migration among the top stiffness-induced pathways in LSEC.
Association between changes in FA composition and the mRNA profile in response to stiffness in LSEC has yet to be explored. To gain a comprehensive, non-biased picture of this stiffness-induced signaling program, we performed RNA sequencing (RNAseq) analysis from primary human LSEC (Figure 4A) and murine LSEC (Supplementary Figure 9A) seeded on soft (0.2kPa) and hard (32kPa) collagen-I-coated gels (for 6 and 12 hours, respectively). A list of the up-/downregulated genes can be found in Supplementary Tables 3 and 4. Ingenuity pathway analysis (IPA) identified granulocyte and agranulocyte migration/diapedesis as two of the top pathways with the chemokine CXCL1 being among the top up-regulated genes (Figure 4B). In addition, these pathways were the top two hits in IPA of RNAseq data from murine LSEC (Supplementary Figure 9B). We confirmed the RNAseq results by qPCR, which demonstrated a 2-fold increase of CXCL1 expression in response to stiffness (Figure 4C). To test the specificity of CXCL1 involvement in LSEC, we compared RPKM values (reads per kilobase million) of different CXCL chemokines in LSEC with those in HSC and hepatocytes; CXCL1 expression in LSEC was more than 8-times and 24-times higher than that in HSC and hepatocytes, respectively (Figure 4D). These findings show that CXCL chemokines with CXCL1 as a prototype are among the top up-regulated genes in response to stiffness and are therefore part of the mechanotransduction-induced angiocrine signaling program in LSEC.
Fig. 4. Neutrophil migration pathway with CXCL1 is one of the top pathways affected by stiffness.

A. Heatmap of RNAseq data with up- and downregulated genes (logFC≥0.8, FDR<0.05) in primary human LSEC plated on hard (32kPa) vs. soft (0.2kPa) gels. Green color indicates upregulation, red color indicates downregulation. Triplicates are shown for both conditions. B. Ingenuity pathway analysis of RNAseq results reveals neutrophil migration (granulocyte diapedesis) as one of the top affected pathways on hard vs. soft gels, ranked by −log p-value. Insert shows contributing genes to this pathway with CXCL1 being among the top upregulated genes on hard vs. soft gel. C. qPCR from human LSEC plated on hard vs. soft gels confirms RNAseq data with significant upregulation of CXCL1 mRNA expression on hard gel (n=3, **p<0.01). D. Comparison of CXCL1, 2, 6 and 8 expression among different human liver cell types based on RPKM levels shows highest expression of CXCL 1, 6 and 8 in LSEC (n=3, **p<0.01, ***p<0.001).
Mechanotransduction—induced angiocrine signaling is promoted through glycolysis-mediated actin polymerization.
Given a direct effect of stiffness-induced glycolysis on actin assembly, we sought to investigate whether CXCL1 expression is mediated through glycolysis and increased actin polymerization. Inhibition of the two glycolytic enzymes PFKFB3 and Hexokinase 2 (HK2) – either by siRNA (PFKFB3 siRNA, HK2 siRNA) or competitive inhibitors (3PO blocking PFKFB3 and 2-DG inhibiting HK2) – resulted in a significant reduction of CXCL1 mRNA levels (Figure 5A and B). Such effect was not seen with inhibition of mitochondrial respiration (Figure 5A). Inhibition of glycolysis further attenuated mechanotransduction-induced increase in CXCL1 expression (Figure 5C). Disruption of actin polymerization by cytochalasin D resulted in a similar CXCL1 reduction (Figure 5D). These findings indicate that CXCL1 expression is mediated through glycolysis and actin polymerization.
Fig. 5. Inhibition of glycolysis attenuates mechanotransduction-induced CXCL1 production.
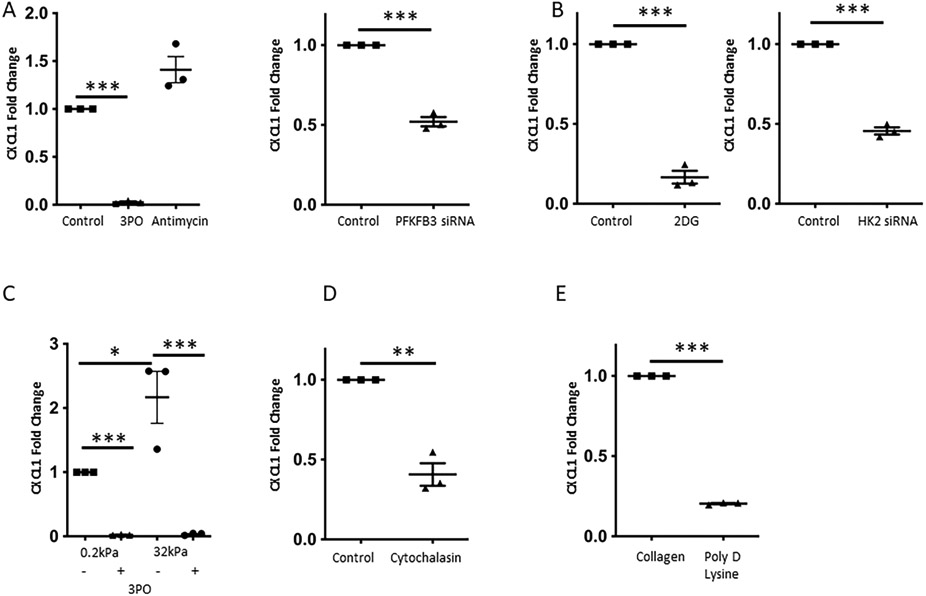
A. Increase in CXCL1 mRNA levels is attenuated upon inhibition of glycolysis with 3PO (15 μM), but not after blocking mitochondrial respiration (Antimycin), on plastic dishes (10MPa), on the left (n=3, ***p<0.001). Knockdown of the glycolytic activator PFKFB3 significantly decreased CXCL1 mRNA expression, on the right (n=3, ***p<0.001). B. Blocking glycolysis by 2-DG, an inhibitor of hexokinase 2 (on the left), and knockdown of hexokinase 2 (on the right) resulted in a significant decrease of CXCL1 mRNA levels (n=3, ***p<0.001). C. 3PO (15 μM) decreased CXCL1 mRNA levels on both soft and hard gels, and significantly attenuated the hard-gel induced CXCL1 increase (n=3, *p<0.05, ***p<0.001). D. Disruption of the cytoskeleton by using Cytochalasin resulted in a decrease of CXCL1 mRNA levels (n=3, **p<0.01). E. Poly-D-lysine coating promoting non-Integrin mediated cell attachment significantly decreased CXCL1 mRNA expression compared to collagen I coating, which promotes Integrin mediated cell attachment (n=3, ***p<0.001). All experiments performed with human LSEC.
To gain further insights how CXCL1 is affected by stiffness-induced FA changes and given our previous findings of association between glycolytic enzymes and Integrin beta 1, we analyzed CXCL1 mRNA levels upon interference with Integrin signaling. Integrin signaling pathway was inhibited using Poly-D-Lysine coating and the monoclonal antibody MAb13. Both interventions resulted in a significant decrease of CXCL1 expression (Figure 5E, Supplementary Figure 9C). On gels with incremental stiffness, Poly-D-Lysine coating attenuated mechanotransduction-induced CXCL1 expression by 52% (Supplementary Figure 9D). These findings led us to the conclusion that mechanotransduction-induced CXCL1-expression is mediated through Integrin signaling-promoted glycolysis.
Glycolysis mediates CXCL1 expression through NFkB and epigenetic modifications at H3K27.
The mechanisms of mechanocrine CXCL1 expression are largely unknown. We therefore explored upstream regulators of the stiffness-induced genes identified in our RNAseq experiment (human LSEC). Upstream analysis using IPA unexpectedly revealed NFkB and TNF as the most activated regulators with a z-score of 3.389 and 3.198, respectively (Figure 6A). Similar upregulation of TNF-NFkB was seen when the murine RNAseq dataset was analyzed (Supplementary Figure 10A). TNF activation of the transcriptional factor NFkB plays a key role in inflammation through its ability to induce transcription of pro-inflammatory genes.(16, 17) Our non-biased RNAseq approach has led us to the hypothesis that stiffness and TNF can induce a similar angiocrine and pro-inflammatory signaling program in LSEC. Indeed, NFkB activation has been proposed previously in FA-mediated response to mechanical stimuli.(18) Therefore we sought to further investigate whether NFkB activation is involved in the process of stiffness-induced glycolysis-mediated CXCL1 expression in LSEC. For this, we used both gels with incremental stiffness and treatment with TNF as stimulating factors. Nuclear translocation of NFkB was significantly increased on hard compared to soft environment (Figure 6B, Supplementary Figure 10C). 3PO treatment largely attenuated TNF-induced NFkB translocation to the nucleus in LSEC seeded on plastic dish (10mPA, Figure 6C). Congruently, TNF-induced CXCL1 expression was significantly decreased, when LSEC were pre-treated with the glycolysis inhibitor 3PO (Supplementary Figure 10B). These findings show that stiffness-induced glycolysis might mediate CXCL1 expression through NFkB.
Fig. 6. Glycolysis promotes CXCL1 expression through NFkB and epigenetic modifications at H3K27 mediated by super-enhancer formation.

A. Upstream regulator analysis of RNAseq data from human LSEC plated on hard (32kPa) vs. soft (0.2kPa) collagen-I coated gels revealed TNF-NFkB as the top pathway involved. B. Nuclear NFkB protein levels are significantly increased on hard compared to soft gels without an increase in cytosolic NFkB (representative western blot on the left, quantification on the right, n=3, *p<0.05. C. TNF-induced nuclear NFkB translocation is significantly attenuated by co-treatment with 3PO (representative immunofluorescence on the left, quantification on the right, n=10, ***p<0.001). D. H3K27 acetylation of the CXCL1 promoter is significantly decreased upon treatment of human LSEC with 3PO and Cytochalasin (n=3, ***p<0.001). E. Circularized chromosome conformation capture (4C) of the CXCL1 promoter (Chr4q13.3) identifies a superenhancer at a genomic site 130-170kb upstream. The interaction between CXCL1 promoter and this genomic site is largely attenuated by inhibition of glycolysis with 3PO (15 μM), n=1. AU arbitrary unit. All experiments performed with human LSEC.
To gain further insights into how CXCL1 is transcriptionally regulated, we performed targeted chromatin-immunoprecipitation analysis for the gene activation mark histone 3 K27 acetylation (H3K27Ac). H3K27ac has been previously implicated in mechanotransduction as activation mark for various fibrosis-related genes.(19),(20) We found that both 3PO and cytoskeletal disruption significantly decreased H3K27Ac at the CXCL1 promoter region (Figure 6D). We hypothesize that glycolysis-mediated cytoskeleton assembly enables histone modifications at the CXCL1 promoter region resulting in CXCL1 gene expression. We next explored the mechanisms of such glycolysis-mediated histone modifications.
4C identifies a glycolysis-mediated CXCL super-enhancer at a genomic site located at 130-170kb upstream of CXCL1 promoter.
Advances in genome architecture have revealed that remote enhancer elements are critical regulators for geographically disparate promoters in part by binding transcription factors and histone modifying enzymes and approximating them to target promoters by generating chromatin loops.(21) Groups of putative enhancers that are in close proximity with unusually high levels of transcriptional coactivator binding are considered super-enhancers (enhancer clustering).(22) Super-enhancers regulate genes with especially important roles in cell identity.(23, 24) Our previous data showed H3K27Ac marks being deposited for the majority of the CXCL family genes including CXCL1, 2, 6 and 8. Therefore, we sought to explore possible super-enhancers that regulate the group of chemokines on Chr4q13.3 gene locus. Circularized chromosome conformation capture (4C) allows investigation of spatial organization of nuclear chromatin in proximity of a specific gene locus (here Chr4q13.3) through next generation sequencing of chromatin enhancer regions that are identified to bind to this gene locus. By 4C, we detected significant chromosomal interaction between CXCL1 promoter and a genomic site located 130-170kb upstream, identified as a possible super-enhancer (Figure 6E). This interaction was considerably attenuated by 3PO. Of note – congruently with our data showing involvement of NFkB as key transcription factor – ChIPseq of LSEC pre-treated with TNF revealed induction of H3K27 acetylation and increased binding of p65, one NFkB component, at this superenhancer region and at the CXCL1 promoter (Supplementary Figure 11). Based on these findings, we hypothesize that glycolysis mediates CXCL1 expression through spatial organization of nuclear chromatin, which enables interaction with a super-enhancer resulting in histone acetylation and NFkB binding at the CXCL1 promoter region.
Glycolysis and CXCL1-mediated neutrophil infiltration, portal hypertension and liver fibrosis in vivo
Endothelial cell-specific inhibition of glycolysis attenuates neutrophil infiltration and development of portal hypertension in vivo.
CXCL1 has been indicated in neutrophil attraction, thrombus formation and pathophysiology of portal hypertension. Whether inhibition of stiffness-induced glycolysis-mediated CXCL1 expression results in protection from neutrophil infiltration and portal hypertension has yet to be determined. For this purpose, we performed partial ligation of the inferior vena cava (pIVCL) for 2 days to induce portal hypertension and neutrophil recruitment as previously described(25) in mice with Tamoxifen-inducible endothelial cell specific HK2 knockdown (VCad-Cre-ERT2 mice crossed with HK2 floxed mice). pIVCL is a murine model, where the diameter of the inferior vena cava is reduced by 70% resulting in increased liver stiffness and development of portal hypertension.(25) Involvement of Hexokinase 2 in stiffness-induced CXCL1 expression has been supported by our in vitro data. In addition, its key and unique role (in contrast to PFKP) for vascular development has been previously demonstrated.(14) We found that pIVCL-induced portal hypertension was significantly attenuated in mice with endothelial cell specific HK2 knockdown (Figure 7A). In parallel, and congruently to our in vitro data, increase in CXCL1 mRNA expression (Figure 7B) and CXCL1 serum levels (Supplementary Figure 12A) was significantly attenuated. Finally, pIVCL-induced neutrophil migration was blocked in mice with HK2 knockdown assessed by using MPO mRNA expression (Figure 7C) and MPO immunohistochemistry (Figure 7D). Similar findings were seen in a second animal model for early liver fibrosis (CCl4) when targeting glycolysis with a compound (3PO). Co-treatment with the glycolysis inhibitor 3PO resulted in a significant attenuation of liver fibrosis (particularly in the perisinusoidal area) and neutrophil infiltration in mice undergoing 3-week CCl4 injections (Figure 8A, Supplementary Figure 12B and C). Based on these findings, we conclude that glycolysis is required for CXCL1-mediated neutrophil infiltration, portal hypertension and early fibrosis in vivo. Thus, glycolytic enzymes could represent promising (and druggable) targets in early liver disease in order to prevent inflammation and portal hypertension.
Fig. 7. Endothelial cell-specific inhibition of glycolysis attenuates neutrophil infiltration and development of portal hypertension in vivo.

A. Portal pressure measurement shows attenuated portal hypertension in HK2 fl/fl Cdh5 Cre-ERT2 mice (=endothelial cell specific HK2 knockdown) after pIVCL compared with HK2 fl/fl mice (n=3-9, *p<0.05). B. CXCL1 mRNA expression is significantly increased after pIVCL in HK2 fl/fl, while this increase is attenuated in HK2 fl/fl Cdh5 Cre-ERT2 mice (n=5-11, *p<0.05, **p<0.01). C. MPO mRNA expression is significantly increased after pIVCL in HK2 fl/fl, while this increase is attenuated in HK2 fl/fl Cdh5 Cre-ERT2 mice (n=5-11, *p<0.05). D. Immunohistochemistry for MPO shows attenuated neutrophil infiltration in HK2 fl/fl Cdh5 Cre-ERT2 mice after pIVCL compared with HK2 fl/fl mice (n=7-14, ***p<0.001). Left panel shows representative pictures (arrowheads indicate neutrophils), right panel shows quantification.
Fig 8. Inhibition of glycolysis by 3PO attenuates liver fibrosis in a 3-week CCl4 model and human liver samples from patients with cirrhosis indicate involvement of CXCl1.

A. Sirius red staining shows significant attenuation of liver fibrosis in 3PO treated mice after 3-week CCl4 injection compared to non 3PO treated mice (n=3-9, **p<0.01). Right panel shows quantification. B. RNA in situ hybridization of human liver samples from patients with cirrhosis shows significantly increased expression of CXCL1 mRNA in cirrhotic vs non-cirrhotic liver, particularly in LYVE1 positive cells. Right panel shows quantification.
CXCL1 is significantly upregulated in patients with end-stage liver disease compared to controls.
CXCL1 has been previously shown to play an important role in alcoholic hepatitis.(26) Indeed, LSEC are an important source of CXCL1 expression in human liver, regulated by the TNFa/NFkB signaling.(26) To further investigate its role in end-stage liver disease, we compared tissue from healthy livers (liver donors) with end-stage liver disease (liver explants) with regards to their CXCL1 expression (Supplementary Table 5). To specify the source of CXCL1, we performed co-staining with the LSEC marker LYVE1. CXCL1 expression was generally increased in cirrhotic compared to healthy livers, and co-localized within LYVE1-positive LSEC (Figure 8B), to a similar extent of what was seen in the 3-week CCl4 murine model (Supplementary Figure 12D). Thus, CXCL1 appears to play an important role in the pathogenesis of end-stage liver disease in vivo.
DISCUSSION
A link between mechanosensing, angiocrine signaling, and a potential role of glycolysis in this process in the context of liver fibrosis have not been previously explored. Our experimental design involved the use of silicon-based gels with incremental stiffness mimicking healthy and fibrotic liver, high throughput experiments such as mass spectrometry of isolated FA proteins and RNAseq analyses, transmission electron microscopy to study nuclear pores, novel techniques to study epigenetic regulatory mechanisms such as 4C, the use of genetically modified mice, pharmacological inhibition of glycolysis in vivo, and finally human liver samples. Figure 9 summarizes our key findings, which are discussed in the following section in more detail.
Fig. 9. Summary cartoon.

The glycolytic enzyme PFKP is recruited to FA in response to stiffness. This results in the formation of stress fibers (actin polymerization). Actin tethering increases nuclear pore size enabling translocation of the transcription factor NFkB. Through 3D conformational changes, enhancers and promoter of stiffness-inducible genes (here CXCL1) get in close proximity resulting in histone modification and gene transcription. CXCL1 induces neutrophil migration and finally portal hypertension, early liver fibrosis.
First, we show that the glycolytic key enzyme PFKP is recruited to FA, which results in increased glycolysis in response to stiffness through Integrin signaling. Previous publications have demonstrated the importance of glycolysis in EC metabolism.(13, 14) Integrins and FA have long been known to be key players in mechanosensing, cell attachment and motility.(8) Despite the importance of glycolysis and FA in EC migration, this study is the first of its kind directly linking glycolysis and glycolytic enzymes to FA and Integrin signaling using mass spectrometry of isolated FA proteins. Mechanotransduction-induced glycolysis promotes actin polymerization and stabilizes FA. Of note, involvement of glycolysis is rather specific since inhibition of oxidative phosphorylation as an alternative source of ATP did not have any impact on FA downstream signaling and actin polymerization. Such preference of glycolysis in EC in contrast to mitochondrial respiration has been previously proposed by De Bock and colleagues.(13) Rapid, dynamic changes in FA and the fact that mechanosensing occurs in rather small subcellular compartments (filopodia/lamellipodia) favor glycolysis over mitochondrial respiration as primary energy source. Our data further indicates recruitment of glycolytic enzymes through binding to Integrin β1. Still, further work is needed to clarify the exact mechanisms of glycolytic enzyme recruitment to FA.
Next, we demonstrate that stiffness-induced glycolysis promotes actin polymerization and increases nuclear pore size. Several previous findings support this concept. It has been known for over three decades that PFK can bind to actin filaments.(27) Glycolytic enzymes positively affect stress fiber formation and secure the integrity of the cellular cytoskeleton.(28) The cytoskeleton in turn can mediate traction forces from the cellular environment to the nucleus. Elosegui and colleagues have recently revealed that force leads to nuclear flattening and increased nuclear import of mechanosensitive transcription factors due to decreased mechanical restriction to molecular transport in nuclear pores.(15) Changes in nuclear pore size and nuclear translocation of transcription factors in response to stiffness are dependent on cytoskeleton integrity and stress fiber formation.(15, 29-31) Similar findings have been reported by Guixé-Muntet and colleagues in chronic liver disease.(32) Our data reveal that these mechanisms of mechanosensing rely on stiffness-induced glycolytic activity.
Non-biased RNAseq revealed that stiffness induces an angiocrine program in LSEC with CXCL1 being one of the most upregulated genes. CXCL1 has been previously shown to be involved in vascular recruitment of neutrophils and intrasinusoidal aggregation of platelets in liver disease.(5) The obvious question is how metabolism-driven changes in actin cytoskeleton and increase in nuclear pore size lead to this angiocrine gene regulation. Our findings give some important insight. We demonstrate that glycolysis might mediate spatial organization of the nuclear chromatin enabling interaction with superenhancers that results in H3K27 acetylation and NFkB binding at the CXCL1 promoter region. Although exact mechanisms have yet to be explored, stiffness-induced glycolysis appears to result in generation of chromatin loops that approximate histone-modifying enzymes to the CXCL gene promoter. Such mechanism has been recently explored in alcoholic hepatitis revealing NFkB-mediated CXCL1 as one of its key pathways.(26) Targeting angiocrine signaling by interfering with these two processes – LSEC metabolism and/or spatial organization of nuclear chromatin, super-enhancers – might potentially affect not only CXCL1, but rather the whole pathogenic angiocrine program. Such treatment might be more effective than targeting a single chemokine, as recently shown by our group using iBET151, a pan-BET inhibitor that interferes with super-enhancer function. iBET151 was able to attenuate CXCL expression, neutrophilic infiltration and finally liver inflammation in a murine model of human alcoholic hepatitis.(26)
Finally, we demonstrate significance of glycolysis in two short-term in vivo models for portal hypertension and early liver fibrosis. This has direct clinical implications: Portal hypertension represents a major problem in patients with liver disease and considerably accounts for morbidity and mortality. While non-selective betablockers are used in patients with already established portal hypertension (for primary or secondary prevention of variceal bleeding), there is currently no treatment available that directly targets development of portal hypertension. However – given median survival time of only 2 years in case of development of portal hypertension with decompensation of cirrhosis – novel treatment options are urgently needed. Glycolytic enzymes are “druggable” targets and such approach has already been tested in oncology. 3PO has been previously used for other diseases such as lung fibrosis further proposing its use in end-stage liver disease.(33) Here, we expand our knowledge on the role of glycolysis and show that – particularly if selectively blocked in endothelial cells – glycolysis, such as the enzyme hexokinase 2, could be promising targets in order to prevent development of portal hypertension.
Our study still has several limitations: First) Some of our experiments were carried out on plastic dish as a model for extreme stiffness (10MPa) due to limited cell attachment on gels. Particularly, cells on soft gel showed decreased cell attachment. Complete disattachment (for example by using co-treatment with integrin antibodies) would have confounded our results. Nevertheless, plastic dish and even coverslips have previously served as a model for extreme stiffness in key studies on mechanosensing.(34) Second) Mice used in our study displayed rather low portal pressure gradients.(35, 36) With 48h, the pIVCL experiment was considerably shorter than the originally described experiment by our group in 2015, which most probably contributed to the lower portal pressure levels.(25) However, the difference between intervention and sham group was still statistically significant, clearly indicating an increase in portal pressure after the intervention, which is attenuated in HK2 knockout mice. In addition, sham results in our study were comparable to a similar model with a sacrifice after 24h.(5) Third) Epigenetic 4C experiments are hypothesis-generating rather than hypothesis-testing studies. Our data only suggest the involvement of super-enhancer formation resulting in histone acetylation and NFkB binding at the CXCL1 promoter region. However, our group has recently shown in detail how TNF and NFkB are involved in super-enhancer modulation and CXCL chemokine expression in alcoholic hepatitis further supporting this hypothesis.(26) Fourth) Capillarization and dedifferentiation of human LSEC might occur over time. Nevertheless, we used them at early passages in all our experiments. In addition, characterization by immunofluorescence and western blotting revealed enrichment of typical LSEC markers such as LYVE1, but absence of markers for capillarization (CD34), in clear contrast to endothelial cells. Key experiments were replicated with freshly isolated mouse LSEC showing consistent results. Furthermore, such consistency between human LSEC and freshly isolated mouse LSEC has been shown in previous publications.(26, 37-39) Finally) We used short rather than long-term models for liver fibrosis and portal hypertension. This approach was chosen for the following reasons: CXCL1 induces neutrophil inflammation being known as an early-stage inflammatory response. Our aim was to study initiating processes in mechanotransduction and our in vitro experiments have shown that glycolysis and focal adhesion dynamics are important within only few hours after seeding cells on gels with incremental stiffness. Short-term pIVCL models (such as 24h) have been previously used to study the role of portal hypertension.(5) Using two short-term models (pIVCL and the 3-week CCl4 model) we were able to show consistent involvement of glycolysis. However, our findings cannot be extrapolated 1:1 to end-stage liver diseases as our study focuses on early injury and not late-stage disease models.
In conclusion, we reveal glycolytic enzymes and glycolysis as novel key players in FA in response to stiffness. We show that increased glycolytic activity results in increased actin polymerization, which increases nuclear pores size and enables NFkB-mediated CXCL1 expression. The latter was identified by high throughput experiments as a key downstream target of stiffness. Glycolytic enzymes might be a novel druggable target in liver disease – at least in its early stages – an area with an ongoing unmet need for effective therapies. Further studies are needed to evaluate their potential in chronic end-stage liver diseases.
Supplementary Material
Highlights.
Increased stiffness results in recruitment of glycolytic enzymes to focal adhesions in liver sinusoidal endothelial cells (LSEC), paralleling an increase in glycolysis.
Inhibition of glycolysis blocks a stiffness-induced CXCL1-dominant angiocrine program.
Glycolysis promotes CXCL1 expression through nuclear pore changes and increases in NFkB translocation.
LSEC-selective deletion of glycolytic enzymes results in attenuation of portal hypertension in vivo.
Treatment with the glycolysis inhibitor 3PO attenuates early liver fibrosis in vivo.
Grant support:
This study was supported by the National Institutes of Health grants DK59615 and AA021171 (V.H.S.), the Swiss National Science Foundation grant P2ZHP3_168561 (T.G.) and a Novartis Research Foundation grant (T.G.). This research was further supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award Number P30DK084567 (to the Mayo Clinic Center of Signaling in Gastroenterology). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest: The authors disclose no conflicts.
Data availability statement:
Data supporting the findings of this study are available within the article and its supplementary material files. RNAseq data generated in this study have been deposited in NCBI’s gene expression omnibus.
REFERENCES
- 1.Greuter T, Shah VH. Hepatic sinusoids in liver injury, inflammation, and fibrosis: new pathophysiological insights. J Gastroenterol. 2016;51(6):511–9. [DOI] [PubMed] [Google Scholar]
- 2.Dauphinee SM, Karsan A. Lipopolysaccharide signaling in endothelial cells. Lab Invest. 2006;86(1):9–22. [DOI] [PubMed] [Google Scholar]
- 3.Schrage A, Wechsung K, Neumann K, Schumann M, Schulzke JD, Engelhardt B, et al. Enhanced T cell transmigration across the murine liver sinusoidal endothelium is mediated by transcytosis and surface presentation of chemokines. Hepatology. 2008;48(4):1262–72. [DOI] [PubMed] [Google Scholar]
- 4.Kostallari E, Shah VH. Angiocrine signaling in the hepatic sinusoids in health and disease. Am J Physiol Gastrointest Liver Physiol. 2016;311(2):G246–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hilscher MB, Sehrawat T, Arab JP, Zeng Z, Gao J, Liu M, et al. Mechanical Stretch Increases Expression of CXCL1 in Liver Sinusoidal Endothelial Cells to Recruit Neutrophils, Generate Sinusoidal Microthombi, and Promote Portal Hypertension. Gastroenterology. 2019;157(1):193–209.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wells RG. Tissue mechanics and fibrosis. Biochim Biophys Acta. 2013;1832(7):884–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duscher D, Maan ZN, Wong VW, Rennert RC, Januszyk M, Rodrigues M, et al. Mechanotransduction and fibrosis. J Biomech. 2014;47(9):1997–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Humphrey JD, Dufresne ER, Schwartz MA. Mechanotransduction and extracellular matrix homeostasis. Nat Rev Mol Cell Biol. 2014;15(12):802–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ross TD, Coon BG, Yun S, Baeyens N, Tanaka K, Ouyang M, et al. Integrins in mechanotransduction. Curr Opin Cell Biol. 2013;25(5):613–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.del Rio A, Perez-Jimenez R, Liu R, Roca-Cusachs P, Fernandez JM, Sheetz MP. Stretching single talin rod molecules activates vinculin binding. Science. 2009;323(5914):638–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang X, Cook PC, Zindy E, Williams CJ, Jowitt TA, Streuli CH, et al. Integrin α4β1 controls G9a activity that regulates epigenetic changes and nuclear properties required for lymphocyte migration. Nucleic Acids Res. 2016;44(7):3031–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Atherton P, Stutchbury B, Jethwa D, Ballestrem C. Mechanosensitive components of integrin adhesions: Role of vinculin. Exp Cell Res. 2016;343(1):21–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013;154(3):651–63. [DOI] [PubMed] [Google Scholar]
- 14.Yu P, Wilhelm K, Dubrac A, Tung JK, Alves TC, Fang JS, et al. FGF-dependent metabolic control of vascular development. Nature. 2017;545(7653):224–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elosegui-Artola A, Andreu I, Beedle AEM, Lezamiz A, Uroz M, Kosmalska AJ, et al. Force Triggers YAP Nuclear Entry by Regulating Transport across Nuclear Pores. Cell. 2017;171(6):1397–410.e14. [DOI] [PubMed] [Google Scholar]
- 16.Baldwin AS. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–83. [DOI] [PubMed] [Google Scholar]
- 17.Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107(1):7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Young SR, Gerard-O'Riley R, Harrington M, Pavalko FM. Activation of NF-kappaB by fluid shear stress, but not TNF-alpha, requires focal adhesion kinase in osteoblasts. Bone. 2010;47(1):74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stein C, Bardet AF, Roma G, Bergling S, Clay I, Ruchti A, et al. YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers. PLoS Genet. 2015;11(8):e1005465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dou C, Liu Z, Tu K, Zhang H, Chen C, Yaqoob U, et al. P300 Acetyltransferase Mediates Stiffness-Induced Activation of Hepatic Stellate Cells Into Tumor-Promoting Myofibroblasts. Gastroenterology. 2018;154(8):2209–21.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao Z, Tavoosidana G, Sjölinder M, Göndör A, Mariano P, Wang S, et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat Genet. 2006;38(11):1341–7. [DOI] [PubMed] [Google Scholar]
- 22.Pott S, Lieb JD. What are super-enhancers? Nat Genet. 2015;47(1):8–12. [DOI] [PubMed] [Google Scholar]
- 23.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153(2):307–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155(4):934–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simonetto DA, Yang HY, Yin M, de Assuncao TM, Kwon JH, Hilscher M, et al. Chronic passive venous congestion drives hepatic fibrogenesis via sinusoidal thrombosis and mechanical forces. Hepatology. 2015;61(2):648–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu M, Cao S, He L, Gao J, Arab JP, Cui H, et al. Super enhancer regulation of cytokine-induced chemokine production in alcoholic hepatitis. Nat Commun. 2021;12(1):4560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roberts SJ, Somero GN. Properties of the interaction between phosphofructokinase and actin. Arch Biochem Biophys. 1989;269(1):284–94. [DOI] [PubMed] [Google Scholar]
- 28.Gizak A, Wiśniewski J, Heron P, Mamczur P, Sygusch J, Rakus D. Targeting a moonlighting function of aldolase induces apoptosis in cancer cells. Cell Death Dis. 2019;10(10):712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Das A, Fischer RS, Pan D, Waterman CM. YAP Nuclear Localization in the Absence of Cell-Cell Contact Is Mediated by a Filamentous Actin-dependent, Myosin II- and Phospho-YAP-independent Pathway during Extracellular Matrix Mechanosensing. J Biol Chem. 2016;291(12):6096–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, et al. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell. 2013;154(5):1047–59. [DOI] [PubMed] [Google Scholar]
- 31.Elosegui-Artola A, Oria R, Chen Y, Kosmalska A, Pérez-González C, Castro N, et al. Mechanical regulation of a molecular clutch defines force transmission and transduction in response to matrix rigidity. Nat Cell Biol. 2016;18(5):540–8. [DOI] [PubMed] [Google Scholar]
- 32.Guixé-Muntet S, Ortega-Ribera M, Wang C, Selicean S, Andreu I, Kechagia JZ, et al. Nuclear deformation mediates liver cell mechanosensing in cirrhosis. JHEP Rep. 2020;2(5):100145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xie N, Tan Z, Banerjee S, Cui H, Ge J, Liu RM, et al. Glycolytic Reprogramming in Myofibroblast Differentiation and Lung Fibrosis. Am J Respir Crit Care Med. 2015;192(12):1462–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu L, You Z, Yu H, Zhou L, Zhao H, Yan X, et al. Mechanotransduction-modulated fibrotic microniches reveal the contribution of angiogenesis in liver fibrosis. Nat Mater. 2017;16(12):1252–61. [DOI] [PubMed] [Google Scholar]
- 35.Garcia-Pras E, Gallego J, Coch L, Mejias M, Fernandez-Miranda G, Pardal R, et al. Role and therapeutic potential of vascular stem/progenitor cells in pathological neovascularisation during chronic portal hypertension. Gut. 2017;66(7):1306–20. [DOI] [PubMed] [Google Scholar]
- 36.Klein S, Rick J, Lehmann J, Schierwagen R, Schierwagen IG, Verbeke L, et al. Janus-kinase-2 relates directly to portal hypertension and to complications in rodent and human cirrhosis. Gut. 2017;66(1):145–55. [DOI] [PubMed] [Google Scholar]
- 37.Furuta K, Guo Q, Pavelko KD, Lee JH, Robertson KD, Nakao Y, et al. Lipid-induced endothelial vascular cell adhesion molecule 1 promotes nonalcoholic steatohepatitis pathogenesis. J Clin Invest. 2021;131(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang L, Yang J, Liu W, Tang X, Chen J, Zhao D, et al. Liver sinusoidal endothelial cell lectin, LSECtin, negatively regulates hepatic T-cell immune response. Gastroenterology. 2009;137(4):1498–508.e1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nahmias Y, Casali M, Barbe L, Berthiaume F, Yarmush ML. Liver endothelial cells promote LDL-R expression and the uptake of HCV-like particles in primary rat and human hepatocytes. Hepatology. 2006;43(2):257–65. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting the findings of this study are available within the article and its supplementary material files. RNAseq data generated in this study have been deposited in NCBI’s gene expression omnibus.