

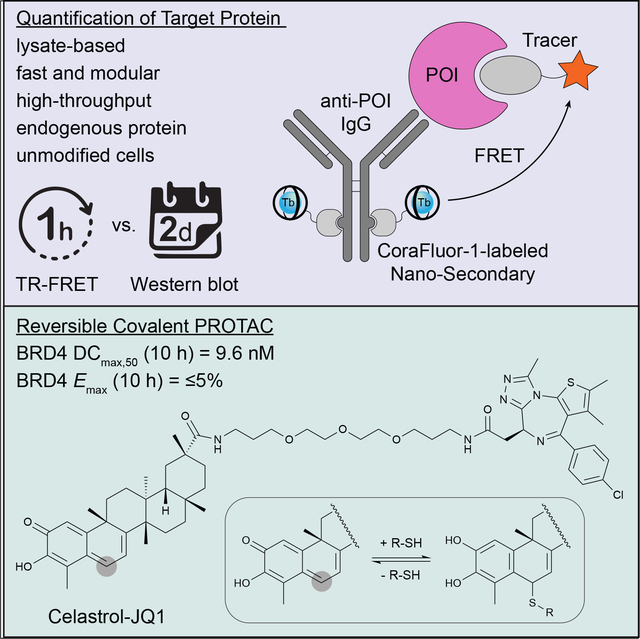
Summary
We describe a generalizable time-resolved Förster resonance energy transfer (TR-FRET)-based platform to profile the cellular action of heterobifunctional degraders (or proteolysis-targeting chimeras; PROTACs), capable of both accurately quantifying protein levels in whole-cell lysates in less than 1 h and measuring small-molecule target engagement to endogenous proteins, here specifically for human bromodomain-containing protein 4 (BRD4). The detection mix consists of a single primary antibody targeting the protein of interest, a luminescent donor-labeled anti-species nanobody, and a fluorescent acceptor ligand. Importantly, our strategy can readily be applied to other targets of interest and will greatly facilitate the cell-based profiling of small molecule inhibitors and PROTACs in high-throughput format with unmodified cell lines. We furthermore validate our platform in the characterization of celastrol, a p-quinone methide-containing pentacyclic triterpenoid, as a broad cysteine-targeting E3 ubiquitin ligase warhead for potent and efficient targeted protein degradation.
Keywords: TR-FRET, small molecules, targeted protein degradation, PROTAC, degrader, high-throughput, protein quantification, reversible, covalent, celastrol
In brief:
Payne et. al. describe a generalizable high-throughput assay strategy for the direct, rapid quantification of endogenous target proteins in lysates from unmodified cells in one hour. Using this simple mix-and-read platform, the authors characterize celastrol as a potent E3-ligase recruiting element for the development of proteolysis-targeting chimeras (PROTACs).
Graphical Abstract
Introduction
Over the past decade, small molecule-induced degradation of specific protein targets has emerged as a novel modality in biomedical research that holds promise to revolutionize drug development, providing access to new classes of therapeutics that can act on disease-relevant proteins previously considered to be undruggable (Burslem and Crews, 2020; Chamberlain and Hamann, 2019). The concept of targeted protein degradation (TPD) is based on the ligand-induced recruitment of a protein of interest (POI) to an E3 ubiquitin ligase complex for proximity-induced ubiquitination and subsequent proteasomal degradation. Most rational TPD development strategies utilize PROTACs, which are heterobifunctional molecules composed of two individual ligands, one for binding the E3 ligase complex and one for targeting the POI, tethered by a linker (Hanzl and Winter, 2020; Maniaci and Ciulli, 2019).
Despite > 600 E3 ligases in the human proteome, early PROTAC development approaches have focused on a multitude of POIs while relying on only a few E3 ligases (e.g. von Hippel-Lindau (VHL) and cereblon) that are targeted by a small number of ligand classes (Buckley et al., 2012; Winter et al., 2015). Although the specific POIs vary between studies, BRD4 is often used as a proof-of-concept in PROTAC development programs that explore new E3 ligase binders and/or linker elements (Belcher et al., 2021; Schapira et al., 2019). The reason for this is likely a combination of several factors, including historic reference data, ubiquitous expression, robustness of response, validated orthogonal inhibitor classes, and more recently the availability of commercial building blocks.
Independent of the nature of the POI and E3 ligase, efficient optimization of PROTACs depends on the availability of robust assay systems that enable the facile, reliable measurement of both biochemical ligand affinities and time- and dose-dependent cellular levels of the POI in response to compound treatment. POI quantification is most commonly done by Western blot analysis, which is inherently time-consuming and low-throughput. Although various assay technologies, including in-cell Western, ELISA, AlphaLISA, and luciferase reporter systems, have been developed to increase accuracy and throughput, many depend on the expression of the POI as a fusion protein and/or require expensive specialized equipment and consumables (Simard et al., 2021).
To address these shortcomings, we have developed a set of complementary assay strategies based on a common TR-FRET assay platform that greatly facilitates both the characterization of ligand-target engagement and the quantification of endogenous target protein levels directly in cell lysates in high-throughput format. We furthermore employ this approach to identify and characterize celastrol, a triterpene natural product that reversible covalently binds cysteine side chains, as an E3 ligase recruiting element for the development of next-generation PROTACs.
Results
Assay concept and reagent validation
In recent years, homogenous assay platforms based on TR-FRET measurements have emerged as attractive alternatives to ELISAs and have been successfully employed in PROTAC development for POI quantification and ligand characterization (Simard et al., 2021; Sy et al., 2016; Yue et al., 2020). Similar to sandwich ELISAs, TR-FRET assays generally employ a matched pair of antibodies for POI quantification, yet do not require antibody immobilization or wash steps (Figure S1).
TR-FRET assays are also frequently used to determine the affinity of small molecules for respective POIs (Figure S1). This format generally employs a fluorescent small-molecule tracer in combination with a recombinantly expressed protein tagged with a TR-FRET donor (Busby et al., 2020; Zwier et al., 2010). While the identification of a linker attachment site for tracer synthesis can be difficult, PROTAC development campaigns by default have solved this problem early on. In fact, several recent studies have shown the superior performance of TR-FRET-based assays for the characterization of PROTAC binding affinities, kinetics, and ternary complex formation (Lin and Chen, 2021; Nowak et al., 2018; Payne et al., 2021).
We hypothesized that rather than utilizing orthogonally labeled matched antibody pairs, which are often difficult to identify, and recombinant POIs, the combination of a tracer with a single antibody directed against the native protein would offer a particularly attractive approach to support PROTAC campaigns by providing a flexible assay platform capable of both ligand affinity profiling and POI quantification directly in cell lysate (Figure 1A). To eliminate the need for direct covalent labeling of the primary antibody, we here developed an approach employing single-domain nanobodies (Nano-Secondaries), which we labeled with CoraFluor-1, a TR-FRET donor complex with excellent stability and photophysical properties (Payne et al., 2021; Payne and Mazitschek, 2022).
Figure 1.
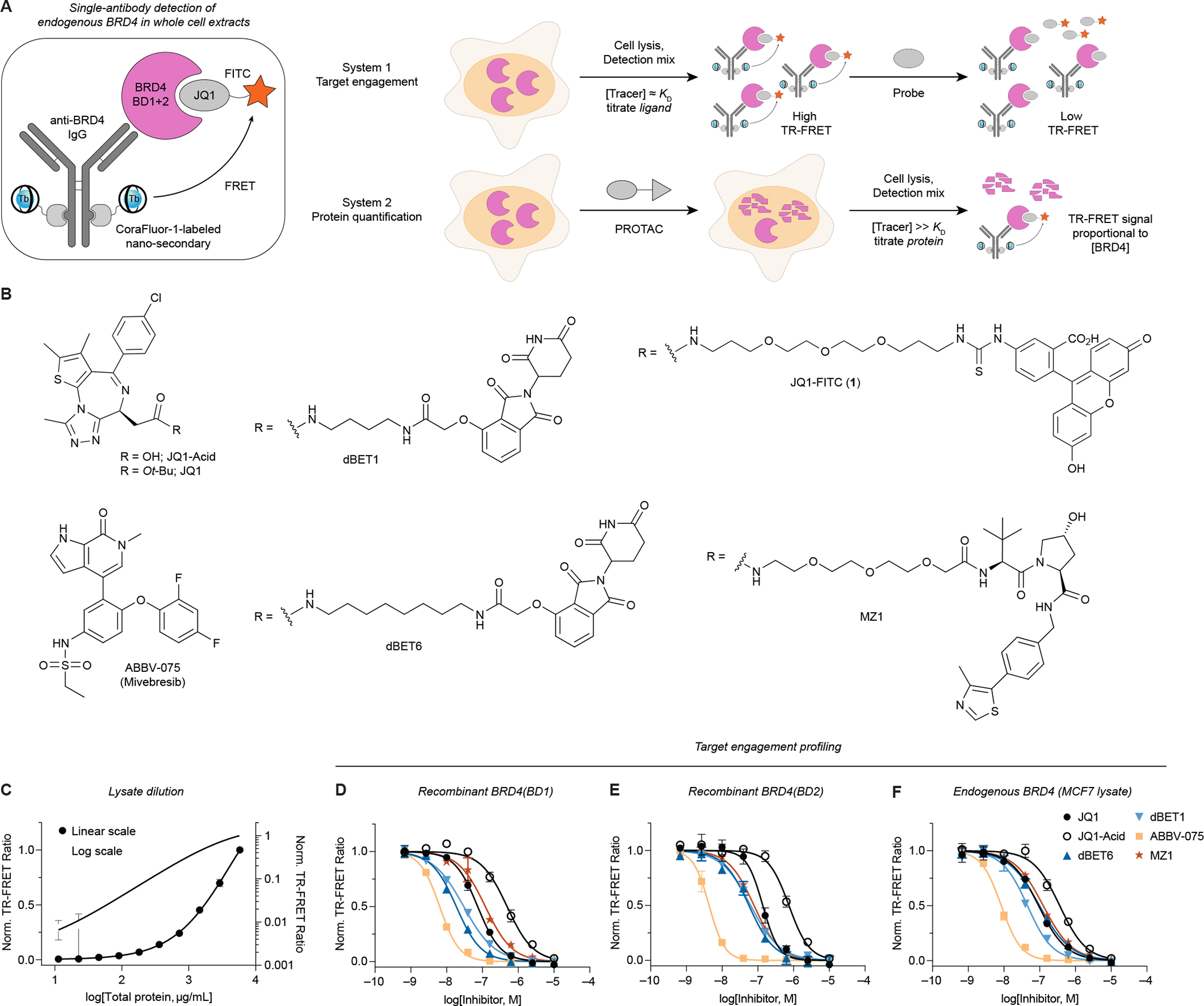
A single-antibody TR-FRET platform to quantitatively measure small molecule target engagement and endogenous protein levels in whole-cell extracts. (A) Quantification of both small molecule target engagement and protein levels with endogenous POIs, here for BRD4. The detection mix consists of a primary antibody, CoraFluor-1-labeled Nano-Secondary, and a fluorescent JQ1-based tracer. (B) Chemical structures of bromodomain inhibitors, degraders, and tracers. (C) TR-FRET-based BRD4 quantification (see STAR Methods) in serially diluted MCF7 cell lysate shows linearity over approximately three orders of magnitude. (D-F) Dose-titration of small molecule inhibitors and degraders in TR-FRET ligand displacement assays with (D-E) recombinant BRD4(BD1) and BRD4(BD2) domains, and (F) endogenous BRD4 in MCF7 cell lysate. Data in (C-F) are expressed as mean ± SD of 2 technical replicates and are representative of at least 2 independent experiments.
Because of its extensive use in PROTAC development, we have selected BRD4 for our proof-of-concept studies. Based on the potent prototype BRD4 inhibitor JQ1 (KD,BD1/2 = 50/90 nM), we synthesized JQ1-FITC (1, Figure 1B) and validated its applicability with individual recombinant BRD4 bromodomains (Figure S2) (Filippakopoulos et al., 2010; Jung et al., 2014).
Quantifying BRD4 levels in response to degrader treatment
Following validation of the target engagement assay for recombinant proteins, we next sought to apply the system to measure endogenous BRD4. For ligand displacement assays, the optimal tracer concentration is ~KD,app to balance sensitivity and signal intensity. In contrast, in protein quantification experiments, a titration regime is desired (i.e. [tracer] ≫KD,app) to maximize occupancy and reduce sensitivity to competitors (Jarmoskaite et al., 2020; Wilson and Soh, 2020). For proof-of-concept, we selected the well-established BRD4 degrader dBET6 as a positive control (Winter et al., 2017).
Titration of MCF7 lysate against fixed concentrations of primary anti-BRD4 antibody, CoraFluor-1-labeled Nano-Secondary, and JQ1-FITC demonstrated linearity over several orders of magnitude (Figure 1C). Under ligand displacement conditions ([JQ1-FITC] ≈ KD,app), we determined the binding affinities of various ligands for endogenous BRD4, which closely matched reference data (Figure 1D–F, Table S1) (Winter et al., 2017).
Next, under titration conditions ([JQ1-FITC] ≫ KD,app), we quantified endogenous BRD4 levels in MCF7 and MDA-MB-231 cells following treatment with JQ1 or dBET6 at varying concentrations for 5 h, followed by a 1 h washout to remove excess ligand before TR-FRET quantification (Donovan et al., 2020). As expected, we observed a dose-dependent decrease in TR-FRET signal in cells treated with dBET6, but not JQ1, indicating potent degradation of BRD4 (Figure 2A–B, Table S2). The total time from lysis to TR-FRET measurement was ~1.5 h. Western blot analysis on the same samples, which required approximately 2 days, provided near-identical results (Figure 2A–B).
Figure 2.
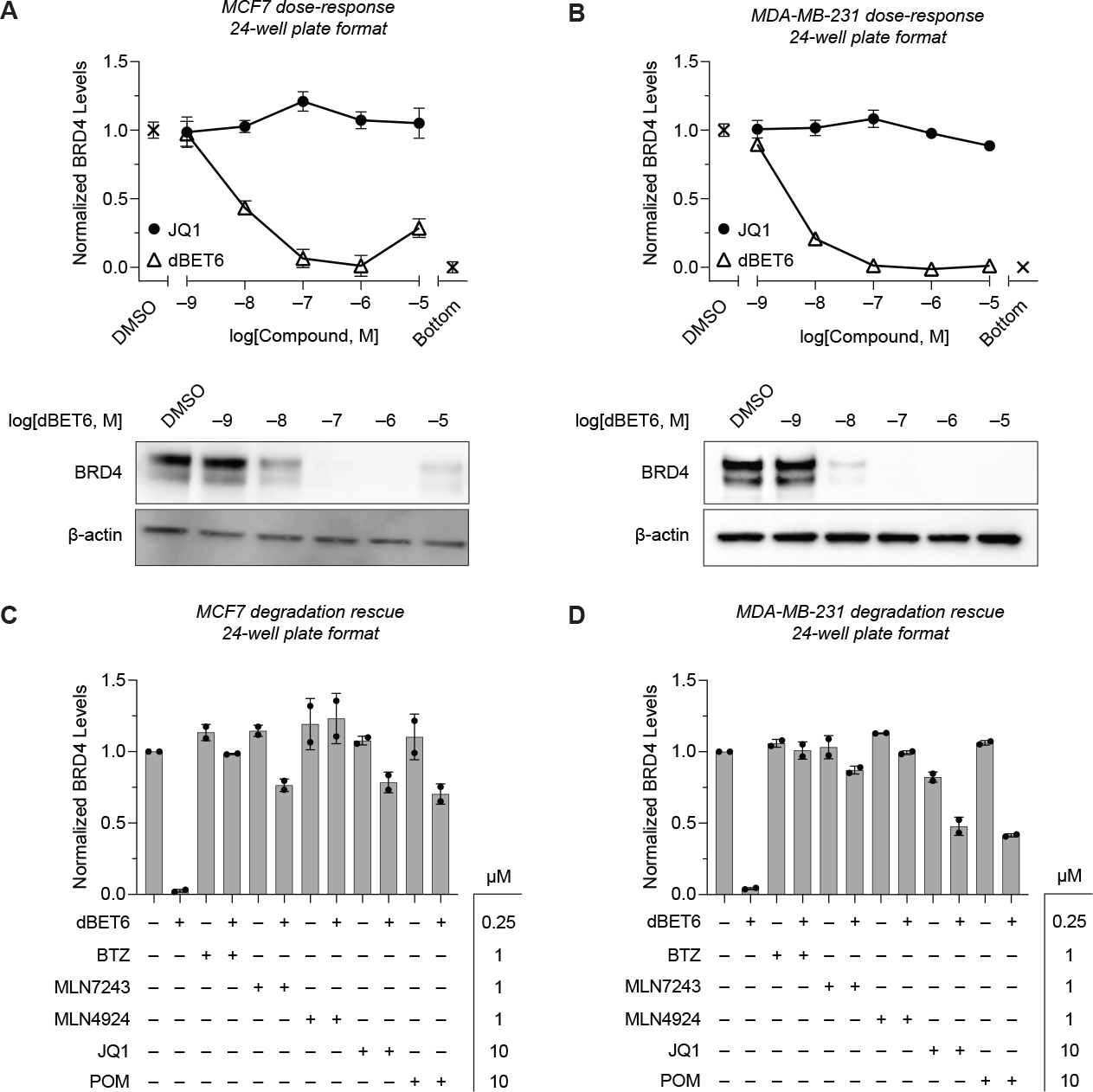
TR-FRET-based quantification of BRD4 levels in unmodified cell lines after degrader treatment. BRD4 protein levels in cell lysate after 5 h treatment with dBET6 or JQ1 were measured with TR-FRET assay as described in Figure 1A. Assays were run in a 24-well plate format with either (A) MCF7 or (B) MDA-MB-231 cells. Western blot analyses on the same samples are shown in the bottom panels. (C-D) Quantitative profiling of BRD4 degradation rescue by co-treatment of (C) MCF7 or (D) MDA-MB-231 cells with dBET6 and BTZ, MLN7243, MLN4924, JQ1, or POM after 5 h shows efficient attenuation of degradation. Data in (A-D) are expressed as mean ± SD of 2 technical × 2 biological replicates. Unprocessed Western blot data is shown in Figure S4.
We also tested the ability of our TR-FRET assay to quantify the rescue of dBET6-induced BRD4 degradation by direct competition with JQ1 and pomalidomide (POM), and by co-treatment with selective inhibitors of the ubiquitin-proteasome system (UPS), including bortezomib (BTZ, 20S proteasome inhibitor), MLN7243 (E1 ubiquitin-activating enzyme inhibitor), and MLN4924 (NEDD8 inhibitor) (Figure 2C–D). In both cell lines, BRD4 degradation was attenuated by all compounds, consistent with previous reports (Winter et al., 2017).
Assay miniaturization to 96-well plate format
PROTAC development and characterization demands the combinatorial analysis of multiple variables, including incubation time and compound concentration, which are ideally performed with multiple replicates in parallel to ensure consistency. Accordingly, the number of required data points can quickly grow exponentially. Therefore, rapid, scalable, and quantitative assays – especially in unmodified cell lines – are highly desirable (Simard et al., 2021). We thus aimed to further miniaturize our assay platform to a 96-well plate format.
Treatment of MDA-MB-231 cells (20,000 cells/well) with varying concentrations of dBET6 and JQ1 for 5 h, followed by a wash step, cell lysis, and incubation with the TR-FRET detection mix showed dose-dependent decrease of BRD4 with dBET6 treatment, while no changes were observed with JQ1 (Figure 3A). Assessment of assay robustness yielded a Z′-factor of 0.75 (Figure S3) (Zhang et al., 1999). Furthermore, to provide an optional mean for data normalization in high-throughput, we employed CellTiter-Glo 2.0 cell viability assay after TR-FRET analysis (Figure S3) (Kepp et al., 2011).
Figure 3.
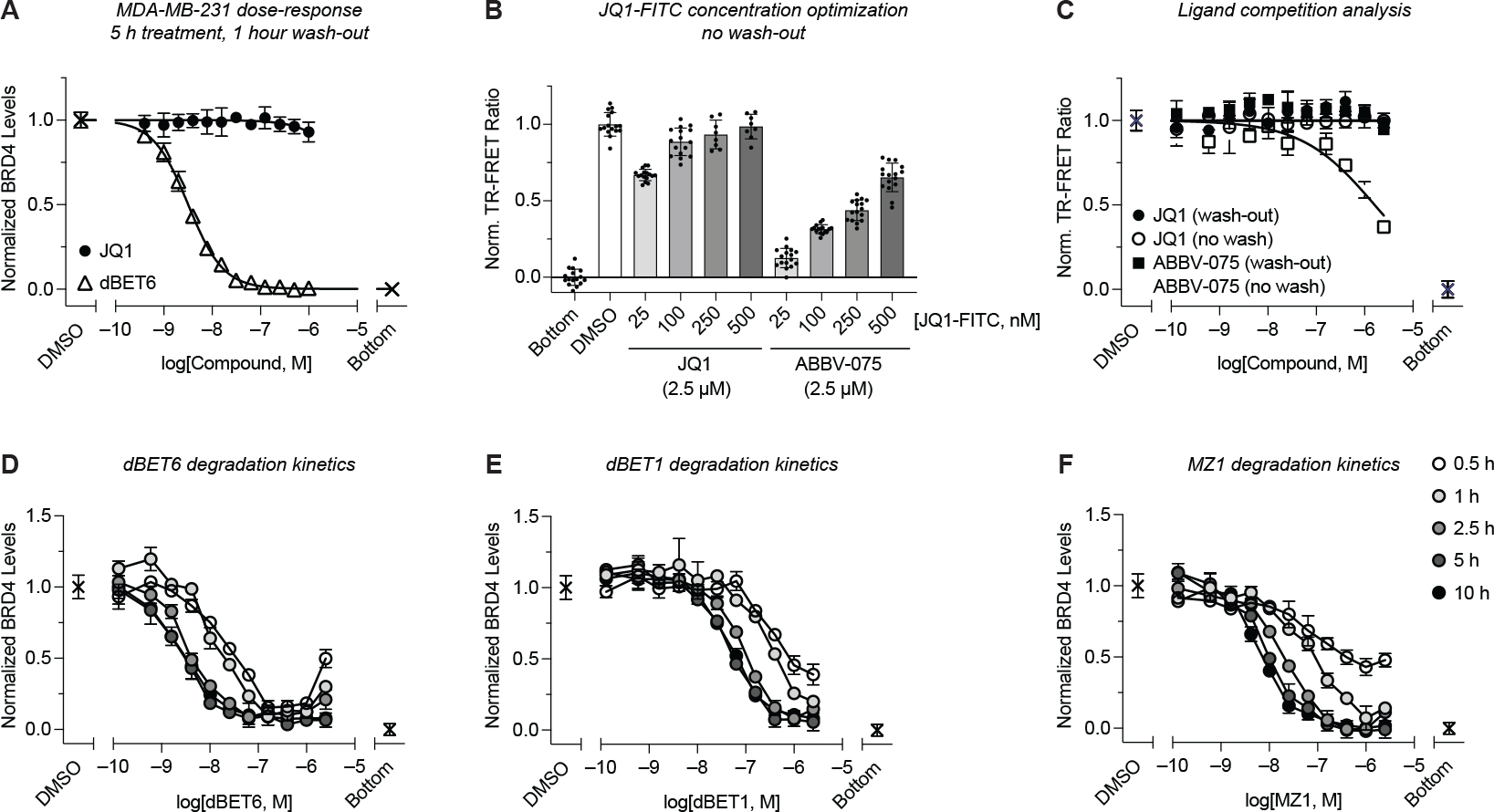
Assay miniaturization to 96-well plate format and kinetic profiling of BRD4 degradation. (A) MDA-MB-231 cells in 96-well plates were treated with a dose-titration of dBET6 or JQ1 for 5 h followed by a 1 h wash-out. BRD4 levels were quantified via subsequent addition of lysis buffer and detection mix as in Figure 1A (n = 3). (B) Comparative analysis of ligand competition across a dose range of JQ1-FITC after treatment of MDA-MB-231 cells in 96-well plates with JQ1 or ABBV-075 and no compound wash-out (n = 8). (C) As in (B), with constant 250 nM JQ1-FITC in the detection mix and dose-titrations of JQ1 and ABBV-075, with or without compound wash-out (n = 2). (D-F) Quantitative kinetic profiling of the time- and dose-dependent BRD4 degradation by dBET6, dBET1, and MZ1 after treatment in 96-well plates and quantification under no-wash conditions with 250 nM JQ1-FITC (n = 2). Data in (A-B) are expressed as mean ± SD of n technical replicates and are representative of at least two independent experiments. Data in (C-F) are expressed as mean ± SD of 2 technical × 2 biological replicates.
Next, we evaluated the sensitivity of our approach to ligand competition, which could result in the underestimation of POI concentrations. This undesired effect is expected to be most pronounced for high-affinity ligands at high cellular concentrations. We treated cells with BRD4 ligands having equal or higher potency than JQ1-FITC, with or without compound wash-out (Figure 3B–C). In this experiment, we chose non-PROTAC ligands to ensure constant BRD4 concentration. Notably, for JQ1-based ligands, concentrations of JQ1-FITC ≥100 nM in the detection mix were sufficient to prevent ligand competition, even without compound wash-out. For more potent ligands, such as ABBV-075 (KD,BD1/2 = 13.8/2.5 nM), competition with JQ1-FITC was observed at high ligand concentrations without a wash step, resulting in underestimation of BRD4 concentrations. However, this effect was fully resolved by compound wash-out (Figure 3B–C) (Faivre et al., 2020).
To demonstrate assay capacity, we profiled the time- and dose-dependent degradation of BRD4 by dBET6, dBET1, and MZ1 at five time points over eleven concentrations (Figure 3D–F) (Winter et al., 2015; Zengerle et al., 2015). dBET6 was the fastest acting degrader, with >75% reduction in BRD4 levels in the 0.1–1 μM range after only 30 min (Figure 3D). MZ1 exhibited slightly slower degradation kinetics, yet similar DC50 and Emax values as dBET6 at time points >2.5 h, with both compounds showing superior potency compared to dBET1 (Figure 3D–F, Table S2). Importantly, inherent to our platform is the quantitative measurement of BRD4 levels relative to background signal (see STAR Methods), allowing for facile determination of both Emax and DC50 across these time-course experiments.
Characterization of a celastrol-derived BRD4 degrader
Recently, the targeting of other E3 ubiquitin ligase complexes, apart from cereblon and VHL, with (reversible) covalent ligands that react with cysteine side chains has gained increasing attention. Of the predicted > 600 E3 ligases, a substantial fraction feature solvent-exposed cysteine residues that can potentially be exploited with corresponding thiophilic biasing elements (Belcher et al., 2021; Kannt and Dikic, 2021). While selective recruitment of E3 ligases can potentially provide cell type- or tissue-specific degradation, we reasoned that targeting multiple ligase complexes with a “promiscuous” reversible covalent ligand could provide a means for efficient TPD. The triterpenoid celastrol (CS, Figure 4A) forms reversible covalent adducts with multiple cysteine nucleophiles (Liu et al., 2015; Salminen et al., 2010). Recently, we demonstrated that CS also targets Keap1 (Kelch-like ECH-associated protein 1), a redox-regulated member of the CRL3 (Cullin-RING E3 ligase) complex that controls the homeostatic abundance of the transcription factor Nrf2 (Nuclear factor-erythroid factor 2-related factor 2) (Cuadrado et al., 2019; Payne et al., 2021). Furthermore, a previous report has demonstrated the promise of PROTACs (CDDO-JQ1) derived from bardoxolone methyl (CDDO-Me), a synthetic triterpenoid that binds Keap1 with high affinity (Figure 4A) (Tong et al., 2020). Thus, we aimed to evaluate the capacity of CS to function as a recruiting element for E3 ligase activity in a similar manner.
Figure 4.
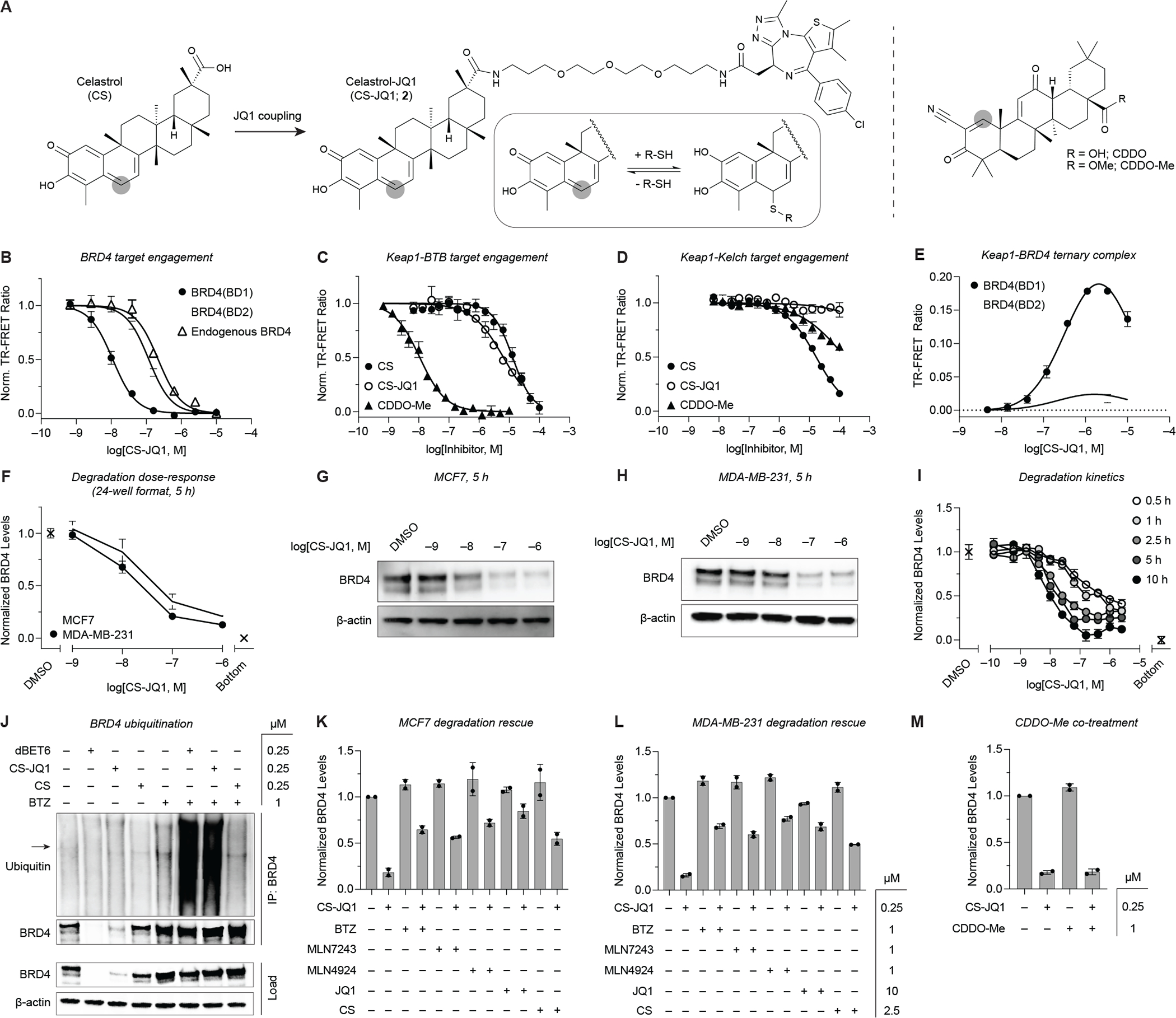
Celastrol is an E3 ubiquitin ligase recruiter suitable for PROTAC applications. (A) Chemical structures of CS, CS-JQ1 (2), CDDO, and CDDO-Me. Thiophilic sites are highlighted with grey circles. (B-D) Target binding of the various compounds for (B) recombinant BRD4(BD1), BRD4(BD2), and endogenous BRD4 in MCF7 cell extract (n = 2), (C) Keap1-BTB domain (n = 4), and (D) Keap1-Kelch domain (n = 2) (see STAR Methods). (E) CS-JQ1-induced ternary complex formation between full-length Keap1 and individual BRD4 bromodomains (n = 2). (F) TR-FRET quantification of BRD4 levels in MCF7 and MDA-MB-231 cells after treatment with a dose-titration of CS-JQ1 for 5 h in 24-well plates (n = 2). (G-H) Western blot analysis of the same samples used for TR-FRET quantification in (F). (I) Quantitative profiling of the time- and dose-dependent degradation of BRD4 in MDA-MB-231 cells by CS-JQ1 in 96-well plates (n = 2). (J) Ubiquitination of BRD4 induced by dBET6 and CS-JQ1 in MDA-MB-231 cells. Arrow in Ubiquitin blot represents BRD4 molecular weight (IP = immunoprecipitation). (K-L) Rescue of CS-JQ1-induced BRD4 degradation by BTZ, MLN7243, MLN4924, JQ1, and CS in (K) MCF7 or (L) MDA-MB-231 cells (n = 2). (M) Co-treatment of MDA-MB-231 cells with CS-JQ1 and CDDO-Me does not attenuate BRD4 degradation, indicating potential activity mediated through additional E3 ligase complexes other than Keap1/CRL3 (n = 2). Data in (B-E) are expressed as mean ± SD of n technical replicates and are representative of at least two independent experiments. Data in (F, I-M) are expressed as mean ± SD of 2 technical × 2 biological replicates. Unprocessed Western blot data is shown in Figure S4.
We synthesized a prototype PROTAC candidate CS-JQ1 (2, Figure 4A) and validated target engagement with BRD4 (Figure 4B, Table S1). Interestingly, however, CS-JQ1 lost the ability to bind the Keap1 Kelch domain, while exhibiting slightly improved affinity for the BTB domain (Figure 4C–D, Table S3). In contrast, functionalization of CDDO results in substantially decreased affinity for the BTB domain (Payne et al., 2021). Furthermore, CS-JQ1 could induce ternary complex formation between full-length Keap1 and individual BRD4 bromodomains (Figure 4E).
Next, MCF7 and MDA-MB-231 cells were treated for 5 h with varying concentrations of CS-JQ1, followed by BRD4 quantification by TR-FRET and Western blot (Figure 4F–H). The experiments were performed in 24-well format to allow for quantification of the same samples by both techniques. CS-JQ1 caused dose-dependent and efficient degradation of BRD4 in both cell lines. Next, we profiled BRD4 degradation kinetics by TR-FRET. CS-JQ1 and MZ1 exhibited similar time-dependence in terms of DC50, while CS-JQ1 displayed slightly slower kinetics for reaching more pronounced Emax values than the VHL and cereblon-based degraders (Figure 4I, Table S2). To validate that CS-JQ1 is acting as PROTAC and not through alternative pathways, we confirmed the ability of CS-JQ1 and dBET6 (positive control) to induce BRD4 ubiquitination in MDA-MB-231 cells (Figure 4J). In addition, we tested the capacity of UPS-pathway inhibitors BTZ, MLN7243, and MLN4924 to rescue BRD4 degradation (Figure 4K–L). In both MCF7 and MDA-MB-231 cells, co-treatment with either UPS-pathway inhibitor restored BRD4 levels comparable to dBET6.
Furthermore, co-treatment with JQ1 or CS similarly reduced CS-JQ1 degradation efficiency (Figure 4K–L). In contrast, however, BRD4 degradation was not altered in the presence CDDO-Me (BTB KD = 24 nM, Figure 4C), suggesting that CS and CDDO target overlapping but separate sets of E3 ligases. Together these data provide evidence that CS-JQ1 primarily functions as a PROTAC with a mechanism distinct from CDDO-derived degraders.
Discussion
Quantifying cellular protein levels in response to various stimuli is often a central yet resource-intensive aspect in chemical biology research. To overcome the inherent limitations of Western blot and ELISA, bioluminescence-based assay formats have been recently developed to simplify workflow, increase throughput, and improve sensitivity (Schwinn et al., 2018). While such approaches can even enable real-time detection of POI levels in living cells, they are not compatible with unmodified cell lines but require the expression of the POI as a (split-)luciferase fusion protein. This is usually accomplished ectopically or by genomic editing of a target cell line. The latter constitutes a lengthy process that generally takes weeks to months and needs to be performed for each cell line of interest.
In contrast, the approach presented here is directly applicable to unmodified cell lines, demonstrates excellent robustness and low technical variability, and is readily adaptable to high-throughput formats, enabling quantitative measurements within 1 h. Another significant improvement is the adaptation of TR-FRET donor-labeled Nano-Secondaries, which circumvent the need for labeling individual primary antibodies and avoid the formation of higher-order immune complexes, which can cause nonlinear signal responses.
Importantly, as a collateral benefit, our approach provides direct access to a ligand displacement platform for profiling POI ligands using endogenous protein, further guiding the development of PROTAC structure-catalytic efficiency relationships. Owing to the inherent characteristics of TR-FRET, POI-selectivity of the tracer is not required, provided that the employed antibody is selective for the POI.
We further validate our approach in the discovery of CS as a promising new ligand for the recruitment of E3 ligase activity to neosubstrates. Building on our earlier work and inspired by the previous report of a potent PROTAC derived from CDDO and JQ1, we show that the analogous CS-derived degrader exhibits potent activity in reducing cellular BRD4. Additional mechanistic studies confirmed UPS dependence, providing support that CS-JQ1 functioning as a PROTAC. Interestingly, co-treatment with CDDO-Me did not reduce the efficacy of CS-JQ1. Since the binding affinity of CS and CS-JQ1 for Keap1-BTB was determined in a ligand displacement assay using a CDDO-based tracer, it is reasonable to assume that CDDO-Me would efficiently displace CS-JQ1.
Limitations of the study
While the protein quantification approach developed here overcomes many inherent shortcomings of existing platforms, excess ligand can potentially compete with the tracer, resulting in decreased TR-FRET signal and reporting an apparent lower POI concentration. For potent, reversible ligands, this issue can be avoided by including a wash step, optionally in the presence of cycloheximide to suppress de novo POI synthesis. Similarly, ligands with slow off-rates may require longer incubation before TR-FRET readout to ensure equilibrium binding. We have outlined a general strategy to identify such compounds. Indeed, once more potent ligands are identified, they can be adapted as fluorescent tracers to minimize the effect of ligand competition. However, for irreversible ligands our strategy might significantly underestimate the actual protein concentration, as it will only report the remaining fraction of functional protein. To accurately estimate total (active+inactive) POI levels, other techniques will be better suited.
Furthermore, while CDDO-Me co-treatment did not attenuate CS-JQ1-mediated BRD4 degradation, we cannot rule out that CS-JQ1 still mediates its activity through Keap1 by binding a previously unrecognized site (distinct from the BTB domain). However, it is more likely that CS-JQ1 is dominantly acting by engaging with one or more Keap1-independent E3 ligase complexes. Future studies, which are beyond the scope of this report, are directed towards identifying the principle E3 ligase(s) exploited by celastrol-based PROTACs.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ralph Mazitschek (ralph@broad.harvard.edu).
Materials availability
This study did not generate unique reagents.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
MCF7 cells (ATCC HTB-22; H. sapiens female) were propagated in RPMI-1640 medium supplemented with 10% FBS, 1% pen-strep, and 1% L-glutamine at 37°C and 5% CO2. MDA-MB-231 cells (ATCC HTB-26; H. sapiens female) were propagated in DMEM medium supplemented with 10% FBS, and 1% pen-strep at 37°C and 5% CO2.
METHOD DETAILS
Preparation of MCF7 cell extracts
A cell pellet from one 15 cm dish (~25 M cells) of MCF7 cells was allowed to thaw on ice and cells were suspended in 400 μL lysis buffer (25 mM HEPES, 150 mM NaCl, 0.2% (v/v) Triton X-100, 0.02% (v/v) TWEEN-20, pH 7.5 supplemented with 2 mM DTT, 250 U Benzonase (Sigma E1014) and 1 mM AEBSF hydrochloride (Combi-Blocks SS-7834)). Optionally, Roche cOmplete, Mini, EDTA-free protease inhibitor cocktail (Sigma 11836170001) can be used in place of, or in combination with, AEBSF hydrochloride. Cells were homogenized via passage through a 27.5-gauge needle 5 times, and the resulting mixture was incubated with slow, end-over-end mixing at 4°C for 30 min. The lysate was clarified via centrifugation at 16,100 × g for 20 min at 4°C then 800 μL (1:3 dilution) dilution buffer (25 mM HEPES, 150 mM NaCl, 0.005% (v/v) TWEEN-20, pH 7.5) was added and the lysate was re-clarified at 16,100 × g for 20 min at 4°C. Total protein was quantified via detergent-compatible Bradford assay (ThermoFisher 23246). Expected total protein ranges were 4–7 mg/mL for a 15 cm dish pellet. The lysate was used fresh or flash-frozen in liquid nitrogen and stored at −80°C in single-use aliquots.
TR-FRET measurements
Unless otherwise noted, experiments were performed in white, 384-well microtiter plates (Corning 3572) in 30 μL assay volume. TR-FRET measurements were acquired on a Tecan SPARK plate reader with SPARKCONTROL software version V2.1 (Tecan Group Ltd.), with the following settings: 340/50 nm excitation, 490/10 nm (Tb), and 520/10 nm (FITC) emission, 100 μs delay, 400 μs integration. The 490/10 and 520/10 emission channels were acquired with a 50% mirror and a dichroic 510 mirror, respectively, using independently optimized detector gain settings unless specified otherwise. The TR-FRET ratio was taken as the 520/490 nm intensity ratio on a per-well basis.
Antibody and nanobody labeling
Nano-Secondary alpaca anti-rabbit IgG (ChromoTek shurbGNHS-1), GST VHH (ChromoTek st-250), anti-6xHis IgG (Abcam 18184), and anti-GST IgG (Abcam 19256) were labeled with CoraFluor-1-Pfp as previously described (Payne et al., 2021). The following extinction coefficients were used to calculate protein concentration and degree-of-labeling (DOL): ChromoTek shurbGNHS-1 E280 = 24,075 M−1cm−1, ChromoTek st-250 E280 = 28,545 M−1cm−1, IgG E280 = 210,000 M−1cm−1, CoraFluor-1-Pfp E340 = 22,000 M−1cm−1. Nanobody conjugates were diluted with 50% glycerol and stored at −20°C. IgG conjugates were diluted with 50% glycerol, snap-frozen in liquid nitrogen, and stored at −80°C.
Determination of apparent equilibrium dissociation constant (KD,app) of JQ1-FITC toward individual recombinant bromodomains and endogenous BRD4 in MCF7 lysate
Recombinant BRD4(BD1) and BRD4(BD2) were purchased from BPS Biosciences, Inc and Epicypher, Inc (GST-BRD4(BD1), 31040; GST-BRD4(BD2), 15–0013, respectively). Recombinant bromodomains were diluted to 0.5 nM in assay buffer (25 mM HEPES, 150 mM NaCl, 0.5 mg/mL BSA, 0.005% TWEEN-20, pH 7.5) with 2 nM CoraFluor-1-labeled anti-GST VHH, then JQ1-FITC was added in serial dilution (1:1.6 titration, 15-point, cmax = 100 nM) using a HP D300 digital dispenser (Hewlett-Packard) and allowed to equilibrate for 2 h at room temperature before TR-FRET measurements were taken. Nonspecific signal was determined with 50 μM JQ1-Acid, and data were fitted to a One Site – Specific Binding model in Prism 9.
For the profiling of endogenous BRD4, MCF7 cell lysate as prepared above was diluted to 0.8 mg/mL total protein in 1:3 lysis buffer:dilution buffer with 0.5 nM rabbit anti-BRD4 antibody (Cell Signaling Technology; E2A7X) and 1 nM CoraFluor-1-labeled anti-rabbit Nano-Secondary. JQ1-FITC was added in serial dilution (1:1.7 titration, 15-point, cmax = 200 nM) using a HP D300 digital dispenser and allowed to equilibrate for 2 h at room temperature before TR-FRET measurements were taken. Nonspecific signal was determined with 50 μM JQ1-Acid, and data were fitted to a One Site – Specific Binding model in Prism 9.
TR-FRET ligand displacement assays
The following assay parameters have been used: (i) 4 nM GST-BRD4(BD1), 4 nM CoraFluor-1-labeled anti-GST VHH, 20 nM JQ1-FITC in assay buffer, (ii) 4 nM GST-BRD4(BD2), 4 nM CoraFluor-1-labeled anti-GST VHH, 20 nM JQ1-FITC in assay buffer, (iii) MCF7 cell lysate at 0.8 mg/mL total protein, 0.5 nM rabbit anti-BRD4 antibody, 1 nM CoraFluor-1-labeled anti-rabbit Nano-Secondary, 20 nM JQ1-FITC. In all cases, test compounds were added in serial dilution (1:4 titration, 9-point, cmax = 10 μM) using a HP D300 digital dispenser and allowed to equilibrate for 2 h at room temperature before TR-FRET measurements were taken. The assay floor (background) was defined with the 10 μM ABBV-075 dose, and the assay ceiling (top) was defined via a no-inhibitor control.
TR-FRET ligand displacement assays with 6xHis-GST-Keap1 construct were performed as previously described (Payne et al., 2021). Data were background corrected, normalized, and fitted to a four-parameter dose response model using Prism 9.
Calculation of inhibitor KD,app values from measured TR-FRET IC50
For TR-FRET ligand displacement assays with recombinant proteins and endogenous BRD4 in whole-cell extract, we have determined the KD,app of the respective fluorescent tracer under each assay condition. Inhibitor KD,app values have been calculated using Cheng-Prusoff principles, outlined in Equation 1 below:
| (1) |
Where IC50 is the measured IC50 value, [S] is the concentration of the fluorescent tracer, and KX is the KD,app of the fluorescent tracer for a given condition (Cheng and Prusoff, 1973).
TR-FRET assay to measure ternary complex formation between full-length, wildtype Keap1 and individual recombinant bromodomains
Keap1 (tag-free; 11981-HNCB; Sino Biological) was diluted to 40 nM in assay buffer (supplemented with 1 mM DTT) containing 40 nM FITC-Ahx-LDEETGEFL-CONH2 tracer (Payne et al., 2021), 20 nM CoraFluor-1-labeled anti-GST antibody, and either 40 nM GST-BRD4(BD1) or GST-BRD4(BD2). CS-JQ1 was added in serial dilution (1:4 titration, 9-point, cmax = 10 μM) using a HP D300 digital dispenser and allowed to equilibrate for 4 h at room temperature before TR-FRET measurements were taken with identical detector gain settings. Data were background-subtracted from wells containing no CS-JQ1.
TR-FRET BRD4 quantification assay in 24-well plate format
MCF7 or MDA-MB-231 cells were seeded into 24-well plates (Corning 353047) at a density of 200,000 cells/well in 0.5 mL cell culture medium and allowed to adhere overnight. For dose-response profiling, a HP D300 digital dispenser was used to dispense serial dilutions of test compounds (1:10 titration, 6-point, cmax = 1 or 10 μM) normalized to 0.1% DMSO. Cells were incubated for 5 h at 37°C and 5% CO2 then media was replaced with pre-warmed cell culture medium (1 mL/well) and residual test compound was washed out for 1 h at 37°C and 5% CO2. After, media was aspirated and cells were washed with PBS (2 mL/well), followed by the addition of ice-cold lysis buffer (200 μL/well). The plate was shaken at 200 rpm on an orbital shaker (IKA KS 260 basic) for 10 min, then lysate was transferred to 1.5 mL microcentrifuge tubes and further incubated with slow, end-over-end mixing for 10 min at 4°C. The lysate was clarified via centrifugation at 16,100 × g for 10 min at 4°C then total protein concentration was measured using a detergent-compatible Bradford assay (ThermoFisher 23246). Expected total protein ranges were 0.4–0.6 mg/mL. Lysate was transferred to a 384-well plate (30 μL × 3 TR-FRET replicates) then 5 μL of 7× detection mix (3.5 nM rabbit anti-BRD4 antibody, 7 nM CoraFluor-1-labeled anti-rabbit Nano-Secondary, 700 nM JQ1-FITC, prepared in dilution buffer) was added to each well and allowed to equilibrate for 1 h before TR-FRET measurements were taken. TR-FRET ratios were background-subtracted from wells containing lysis buffer, 0.5 mg/mL BSA, and detection mix, then normalized to total protein concentration. The average TR-FRET intensity was normalized to DMSO for each biological replicate before being analyzed in Prism 9.
For degradation rescue experiments, a HP D300 digital dispenser was used to dispense rescue compounds (see respective figure panels for concentrations) normalized to 0.2% DMSO and were pre-incubated for 30 min at 37°C and 5% CO2 before degraders (250 nM) were added followed by treatment for 5 h at 37°C and 5% CO2. Cell treatment, lysate preparation, and TR-FRET analysis were performed as described above.
Note: The use of 0.5 nM anti-BRD4 antibody establishes an upper limit of detection of 0.5–1 nM BRD4 in the cell lysate. To ensure that BRD4 concentrations do not exceed this upper limit, a lysate dilution series (as in Figure 1C) should always be performed for each cell line of interest. The concentration of anti-BRD4 antibody can be further adjusted based on the dynamic range observed for each cell line. In our experience, the BRD4 concentration remained below 1 nM in various lysates at 1–2 mg/mL total protein concentration.
TR-FRET BRD4 quantification assay in 96-well plate format (1 h wash-out)
MDA-MB-231 cells were seeded into 96-well plates (Corning 3903 or Corning 3904) at a density of 20,000 cells/well in 100 μL cell culture medium and allowed to adhere overnight. A HP D300 digital dispenser was used to dispense serial dilutions of test compounds (1:2 titration, 12-point, cmax = 1 μM) normalized to 0.1% DMSO. Cells were incubated for 5 h at 37°C and 5% CO2 then media was replaced with pre-warmed cell culture medium (150 μL/well) and residual test compound was washed out for 1 h at 37°C and 5% CO2. After, media was aspirated and cells were washed with PBS (200 μL/well), followed by the addition of ice-cold lysis buffer (60 μL/well). The plate was shaken at 1,000 rpm on an orbital shaker (Boekel Scientific Jitterbug, model 130000) for 10 min. After, 10 μL of 7× control detection mix (7 nM CoraFluor-labeled anti-rabbit Nano-Secondary, 700 nM JQ1-FITC final concentrations, prepared in dilution buffer) was added to four of the eight DMSO-treated control wells, and 10 μL of complete detection mix (3.5 nM rabbit anti-BRD4 antibody, 7 nM CoraFluor-1-labeled anti-rabbit Nano-Secondary, 700 nM JQ1-FITC final concentrations, prepared in dilution buffer) was added to all other wells and allowed to equilibrate for 1 h. The plate was centrifuged at 2,000 × g for 1 min then lysate was transferred to a 384-well plate (30 μL × 2 TR-FRET replicates) using an adjustable electronic multichannel pipette (Matrix Equalizer, ThermoFisher 2231), and TR-FRET measurements were taken. Optionally, after TR-FRET measurements, 5 μL/well of CellTiter-Glo 2.0 reagent (Promega G9241) was added to the wells of the 384-well plate and allowed to equilibrate for 10 minutes before luminescence intensity was recorded on a Tecan SPARK plate reader (luminescence module, no attenuation, 250 ms integration time, output Counts/s). TR-FRET ratios were background-subtracted from the four (eight TR-FRET replicates) DMSO-treated wells containing control detection mix. The average TR-FRET intensity was then normalized to the four (eight TR-FRET replicates) DMSO-treated wells containing complete detection mix for each biological replicate, then data were fitted to a four-parameter dose-response model using Prism 9.
TR-FRET BRD4 quantification assay in 96-well plate format (no wash-out)
MDA-MB-231 cells were seeded into 96-well plates (Corning 3903 or Corning 3904) at a density of 20,000 cells/well in 100 μL cell culture medium and allowed to adhere overnight. A HP D300 digital dispenser was used to dispense serial dilutions of test compounds (1:2.5 titration, 12-point, cmax = 2.5 μM) normalized to 0.25% DMSO. Cells were incubated for 30 min, 1 h, 2.5 h, 5 h, or 10 h at 37°C and 5% CO2 then media aspirated and cells were washed twice with PBS (200 μL/well), followed by the addition of ice-cold lysis buffer (60 μL/well). The plate was shaken at 1,000 rpm on an orbital shaker (Boekel Scientific Jitterbug, model 130000) for 10 min. After, 10 μL of 7× control detection mix (7 nM CoraFluor-labeled anti-rabbit Nano-Secondary, 1,750 nM JQ1-FITC final concentrations, prepared in dilution buffer) was added to four of the eight DMSO-treated control wells, and 10 μL of complete detection mix (3.5 nM rabbit anti-BRD4 antibody, 7 nM CoraFluor-1-labeled anti-rabbit Nano-Secondary, 1,750 nM JQ1-FITC final concentrations, prepared in dilution buffer) was added to all other wells and allowed to equilibrate for 1 h. The plate was centrifuged at 2,000 × g for 1 min then lysate was transferred to a 384-well plate (30 μL × 2 TR-FRET replicates) using an adjustable electronic multichannel pipette (Matrix Equalizer, ThermoFisher 2231), and TR-FRET measurements were taken. TR-FRET ratios were background-subtracted from the four (eight TR-FRET replicates) DMSO-treated wells containing control detection mix. The average TR-FRET intensity was then normalized to the four (eight TR-FRET replicates) DMSO-treated wells containing complete detection mix for each biological replicate.
Immunoblotting
Proteins in lysates (10 μg) were analyzed by electrophoresis on 3–8% SDS-polyacrylamide gels (ThermoFisher) and subsequently transferred to a nitrocellulose membrane (Bio-Rad). All antibodies were purchased from Cell Signaling Technology. Ponceau staining and β-actin probing (8H10D10, 1:1,000) were used to verify equal protein loading on the blot. The membrane was blocked using 5% nonfat milk powder in TBS-T (Tris-buffered saline; 0.1% TWEEN-20) at room temperature for 1 h and then incubated with an anti-rabbit IgG BRD4 antibody (E2A7X, 1:750) in 2.5% nonfat milk overnight at 4°C. The membrane was then incubated with an anti-rabbit IgG HRP-linked antibody (1:5,000 in 2.5% nonfat milk; 7074S). The proteins were detected using SuperSignal™ West Femto Maximum Sensitivity Substrate (ThermoFisher).
Immunoprecipitation experiments
Antibodies against BRD4 (E2A7X, 1:750), Ubiquitin (P4D1, 1:750), and β-actin (8H10D10, 1:3,000), as well as secondary anti-rabbit (7074S, 1:5,000) or anti-mouse (7076S, 1:5,000) HRP-linked antibodies (Cell Signaling Technology), were used in this experiment. MDA-MB-231 cells were plated in 10 cm dishes and treated with the corresponding compounds (~70% confluency) for 5 h. When indicated, cells were pre-treated with Bortezomib for 1 h. For immunoprecipitation assays, cells were resuspended in a lysis buffer (25 mM HEPES, 150 mM NaCl, 0.2% Triton X-100, 0.02% TWEEN-20, 2 mM DTT, pH 7.5) supplemented with protease inhibitors (Roche) and 20 mM N-ethylmaleimide (Millipore-Sigma). Cell homogenization and protein quantification was performed as above in “Preparation of MCF7 cell extracts.” The immunoprecipitation was performed using the Dynabeads™ Protein G Immunoprecipitation Kit (ThermoFisher), following the manufacturer’s instructions. 20 μg of protein was used for the input loading control while BRD4 was isolated from 1,000 μg of cell lysate with anti-BRD4 antibody (E2A7X, 1:50). Immunoblotting was performed as above.
Chemical synthesis
Reagents & Equipment
Reagents and ligands were purchased from Chem-Impex International, Millipore-Sigma, TCI America, Beantown Chemical, Combi-Blocks, MedChemExpress, Ontario Chemicals, and BOC Sciences and used as received. FITC-Ahx-LDEETGEFL-CONH2 (FITC-KL9) peptide tracer was custom synthesized by Genscript (Piscataway, New Jersey). CDDO-FITC fluorescent tracer was prepared as previously described (Payne et al., 2021). Column purifications were performed on a Biotage Isolera 4 Purification System equipped with a 200– 400 nm diode array detector. For normal phase flash purifications, Sorbtech Purity Flash Cartridges were used (CFC-52300–012-18 and CFC-52500–025-12). For reverse phase flash purifications, Biotage Sfär Bio C18 Duo 300 Å, 20 μm cartridges were used (FSBD-0411–001). Analytical LC/MS was performed on a Waters 2545 HPLC equipped with a 2998 diode array detector, a 2424 evaporative light scattering detector, a 2475 multichannel fluorescence detector, and a Waters 3100 ESI-MS module, using a XTerraMS C18 5 μm, 4.6 × 50 mm column at a flow rate of 5 mL/min with a linear gradient (95% A: 5% B to 100% B 90 sec and 30 sec hold at 100% B, solvent A = water + 0.1% formic acid, solvent B = acetonitrile + 0.1% formic acid). LC/MS data analysis was performed using Waters Masslynx V4.1 SCN 846 software. Proton and carbon nuclear magnetic resonance (1H and 13C NMR spectra) were recorded on a Bruker Avance III 400 spectrometer using Topspin 3.2 software and data were analyzed using MestreNova (version 12.0.1–20560, Mestrelab Research). Chemical shifts for protons are reported in parts per million (ppm) and are referenced to residual solvent peaks. Data is reported as follows: chemical shift, multiplicity (s = singlet, br s, = broad singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet), proton coupling constants (J, Hz), and integration.
Synthetic procedures (see Data S1 for NMR spectra) 5-(3-(1-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)-2-oxo-7,10,13-trioxa-3-azahexadecan-16-yl)thioureido)-2-(6-hydroxy-3-oxo-3H-xanthen-9-yl)benzoic acid – JQ1-FITC – (1)
1-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)-2-oxo-7,10,13-trioxa-3-azahexadecan-16-aminium chloride (6.0 mg, 9.4 μmol, 1.1 eq) was dissolved in DMF (200 μL) with DIPEA (15 μL, 11 mg, 85 μmol, 10 eq) then 5(6)-fluorescein isothiocyanate (FITC; 3.3 mg, 8.5 μmol, 1 eq) was added and the reaction mixture was briefly vortexed and allowed to stand at room temperature for 10 min. The reaction mixture was directly purified via reverse phase flash chromatography (λ 220 nm, 250 nm; gradient: 5% ACN/H2O + 0.1% formic acid for 3 CV, 5% to 70% ACN/H2O + 0.1% formic acid over 14 CV, 70% ACN/H2O to 100% ACN + 0.1% formic acid over 1 CV, 100% ACN + 0.1% formic acid for 3 CV). Yield = 4.0 mg, 48% as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (br s, 3H), 8.42 – 8.32 (m, 1H), 8.30 – 8.15 (m, 2H), 7.75 (d, J = 7.7 Hz, 1H), 7.48 (d, J = 8.5 Hz, 2H), 7.41 (d, J = 8.5 Hz, 2H), 7.16 (d, J = 8.3 Hz, 1H), 6.66 (s, 2H), 6.62 – 6.52 (m, 4H), 4.50 (t, J = 7.1 Hz, 1H), 3.53 – 3.42 (m, 12H), 3.29 – 3.08 (m, 6H), 2.59 (s, 3H), 2.40 (s, 3H), 1.83 – 1.76 (m, 2H), 1.71 – 1.65 (m, 2H), 1.61 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 169.46, 168.58, 163.04, 159.63, 155.12, 151.93, 149.83, 146.83, 141.50, 136.76, 135.24, 132.27, 130.71, 130.14, 129.84, 129.57, 129.07, 128.49, 125.01, 124.06, 112.68, 109.79, 102.24, 69.78, 69.58, 68.20, 68.05, 53.90, 37.65, 35.79, 29.46, 28.58, 14.08, 12.70, 11.32. MS (ESI+/−) m/z (M+H)+ 992.45, m/z (M−H)− 990.25, [calculated C50H50ClN7O9S2: 991.28].
(2R,4aS,6aS,12bR,14aS,14bR)-N-(1-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)-2-oxo-7,10,13-trioxa-3-azahexadecan-16-yl)-10-hydroxy-2,4a,6a,9,12b,14a-hexamethyl-11-oxo-1,2,3,4,4a,5,6,6a,11,12b,13,14,14a,14b-tetradecahydropicene-2-carboxamide – Celastrol-JQ1 – (2)
Celastrol (10.0 mg, 22 μmol, 1 eq) was dissolved in DMF (500 μL) then PyBOP (12.1 mg, 23 μmol, 1.05 eq) and 1-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)-2-oxo-7,10,13-trioxa-3-azahexadecan-16-aminium chloride (14.9 mg, 23 μmol, 1.05 eq) were added followed by DIPEA (19 μL, 14 mg, 111 μmol, 5 eq) and the reaction mixture was briefly vortexed then allowed to stand at room temperature for 10 min. The reaction mixture was diluted into EtOAc (50 mL) and the organic layer was washed 2 × equal volume 0.2 N HCl, 2 × H2O, 1 × saturated brine solution. The organic layer was then dried over Na2SO4, filtered, and concentrated. The crude product was purified via reverse phase flash chromatography (λ 250 nm, 400 nm; gradient: 5% ACN/H2O for 3 CV, 5% ACN/H2O to 100% ACN over 18 CV, 100% ACN for 3 CV). Yield = 11.0 mg, 48% as an orange powder. 1H NMR (400 MHz, CDCl3) δ 7.40 (d, J = 8.2 Hz, 2H), 7.32 (d, J = 8.3 Hz, 2H), 7.00 (d, J = 6.8 Hz, 2H), 6.88 (t, J = 5.0 Hz, 1H), 6.63 (t, J = 4.4 Hz, 1H), 6.52 (s, 1H), 6.33 (d, J = 7.2 Hz, 1H), 4.64 (t, J = 7.0 Hz, 1H), 3.68 – 3.63 (m, 4H), 3.63 – 3.57 (m, 6H), 3.53 (t, J = 5.9 Hz, 3H), 3.45 – 3.31 (m, 3H), 3.22 (s, 2H), 2.66 (s, 3H), 2.53 – 2.43 (m, 1H), 2.40 (s, 3H), 2.20 (s, 3H), 2.17 (s, 3H), 2.11 – 1.75 (m, 10H), 1.67 (br s, 6H), 1.59 – 1.48 (m, 5H), 1.42 (s, 3H), 1.12 (s, 3H), 1.09 (s, 3H), 0.63 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 178.48, 177.66, 170.61, 170.51, 164.94, 163.95, 155.85, 149.99, 146.14, 136.89, 136.82, 134.24, 132.29, 131.06, 130.92, 130.60, 129.98, 128.84, 127.49, 119.67, 118.12, 117.16, 71.34, 70.65, 70.55, 70.37, 69.96, 54.60, 45.22, 44.55, 43.16, 40.22, 39.53, 39.01, 38.30, 37.97, 36.55, 35.17, 33.92, 33.68, 31.78, 31.23, 31.10, 30.96, 30.12, 29.83, 29.45, 29.18, 28.85, 28.71, 21.88, 18.38, 14.56, 13.25, 12.00, 10.42. MS (ESI+/−) m/z (M+H)+ 1035.79, m/z (M−H)− 1033.47, [calculated C58H75ClN6O7S: 1034.51].
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details of experiments can be found in the figure legends. For cell-based assays, n biological replicates have been defined as independent cell treatments, performed at different times with biologically distinct samples. For biochemical assays, the n numbers denoted in the figure legends refer to the number of technical replicates used to calculate mean ± SD for a given data point within an experiment. Experiments have been independently repeated ≥2 times, and representative results are shown. No statistical methods were used to predetermine sample size and investigators were not blinded to outcome assessment. Graphpad Prism 9 was used for statistical analyses using default settings (nonlinear regression analysis, standard deviations, and confidence intervals).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-6xHis | Abcam | Cat# ab18184, RRID:AB_444306 |
| Anti-GST | Abcam | Cat# ab19256, RRID:AB_444809 |
| Anti-rabbit nano-secondary | ChromoTek | Cat# shurbGNHS-1, RRID:AB_2864267 |
| GST-VHH | ChromoTek | Cat# ST-250, RRID:AB_2631368 |
| Anti-BRD4 | Cell Signaling Technology | Cat# E2A7X, RRID:AB_2687578 |
| Anti-beta actin HRP | Cell Signaling Technology | Cat# 8H10D10, RRID:AB_2566811 |
| Anti-rabbit HRP | Cell Signaling Technology | Cat# 7074, RRID:AB_2099233 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant GST-BRD4(BD1) | BPS Bioscience | Cat# 31040 |
| Recombinant GST-BRD4(BD2) | EpiCypher | Cat# 15-0013 |
| Recombinant His/GST-Keap1 | Sino Biological | Cat# 11981-H20B |
| Recombinant Keap1 (tag-free) | Sino Biological | Cat# 11981-HNCB |
| FITC-Ahx-LDEETGEFL-CONH2 peptide | Genscript | Custom peptide synthesis |
| Celastrol | Ontario Chemicals | Cat# C1307 |
| JQ1-Acid | Dr. Jun Qi | N/A |
| JQ1 | Medchemexpress | Cat# HY-13030 |
| dBET1 | Medchemexpress | Cat# HY-101838 |
| dBET6 | Medchemexpress | Cat# HY-112588 |
| MZ1 | Medchemexpress | Cat# HY-107425 |
| ABBV-075 (Mivebresib) | Medchemexpress | Cat# HY-100015 |
| CDDO-Me | Medchemexpress | Cat# HY-13324 |
| AEBSF hydrochloride | Combi Blocks | Cat# JL-8396 |
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail | Millipore Sigma | Cat# 11836170001 |
| Critical Commercial Assays | ||
| Pierce™ Detergent Compatible Bradford Assay Kit | ThermoFisher Scientific | Cat# 23246 |
| Experimental Models: Cell Lines | ||
| MCF7 | ATCC | Cat# HTB-22, RRID:CVCL_0031 |
| MDA-MB-231 | ATCC | Cat# HTB-26, RRID:CVCL_0062 |
| Software and Algorithms | ||
| Prism 9 | Graphpad | RRID:SCR_002798 |
| MestreNova 14.2.2 | Mestrelab Research | N/A https://mestrelab.com/download/mnova/ |
| Masslynx V4.1 SCN 846 | Waters | N/A www.waters.com |
| Topspin 3.2 | Bruker | N/A https://www.bruker.com/en/products-and-solutions/mr/nmr-software/topspin.html |
| ND-1000 3.8.1 | ThermoFisher Scientific | N/A www.thermofisher.com |
| SPARKCONTROL V2.1 | Tecan Group | https://lifesciences.tecan.com/multimode-plate-reader |
Highlights.
Dual-mode TR-FRET assay for target protein abundance and ligand affinity profiling
High-throughput quantification of unmodified endogenous proteins in cell lysate
Celastrol is a reversible covalent warhead for redirecting E3 ligase activity
Significance.
The ability to quantitatively measure target engagement of small molecules or changes in the abundance of target proteins of interest in response to drug treatment is critical to many biomedical research programs and at the heart of targeted protein degradation. Ideally, these measurements are performed with endogenous protein and in disease-relevant cell line(s). We here report the development of a generalizable, sensitive, and flexible assay system that greatly facilitates the measurement of both ligand binding affinities and target protein abundance in high-throughput. Unlike other assay approaches, which are low-throughput or depend on epitope-tagged fusion proteins – either expressed recombinantly or in genetically modified human cell lines – the presented platform is highly modular and uses endogenous proteins in lysates from unmodified cell lines. We validate the robustness by characterizing the biochemical and cellular activity of prototype small molecule degraders targeting BRD4. Furthermore, we identify the natural product celastrol, a triterpenoid that reversibly reacts with cysteine residues, as a promising ligand for PROTAC development that expands the scope of accessible E3-ligases for targeted protein degradation.
Acknowledgements
This work was supported by NSF 1830941 (R.M.), and NIH 1R01NS064983 (B.A.T. and R.M.). N.C.P. was supported by a National Science Foundation Graduate Research Fellowship (DGE1745303). We thank Dr. Stephen J. Haggarty for generously providing access to tissue culture space. We thank Dr. Jun Qi for donating JQ1-Acid.
Footnotes
Competing interests statement
R.M. is a scientific advisory board (SAB) member and equity holder of Regenacy Pharmaceuticals, ERX Pharmaceuticals, and Frequency Therapeutics. R.M. and N.C.P. are inventors on patent applications related to the CoraFluor TR-FRET probes used in this work. R.M., N.C.P., and B.A.T. are inventors on patent applications related to the celastrol-based degraders developed in this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Belcher BP, Ward CC, and Nomura DK (2021). Ligandability of E3 Ligases for Targeted Protein Degradation Applications. Biochemistry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley DL, Van Molle I, Gareiss PC, Tae HS, Michel J, Noblin DJ, Jorgensen WL, Ciulli A, and Crews CM (2012). Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1alpha interaction. J Am Chem Soc 134, 4465–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burslem GM, and Crews CM (2020). Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 181, 102–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busby SA, Carbonneau S, Concannon J, Dumelin CE, Lee Y, Numao S, Renaud N, Smith TM, and Auld DS (2020). Advancements in Assay Technologies and Strategies to Enable Drug Discovery. ACS Chem Biol 15, 2636–2648. [DOI] [PubMed] [Google Scholar]
- Chamberlain PP, and Hamann LG (2019). Development of targeted protein degradation therapeutics. Nat Chem Biol 15, 937–944. [DOI] [PubMed] [Google Scholar]
- Cheng Y, and Prusoff WH (1973). Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- Cuadrado A, Rojo AI, Wells G, Hayes JD, Cousin SP, Rumsey WL, Attucks OC, Franklin S, Levonen AL, Kensler TW, et al. (2019). Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat Rev Drug Discov 18, 295–317. [DOI] [PubMed] [Google Scholar]
- Donovan KA, Ferguson FM, Bushman JW, Eleuteri NA, Bhunia D, Ryu S, Tan L, Shi K, Yue H, Liu X, et al. (2020). Mapping the Degradable Kinome Provides a Resource for Expedited Degrader Development. Cell 183, 1714–1731 e1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faivre EJ, McDaniel KF, Albert DH, Mantena SR, Plotnik JP, Wilcox D, Zhang L, Bui MH, Sheppard GS, Wang L, et al. (2020). Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature 578, 306–310. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. (2010). Selective inhibition of BET bromodomains. Nature 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzl A, and Winter GE (2020). Targeted protein degradation: current and future challenges. Curr Opin Chem Biol 56, 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarmoskaite I, AlSadhan I, Vaidyanathan PP, and Herschlag D (2020). How to measure and evaluate binding affinities. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung M, Philpott M, Muller S, Schulze J, Badock V, Eberspacher U, Moosmayer D, Bader B, Schmees N, Fernandez-Montalvan A, et al. (2014). Affinity map of bromodomain protein 4 (BRD4) interactions with the histone H4 tail and the small molecule inhibitor JQ1. J Biol Chem 289, 9304–9319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannt A, and Dikic I (2021). Expanding the arsenal of E3 ubiquitin ligases for proximity-induced protein degradation. Cell Chem Biol 28, 1014–1031. [DOI] [PubMed] [Google Scholar]
- Kepp O, Galluzzi L, Lipinski M, Yuan J, and Kroemer G (2011). Cell death assays for drug discovery. Nat Rev Drug Discov 10, 221–237. [DOI] [PubMed] [Google Scholar]
- Lin W, and Chen T (2021). General Stepwise Approach to Optimize a TR-FRET Assay for Characterizing the BRD/PROTAC/CRBN Ternary Complex. ACS Pharmacol Transl Sci 4, 941–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lee J, Salazar Hernandez MA, Mazitschek R, and Ozcan U (2015). Treatment of obesity with celastrol. Cell 161, 999–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniaci C, and Ciulli A (2019). Bifunctional chemical probes inducing protein-protein interactions. Curr Opin Chem Biol 52, 145–156. [DOI] [PubMed] [Google Scholar]
- Nowak RP, DeAngelo SL, Buckley D, He Z, Donovan KA, An J, Safaee N, Jedrychowski MP, Ponthier CM, Ishoey M, et al. (2018). Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat Chem Biol 14, 706–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne NC, Kalyakina AS, Singh K, Tye MA, and Mazitschek R (2021). Bright and stable luminescent probes for target engagement profiling in live cells. Nat Chem Biol 17, 1168–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne NC, and Mazitschek R (2022). Resolving the deceptive isoform and complex selectivity of HDAC1/2 inhibitors. Cell Chemical Biology. Advance online publication doi: 10.1016/j.chembiol.2022.03.002 [DOI] [PubMed] [Google Scholar]
- Salminen A, Lehtonen M, Paimela T, and Kaarniranta K (2010). Celastrol: Molecular targets of Thunder God Vine. Biochem Biophys Res Commun 394, 439–442. [DOI] [PubMed] [Google Scholar]
- Schapira M, Calabrese MF, Bullock AN, and Crews CM (2019). Targeted protein degradation: expanding the toolbox. Nat Rev Drug Discov 18, 949–963. [DOI] [PubMed] [Google Scholar]
- Schwinn MK, Machleidt T, Zimmerman K, Eggers CT, Dixon AS, Hurst R, Hall MP, Encell LP, Binkowski BF, and Wood KV (2018). CRISPR-Mediated Tagging of Endogenous Proteins with a Luminescent Peptide. ACS Chem Biol 13, 467–474. [DOI] [PubMed] [Google Scholar]
- Simard JR, Lee L, Vieux E, Improgo R, Tieu T, Phillips AJ, Fisher SL, Pollock RM, and Park E (2021). High-Throughput Quantitative Assay Technologies for Accelerating the Discovery and Optimization of Targeted Protein Degradation Therapeutics. SLAS Discov 26, 503–517. [DOI] [PubMed] [Google Scholar]
- Sy M, Nonat A, Hildebrandt N, and Charbonniere LJ (2016). Lanthanide-based luminescence biolabelling. Chem Commun (Camb) 52, 5080–5095. [DOI] [PubMed] [Google Scholar]
- Tong B, Luo M, Xie Y, Spradlin JN, Tallarico JA, McKenna JM, Schirle M, Maimone TJ, and Nomura DK (2020). Bardoxolone conjugation enables targeted protein degradation of BRD4. Sci Rep 10, 15543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BD, and Soh HT (2020). Re-Evaluating the Conventional Wisdom about Binding Assays. Trends Biochem Sci 45, 639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, and Bradner JE (2015). DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter GE, Mayer A, Buckley DL, Erb MA, Roderick JE, Vittori S, Reyes JM, di Iulio J, Souza A, Ott CJ, et al. (2017). BET Bromodomain Proteins Function as Master Transcription Elongation Factors Independent of CDK9 Recruitment. Mol Cell 67, 5–18 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue H, Nowak RP, Overwijn D, Payne NC, Fischinger S, Atyeo C, Baden LR, Nilles EJ, Karlson EW, Yu XG, et al. (2020). Rapid ‘mix and read’ assay for scalable detection of SARS-CoV-2 antibodies in patient plasma. medRxiv. [Google Scholar]
- Zengerle M, Chan K-H, and Ciulli A (2015). Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chemical Biology 10, 1770–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, and Oldenburg KR (1999). A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen 4, 67–73. [DOI] [PubMed] [Google Scholar]
- Zwier JM, Roux T, Cottet M, Durroux T, Douzon S, Bdioui S, Gregor N, Bourrier E, Oueslati N, Nicolas L, et al. (2010). A fluorescent ligand-binding alternative using Tag-lite(R) technology. J Biomol Screen 15, 1248–1259. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.