

SUMMARY
Lactate accumulates to a significant amount in GBMs, the most common primary malignant brain tumor with an unfavorable prognosis. However, it remains unclear whether lactate is metabolized by GBMs. Here, we demonstrated that lactate rescued patient-derived xenograft (PDX) GBM cells from nutrient deprivation mediated cell death. Transcriptome analysis, ATAC-seq., and CHIP-seq. showed that lactate entertained a signature of oxidative energy metabolism. LC/MS analysis demonstrated that U-13C-Lactate elicited substantial labeling of TCA-cycle metabolites, acetyl-CoA and histone protein acetyl-residues in GBM cells. Lactate enhanced chromatin accessibility and histone acetylation in a manner dependent on oxidative energy metabolism and the ATP-citrate lyase (ACLY). Utilizing orthotopic PDX models of GBM, a combined tracer experiment unraveled that lactate carbons were substantially labeling the TCA cycle metabolites. Finally, pharmacological blockage of oxidative energy metabolism extended overall survival in two orthotopic PDX models in mice. These results establish lactate metabolism as a novel druggable pathway for GBM.
Graphical Abstract
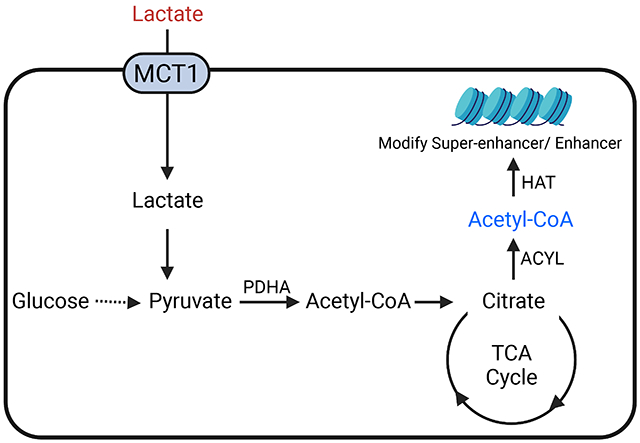
In brief
Torrini and Nguyen et al. reveal an important role of lactate in a broad range of glioblastoma (GBM). They demonstrate that lactate is actively metabolized in a manner reliant on cellular respiration and that lactate affects gene expression through modulation of the epigenome. Thus, targeting oxidative metabolism and lactate metabolism may be a novel therapeutic approach for GBM.
INTRODUCTION
Glioblastoma (GBM) is the most common primary malignant brain tumor with an unfavorable prognosis and a reprogrammed metabolism (Cancer Genome Atlas Research et al., 2015; Parsons et al., 2008). Tumor growth is ultimately determined by nutrients and how cancer cells metabolize and harness them for biosynthesis and energy production (Faubert et al., 2017). In general, it assumed that akin to other malignancies GBMs consume ample amounts of glucose and metabolize it via pyruvic acid to lactate, a phenomenon, which is also referred as the Warburg effect and goes back to the 1920s when Otto Warburg made this initial observation. However, it is important to note that activity of oxidative metabolism is necessary to enable the biosynthesis of fatty acids and cholesterol from glucose carbons given that acetyl-CoA (from glucose carbons) is generated and converted to citric acid in the mitochondrial matrix first (DeBerardinis and Chandel, 2020). In turn, mitochondrial citrate is transported to the cytosol to generate cytosolic acetyl-CoA via the ATP citrate lyase (ACLY) reaction (Carrer et al., 2019; Trefely and Wellen, 2018; Wellen et al., 2009). In addition to being necessary for biosynthesis, acetyl-CoA serves as substrate for post-translational modifications of histones, e.g. acetylation. In this vein, prior research has pointed out that glucose carbons regulate histone acetylation and thereby affect gene expression, but whether lactate may regulate the epigenome has not been demonstrated yet.
While as indicated glioblastomas mostly utilize glucose, there are other carbon sources at their disposal. Originally considered as a waste product of glucose catabolism, lactate accumulates to a significant amount in tumor tissue (up to 40 mM) (Faubert et al., 2017; Hui et al., 2017). Prior and recent single cell sequencing studies confirm the notion that the core of GBMs is glycolytic, whereas the periphery is more oxidative (Darmanis et al., 2017). Moreover, mass spectrometry imaging data from human glioblastoma tissues shows that lactate along with fatty acid is substantially enriched in the infiltrative margin (Kampa et al., 2020; Randall et al., 2020). This concept enables the tumor core to rely on glucose, while lactate that in turn along with fatty acids may serve as a substrate for tumor cells in the periphery. However, it remains to be proven whether indeed lactate serves a fuel for GBMs and thus in turn would establish lactate metabolism as therapeutic vulnerability. Lactate uptake appears to be primarily mediated by the MCT1 transporter though it may be possible that it does not constitute the only means to transport lactate into tumor cells (Faubert et al., 2017).
We launched our studies with the central hypothesis that lactate is heavily metabolized by GBM cells in the TCA-cycle to promote their survival and that lactate through conversion to acetyl-CoA via pyruvic acid is an epigenetic metabolite to regulate pro-survival gene expression. Moreover, pharmacological targeting of the TCA-cycle abrogated these effects and extended animal survival in an orthotopic PDX model in mice. Therefore, the interference with lactate metabolism may be a suitable target for GBM therapy.
RESULTS
Lactate rescues a broad array of different GBM model systems from nutrient deprived mediated reduction in cellular viability
In prior studies the transporter MCT1 has been shown to be involved in lactate uptake (Faubert et al., 2017) and we found that MCT1 mRNA levels are increased in glioblastoma tissues as compared to normal brain (Figure 1A). Following interrogation of MCT1 expression in a cell line database, comprising over 1739 different cell cultures, we found several cell lines with high levels of MCT1. Amongst those several central nervous system tumor cell lines were identified, e.g. KNS42, a pediatric GBM cell line (Figure 1B). Consistently, various GBM cell cultures revealed increased levels of MCT1 as compared to human astrocytes (Figure 1C). Next, we posed the questions whether lactate is capable of rescuing KNS42 from nutrient deprived culturing conditions by employing cellular viability assays (Figure 1D). We chose a variety of different nutrient conditions, including close to normal physiological conditions (5 mM glucose, 1 mM glutamine) as well as glucose deprived conditions, which are known to be encountered in malignant tumors (Burgess and Sylven, 1962; Flavahan et al., 2013; Yun et al., 2009). Close to physiological culturing conditions were highly reminiscent of the high-glucose environment, suggesting no further benefit elicited by high glucose levels (Figure S1C). In contrast, low glucose conditions resulted in a substantial loss in cellular viability as compared to the physiological glucose culturing conditions (Figure 1D). Strikingly, 10-15 mM lactate was capable of essentially full reversing the loss in viability by low glucose conditions (Figure 1D). Next, we wanted to extend these findings to a larger set of GBM model system and continued our studies in well-known established GBM cell lines, U251, U87 and DBTRG. Akin to the KNS42 line, lactate rescued from nutrient-deprived mediated reduction in viability (Figure 1E, Figure S1A). In addition, stem-like GBM cells, NCH644, were protected as well (Figure 1F). In GBM12, GBM22 and GBM43 PDX lines tested we confirmed the earlier observations that lactate enhances cellular viability following nutrient-deprived culturing conditions, suggesting that these findings are likely applicable to a broad range of different model systems (Figure 1G). Considering IDH1 and EGFR mutational status our data suggested that in the presence of these mutations lactate rescues from glucose deprivation mediated reduction of cellular viability as well (Figure 1E; Figure S1A). We confirmed our findings using another proliferation assay (crystal violet) and included a time course. These results demonstrated that lactate drives cellular proliferation and compensates for the loss of glucose (Figure S1B). Since the brain interstitial fluid (ISF) concentration of glutamine is about 80 μM (Dolgodilina et al., 2016) we assessed whether or not lactate is capable of rescuing GBM cells from glucose-deprivation. While the effect of lactate on the cellular viability was slightly reduced in glutamine-reduced conditions, there was still a substantial rescue from glucose-deprivation mediated reduction of viability by lactate (Figure S1D). Regarding the formulation, both lactic acid and lactate essentially equally rescued from glucose-deprived reduction of viability in GBM cells (Figure S1E). Other acids have been described to have tumor promoting properties, such as acetate (Liu et al., 2018; Mashimo et al., 2014). We assessed whether acetate rescued from nutrient-deprived culturing conditions. Our results show that acetate did not have an impact on cellular viability neither in established nor in PDX lines (Figure S1F).
Figure 1. Lactate facilitates survival of GBM model systems under nutrient deprived conditions in a manner dependent on the MCT1 transporter.
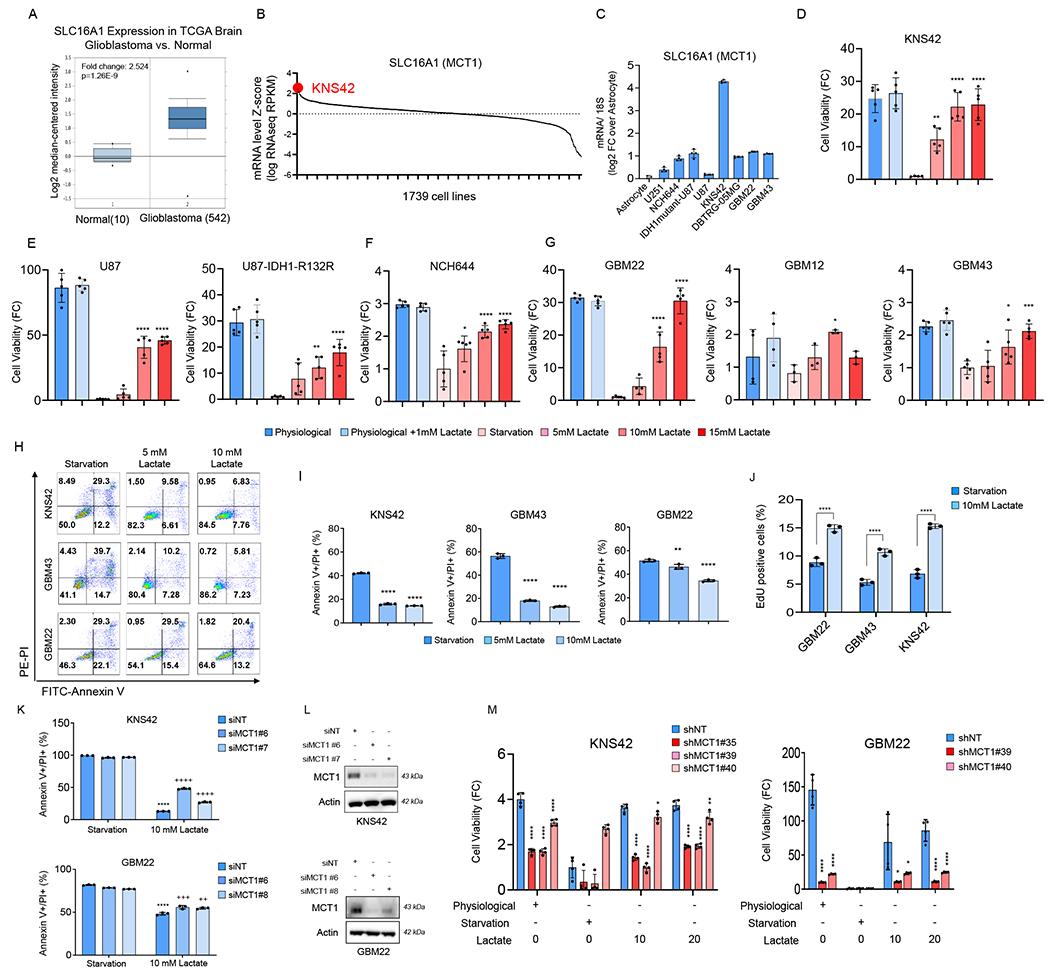
(A) Shown is the mRNA levels of SLC16A1 (MCT1) in the TCGA glioblastoma database. Data was extracted from oncomine.org. Middle lines in boxplot: median; box ranges: upper: 75th percentile; lower: 25th percentile; error bars: 10th and 90th percentile.
(B) SLC16A1 (MCT1) mRNA expression in the cBioPortal cancer genomics database of 1739 cell lines. KNS42, a pediatric GBM cell line (highlighted in red), is among the top of cell lines with high expression of MCT1 mRNA levels. A complete list of cell lines with the relative SLC16A1 mRNA levels is shown in the Table S1.
(C) Real time PCR analysis of SLC16A1 (MCT1) mRNA levels in astrocyte, established glioblastoma, patient-derived xenograft glioblastoma, and stem-like glioblastoma cells (n=4). FC: fold change. 18S is used as a housekeeping gene.
(D-G) GBM cells were cultured in different media conditions: physiological media, starvation media, and lactate media for 72h and cell viability analysis was assessed by using CellTiter-Glo (n=5).
(H, I) GBM cells were cultured in starvation media, 5 mM lactate media, or 10 mM lactate media for 48h and the cells were stained with annexin V/propidium iodide staining (n=3). The quantification was shown in (I).
(J) Shown is the quantification of EdU positive (S-phase) staining in GBM cells cultured in starvation media or 10 mM lactate media for 48h (n=3).
(K) KNS42 and GBM22 cells were transfected with non-targeting siNT or MCT1 specific siRNAs, cultured in starvation media or 10 mM lactate media for 48h and the cells were stained with annexin V/propidium iodide staining (n=3). *Statistical analysis was performed between starvation media and lactate media, +statistical analysis was performed between siNT and siMCT1.
(L) Western blot of GBM cells that were transfected with non-targeting siNT or MCT1 specific siRNA.
(M) KNS42 and GBM22 cells were transduced with non-targeting shNT or MCT1 specific shRNAs, cultured in physiological media, starvation media, or lactate media for 48h and analysis of cell viability was performed (n=4).
Statistical significance was assessed by a two-tailed student’s t-test in (J) or ANOVA with Dunnett’s multiple comparison test in (D-G, I, K, M). Data are shown as mean ± SD. *p<0.05, **p<0.01, ***/ ****p<0.001. Media condition: physiological media (5 mM glucose, 1 mM glutamine), starvation media (0.5 mM glucose, 0.5 mM glutamine), 5 mM lactate media (0.5 mM glucose, 0.5 mM glutamine, 5 mM lactate), 10 mM lactate media (0.5 mM glucose, 0.5 mM glutamine, 10 mM lactate), 15 mM lactate media (0.5 mM glucose, 0.5 mM glutamine, 15 mM lactate). See also Figure S1.
Next, we wondered whether lactate affects survival of cells as well. The results of these studies demonstrate a substantial rescue from nutrient-deprived mediated induction of a cell death with apoptotic features by lactate in a slightly concentration dependent manner (Figure 1H and 1I). Concerning the cell cycle, we found that lactate increased the number of cells in the S-phase (Figure 1J; Figure S1G). As indicated, the transportation of lactate over the plasma membrane is known to be mediated by MCT1 (Faubert et al., 2017). Therefore, we silenced the expression of MCT1 by siRNA and shRNA in KNS42 and GBM22 cells. We found that suppression of MCT1 mitigated the lactate mediated rescue from nutrient-deprivation both in GBM22 and KNS42 GBM cells (Figure 1K–1M; Figure S1H–S1J).
Lactate activates oxidative metabolism and thereby facilitates promotion of proliferation and survival of GBM cells
To determine how lactate drives survival of GBM cell cultures, we took a two-pronged strategy by performing a broad polar metabolite in KNS42 cells and transcriptome analysis in GBM22, GBM43 and KNS42 cells, utilizing nutrient-deprived conditions in the presence or absence of lactate (Figure 2A and 2B; Table S1; Figure S2A–S2D). Concerning the metabolite screen, we found an elevation of metabolites related to the TCA-cycle in the lactate containing condition (Figure 2A and 2B; Table S1; Figure S2A–S2D). Metabolite set enrichment analysis revealed an enrichment for the TCA-cycle (Figure 2C and 2D; Figure S2C and S2D) following lactate exposure. We performed gene set enrichment analysis and noted that lactate entertained an increase of pathways related to mitochondrial oxidative metabolism (Figure 2A and 2B; Figure S2B). Next, we confirmed whether indeed oxidative metabolism is a prerequisite for lactate to elicit a pro-survival effect and to impact oxidative metabolism. To this purpose, we performed extracellular flux analysis on the seahorse analyzer. We found that lactate facilitates an increase in oxygen consumption rate as compared to glucose only conditions (Figure 2E). Moreover, we treated KNS42 cells with the complex I inhibitor, IACS-010759 (Molina et al., 2018) in the presence of lactate. As expected, we found a substantial suppression of the oxygen consumption rate following administration of IACS-010759 (Figure 2F). Consistently, lactate renders GBM cultures more sensitive to the cytotoxic actions by inhibitors of oxidative phosphorylation and the electron transport chain, respectively (Figure 2G and 2H; Figure S3A and S3B). In the presence of lactate, KNS42 cells became highly susceptible to increasing concentrations of IACS-010759 and oligomycin, whereas conditions devoid of lactate displayed a significantly more resistant phenotype against these compounds (Figure 2G and 2H; Figure S3A and S3B). In contrast, lactate containing conditions were not efficient to sensitize for etoposide, suggesting specificity of the findings related to oxidative energy metabolism (Figure S3A). We also noted that lactate mediated transcriptional up-regulation of respiratory complexes was mitigated by IACS-010759 (Figure S3C).
Figure 2. Lactate mediated rescue of GBM viability is dependent on a functional electron transport chain/oxidative phosphorylation.
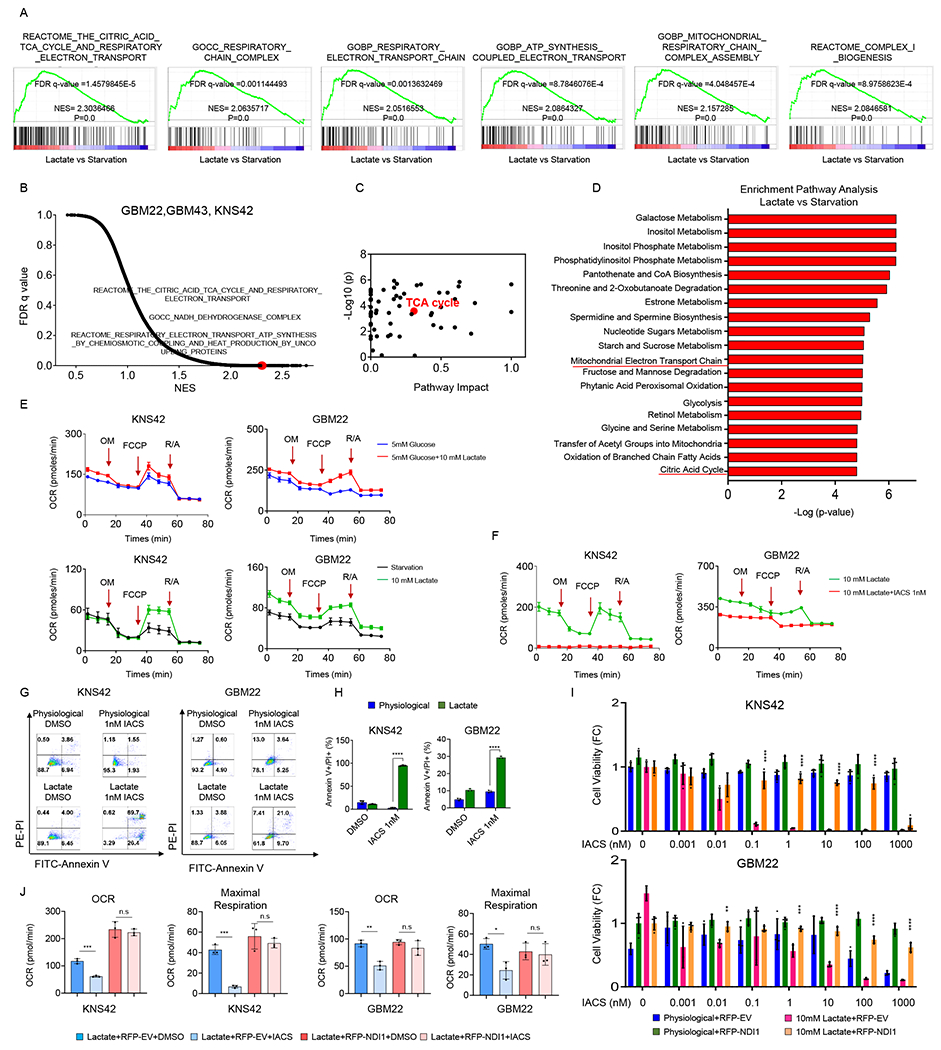
(A, B) GBM cells were exposed to the starvation media (0.5 mM glucose, 0.5 mM glutamine) or lactate media (0.5 mM glucose, 0.5 mM glutamine, 10 mM lactate) overnight and were subjected to transcriptomic analysis and followed by GSEA. Shown are the enrichment plots of TCA cycle, mitochondrial respiration complex, and electron transport chain. NES: normalized enrichment score. FDR: false discovery rate.
(C, D) KNS42 cells were exposed to the starvation media or lactate media overnight. Cells were then harvested and processed for polar metabolite LC/MS analysis. Metabolite set enrichment analysis was performed using MetaboAnalyst. Shown is the pathway impact and the TCA cycle is highlighted in red.
(E) KNS42 and GBM22 cells were starved for 30 minutes (0 mM glucose, 0 mM glutamine, 0 mM lactate), exposed to 5mM glucose or 5mM glucose+10 mM lactate, and subjected to extracellular flux analysis on the Seahorse XFp instrument to perform a mitochondrial stress assay. OM; oligomycin. FCCP; Carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone. R/A; Rotenone and antimycin (n=3).
(F) KNS42 and GBM22 cells were starved for 30 minutes, exposed to 10 mM lactate in the presence or absence of 1nM IACS-010759, a clinically validated respiratory complex I inhibitor, and subjected to extracellular flux analysis to perform a mitochondrial stress assay (n=3).
(G, H) KNS42 and GBM22 cells were exposed to physiological media (5 mM glucose, 1mM glutamine) or lactate media (0.5 mM glucose, 0.5 mM glutamine, 10 mM lactate), treated with 1nM IACS-010759 for overnight (KNS42)/ 7h (GBM22) and the cells were stained with annexin V/propidium iodide staining (n=3). The quantification was shown in (H).
(I) KNS42 and GBM22 cells transduced with empty vector (EV) or a vector containing NDI1 cDNA, exposed to physiological media or lactate media, treated with increasing concentration of IACS-010759 for 24h and cell viability was analyzed (n=4).
(J) KNS42 and GBM22 cells transduced with empty vector (EV) or a vector containing NDI1 cDNA, exposed lactate media (0.5 mM glucose, 0.5 mM glutamine, 10 mM lactate), treated with 1nM IACS-010759 for 7h, and subjected to extracellular flux analysis on the Seahorse XFp instrument to perform a mitochondrial stress assay (n=3).
Statistical significance was assessed by a two-tailed student’s t-test. Data are shown as mean ± SD. *p<0.05, **p<0.01, ***/ ****p<0.001. n.s: not significant. See also Figure S2, S3 and Table S1.
To further refine the specificity of these findings we took advantage of yeast Ndi1, which encodes for NADH dehydrogenase and facilitates a rescue from complex I inhibition mediated by IACS-010759 in human GBM cells. Consistently, inhibition of lactate mediated rescue from glucose deprived mediated reduction in cellular viability by complex I inhibition was almost fully rescued by Ndi1 overexpression (Figure 2I). Furthermore, IACS-010759 mediated reduction of oxygen consumption rate as well as maximal respiration was reversed in the presence of Ndi1 (Figure 2J). Overall, these findings support a central role of complex I (NADH dehydrogenase) in lactate metabolism. A fundamental component of cellular respiration is the complex I substrate, NADH2 which will be oxidized to NAD in the presence of sufficient molecular oxygen. Therefore, in the context of activation of oxidative energy metabolism through lactate it is of interest to assess the NAD/NADH2 ratio (Sullivan et al., 2015). We found that lactate only had a small impact on the NAD/NADH ratio, which was not consistent amongst the cell cultures tested (Figure S3D).
Lactate provides carbons to support the intermediary metabolism of GBM cells
We hypothesized that lactate is taken up and readily converted to pyruvic acid through lactate dehydrogenase and subsequently converted to citric acid via acetyl-CoA in two enzymatic catalyzed reactions by pyruvate dehydrogenase and citrate synthase, respectively. Alternatively, lactate may be converted from pyruvate to oxaloacetate via the pyruvate carboxylase reaction to yield the m+3 citric acid isotopologue (Figure 3B). To this purpose, we utilized U-13C-lactate to trace its carbons through the intermediary metabolism in a broad variety of different GBM cells (Figure 3A). We found substantial labeling of citrate, succinate, fumarate and malate. Moreover, we noted labeling of amino acids, including aspartate, alanine and glutamate (Figure S4A). Next, we sought to elucidate the impact of other fuel sources on the metabolization of lactate in GBM cells. To this purpose, we focused on glucose and glutamine. We found that as glucose and/or glutamine concentrations increased less labeling of TCA-cycle metabolites, amino acids and glutathione from lactate occurred (Figure 3C; Figure S4B). However, it is noteworthy that despite the presence of glucose and/or glutamine, lactate carbons still substantially contributed to the intermediary metabolism of GBM cells (Figure 3C). Lactate derived carbons may also be used for gluconeogenesis. Indeed, we found labeling of phospho-enol pyruvate and 3-phosphoglycerate, while in contrast no labeling of glucose-6-phosphate was detected, suggesting that lactate provides carbons efficiently to the lower portion of glycolysis in GBM cells (Figure S4B). Consistently, GBM cells express detectable levels of key enzymes, involved in gluconeogenesis (i.e. PC, PCK1) (Figure S4C and S4D). Given the labeling of acetyl-CoA by lactate we speculated whether fatty acid synthesis is in part supported by lactate and found that palmitic acid was partially labeled by lactate (Figure S4E). Overall, these findings implicate that lactate carbons are heavily used by a broad range of malignant glioma cells to fuel various metabolic pathways that are essential for growth and resistance to cell death.
Figure 3. U-13C lactate substantially labels the intermediary metabolism of GBM cultures and PDHA1 is involved in lactate metabolism/survival pathways.
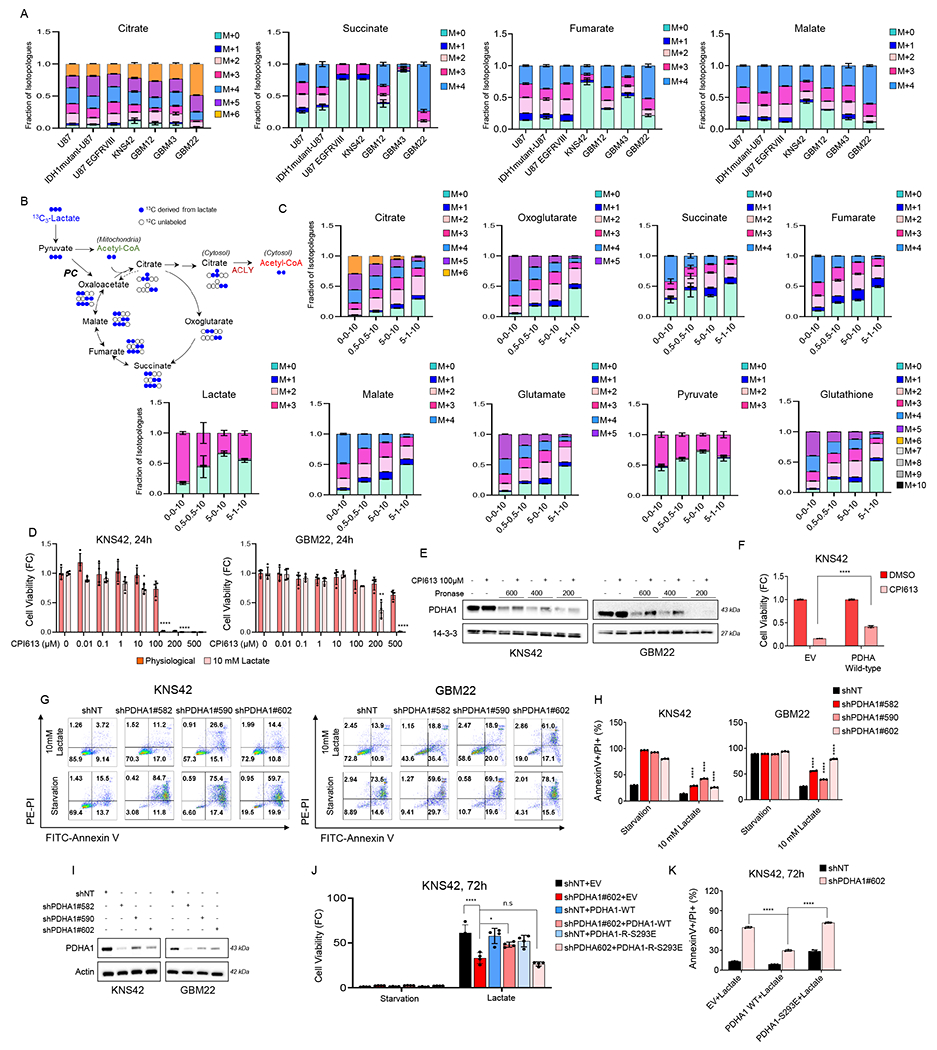
(A) Established GBM cells, pediatric GBM cells, and patient-derived GBM cells were incubated in DMEM depleted of phenol red, glucose, and glutamine, supplemented with 1.5% dialyzed FBS in presence or absence of 10 mM U-13C-labelled lactate for 16h and were processed for LC/MS polar metabolite analysis. Shown are fraction of the isotopologues for each metabolite (n = 3).
(B) Shown are the 13C-sodium lactate carbons and how they are transferred among molecules of the TCA cycle. Lactate is metabolized to pyruvate (m + 3). Lactate is either oxidized in the TCA cycle, resulting in (m + 2) citric acid. If lactate is used for anaplerosis, citric acid (m + 3) is produced. Lactate carbons also label acetyl-CoA via the ATP-citrate lyase (ACLY) reaction in the cytosol.
(C) KNS42 cells were exposed to different media conditions containing U-13C-lactate overnight and were performed LC/S analysis. Media condition: 0-0-10: 0 mM glucose, 0 mM glutamine, 10 mM lactate, 0.5-0.5-10: 0.5 mM glucose, 0.5 mM glutamine, 10 mM lactate, 5-0-10: 5 mM glucose, 0 mM glutamine, 10 mM lactate, 5-1-10: 5 mM glucose, 1 mM glutamine, 10 mM lactate. Shown are fraction of the isotopologues for each metabolite (n=3).
(D) KNS42 and GBM22 cells were exposed to physiological media (5 mM glucose, 1 mM glutamine) or lactate media (0.5 mM glucose, 0.5 mM glutamine, 10 mM lactate) and were treated with increasing concentration of CPI613 for 24h and cell viability analysis was performed (n=4).
(E) DARTS assay identifies PDHA1 as a CPI-613 binding protein. Standard western blot of GBM cell extracts were used in the presence or absence of Pronase. 14-3-3 is used as a loading control.
(F) Stably transduced empty vector (EV) and PDHA wild-type KNS42 cells were exposed to 10 mM lactate and were treated with CPI613 for 24h and the cells were stained with annexin V/propidium iodide staining (n=3). Shown is the quantification of viable cells. FC: fold change.
(G, H) Stably transduced non-targeting shRNA or PDHA specific shRNA KNS42 and GBM22 cells were cultured in starvation media or 10 mM lactate media and cells were labeled with Annexin/PI dye and analyzed by flow cytometry. Quantification is shown in (H) (n=3).
(I) Standard western blot of KNS42 and GBM22 cells transduced with non-targeting shNT or PDHA1 specific shRNA.
(J-K) Stably transduced non-targeting shNT or PDHA1 specific shRNA KNS42 cells were transduced with empty vector (EV), PDHA1-R wild-type (PDHA1 cDNA resistant against PDHA1 shRNA), or PDHA1-R S293E, cultured in starvation media or 10 mM lactate media, and cell viability analysis or Annexin/PI staining were performed (n=3).
Statistical significance was assessed by a two-tailed student’s t-test. Data are shown as mean ± SD. *p<0.05, **p<0.01, ****p<0.001. n.s: not significant. See also Figure S4.
We evaluated a novel inhibitor of PDHA1 that has reached clinical phase III trials, called CPI-613 (devimistat) (Zachar et al., 2011). To this purpose, we treated GBM22 and KNS42 cells with increasing concentrations of CPI-613 in the presence or absence of lactate. As anticipated, exposure to lactate facilitated the potency of CPI-613 to reduce the cellular viability of GBM cells, in keeping with the observations, involving blockers of the respiratory chain complexes (Figure 3D). Moreover, we performed the drug affinity responsive target stability (DARTS) assay to confirm that CPI-613 binds to PDHA1 in both KNS42 and GBM22 cells (Figure 3E). Consistently, over-expression of PDHA1 partially rescued for the cytotoxicity mediated by CPI-613 (Figure 3F). To further narrow down the specific role of PDHA1 in lactate metabolism, we specifically silenced the expression of this enzyme in KNS42 and GBM22 cells by either shRNA or siRNA. Our data shows that silencing of PDHA1 attenuates lactate mediated rescue from glucose deprivation (Figure 3G–3I; Figure S4F). Since shRNAs may display off-targets, we performed cDNA rescue experiments, involving a shRNA-resistant PDHA1 wild-type (active) or S293E mutated (partially inactive enzyme) cDNA (Figure 3J and 3K). While PDHA1 wild-type cDNA was capable of reversing cell death/reduction in cellular viability, the catalytically inactive mutant failed to rescue the effects of the PDHA1 shRNA, suggesting specificity of our findings related to the relevant target (Figure 3J and 3K). In addition, we confirmed our findings related to PDHA with siRNA, which resembled the effects seen with the shRNA (Figure S4G–4J). Moreover, we further assessed the role of PC by employing siRNA and demonstrated that loss of PC dampens lactate mediated rescue from glucose deprivation (Figure S4G–4J).
We extended our studies to in vivo experiments. Due to their rapid growth, we implanted the U87-EGFRvIII cells in the subcutis of nude mice and following establishment of tumors we injected uniformly labeled lactate and harvested the tumors for LC/MS analysis (Figure S5A). We found that akin to the in vitro findings lactate substantially labeled metabolites of the TCA-cycle, suggesting that indeed lactate carbons are readily utilized by the intermediary metabolism of GBM models in vivo (Figure S5B). In addition, we determined the absolute concentrations of lactate in tumors and normal tissue (Figure S5C). As anticipated, the heterotopic xenograft tumor displayed higher levels of lactate as compared to normal tissue (Figure S5C). Next, we assessed whether injection of GBM cells in an orthotopic location (right striatum) in nude mice would also reveal labeling of TCA-cycle metabolites from lactate (Figure 4A and 4B). Moreover, we were wondering about the relative contributions of glucose and lactate carbons to the TCA-cycle. To this purpose, we injected U87-EGFR-vIII GBM cells orthotopically. When tumors were established based on MRI imaging, we injected glucose and lactate tracers simultaneously and after 20 minutes the brain tumors and adjacent normal brain tissue were collected for LC/MS analysis (Figure S5D and S5E). The choice of uniformly labeled glucose and single carbon labeled lactate coupled with a short interval between injection and harvesting of the tumors enabled us to distinguish the carbon source of TCA-cycle metabolite labeling (Figure S5E). We found that lactate carbons in tumor tissue was derived from both glucose and lactate. Concerning citric acid, we found that glucose carbons contributed significantly less to citric acid in tumor tissue as compared to normal brain. Remarkably, lactate contributed its carbons to a much larger extent to citric acid as compared to glucose in brain tumor tissue. Consistently, the ratio between lactate derived citric acid and glucose derived citric acid is significantly elevated in brain tumor tissue as compared to normal brain. Similar observations were made with other TCA-cycle metabolites (Figure S5E). It is interesting to note that lactate also substantially labeled glutamic acid, supporting a potential cross talk between glutamine and lactate metabolism. Orthotopic PDX GBM models in mice remain the current standard model for resembling human disease properly and for preclinical testing of therapeutic interventions, respectively. To this end, we utilized two PDX models, the GBM12 and GBM22 (Figure 4C and 4D; Figure S5F). We observed the reduced incorporation of glucose carbons in citric acid in tumor tissue as compared to normal tissue and the relative increase of lactate derived carbons over glucose carbons in citric acid in tumor tissue (Figure 4D). All in all, these results underscore a significant role of lactate in GBM metabolism and that lactate is fundamentally important to maintain an active TCA-cycle in these malignancies.
Figure 4. Lactate predominantly labels the TCA-cycle in orthotopic PDX models in mice and pharmacological targeting of the TCA-cycle extends animal survival.
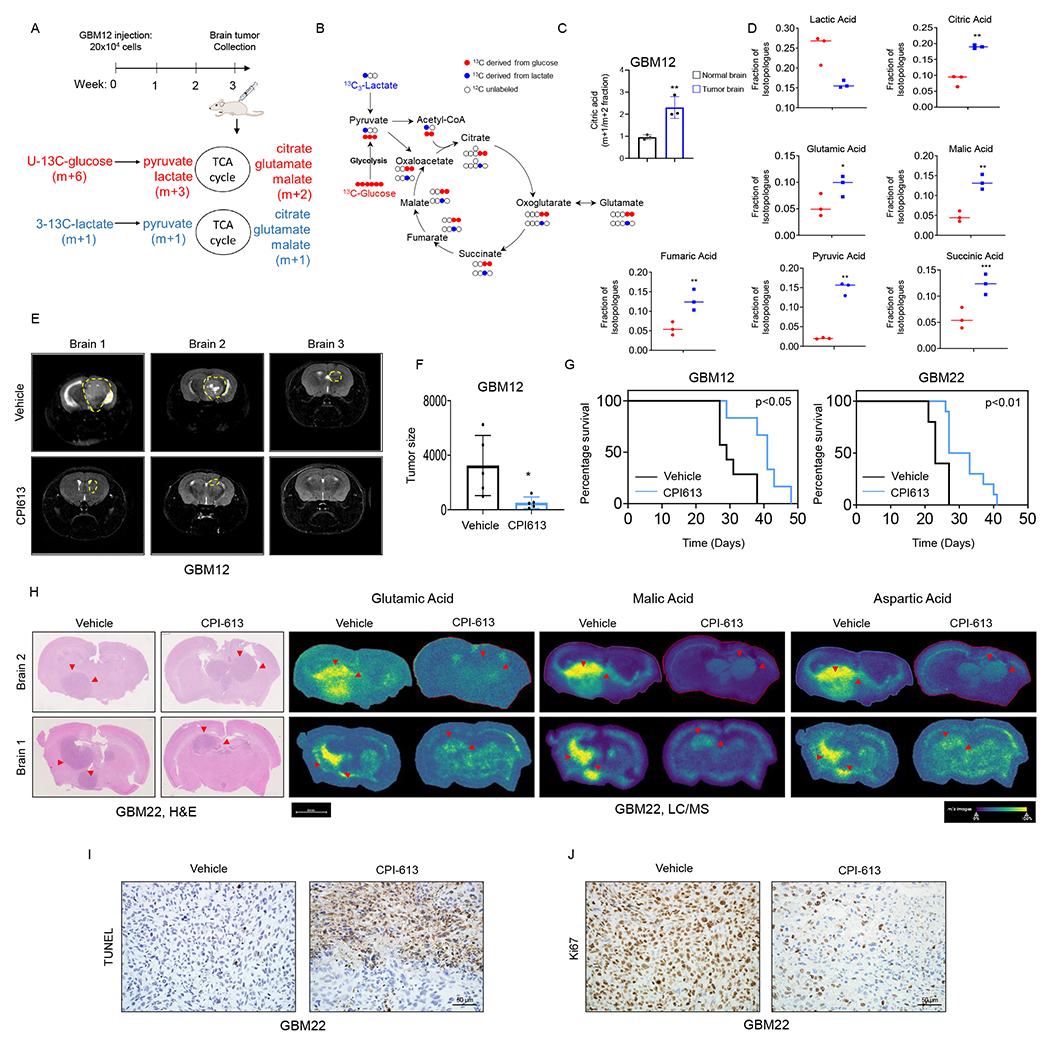
(A) Schematic representation of the intracranial model of glioblastoma. GBM cells were implanted into the right striatum of nude mice for three weeks. An equal amount of both 3-13C-labelled lactate (m+1) and U-13C-labelled glucose (m+6) were injected i.p 30 min prior to brain tumor collection.
(B) Shown is the U-13C-glucose carbons (red) and 3-13C-lactate carbons (blue) and how they are transferred among molecules of the TCA cycle. Glucose is metabolized to pyruvic acid labelling three carbons, while lactate is metabolized to pyruvic acid labelling just one carbon. Glucose derived carbons label the TCA cycle metabolites in a (m + 2) pattern, while lactate labelling occurs in (m+1).
(C) GBM12 cells were implanted into the right striatum of Nu/Nu mice for three weeks. An equal amount of both 3-13C-labelled lactate (m+1) and U-13C-labelled glucose (m+6) were injected i.p 30 min prior to brain tumor collection for LC/MS analysis.
(D) Shown are the levels of TCA cycle-intermediates isotopologue m+1 (derived from lactate, blue dot graph) and TCA cycle-intermediates isotopologue m+2 (derived from glucose, red dot graphs).
(E, F) Two groups of mice implanted with GBM12 cells were randomly assigned: vehicle and CPI613 (50mg/kg) treatment was initiated after seven days of the implantation. Mice were treated four times per week by i.p. injection. Shown is the tumor size of representative MRI images using a Bruker BioSpecTM, 9.4 Tesla MR Imager. The quantification is shown in (F) (n=5).
(G) GBM cells were implanted into the right striatum of Nu/Nu mice and treated with vehicle or CPI613 (50mg/kg) four times a week to assess tumor growth-related symptoms/survival.
(H) Representative hematoxylin and eosin staining and MALDI-TOF imaging of the brain tissues treated with vehicle or CPI613.
(I, J) TUNEL and Ki67 staining of the vehicle or CPI613 treated GBM22 tumors treatment in (G). Scale bar: 50μm.
Statistical significance was assessed by a two-tailed student’s t-test. Kaplan–Meier curves of animal’s survival are provided and the log-rank test was used to assess statistical significance. Data are shown as mean ± SD. *p<0.05, **p<0.01. See also Figure S5.
To assess the efficacy of CPI-613 in vivo we harnessed two orthotopic PDX models in mice (Figure 4E–4G). Following intracranial injection and establishment of GBM22 PDX tumors in mice, two groups were randomly assigned. One group was treated with vehicle, while a second group received CPI-613 four times a week. Notably, mice receiving CPI-613 displayed a reduced tumor size and an extended overall survival as compared to animals treated with vehicle without induction of toxicity (Figure 4E–4G; Figure S5G and S5H). Given that CPI-613 is expected to affect TCA-cycle metabolite, we determined relevant levels through MALDI-TOF from our in vivo experiments. To this end, we found that CPI-613 reduced the levels of malate, succinate, fumarate and TCA-cycle associated amino acids, aspartate and glutamate (Figure 4H; Figure S5I). We performed TUNEL-staining and found that CPI-613 induced cell death in vivo (Figure 4I). Conversely, CPI-613 reduced the proliferation of tumor cells (Figure 4J).
Lactate labels acetyl-CoA and subsequent histone acetyl-residues in a manner dependent on ATP citrate lyase (ACLY)
While our data showed that lactate prominently labeled the TCA-cycle, we sought to determine whether acetyl-CoA were labeled by lactate as well given that acetyl-CoA plays a fundamental role in post translational histone modification executed by HAT enzymes (Figure 5A–5L). To this purpose, KNS42 and GBM22 cells were exposed to U-13C labeled lactate and harvested for LC/MS analysis focused on isotopologues of acetyl-CoA. We found that the majority of acetyl-CoA was labeled by lactate carbons and that the total levels of acetyl-CoA were elevated as well (Figure 5A–5D). Our expectation was that lactate was metabolized to pyruvic acid (LDH reaction) and then within the mitochondrial matrix reacted to acetyl-CoA via oxidative decarboxylation through the pyruvate dehydrogenase reaction, which would yield the m+2 acetyl-CoA isotopologue (Figure 5D). In line with this hypothesis, the m+2 acetyl-CoA isotopologue revealed the strongest labeling pattern derived from lactate carbons (Figure 5D).
Figure 5. Lactate labels acetyl-CoA and acetyl-residues of histone proteins in GBM models.
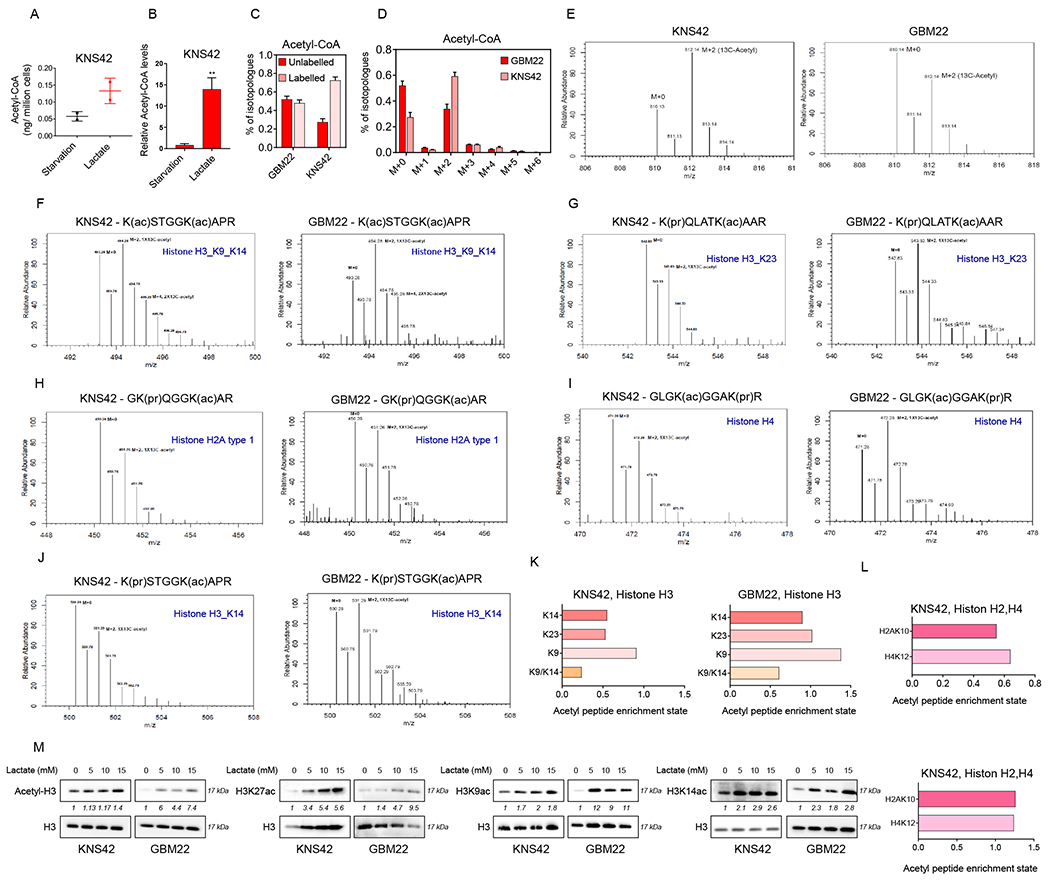
(A, B) KNS42 cells were incubated overnight in starvation media (0.5 mM glucose, 0.5 mM glutamine) or lactate media (0.5 mM glucose, 0.5 mM glutamine, 10 mM lactate) and processed for LC/MS analysis to measure the absolute and relative acetyl-CoA levels.
(C, D) KNS42 and GBM22 cells were incubated overnight in the presence of 10 mM U-13C-labelled lactate and processed for LC/MS analysis. Shown are labeled and unlabeled fractions of Acetyl-CoA.
(E-L) KNS42 and GBM22 cells incubated with 10 mM of U-13C-labelled lactate overnight, the cells were used to extract histones (acidic extraction) to evaluate the incorporation of labeled carbons in histone peptides. Shown are the spectra of different histone peptides and the related acetylation sites which have incorporated the labelled lactate carbons. Shown in (K, L) are the fractions of enriched acetyl-peptides derived from U-13C-lactate carbons for the histone 3 (H3), histone 2A (H2A) and histone 4 (H4) at different lysine.
(M) Standard western blot of GBM cells were incubated with different concentration of lactate for 24 h. H3 is used as a loading control. See also Figure S6.
Next, we determined whether lactate carbons label the acetyl-residues in histone proteins by performing a tracing experiment with uniformly labeled lactate and subsequent acid-based extraction of histone followed by LC/MS analysis. In keeping with our assumptions, we found that lactate carbons significantly labeled histone proteins, including histones H2A, H3 and H4 (Figure 5E–5L). Specifically, activating histone marks, such H3K9 and H3K14, were identified to be labeled by lactate carbons. To further extent the notion that lactate affects histone acetylation at key residues we performed western blotting on lysates from several GBM cell cultures (Figure 5M). We focused mainly on acetylated H3K27, H3K9 and H3K14 given that these histone marks are well-studied and associated with open chromatin. Moreover, the H3K27ac modification highlights active promoters as well as cis-regulatory elements, such as enhancers/super-enhancers. First, we focused on the H3K27ac mark and assessed its levels with increasing concentrations of lactate either in the presence or absence of glucose and glutamine (Figure S6A). We found that irrespective of glucose levels lactate increased the acetylation of H3K27 in a dose dependent manner. Moreover, even the addition of glutamine did not suppress the effects of lactate on H3K27ac. In alignment with these findings, we found that histone acetyltransferases were up-regulated by lactate (Figure S6B).
Lactate modulates the binding of H3K27ac to promoter regions and cis-regulatory elements and affects chromatin accessibility.
To gain a more detailed understanding how lactate regulates the epigenome we performed genome wide chromatin immunoprecipitation sequencing (H3K27ac) with three defined conditions (starvation, lactate and physiological conditions) (Figure 6A–6G; Figure S7A). Following sequencing and mapping to the genome, peaks from the three different conditions were called and annotated, e.g. promoter, exon, intron and intergenic regions (Figure 6A–6D; Figure S6C). First, we narrowed the focus to peaks located around the transcriptional start site and found that lactate facilitated the binding of H3K27ac to the transcriptional start sites in cluster 1 and 2 without obvious changes in the distribution pattern of the peaks (enrichment plot) (Figure 6A; Figures S6C). The three conditions resulted in a different total number of peaks with the highest number identified in the lactate containing conditions (Figure 6A; Figure S6C). Consistently, peak enrichments were detected near individual genes related to oxidative phosphorylation, proliferation, lactate uptake and histone acetyl-transferases (HATs) (Figure S6D–S6I). A Chip-qPCR assay confirmed the increased presence of H3K9ac and H3K27ac at the PDGFRA promoter following lactate exposure, in keeping with the CHIP-seq. results (Figure S6H). Pathway analysis of the lactate related peaks surrounding the transcriptional start sites were especially enriched for genes involved in mitochondrial energy metabolism in keeping with our transcriptional analysis that uncovered the electron transport chain and oxidative phosphorylation as critical pathways to be activated by lactate (Figure 6E). Extending the analysis beyond the transcriptional start sites demonstrated that the distribution pattern of the peaks changed in a manner dependent on the treatment conditions, showing a relative increase in peaks located in exons, introns and intergenic regions in the lactate containing condition as compared to the starvation condition (Figure 6C and 6D). However, taken the absolute values into account the lactate containing conditions revealed the highest number of peaks in each designated category compared to both the physiological as well as the starvation conditions (Figure 6C). Next, we assessed cis-regulatory elements given that such regions have been shown to be central for tumor growth, development and resistance to therapy (Figure 6E–6G). Based on our super-enhancer analysis we found an increase in super-enhancer formation following lactate exposure and pathway analysis, using GREAT, indicated enrichment related to mitochondria in keeping with the earlier findings above (Figure 6F). The remaining open question to answer was whether these epigenetic changes driven by lactate would be associated with an increase in accessibility of the chromatin. To this purpose, we proceeded with ATAC-seq. under starvation conditions in the presence or absence of lactate. As anticipated, we found significant enhanced accessibility in samples exposed to lactate (Figure 6H).
Figure 6. Lactate enhances the accessibility of chromatin and regulates the enhancer landscape.
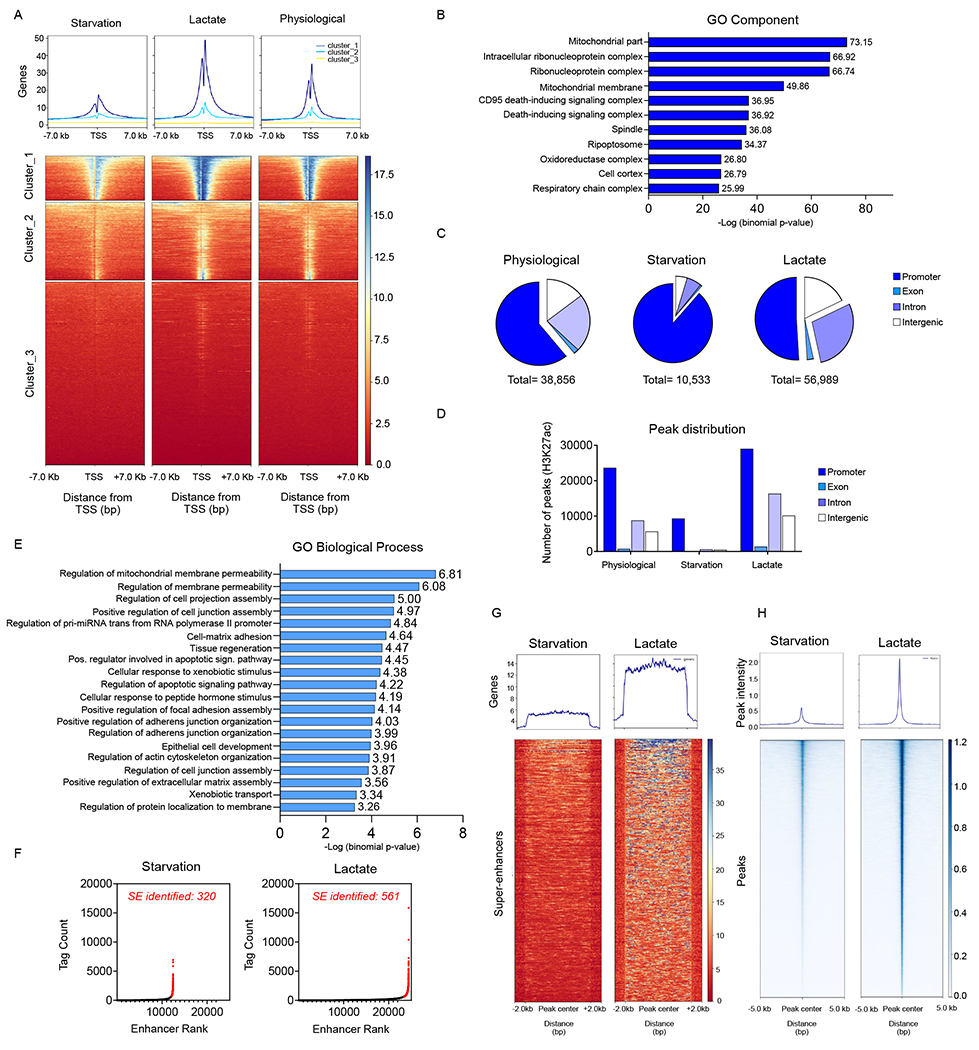
(A) KNS42 were exposed to the media conditions as indicated: starvation media (0.5 mM glucose, 0.5 mM glutamine), lactate media (0.5 mM glucose, 0.5 mM glutamine, 10 mM lactate), physiological media (5 mM glucose, 1mM glutamine) overnight and harvested for ChIP with H3K27ac antibody followed by massive parallel DNA-sequencing (ChIP-seq). The CHIP H3K27ac signal enrichments of normalized reads at the transcriptional start sites as well as the metagene plots are shown (presented as clusters with the highest to lowest peak intensities).
(B) The gene ontology cluster analysis shows enrichment for mitochondrial related genes (cluster 1 from (A)).
(C, D) MACS2 peak calling was performed on the three individual samples in (A), annotated to their localization (promoter, exon, intron or intergenic) and quantified in relative and absolute number.
(E-G) Enhancer and super-enhancer analysis was performed in KNS42 exposed to the starvation and lactate media using a modified ROSE algorithm. GO biological process analysis on the super-enhancer regions is shown in (E). The super-enhancers (SE) are highlighted in a hockey-stick plot is shown in (F). A heat map and a metagene plot of the super-enhancer regions are presented in (G).
(H) ATAC-seq. analysis was performed to compare chromatin accessibility in KNS42 were exposed to the starvation media and lactate media. Shown are the heatmaps for the peak intensity detected. See also Figure S6.
Lactate drives histone-acetylation in a manner dependent on ACLY
To pinpoint the involved pathway how lactate facilitates histone acetylation, we hypothesized that this involves oxidative metabolism and in particular ACLY. In this context, ACLY holds a central role since it supplies cytosolic/nuclear acetyl-CoA to be utilized as a substrate by histone-acetyltransferases (Wellen et al., 2009). Therefore, we took a two-pronged strategy to block the activity of this enzyme, including genetic and a pharmacological approach. We found that BMS303141, an inhibitor of ACLY, reduced the viability of GBM cells in lactate culturing conditions (Figure 7A). Next, we utilized two shRNA constructs to specifically silence ACLY expression in KNS42 and GBM22 cells to study the impact of ACLY on lactate mediated support of cellular viability (Figure 7B and 7C). As expected, lactate mediated rescue from glucose deprivation was mitigated by silencing of ACLY (Figure 7B and 7C). To account for the specificity of these findings, we performed a cDNA rescue experiment, involving the shACLY#3 constructs since this shRNA targets an untranslated region of the ACLY mRNA (Figure 7D and 7E; Figure S7A and S7B). Consistently, this shRNA construct did not affect the expression of the ACLY cDNA in GBM cells. In keeping with the expression lentiviral mediated ectopic expression of ACLY reversed the effect of the ACLY shRNA and almost fully restored the effect of lactate mediated rescue from glucose deprivation (Figure 7E; Figure S7A). Next, we investigated whether ACLY is involved in lactate mediated histone acetylation in GBM cells. To this end, we found that the ATP-citrate lyase inhibitor broadly counteracted lactate mediated acetylation of total acetylated H3, H3K9, and H3K27 in KNS42 and GBM22 cells (Figure 7F). In addition, we assessed histone acetylation following lactate exposure in the context of shRNA-mediated suppression of ACLY. In agreement with the viability data, we found that suppression of ACLY counteracted lactate mediated H3 total histone acetylation as well as the specific lysine residues K9 and K27 (Figure 7G). These observations were independently confirmed by using siRNA (Figure S7C). While cells transfected with non-targeting siRNA revealed a sharp increase in both acetylation of H3K9 and H3K27, this effect was substantially attenuated following silencing of ACLY (Figure S7C). To further enhance the specificity of these findings and to rule out major off-target effects we performed a cDNA rescue in analogy to the viability experiment (Figure 7H). We found that lactate mediated acetylation of histone H3 residues was reduced following silencing of ACLY and that lentiviral mediated restoration of ACLY expression with ACLY silencing restored lactate mediated histone acetylation (Figure 7H). Next, we determined how the substrate of histone acetyltransferases, acetyl-CoA, was affected by silencing of ACLY. We found that both total levels of acetyl-CoA and lactate labeled acetyl-CoA (m+2 isotopologue) were significantly reduced after interference with ACLY expression, in keeping with the histone acetylation results (Figure 7I). These findings position ACLY as a central molecule to mediate lactate mediated histone acetylation and survival following lactate exposure.
Figure 7. ATP-citrate lyase (ACLY) is a key enzyme in lactate metabolism and facilitates histone acetylation in GBM models.
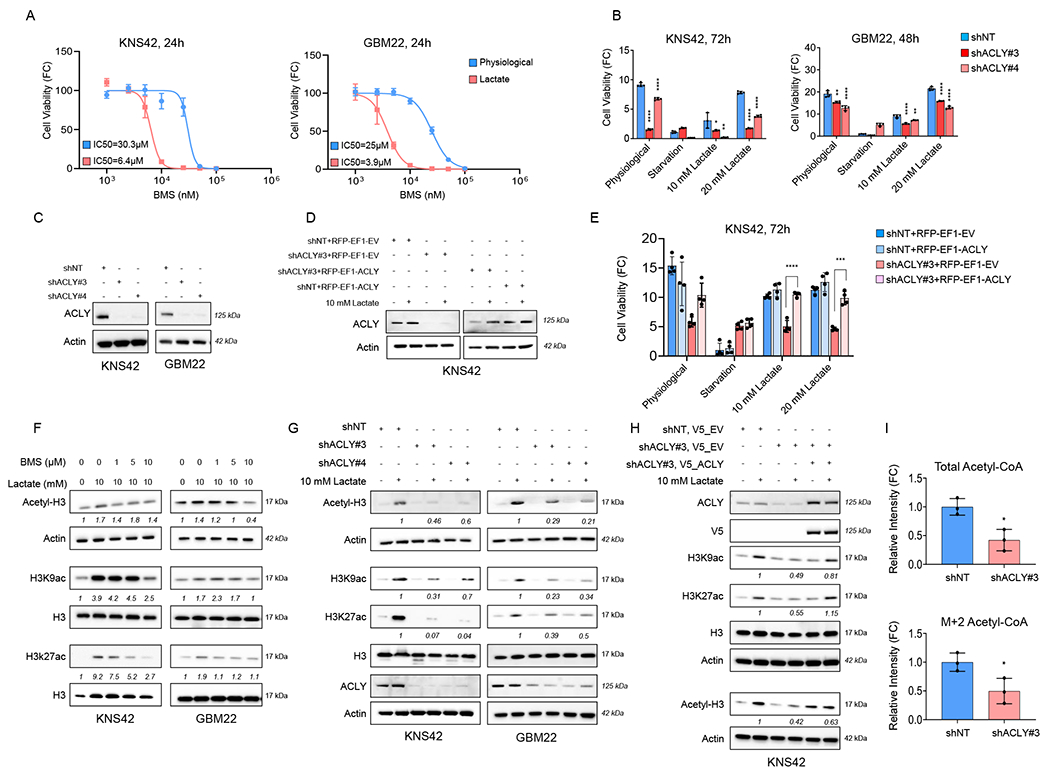
(A) GBM cells were cultured in physiological media (5 mM glucose, 1mM glutamine) and lactate media (0.5 mM glucose, 0.5 mM glutamine, 10 mM lactate) and treated with increasing concentrations of BMS303141 for 24h and cell viability analysis was performed. FC: fold change (n=4).
(B) GBM cells were transduced with non-targeting shNT or ACLY specific shRNAs, exposed to different media conditions: physiological, starvation media, and lactate media, and cell viability analysis was performed (n=4). *the statistical analysis was compared between shNT and shACLY.
(C) Standard western blot of KNS42 and GBM22 cells transduced with non-targeting shNT or ACLY specific shRNA. Actin is used as a loading control.
(D) Standard western blot of stably transduced shNT or shACLY KNS42 cells further infected with empty vector (EV) or ACLY cDNA in the presence or absence of 10 mM lactate.
(E) Stably transduced shNT or shACLY (3’UTR) KNS42 cells were transduced with empty vector (EV) or ACLY cDNA, exposed to different media culture: physiological media, starvation media, and lactate media, and cell viability analysis was performed (n=4).
(F) Standard western blot of KNS42 and GBM22 cells cultured in the presence or absence of 10 mM lactate media and treated with increasing concentrations of BMS303141 for 24h.
(G) Standard western blot of stably transduced shNT or shACLY in KNS42 and GBM22 cells cultured in the presence or absence of 10 mM lactate media.
(H) Stably transduced shNT or shACLY (3’UTR) KNS42 cells were transduced with empty vector (EV) or ACLY cDNA, exposed to different media culture: physiological media, starvation media, and lactate media. Thereafter, the cells were collected for standard western blot with indicated antibodies.
(I) Stably transduced shNT or shACLY in KNS42 cells were incubated with 10 mM U-13C-labelled lactate overnight and processed for LC/MS analysis (n=3).
Statistical significance was assessed by a two-tailed student’s t-test in (E, I) or ANOVA with Dunnett’s multiple comparison test in (B). Data are shown as mean ± SD and ±SEM in (A). *p<0.05, **p<0.01, ***/ ****p<0.001. See also Figure S7.
Next, we further interrogated the required pathways and processes that lead to histone acetylation by lactate. Given the involvement of oxidative metabolism in lactate metabolism, we interfered with cellular respiration and PDHA1 by silencing relevant genes or treatment with pharmacological inhibitors in the presence or absence of lactate (Figure S7D–S7H). In the presence of lactate, we found that IACS reduced histone acetylation and that this effect was partially rescued by ectopic expression of Ndi1 (Figure S7E). Concerning PDHA1, both CPI-613 and shRNA specific silencing of PDHA1 reduced histone acetylation under lactate culturing conditions (Figure S7F and S7G). The specificity of the shRNA experiment was further confirmed through ectopic expression of a shRNA resistant PDHA1 cDNA, which counteracted the effects of the PDHA1 shRNA on histone acetylation, whereas a shRNA resistant PDHA1 S293E mutant was unable to do so (Figure S7H). Overall, these results suggest that functional oxidative metabolism is a perquisite for lactate to act as a modifier of histone acetylation.
DISCUSSION
While interference with genetic changes is currently the foremost strategy to block tumor growth, it has become obvious in the recent years that there is a significant potential to target tumor cell metabolism for therapy (Fu et al., 2015; Ghosh et al., 2015; Sancho et al., 2015). Recent strategies include interference with glycolytic metabolism (Warburg effect) (e.g. by the compound 2-DG), the electron transport chain, glutamine and branched chain amino acid metabolism and fatty acid oxidation and synthesis (Leone et al., 2019). In this vein, a better understanding of energetic fuel requirements are a mean to the identify metabolic dependencies and thus potentially novel therapeutic approaches to treat recalcitrant solid malignancies such as glioblastoma WHO IV (Chowdhry et al., 2019; Field et al., 2020; Kofuji et al., 2019; Leone et al., 2019; Masui et al., 2020; Michealraj et al., 2020; Ngo et al., 2020; Vander Heiden et al., 2009). Undoubtedly, glucose is a central fuel for all tumors, which is either oxidized or converted to lactate (Carrer et al., 2019; Lee et al., 2018). In the present study, we extend these earlier findings and demonstrate a significant role of lactate to be a fuel source of oxidative energy metabolism in model system of glioblastoma. Our findings highlight a significant role of lactate to preserve GBM cell proliferation and survival even in the presence of mutated IDH1 R132H, which was associated to have an impact on cellular respiration before (Fu et al., 2015; Karpel-Massler et al., 2017). It is noteworthy that acetate was not able to rescue GBM cells from nutrient-deprived mediated reduction in viability, further emphasizing the impact of lactate. By preforming carbon tracing experiments in vivo we demonstrated further that in orthotopic GBM PDX models lactate labels substantially the TCA-cycle. Moreover, combined tracer experiments, involving labeled glucose and lactate suggest that at least in some orthotopic GBM PDX models lactate appears to be the most predominant carbon substrate to fuel the TCA-cycle, indicating a pivotal role of lactate in GBM metabolism and suggesting both lactate and more broadly the TCA-cycle as a potential metabolic liability in GBMs. Others have made similar observations in preclinical models of non-small cell lung cancer (Faubert et al., 2017). However, our studies are the first to demonstrate such observations in glioblastoma, which are biologically highly distinct from lung cancer related to several tumor specific properties, especially migratory, metastatic behavior and the microenvironment, in particular related to the oxygen tension. These observations are remarkable for a number of reasons: i) it suggests that oxidative metabolism plays a vital role in glioma growth, ii) it challenges the notion that lactate is a waste product of metabolism given that it actually is substantially utilized in GBM metabolism, iii) it opens up therapeutic opportunities. Regarding the latter, we took advantage of the clinically validated CPI-613 drug compound (Alistar et al., 2017; Pardee et al., 2018; Philip et al., 2019). We found that this compound affected animal survival in PDX models, opening the opportunity for potential additional studies, involving the standard of care.
We noted prominent labeling of acetyl-CoA by lactate carbons and therefore hypothesized that lactate derived acetyl-CoA could serve as substrate for HATs. Remarkably, we found substantial labeling of histone acetyl residues derived from lactate carbons, increased global binding of H3K27ac marks to regions around the transcriptional start sites as well as to cis-regulatory elements (CHIP-seq) and enhanced accessibility of chromatin (ATAC-seq.) (Buenrostro et al., 2013; Buenrostro et al., 2015) coupled with integral changes in gene expression, positioning lactate as a significant modifier of the epigenome and gene expression in model systems of GBM. Interference with this metabolic process at multiple levels counteracts histone acetylation, reverses pro-survival expression changes and ultimately ablates growth of GBM cells. These findings extent earlier results, demonstrating that glucose modifies the epigenome of cancer cells in an ACLY dependent manner to facilitate growth and survival of tumor cells, including GBM model systems. While other acids, such as acetate, have been shown to modulate gene expression and the epigenome, at least, in our model systems tested, acetate was insufficient to reverse cell death precipitated by low glucose levels. Other model systems suggested that acetate enhances ability of T-cells to inhibit tumor formation by regulation of their histone landscape.
In summary, we have identified lactate as a fuel source to drive GBM survival by modulating the epigenome of GBMs and that this novel axis can be therapeutically targeted, involving a promising drug compound currently in clinical trials for other solid and non-solid malignancies.
Limitations of the Study
While our study provides evidence that GBM cells metabolize lactate, which will lead to the modulation of the epigenome, there are several aspects that remain to be determined. It remains to be shown whether lactate would affect lactylation and methylation of histones. Based on research by others it appears likely that a mixture of different post-translational histone modifications could affect gene expression following lactate exposure. In addition, a more rigorous characterization of the requirements with regards to lactate transport over the mitochondrial membrane appears to be necessary, which may involve mitochondrial localization of MCT1 and related chaperones. In addition, it is necessary to investigate potential clinical applications further with regards to the administration of CPI-613 and whether or not it will enhance the efficacy of the standard of care (temozolomide +/− radiation).
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Markus Siegelin (ms4169@cumc.columbia.edu).
Materials availability
Plasmids, cell lines and other unique/stable reagents generated in this study are available from the lead contact without restriction.
Data and code availability
The unprocessed and uncompressed imaging data have been deposited to ‘Mendeley Data and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| MCT1/SLC16A1 | Cell signaling | Cat# 85680 |
| ATP-Citrate Lyase | Cell signaling | Cat# 4332 |
| PCK1 (D12F5) | Cell signaling | Cat# 12940 |
| Acetyl-Histone H3 (Lys9) (C5B11) | Cell signaling | Cat# 9649 |
| Acetyl-Histone H3 (Lys27) | Cell signaling | Cat# 4353 |
| Histone H3 (1B1B2) | Cell signaling | Cat# 14269 |
| PDHA1 | Invitrogen | Cat# 8D10E6 |
| Acetyl-Histone H3 | Sigma | Cat# 06-599 |
| Histone H3K14ac | Active Motif | Cat# 61433 |
| PCB | Abcam | Cat# ab115579 |
| 14-3-3 (C20) | Santa Cruz | Cat# sc-628 |
| V5-Tag (E9H8O) | Cell signaling | Cat# 80076 |
| β-actin | Sigma | Cat# A1978, clone AC15 |
| Flag | Sigma | Cat# F3165 |
| anti-rabbit IgG (H + L) secondary antibody, HRP | Thermo Fisher | Cat# 31460 |
| anti-mouse IgG (H + L) secondary antibody, HRP | Thermo Fisher | Cat# 31430 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| BMS-303141 | MedChemExpress | Cat# HY-16107 |
| Oligomycin A | Selleckchem | Cat# S1478 |
| IACS-010759 | Selleckchem | Cat# S8731 |
| Devimistat (CPI-613) | Selleckchem | Cat# S2776 |
| Pronase | Sigma-Aldrich | Cat# 10165921001 |
| Sodium Acetate | Sigma-Aldrich | Cat# S2889 |
| L-(+)-Lactic acid >98% | Sigma-Aldrich | Cat# L1750 |
| Sodium L-lactate | Sigma-Aldrich | Cat# L7022 |
| D-Glucose (U-13C6, 99%) | Cambridge Isotope Laboratories, Inc | Cat# CLM-1396-2 |
| Sodium L-Lactate (3-13C, 98%) 20% w/w in H2O | Cambridge Isotope Laboratories, Inc | Cat# CLM-1578-PK |
| Sodium L-Lactate (Microbiological/Pyrogen Tested) (13C3, 98%) 20% w/w in H2O | Cambridge Isotope Laboratories, Inc | Cat# CLM-1579-MPT-PK |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| NuPAGE™ 4 to 12%, Bis-Tris, 1.0–1.5 mm, Mini Protein Gels | Thermo Fisher | Cat# NP0321BOX |
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | Cat# G7571 |
| Crystal Violet Assay Kit | Abcam | Cat# ab232855 |
| 16% Formaldehyde (w/v), Methanol-free | Thermo Fisher | Cat# 28908 |
| Lipofectamine RNAiMAX Transfection Reagent | Invitrogen | Cat# 13778075 |
| QuikChange II XL Site-Directed Mutagenesis Kit | Agilent | Cat# 200521 |
| Seahorse XFp Glycolysis Stress Test Kit | Agilent | Cat# 103017-100 |
| Seahorse XFp Cell Mito Stress Test Kit | Agilent | Cat# 103010-100 |
| Annexin V Apoptosis Detection Kit | BD Biosciences | Cat# 556547 |
| Propidium Iodide (PI)/RNase Staining Solution | Cell signaling | Cat# 4087S |
| Click-iT EdU Alexa Flour 488 Flow Cytometry Assay Kit | Thermo Fisher | Cat# C10632 |
| miRNeasy Mini kit | Qiagen | Cat# 217004 |
| First Strand cDNA Synthesis Kit | Origene | Cat# NP100042 |
| PerfeCta® SYBR® Green FastMix | VWR | Cat# 101414-270 |
| SimpleChIP® Enzymatic Chromatin IP Kit | Cell signaling | Cat# 9003 |
| ATAC-Seq Kit | Active Motif | Cat# 53150 |
|
| ||
| Deposited Data | ||
|
| ||
| Microarray data | GEO database | ID: GSE145699 |
| ChIP-seq data | GEO database | ID: GSE151852 |
| ATAC-seq data | GEO database | ID: GSE162831 |
| Raw Data Western Blot | Mendeley | http://dx.doi.org/10.17632/pd854w5cyy.1 |
|
| ||
| Experimental Models: Cell Lines | ||
|
| ||
| KNS42 | Japanese Collection of Research Bioresources Cell Bank | Cat# IFO50356 |
| U87-MG | American Type Culture Collection | Cat# HTB-14 |
| DBRTG-05MG | American Type Culture Collection | Cat# CRL-2020 |
| U251 | Sigma | Cat# CB_09063001 |
| NCH644 | Cell Line Services | Cat# 300124 |
| IDH1(R132H) U87 | American Type Culture Collection | Cat# HTB-14IG |
| U87-EGFRVIII | Ludwig Institute for Cancer Research | Frank Furnari |
| GBM22 cells | Mayo Clinic | Jann Sarkaria |
| GBM43 cells | Mayo Clinic | Jann Sarkaria |
| GBM12 cells | Mayo Clinic | Jann Sarkaria |
| Astrocyte | ScienceCell Research Laboratories | Cat# 1800 |
|
| ||
| Oligonucleotides | ||
|
| ||
| ON-TARGETplus Non-targeting Pool | Dharmacon | Cat# D-001810-10 |
| ON-TARGETplus Human SLC16A1siRNA#6 | Dharmacon | Cat# J-007402-06 |
| ON-TARGETplus Human SLC16A1siRNA#7 | Dharmacon | Cat# J-007402-07 |
| ON-TARGETplus Human SLC16A1siRNA#8 | Dharmacon | Cat# J-070402-08 |
| ON-TARGETplus Human PDHA1 siRNA | Dharmacon | Cat# L-010329-00-0005 |
| ON-TARGETplus Human PC siRNA | Dharmacon | Cat# L008950-00-0005 |
| ON-TARGETplus Human ACLY siRNA#1 | Dharmacon | Cat# J-004915-05 |
| ON-TARGETplus Human ACLY siRNA#2 | Dharmacon | Cat# J-004915-06 |
| ON-TARGETplus Human ACLY siRNA#3 | Dharmacon | Cat# J-004915-07 |
| ON-TARGETplus Human ACLY siRNA#4 | Dharmacon | Cat# J-004915-08 |
| shSLC16A1#35 | Sigma | Cat# TRCN0000038535 |
| shSLC16A1#39 | Sigma | Cat# TRCN0000038339 |
| shSLC16A1#40 | Sigma | Cat# TRCN0000038340 |
| shPDHA1#582 | Sigma | Cat# TRCN0000028582 |
| shPDHA1#590 | Sigma | Cat# TRCN0000028590 |
| shPDHA1#602 | Sigma | Cat# TRCN0000028602 |
| shACLY#3 | Sigma | Cat# TRCN0000078283 |
| shACLY#4 | Sigma | Cat# TRCN0000078284 |
|
| ||
| Recombinant DNA | ||
|
| ||
| pLV-EF1-NDI1-IRES-RFP-WPRE | Northwestern University | Navdeep Chandel |
| pLV-EF1-ORF-IRES-RFP-WPRE | Northwestern University | Navdeep Chandel |
| pCDH-3xFlag | Wake Forest School of Medicine, Winston-Salem, NC | Hui-Kuan Lin |
| pCDH-PDHA-WT-Flag-Blasticidin | Wake Forest School of Medicine, Winston-Salem, NC | Hui-Kuan Lin |
| pLX304 | Genecopoeia | EX-NEG-LX304 |
| pLX304_ACLY_V5 | Genecopoeia | EX-OL00009-LX304 |
|
| ||
| Software and Algorithms | ||
|
| ||
| Galaxy software | https://usegalaxy.org/ | N/A |
| Integrated Genome Browser 9.1.8 | BioViz.org. | N/A |
| GraphPad Prism 9 | GraphPad | N/A |
| MataboAnalysis 5.0 | https://www.metaboanalyst.ca/home.xhtml | N/A |
| Basepair | Basepair Tech | N/A |
| CompuSyn | ComboSyn, Inc. | N/A |
| FlowJo software (version 10.8.1) | BD | N/A |
| Azure (C300) imaging system | Azure Biosystems | N/A |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell cultures and growth conditions
KNS42 cells were obtained from the Japanese Collection of Research Bioresources Cell Bank (JCRB, IFO50356). U87MG, DBTRG05MG and U251 human glioblastoma cell lines were purchase from the American Type Culture Collection (Manassas, VA). NCH644 stem cell-like glioma cells were obtained from Cell Line Services (CLS, Heidelberg, Germany). U87-EGFRvIII cells were kindly provided by Dr. Frank Furnari (Ludwig Institute for Cancer Research, La Jolla, CA). Patient-derived glioblastoma cell lines, GBM22, GBM43, GBM12 were obtained from Dr. Jann Sarkaria (Mayo Clinic, Rochester, MN) 2018 and 2021. GBM12 is a patient-derived xenograft line derived from a male patient (glioblastoma WHO grade IV, IDH-wildtype, mesenchymal molecular subtype, EGFR amplified, PTEN wt, TP53 mutated (splice mutation), MYC non-amplified). GBM22 is a patient-derived xenograft line derived from a male patient (Gliosarcoma WHO grade IV, IDH-wildtype, classical molecular subtype, IDH1 wt, EGFR gain, PTEN wt, TP53 mutated, MYC amplified). GBM43 is a patient-derived xenograft line derived from a male patient (Glioblastoma WHO grade IV, IDH-wildtype, classical molecular subtype, IDH wt, EGFR gain, PTEN wildtype, TP53 mutated, MYC non-amplified). The GBM cell lines were cultured in Dulbecco’s modified Eagle’s medium (Fisher Scientific, MT10013CV), supplemented with 10% fetal bovine serum (Gemini) and 100 μg/ml Primocin (Invivogen, ant-pm-1) and were maintained at 37°C in an atmosphere containing 5% CO2.
METHOD DETAILS
Cell viability
Cell viability was performed in a 96-well plate with density 3,000 cells per well, incubated with different media conditions for 24h or 72h, and was analyzed with CellTiter-Glo® Luminescent Cell Viability Assay (Promega, G7571) or Crystal Violet Assay Kit (Abcam, ab232855) according to the manufacturer’s instruction. For the crystal violet staining, cells were fixed in 3.7% PFA (Thermo Fisher, 28908) and staining with 0.05% crystal violet for 30min followed by three washes in water. The stained-wells were dried for few minutes and solubilized with 50% methanol. The plate was analyzed in a plate reader at 545 OD.
Small interference RNA transfection
GBM cells were seeded with density 30,000 cells per well in a 12 well plate for 24h prior to the transfection. Lipofectamine RNAiMAX (Invitrogen, 13778075) was used according to manufacturers’ instructions. siRNAs were purchased from Dharmacon: siNT (D-001810-10), siSLC16A1#6 (J-007402-06), siSLC16A1#7(J-007402-07), siSLC16A1#8 (J-070402-08), siPDHA1 (L-010329-00-0005), siPC (L008950-00-0005), siACLY#1 (J-004915-05 ), siACLY#2 (J-004915-06 ), siACLY#3 (J-004915-07), siACLY#4 (J-004915-08).
Plasmid constructs
Lentivirus production was performed by transfection of pMD2.G, psPAX2 and the relevant lentivirus transfer plasmid into 293T cells. The viral supernatant was collected over 72h. The viral particles were filtered and concentrated before they were used for transduction of target GBM cells. For knock down experiments, cells were infected in the presence of 8 μg/mL polybrene (Santa Cruz Biotechnology, sc-134220) and were selected with puromycin (Santa Cruz Biotechnology, sc-108071) to obtain stable cell lines expressing the desired constructs. Plasmid constructs were purchased from Sigma: shSLC16A1#35 (TRCN0000038535), shSLC16A1#39 (TRCN0000038339), shSLC16A1#40 (TRCN0000038340), shPDHA1#582 (TRCN0000028582), shPDHA1#590 (TRCN0000028590), shPDHA1#602 (TRCN0000028602), shACLY#3 (TRCN0000078283), shACLY#4 (TRCN0000078284). shScramble was purchased from Addgene (Cat#1864). pLV-EF1-NDI1-IRES-RFP-WPRE (Saccaromyces.Cerevisiae) and pLV-EF1-ORF-IRES-RFP-WPRE (empty vector) (Saccaromyces Cerevisiae) were kindly provided by Dr. Chandel (Northwestern University, Chicago, IL) (Wheaton et al., 2014). pCDH-3xFlag (empty vector) and pCDH-PDHA-WT-Flag-Blasticidin were provided by Dr. Lin (Wake Forest School of Medicine, Winston-Salem, NC) (Cai et al., 2020). pLX304 control (EX-NEG-LX304) and pLX304_ACLY_V5 (EX-OL00009-LX304) were purchased from Genecopoeia (Rockville, MD).
Site directed mutagenesis
Mutated PDHA constructs was generated by using QuikChange II XL Site-Directed Mutagenesis Kit (Agilent, 200521) according to the manual guidelines. Primer sequences are in the Table S2.
Seahorse mito stress assay
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were evaluated using the 8-well Seahorse XFp Extracellular Flux Analyzers (Agilent Inc.) according to the manufacturer instructions. Briefly, GBM cell lines were seeded at 80-90% density for 24h prior the assay. Cells were starved for 30 minutes. Thereafter, cells were either left in Seahorse XF basic medium without glucose and glutamine in the presence or absence of 10 mM lactate (0-0-10mM) or in the Seahorse XF basic medium supplemented with 5mM glucose (5-0-0). The assay medium was adjusted to the final pH of 7 prior to incubate GBM cells for 1h in a CO2free incubator. Then, cells were exposed to extracellular flux analysis on the Seahorse XFp instrument to perform a mitochondrial stress assay and assess the different oxygen consumption rate (OCR). The following compounds were injected in a sequential order: 2.5 μM oligomycin, 2 μM FCCP, and 0.5 μM rotenone/antimycin. The results are automatically generated in the Seahorse XF Mito Stress Test Report.
Flow cytometry
Apoptosis evaluation was assessed by Annexin V/ propidium iodide staining using the Annexin V Apoptosis Detection Kit (BD Biosciences, 556547) according to the manufacturer’s instruction. For PI staining, cells were collected in 200 μL ice-cold PBS in flow cytometry tubes and were fixed with 800μl ice-cold 85% ethanol overnight at 4°C. Cells were centrifuged at 1400 rpm, the supernatant was removed and the cells were stained with 200 μL PI/RNase staining solution (Cell Signaling Technology, 4087S) prior to the flow cytometric analysis (LSRII). For the EdU/ PI staining, the Click-iT EdU Alexa Flour 488 Flow Cytometry Assay kit (Thermo Fisher Scientific, C10632) was used. The EdU dye was added to the culture media 1h prior to collect the cells according to the manufacturer’s guidelines. Data were analyzed using FlowJo software (version 10.8.1; Tree Star, Ashland, OR).
Western blot analysis
Whole cell lysates were collected using 1X Laemmli Buffer and denatured for 5 min at 95°C. Protein were loaded on gradient precast gels purchased by Invitrogen (NP0321BOX). Western blot apparatus system from Invitrogen was utilized. Primary antibody used: MCT1/SLC16A1 (CST, 85680), ATP-Citrate Lyase (CST, 4332), PCK1 (D12F5) (CST, 12940), H3K9ac (CST, 9649), H3K27ac (CST, 4353), H3 (CST, 14269), PDHA1 (Invitrogen, 8D10E6), Acetyl-H3 (Sigma-Aldrich, 06-599), H3K14ac (Active Motif, 61433), PCB (Abcam, ab115579), 14-3-3 (C20) (Santa Cruz, sc-628), V5-Tag (E9H8O) (CST, 80076), β-actin (Sigma-Aldrich A1978, clone AC15), and Flag (Sigma-Aldrich, F3165). The secondary antibodies were used: anti-rabbit IgG (H + L) secondary antibody, HRP (Thermo Fisher 31460; 1: 3000) and anti-mouse IgG (H + L) secondary antibody, HRP (Thermo Fisher 31430; 1: 3000). The western blots were acquired by using the Azure (C300) imaging system (Azure Biosystems).
Real-time PCR analysis
RNA was extracted with the miRNAeasy Mini Kit (QIAGEN, 217004). 500ng of total RNA was reverse transcribed using cDNA synthesis kit (Origene). Gene expression was evaluated by qPCR using SYBR green RT-PCR kit (Applied Biosystems) and the reaction was run at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s, 60°C for 30 s, and 72°C for 30 s, on a qPCR Instrument (Quantabio). Primer sequences are in the Table S2.
Microarray and gene set enrichment analysis
Transcriptomic analysis and gene set enrichment analysis (GSEA) was performed as previously described (Nguyen et al., 2020) and was deposited in GEO (GSE145699).
Liquid chromatography and mass spectrometry (LC/MS) and isotope tracing
Histones were purified according to the acid extraction protocol (Sidoli et al., 2016). KNS42 cells were cultured in 15 cm dishes and nuclei were harvested on ice-cold NIB buffer (15 mM Tris-HCL pH7.5, 15 mM NaCl, 250 mM sucrose 5 mM MgCl2, 1 mM CaCl2, 60 mM KCl and 1mM DTT, 10 mM sodium butyrate, 1X protease inhibitors, 0.1% NP-40), followed by a 10 min incubation on ice with vortexing every 3 min. Nuclei were pelleted at 700xg for 5 min at 4 °C and washed with NIB buffer without NP-40. The nuclear pellets were then resuspended in 0.2M H2SO4 and incubated for 4 h at 4°C. After centrifugation, histones were precipitated from the supernatant by addition of 20% TCA for 1h. The pellet was washed with acetone containing 0.1% HCl, and then with 100% acetone. Histones were dried at RT and resuspended in distilled water and analyzed by LC/MS. For the in vitro stable isotope tracing experiments, GBM cells were grown in DMEM high glucose, 10% FBS and primocin media condition to 80-90% confluence. Then, they were washed with PBS before a 24h incubation with a media consisting of DMEM without glucose, glutamine, and phenol red in the presence of 10% dialyzed FBS (Thermo Fisher) and 10mM solution of [U-13C] sodium L-lactate (Cambridge Isotope Laboratories, Inc; CLM-29731). Thereafter, polar metabolites were extracted using 80% ice-cold HPLC grade methanol and the supernatant was processed for analysis by LC/MS.
Chromatin immunoprecipitation (CHIP) sequencing and ATAC-sequencing
ChIP assays were performed according to the manual instructions (SimpleChIP® Enzymatic Chromatin IP Kit, CST, 9003). Each immunoprecipitation required 4x106 GBM cells, which were cross-linked using 1% formaldehyde in culture medium for 10min at RT and quenched with glycine for 5 min. Chromatin Immunoprecipitations were performed using H3K27ac antibody (CST, 4535), H3K9ac antibody (CST, 9649), Rabbit IgG antibody (CST, 2729). DNA was eluted, purified and analyzed by RT-PCR (QuantaBio). CHIP H3K27ac or H3K9ac samples were analyzed by next generation sequencing (Illumina HiSeQ instrument; HiSeq 4000; single read 50 bp (SR50)). ChIP-seq data were deposited in GEO (GSE151852).
For the ATAC-seq, cells were grown in different media conditions with or without L-lactate for 24h. Thereafter, cells were harvested and subjected to ATAC-seq kit according to the manual instructions (Active Motif; 53150). Purified tagmented DNA was processed for PCR amplification to create a library for the subsequent Next Generation Sequencing of ATAC samples. ATACseq data were deposited in GEO (GSE162831).
Orthotopic glioblastoma models
500,000 U87-EGFRVIII, GBM12 or GBM22 PDX GBM cells were prepared in PBS for stereotactic injection into 6–8 weeks old Nu/Nu mice as previously described (Nguyen et al., 2020). A Hamilton syringe was introduced into a burr hole 3 mm lateral, 1 mm anterior of the bregma and 3 mm down into the striatum using a stereotactic device. Tumor cells were then stereotactically injected at a rate of 1 μL/min. For monitoring the tumor growth, MRI imaging was performed using a Bruker BioSpec™, 9.4 Tesla imaging device. Mice were injected intraperitoneally four times a week with CPI613 or vehicle at the dosage of 50mg/kg. For intraperitoneal application CPI-613 (APExBIO) was dissolved in 10% DMSO, 32% Kolliphor (Kolliphor® EL- Sigma Aldrich) 8% Ethanol (Pharmco-Aaper, Brookfield, CT) and 50% PBS. Animals were monitored for tumor growth-related symptoms, such as neurological deficits, weight loss or death of the animal, which was considered as the primary endpoint. All procedures were conducted in accordance with Animal Welfare Regulations and were approved by the Institutional Animal Care and Use Committee (IACUC) at the Columbia University Medical Center (AC-AABC6505, AC-AAAV7451, and AC-AABI3633).
Liquid chromatography and mass spectrometry (LC/MS) extraction from tumors
A 20% (w/w) solution of [U-13C] sodium L-lactate in H2O was purchased from CIL (Cambridge Isotope Laboratories, Inc; CLM-29731). The labeled-lactate solution was injected intraperitoneal into 6–8 weeks old Nu/Nu at doses 2 g/kg which were sacrificed 30 min after the i.p. injection. Tumor tissue and the other organs were rapidly harvested and weighted on an analytic balance. 10-20 mg of brain tumor or other organs were placed in tubes containing 80% ice-cold HPLC grade methanol and dissociated. The homogenized tissue was centrifuged at 14000g for 10min at 4°C and the supernatant was processed for polar metabolite analysis by LC/MS.
Mass spectrometry imaging (MSI)
Formalin fixed, paraffin embedded mouse brain sections were deparaffinized by submerging in xylene for 3 min and allowed to dry. A digital image of the slide was acquired using an Epson V600 flatbed document scanner for alignment and region-of-interest definition in the mass spectrometer. MALDI matrix (1,5-diaminonapthalene [DAN]) was prepared at a concentration of 10 mg/mL in 50% acetonitrile. Matrix was deposited as a uniform coating using and HTX M5 Robotic Reagent Sprayer (HTX Technologies, Chapel Hill, NC). Image acquisition was performed using FlexImaging 5.1 in negative ion mode using a Bruker timsTOF fleX MALDI QTOF mass spectrometer (Bruker Daltonics, Billerica, MA). Digital microscopy images were acquired of the sections at 20X magnification using a Hamamatsu NanoZoomerSQ Slide Scanner (Hamamatsu Corporation, Bridgewater, NJ). Images were viewed and annotated for tumor regions using NDP.view2 (Hamamatsu). Annotated images were merged with the images of the sections used for data collection using GIMP (www.gimp.org) and the annotated regions of interest extracted for data analysis. Metabolite identification was performed by loading the image data into MetaboScape 2021b (Bruker) and searching against the HMDB database.
Quantification and statistical analysis
Statistical significance was assessed by two-tailed student’s t-test or ANOVA (for multiple comparisons) using Prism version 8 (GraphPad, La Jolla, CA). A p ≤ 0.05 was considered statistically significant.
Supplementary Material
Highlights.
Lactate is metabolized in glioblastoma, which is dependent on cellular respiration.
Lactate provides carbons to acetyl-residues of histones.
Lactate modifies the enhancer and super-enhancer landscape of GBM cells.
ACKNOWLEDGEMENTS
M.D. Siegelin: NIH NINDS R01NS095848, R01NS102366, R01NS113793; Louis V. Gerstner, Jr. Scholars Program (2017-2020), Schaefer Research Scholars Program Awards (2020), American Brain Tumor Association Discovery Grant 2017 (DG1700013) and American Brain Tumor Association Discovery Grant Supported by Sontag Foundation 2021 (DG2100041). Trang T.T. Nguyen: American Brain Tumor Association Basic Research Fellowship in Memory of Katie Monson (BRF1900018). Transcriptome analysis was supported by the CTSA grant UL1-TR001430 to the Boston University Microarray and Sequencing Resource Core Facility. These studies used the resources of the Cancer Center Flow Core Facility funded in part through center grant P30CA013696 and S10RR027050 and CPRIT (Cancer Prevention and Research Institute of Texas) Award RP190617. The graphical abstract was created with BioRender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alistar A, Morris BB, Desnoyer R, Klepin HD, Hosseinzadeh K, Clark C, Cameron A, Leyendecker J, D’Agostino R Jr., Topaloglu U, et al. (2017). Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: a single-centre, open-label, dose-escalation, phase 1 trial. Lancet Oncol 18, 770–778. 10.1016/S1470-2045(17)30314-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218. 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109, 21 29 21–21 29 29. 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess EA, and Sylven B (1962). Glucose, lactate, and lactic dehydrogenase activity in normal interstitial fluid and that of solid mouse tumors. Cancer Res 22, 581–588. [PubMed] [Google Scholar]
- Cai Z, Li CF, Han F, Liu C, Zhang A, Hsu CC, Peng D, Zhang X, Jin G, Rezaeian AH, et al. (2020). Phosphorylation of PDHA by AMPK Drives TCA Cycle to Promote Cancer Metastasis. Mol Cell 80, 263–278 e267. 10.1016/j.molcel.2020.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N., Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, Rheinbay E, Miller CR, Vitucci M, et al. (2015). Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N Engl J Med 372, 2481–2498. 10.1056/NEJMoa1402121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrer A, Trefely S, Zhao S, Campbell SL, Norgard RJ, Schultz KC, Sidoli S, Parris JLD, Affronti HC, Sivanand S, et al. (2019). Acetyl-CoA Metabolism Supports Multistep Pancreatic Tumorigenesis. Cancer Discov 9, 416–435. 10.1158/2159-8290.CD-18-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhry S, Zanca C, Rajkumar U, Koga T, Diao Y, Raviram R, Liu F, Turner K, Yang H, Brunk E, et al. (2019). NAD metabolic dependency in cancer is shaped by gene amplification and enhancer remodelling. Nature 569, 570–575. 10.1038/s41586-019-1150-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmanis S, Sloan SA, Croote D, Mignardi M, Chernikova S, Samghababi P, Zhang Y, Neff N, Kowarsky M, Caneda C, et al. (2017). Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep 21, 1399–1410. 10.1016/j.celrep.2017.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, and Chandel NS (2020). We need to talk about the Warburg effect. Nat Metab 2, 127–129. 10.1038/s42255-020-0172-2. [DOI] [PubMed] [Google Scholar]
- Dolgodilina E, Imobersteg S, Laczko E, Welt T, Verrey F, and Makrides V (2016). Brain interstitial fluid glutamine homeostasis is controlled by blood-brain barrier SLC7A5/LAT1 amino acid transporter. J Cereb Blood Flow Metab 36, 1929–1941. 10.1177/0271678X15609331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D, et al. (2017). Lactate Metabolism in Human Lung Tumors. Cell 171, 358–371 e359. 10.1016/j.cell.2017.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field CS, Baixauli F, Kyle RL, Puleston DJ, Cameron AM, Sanin DE, Hippen KL, Loschi M, Thangavelu G, Corrado M, et al. (2020). Mitochondrial Integrity Regulated by Lipid Metabolism Is a Cell-Intrinsic Checkpoint for Treg Suppressive Function. Cell Metab 31, 422–437 e425. 10.1016/j.cmet.2019.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim Y, Sloan AE, Weil RJ, Nakano I, Sarkaria JN, Stringer BW, et al. (2013). Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat Neurosci 16, 1373–1382. 10.1038/nn.3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Chin RM, Vergnes L, Hwang H, Deng G, Xing Y, Pai MY, Li S, Ta L, Fazlollahi F, et al. (2015). 2-Hydroxyglutarate Inhibits ATP Synthase and mTOR Signaling. Cell Metab 22, 508–515. 10.1016/j.cmet.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh JC, Siegelin MD, Vaira V, Faversani A, Tavecchio M, Chae YC, Lisanti S, Rampini P, Giroda M, Caino MC, et al. (2015). Adaptive mitochondrial reprogramming and resistance to PI3K therapy. J Natl Cancer Inst 107. 10.1093/jnci/dju502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, Esparza LA, Reya T, Le Z, Yanxiang Guo J, et al. (2017). Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118. 10.1038/nature24057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampa JM, Kellner U, Marsching C, Ramallo Guevara C, Knappe UJ, Sahin M, Giampa M, Niehaus K, and Bednarz H (2020). Glioblastoma multiforme: Metabolic differences to peritumoral tissue and IDH-mutated gliomas revealed by mass spectrometry imaging. Neuropathology. 10.1111/neup.12671. [DOI] [PubMed] [Google Scholar]
- Karpel-Massler G, Ishida CT, Bianchetti E, Zhang Y, Shu C, Tsujiuchi T, Banu MA, Garcia F, Roth KA, Bruce JN, et al. (2017). Induction of synthetic lethality in IDH1-mutated gliomas through inhibition of Bcl-xL. Nat Commun 8, 1067. 10.1038/s41467-017-00984-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuji S, Hirayama A, Eberhardt AO, Kawaguchi R, Sugiura Y, Sampetrean O, Ikeda Y, Warren M, Sakamoto N, Kitahara S, et al. (2019). IMP dehydrogenase-2 drives aberrant nucleolar activity and promotes tumorigenesis in glioblastoma. Nat Cell Biol 21, 1003–1014. 10.1038/s41556-019-0363-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JV, Berry CT, Kim K, Sen P, Kim T, Carrer A, Trefely S, Zhao S, Fernandez S, Barney LE, et al. (2018). Acetyl-CoA promotes glioblastoma cell adhesion and migration through Ca(2+)-NFAT signaling. Genes Dev 32, 497–511. 10.1101/gad.311027.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone RD, Zhao L, Englert JM, Sun IM, Oh MH, Sun IH, Arwood ML, Bettencourt IA, Patel CH, Wen J, et al. (2019). Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 366, 1013–1021. 10.1126/science.aav2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Cooper DE, Cluntun AA, Warmoes MO, Zhao S, Reid MA, Liu J, Lund PJ, Lopes M, Garcia BA, et al. (2018). Acetate Production from Glucose and Coupling to Mitochondrial Metabolism in Mammals. Cell 175, 502–513 e513. 10.1016/j.cell.2018.08.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, Nannepaga S, Piccirillo SG, Kovacs Z, Foong C, et al. (2014). Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 159, 1603–1614. 10.1016/j.cell.2014.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masui K, Harachi M, Cavenee WK, Mischel PS, and Shibata N (2020). mTOR complex 2 is an integrator of cancer metabolism and epigenetics. Cancer Lett 478, 1–7. 10.1016/j.canlet.2020.03.001. [DOI] [PubMed] [Google Scholar]
- Michealraj KA, Kumar SA, Kim LJY, Cavalli FMG, Przelicki D, Wojcik JB, Delaidelli A, Bajic A, Saulnier O, MacLeod G, et al. (2020). Metabolic Regulation of the Epigenome Drives Lethal Infantile Ependymoma. Cell. 10.1016/j.cell.2020.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, McAfoos T, Morlacchi P, Ackroyd J, Agip AA, et al. (2018). An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med 24, 1036–1046. 10.1038/s41591-018-0052-4. [DOI] [PubMed] [Google Scholar]
- Ngo B, Kim E, Osorio-Vasquez V, Doll S, Bustraan S, Liang RJ, Luengo A, Davidson SM, Ali A, Ferraro GB, et al. (2020). Limited Environmental Serine and Glycine Confer Brain Metastasis Sensitivity to PHGDH Inhibition. Cancer Discov. 10.1158/2159-8290.CD-19-1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TTT, Zhang Y, Shang E, Shu C, Torrini C, Zhao J, Bianchetti E, Mela A, Humala N, Mahajan A, et al. (2020). HDAC inhibitors elicit metabolic reprogramming by targeting super-enhancers in glioblastoma models. J Clin Invest 130, 3699–3716. 10.1172/JCI129049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardee TS, Anderson RG, Pladna KM, Isom S, Ghiraldeli LP, Miller LD, Chou JW, Jin G, Zhang W, Ellis LR, et al. (2018). A Phase I Study of CPI-613 in Combination with High-Dose Cytarabine and Mitoxantrone for Relapsed or Refractory Acute Myeloid Leukemia. Clin Cancer Res 24, 2060–2073. 10.1158/1078-0432.CCR-17-2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. (2008). An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812. 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip PA, Buyse ME, Alistar AT, Rocha Lima CM, Luther S, Pardee TS, and Van Cutsem E (2019). A Phase III open-label trial to evaluate efficacy and safety of CPI-613 plus modified FOLFIRINOX (mFFX) versus FOLFIRINOX (FFX) in patients with metastatic adenocarcinoma of the pancreas. Future Oncol 15, 3189–3196. 10.2217/fon-2019-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall EC, Lopez BGC, Peng S, Regan MS, Abdelmoula WM, Basu SS, Santagata S, Yoon H, Haigis MC, Agar JN, et al. (2020). Localized Metabolomic Gradients in Patient-Derived Xenograft Models of Glioblastoma. Cancer Res 80, 1258–1267. 10.1158/0008-5472.CAN-19-0638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, Barneda D, Sellers K, Campos-Olivas R, Grana O, et al. (2015). MYC/PGC-1alpha Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab 22, 590–605. 10.1016/j.cmet.2015.08.015. [DOI] [PubMed] [Google Scholar]
- Sidoli S, Bhanu NV, Karch KR, Wang X, and Garcia BA (2016). Complete Workflow for Analysis of Histone Post-translational Modifications Using Bottom-up Mass Spectrometry: From Histone Extraction to Data Analysis. J Vis Exp. 10.3791/54112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, and Vander Heiden MG (2015). Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 162, 552–563. 10.1016/j.cell.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trefely S, and Wellen KE (2018). Metabolite regulates differentiation. Science 360, 603–604. 10.1126/science.aat6663. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, and Thompson CB (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, and Thompson CB (2009). ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080. 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM, Budigner GS, and Chandel NS (2014). Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife 3, e02242. 10.7554/eLife.02242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JK, Markowitz S, Zhou S, et al. (2009). Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 325, 1555–1559. 10.1126/science.1174229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachar Z, Marecek J, Maturo C, Gupta S, Stuart SD, Howell K, Schauble A, Lem J, Piramzadian A, Karnik S, et al. (2011). Non-redox-active lipoate derivates disrupt cancer cell mitochondrial metabolism and are potent anticancer agents in vivo. J Mol Med (Berl) 89, 1137–1148. 10.1007/s00109-011-0785-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The unprocessed and uncompressed imaging data have been deposited to ‘Mendeley Data and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| MCT1/SLC16A1 | Cell signaling | Cat# 85680 |
| ATP-Citrate Lyase | Cell signaling | Cat# 4332 |
| PCK1 (D12F5) | Cell signaling | Cat# 12940 |
| Acetyl-Histone H3 (Lys9) (C5B11) | Cell signaling | Cat# 9649 |
| Acetyl-Histone H3 (Lys27) | Cell signaling | Cat# 4353 |
| Histone H3 (1B1B2) | Cell signaling | Cat# 14269 |
| PDHA1 | Invitrogen | Cat# 8D10E6 |
| Acetyl-Histone H3 | Sigma | Cat# 06-599 |
| Histone H3K14ac | Active Motif | Cat# 61433 |
| PCB | Abcam | Cat# ab115579 |
| 14-3-3 (C20) | Santa Cruz | Cat# sc-628 |
| V5-Tag (E9H8O) | Cell signaling | Cat# 80076 |
| β-actin | Sigma | Cat# A1978, clone AC15 |
| Flag | Sigma | Cat# F3165 |
| anti-rabbit IgG (H + L) secondary antibody, HRP | Thermo Fisher | Cat# 31460 |
| anti-mouse IgG (H + L) secondary antibody, HRP | Thermo Fisher | Cat# 31430 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| BMS-303141 | MedChemExpress | Cat# HY-16107 |
| Oligomycin A | Selleckchem | Cat# S1478 |
| IACS-010759 | Selleckchem | Cat# S8731 |
| Devimistat (CPI-613) | Selleckchem | Cat# S2776 |
| Pronase | Sigma-Aldrich | Cat# 10165921001 |
| Sodium Acetate | Sigma-Aldrich | Cat# S2889 |
| L-(+)-Lactic acid >98% | Sigma-Aldrich | Cat# L1750 |
| Sodium L-lactate | Sigma-Aldrich | Cat# L7022 |
| D-Glucose (U-13C6, 99%) | Cambridge Isotope Laboratories, Inc | Cat# CLM-1396-2 |
| Sodium L-Lactate (3-13C, 98%) 20% w/w in H2O | Cambridge Isotope Laboratories, Inc | Cat# CLM-1578-PK |
| Sodium L-Lactate (Microbiological/Pyrogen Tested) (13C3, 98%) 20% w/w in H2O | Cambridge Isotope Laboratories, Inc | Cat# CLM-1579-MPT-PK |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| NuPAGE™ 4 to 12%, Bis-Tris, 1.0–1.5 mm, Mini Protein Gels | Thermo Fisher | Cat# NP0321BOX |
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | Cat# G7571 |
| Crystal Violet Assay Kit | Abcam | Cat# ab232855 |
| 16% Formaldehyde (w/v), Methanol-free | Thermo Fisher | Cat# 28908 |
| Lipofectamine RNAiMAX Transfection Reagent | Invitrogen | Cat# 13778075 |
| QuikChange II XL Site-Directed Mutagenesis Kit | Agilent | Cat# 200521 |
| Seahorse XFp Glycolysis Stress Test Kit | Agilent | Cat# 103017-100 |
| Seahorse XFp Cell Mito Stress Test Kit | Agilent | Cat# 103010-100 |
| Annexin V Apoptosis Detection Kit | BD Biosciences | Cat# 556547 |
| Propidium Iodide (PI)/RNase Staining Solution | Cell signaling | Cat# 4087S |
| Click-iT EdU Alexa Flour 488 Flow Cytometry Assay Kit | Thermo Fisher | Cat# C10632 |
| miRNeasy Mini kit | Qiagen | Cat# 217004 |
| First Strand cDNA Synthesis Kit | Origene | Cat# NP100042 |
| PerfeCta® SYBR® Green FastMix | VWR | Cat# 101414-270 |
| SimpleChIP® Enzymatic Chromatin IP Kit | Cell signaling | Cat# 9003 |
| ATAC-Seq Kit | Active Motif | Cat# 53150 |
|
| ||
| Deposited Data | ||
|
| ||
| Microarray data | GEO database | ID: GSE145699 |
| ChIP-seq data | GEO database | ID: GSE151852 |
| ATAC-seq data | GEO database | ID: GSE162831 |
| Raw Data Western Blot | Mendeley | http://dx.doi.org/10.17632/pd854w5cyy.1 |
|
| ||
| Experimental Models: Cell Lines | ||
|
| ||
| KNS42 | Japanese Collection of Research Bioresources Cell Bank | Cat# IFO50356 |
| U87-MG | American Type Culture Collection | Cat# HTB-14 |
| DBRTG-05MG | American Type Culture Collection | Cat# CRL-2020 |
| U251 | Sigma | Cat# CB_09063001 |
| NCH644 | Cell Line Services | Cat# 300124 |
| IDH1(R132H) U87 | American Type Culture Collection | Cat# HTB-14IG |
| U87-EGFRVIII | Ludwig Institute for Cancer Research | Frank Furnari |
| GBM22 cells | Mayo Clinic | Jann Sarkaria |
| GBM43 cells | Mayo Clinic | Jann Sarkaria |
| GBM12 cells | Mayo Clinic | Jann Sarkaria |
| Astrocyte | ScienceCell Research Laboratories | Cat# 1800 |
|
| ||
| Oligonucleotides | ||
|
| ||
| ON-TARGETplus Non-targeting Pool | Dharmacon | Cat# D-001810-10 |
| ON-TARGETplus Human SLC16A1siRNA#6 | Dharmacon | Cat# J-007402-06 |
| ON-TARGETplus Human SLC16A1siRNA#7 | Dharmacon | Cat# J-007402-07 |
| ON-TARGETplus Human SLC16A1siRNA#8 | Dharmacon | Cat# J-070402-08 |
| ON-TARGETplus Human PDHA1 siRNA | Dharmacon | Cat# L-010329-00-0005 |
| ON-TARGETplus Human PC siRNA | Dharmacon | Cat# L008950-00-0005 |
| ON-TARGETplus Human ACLY siRNA#1 | Dharmacon | Cat# J-004915-05 |
| ON-TARGETplus Human ACLY siRNA#2 | Dharmacon | Cat# J-004915-06 |
| ON-TARGETplus Human ACLY siRNA#3 | Dharmacon | Cat# J-004915-07 |
| ON-TARGETplus Human ACLY siRNA#4 | Dharmacon | Cat# J-004915-08 |
| shSLC16A1#35 | Sigma | Cat# TRCN0000038535 |
| shSLC16A1#39 | Sigma | Cat# TRCN0000038339 |
| shSLC16A1#40 | Sigma | Cat# TRCN0000038340 |
| shPDHA1#582 | Sigma | Cat# TRCN0000028582 |
| shPDHA1#590 | Sigma | Cat# TRCN0000028590 |
| shPDHA1#602 | Sigma | Cat# TRCN0000028602 |
| shACLY#3 | Sigma | Cat# TRCN0000078283 |
| shACLY#4 | Sigma | Cat# TRCN0000078284 |
|
| ||
| Recombinant DNA | ||
|
| ||
| pLV-EF1-NDI1-IRES-RFP-WPRE | Northwestern University | Navdeep Chandel |
| pLV-EF1-ORF-IRES-RFP-WPRE | Northwestern University | Navdeep Chandel |
| pCDH-3xFlag | Wake Forest School of Medicine, Winston-Salem, NC | Hui-Kuan Lin |
| pCDH-PDHA-WT-Flag-Blasticidin | Wake Forest School of Medicine, Winston-Salem, NC | Hui-Kuan Lin |
| pLX304 | Genecopoeia | EX-NEG-LX304 |
| pLX304_ACLY_V5 | Genecopoeia | EX-OL00009-LX304 |
|
| ||
| Software and Algorithms | ||
|
| ||
| Galaxy software | https://usegalaxy.org/ | N/A |
| Integrated Genome Browser 9.1.8 | BioViz.org. | N/A |
| GraphPad Prism 9 | GraphPad | N/A |
| MataboAnalysis 5.0 | https://www.metaboanalyst.ca/home.xhtml | N/A |
| Basepair | Basepair Tech | N/A |
| CompuSyn | ComboSyn, Inc. | N/A |
| FlowJo software (version 10.8.1) | BD | N/A |
| Azure (C300) imaging system | Azure Biosystems | N/A |