

Abstract
Objective:
Primary Age-Related Tauopathy (PART) refers to tau neurofibrillary tangles restricted largely to the medial temporal lobe in the absence of significant beta-amyloid plaques. PART has been associated with cognitive impairment, but contributions from concomitant Limbic Age-Related TDP-43 Encephalopathy neuropathologic change (LATE-NC) are underappreciated.
Methods:
We compare prevalence of LATE-NC and vascular co-pathologies in age- and Braak-matched patients with PART (n=45, Braak I-IV, Thal phase 0–2) or early-stage Alzheimer’s Disease neuropathologic change (ADNC, n=51, Braak I-IV, Thal 3–5), and examine their influence on clinical and cognitive decline.
Results:
Concomitant LATE-NC and vascular pathology were equally common, and cognition was equally impaired, in PART (MMSE=24.8±6.9) and ADNC (MMSE=24.2±6.0). Patients with LATE-NC were more impaired than those without LATE-NC on the MMSE [by 5.8 points; 95%CI: 3.0–8.6], DRS [17.5 points; 7.1–27.9], CDR-sob [5.2 points; 2.1–8.2], Memory composite [0.8 standard deviations (SDs); 0.1–1.6] and Language composite [1.1 SDs; 0.2–2.0], and more likely to receive a dementia diagnosis [odds ratio 4.8; 1.5–18.0]. Those with vascular pathology performed worse than those without on the DRS [by 10.2 points; 0.1–20.3] and Executive composite [1.3 SDs; 0.3–2.3]. Cognition declined similarly in PART and ADNC over 5 years preceding death; however LATE-NC was associated with more rapid decline on the MMSE (β=1.9 [0.9–3.0]), DRS (β=7.8 [3.4–12.7), CDR-sob (β=1.9 [0.4–3.7]), and Language composite (β=0.5 SD [0.1–0.8]), and vascular pathology with more rapid decline on the DRS (β=5.2 [0.6–10.2]).
Interpretation:
LATE-NC, and to a lesser extent vascular co-pathology, exacerbates cognitive impairment and decline in PART and early-stage ADNC.
Introduction:
Primary Age-related Tauopathy (PART) is the neuropathologic designation for tau neurofibrillary tangles restricted to medial temporal lobe, basal forebrain, olfactory bulb and brainstem in the absence of significant beta-amyloid plaques1. Although a “tangle-only” form of Alzheimer’s disease (AD) has been recognized for decades2–7, a working classification for PART was not proposed until 20141. This current classification requires Braak stage I-IV tau pathology (i.e., transentorhinal to limbic) with no (Thal Phase 0, “definite PART”) or few (Thal Phase 1–2, “possible” PART) amyloid plaques. Several studies suggest that PART, even when restricted to Braak stages I-II, can result in significant cognitive impairment and dementia1,6,8–12; however, this cognitive decline is less severe and slower than in typical late-stage AD (i.e., Braak V-VI, Thal phase 3–5)6,8,10,13. Whether cognitive decline in PART is less severe or similar to decline associated with early-stage AD neuropathologic change (ADNC) (i.e., Braak stage I-IV, Thal phase 3–5) is open to debate8,10.
Individuals with PART are on average older than those with AD1,2,6,14 and may have greater susceptibility to age-related co-pathologies such as Limbic-predominant Aging-related TDP-43 Encephalopathy neuropathologic change (LATE-NC)15–21 or cerebrovascular pathology. Distinct from TDP-43-associated Frontotemporal Lobar Degeneration (FTLD-TDP), LATE-NC is characterized by neuronal nuclear-to-cytoplasmic translocation of TDP-43, with or without hippocampal sclerosis, and occurs largely in advanced age20,22,23. LATE-NC has been associated with cognitive impairment in the presence or absence of significant ADNC20,24,25, but its effects have not been systematically compared in patients with PART versus early-stage ADNC. Cerebrovascular pathology such as infarcts, microinfarcts, and hemorrhages also increase with age and may contribute to cognitive decline, particularly executive dysfunction, in ADNC and PART11,26.
The severity of cognitive impairment often observed in individuals with PART is surprising given the low level and localized nature of the tangle pathology, even considering greater than expected hippocampal CA2 region involvement27 or medial temporal left-right asymmetry28. We therefore hypothesized that other age-related co-pathologies such as LATE-NC or vascular pathology may play a significant role in the cognitive decline of these patients. We also hypothesized that, in the presence of low levels of tau pathology, amyloid may contribute to cognitive decline independently of other co-pathologies. However, few studies have compared profiles of cognitive impairment or rates of cognitive decline in patients with PART or early-stage ADNC who have the same degree of tau pathology (Braak stage I-IV) and differ only in level of amyloid plaque pathology (i.e., Thal phase 0–2 vs. Thal phase 3–5), and none have considered potential differential effects of LATE-NC co-pathology. We address these hypotheses by comparing neuropathological features, clinical and cognitive profiles, and rates of cognitive decline in individuals who were found at autopsy to have PART (Braak stage I-IV, Thal phase 0–2) or early-stage ADNC (Braak stage I-IV, Thal phase 3–5), differing only in their degree of amyloid pathology. The impact of degree of intrinsic tau pathology is considered across both groups. We also examine the prevalence and contributions of LATE-NC and vascular co-pathologies to the clinical and cognitive phenotypes of PART and early-stage ADNC.
Methods:
Standard Protocol Approvals, Registrations, and Patient Consent.
The research protocol was reviewed and approved by the human subjects review board at the University of California, San Diego (UCSD). Informed consent was obtained from all patients or their caregivers consistent with California State law.
Participants.
All individuals autopsied at the UCSD Shiley-Marcos Alzheimer’s Disease Research Center (ADRC) who met criteria for PART (i.e., Braak stage I-IV, low amyloid burden) or early-stage ADNC (i.e., Braak stage I-IV, high amyloid burden) were identified. Cases were initially identified based on Thal phasing29 or CERAD neuritic plaque score30 as a proxy for amyloid burden. Participants were included in these cohorts without regard to LATE-NC or cerebrovascular pathology, but were excluded if they did not have sufficient clinical and cognitive data for analysis or if they had a pathologic diagnosis of FTLD (FTLD-tau or FTLD-TDP43), Lewy Bodies in the brainstem, limbic and neocortical regions, or any other neuropathologic condition that could result in neurologic impairment (e.g. Creutzfeldt-Jakob Disease, Multiple Sclerosis). Lewy Bodies were not systematically assessed in the amygdala, as “amygdala-predominant” Lewy Body Disease is generally only seen in cases with more advanced ADNC than included in this study31, and has not been shown to significantly affect cognition32,33. All cases with PART and an equal number of randomly selected cases with ADNC in the same age and Braak stage range as the PART cases were included in the final sample. Case selection was blind to all other clinical information and presence or degree of LATE-NC or cerebrovascular pathology. During the retrospective pathologic workup, participants were dropped if tissue unavailability precluded full LATE-NC staging or Thal phasing, and several were re-classified from PART to ADNC when retrospective Thal phasing was complete. The final sample, therefore, included 44 PART cases and 52 ADNC cases that were fully staged for all pathologies analyzed and were extremely well matched on age and Braak stage.
Neuropathological Evaluation.
Autopsy was performed using a previously described protocol34. Briefly, the left hemibrain was fixed in 10% buffered formalin for at least 14 days, then 1 cm coronal sections were evaluated grossly and sections taken for paraffin-embedding. Hematoxylin and eosin (H&E) was performed on 5 μm sections of middle frontal cortex (Brodmann areas 8/9), rostral superior temporal cortex, inferior parietal cortex, hippocampus (CA1-CA4 and dentate gyrus), entorhinal cortex, basal ganglia, midbrain with substantia nigra, pons with locus coeruleus, and cerebellar cortex with dentate nucleus.
AD and PART pathology.
Neuritic plaques, diffuse plaques, and neurofibrillary tangles (NFT) were identified on either 10 μm 1% thioflavin-S stained sections using ultraviolet illumination and a 440 μm bandpass excitation filter, or with immunohistochemical staining using antibodies to Aβ (Ab69D/E, polyclonal, Edward Koo, 1:1200) and PHF1 tau (Peter Davies, 1:600). CERAD methods were used to estimate neuritic plaque density30, Thal phases for spread of amyloid pathology29, and Braak stages for NFT pathology35. PART was defined using the preferred published criteria1 as Braak stages I-IV tau pathology with Thal phases 0–2 amyloid. Early stage ADNC was also defined as Braak I-IV tau pathology but with Thal phase 3–5 amyloid.
LATE-NC and Vascular pathology.
TDP-43 pathology was identified by immunohistochemistry for total TDP-43 (Proteintech#10782–2-AP polyclonal, 1:12,000) and staged according to LATE-NC consensus guidelines20 into “amygdala”, “hippocampal”, or “neocortical” stages. Hippocampal sclerosis (HS) was diagnosed independent of TDP-43 pathology by CA1/subiculum neuronal loss out of proportion to hippocampal tau pathology. Vascular pathology assessment included gross and microscopic examination for large and lacunar infarcts, microinfarcts, and gross and microscopic hemorrhages. Arteriolosclerosis, atherosclerosis, and amyloid angiopathy were semi-quantitatively rated as “none”, “mild”, “moderate”, or “severe”.
Clinical and Neuropsychological Evaluation.
Participants had annual standardized clinical, neurological, and neuropsychological evaluations as previously described36,37. Clinical evaluation included review of history with the patient and/or informant, mental status testing, and assessment of functional impairment. Clinical Dementia Rating (CDR) score and sum of its six subdomain scores (i.e., CDR sum of boxes) were computed. Neuropsychological assessment included tests for Global Cognition (Mini-Mental State Exam (MMSE), Mattis Dementia Rating Scale (DRS)); Memory (Visual Reproduction Test, Logical Memory Test, California Verbal Learning Test (CVLT), CERAD Word List); Language (30-item Boston Naming Test, Letter Fluency Test (F-A-S),Category Fluency Test); Executive functions (modified Wisconsin Card Sorting Test, Trail Making Test Parts A and B, Digit Symbol Substitution Test); and Visuospatial abilities (Block Design Test, Visual Reproduction Test copy, Clock Drawing Test).
Consensus clinical diagnoses were made according to published criteria by at least two board-certified neurologists blind to individual cognitive test scores but told whether neuropsychological assessment identified deficits in two or more cognitive domains. Probable or Possible AD or Mild Cognitive Impairment (MCI) was diagnosed according to NINCDS-ADRDA38 or NIA-AA criteria39.
Statistical Analyses:
Demographics and prevalence of concomitant pathologies in the PART and ADNC groups were compared using Student’s t-test or Chi-squared test, as appropriate. The odds ratio of a dementia diagnosis at final visit was examined using logistic regression with terms for all four pathologic factors (i.e. PART vs ADNC group for degree of amyloid pathology; Braak I-II vs III-IV for degree of tau pathology; LATE-NC− vs LATE-NC+ for presence of TDP43 pathology; Vasc− vs Vasc+ for presence of ischemic vascular pathology), adjusted for age at death, sex, education, and interval from last visit to autopsy. The proportion of all dementia diagnoses attributable to each pathologic factor within our sample was calculated using a previously described method for estimating attributable risk.40
Cognitive domain scores were created from the neuropsychological test battery using previously described methods41,42. Principal component analysis with varimax rotation identified 4 orthogonal components conceptually labeled as “Visuospatial”, “Memory”, “Executive”, and “Language”. Component scores were derived and transformed to z-scores using reference values from an independent pool of 497 “robust” controls (without pathologic confirmation) diagnosed as cognitively normal on their first and all subsequent annual evaluations (average 5.2±5.0 evaluations).
Cross-sectional analyses of the pathologic predictors of scores on global cognitive measures (MMSE, DRS, and CDR-sob) at the last clinical evaluation were performed using linear regression adjusting for age at death, sex, education, and interval from last evaluation to autopsy. A term for group (PART vs ADNC, i.e. Thal 0–2 vs 3–5) was included in the base model to examine the effect of amyloid on impairment. Next, terms for tau (Braak I-II vs Braak III-IV), TDP-43 (LATE-NC− vs LATE-NC+), or vascular (Vasc− vs Vasc+) pathology were separately added to the model to examine their effects on impairment. An interaction between group and each of the other pathologic measures was tested to examine if these pathologies have differential effects in PART and ADNC but was dropped if it did not reach statistical significance.
Longitudinal decline on each cognitive measure was examined in the 5-year period prior to death using linear mixed effects models adjusting for age at death, sex, education, score at the final visit, and interval from last visit to autopsy, along with each of their interactions by time. Participant-specific intercepts and slopes that were assumed to follow a normal distribution with unknown variance were included as random effects. Much like the cross-sectional models, terms for group (PART vs ADNC) and its interaction with time were included in the base model to examine the effect of amyloid on decline. Next, terms for tau (Braak I-II vs Braak III-IV), TDP-43 (LATE-NC− vs LATE-NC+), or vascular (Vasc− vs Vasc+) pathology were separately added to the model and allowed to interact with time to examine their effects on decline. An interaction between group and each of these pathologic terms on the slope of decline was tested but dropped from the final model if found to not reach significance.
Results:
PART and ADNC Pathology and Clinical Characteristics.
As expected, the ADNC group had higher CERAD neuritic plaque density scores than the PART group (p=6.8×10−18) and the two groups had comparable Braak staging (Figure 1A, p=0.99) with approximately 65% of both groups at Braak stages I-II and 35% at stages III-IV (see Table 1). PART and ADNC groups did not differ in age at death (p=0.44), sex (43–46% female) (p=0.93), age at final clinical evaluation (p=0.24), years of formal education (p=0.40), or premorbid verbal IQ measured by the ANART (p=0.21). Both groups had mild impairment (e.g., MMSE 24–25), on average, at last clinical evaluation, and were similar in the proportions of individuals clinically classified as cognitively normal, MCI, or dementia (p=0.39), with approximately 50 to 60 percent of both groups diagnosed with dementia. The APOE ε4 allele was more common in the ADNC group than in the PART group (p=0.01).
Figure 1: Comparative distributions of pathologies.
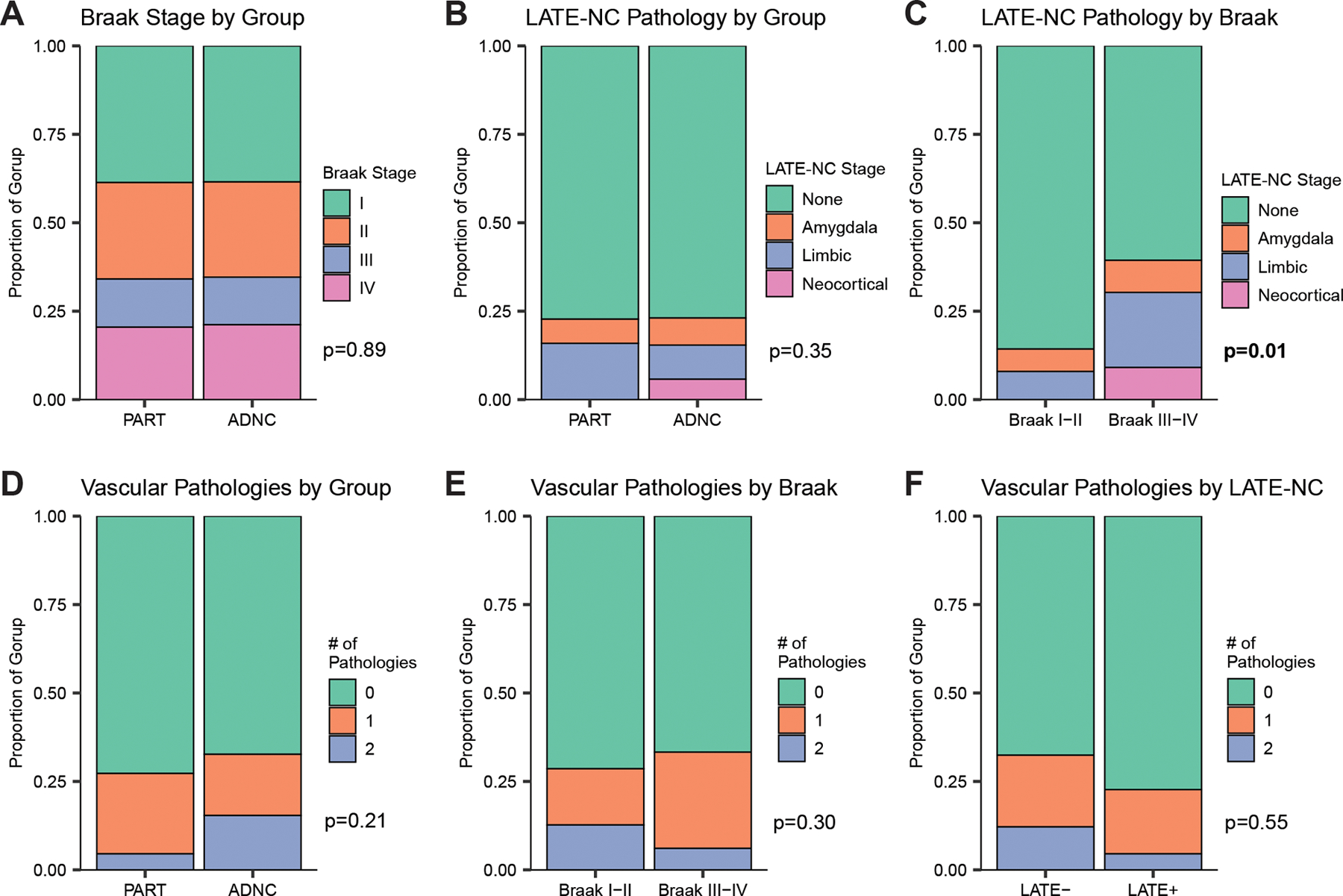
The distribution of Braak stages (A) and stages of concomitant LATE-NC pathology (B) did not differ between participants with PART and ADNC in our sample. However, those with relatively higher Braak stages (i.e. III-IV vs I-II) were more likely to have concomitant LATE-NC pathology (C), even in models adjusted for age and sex. In contrast, the number of concomitant ischemic vascular pathologies (tallied across infarcts, microinfarcts, and hemorrhages/microbleeds) did not differ between PART and ADNC (D), Braak stages (E), or presence of LATE-NC co-pathology (F).
Table 1:
Participant Characteristics
| PART (N=44) | ADNC (N=52) | P value | |
|---|---|---|---|
| Age at Death (y) | 88.0 ± 7.4 | 89.1 ± 6.2 | 0.44 |
| Age at Final Visit | 86.7 ± 7.3 | 88.4 ± 6.4 | 0.24 |
| Final Visit – Autopsy Interval | 1.2 ± 1.4 | 0.9 ± 1.1 | 0.27 |
| Female | 19 (43%) | 24 (46%) | 0.93 |
| Thal Phase: 0 (A0) / 1–2 (A1) / 3 (A2) / 4–5 (A3) |
20 / 24 / 0 / 0 (45% / 55% / 0% / 0%) |
0 / 0 / 13 / 39 (0% / 0% / 25% / 75%) |
(definition) |
| Braak Stage: I-II (B1) / III-IV (B2) / V-VI (B3) |
29 / 15 / 0 (66% / 34% / 0%) |
34 / 18 / 0 (65% / 35% / 0%) |
0.99 |
| Neuritic Plaques: None (C0) / Sparse (C1) / Moderate (C2) / Frequent (C3) |
39 / 5 / 0 / 0 (89% / 11% / 0% / 0%) |
0 / 9 / 38 / 5 (0% / 17% / 73% / 10%) |
6.79 × 10 −18 |
| LATE-NC: None / Amyg / Limbic / Neocortical |
34 / 3/ 7/ 0 (77% / 7% / 16% / 0%) |
40 / 4 / 5 / 3 (77%, 8% / 10% / 6%) |
0.35 |
| Amyloid Angiopathy: None / Mild / Moderate / Severe |
31 / 7 / 5 / 1 (70% / 16% / 11% / 2%) |
18 / 13 / 13 / 8 (35% / 25% / 25% / 15%) |
0.004 |
| Atherosclerosis: None / Mild / Moderate / Severe |
8 / 15 / 14 / 6 (18% / 34% / 32% / 14%) |
7 /12 / 26 / 7 (13% / 23% / 50% / 13%) |
0.35 |
| Arteriolosclerosis: None / Mild / Moderate / Severe |
12 / 13 / 16 / 3 (27% / 30% / 36% / 7%) |
16 / 18 / 16 / 2 (31% / 35% / 31% / 4%) |
0.82 |
| Infarcts | 8 (18%) | 12 (23%) | 0.74 |
| Microinfarcts | 4 (9%) | 10 (19%) | 0.27 |
| Hemorrhage/Microbleeds | 2 (5%) | 3 (6%) | 0.99 |
| Hippocampal Sclerosis | 7 (16%) | 10 (19%) | 0.88 |
LATE-NC and Vascular Co-pathology.
Approximately 24% of individuals in both the PART and ADNC groups had some degree of LATE co-pathology and distributions of LATE-NC stages did not differ between groups (p=0.35) (see Table 1). 77% of both groups had no LATE-NC (Figure 1B, p=0.99). Hippocampal sclerosis was equally common in PART and ADNC at 16–19% of each group (p=0.88). However, in both unadjusted chi-squared analyses and models adjusted for age and sex, LATE pathology was associated with higher Braak stages across both PART and ADNC groups (Figure 1C, p=0.01).
Amyloid angiopathy, which is associated with greater parenchymal amyloid plaques, was more common in ADNC (65%) than in PART (30%) (p=0.004). However, the PART and ADNC groups did not differ in the prevalence or stage of other individual vascular pathologies including atherosclerosis of the circle of Willis, arteriolosclerosis, large or lacunar infarcts, microinfarcts, hemorrhages, or microbleeds. ADNC and PART did not differ in distribution of the number of ischemic vascular pathologies per individual (0–2) tallied across infarcts, microinfarcts, and hemorrhages (Figure 1D, p=0.21). Number of vascular pathologies did not differ between Braak I-II versus Braak III-IV across PART and ADNC groups (Figure 1E, p=0.30), nor in those with versus without concomitant LATE pathology (Figure 1F, p=0.55).
Characteristics by Presence of LATE-NC.
Demographic and pathologic factors were compared in participants with or without LATE-NC (Table 2). The groups were well matched on age (p=0.99) and did not differ in their amyloid staging by CERAD scores (p=0.16) or Thal phasing (p=0.42), nor any of the measures of vascular pathology (p>0.05). However, those with LATE-NC were more likely than those without LATE-NC to have a higher Braak stage (p=0.007). In addition, 55% of the LATE-NC+ group had concomitant hippocampal sclerosis compared to only 7% of the LATE-NC− group (consistent with the well-established association).20
Table 2:
Participant Characteristics by LATE-NC
| LATE-NC − (N=74) | LATE-NC + (N=22) | P value | |
|---|---|---|---|
| Age at Death (y) | 88.4 ± 6.8 | 89.0 ± 6.9 | 0.73 |
| Age at Final Visit | 87.6 ± 6.9 | 87.6 ± 6.9 | 0.99 |
| Final Visit – Autopsy Interval | 0.8 ± 0.8 | 1.3 ± 1.8 | 0.72 |
| Female | 35 (47%) | 8 (36%) | 0.51 |
| Thal Phase: 0 (A0) / 1–2 (A1) / 3 (A2) / 4–5 (A3) |
15 / 19 / 11 / 29 (20% / 26% / 15% / 39%) |
5 / 5 / 2 / 10 (23% / 23% / 9% / 45%) |
0.42 |
| Braak Stage: I-II (B1) / III-IV (B2) / V-VI (B3) |
54 / 20 / 0 (73% / 27% / 0%) |
9 / 13 / 0 (41% / 59% / 0%) |
0.007 |
| Neuritic Plaques: None (C0) / Sparse (C1) / Moderate (C2) / Frequent (C3) |
29 / 11 / 32 / 2 (39% / 15% / 43% / 3%) |
10 / 3 / 6 / 3 (45% / 14% / 27% / 14%) |
0.16 |
| LATE-NC: None / Amyg / Limbic / Neocortical |
74 / 0 / 0 / 0 (100% / 0% / 0% / 0%) |
0 / 7 / 12 / 3 (0%, 32% / 55% / 14%) |
(definition) |
| Amyloid Angiopathy: None / Mild / Moderate / Severe |
42 / 12 / 12 / 8 (57% / 16% / 16% / 11%) |
7 / 8 / 6 / 1 (32% / 36% / 27% / 5%) |
0.07 |
| Atherosclerosis: None / Mild / Moderate / Severe |
10 / 18 / 35 / 11 (14% / 24% / 47% / 15%) |
7 /6 / 9 / 0 (32% / 27% / 41% / 0%) |
0.14 |
| Arteriolosclerosis: None / Mild / Moderate / Severe |
21 / 25 / 23 / 5 (28% / 34% / 31% / 7%) |
7 / 6 / 9 / 0 (32% / 27% / 41% / 0%) |
0.52 |
| Infarcts | 17 (23%) | 3 (14%) | 0.52 |
| Microinfarcts | 12 (16%) | 2 (9%) | 0.62 |
| Hemorrhage/Microbleeds | 4 (5%) | 1 (5%) | 0.99 |
| Hippocampal Sclerosis | 5 (7%) | 12 (55%) | 1.3 × 10 −6 |
Influence of Pathology on Clinical Diagnosis.
In a logistic regression model adjusted for age, sex, and education, and all four pathologic factors, the likelihood of receiving a dementia diagnosis at the clinical evaluation closest to death was the same for those with PART or ADNC (OR 1.25 [95%CI: 0.49–3.24], p=0.65, Figure 2A) (see Table 3). In contrast, those at Braak stage III-IV had a greater likelihood of dementia than those at Braak stage I-II (OR 4.37 [95%CI: 1.52 – 13.53], p=0.007, Figure 2B). Similarly, individuals with concomitant LATE-NC were more likely to receive a dementia diagnosis than those without LATE-NC (OR 4.76 [99%CI: 1.46 – 17.96], p=0.01, Figure 2C). This was not true for those with or without concomitant vascular pathology (OR 0.86 [95%CI: 0.29 – 2.45], p=0.77, Figure 2D). When tested, interaction terms between each pathologic factor and group were not significant, suggesting that these effects are consistent across PART and ADNC.
Figure 2: Clinical diagnoses of participants at the evaluation closest to autopsy by pathology.
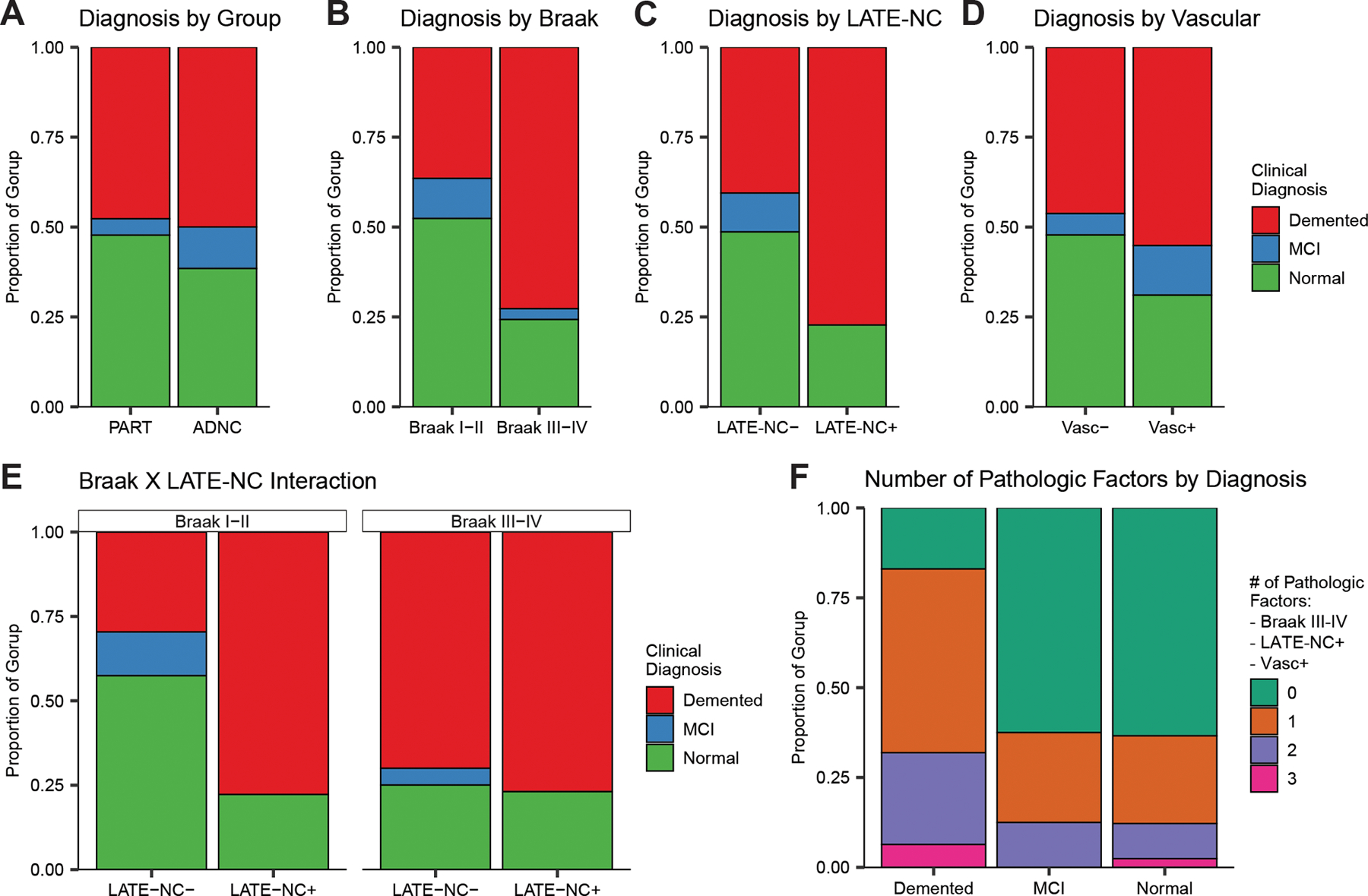
The distributions of clinical diagnoses at the clinical evaluation closest to death presented in the participants divided by each of the four pathologic factors (A-D) suggests while there was no significant difference between individuals with PART vs ADNC and between those with vs without vascular pathology, individuals with higher Braak stages or concomitant LATE-NC were more likely to be demented prior to death. Since concomitant LATE-NC pathology is more likely among those with higher Braak stages (Figure 1C), we examined the interaction of those pathologies with regards to the diagnosis (D) and observed that either higher Braak stages, concomitant LATE-NC, or both appear to increase the likelihood of a dementia diagnosis compared to those with neither pathology. If the number of significant pathologic factors (tallied across higher Braak, concomitant LATE-NC, and concomitant Vascular) are examined by the final clinical diagnosis (F), we can see that those with dementia were far more likely (>75%) to have at least one of these pathologies compared to those with either MCI or Normal cognition (<50%).
Table 3:
Statistics for cognitive performance at last visit by pathologic feature and their interaction with group
| PART vs ADNC | Braak I-II vs Braak III-IV | LATE-NC− vs LATE-NC + | Vasc− vs Vasc+ | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Coefficient (95% CI) | Group P value | Coefficient (95% CI) | Braak P value | Group Interaction P value | Coefficient (95% CI) | LATE P value | Group Interaction P value | Coefficient (95% CI) | Vasc P value | Group Interaction P value | |
| MMSE | −0.67 (−3.25 – 1.91) | 0.606 | −4.38 (−7.01 – −1.75) | 0.001 | 0.115 | −5.8 (−8.62 – −2.98) | <0.001 | 0.769 | −1.20 (−4.08 – 1.67) | 0.408 | 0.053 |
| DRS | −1.33 (−10.56 – 7.91) | 0.776 | −10.81 (−20.53 – −1.08) | 0.03 | 0.256 | −17.51 (−27.89 – −7.14) | 0.001 | 0.614 | −10.20 (−20.32 – −0.08) | 0.048 | 0.118 |
| CDR-sob | 1.12 (−1.50 – 3.75) | 0.396 | 6.88 (4.62 – 9.13) | <0.001 | 0.966 | 5.17 (2.10 – 8.24) | 0.001 | 0.737 | 1.32 (−1.59 – 4.23) | 0.369 | 0.133 |
| Memory | −0.02 (−0.68 – 0.64) | 0.956 | −0.90 (−1.60 – −0.21) | 0.011 | 0.173 | −0.82 (−1.59 – −0.05) | 0.037 | 0.858 | −0.13 (−0.89 – 0.63) | 0.741 | 0.749 |
| Executive | 0.05 (−0.83 – 0.94) | 0.904 | −0.03 (−0.97 – 0.92) | 0.958 | 0.791 | 0.28 (−0.76 – 1.32) | 0.596 | 0.445 | −1.30 (−2.27 – −0.34) | 0.009 | 0.117 |
| Visuospatial | −0.34 (−1.30 – 0.62) | 0.487 | −0.27 (−1.31 – 0.76) | 0.600 | 0.655 | −0.89 (−2.02 – 0.24) | 0.12 | 0.65 | −0.90 (−1.98 – 0.18) | 0.102 | 0.430 |
| Language | −0.50 (−1.28 – 0.28) | 0.205 | −0.62 (−1.46 – 0.22) | 0.145 | 0.057 | −1.08 (−2.00 – −0.16) | 0.021 | 0.627 | 0.39 (−0.51 – 1.29) | 0.391 | 0.153 |
For each cognitive test or composite, the coefficient (99% Confidence Interval) for the cross-sectional performance at the last visit is provided along with the p value for each pathologic term as indicated along the top. For each pathologic term other than the Group term (i.e., PART vs ADNC), the p value for the interaction of the pathologic term with Group is also provided so assess if the effects differ between those with PART or ADNC.
Using this logistic regression, the risk of a dementia diagnosis attributable to each pathologic variable in this sample was calculated by comparing the known number of dementia diagnoses to the simulated number of dementia diagnoses if that pathology was absent from all participants (i.e., the pathologic term set to 0 in the model). Results estimated that 21% of the dementia prevalence in the cohort could be attributed to higher Braak stages and 14% to concomitant LATE-NC. When tested, the interaction of LATE-NC and Braak stage was not significant (p=0.11) suggesting that the two pathologies contribute independently to probability of dementia diagnosis.
Overall, individuals receiving a diagnosis of dementia were more likely than those classified with normal cognition or MCI to have some combination of higher Braak stage, LATE-NC, or vascular pathology (Figure 2F). Nearly 80% of those receiving a diagnosis of dementia had at least one of these pathologic features, whereas this was true for less than 50% of those classified with normal cognition or MCI. Of those with normal cognition or MCI, more than half of those with at least one pathology had only vascular pathology.
Influence of Pathology on Cognitive Test Performance.
In models adjusting for age, sex, education, and interval from last evaluation to death, PART and ADNC groups did not differ on MMSE (p=0.61), DRS (p=0.78), or CDR-sob (p=0.40) scores closest to the time of death (within 1.2 ± 1.4 and 0.9 ± 1.1 years for PART and ADNC groups, respectively), nor on domain-specific composite scores for Memory (p=0.96), Executive function (p=0.90), Visuospatial ability (p=0.49), or Language (p=0.21) (see Table 3). Scores achieved on each of these measures are presented as a function of group (PART vs. ADNC) and each of the various pathologies (Braak stage, LATE-NC, vascular) in Figure 3. There were no significant interactions between Group (PART vs. ADNC) and Braak stage, presence of LATE-NC, or presence of vascular pathology in determining global and domain specific-cognitive scores, indicating comparable effects of these pathologies on cognition in PART and ADNC. Collapsing across groups, individuals at Braak stage III-IV scored worse than those at Braak stage I-II by 4.4 MMSE points (95% CI: 1.8–7.0), 10.8 DRS points (95% CI: 1.1–20.5), 6.9 CDR-sob points (95% CI: 4.6–9.1), and 0.9 standard deviations (SDs) on the Memory composite (95% CI: 0.2–1.6). Patients with concomitant LATE-NC were more impaired by 5.8 MMSE points (95% CI: 3.0–8.6), 17.5 DRS points (95% CI: 7.1–27.9), 5.2 CDR-sob points (95% CI: 2.1–8.2), 0.8 SDs on a Memory composite (95% CI: 0.1–1.6), and 1.1 SDs on a Language composite (95% CI: 0.2–2.0). Those with concomitant vascular pathology performed worse by 10.2 DRS points (95% CI: 0.1–20.3) and 1.3 SDs on an Executive composite (95% CI: 0.3–2.3).
Figure 3: Cognitive performance at the evaluation closest to autopsy by pathologic factor.
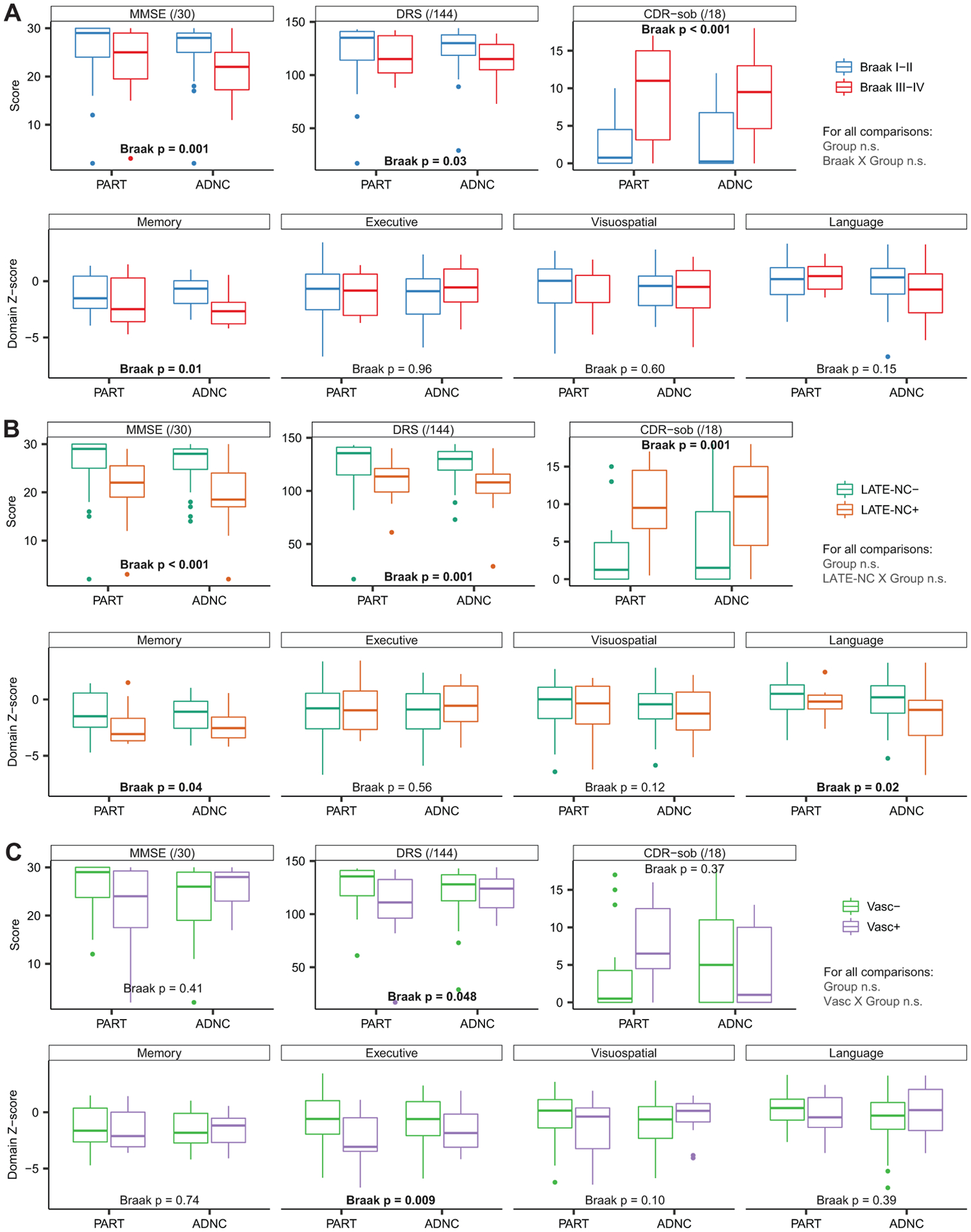
Cognitive performance of the participants at the evaluation closest to death is presented as a function of higher Braak stage (A), concomitant LATE-NC pathology (B), or concomitant vascular pathology (C) divided by group (i.e. PART vs ADNC). There were no significant differences in performance between PART and ADNC on any measures, and no significant interactions of the group term with each of the three other pathologic factors, suggesting that they produce similar effects on cognition across the groups. However, those with higher Braak stages performed worse on each of the three global cognitive measures (MMSE, DRS, CDR-sob), as well as the Memory cognitive composite. Similarly, those with concomitant LATE-NC pathology performed worse on the three global measures and both the Memory and Language composites. Those with concomitant vascular pathology declined more rapidly on the DRS. For effect sizes and p value please see Table 4.
Influence of Pathology on Rate of Cognitive Decline.
PART and ADNC groups did not differ in rates of decline in the 5-year period prior to death on the MMSE, DRS, CDR-sob, or any cognitive composite measure (see Figure 4 and Table 4). After collapsing across PART and ADNC, individuals at Braak stage III-IV declined 1.3 points/year more rapidly on the MMSE (95% CI: 0.3–2.4), 2.8 points/year more rapidly on CDR-sob (95% CI: 1.5–4.2), and 0.4 standard deviations (SDs) per year more rapidly on the Memory composite score (95% CI: 0.1–0.7) than those at Braak stage I-II. Similarly, individuals with concomitant LATE-NC pathology declined 1.9 points/year more rapidly on the MMSE (95% CI: 0.8–3.0), 7.76 points/year more rapidly on the DRS (95% CI: 3.4–12.7), 1.93 points/year more rapidly on CDR-sob (95% CI: 0.4–3.7), and 0.48 SD/year more rapidly on the Language composite score (95% CI: 0.1–0.8) than those without concomitant LATE-NC. Individuals with concomitant vascular pathology declined 5.18 points/year more rapidly on the DRS (95% CI: 0.6–10.2) than those without vascular co-pathology. There were no significant interactions between group and Braak stage, presence of LATE-NC, or presence of vascular pathology on rates of decline on any cognitive measure.
Figure 4: Rates of longitudinal cognitive decline prior to autopsy by pathologic factor.
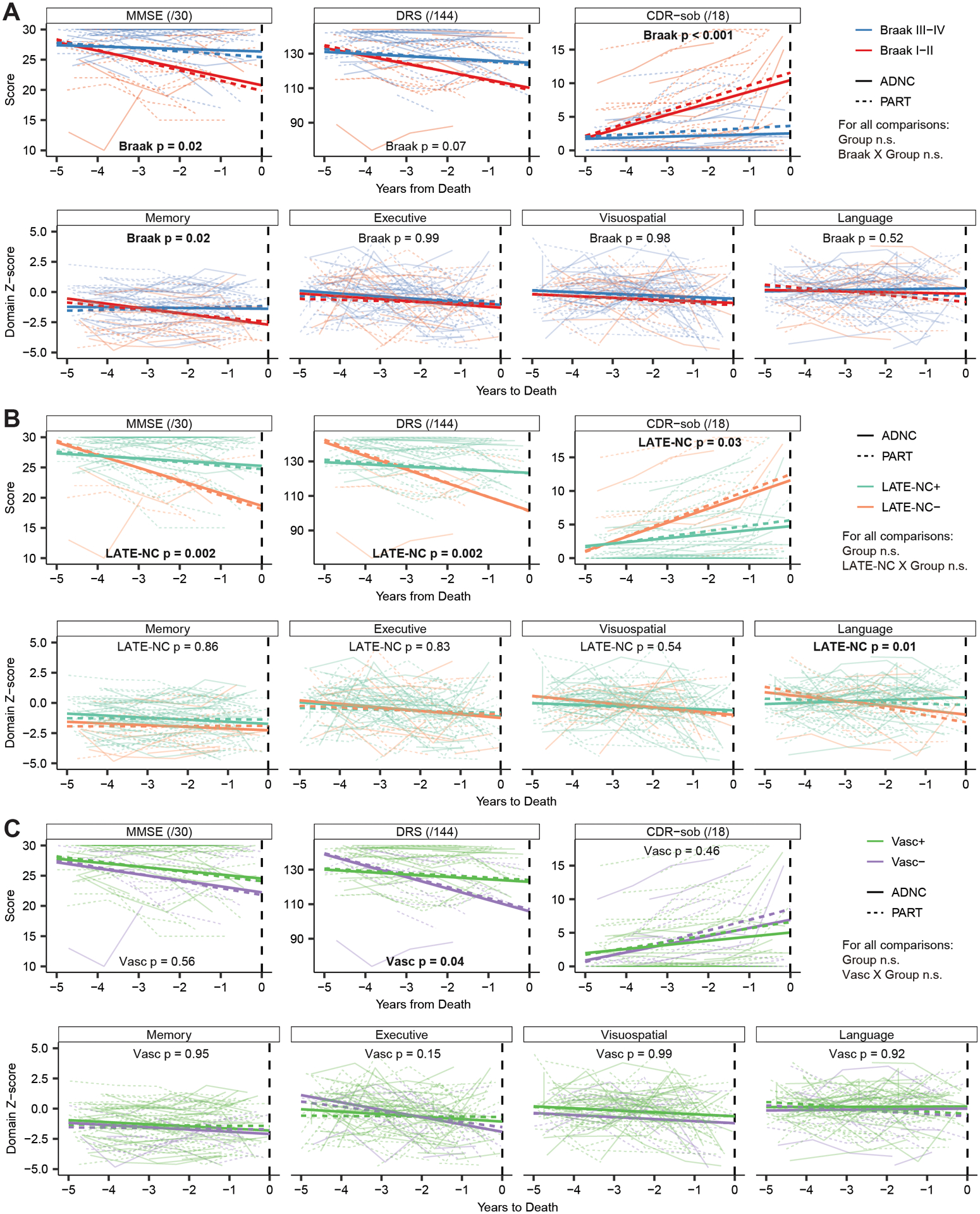
Trajectories of cognitive decline in the 5 years prior to autopsy of the participants are presented as a function of higher Braak stage (A), concomitant LATE-NC pathology (B), or concomitant vascular pathology (C) divided by group (i.e. PART vs ADNC). There were no significant differences in performance between PART and ADNC on any measures, and no significant interactions of the group term with each of the three other pathologic factors, suggesting that they produce similar effects on cognitive delcine across the groups. However, those with higher Braak stages declined more rapidly on the MMSE, CDR-sob, and the Memory composite. Similarly, those with concomitant LATE-NC pathology performed worse on all three global measures and the Language composite. Those with concomitant vascular pathology performed worse on the DRS and the Executive composite.
Table 4:
Statistics for longitudinal slopes of decline by pathologic feature and their interaction with group
| PART vs ADNC | Braak I-II vs Braak III-IV | LATE-NC− vs LATE-NC + | Vasc− vs Vasc+ | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Coefficient (95% CI) | Group P value | Coefficient (95% CI) | Braak P value | Group Interaction P value | Coefficient (95% CI) | LATE P value | Group Interaction P value | Coefficient (95% CI) | Vasc P value | Group Interaction P value | |
| MMSE | −0.16 (−1.11 – 0.79) | 0.752 | −1.31 (−2.40 – −0.32) | 0.019 | 0.289 | −1.89 (−3.04 – −0.84) | 0.002 | 0.706 | −0.34 (−1.52 – 0.74) | 0.562 | 0.309 |
| DRS | 0.12 (−3.77 – 4.08) | 0.952 | −4.07 (−8.42 – −0.06) | 0.066 | 0.258 | −7.76 (−12.74 – −3.36) | 0.002 | 0.546 | −5.18 (−10.18 – −0.63) | 0.037 | 0.194 |
| CDR-sob | 0.61 (−0.81 – 2.08) | 0.439 | 2.77 (1.47 – 4.18) | <0.001 | 0.417 | 1.93 (0.38 – 3.66) | 0.029 | 0.202 | 0.55 (−0.75 – 1.91) | 0.455 | 0.838 |
| Memory | 0.14 (−0.14 – 0.43) | 0.351 | −0.4 (−0.72 – −0.10) | 0.015 | 0.409 | 0.03 (−0.31 – 0.35) | 0.864 | 0.375 | −0.01 (−0.37 – 0.36) | 0.951 | 0.555 |
| Executive | 0.15 (−0.25 – 0.56) | 0.471 | 0.00 (−0.44 – 0.45) | 0.998 | 0.619 | −0.06 (−0.55 – 0.44) | 0.829 | 0.769 | −0.40 (−0.92 – 0.13) | 0.151 | 0.829 |
| Visuospatial | −0.02 (−0.49 – 0.46) | 0.952 | −0.01 (−0.53 – 0.50) | 0.977 | 0.529 | −0.19 (−0.78 – 0.39) | 0.540 | 0.935 | 0.00 (−0.61 – 0.59) | 0.990 | 0.066 |
| Language | −0.21 (−0.52 – 0.09) | 0.194 | −0.12 (−0.46 – 0.22) | 0.517 | 0.539 | −0.48 (−0.84 – −0.13) | 0.013 | 0.757 | 0.02 (−0.39 – 0.41) | 0.921 | 0.742 |
For each cognitive test or composite, the coefficient (99% Confidence Interval) for the annualized rate of decline is provided along with the p value for each pathologic term as indicated along the top. For each pathologic term other than the Group term (i.e., PART vs ADNC), the p value for the interaction of the pathologic term with Group is also provided so assess if the effects differ between those with PART or ADNC.
Discussion:
The present study examined the contributions of concomitant LATE-NC or ischemic vascular neuropathology to the clinical and cognitive deficits associated with tangle pathology restricted to the medial temporal lobe (i.e., Braak I-IV) in the absence (i.e., PART) or presence (i.e., ADNC) of significant amyloid plaques. Our study showed no difference in the prevalence of concomitant LATE-NC pathology (about 24% in each condition) or cerebrovascular pathology (about 25–30% in each condition) in PART and early-stage ADNC. Only cerebral amyloid angiopathy (CAA) occurred more often in ADNC than in PART, in concert with a higher APOE ε4 allele prevalence. There was little difference in the clinical presentations of PART and early-stage ADNC. The groups did not differ in severity of global cognitive impairment or impairment in specific cognitive domains at the last evaluation before death, nor in the proportion of individuals who received a final clinical diagnosis of dementia (50–60% in each group) or MCI (10–25% of each group). Rate of decline in global cognition or specific cognitive domains also did not differ in PART and ADNC.
The presence of concomitant LATE-NC was associated with worse global cognition, worse memory impairment, and a faster rate of decline in language abilities across both PART and ADNC. The effects of LATE-NC on cognition were similar in both groups (i.e., there was no group by LATE-NC interaction effect), and none of the participants with LATE-NC had any clinical features of FTD or neuropathologic features of FTLD. The primary impact of LATE-NC on memory and language is consistent with the predominance of TDP-43 pathology in limbic regions important for these cognitive functions, and is largely consistent with reported effects of LATE-NC in other contexts such as advanced AD and hippocampal sclerosis21,43,44. These findings suggest that undetected LATE-NC may have influenced the severity and nature of cognitive decline reported in previous studies of PART where cognitive decline seemed out of proportion to the degree of tangle pathology1,6,10–12,24.
Concomitant vascular pathology in the form of infarcts, microinfarcts, and hemorrhages was associated with greater executive function deficits in both PART and ADNC. This finding is consistent with a recent multi-center cohort analysis which showed that vascular pathology and age are the strongest predictors of cognitive impairment in patients with PART - beyond degree of tau pathology11. While vascular pathology is a well-known predictor of cognitive impairment, wide variability in the location and extent of associated necrosis makes it difficult to consistently characterize its effects on cognition. The most commonly reported deficit is executive dysfunction attributable to diffuse white matter disruption45. In line with this view, the greatest impact of concomitant vascular co-pathology in PART and ADNC in the present study was on executive functions. We also found that degree of executive dysfunction, rather than its rate of decline, was impacted by concomitant vascular pathology in PART and ADNC. This finding is consistent with an expected step-wise progression of vascular pathology and its impact on cognition. Cerebral amyloid angiopathy was more prevalent in ADNC than in PART, consistent with its known association with amyloid plaque deposition, but the impact of CAA on cognition is marginal46,47 and not likely to contribute to differences in the clinical presentation of ADNC and PART. Overall, we found no difference in the influence of concomitant cerebrovascular pathology on clinical presentation or cognitive decline in PART and ADNC.
Consistent with previous studies1,9, we found that higher Braak stage (i.e., III-IV vs. I-II) was associated with greater memory impairment, worse global cognition (based on tests weighted towards memory function), and faster memory and global cognitive decline in PART and early-stage ADNC. Furthermore, Braak stage influenced memory and its decline in PART and ADNC to a similar degree (i.e., there was no group by interaction effect). The specificity of effects on memory is not surprising given that, by definition, tau pathology in PART and early-stage ADNC is restricted to medial temporal lobe structures critical for memory. The association we find between cognition and Braak stage in PART and AD may explain why previous studies that compared PART with little or no neocortical tau pathology (Braak I-IV) to late stage AD with significant neocortical tau pathology (Braak V-VI) found greater cognitive impairment and faster cognitive decline in AD than in PART6,8,13. When patients with PART or ADNC with nearly identical age, sex, and Braak stage were compared in the present study, no important differences in cognition were found. A previous study using data from the National Alzheimer’s Coordinating Center (NACC) found small and inconsistent differences in cognition between definite PART (i.e., Braak III-IV) and AD restricted to Braak III-IV, but did not take into account the potential presence and influence of LATE-NC or cerebrovascular co-pathologies10.
Similarities in cognitive outcomes of patients with PART or ADNC who differed only in the presence or absence of significant amyloid pathology (i.e., PART: Thal phase 0–2; ADNC: Thal phase 3–5) is consistent with previous findings of a minimal direct effect of amyloid deposition on cognition in AD48. Current conceptualizations of AD suggest, however, that amyloid deposition may accelerate neurofibrillary tangle formation and neuron loss that correlates with cognitive decline49. Thus, memory decline might be expected to be faster in ADNC than in PART. This was not the case in the present study. We found no differences in rates of memory or other cognitive decline for PART and ADNC at any Braak stage (even accounting for cerebrovascular and LATE-NC co-pathology). It should be noted that this conclusion is limited by the modest sample size of the study and the inherent nature of autopsy-based studies which only allow rate of pathology accumulation to be inferred. While we adjust our analyses for the test-autopsy interval, this may not always capture the complexity of multiple co-pathologies that may progress at different rates. This question may be more clearly addressed in future longitudinal studies of PART and low level ADNC using tau and amyloid PET imaging, although such studies will be limited by the inability to account for concomitant pathologies (e.g., Lewy bodies, LATE-NC).
Concomitant LATE-NC (presence vs. absence) and higher Braak stages (Braak III-IV vs. Braak I-II) were associated with similar increases in likelihood of dementia at the last clinical evaluation before death in both PART and ADNC, each accounted for 15–20% of dementia diagnoses, and they had comparable magnitudes of effects on global cognitive decline. This suggests that LATE-NC may have as great an impact as degree of tau pathology on cognitive decline and development of dementia in PART and early-stage ADNC, and is consistent with studies showing that both tau and LATE-NC are major predictors of degree of medial temporal lobe atrophy on MRI in patients with PART or ADNC50. IGiven our results, it may be that a substantial portion of the cognitive impairment and cognitive decline attributed to PART in previous studies with nearly identical inclusion criteria to ours1,10,11 could be due to unassessed LATE-NC.
A limitation of the present study is that there were changes in clinical and neuropathologic practices and criteria over the 35 years during which this clinical-pathological cohort was established. We minimized potential bias this may have caused by conducting immunohistochemical staining for TDP-43 and amyloid on all participants in order to retrospectively apply current consensus neuropathological criteria for AD and LATE-NC. Additionally, the extent of tau pathology in both PART and AD was approximated using the Braak staging scheme that was designed for use in AD. As PART is relatively recently defined, it is possible that Braak staging is not directly applicable to PART. Some studies have circumvented this concern by assessing p-tau staining density11. However, a previous study has shown that the distribution of tau pathology is largely consistent between early AD and PART, suggesting that Braak stage is a sufficient proxy for the extent of tau pathology in both conditions13. Lastly, this study was based on a convenience sample of cases from an ADRC brain that is biased towards Caucasians and those with high levels of education, and away from those with other significant medical comorbidities that may have excluded them from participation in the longitudinal ADRC study. This limits the generalizability of these results to the general population, and in particular the generalizability of attributable risk calculations which are traditionally more meaningful in the context of epidemiologic studies.
Despite these limitations, our findings have important clinical implications. We demonstrate here that LATE-NC contributes to cognitive decline in the context of mild NFT and amyloid plaque pathology. The contributions of undetected LATE-NC to risk of cognitive impairment and dementia in individuals with PART or early-stage ADNC may increase variability in the prognostic value of in vivo tau and amyloid PET imaging or CSF and plasma biomarkers. In addition, undetected LATE-NC \ could dampen therapeutic benefits observed of anti-amyloid or anti-tau medications in clinical trials that include patients with early stage tauopathy or AD. Finally, our findings strongly highlight the need to further investigate mechanisms underlying TDP-43 proteinopathies and to develop biomarkers and treatments specific for LATE-NC.
Acknowledgments:
We would like to thank the faculty, staff, and participants at the UC San Diego Shiley-Marcos Alzheimer’s Disease Research Center. This work was supported by NIH P30-AG062429 and F30-AG063440.
Abbreviations:
- PART
Primary Age-related Tauopathy
- ADNC
Alzheimer’s Disease Neuropathologic Change
- LATE-NC
Limbic Age-related TDP-43 Encephalopathy Neuropathologic Change
- MMSE
Mini Mental State Exam
- DRS
(Mattis) Dementia Rating Scale
- CDR-sob
Clinical Dementia Rating, sum of boxes scale
Footnotes
Potential Conflicts of Interest:
All authors have nothing to report.
References:
- 1.Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 2014;128(6):755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nelson PT, Abner EL, Schmitt FA, et al. Brains with medial temporal lobe neurofibrillary tangles but no neuritic amyloid plaques are a diagnostic dilemma but may have pathogenetic aspects distinct from Alzheimer disease. Journal of neuropathology and experimental neurology 2009;68(7):774–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ikeda K, Akiyama H, Arai T, et al. Clinical Aspects of ‘Senile Dementia of the Tangle Type’ – A Subset of Dementia in the Senium Separable from Late-Onset Alzheimer’s Disease. Dement Geriatr Cogn 1999;10(1):6–11. [DOI] [PubMed] [Google Scholar]
- 4.Itoh Y, Yamada M, Yoshida R, et al. Dementia Characterized by Abundant Neurofibrillary Tangles and Scarce Senile Plaques: A Quantitative Pathological Study. Eur Neurol 1996;36(2):94–97. [DOI] [PubMed] [Google Scholar]
- 5.Bancher C, Jellinger KA. Neurofibrillary tangle predominant form of senile dementia of Alzheimer type: a rare subtype in very old subjects. Acta Neuropathol 1994;88(6):565–570. [DOI] [PubMed] [Google Scholar]
- 6.Jellinger KA, Attems J. Neurofibrillary tangle-predominant dementia: comparison with classical Alzheimer disease. Acta Neuropathol 2007;113(2):107–117. [DOI] [PubMed] [Google Scholar]
- 7.Jellinger KA, Bancher C. Senile Dementia with Tangles (Tangle Predominant Form of Senile Dementia). Brain Pathol 1998;8(2):367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teylan M, Besser LM, Crary JF, et al. Clinical diagnoses among individuals with primary age-related tauopathy versus Alzheimer’s neuropathology. Laboratory Investigation J Technical Methods Pathology 2019;99(7):1049–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jefferson-George KS, Wolk DA, Lee EB, McMillan CT. Cognitive decline associated with pathological burden in primary age-related tauopathy. Alzheimer’s Dementia 2017;13(9):1048–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Besser LM, Mock C, Teylan MA, et al. Differences in Cognitive Impairment in Primary Age-Related Tauopathy Versus Alzheimer Disease. J Neuropathology Exp Neurology 2019;78(3):219–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iida MA, Farrell K, Walker JM, et al. Predictors of cognitive impairment in primary age-related tauopathy: an autopsy study. Acta Neuropathologica Commun 2021;9(1):134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Besser LM, Crary JF, Mock C, Kukull WA. Comparison of symptomatic and asymptomatic persons with primary age-related tauopathy. Neurology 2017;89(16):1707–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bell WR, An Y, Kageyama Y, et al. Neuropathologic, genetic, and longitudinal cognitive profiles in primary age-related tauopathy (PART) and Alzheimer’s disease. Alzheimer’s Dementia 2019;15(1):8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jellinger KA, Alafuzoff I, Attems J, et al. PART, a distinct tauopathy, different from classical sporadic Alzheimer disease. Acta Neuropathol 2015;129(5):757–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smirnov DS, Galasko D, Hiniker A, et al. Age-of-Onset and APOE -Related Heterogeneity in Pathologically Confirmed Sporadic Alzheimer Disease. Neurology 2021; 10.1212/WNL.0000000000011772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smirnov DS, Galasko D, Hiniker A, et al. Age-of-Onset and APOE-Related Heterogeneity in Pathologically Confirmed Sporadic Alzheimer Disease. Neurology, In Press [date unknown]; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nelson PT, Trojanowski JQ, Abner EL, et al. “New Old Pathologies”: AD, PART, and Cerebral Age-Related TDP-43 With Sclerosis (CARTS). Journal of neuropathology and experimental neurology 2016;75(6):482–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murray ME, Cannon A, Graff-Radford NR, et al. Differential clinicopathologic and genetic features of late-onset amnestic dementias. Acta Neuropathologica 2014;128(3):411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katsumata Y, Abner EL, Karanth S, et al. Distinct clinicopathologic clusters of persons with TDP-43 proteinopathy. Acta Neuropathol 2020;140(5):659–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nelson PT, Dickson DW, Trojanowski JQ, et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 2019;142(6):1503–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilson RS, Yu L, Trojanowski JQ, et al. TDP-43 pathology, cognitive decline, and dementia in old age. JAMA neurology 2013;70(11):1418–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Josephs K, Whitwell J, Knopman D, et al. Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology 2008;70(19 Pt 2):1850–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uchino A, Takao M, Hatsuta H, et al. Incidence and extent of TDP-43 accumulation in aging human brain. Acta Neuropathologica Communications 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nelson PT. LATE Neuropathologic Changes with Little or No Alzheimer Disease is Common and is Associated with Cognitive Impairment but Not Frontotemporal Dementia. J Neuropathology Exp Neurology 2021;80(7):649–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kapasi A, Yu L, Boyle PA, et al. Limbic-predominant age-related TDP-43 encephalopathy, ADNC pathology, and cognitive decline in aging. Neurology 2020;95(14):e1951–e1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bennett DA, Buchman AS, Boyle PA, et al. Religious Orders Study and Rush Memory and Aging Project. J Alzheimer’s Dis 2018;Preprint(Preprint):1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walker JM, Richardson TE, Farrell K, et al. Early Selective Vulnerability of the CA2 Hippocampal Subfield in Primary Age-Related Tauopathy. J Neuropathology Exp Neurology 2021;80(2):102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walker JM, Fudym Y, Farrell K, et al. Asymmetry of Hippocampal Tau Pathology in Primary Age-Related Tauopathy and Alzheimer Disease. J Neuropathology Exp Neurology 2021;80(5):436–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thal DR, Rüb U, Orantes M, Braak H. Phases of A&bgr;-deposition in the human brain and its relevance for the development of AD. Neurology 2002;58(12):1791–1800. [DOI] [PubMed] [Google Scholar]
- 30.Mirra S, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD): Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991;41(4):479–479. [DOI] [PubMed] [Google Scholar]
- 31.Uchikado H, Lin W-L, DeLucia MW, Dickson DW. Alzheimer Disease With Amygdala Lewy Bodies: A Distinct Form of α-Synucleinopathy. J Neuropathology Exp Neurology 2006;65(7):685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nelson PT, Abner EL, Schmitt FA, et al. Modeling the association between 43 different clinical and pathological variables and the severity of cognitive impairment in a large autopsy cohort of elderly persons. Brain pathology (Zurich, Switzerland) 2010;20(1):66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies. Neurology 2017;89(1):88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Terry RD, Katzman RK. Senile dementia of the Alzheimer type. Ann Neurol 1983;14(5):497–506. [DOI] [PubMed] [Google Scholar]
- 35.Braak H, Alafuzoff I, Arzberger T, et al. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 2006;112(4):389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Galasko D, Hansen LA, Katzman R, et al. Clinical-Neuropathological Correlations in Alzheimer’s Disease and Related Dementias. Archives of Neurology 1994;51(9):888–895. [DOI] [PubMed] [Google Scholar]
- 37.Salmon D, Butters N. Neuropsychological assessment of dementia in the elderly. Principles of geriatric neurology. Philadelphia: FA Davis 1992;144 [Google Scholar]
- 38.McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer’s disease Report of the NINCDS‐ADRDA Work Group* under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984;34(7):939–939. [DOI] [PubMed] [Google Scholar]
- 39.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dementia 2011;7(3):263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boyle PA, Yu L, Leurgans SE, et al. Attributable risk of Alzheimer’s dementia attributed to age‐related neuropathologies. Ann Neurol 2019;85(1):114–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smirnov DS, Galasko D, Edland SD, et al. Cognitive decline profiles differ in Parkinson disease dementia and dementia with Lewy bodies. Neurology 2020;94(20):e2076–e2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smirnov DS, Salmon DP, Galasko D, et al. Association of Neurofibrillary Tangle Distribution With Age at Onset–Related Clinical Heterogeneity in Alzheimer Disease. Neurology 2022;98(5):e506–e517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu L, Schneider JA, Kapasi A, et al. Limbic-predominant Age-related TDP-43 Encephalopathy and Distinct Longitudinal Profiles of Domain-specific Literacy. Alz Dis Assoc Dis 2020;34(4):299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Teylan MA, Mock C, Gauthreaux K, et al. Symptomatic presentation and cognitive performance in autopsy‐confirmed, limbic‐predominant, age‐related TDP‐43 encephalopathy with comorbid Alzheimer’s disease. Alzheimer’s Dementia 2021;17:e052215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reed BR, Mungas DM, Kramer JH, et al. Profiles of neuropsychological impairment in autopsy-defined Alzheimer’s disease and cerebrovascular disease. Brain 2007;130(3):731–739. [DOI] [PubMed] [Google Scholar]
- 46.Boyle PA, Yu L, Nag S, et al. Cerebral amyloid angiopathy and cognitive outcomes in community-based older persons. Neurology 2015;85(22):1930–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Planton M, Raposo N, Albucher J-F, Pariente J. Cerebral amyloid angiopathy-related cognitive impairment: The search for a specific neuropsychological pattern. Rev Neurol 2017;173(9):562–565. [DOI] [PubMed] [Google Scholar]
- 48.Aschenbrenner AJ, Gordon BA, Benzinger TLS, et al. Influence of tau PET, amyloid PET, and hippocampal volume on cognition in Alzheimer disease. Neurology 2018;91(9):e859–e866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jack CR, Bennett DA, Blennow K, et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dementia 2018;14(4):535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Josephs KA, Martin PR, Weigand SD, et al. Protein contributions to brain atrophy acceleration in Alzheimer’s disease and primary age-related tauopathy. Brain 2020;143(11):3463–3476. [DOI] [PMC free article] [PubMed] [Google Scholar]