

Summary
The β2-adrenergic receptor (β2AR), a prototypic G protein-coupled receptor (GPCR), is a powerful driver of bronchorelaxation, but the effectiveness of β-agonist drugs in asthma is limited by desensitization and tachyphylaxis. We find that during activation, the β2AR is modified by S-nitrosylation, which is essential for both classic desensitization by PKA as well as desensitization of NO-based signaling that mediates bronchorelaxation. Strikingly, S-nitrosylation alone can drive β2AR internalization in the absence of traditional agonist. Mutant β2AR refractory to S-nitrosylation (Cys265Ser) exhibits reduced desensitization and internalization, thereby amplifying NO-based signaling, and mice with Cys265Ser mutation are resistant to bronchoconstriction, inflammation, and the development of asthma. S-nitrosylation is thus a central mechanism in β2AR signaling that may be operative widely among GPCRs and targeted for therapeutic gain.
Keywords: S-nitrosylation, asthma, β2-adrenergic receptor, beta-agonist, nitric oxide, desensitization, receptor internalization, airway hyperreactivity, caveolae
eTOC blurb
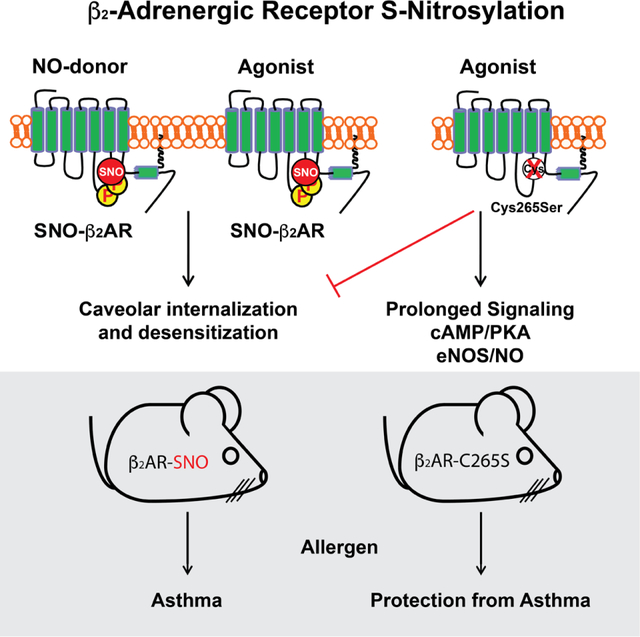
Post-translational modification of the β2AR by S-nitrosylation, resulting from receptor activation or direct modification, promotes receptor desensitization and caveolar internalization. Preventing β2AR S-nitrosylation leads to sustained receptor signaling, thereby increasing cAMP, Erk and NO. Mice with mutant β2AR refractory to S-nitrosylation are resistant to bronchoconstriction, inflammation and development of asthma, suggesting that this newly-identified desensitization mechanism may be targeted therapeutically in obstructive airway diseases.
Graphical Abstract
Introduction
Asthma is an allergic lung disease where chronic inflammation leads to airway remodeling and hyperreactivity that reduces lung function (Papi et al., 2018). The β2-adrenergic receptor (β2AR) is a G protein-coupled receptor (GPCR) that promotes airway relaxation, and β2AR agonists find extensive use as asthma therapeutics (Matera et al., 2020; Wendell et al., 2020). However, overstimulation of β2AR by β-agonists drugs can be detrimental and even lead to death, limiting their utility as front-line therapy (Kersten et al., 2017). Mechanistically, ‘overstimulation’ results in desensitization and downregulation of the β2AR (Wendell et al., 2020), serving as a model for GPCR aberrancy in disease. The molecular basis of receptor desensitization in the context of disease is not well understood.
Activation of the β2AR stimulates production of nitric oxide (NO) (Figueroa et al., 2009), which is implicated in airway relaxation (Antosova et al., 2017). However, the classic NO/guanylyl cyclase/protein kinase G (PKG) pathway that mediates relaxation of vascular smooth muscle has less relevance to airway smooth muscle (Gaston et al., 1994). Additionally, NO directly regulates proteins through S-nitrosylation, the post-translational modification of cysteine residues to form S-nitrosothiols (SNOs) (Hess et al., 2005). Protein S-nitrosylation may involve multienzyme machinery (Jia et al., 2014; Nakamura et al., 2021; Que et al., 2005; Seth et al., 2018; Stomberski et al., 2019) that conjugates and removes NO from proteins, including key GPCR regulatory components (G protein-coupled receptor kinase 2 (GRK2) (Whalen et al., 2007) and β-arrestins 1 and 2 (Hayashi et al., 2018; Ozawa et al., 2008)), which may influence airway responsivity (Que et al., 2005). These data raise the idea that effects of NO/SNO on airway tone may be mediated through regulation of the β2AR itself.
The β2AR co-localizes with eNOS (NO synthase type 3) to plasma membrane caveolae (Schwencke et al., 1999; Shaul et al., 1996), and active β2AR stimulates the production of NO (Figueroa et al., 2009). Further, β2AR signaling has been reported be inhibited by NO (Abi-Gerges et al., 2002; Adam et al., 1999), but the underlying mechanisms have not been identified. β2AR has 13 cysteines, including multiple intracellular residues potentially available for S-nitrosylation (Gether et al., 1997a; Wanschel et al., 2014). We therefore reasoned that the β2AR might be regulated by S-nitrosylation to desensitize the receptor, and demonstrate that β2AR stimulation is in fact coupled to S-nitrosylation of three intracellular Cys residues, including one (C265) that serves in allosteric control of receptor signaling, desensitization and internalization. Mutant mice refractory to S-nitrosylation are profoundly resistant to developing asthma. Our findings reveal general receptor signaling principles, including previously unappreciated roles for NO/SNO in receptor signaling, trafficking and desensitization. They also uncover a major role for NO/SNO in relaxation of airways that has been overlooked and expound a paradigm for how β2AR function might be targeted in asthma or other obstructive airway disease.
Results
β2AR is S-nitrosylated during activation
Treatment of HEK293 cells with the cell-permeable SNO donor ethyl ester CysNO (ECNO) promoted the S-nitrosylation of human β2AR (Figure 1A). As measures of specificity of S-nitrosylation in the SNO-RAC assay (Forrester et al., 2009), no SNO signal was generated in the absence of ascorbate (control) or when SNO is removed by UV photolysis prior to assay (Foster and Stamler, 2004). Importantly, activation of the β2AR with isoproterenol (ISO) also increased SNO-β2AR, through the activity of eNOS native to HEK293 cells (Hayashi et al., 2018; Ozawa et al., 2008), since pretreatment with the NOS inhibitor L-NMMA eliminated the agonist-stimulated SNO-β2AR signal (Figure 1B). ISO-stimulated SNO-β2AR was maximal after 10 minutes (Supplemental Figure 1A), and multiple β2AR agonists and partial agonists promoted SNO-modification of the receptor (Supplemental Figure 1B). We used mass spectrometry to identify sites on the β2AR modified by SNO after ECNO treatment and identified peptides containing three distinct sites: Cys265, Cys341 and Cys378 (Supplemental Figure 1C,D). Mutations in the β2AR of all Cys residues in the carboxyl tail region (Cys 328, 341, 378 and 406) plus Cys265 in the third loop near TM6 (five sites), or of the three sites from mass spectrometry (Cys265, Cys341 and Cys378), reduced ISO-stimulated SNO-β2AR to background levels (Figure 1C, Supplemental Figure 1E,F). Mutation of just one residue, Cys265, dramatically reduced ISO-stimulated SNO-β2AR (Figure 1D) without affecting expression level (Supplemental Figure 1G,H), establishing this as the primary site of modification. S-nitrosylation of native β2AR was also detected in mouse lungs (as will be described below) and in human and mouse heart samples (Figure 1 E,F), and the SNO level was increased in ischemic heart failure (Figure 1E) or by ISO infusion in vivo (Figure 1F). In the same human heart samples, SNO-β1AR was detected in normal heart, and SNO levels increased in both ischemic and non-ischemic heart failure (Supplemental Figure 1I). Additionally, angiotensin II receptor 1 (ATR1) was also S-nitrosylated basally in HEK cells, and this level was increased by AngII treatment (Supplemental Figure 1J). Thus, multiple GPCRs are S-nitrosylated following agonist stimulation or disease, and β2AR is modified by eNOS-derived SNO, primarily at Cys265, in both model cells and in vivo. Altogether, these data suggest a widespread role for S-nitrosylation in normal GPCR function in health and disease.
Figure 1. β2AR is S-nitrosylated primarily at Cys265 in response to NO donor or activation by agonist to regulate receptor desensitization.
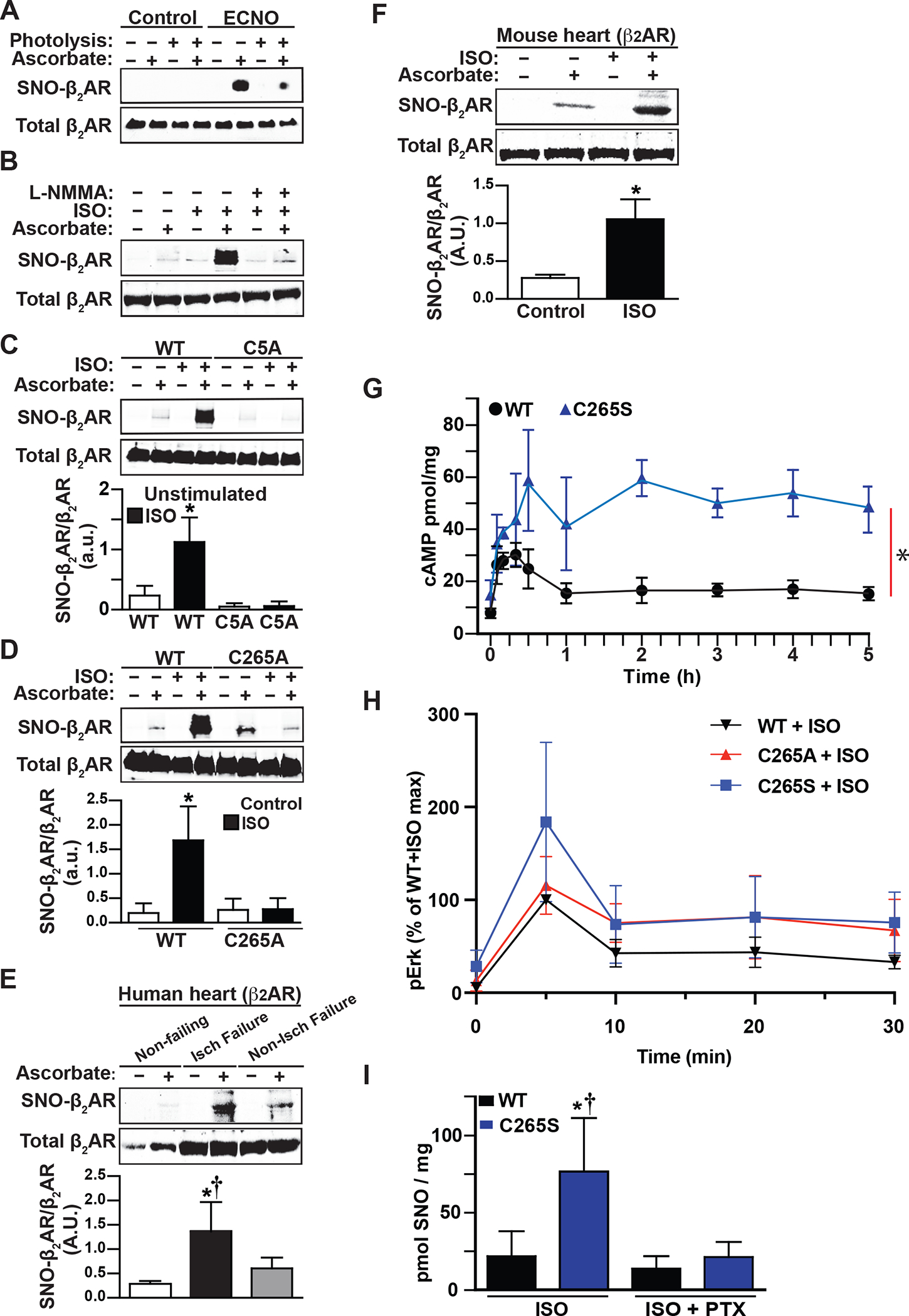
A. SNO-β2AR is detected by immunoblotting with anti-Flag antibody after isolation of SNO-proteins (by SNO-RAC) from HEK293 W9 cells stably expressing Flag-β2AR, untreated or treated with ECNO (100 μM, 10 min) prior to harvesting cells. Ascorbate (−) and UV photolysis during Cys blocking, which removes SNO from Cys residues, serve as specificity controls. Total β2AR control is from Flag immunoblot of lysate. Blot is representative of 3 replicates. B. SNO-β2AR detected by SNO-RAC of lysates from HEK293 cells stably expressing Flag-β2AR, untreated or treated with isoproterenol (ISO, 10 μM, 10 min). The NOS inhibitor L-NMMA was incubated with cells for 2h prior to stimulation. Total β2AR control is from Flag immunoblot of lysate. Blot is representative of 5 replicates. C. SNO-β2AR detected by SNO-RAC of lysates from HEK293 cells stably expressing Flag-β2AR WT (W9) or Flag-β2AR 5-Cys mutant (C5A), untreated or treated with ISO (10 μM, 10 min) prior to harvesting cells. Total β2AR control is from Flag immunoblot of lysate. Blot shown is representative of 5 replicates, which were quantified by densitometry and graphed as mean±SD. p<0.0001 by one-way ANOVA for treatment (F (3,16) = 29.22); * p<0.0001 basal vs ISO by Tukey test. D. SNO-β2AR detected by SNO-RAC of lysates from HEK293 cells stably expressing Flag-β2AR WT (W9) or Flag-β2AR C265A mutant, untreated or treated with isoproterenol (ISO, 10 μM, 10 min) prior to harvesting cells. Total β2AR control is from Flag immunoblot of lysate. Blot is representative of 6 replicates. p<0.0001 by one-way ANOVA for treatment (F (3,20) = 22.09); * p<0.0001 basal vs ISO by post-hoc Tukey multiple comparisons test. E. SNO-β2AR detected by SNO-RAC of lysates from human heart samples, from normal non-failing heart, ischemic failing heart, and non-ischemic failing heart. Total β2AR control is from anti-β2AR immunoblot of lysate. Representative blot is shown, of 3 non-failing, 3 ischemic and 3 non-ischemic failing heart samples tested, which are all quantified in the bar graph. * p<0.05 vs non-failing and non-ischemic failing heart by one-way ANOVA with Tukey post-hoc test. F. SNO-β2AR detected by SNO-RAC of lysates from mouse heart after 10 min i.v. infusion of saline control or isoproterenol (ISO). Total β2AR control is from anti-β2AR immunoblot of lysate. Blot shown is representative of 3 replicates, which are all quantified in the bar graph. * p<0.05 by t-test. G. Time course of ISO (10 μM)-stimulated cAMP accumulation in HEK293 cells stably expressing WT Flag-β2AR (W9) or Flag-β2AR-C265S mutation for the indicated times, measured using ELISA assay from cell lysates. Data are shown as mean±SD for 3 assays. * p=0.0033 in RM-ANOVA by genotype (F (1, 4) = 39.5). H. Time course of ISO (10 μM)-stimulated Erk activation in HEK293 cells stably expressing Flag-β2AR WT (W9) or Flag-β2AR-C265S or C265A mutations for the indicated times, measured using immunoblotting for phospho-Thr202/Tyr204-Erk1/2 from cell lysates, normalized to total Erk. Cells were preincubated with 100 ng/ml PTX overnight, or untreated. Data are plotted as mean±SD for 4 assays; representative pErk/Erk blots are shown as Supplemental Fig 2E and PTX sensitivity is plotted in Supplemental Fig 2F. I. Total protein SNO levels in HEK293 cells expressing WT or β2AR C265S, pretreated with PTX (100 ng/ml for 16h) and stimulated with ISO (10 μM for 5 min), measured by Hg-coupled photolysis/chemiluminescence. Data are shown as mean±SD for 5 assays; representative Nitrolyte traces are shown as Supplemental Fig 2G. * p<0.05 vs WT, † p<0.05 vs C265S+PTX using one-way ANOVA (F (3,16) = 10.34) and post-hoc Tukey multiple comparisons test.
β2AR S-nitrosylation at Cys265 regulates receptor signaling
The β2AR signals through cAMP (via Gs) and Erk (via Gi/β-arrestin), but also through activation of eNOS (Queen and Ferro, 2006). To understand the importance of Cys265 modification by SNO in β2AR function, we assessed signaling by C265 mutant β2ARs. While ISO-stimulated cAMP accumulation at 10 min of stimulation appeared normal (Supplemental Figure 2A,B), cAMP signaling by C265S (and C265A) mutants remained elevated through 30 min (Supplemental Figure 2C). On further analysis, cAMP accumulation by C265S β2AR remained markedly elevated through 5 hours (Figure 1G). Consistent with this, a pan-PKA-substrate phospho-site antibody detected prolonged elevation of PKA substrate phosphorylation following activation of C265S β2AR versus transient response of WT receptor (Supplemental Figure 2D). Similarly, Erk activation was prolonged after ISO-stimulation of C265S (and C265A) mutant receptors (Figure 1H, Supplemental Figure 2E). Using pertussis toxin (PTX), which prevents receptor activation of Gi pathways, we determined that ~80% of Erk activation by mutants was dependent on Gi proteins, in contrast to the much lower (~30–40%) Gi-dependence of WT receptor (reflecting WT dependence on βarrestin) (Lefkowitz et al., 2002) (Supplemental Figure 2E,F). Also, ISO-stimulated cellular S-nitrosylation was elevated in cells expressing C265S β2AR, demonstrating increased β2AR signaling through Gi to eNOS/NO that is sensitive to PTX (Figure 1I, Supplemental Figure 2G). This increased cAMP accumulation was not due to reduced phosphodiesterase, as basal PDE activity was higher in C265S-expressing cells (Supplemental Figure 2H). Altogether, these data demonstrate that S-nitrosylation of β2AR at Cys265 is important for termination of receptor signaling to PKA, Erk and eNOS. Furthermore, the sustained production of cAMP suggests that classic PKA phosphorylation of the β2AR is not sufficient to promote desensitization.
β2AR S-nitrosylation regulates receptor internalization through a PKA-caveolae pathway
Sustained signaling by Cys265-mutant β2ARs suggested that S-nitrosylation at Cys265 enables desensitization and/or trafficking from the cell surface. ISO-stimulated internalization of β2AR-C265A mutant from the cell surface was impaired (Figure 2A). Agonist-activated β2AR is reported to be internalized in clathrin-coated pits primarily through a pathway dependent on receptor phosphorylation by GRKs and binding to β-arrestins that scaffold adaptin-AP2 and clathrin (Ferguson et al., 1996; Laporte et al., 1999; Zhang et al., 1996). When β2AR C265A mutant was stimulated by ISO, receptors moved into intracellular vesicles and also recruited β-Arrestin2-GFP to the plasma membrane (Figure 2B, Supplemental Figure 3A), indicating that the mutation did not prevent agonist-activated β-Arrestin2 recruitment or the receptor phosphorylation that this requires.
Figure 2. β2AR S-nitrosylation drives agonist-independent receptor internalization through the caveolae pathway, but not the GRK/β-arrestin2/clathrin pathway.
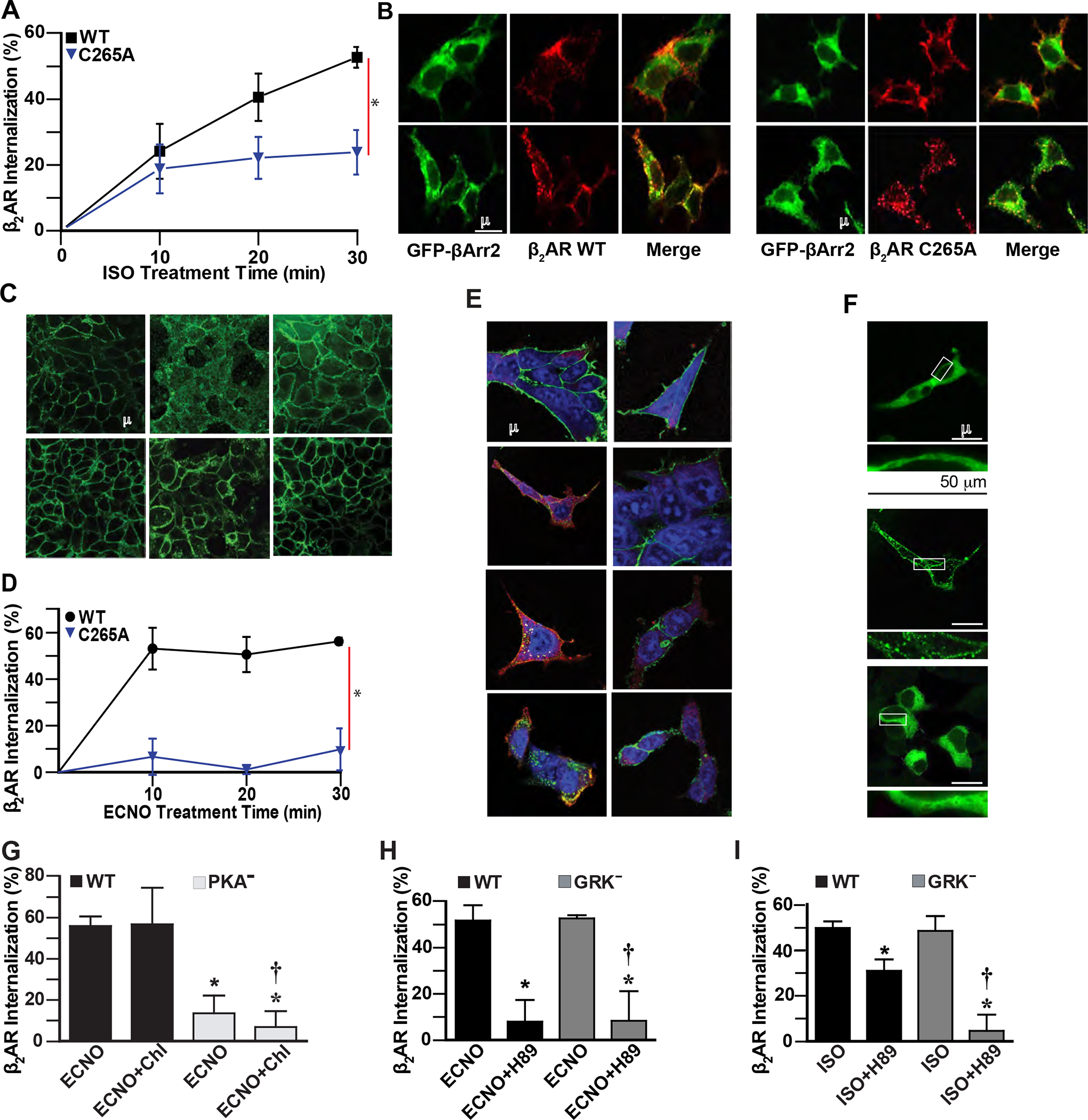
A. ISO (10 μM) induces time-dependent internalization of cell surface WT Flag-β2AR in HEK293 cells, but internalization of C265A β2AR is reduced, quantified by flow cytometry. Data are shown as mean±SD for 9 assays. * p<0.0001 for cumulative response (area under curve) for WT vs mutant using two-tail t-test test. B. ISO (10 μM, 10 min) induces colocalization of HA-β2AR or HA-β2AR C265A with β-Arrestin2-GFP (green) in HEK293 cells, detected by confocal microscopy (yellow) after labeling with anti-HA antibody/anti-rabbit AlexaFluor-568 (β2AR, red). Data are representative of 12 cells surveyed in 3 experiments. C. ECNO treatment (100 μM, 10 min) or overexpression of eNOS induces internalization of cell surface WT but not C5A mutant Flag-β2AR in HEK293 cells, detected by confocal microscopy using labeling with anti-Flag antibody and anti-mouse AlexaFluor-488. Images are representative of 5 assays. D. ECNO (100 μM) induces time-dependent internalization of cell surface WT but not C265A Flag-β2AR in HEK293 cells, quantified by flow cytometry after labeling with anti-Flag M1 and AlexaFluor-647. Data are shown as mean±SD for 3 assays. * p<0.0001 for cumulative response (area under curve) for WT vs drug-treated using two-tail t-test test. E. ISO (10 μM) treatment for the indicated times induces colocalization (yellow) of HA-β2AR and Flag-β-Arrestin2 in HEK293 cells, while ECNO (100 μM) treatment does not, detected by confocal microscopy after labeling with anti-HA antibody/anti-rabbit AlexaFluor-488 (β2AR, green), anti-Flag antibody/anti-goat AlexaFluor-568 (β-Arrestin2, red), and Draq5 (nuclei, blue). Data are representative of 15 cells surveyed in 3 experiments. F. ISO (10 μM, 10 min) induces translocation of β-Arrestin2-GFP (green) in HEK293 cells expressing WT β2AR, but treatment with ECNO (50 μM, 20 min) does not, detected by confocal microscopy. Lower panels are magnified versions of the boxed regions. Data are representative of 15 cells surveyed in 3 experiments. G. ECNO-driven (50 μM, 20 min) internalization of WT β2AR is not inhibited by the clathrin inhibitor chlorpromazine (Chl, 10 μM 1h), while the β2AR deficient in PKA phosphorylation sites (PKA−) is not internalized by ECNO, as measured by flow cytometry. Data are shown as mean±SD for 3 assays in triplicate. p<0.0001 by one-way ANOVA for treatment (F (3,32) = 75.29); * p<0.0001 vs WT ECNO and † p<0.0001 vs WT ECNO Chl by Tukey test. H. ECNO-driven (50 μM, 20 min) internalization of WT β2AR and GRK− mutant β2AR is inhibited by the PKA inhibitor H-89 (10 μM, 15 min prior to ECNO), as measured by flow cytometry. Data are shown as mean±SD for 6 assays. p<0.0001 by one-way ANOVA for treatment (F (3,20) = 84.55); * p<0.0001 vs WT ISO and † p<0.0001 vs GRK− ISO by Tukey test. I. ISO-driven (10 μM, 20 min) internalization of WT β2AR is partially inhibited by the PKA inhibitor H-89 (10 μM, 15 min prior to agonist), while internalization of GRK− mutant β2AR is completely inhibited, as measured by flow cytometry. Data are shown as mean±SD for 6 assays. p<0.0001 by one-way ANOVA for treatment (F (3,20) = 18.62); * p<0.0001 vs WT ISO and † p<0.0001 vs GRK− ISO by Tukey test.
Surprisingly, the NO-donor ECNO (or NO derived from eNOS) promoted internalization of WT β2AR from the cell surface in the absence of agonist, and this internalization was absent in the 5-cysteine or C265A mutants (Figure 2C,D). Dual labeling of β2AR and β-Arrestin2 showed that, in contrast to ISO stimulation, ECNO-driven receptor internalization did not lead to association of β-Arrestin2 with β2AR (Figure 2E). ECNO treatment was unable to recruit β-Arrestin2-GFP to β2AR at the plasma membrane or intracellular vesicles (Figure 2F), suggesting that NO/SNO induces an alternative pathway for β2AR internalization. To investigate this matter more rigorously, we used double-knockout (DKO) MEF cells lacking β-Arrestin1 and β-Arrestin2 (Kohout et al., 2001) to measure β2AR internalization, and found that these cells lacking β-Arrestins effectively internalized β2AR after stimulation with ECNO or ISO. Thus, S-nitrosylation-dependent receptor internalization does not require β-Arrestins (Supplemental Figure 3B). To confirm a role for endogenously derived NO in β-Arrestin-independent internalization, we expressed exogenous eNOS in these cells. Notably, eNOS induced receptor removal from the cell surface (Supplemental Figure 3B). Furthermore, the NOS inhibitor L-NMMA inhibited ISO-stimulated β2AR internalization (Supplemental Figure 3C). Taken together, these data demonstrate that endogenous NOS and NO can regulate β2AR internalization independently of β-Arrestins.
Because β-Arrestin2 recruitment to β2AR requires receptor phosphorylation by GRKs, we examined a β2AR mutant that lacks GRK phosphorylation sites (GRK− mutant) (Seibold et al., 1998). ECNO treatment stimulated equivalent internalization of the WT and GRK− β2AR (Supplemental Figure 3D). In contrast to the β2AR, the ISO-stimulated β1AR is known to internalize through two distinct pathways, a PKA/caveolae pathway as well as the GRK/β-arrestin/clathrin pathway (Rapacciuolo et al., 2003). Since β2AR localizes in caveolae in cell types that have these domains (Ostrom and Insel, 2004; Xiang et al., 2002) we sought to determine if ECNO-treated β2AR might also utilize a caveolae pathway. Indeed, ECNO-induced internalization of both WT and GRK− β2AR were inhibited equally by pretreatment with the caveolae inhibitors β-methyl-D-cyclodextrin (βMCD), genistein (Gen) or filipin III (Fili) (Supplemental Figure 3D,E.F). ECNO-stimulated internalization of the PKA− mutant β2AR, which lacks the four consensus PKA phosphorylation sites (Friedman et al., 2002), was greatly diminished, and was unaffected by treatment with chlorpromazine (Chl), an inhibitor of the clathrin internalization pathway (Figure 2G), while ECNO-stimulated internalization of both WT and GRK− mutant β2AR were both blocked by the PKA inhibitor H-89 (Figure 2H). Taken together, these data indicate that NO stimulates β2AR internalization through a caveolae pathway and that PKA phosphorylation of the receptor is required for NO-stimulated transit through caveolae.
Inhibition of the GRK/β-arrestin/clathrin pathway using the GRK− mutant β2AR renders ISO-stimulated GRK− β2AR internalization sensitive to the caveolae inhibitors βMCD, Gen and Fili (Supplemental Figure 3G,H,I). In the same way, inhibition of the caveolae pathway using the PKA− mutant β2AR renders ISO-stimulated receptor internalization sensitive to the clathrin inhibitor Chl (Supplemental Figure 3J), while inhibition of PKA using the inhibitor H-89 reduces internalization of the WT β2AR but ablates internalization of the GRK− mutant β2AR (Figure 2I). Altogether, these data show that S-nitrosylation at Cys265 drives β2AR internalization through a PKA-caveolae pathway, even in the absence of a receptor agonist, and mutation of Cys265 to prevent this modification reduces receptor internalization from the cell surface to prolong signaling activity. Thus, S-nitrosylation of Cys265 is necessary and sufficient (absent agonist) for receptor internalization via caveolae.
β2AR S-nitrosylation and PKA-phosphorylation are interdependent
Notably, removal of PKA phosphorylation sites from β2AR significantly reduced the ability of ECNO to S-nitrosylate the receptor (Figure 3A). Moreover, ISO-stimulated S-nitrosylation of WT β2AR was inhibited by treatment with the PKA inhibitor H-89 or KT-5720, or with the Gi inhibitor PTX that uncouples receptor from eNOS (Figure 3B, Supplemental Figure 4A). Thus remarkably, S-nitrosylation of β2AR is dependent on its phosphorylation by PKA. PKA phosphorylates two pairs of serines on the β2AR, at Ser261/262 in the third intracellular loop adjacent to the Cys265 SNO site and at Ser345/346 in the carboxyl terminal tail (Hausdorff et al., 1989), and PKA phosphorylation at Ser261/262 enables β2AR activation of Gi proteins (Daaka et al., 1997). ECNO-treatment induces significant phosphorylation of WT β2AR at Ser261/262, but phosphorylation is reduced in the C265A mutant receptor (Figure 3C). ECNO or ISO do not stimulate phosphorylation at Ser345/346 over basal on the WT receptor, but phosphorylation levels with or without stimulation are significantly reduced in the C265A mutant receptor (Supplemental Figure 4B). Thus, β2AR phosphorylation by PKA is dependent on S-nitrosylation of Cys265.
Figure 3. β2AR S-nitrosylation and PKA phosphorylation are co-dependent.
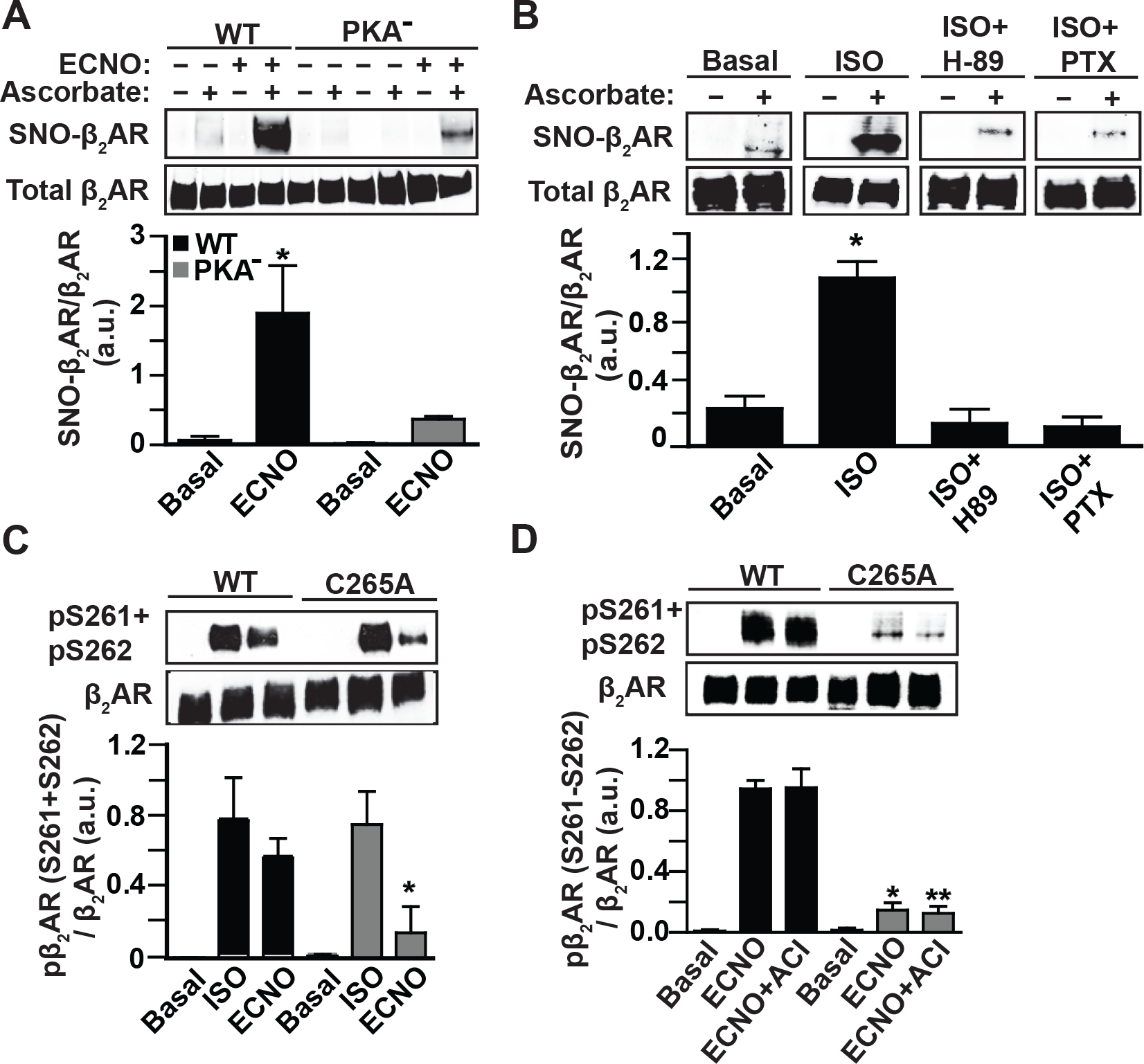
A. SNO-β2AR detected by SNO-RAC of lysates from HEK293 cells stably expressing Flag-β2AR WT or Flag-β2AR PKA− mutant, untreated or treated with ECNO (100 μM, 10 min) prior to harvesting cells. Total β2AR control is from Flag immunoblot of lysate. Blots are representative of 4 replicates. p<0.0001 by one-way ANOVA for treatment (F (3,16) = 39.37); * p<0.0001 basal vs ECNO by Tukey test. B. SNO-β2AR detected by SNO-RAC of HEK293 cells stably expressing Flag-β2AR, in cells pretreated with PKA inhibitor H-89 (10 μM, 15 min prior to ISO) or the Gi inhibitor PTX (100 ng/ml, 16h), untreated or treated with ISO (10 μM, 10 min), prior to harvesting cells. Total β2AR control is from Flag immunoblot of lysate. Blots are representative of 3 replicates that were quantified by densitometry and plotted as mean±SD. p<0.0001 by one-way ANOVA for treatment (F (3, 8) = 84.86); * p<0.0001 basal/H-89/PTX samples vs ISO by Tukey test. C. PKA phosphorylation of Flag-β2AR WT or C265A mutant in stable HEK293 cells, stimulated with ISO (10 μM, 10 min) or ECNO (100 μM, 10 min), detected by immunoblotting of cell lysates using anti-phospho-Ser261/Ser262-β2AR antiserum. Blots are representative of 3 replicates that were quantified by densitometry and plotted as mean±SD. p<0.0001 by one-way ANOVA for treatment F (5, 12) = 18.56; * p<0.0382 WT+ECNO vs C265A+ECNO by Tukey test. D. Protein kinase A-mediated phosphorylation of the Flag-β2AR WT or C265A mutant in stable HEK293 cells, pretreated with adenylyl cyclase inhibitor cocktail (membrane AC inhibitor 2’,5’-dideoxyadenosine 3’-triphosphate 10 μM plus soluble AC inhibitor KH7 50 μM, preincubated 1h), stimulated with ECNO (100 μM, 10 min), detected by immunoblotting of cell lysates using anti phospho-Ser261/Ser262-β2AR antiserum. Blot shown is representative of 3 replicates that were quantified by densitometry and plotted as mean±SD. p<0.0001 by one-way ANOVA for treatment (F (5,12) = 161.9); * p<0.0001 vs WT ECNO and ** p<0.0001 vs WT ECNO+ACI by Tukey test.
ECNO-mediated PKA phosphorylation of WT β2AR in the absence of ISO (Figure 3D) suggests a G protein-independent activity. Interestingly, PKA is directly activated by S-nitrosylation (Burgoyne and Eaton, 2009), so we tested whether SNO-PKA, rather than the traditional cAMP pathway, might mediate β2AR phosphorylation. ECNO treatment led to increased phospho-β2AR at Ser261/262 and Ser345/346 that was not reduced by pretreatment with adenylyl cyclase inhibitors (Figure 3D, Supplemental Figure 4C,D), suggesting that NO activates PKA independently of β2AR activation. Altogether, these data show that Cys265 S-nitrosylation is critical for PKA activation by the β2AR leading to phosphorylation of the receptor.
Since PKA phosphorylation of β2AR at Ser261/262 is known to be necessary for β2AR switching its coupling from Gs to Gi (Daaka et al., 1997), we wondered if G protein coupling was altered by β2AR S-nitrosylation. Using BRET-based assays for Gs and Gi coupling to the β2AR, we found no differences of C265A or C265S mutant receptors compared to WT β2AR (Supplemental Figure 4E), indicating that SNO-β2AR likely does not alter G protein coupling directly.
Overall, our results suggest a model for β2AR function (Supplemental Figure 4F) where β2AR stimulation promotes Gs-PKA activation that phosphorylates the receptor to promote Gi coupling and activation of eNOS, leading to feedback S-nitrosylation of the receptor. Receptor S-nitrosylation and PKA phosphorylation together induce trafficking of the receptor through caveolae, distinct from the GRK/arrestin/clathrin-mediated internalization expected for β2AR. β2AR S-nitrosylation in the absence of receptor activation by agonist promotes agonist-independent receptor phosphorylation by PKA and internalization through the caveolae pathway. In the absence of β2AR S-nitrosylation (due to NOS inhibition, Gi or PKA inhibition, or to PKA-site or SNO-site mutations), the agonist-activated receptor fails to internalize through this caveolae pathway, leading to prolonged signaling by Gs-cAMP/PKA, and also by Gi-eNOS/NO. Prolonged signaling by Gs and Gi in mutant receptors suggests that in the wildtype β2AR, S-nitrosylation functions as a feedback regulatory mechanism to desensitize the receptor to Gi-eNOS signaling, in coordination with traditional feedback desensitization through Gs-PKA signaling.
β2AR S-nitrosylation masks NO-based bronchodilation in vivo
β2AR is expressed in the lung, and native β2AR in mouse lung is S-nitrosylated under basal conditions (Figure 4A, Supplemental Figure 1J). To understand the role of β2AR S-nitrosylation in vivo, we generated knock-in mice bearing the β2AR C265S mutation (Supplemental Figure 5A,B,C). ADRB2C265S/C265S (C265S) mice survive and breed normally, and have normal level of expression of β2AR in the lung (Supplemental Figure 5D,E). When subjected to airway challenge with methacholine, a muscarinic agonist that constricts airways, C265S mice are substantially protected from airway constriction (Figure 4B). We predicted that this airway protection by the C265S β2AR would be dependent on prolonged PTX-sensitive Gi signaling to eNOS, and pretreatment of mice with PTX eliminated the protective effect of C265S on methacholine-induced airway resistance (PTX also exacerbated airway constriction in WT mice) (Figure 4C). Further, inhibition of NOS, either pharmacologically with L-NAME or genetically by crossing with mice deficient in eNOS (eNOS−/−), eliminated the protective effect of C265S on methacholine-induced airway constriction (Figure 4C). Taken together, these data demonstrate a major, previously unappreciated role for NO in β2AR-mediated airway relaxation. However, this Gi-stimulated eNOS-mediated airway relaxation is masked by feedback S-nitrosylation of the β2AR.
Figure 4. Essential role of endogenous NO in airway relaxation is counteracted by β2AR S-nitrosylation.
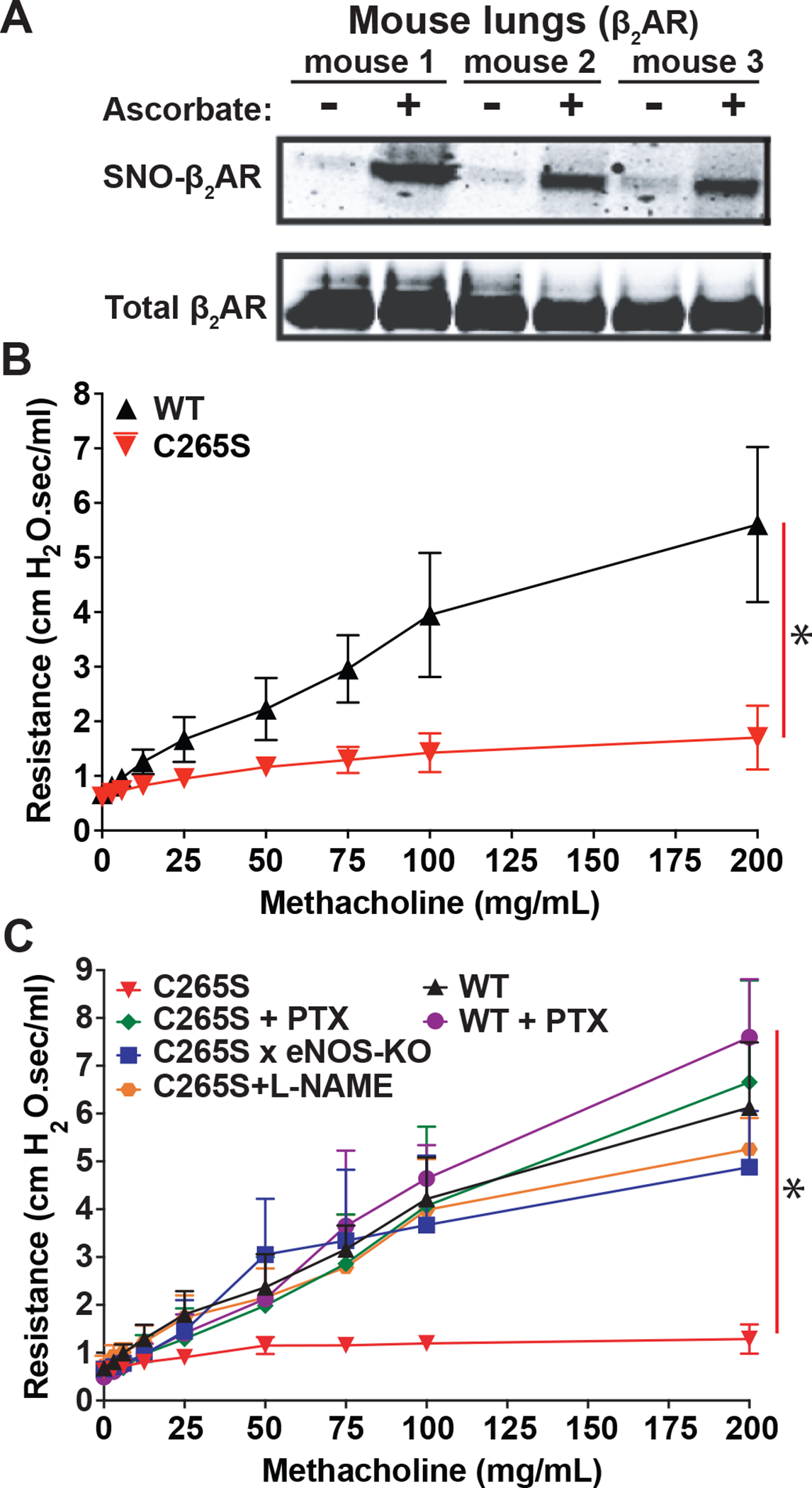
A. Native SNO-β2AR detected by SNO-RAC of lysates from WT mouse lung (n=3 mice). Total β2AR control is from immunoblot of lung lysate. B. Respiratory system resistance in mice bearing WT β2AR vs. β2AR C265S knock-in (C265S), measured by flexiVent after the indicated progressive dose of aerosolized methacholine. Data are shown as mean±SD for 16 mice each. * p<0.0001 for cumulative response (area under curve) by genotype using two-tail t-test. C. Airway resistance in mice bearing WT β2AR vs. β2AR C265S knock-in (C265S), after pretreatment with PTX (100 μg/kg i.p. 18h prior to assay) or L-NAME (100 mg/kg i.p. 1h prior to assay), or in mice bred to have β2AR C265S plus eNOS deficiency. Resistance was measured by flexiVent after the indicated dose of methacholine. Data are shown as mean±SD for 8 mice each. p<0.0001 in a two-way repeated measures ANOVA among genotypes and treatment over dose (F (5, 42) = 10.37); * p<0.0002 for cumulative response (area under curve) using Tukey test, for all groups vs C265S.
To extend these findings to other physiological systems, we assessed contraction and relaxation of isolated aorta in vitro. Contraction in response to phenylephrine (through Gq-coupled α1-adrenergic receptor) is markedly reduced in C265S mice, while ISO-stimulated relaxation is essentially unchanged (Supplemental Figure 5F,G).
β2AR S-nitrosylation promotes asthmatic response
Airway hyperreactivity is one hallmark of asthma, so we reasoned that C265S mice might be protected from hyper-reactivity in an asthma model. We sensitized mice to house dust mite allergens (Dermatophagoides pteronyssinus, D.p. or Dermatophagoides farina, D.f.), and tested the sensitivity of airways to bronchoconstriction by methacholine. While WT mice exhibited increased methacholine-induced airway resistance after exposure to D.p. or D.f. allergens, C265S mice exhibited minimal airway bronchoconstriction to methacholine, with or without allergen pre-exposure (Figure 5A,B). Cyclic AMP levels in lung homogenates of naive mice were elevated in C265S vs WT, replicating cellular data (Figure 1G), and remained high after allergen exposure (Figure 5C), consistent with blocked desensitization. Lung expression of muscarinic receptors as assessed by qRT-PCR, and M1, M2, M3 and M5 receptor mRNA levels failed to increase after allergen sensitization of C265S β2AR mice (Supplemental Figure 6A–D). Other muscarinic pathway components (Cpi17, RhoA, Prkcd) showed a similar pattern, while CD38 expression dropped in allergen-treated C265S β2AR samples (Supplemental Figure 6E–H). Thus, the protective effect of C265S β2AR expression appears to result from both direct signaling effects (chronically elevated cAMP and NO) and beneficial compensatory changes (reduction of muscarinic signaling components that lead to bronchoconstriction). Understanding of this well-described cross-talk between muscarinic and adrenergic systems is poor, but evidently may reflect S-nitrosylation status of β2AR based on evidence in C265A mice.
Figure 5. β2AR C265S protects airway function in allergic asthma model.
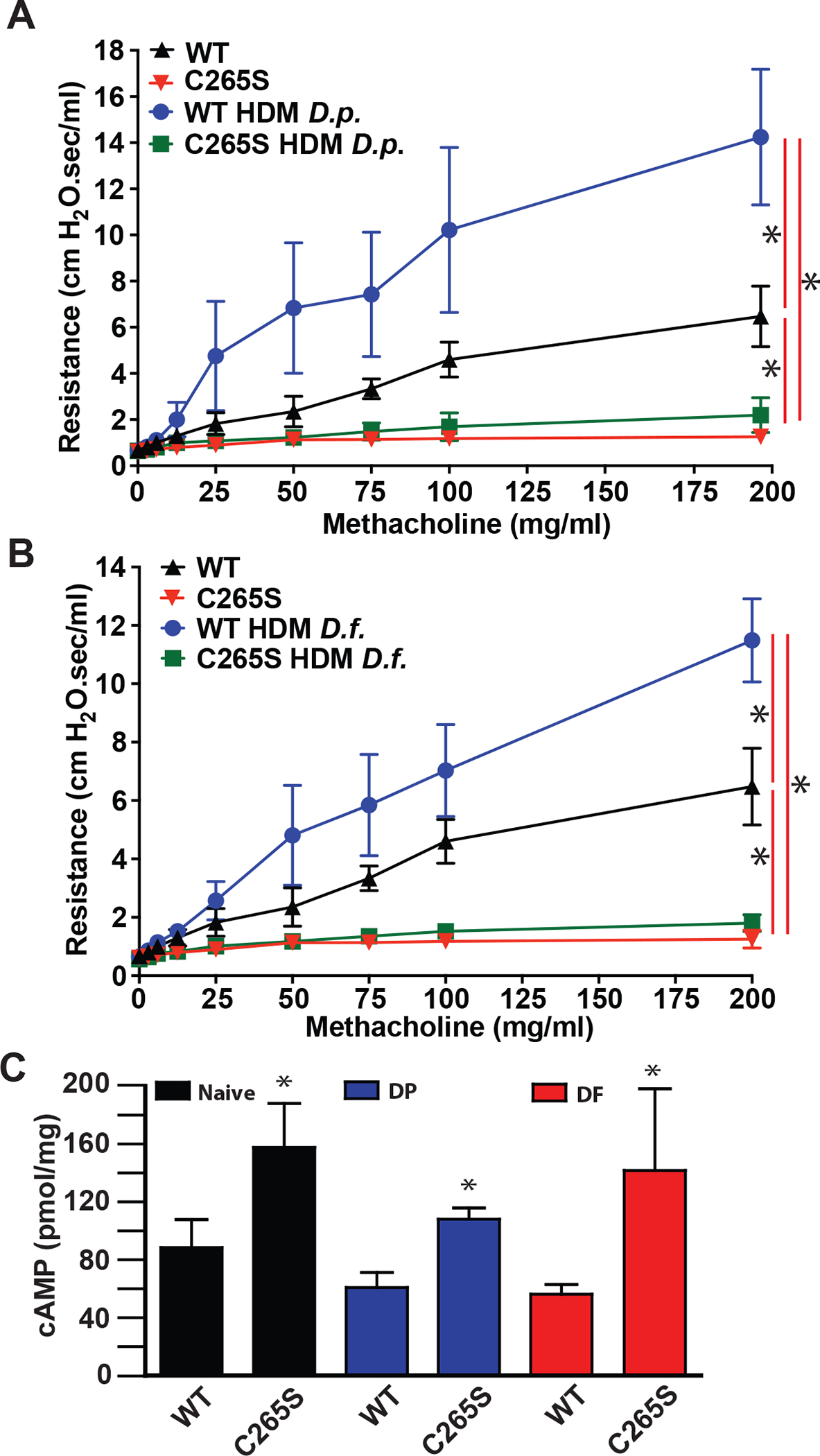
A. Respiratory system resistance in mice bearing β2AR WT vs. β2AR C265S knock-in (C265S), following pretreatment with house dust mite D. pteronyssinus allergen (D.p.; 100 μg i.p. on day 0, 30 μg intranasal instillation on days 1–14, tested on day 15), measured by flexiVent after the indicated dose of aerosolized methacholine. Data are shown as mean±SD for 9 mice each. p<0.0001 in a two-way repeated measures ANOVA among genotypes and treatment over dose (F (3, 32) = 74.42); * p<0.0001 for cumulative response (area under curve) using Tukey test. B. Airway resistance in WT vs. C265S mice, following pretreatment with house dust mite D. farinae allergen (D.f.; treatment as in (A)), measured by flexiVent after the indicated dose of aerosolized methacholine. Data are shown as mean±SD for 9 mice each. p<0.0001 in a two-way repeated measures ANOVA among genotypes and treatment over dose (F (3, 32) = 156.1); * p<0.0001 for cumulative response (area under curve) using Tukey test. C. Cyclic AMP levels in whole lung lysate from WT or C265S mice before vs after treatment with house dust mite allergens, measured by ELISA. Data are shown as mean±SD for 4 mice each. * p<0.05 using two-tailed t-test comparing WT and C265S for each treatment (naïve, D.p., D.f.).
Asthma is characterized by elevated cytokines and immune cell infiltration into the lung and these have been linked to β2AR activity and cAMP (Papi et al., 2018; Wendell et al., 2020). Asthma-associated inflammatory cytokines, including IL4, IL5, IL9, IL10, IL13 and interferon-γ, all failed to increase after allergen exposure in C265S mouse lungs (Supplemental Figure 7A–F). Quantification of infiltrated immune cells from bronchoalveolar lavage fluid following allergen exposure revealed that expected increases in total white blood cells, neutrophils, lymphocytes, and eosinophils are all blunted in C265S mice (Figure 6A–D). Chronic inflammation in asthma leads to airway remodeling, where lung structure is altered by immune- and injury-driven proliferation of smooth muscle, epithelia, fibroblasts and extracellular matrix to generate thicker airway walls and narrowed airway lumen (Papi et al., 2018; Wendell et al., 2020). Lungs from allergen-treated C265S mice failed to show elevated levels of epithelial cell adhesion molecule (EpCAM) or immune cell marker myeloperoxidase (MPO) (Figure 6E). Lung sections stained with Masson’s Trichrome revealed thickened airway walls with fibrosis in allergen-sensitized WT mice, but C265S mice were largely protected from these changes (Figure 6F). When lung sections were stained with H&E, thickened airways and focal inflammation within alveoli that were evident in allergen-sensitized WT mice were notably absent from treated C265S mice (Figure 6G). Altogether, these data demonstrate that S-nitrosylation of the β2AR at Cys265 subsequent to its activation, or through heterologous production of NO, is a critical part of classic desensitization of this receptor, and in an asthma model in vivo, this receptor downregulation is ultimately maladaptive and contributes to multiple aspects of allergic asthma pathology.
Figure 6. β2AR C265S protects against immune, inflammatory and remodeling effects in asthma model.
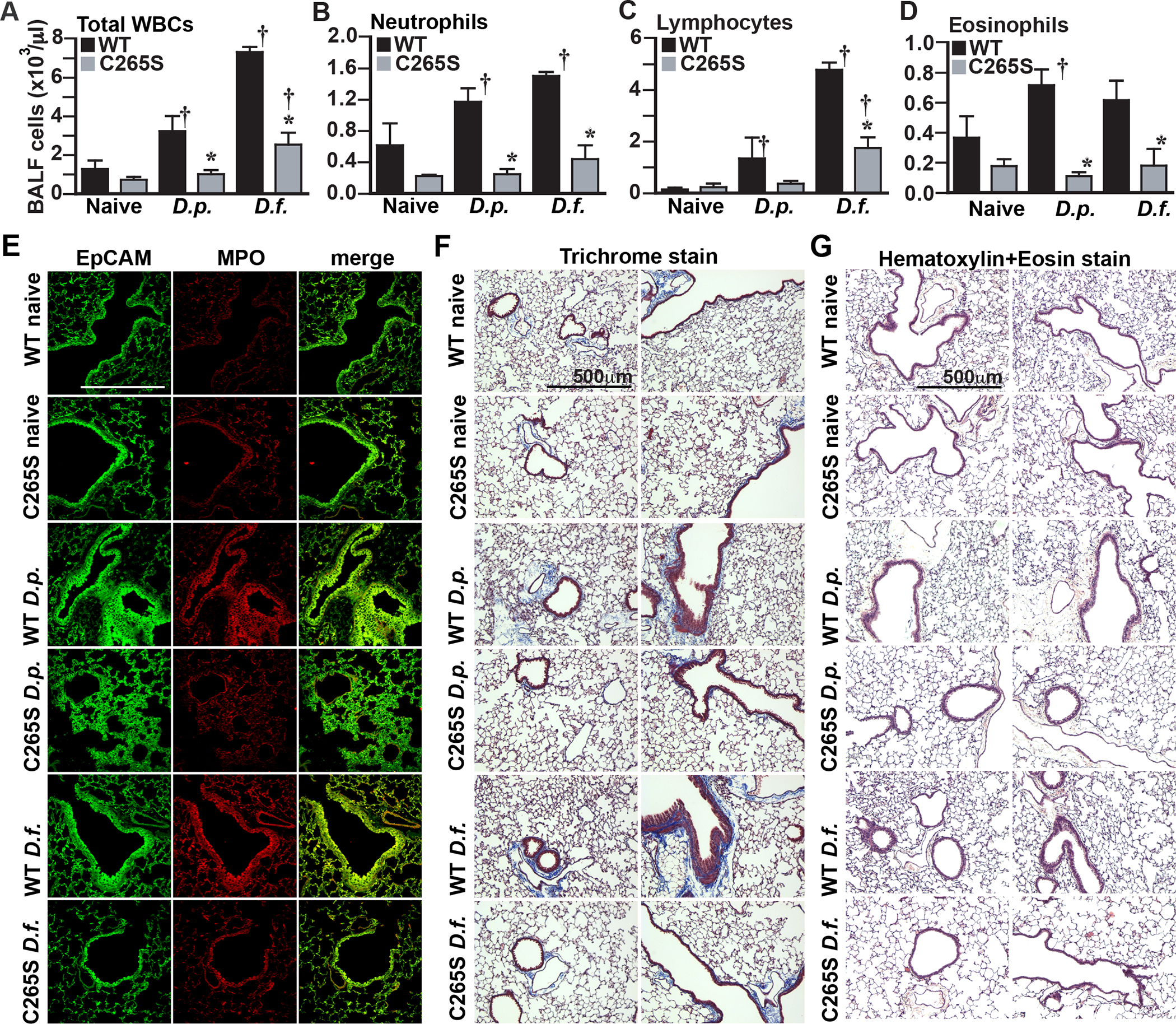
A.-D. Total infiltrating immune cell numbers in bronchoalveolar lavage fluid from WT and C265S mice treated with house dust mite allergens, measured by HEMAVENT 950 cytometry. (A) Total infiltrating white blood cells (WBC), (B) neutrophils, (C) lymphocytes, (D) eosinophils. Data is shown as mean±SD from samples from 3 mice. p<0.0001 for all panels A-D by one-way ANOVA; * p<0.005 vs corresponding WT treatment control, or † p<0.05 from corresponding naïve control, by Tukey test. E. Immunofluorescent staining for epithelial cell adhesion molecule (EpCAM, green) and myeloperoxidase (MPO, red), and merge for colocalization (yellow), in lung sections from WT and C265S mice treated with the indicated house dust mite allergens. Immune cell infiltration at sites of hyperplasia (visible as yellow) are prominent in allergen-treated WT mice, but largely absent from C265S mice. Confocal photos shown are representative of 4 individual mice assessed in each condition. F. Masson’s Trichrome staining in sections of lungs from WT and C265S mice treated with house dust mite allergens. Bronchial muscle thickening, epithelial hyperplasia and luminal occlusion are prominent in allergen-treated WT mice, but largely absent from C265S mice. Photos shown are representative of 4 individual mice assessed in each condition. G. H&E staining in sections of lungs from WT and C265S mice treated with house dust mite allergens. Epithelial hyperplasia, immune cell infiltration and luminal occlusion are prominent in allergen-treated WT mice, but largely absent from C265S mice. Photos shown are representative of 4 individual mice assessed in each condition.
Discussion
The classic GPCR signaling paradigm entails activation of G protein, synthesis of cAMP, and PKA-driven phosphorylation events, including desensitization and internalization of the receptor. For airway β2AR, transduction results in cAMP/PKA-mediated relaxation of airway smooth muscle, providing a mainstay treatment for airway diseases. In this report, we show that prototypic signaling through the β2AR involves an essential, previously unappreciated role for nitric oxide. Notably, S-nitrosylation by NO is required for both classic desensitization by PKA as well as desensitization of previously unappreciated NO-based signaling that mediates airway relaxation. Mutant receptor (C265S) resistant to S-nitrosylation cannot be desensitized and thus maintains signaling to cAMP/PKA, but also to eNOS/NO necessary for airway relaxation. Mice bearing the β2AR-C265S mutation thus lose the ability to bronchoconstrict, leading to marked protection from asthma. We conclude that β2AR S-nitrosylation is a central regulatory mechanism that limits receptor signaling and contributes to the asthmatic phenotype, and that targeting this mechanism to extend β2AR activity can ameliorate airway disease.
The β2AR is modified by S-nitrosylation in response to agonists and exogenous sources of NO and exhibits endogenous S-nitrosylation in situ. The major SNO site, Cys265, is located in the receptor third intracellular loop at transmembrane span 6 (TM6), a region critically important for active state conformation changes that mediate G protein coupling (Gether et al., 1997a; Gether et al., 1997b; Kobilka, 2002) and adjacent to one of two clusters of PKA phosphorylation sites (Ser261,262) (Hausdorff et al., 1989). Indeed, Cys265 is commonly used to label β2AR for conformation measurements relating to TM6 (Gether et al., 1997b; Kobilka, 2002). Additionally, Cys265 is a site for dynamic β2AR palmitoylation, although on a distinctly slower timescale (hours) and without obvious effects (Adachi et al., 2016). Thus, a regulatory role for Cys265 has not been previously considered. Strikingly evident in our assays is that receptor S-nitrosylation can act as an allosteric promoter of receptor internalization in the absence of orthosteric ligand. This form of receptor regulation has not been described previously and suggests fundamentally distinct approaches to regulate GPCR function. A recent report that thiol alkylators can modify the P2Y6 receptor to drive its internalization and ubiquitin-mediated downregulation (Nishiyama et al., 2022) reinforces our contention that endogenous nitrosylation, as shown here for multiple GPCRs (Figure 1E,F; Figure 4A, Supplemental Figure 1I,J), may represent a general mode of receptor regulation. Inasmuch as signaling by GPCRs may occur within endosomes (Irannejad et al., 2013), the signaling dynamics of receptors internalized allosterically should be further explored.
Our studies with β2AR-C265S mutant, and with receptor mutants unable to be phosphorylated by PKA (PKA−), or by GRKs (GRK−) to engage the GRK/arrestin/clathrin internalization pathway, reveal that SNO-β2AR can unexpectedly engage a distinct PKA/caveolae internalization pathway that has been described for other GPCRs, including the β1AR (Rapacciuolo et al., 2003), but has not been appreciated previously for β2AR. That is, in cells where β2AR activates eNOS, distinct caveolae and clathrin internalization pathways are dynamically engaged in response to feedback regulation by NO or GRKs, respectively. In the β2AR-C265S mutant or PKA− mutant, the inability to be S-nitrosylated or PKA phosphorylated thus allows sustained signaling to Gs/cAMP/PKA and β-Arrestin2/Erk, but critically also to Gi/eNOS/NO. However, while signaling to cAMP may be self-limiting given the multiple mechanisms capable of diminishing β2AR responsiveness over time (e.g. cAMP phosphodiesterases, PKA desensitization of Gs and adenylyl cyclase), sustained NO production via eNOS is independent of cAMP/PKA. That distinct Gs/cAMP/PKA and Gi/eNOS/NO pathways are operative is most clearly evident in β2AR C265S mice where elevations are seen in both cAMP and NO bioactivity (due to prolonged Gs/Gi signaling) but where protection from bronchoconstriction is abolished by PTX or eNOS inhibition. NO mediated receptor S-nitrosylation is thus a previously unappreciated regulatory mechanism that proceeds from receptor activation and feeds back to desensitize a critical pathway leading to NO production. Ligands (long and short acting β2-agonists) that bias internalization to one pathway over another or that differentially couple β2AR to eNOS and thus to receptor S-nitrosylation might be exploited for therapeutic benefit.
β2AR-C265S mice are not only protected from bronchoconstriction, but also from airway inflammation, consistent with a known role for the β2AR in asthmatic airway inflammation (Nguyen et al., 2009), and implicating β2AR S-nitrosylation in the phlogistic effect of β2AR stimulation. This is a provocative finding, since prolonged or repeated use of β2AR agonists in asthma has been associated with poor outcomes (including death) in patients and in animal models (Amrani and Bradding, 2017; Knight et al., 2015; Lin et al., 2012; Vitale et al., 2017) and NO derived from inflammatory cells is markedly elevated in asthma (Kharitonov et al., 1994). Further, in mouse models the development of asthma actually requires epinephrine/β2AR stimulation, and asthma development can be attenuated with β-blockers, which prevent receptor desensitization (Nguyen et al., 2017; Nguyen et al., 2009; Thanawala et al., 2013; Thanawala et al., 2015). Importantly, C265S mutant mice resistant to desensitization are resistant to the development of allergic asthma, including airway hyper-reactivity, immune inflammation and chronic remodeling of the lung.
NO bioactivity plays multiple roles in lung function (Antosova et al., 2017), but there has been little clarity as to its role in asthma. Asthmatics have elevated iNOS activity in immune cells (Kharitonov et al., 1994), and elevated NO gas in exhaled air is used as a biomarker of lung inflammation (Hoyte et al., 2018). However, iNOS knockout mice are not protected from asthma (Landgraf et al., 2005). Likewise, NO can relax airways (Antosova et al., 2017), but eNOS knockouts have no asthmatic phenotype (Mathrani et al., 2007). Thus, the prevailing view is that NO plays a minor role in asthma. Our data suggest otherwise and provide a fresh perspective whereby signaling through the β2AR includes a major role for NO/SNO in relaxing airways that is blocked upon β2AR desensitization. In this model, desensitization of the β2AR involves an essential role for feedback S-nitrosylation that mediates airway constriction and inflammation in asthmatic mice. Thus, desensitization of the β2AR is causal in asthma and may likely involve SNOs that can both bronchodilate and S-nitrosylate the receptor. Our data are consistent with reports that endogenous SNOs are potent relaxants in human airways (Gaston et al., 1993) and that the SNO-metabolizing enzyme GSNO reductase (GSNOR) regulates β2AR signaling and desensitization (Que et al., 2005). Intriguingly, gene association studies have identified β2AR (ADRB2) and GSNOR (ADH5) gene polymorphisms in asthma patients (Choudhry et al., 2010; Moore et al., 2009; Wu et al., 2007), and ADRB2/ADH5 interaction predicts response to β-agonists (Moore et al., 2009), suggesting a mechanistic link involving β2AR S-nitrosylation. Collectively, these data suggest reevaluation of the role of NO in asthma and an additional therapeutic approach to airway disease.
Limitations of the Study.
Although we identified three sites in the β2AR that can be S-nitrosylated in model cells, we characterized only the primary site, Cys265. The effect of S-nitrosylation of the other two sites individually and collectively remains unknown, as do conditions that may favor their modification (O’Dowd et al., 1989). Distinct pathways, such as regulation of PP2-dependent β2AR dephosphorylation that alters receptor re-sensitization, can also lead to asthma protection (Gupta et al., 2015), but whether and how this intersects with β2AR regulation by SNO (or particularly receptor de-nitrosylation) remain unexamined. The β2AR has been reported to signal in endosomes after clathrin pathway internalization (Irannejad et al., 2013), but any β2AR endosomal signaling after caveolar internalization or after β2AR S-nitrosylation remains undefined. The β2AR C265S mouse is a constitutive knock-in, so that prolonged or otherwise altered signaling by the mutant receptor through development may lead to adaptive changes. Among the changes we do see are altered expression of multiple signaling proteins in pro-contractile pathways, but the mechanisms linking β2AR S-nitrosylation and these changes remain uncharacterized. We utilized a mouse model of allergic asthma, which may differ from asthma in humans and other mouse species, and treatment of mice with PTX or L-NAME will have effects outside airways.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jonathan Stamler (jss156@case.edu).
Materials Availability
Plasmids and animal models generated in this study will be shared by the lead contact upon request, subject to an institutional MTA.
Data and code availability
Raw image data for all Figures have been deposited with Mendeley and are publicly available as of the date of publication. The accession number is listed in the Key Resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-Flag M1 (mouse monoclonal) | Sigma-Aldrich | Cat# F3040 RRID:AB_439712 |
| anti-Flag M2 (mouse monoclonal) | Sigma-Aldrich | Cat# F3165 RRID:AB_439685 |
| anti-HA-7 (mouse monoclonal) | Sigma-Aldrich | Cat# H9658 RRID:AB_260092 |
| anti-Erk1/2 | Cell Signaling Technology | Cat# 4695 RRID:AB_390779 |
| anti-Erk1/2 pThr202+pTyr204 | Cell Signaling Technology | Cat# 9101 RRID:AB_331646 |
| anti-Protein kinase A - catalytic subunit | Cell Signaling Technology | Cat# 5842 RRID:AB_10706172 |
| anti-Protein kinase A - catalytic subunit, pThr197 | Cell Signaling Technology | Cat# 5661 RRID:AB_10707163 |
| anti-Pan-PKA substrate pSer/pThr | Cell Signaling Technology | Cat# 9621 RRID:AB_330304 |
| anti-β2 adrenergic receptor (H-20) | Santa Cruz Biotechnology | Cat# sc-569 Lot# L0415 RRID:AB_630926 |
| anti-β2AR pSer261/pSer262 | Robert Lefkowitz (Nobles et al., 2011) | N/A |
| anti-β2AR pSer345/pSer346 | Robert Lefkowitz (Nobles et al., 2011) | N/A |
| anti-β1AR (A-20) | Santa Cruz Biotechnology | Cat# sc-567 RRID:AB_2225390 |
| anti-Phospholipase C-beta1 (D8) | Santa Cruz Biotechnology | Cat# sc-5291 RRID:AB_628118 |
| anti-Phospholipase C-beta3 (D-7) | Santa Cruz Biotechnology | Cat# sc-133231 RRID:AB_2299534 |
| anti-EpCAM (CD326) - Alexafluor488 Conjugate | BioLegend | Cat# 118210 RRID:AB_1134099 |
| anti-myeloperoxidase | Abcam | Cat# ab45977 RRID:AB_944318 |
| anti-GAPDH (6C5) | Millipore (Sigma-Aldrich) | Cat# MAB374 RRID:AB_2107445 |
| Donkey anti-rabbit-Alexafluor594 | Molecular Probes, ThermoFisher | Cat# A32754 RRID:AB_2762827 |
| Goat anti-mouse-Alexafluor568 | Molecular Probes, ThermoFisher | Cat# A11031 RRID:AB_144696 |
| Goat anti-rabbit-Alexafluor488 | Molecular Probes, ThermoFisher | Cat# A32731 RRID:AB_2633280 |
| Goat anti-rabbit-Alexafluor647 | Abcam | Cat# ab150083 RRID:AB_2714032 |
| Bacterial and Virus Strains | ||
| N/A | ||
| Biological Samples | ||
| Human heart failure patient ventricular tissue | Biorepository and Precision Pathology Center, Duke University | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Thiopropyl-Sepharose 6B | Sigma-Aldrich | T8387 |
| S-Methyl methanethiosulfonate (MMTS) | Sigma-Aldrich | 64306 |
| L-NG-monomethyl arginine (L-NMMA) | Cayman Chemical | 80200 |
| L-NG-Nitroarginine methyl ester (L-NAME) | Cayman Chemical | 80210 |
| (−)-Isoproterenol (+)-bitartrate salt | Sigma-Aldrich | I2760 |
| Salbutamol | Sigma-Aldrich | S8260 |
| Salmeterol | Sigma-Aldrich | S5068 |
| Pindolol | Sigma-Aldrich | P0778 |
| Procaterol | Sigma-Aldrich | P9180 |
| Fenoterol | Sigma-Aldrich | F1016 |
| Epinephrine | Sigma-Aldrich | E4250 |
| Isoetharine | Sigma-Aldrich | I3639 |
| Pertussis toxin | Sigma-Aldrich | P7208 |
| DRAQ5 | Thermo-Fisher | 62251 |
| β-methyl cyclodextrin | Sigma-Aldrich | C4555 |
| Chlorpromazine hydrochloride | Sigma-Aldrich | C0982 |
| H-89 (N-[2-(p-Bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide dihydrochloride) | Sigma-Aldrich | B1427 |
| Genistein | Sigma-Aldrich | G6649 |
| Filipin III | Sigma-Aldrich | SAE0087 |
| Methacholine chloride | Sigma-Aldrich | 1396364 |
| 2’,5’-dideoxyadenosine 3’-triphosphate | Sigma-Aldrich | D0939 |
| KH7 ((E)-2-(1H-Benzo[d]imidazol-2-ylthio)-N’-(5-bromo-2-hydroxybenzylidene)propanehydrazide) | Sigma-Aldrich | K3394 |
| house dust mite Dermatophagoides pteronyssinus allergen | Stallergenes-Greer | B70 |
| house dust mite Dermatophagoides farinae allergen | Stallergenes-Greer | B64 |
| X-tremeGENE 9 transfection reagent | Sigma-Aldrich | 6365779001 |
| X-tremeGENE HP transfection reagent | Sigma-Aldrich | 6366244001 |
| KT-5720 | Tocris | 1288 |
| SQ-22536 | Tocris | 1435 |
| Phenylephrine hydrochloride | Sigma-Aldrich | 1533002 |
| [3H]-alprenolol; Dihydroalprenolol Hydrochloride, Levo-[Ring, Propyl-3H(N)] | PerkinElmer | NET720250UC |
| Critical Commercial Assays | ||
| PARAMETER cAMP ELISA kit | R&D Systems | KGE012 |
| GloSensor cAMP biosensor reagent | Promega | E1290 |
| Aurum Total RNA Fatty and Fibrous Tissue Kit | Bio-Rad | 7326830 |
| iScript cDNA Synthesis Kit | Bio-Rad | 1708890 |
| Fast SYBR Green Master Mix | Thermo-Fisher | 4385612 |
| TaqMan Fast Advanced Master Mix | Thermo-Fisher | 4444556 |
| cAMP Phosphodiesterase activity kit (Fluorometric) | Biovision | K2013-100 |
| Deposited Data | ||
| Image raw data files | Mendeley Data | http://dx.doi.org/10.17632/p2zycrb6dr |
| Experimental Models: Cell Lines | ||
| HEK293 | ATCC | Cat# CRL-1573 RRID:CVCL_0045 |
| HEK293 W9, stably expressing human Flag-β2AR | Robert Lefkowitz, Duke University | Shenoy et al., 2006 PMID 16280323 |
| HEK293, stably expressing HA-ATR1 | this study | N/A |
| Arrb1-/- mouse embryonic fibroblast (MEF) cell line | Robert Lefkowitz, Duke University | Kohout et al., 2001, PMID 11171997 |
| Arrb2-/- mouse embryonic fibroblast (MEF) cell line | Robert Lefkowitz, Duke University | Kohout et al., 2001, PMID 11171997 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Adrb2Cys265Ser/Cys265Ser knock-in | this study | N/A |
| Mouse: Nos3 knockout: B6.129P2-Nos3tm1Unc/J | The Jackson Laboratories | Cat# Jax:002684 RRID:IMSR_JAX:002684 |
| Mouse: FLP deleter strain: B6.Cg-Tg(ACTFLPe)9205Dym/J | The Jackson Laboratories | Cat# Jax:005703 RRID:IMSR_JAX:005703 |
| Mouse: FVB.129-Adrb2tm1Bkk/J | The Jackson Laboratories | Cat# Jax: 031496 RRID:IMSR_JAX:031496 |
| Oligonucleotides | ||
| See Supplemental Table I | this study | N/A |
| Recombinant DNA | ||
| GloSensor-20F cAMP biosensor | Promega | E1171 |
| CAMYEL cAMP biosensor | Paul Sternweis, UT Southwestern, Jiang et al., 2007 PMID 17283075 | N/A |
| Flag-β2AR (human) | Robert Lefkowitz, Oppermann et al., 1996 PMID 8755530 |
N/A |
| Flag-β2AR (human), PKA− mutant | Robert Lefkowitz, Seibold et al., 1998 PMID 9516468 |
N/A |
| Flag-β2AR (human), GRK− mutant | Robert Lefkowitz, Seibold et al., 1998 PMID 9516468 |
N/A |
| GFP-β-Arrestin2 | Robert Lefkowitz, Barak et al, 1997 PMID 9346876 |
N/A |
| Venus 156-239-Gβ1, Venus 1-155-Gγ2 | Nevin Lambert, Hollins et al., 2009 PMID 19258039 |
N/A |
| β2AR-NanoLuc | Nevin Lambert, Okashah et al., 2019 PMID 31142646 |
|
| HA-ATR1 (human) | Robert Lefkowitz, Tohgo et al, 2002 PMID: 11777902 |
N/A |
| Software and Algorithms | ||
| Graphpad Prism (v7, v9) | Graphpad Software | version 7, version 9 |
| Other | ||
| N/A | ||
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Mice with Adrb2 gene bearing C265S mutation were prepared by homologous recombination by InGenious Targeting Laboratories (Ronkonkoma, NY) (see Supplemental Figure 5A). A 9.3kb gene fragment was subcloned from a mouse C57BL/6 BAC clone (RP23:376J14, H1H), and an FRT/loxP-flanked NEO selection marker gene was inserted downstream of the coding region. The C265S mutation was created by overlap PCR, and this 1.9kb fragment was inserted into the targeting construct using ClaI and MfeI restriction enzymes. G418-resistant C57BL/6 ES cell clones were verified by PCR, Southern blot (see Supplemental Figure 5B), DNA sequencing, and karyotyping before injection into mouse Balb/c blastocysts. Chimeric founder mice were bred to C57BL/6J mice for stable transmission, and the resulting heterozygote mice were bred to the Flp-deleter strain (on C57BL/6J background) to remove the NEO cassette. Heterozygote Abrb2+/C265S mice were interbred to obtain wildtype (WT) and Adrb2C265S/C265S (C265S) mice on the C57BL/6J background for study. Male mice from 2 to 4 months of age were tested in all experiments. Mice were genotyped by PCR from tail-tip genomic DNA using the primers ADRB2-NDEL1: 5’-CCTGACTCACGGAAACAGAGTTATGGG and ADRB2-NDEL2 5’-GGGGAAGCCTGGTTTACACAGAAG (see Supplemental Table I), yielding a 372bp product from the WT allele and a 552bp product from the mutated allele (see Supplemental Figure 5C).
For some experiments, C265S mice were bred with eNOS−/− mice on the C57BL/6J background (Jax) to generate double homozygotes, and Adrb2−/− mice (Jax) were used for tissue harvest.
All animal work was performed under protocols approved by the CWRU IACUC and conformed to standards in the Guide for the Care and Use of Laboratory Animals.
Cell lines and culture
The HEK293 stable cell line expressing Flag-β2AR (W9) was obtained from Robert Lefkowitz, and wildtype HEK293 cells were from ATCC (Manassas, Virginia). HEK293 cells were maintained in high-glucose DMEM media (Life Technologies) with 10% fetal bovine serum and antibiotics at 37°C under 5% CO2 in a humidified, water-jacketed incubator. HEK293 cells were transfected with plasmid DNAs using the X-tremeGENE 9 transfection reagent (Roche); for stable transfection, cells were subsequently selected for growth in 400 μg/ml G418 (Life Technologies). Cells were serum-starved with DMEM media for 1h prior to treatment with ISO (10 μM for 5 min), ECNO (100 μM for 10 min), and in some assays, cells were pretreated with PTX (100 ng/ml) for 18h or with inhibitors as described for each assay.
Mouse embryonic fibroblast (MEF) cell lines from β-Arrestin1/β-Arrestin2 double-knockout mice and from control WT mice were obtained from Robert Lefkowitz (Duke University). MEFs were maintained in MEM media (Life Technologies) with 10% fetal bovine serum and antibiotics at 37°C under 5% CO2 in a humidified, water-jacketed incubator. MEFs were transfected with Flag-β2AR using X-tremeGENE HP transfection reagent (Roche).
Human tissue
Deidentified human ventricular tissue samples from normal and heart failure patients were obtained from the Duke University Biorepository and Precision Pathology Center (Durham, NC). Because these tissues were exempted from requiring a human studies protocol by the University Hospitals Cleveland Medical Center IRB (Exemption EM-15-35), no PHI (including age or sex) was provided with the tissue samples, only disease status.
METHOD DETAILS
Plasmids and mutagenesis
β-Arrestin2-GFP, Flag-β-Arrestin2, Flag- and HA-tagged human ATR1 and Flag- and HA-tagged human β2ARs, and PKA− and GRK− phosphorylation-site mutant β2ARs were the kind gift of Robert Lefkowitz (Duke University). Flag-tagged β2AR mutants were created using the QuikChange II site-directed mutagenesis kit (Agilent), using specific primers (see Supplemental Table I). The split-fluor Gβγ (Venus 156-239-Gβ1, Venus 1-155-Gγ2) and β2AR-NanoLuc plasmids have been described previously (Masuho et al., 2015; Okashah et al., 2019).
SNO-RAC assay and western blotting
SNO-protein resin-assisted capture (SNO-RAC) was performed as described previously (Forrester et al., 2009). Briefly, frozen cell pellets were lysed in HEN buffer (20mM HEPES, 1mM EDTA, 0.1 mM Neocuproine,) with protease inhibitors (Roche) by sonication, and incubated with the free-sulfhydryl blocking agent S-methyl methanethiosulfonate (MMTS, 0.1%, Sigma). Proteins were washed free of MMTS by repeated acetone precipitation, and SNO removed from proteins by addition of 30 mM ascorbate in HEN buffer (or left on proteins by omitting ascorbate, as a negative control). All manipulations through the ascorbate step were performed in the dark in opaque tubes to minimize light exposure that can remove SNO from proteins, except in experiments where lysates in MMTS were exposed to UV light to remove SNO by photolysis (by incubating lysate in a glass vial on a Stratalinker (Stratagene) for 5 min). Samples were mixed with thiopropyl-Sepharose beads (GE Cytiva) to capture free thiols (sites previously bound to SNO that were freed by ascorbate), and the beads extensively washed. For western blotting identification, bead-bound proteins were eluted and separated by SDS-PAGE on 10% acrylamide gels (Criterion, Bio Rad, Hercules, CA), and detected with anti-tag (Flag or HA) or anti-target antibodies (see Key Resources).
For proteomic analysis, ECNO-treated HEK293 cells were lysed and fractionated into cytosolic and membrane fractions, and the membrane fraction was processed for SNO-RAC, with simultaneous MMTS blocking and membrane extraction with SDS detergent. SNO-RAC beads were digested with trypsin and washed, and bound peptides eluted and cleaned using C18 Ziptip prior to Orbitrap LC/MS/MS at the CWRU proteomic core facility. Peptides were identified by MASCOT search against the NCBI human NR database.
For standard western blotting of cell lysates and mouse tissues, lysates were prepared in RIPA buffer (Sigma) supplemented with protease inhibitors (Roche). Lysate proteins were quantified using BCA (Bio Rad) and separated by SDS-PAGE on 10% acrylamide gels (Criterion, Bio Rad, Hercules, CA) for detection with anti-tag (Flag or HA) or anti-target antibodies (see Key Resources). For immunoblot detection, transferred nitrocellulose filters were blocked with LI-COR blocking buffer, exposed sequentially to primary and Alexafluor-labeled secondary antisera, and detected on a LI-COR Odyssey imager.
Measurement of cAMP, Phosphodiesterase, Protein kinase A and Erk1/2 activity
PARAMETER mouse/rat cAMP ELISA kit (R&D Systems) was used to measure cAMP accumulation in cell lysates, according to the manufacturer’s instructions. cAMP was measured in live cells using HEK293 cells transiently expressing the CAMYEL cAMP BRET biosensor, as described (Jiang et al., 2007), after transfection with Flag-β2AR, and was also measured in live cells using HEK293 cells stably expressing the luciferase-based cAMP biosensor GloSensor-20F (Fan et al., 2008) (Promega), which were transfected with Flag-β2AR, then split the next day onto a 96-well plate at 80,000 cells/well. One day later, cells were treated with the GloSensor reagent [Promega; 4% (v/v)] at room temperature for 2 h (and with any inhibitors, as indicated in appropriate figure legends) prior to stimulation with ISO at various doses for 5 min, and cAMP quantified using a NOVOstar microplate reader (BMG Labtech) or a GloMax plate reader (Promega).
cAMP Phosphodiesterase activity was quantified using the Fluorometric cAMP phosphodiesterase activity kit according to the manufacturer’s protocol (Biovision, Milpitas, CA) and read on a GloMax plate reader (Promega).
ISO-stimulated phosphorylation of PKA substrates was detected by western blot of HEK293 cell lysates using phospho-(Ser/Thr) PKA substrate antibody (Cell Signaling). ISO-stimulated phosphorylation of Erk1/2 was detected by western blot of HEK293 cell lysates using phospho-Thr202/Tyr204 Erk1/2 antibody (Cell Signaling).
β2AR phosphorylation by PKA was assessed by western blot of cell lysates using phospho-Ser262/263 and phospho-Ser355/356 antisera.
β2AR internalization
Removal of β2AR from the cell surface upon stimulation was measured using flow cytometry according to a published protocol (Barak et al., 1994). Briefly, serum-starved cells were stimulated with agonist or inhibitors as indicated in specific figures, and cells moved to ice. Cells were fixed in 4% paraformaldehyde without permeabilization, and surface β2AR were labeled with M1 Flag antibody (Sigma) in the presence of calcium for 1h on ice, then by anti-mouse secondary antibody conjugated to AlexaFluor-648. Fixed, labeled cells were washed again prior to flow cytometry. Surface labeling is expressed as % loss compared to untreated cells, where specific labeling is gated by comparing cells lacking Flag-β2AR to receptor-expressing cells.
β2AR immunocytochemistry and GFP-β-Arrestin2 translocation visualization
Flag-tagged β2AR and mutants stably expressed in HEK293 cells were detected using antibodies. Cells were grown on 4-chamber slides. After serum-starving for 4h, cells were stimulated with isoproterenol (10 μM) or ECNO (100 μM) for 10 min unless otherwise indicated, then immediately fixed in 4% paraformaldehyde. Permeabilized cells were stained for β2AR using Flag M2 antibody and anti-mouse-Alexa-568 or anti-rabbit-Alexa-488 secondary antibodies, and imaged on a Zeiss confocal microscope. For detection of co-localization of β2AR and GFP-β-Arrestin2, HEK293 cells were transiently co-transfected with Flag-β2AR and GFP-β-arrestin2. Cells were serum-starved, stimulated, fixed, stained and imaged as above.
β2AR coupling to Gs and Gi proteins
Coupling of β2AR to Gs and Gi proteins was measured using BRET-based proximity essentially as described (Okashah et al., 2019) but in standard HEK293 cells. Briefly, β2AR and C265S β2AR were fused at the carboxy terminal to Rluc8. Gβ1 and Gγ2 fused to complementing halves of Venus allowing bimolecular fluorescence complementation have been described (Hollins et al., 2009; Hynes et al., 2004). To measure Gs coupling, Gs-alpha was also overexpressed, and to measure Gi coupling, Gi1-alpha was overexpressed, together with receptor-Rluc8 and Venus-βγ (bimolecular complemented (Venus 156–239)-Gβ1 and (Venus 1–155)-Gγ2) (Masuho et al., 2015). This overexpression overwhelms the native G protein background to give a predominant signal via the G protein of choice in proximity to the receptor (Masuho et al., 2015).
SNO production by HEK293 cells
Receptor-stimulated SNO production was measured by photolysis-chemiluminescence as described (Hausladen et al., 2007; Zhou et al., 2019). WT or mutant receptor-expressing HEK293 cells were pretreated with PTX for 18h or not as indicated, stimulated with isoproterenol (10 μM, 10 min) and harvested. Cell pellets were lysed in HEN buffer with protease inhibitors by sonication, protein determined, and SNO-protein content quantified by UV photolysis of SNO proteins and detection of the released NO by ozone chemiluminescence.
Airway resistance in vivo
Airway responsiveness to inhaled methacholine was measured as an increase in respiratory system resistance (Rrs) using the flexiVent small animal ventilator (SCIREQ Inc., Montreal, Canada), as previously described (Que et al., 2005; Raffay et al., 2016). Mice were anesthetized (i.p. ketamine/xylazine; Phizer, St. Joseph, MO; Lloyd Laboratories, Shenandoah, IA), placed supine on a heated surgical bed, and tracheostomized with an 18-gauge blunt-tip cannula via a midline incision. Animals were paralyzed (i.p. pancuronium bromide; Sigma) to block spontaneous breathing and ventilated at default settings: tidal volume of 10 mL/kg, a positive end expiratory pressure of 3 cm H2O, a rate of 150 breaths/min, and an FiO2 of 50%. Following two recruitment breaths of deep inspiration up to 30 cm H2O for 3 seconds, increasing doses of methacholine solution (0, 3, 6, 12.5, 25, 50, 75, 100, 200 mg/mL in saline; Sigma) were aerosolized over 10 seconds using an ultrasonic nebulizer (Aeroneb, SCIREQ) diverted into the ventilator’s inspiratory flow. Serial measurements of Rrs were calculated by a 2.5 Hz single-frequency forced oscillation maneuver (Snapshot 150) using computer software (flexiWare 5.1, Version 7.2, SCIREQ), and an average was reported for each methacholine dose. Animals were monitored by continuous electrocardiogram; those that did not complete the full methacholine dose response due to severe bradycardia (<60 bpm), asystole, or sustained cannula blockage with excessive secretions were excluded from the analysis.
Drug treatments were as follows: L-NAME, 100 μl total volume of 100 mg/kg in PBS was injected i.p. 1h before analysis; PTX, 100 μl total volume containing ~15 μl of 200 μg/ml PTX (Sigma) (adjusted to 100 μg/kg) and ~85 μl sterile PBS was injected i.p. 18h before analysis, as described (McGraw et al., 2007).
Aorta contraction and relaxation
Thoracic aorta was dissected from wildtype and C265S mice, cut into 3 mm rings (2 per mouse), and strung on a tension hook in an organ bath system (Radnoti, Inc), essentially as described (Zhang et al., 2022). Rings were maintained at 37°C in Krebs buffer and bubbled with 20% O2/5% CO2/balance nitrogen. Contraction (transducer tension) was increased by treatment with 1 μM phenylephrine, and after adjusting tension to a standard 1g, relaxation was induced by treatment with increasing concentrations of ISO, as indicated in the legend.
Isoproterenol infusion in vivo
Mice were anesthetized with ketamine and xylazine (ketamine 80 mg/kg, xylazine 8 mg/kg body weight, i.p.), an abdominal mid-line incision was made to expose the inferior vena cava (IVC), and a micro-catheter was inserted. Using an infusion pump (Harvard Apparatus, Pump 11 Elite), isoproterenol (50 ng/mL) was infused for 10 min with a flow rate at 20 μL/min, and the heart was harvested immediately and snap-frozen.
Human dust mite allergen model of allergic asthma
Dermatophagoides pteronyssinus and Dermatophagoides farinae allergens (Stallergenes-Greer, Lenoir NC) were dissolved in sterile PBS at 3mg/mL. On day 0, mice were injected (i.p.) with 100 μg of one allergen (either D.p. or D.f. as indicated). Mice were anesthetized with isofluorane gas and 30 μg of the same allergen (10 μl) was instilled intranasally daily for the next 14 days (alternating nostrils each day). On day 15 there was no allergen instillation, and mouse airways were analyzed on the flexiVent as described above, prior to harvest of tissues.
RNA isolation and Realtime reverse transcription-PCR
Total RNA from lungs was isolated by using Aurum™ Total RNA Fatty and Fibrous Tissue Kit and reverse transcribed into cDNA using iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). Quantitative real-time PCR was carried out using Fast SYBR Green or TaqMan Fast Advanced Master Mix with gene-specific primers (see Supplemental Table I), and the reactions were performed using the StepOnePlus Real-Time PCR System (Applied Biosystems Inc., Foster City, CA, USA). Endogenous beta-actin (Actb) was used to normalize gene expression. Relative mRNA expression levels (fold changes) between groups were calculated using the ΔΔCt method.
Bronchoalveolar Lavage Fluid (BALF) collection and assay
After terminal anesthesia by ketamine/xylazine overdose, animals were tracheostomized and two aliquots of 1mL PBS/EDTA were instilled into the lungs with a 10 second dwell time. BALF was collected and the volume retrieved was noted, as previously described (Auten et al., 2007). Cells in BALF were isolated by centrifugation at 1000 g for 10 minutes and re-suspended in ice-cold PBS for inflammatory cell differential counts. BALF with gross hemorrhage or with retrieval of less than 70% of instillate volume was excluded from the analysis. Immune cell types were counted using the HEMAVET 950FS (Drew Scientific) using the manufacturer’s protocol.
Histology and immunohistochemistry
As previously described (Raffay et al., 2016), after terminal anesthesia by ketamine/xylazine overdose, animals were perfused in-situ by right ventricular puncture with PBS (pH 7.4) then 10% formalin, cannulated by tracheostomy and lungs inflation-fixed with 10% formalin at 25 cm H2O. The trachea was ligated and the lungs were post-fixed in 10% formalin at 4° C for >24 hours. Tissue was paraffin embedded and 5 μm lung sections were processed for histology or immunostaining. Hematoxylin/Eosin and Masson’s Trichrome staining were performed by the CWRU Tissue Resource Core. Immunofluorescent tissue visualization for EpCAM and MPO by confocal microscopy was performed essentially as previously described (Johnson et al., 2015, 2018). Tissue slices on slides were deparaffinized and rehydrated prior to antigen retrieval in boiling citrate. Slides were blocked with serum and incubated overnight. Slides were washed and incubated with AlexaFluor 488-conjugated rat anti-mouse EpCAM (BioLegend) and rabbit anti-mouse MPO (Abcam) antibodies. Slides were then washed and stained with AlexaFluor 647-conjugated goat anti-rabbit IgG secondary antibody (Abcam). Slides were imaged on a Leica SP5 Scanning Confocal Microscope.
Quantification and statistical analysis
Statistical analysis was performed using Graphpad Prism v8. Specific tests used, number of independent samples or replicates, and significance of results are indicated in figure legends, and all quantitative data are plotted as mean ± SD. Significance level was set at p < 0.05. All data are available in the figures; no large datasets or executable codesets were generated in this project.
Supplementary Material
Highlights.
β2AR is S-nitrosylated at Cys265, mediating receptor desensitization
S-nitrosylation drives β2AR internalization via caveolae
β2AR lacking S-nitrosylation exhibits prolonged signaling to cAMP, Erk and NO
Mice with β2AR refractory to S-nitrosylation are protected from asthma
Acknowledgements
The authors thank Puneet Seth for technical assistance, and Robert Lefkowitz and Howard Rockman (Duke University) and Walter Koch (Temple University) for helpful discussions.
Funded by NIH grants P01 HL075443, P01 HL128192, P01 HL158507, R01 HL126900, and R01 DK119506
Footnotes
Declaration of Interests
JSS has patents relating to nitrosylation, is a founder of SNO bio, and serves as a paid consultant to EFC, which is developing NO-based technology. CWRU and UHCMC have management plans in place. The other authors declare no conflicts.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data and Materials Availability:
All data is available in the main text, Supplemental Materials or Mendeley Data. Requests for research materials should be addressed to JSS, jss156@case.edu
References
- Abi-Gerges N, Szabo G, Otero AS, Fischmeister R, and Mery PF (2002). NO donors potentiate the beta-adrenergic stimulation of I(Ca,L) and the muscarinic activation of I(K,ACh) in rat cardiac myocytes. J Physiol 540, 411–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi N, Hess DT, McLaughlin P, and Stamler JS (2016). S-Palmitoylation of a Novel Site in the beta2-Adrenergic Receptor Associated with a Novel Intracellular Itinerary. J Biol Chem 291, 20232–20246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam L, Bouvier M, and Jones TL (1999). Nitric oxide modulates beta(2)-adrenergic receptor palmitoylation and signaling. J Biol Chem 274, 26337–26343. [DOI] [PubMed] [Google Scholar]
- Amrani Y, and Bradding P (2017). beta2-Adrenoceptor Function in Asthma. Adv Immunol 136, 1–28. [DOI] [PubMed] [Google Scholar]
- Antosova M, Mokra D, Pepucha L, Plevkova J, Buday T, Sterusky M, and Bencova A (2017). Physiology of nitric oxide in the respiratory system. Physiol Res 66, S159–S172. [DOI] [PubMed] [Google Scholar]
- Auten RL, Mason SN, Whorton MH, Lampe WR, Foster WM, Goldberg RN, Li B, Stamler JS, and Auten KM (2007). Inhaled ethyl nitrite prevents hyperoxia-impaired postnatal alveolar development in newborn rats. Am J Respir Crit Care Med 176, 291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak LS, Tiberi M, Freedman NJ, Kwatra MM, Lefkowitz RJ, and Caron MG (1994). A highly conserved tyrosine residue in G protein-coupled receptors is required for agonist-mediated beta 2-adrenergic receptor sequestration. J Biol Chem 269, 2790–2795. [PubMed] [Google Scholar]
- Burgoyne JR, and Eaton P (2009). Transnitrosylating nitric oxide species directly activate type I protein kinase A, providing a novel adenylate cyclase-independent cross-talk to beta-adrenergic-like signaling. J Biol Chem 284, 29260–29268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhry S, Que LG, Yang Z, Liu L, Eng C, Kim SO, Kumar G, Thyne S, Chapela R, Rodriguez-Santana JR, et al. (2010). GSNO reductase and beta2-adrenergic receptor gene-gene interaction: bronchodilator responsiveness to albuterol. Pharmacogenet Genomics 20, 351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daaka Y, Luttrell LM, and Lefkowitz RJ (1997). Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature 390, 88–91. [DOI] [PubMed] [Google Scholar]
- Fan F, Binkowski BF, Butler BL, Stecha PF, Lewis MK, and Wood KV (2008). Novel genetically encoded biosensors using firefly luciferase. ACS Chem Biol 3, 346–351. [DOI] [PubMed] [Google Scholar]
- Ferguson SS, Downey WE 3rd, Colapietro AM, Barak LS, Menard L, and Caron MG (1996). Role of beta-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science 271, 363–366. [DOI] [PubMed] [Google Scholar]
- Figueroa XF, Poblete I, Fernandez R, Pedemonte C, Cortes V, and Huidobro-Toro JP (2009). NO production and eNOS phosphorylation induced by epinephrine through the activation of beta-adrenoceptors. Am J Physiol Heart Circ Physiol 297, H134–143. [DOI] [PubMed] [Google Scholar]
- Forrester MT, Thompson JW, Foster MW, Nogueira L, Moseley MA, and Stamler JS (2009). Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat Biotechnol 27, 557–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster MW, and Stamler JS (2004). New insights into protein S-nitrosylation. Mitochondria as a model system. J Biol Chem 279, 25891–25897. [DOI] [PubMed] [Google Scholar]
- Friedman J, Babu B, and Clark RB (2002). Beta(2)-adrenergic receptor lacking the cyclic AMP-dependent protein kinase consensus sites fully activates extracellular signal-regulated kinase 1/2 in human embryonic kidney 293 cells: lack of evidence for G(s)/G(i) switching. Mol Pharmacol 62, 1094–1102. [DOI] [PubMed] [Google Scholar]
- Gaston B, Drazen JM, Jansen A, Sugarbaker DA, Loscalzo J, Richards W, and Stamler JS (1994). Relaxation of human bronchial smooth muscle by S-nitrosothiols in vitro. J Pharmacol Exp Ther 268, 978–984. [PubMed] [Google Scholar]
- Gaston B, Reilly J, Drazen JM, Fackler J, Ramdev P, Arnelle D, Mullins ME, Sugarbaker DJ, Chee C, Singel DJ, et al. (1993). Endogenous nitrogen oxides and bronchodilator S-nitrosothiols in human airways. Proc Natl Acad Sci U S A 90, 10957–10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gether U, Ballesteros JA, Seifert R, Sanders-Bush E, Weinstein H, and Kobilka BK (1997a). Structural instability of a constitutively active G protein-coupled receptor. Agonist-independent activation due to conformational flexibility. J Biol Chem 272, 2587–2590. [DOI] [PubMed] [Google Scholar]
- Gether U, Lin S, Ghanouni P, Ballesteros JA, Weinstein H, and Kobilka BK (1997b). Agonists induce conformational changes in transmembrane domains III and VI of the beta2 adrenoceptor. EMBO J 16, 6737–6747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta MK, Asosingh K, Aronica M, Comhair S, Cao G, Erzurum S, Panettieri RA Jr., and Naga Prasad SV (2015). Defective Resensitization in Human Airway Smooth Muscle Cells Evokes β-Adrenergic Receptor Dysfunction in Severe Asthma. PLOS ONE 10, e0125803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausdorff WP, Bouvier M, O’Dowd BF, Irons GP, Caron MG, and Lefkowitz RJ (1989). Phosphorylation sites on two domains of the beta 2-adrenergic receptor are involved in distinct pathways of receptor desensitization. J Biol Chem 264, 12657–12665. [PubMed] [Google Scholar]
- Hausladen A, Rafikov R, Angelo M, Singel DJ, Nudler E, and Stamler JS (2007). Assessment of nitric oxide signals by triiodide chemiluminescence. Proc Natl Acad Sci U S A 104, 2157–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H, Hess DT, Zhang R, Sugi K, Gao H, Tan BL, Bowles DE, Milano CA, Jain MK, Koch WJ, et al. (2018). S-Nitrosylation of beta-Arrestins Biases Receptor Signaling and Confers Ligand Independence. Mol Cell 70, 473–487 e476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess DT, Matsumoto A, Kim SO, Marshall HE, and Stamler JS (2005). Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol 6, 150–166. [DOI] [PubMed] [Google Scholar]
- Hollins B, Kuravi S, Digby GJ, and Lambert NA (2009). The c-terminus of GRK3 indicates rapid dissociation of G protein heterotrimers. Cell Signal 21, 1015–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyte FCL, Gross LM, and Katial RK (2018). Exhaled Nitric Oxide: An Update. Immunol Allergy Clin North Am 38, 573–585. [DOI] [PubMed] [Google Scholar]
- Hynes TR, Tang L, Mervine SM, Sabo JL, Yost EA, Devreotes PN, and Berlot CH (2004). Visualization of G protein betagamma dimers using bimolecular fluorescence complementation demonstrates roles for both beta and gamma in subcellular targeting. J Biol Chem 279, 30279–30286. [DOI] [PubMed] [Google Scholar]
- Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, Steyaert J, Rasmussen SGF, Sunahara RK, El-Samad H, Huang B, et al. (2013). Conformational biosensors reveal GPCR signalling from endosomes. Nature 495, 534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J, Arif A, Terenzi F, Willard B, Plow EF, Hazen SL, and Fox PL (2014). Target-selective protein S-nitrosylation by sequence motif recognition. Cell 159, 623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang LI, Collins J, Davis R, Lin KM, DeCamp D, Roach T, Hsueh R, Rebres RA, Ross EM, Taussig R, et al. (2007). Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G(13) pathway. Journal of Biological Chemistry 282, 10576–10584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JL, Jones MB, and Cobb BA (2015). Bacterial capsular polysaccharide prevents the onset of asthma through T-cell activation. Glycobiology 25, 368–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JL, Jones MB, and Cobb BA (2018). Polysaccharide-experienced effector T cells induce IL-10 in FoxP3+ regulatory T cells to prevent pulmonary inflammation. Glycobiology 28, 50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten ET, Koppelman GH, and Thio BJ (2017). Concerns with beta2-agonists in pediatric asthma - a clinical perspective. Paediatr Respir Rev 21, 80–85. [DOI] [PubMed] [Google Scholar]
- Kharitonov SA, Yates D, Robbins RA, Logan-Sinclair R, Shinebourne EA, and Barnes PJ (1994). Increased nitric oxide in exhaled air of asthmatic patients. Lancet 343, 133–135. [DOI] [PubMed] [Google Scholar]
- Knight JM, Mak G, Shaw J, Porter P, McDermott C, Roberts L, You R, Yuan X, Millien VO, Qian Y, et al. (2015). Long-Acting Beta Agonists Enhance Allergic Airway Disease. PLoS One 10, e0142212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobilka BK (2002). Agonist-induced conformational changes in the beta2 adrenergic receptor. J Pept Res 60, 317–321. [DOI] [PubMed] [Google Scholar]
- Kohout TA, Lin FS, Perry SJ, Conner DA, and Lefkowitz RJ (2001). beta-Arrestin 1 and 2 differentially regulate heptahelical receptor signaling and trafficking. Proc Natl Acad Sci U S A 98, 1601–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf RG, Russo M, and Jancar S (2005). Acute inhibition of inducible nitric oxide synthase but not its absence suppresses asthma-like responses. European Journal of Pharmacology 518, 212–220. [DOI] [PubMed] [Google Scholar]
- Laporte SA, Oakley RH, Zhang J, Holt JA, Ferguson SS, Caron MG, and Barak LS (1999). The beta2-adrenergic receptor/betaarrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc Natl Acad Sci U S A 96, 3712–3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz RJ, Pierce KL, and Luttrell LM (2002). Dancing with different partners: protein kinase a phosphorylation of seven membrane-spanning receptors regulates their G protein-coupling specificity. Mol Pharmacol 62, 971–974. [DOI] [PubMed] [Google Scholar]
- Lin R, Degan S, Theriot BS, Fischer BM, Strachan RT, Liang J, Pierce RA, Sunday ME, Noble PW, Kraft M, et al. (2012). Chronic treatment in vivo with beta-adrenoceptor agonists induces dysfunction of airway beta(2) -adrenoceptors and exacerbates lung inflammation in mice. Br J Pharmacol 165, 2365–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuho I, Ostrovskaya O, Kramer GM, Jones CD, Xie K, and Martemyanov KA (2015). Distinct profiles of functional discrimination among G proteins determine the actions of G protein-coupled receptors. Sci Signal 8, ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matera MG, Page CP, Calzetta L, Rogliani P, and Cazzola M (2020). Pharmacology and Therapeutics of Bronchodilators Revisited. Pharmacological Reviews 72, 218. [DOI] [PubMed] [Google Scholar]
- Mathrani VC, Kenyon NJ, Zeki A, and Last JA (2007). Mouse models of asthma: can they give us mechanistic insights into the role of nitric oxide? Curr Med Chem 14, 2204–2213. [DOI] [PubMed] [Google Scholar]
- McGraw DW, Elwing JM, Fogel KM, Wang WC, Glinka CB, Mihlbachler KA, Rothenberg ME, and Liggett SB (2007). Crosstalk between Gi and Gq/Gs pathways in airway smooth muscle regulates bronchial contractility and relaxation. J Clin Invest 117, 1391–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore PE, Ryckman KK, Williams SM, Patel N, Summar ML, and Sheller JR (2009). Genetic variants of GSNOR and ADRB2 influence response to albuterol in African-American children with severe asthma. Pediatr Pulmonol 44, 649–654. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Oh CK, Liao L, Zhang X, Lopez KM, Gibbs D, Deal AK, Scott HR, Spencer B, Masliah E, et al. (2021). Noncanonical transnitrosylation network contributes to synapse loss in Alzheimer’s disease. Science 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LP, Al-Sawalha NA, Parra S, Pokkunuri I, Omoluabi O, Okulate AA, Windham Li E, Hazen M, Gonzalez-Granado JM, Daly CJ, et al. (2017). beta2-Adrenoceptor signaling in airway epithelial cells promotes eosinophilic inflammation, mucous metaplasia, and airway contractility. Proc Natl Acad Sci U S A 114, E9163–E9171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LP, Lin R, Parra S, Omoluabi O, Hanania NA, Tuvim MJ, Knoll BJ, Dickey BF, and Bond RA (2009). Beta2-adrenoceptor signaling is required for the development of an asthma phenotype in a murine model. Proc Natl Acad Sci U S A 106, 2435–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama K, Nishimura A, Shimoda K, Tanaka T, Kato Y, Shibata T, Tanaka H, Kurose H, Azuma Y-T, Ihara H, et al. (2022). Redox-dependent internalization of the purinergic P2Y6 receptor limits colitis progression. Science Signaling 15, eabj0644. [DOI] [PubMed] [Google Scholar]
- O’Dowd BF, Hnatowich M, Caron MG, Lefkowitz RJ, and Bouvier M (1989). Palmitoylation of the human beta 2-adrenergic receptor. Mutation of Cys341 in the carboxyl tail leads to an uncoupled nonpalmitoylated form of the receptor. J Biol Chem 264, 7564–7569. [PubMed] [Google Scholar]
- Okashah N, Wan Q, Ghosh S, Sandhu M, Inoue A, Vaidehi N, and Lambert NA (2019). Variable G protein determinants of GPCR coupling selectivity. Proc Natl Acad Sci U S A 116, 12054–12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrom RS, and Insel PA (2004). The evolving role of lipid rafts and caveolae in G protein-coupled receptor signaling: implications for molecular pharmacology. Br J Pharmacol 143, 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa K, Whalen EJ, Nelson CD, Mu Y, Hess DT, Lefkowitz RJ, and Stamler JS (2008). S-nitrosylation of beta-arrestin regulates beta-adrenergic receptor trafficking. Mol Cell 31, 395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papi A, Brightling C, Pedersen SE, and Reddel HK (2018). Asthma. The Lancet 391, 783–800. [DOI] [PubMed] [Google Scholar]
- Que LG, Liu L, Yan Y, Whitehead GS, Gavett SH, Schwartz DA, and Stamler JS (2005). Protection from experimental asthma by an endogenous bronchodilator. Science 308, 1618–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queen LR, and Ferro A (2006). Beta-adrenergic receptors and nitric oxide generation in the cardiovascular system. Cell Mol Life Sci 63, 1070–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffay TM, Dylag AM, Di Fiore JM, Smith LA, Einisman HJ, Li Y, Lakner MM, Khalil AM, MacFarlane PM, Martin RJ, et al. (2016). S-Nitrosoglutathione Attenuates Airway Hyperresponsiveness in Murine Bronchopulmonary Dysplasia. Mol Pharmacol 90, 418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapacciuolo A, Suvarna S, Barki-Harrington L, Luttrell LM, Cong M, Lefkowitz RJ, and Rockman HA (2003). Protein kinase A and G protein-coupled receptor kinase phosphorylation mediates beta-1 adrenergic receptor endocytosis through different pathways. J Biol Chem 278, 35403–35411. [DOI] [PubMed] [Google Scholar]
- Schwencke C, Okumura S, Yamamoto M, Geng YJ, and Ishikawa Y (1999). Colocalization of beta-adrenergic receptors and caveolin within the plasma membrane. J Cell Biochem 75, 64–72. [DOI] [PubMed] [Google Scholar]
- Seibold A, January BG, Friedman J, Hipkin RW, and Clark RB (1998). Desensitization of beta2-adrenergic receptors with mutations of the proposed G protein-coupled receptor kinase phosphorylation sites. J Biol Chem 273, 7637–7642. [DOI] [PubMed] [Google Scholar]
- Seth D, Hess DT, Hausladen A, Wang L, Wang YJ, and Stamler JS (2018). A Multiplex Enzymatic Machinery for Cellular Protein S-nitrosylation. Mol Cell 69, 451–464 e456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaul PW, Smart EJ, Robinson LJ, German Z, Yuhanna IS, Ying Y, Anderson RG, and Michel T (1996). Acylation targets emdothelial nitric-oxide synthase to plasmalemmal caveolae. J Biol Chem 271, 6518–6522. [DOI] [PubMed] [Google Scholar]
- Stomberski CT, Hess DT, and Stamler JS (2019). Protein S-Nitrosylation: Determinants of Specificity and Enzymatic Regulation of S-Nitrosothiol-Based Signaling. Antioxid Redox Signal 30, 1331–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanawala VJ, Forkuo GS, Al-Sawalha N, Azzegagh Z, Nguyen LP, Eriksen JL, Tuvim MJ, Lowder TW, Dickey BF, Knoll BJ, et al. (2013). beta2-Adrenoceptor agonists are required for development of the asthma phenotype in a murine model. Am J Respir Cell Mol Biol 48, 220–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanawala VJ, Valdez DJ, Joshi R, Forkuo GS, Parra S, Knoll BJ, Bouvier M, Leff P, and Bond RA (2015). beta-Blockers have differential effects on the murine asthma phenotype. Br J Pharmacol 172, 4833–4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale C, Maglio A, Pelaia C, and Vatrella A (2017). Long-term treatment in pediatric asthma: an update on chemical pharmacotherapy. Expert Opin Pharmacother 18, 667–676. [DOI] [PubMed] [Google Scholar]
- Wanschel AC, Caceres VM, Moretti AI, Bruni-Cardoso A, de Carvalho HF, de Souza HP, Laurindo FR, Spadari RC, and Krieger MH (2014). Cardioprotective mechanism of S-nitroso-N-acetylcysteine via S-nitrosated betadrenoceptor-2 in the LDLr−/− mice. Nitric Oxide 36, 58–66. [DOI] [PubMed] [Google Scholar]
- Wendell SG, Fan H, and Zhang C (2020). G Protein-Coupled Receptors in Asthma Therapy: Pharmacology and Drug Action. Pharmacol Rev 72, 1–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen EJ, Foster MW, Matsumoto A, Ozawa K, Violin JD, Que LG, Nelson CD, Benhar M, Keys JR, Rockman HA, et al. (2007). Regulation of beta-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell 129, 511–522. [DOI] [PubMed] [Google Scholar]
- Wu H, Romieu I, Sienra-Monge JJ, Estela Del Rio-Navarro B, Anderson DM, Jenchura CA, Li H, Ramirez-Aguilar M, Del Carmen Lara-Sanchez I, and London SJ (2007). Genetic variation in S-nitrosoglutathione reductase (GSNOR) and childhood asthma. J Allergy Clin Immunol 120, 322–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Rybin VO, Steinberg SF, and Kobilka B (2002). Caveolar localization dictates physiologic signaling of beta 2-adrenoceptors in neonatal cardiac myocytes. J Biol Chem 277, 34280–34286. [DOI] [PubMed] [Google Scholar]
- Zhang J, Ferguson SS, Barak LS, Menard L, and Caron MG (1996). Dynamin and beta-arrestin reveal distinct mechanisms for G protein-coupled receptor internalization. J Biol Chem 271, 18302–18305. [DOI] [PubMed] [Google Scholar]
- Zhang R, Hausladen A, Qian Z, Liao X, Premont RT, and Stamler JS (2022). Hypoxic vasodilatory defect and pulmonary hypertension in mice lacking hemoglobin beta-cysteine93 S-nitrosylation. JCI Insight 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HL, Zhang R, Anand P, Stomberski CT, Qian Z, Hausladen A, Wang L, Rhee EP, Parikh SM, Karumanchi SA, et al. (2019). Metabolic reprogramming by the S-nitroso-CoA reductase system protects against kidney injury. Nature 565, 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw image data for all Figures have been deposited with Mendeley and are publicly available as of the date of publication. The accession number is listed in the Key Resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-Flag M1 (mouse monoclonal) | Sigma-Aldrich | Cat# F3040 RRID:AB_439712 |
| anti-Flag M2 (mouse monoclonal) | Sigma-Aldrich | Cat# F3165 RRID:AB_439685 |
| anti-HA-7 (mouse monoclonal) | Sigma-Aldrich | Cat# H9658 RRID:AB_260092 |
| anti-Erk1/2 | Cell Signaling Technology | Cat# 4695 RRID:AB_390779 |
| anti-Erk1/2 pThr202+pTyr204 | Cell Signaling Technology | Cat# 9101 RRID:AB_331646 |
| anti-Protein kinase A - catalytic subunit | Cell Signaling Technology | Cat# 5842 RRID:AB_10706172 |
| anti-Protein kinase A - catalytic subunit, pThr197 | Cell Signaling Technology | Cat# 5661 RRID:AB_10707163 |
| anti-Pan-PKA substrate pSer/pThr | Cell Signaling Technology | Cat# 9621 RRID:AB_330304 |
| anti-β2 adrenergic receptor (H-20) | Santa Cruz Biotechnology | Cat# sc-569 Lot# L0415 RRID:AB_630926 |
| anti-β2AR pSer261/pSer262 | Robert Lefkowitz (Nobles et al., 2011) | N/A |
| anti-β2AR pSer345/pSer346 | Robert Lefkowitz (Nobles et al., 2011) | N/A |
| anti-β1AR (A-20) | Santa Cruz Biotechnology | Cat# sc-567 RRID:AB_2225390 |
| anti-Phospholipase C-beta1 (D8) | Santa Cruz Biotechnology | Cat# sc-5291 RRID:AB_628118 |
| anti-Phospholipase C-beta3 (D-7) | Santa Cruz Biotechnology | Cat# sc-133231 RRID:AB_2299534 |
| anti-EpCAM (CD326) - Alexafluor488 Conjugate | BioLegend | Cat# 118210 RRID:AB_1134099 |
| anti-myeloperoxidase | Abcam | Cat# ab45977 RRID:AB_944318 |
| anti-GAPDH (6C5) | Millipore (Sigma-Aldrich) | Cat# MAB374 RRID:AB_2107445 |
| Donkey anti-rabbit-Alexafluor594 | Molecular Probes, ThermoFisher | Cat# A32754 RRID:AB_2762827 |
| Goat anti-mouse-Alexafluor568 | Molecular Probes, ThermoFisher | Cat# A11031 RRID:AB_144696 |
| Goat anti-rabbit-Alexafluor488 | Molecular Probes, ThermoFisher | Cat# A32731 RRID:AB_2633280 |
| Goat anti-rabbit-Alexafluor647 | Abcam | Cat# ab150083 RRID:AB_2714032 |
| Bacterial and Virus Strains | ||
| N/A | ||
| Biological Samples | ||
| Human heart failure patient ventricular tissue | Biorepository and Precision Pathology Center, Duke University | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Thiopropyl-Sepharose 6B | Sigma-Aldrich | T8387 |
| S-Methyl methanethiosulfonate (MMTS) | Sigma-Aldrich | 64306 |
| L-NG-monomethyl arginine (L-NMMA) | Cayman Chemical | 80200 |
| L-NG-Nitroarginine methyl ester (L-NAME) | Cayman Chemical | 80210 |
| (−)-Isoproterenol (+)-bitartrate salt | Sigma-Aldrich | I2760 |
| Salbutamol | Sigma-Aldrich | S8260 |
| Salmeterol | Sigma-Aldrich | S5068 |
| Pindolol | Sigma-Aldrich | P0778 |
| Procaterol | Sigma-Aldrich | P9180 |
| Fenoterol | Sigma-Aldrich | F1016 |
| Epinephrine | Sigma-Aldrich | E4250 |
| Isoetharine | Sigma-Aldrich | I3639 |
| Pertussis toxin | Sigma-Aldrich | P7208 |
| DRAQ5 | Thermo-Fisher | 62251 |
| β-methyl cyclodextrin | Sigma-Aldrich | C4555 |
| Chlorpromazine hydrochloride | Sigma-Aldrich | C0982 |
| H-89 (N-[2-(p-Bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide dihydrochloride) | Sigma-Aldrich | B1427 |
| Genistein | Sigma-Aldrich | G6649 |
| Filipin III | Sigma-Aldrich | SAE0087 |
| Methacholine chloride | Sigma-Aldrich | 1396364 |
| 2’,5’-dideoxyadenosine 3’-triphosphate | Sigma-Aldrich | D0939 |
| KH7 ((E)-2-(1H-Benzo[d]imidazol-2-ylthio)-N’-(5-bromo-2-hydroxybenzylidene)propanehydrazide) | Sigma-Aldrich | K3394 |
| house dust mite Dermatophagoides pteronyssinus allergen | Stallergenes-Greer | B70 |
| house dust mite Dermatophagoides farinae allergen | Stallergenes-Greer | B64 |
| X-tremeGENE 9 transfection reagent | Sigma-Aldrich | 6365779001 |
| X-tremeGENE HP transfection reagent | Sigma-Aldrich | 6366244001 |
| KT-5720 | Tocris | 1288 |
| SQ-22536 | Tocris | 1435 |
| Phenylephrine hydrochloride | Sigma-Aldrich | 1533002 |
| [3H]-alprenolol; Dihydroalprenolol Hydrochloride, Levo-[Ring, Propyl-3H(N)] | PerkinElmer | NET720250UC |
| Critical Commercial Assays | ||
| PARAMETER cAMP ELISA kit | R&D Systems | KGE012 |
| GloSensor cAMP biosensor reagent | Promega | E1290 |
| Aurum Total RNA Fatty and Fibrous Tissue Kit | Bio-Rad | 7326830 |
| iScript cDNA Synthesis Kit | Bio-Rad | 1708890 |
| Fast SYBR Green Master Mix | Thermo-Fisher | 4385612 |
| TaqMan Fast Advanced Master Mix | Thermo-Fisher | 4444556 |
| cAMP Phosphodiesterase activity kit (Fluorometric) | Biovision | K2013-100 |
| Deposited Data | ||
| Image raw data files | Mendeley Data | http://dx.doi.org/10.17632/p2zycrb6dr |
| Experimental Models: Cell Lines | ||
| HEK293 | ATCC | Cat# CRL-1573 RRID:CVCL_0045 |
| HEK293 W9, stably expressing human Flag-β2AR | Robert Lefkowitz, Duke University | Shenoy et al., 2006 PMID 16280323 |
| HEK293, stably expressing HA-ATR1 | this study | N/A |
| Arrb1-/- mouse embryonic fibroblast (MEF) cell line | Robert Lefkowitz, Duke University | Kohout et al., 2001, PMID 11171997 |
| Arrb2-/- mouse embryonic fibroblast (MEF) cell line | Robert Lefkowitz, Duke University | Kohout et al., 2001, PMID 11171997 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Adrb2Cys265Ser/Cys265Ser knock-in | this study | N/A |
| Mouse: Nos3 knockout: B6.129P2-Nos3tm1Unc/J | The Jackson Laboratories | Cat# Jax:002684 RRID:IMSR_JAX:002684 |
| Mouse: FLP deleter strain: B6.Cg-Tg(ACTFLPe)9205Dym/J | The Jackson Laboratories | Cat# Jax:005703 RRID:IMSR_JAX:005703 |
| Mouse: FVB.129-Adrb2tm1Bkk/J | The Jackson Laboratories | Cat# Jax: 031496 RRID:IMSR_JAX:031496 |
| Oligonucleotides | ||
| See Supplemental Table I | this study | N/A |
| Recombinant DNA | ||
| GloSensor-20F cAMP biosensor | Promega | E1171 |
| CAMYEL cAMP biosensor | Paul Sternweis, UT Southwestern, Jiang et al., 2007 PMID 17283075 | N/A |
| Flag-β2AR (human) | Robert Lefkowitz, Oppermann et al., 1996 PMID 8755530 |
N/A |
| Flag-β2AR (human), PKA− mutant | Robert Lefkowitz, Seibold et al., 1998 PMID 9516468 |
N/A |
| Flag-β2AR (human), GRK− mutant | Robert Lefkowitz, Seibold et al., 1998 PMID 9516468 |
N/A |
| GFP-β-Arrestin2 | Robert Lefkowitz, Barak et al, 1997 PMID 9346876 |
N/A |
| Venus 156-239-Gβ1, Venus 1-155-Gγ2 | Nevin Lambert, Hollins et al., 2009 PMID 19258039 |
N/A |
| β2AR-NanoLuc | Nevin Lambert, Okashah et al., 2019 PMID 31142646 |
|
| HA-ATR1 (human) | Robert Lefkowitz, Tohgo et al, 2002 PMID: 11777902 |
N/A |
| Software and Algorithms | ||
| Graphpad Prism (v7, v9) | Graphpad Software | version 7, version 9 |
| Other | ||
| N/A | ||
All data is available in the main text, Supplemental Materials or Mendeley Data. Requests for research materials should be addressed to JSS, jss156@case.edu