

Abstract
We describe an innovative use for the recently reported Fast Lipid Analysis Technique (FLAT) that allows for the generation of MALDI tandem mass spectrometry data suitable for lipid A structure analysis directly from a single Gram-negative bacterial colony. We refer to this tandem MS version of FLAT as FLATn. Neither technique requires sophisticated sample preparation beyond selection of a single bacterial colony, which significantly reduces overall analysis time (~ one hour), as compared to conventional methods. Moreover, the tandem mass spectra generated by FLATn provides comprehensive information of fragments of lipid A, e.g., ester bonded acyl chain dissociations, cross-ring cleavages, and glycosidic bond dissociations all of which allows facile determination of novel lipid A structures or confirmation of expected structures. In addition to generating tandem mass spectra directly from single colonies, we also show that FLATn can be used to analyze lipid A structures taken directly from a complex biological clinical sample without the need for ex vivo growth. From a urine sample from a patient with an E.coli infection, FLATn identified the organism and demonstrated that this clinical isolate carried the mobile colistin resistance-1 gene (mcr-1) that results in the addition of a phosphoethanolamine moiety and subsequently resistance to the antimicrobial, colistin (polymyxin E). Moreover, FLATn allowed for determination of the existence of a structural isomer in E.coli lipid A that had either a 1 or 4’-phosphate group modification by phosphoethanolamine generated by a change of bacterial culture conditions.
Keywords: Lipid A, Gram-negative bacteria, Tandem Mass Spectrometry, FLATn, Structural Analysis
Introduction
Gram-negative bacteria cause a variety of human infectious diseases, including urinary tract infections, pneumonia, and bloodstream infections (sepsis).1 Thus, understanding the pathogenic microbial molecules responsible for such maladies is important. One well-known molecule present in the outer membrane of Gram-negative bacteria is lipopolysaccharide (LPS).2 LPS consists of three structural elements: Oantigen, the most exterior region and composed of repeating saccharide units, followed by a carbohydrate core region, and anchored in the membrane by lipid A.3 Most Gram-negative bacteria share a canonical lipid A base structure consisting of two glucosamine backbones connected by a β(1’,6)-glycosidic bond with each sugar having a terminal phosphate group at the 1 and 4’ carbon position of the diglucosamine backbone, respectively.4 Fatty acid moieties are linked to the sugar backbone at the O-3’, N-2’, O-3, or N-2 positions by ester or amide bonds and can vary in length and number of acyl chains, both of which can be influenced by environment conditions during growth.5
Structural modification of lipid A plays a critical role in resistance to antibiotics, such as polymyxins including colistin (polymyxin E).6 Modification of lipid A by attachment of positively-charged groups, such as phosphoethanolamine (PEtN) and 4-amino-4-deoxy-l-arabinopyranose (Ara4N) decrease the overall negative charge of the bacterial membrane, thereby resulting in resistance to cationic antimicrobial peptides, including colistin.7,8 Thus, structural determination is a critical component to provide a better understanding of the lipid A structure activity relationship (SAR) with the host innate immune system.
Historically, lipid A chemical structure was solved by tandem mass spectrometry (MS/MS)-based analysis (alone or together with nuclear magnetic resonance), collision induced dissociation (CID)9–13 using either standard MS2 approaches in quadrupole based mass analyzers, or MSn 14–16 approaches in ion trapping devices. Due to the presence of salts, which can hamper ionization and interpretation in crude preparations of lipid A, highly purified lipid A, free of nucleic acid and protein contamination has been essential to generate high quality MSn data. However, current LPS/lipid A extraction protocols are labor-intensive and require the use of harmful organic chemicals, including phenol and chloroform to produce milligram quantities of lipid A for tandem MS analysis.17 Routine lipid A structural analysis has typically required a minimum 10 milligrams to fully solve a structure of a single lipid A molecule.18 Therefore, enabling rapid analysis without labor- and time-intense purification methods would significantly decrease the bottleneck of lipid A structure analysis.
For rapid lipid-based identification of bacterial species, our group previously developed a method for on-surface and/or on-tissue release of lipid A from LPS allowing detection of lipid A by matrix-assisted laser desorption ionization (MALDI) MS in the negative ion mode.19,20 This new method, termed Fast Lipid Analysis Technique (FLAT) selectively cleaves the glycosidic linkage between the first core sugar, 3-deoxy-D-manno-octulsonic acid (Kdo), and the non-reducing glucosamine (6’ position) of lipid A and core. Since FLAT requires only a single colony and a single step to generate lipid A from intact bacteria, the entire process takes approximately one hour.19 In this study, we demonstrate that a MALDI trapped ion-mobility time-of-flight (timsTOF) mass spectrometer can produce detailed structural information, including cross-ring fragmentation patterns that allowed for the rapid definition of the chemical structure of lipid A. We refer to this tandem MS adoption of FLAT as FLATn and demonstrate that FLATn can produce all the key fragment ions needed to interpret lipid A structure from a single colony using various Gram-negative bacterial backgrounds. With the ability to expedite the structural analysis of lipid A, we can accelerate the rate at which we associate a given structure with an observed activity,21–24 which is key to development of objective SAR predictions for lipid A’s interaction with the MD2/TLR4 complex that can be either antagonistic or agonistic for future studies.
Experimental Section
A single colony (<2 mm) of each bacterial strain (E. coli, P. aeruginosa, mcr-1 A. baumannii, mcr-1 E. coli, mcr-1 K. pneumoniae, and mcr-1 P. aeruginosa) was picked from lysogeny broth (LB, Becton-Dickenson, Hunt Valley, MD, USA). agar plate using a 0.2 – 10 μL capacity pipette tip after 18 hours of growth at 37°C. The single colony was directly smeared on the ITO-glass slide. To confirm reproducibility, three individual, well separated colonies were chosen and used for FLATn from each bacterial plate. In order to determine compatibility of FLATn for liquid cultured samples, E. coli was used for control experiment. In detail, the single colony of E. coli was picked from the LB agar plate using a disposable inoculation loop. The colony was resuspended in deionized water (200 μL). Then, the colony was spun down (8000 × g, 5 min), supernatant was discarded by a pipette, and the remaining 10 μL of slurry was used for MS analysis. For each FLATn, only one microliter of slurry was used. Next, FLAT was conducted according to the previous literature.19,20 Briefly, 1 μL of the prepared citrate buffer solution (0.2 M citric acid, 0.1 M trisodium citrate, pH 3.5) was deposited onto the sample spot on the ITO slide. The plate was incubated in a humidified, closed glass chamber for 30 min at 110°C. After heating, the ITO slide was removed from the chamber, cooled, and the plate was thoroughly washed with water using a pipettor several times and left to dry on the laboratory bench.
For liquid culture, single colonies were picked from agar plates of E. coli, P. aeruginosa, mcr-1 A. baumannii, mcr-1 E. coli, mcr-1 K. pneumoniae, and mcr-1 P. aeruginosa and inoculated into 5 mL of LB broth (Becton-Dickenson, Hunt Valley, MD, USA). Cultures were incubated at 37°C with 200 rpm shaking to mid-log phase (approximately 18 hours). For isolation of bacteria from media, 1.0 mL of liquid culture of E. coli, P. aeruginosa, mcr-1 A. baumannii, mcr-1 E. coli, mcr-1 K. pneumoniae, and mcr-1 P. aeruginosa were harvested into a microfuge tube, pelleted (8000 × g, 5 min), and 900 μL of supernatant discarded. To remove remaining growth media, 900 μL of deionized water was added into the tube and the cell harvesting procedure was conducted one more time and as described above. The remaining 100 μL of bacteria solution were used to resuspend the bacterial pellet and 1 μL of this suspension was used for analysis. The sample was deposited on the ITO glass slide and allowed to dry. FLAT was conducted according to the aforementioned method.
A urine specimen was obtained from the University of Maryland Medical Center clinical microbiology laboratory. The sample was confirmed to be positive by the clinical laboratory for a urinary tract infection (UTI) after quantification via culture contained greater than 100,000 cfu/mL (Institutional Review Board (IRB) HP-00064919). Lipid analysis via MALDI-TOF MS identified the presence of E. coli with an addition of a phosphoethanolamine indicative of the presence of an mcr-1 gene.25 Pathogen identification of the urine specimen was confirmed by the clinical microbiology laboratory via standard culture methods. The presence of the mcr-1 gene was confirmed via polymerase chain reaction (PCR).25 For urine analysis, the sample was concentrated before use as follows: 1.0 mL of human urine were harvested into a microfuge tube, pelleted (8000 × g, 5 min), and 990 μL of supernatant discarded. The remaining 10 μL of bacteria solution was used to re-suspend the bacterial pellet and 1 μL of this suspension was used for analysis. The sample was deposited on the ITO glass slide and allowed to dry. FLAT was conducted according to the aforementioned method.
Expanded materials, bacteria strains, bacterial growth conditions, instrumentation information, and data processing are available in the Supporting Information.
Results
Determination of structure of canonical lipid A from a single bacterial colony
FLAT is compatible with various MALDI MS instruments with different configurations for vacuum pressure (high-vacuum or intermediate-pressure) and different laser wavelengths (337 nm from N2 versus 355 nm from Nd:YAG).20 Previously, we used the Gram-negative organisms, Escherichia coli (E. coli) and Pseudomonas aeruginosa (P. aeruginosa) to examine the efficiency of FLAT to produce sufficient lipid A signal from a single colony of bacteria. However, the extent to which lipid A fragment ions are important for structure determination could reproducibly be generated from a single colony was not determined. Using FLATn, we found that there was sufficient biomass in a single colony to produce lipid A signature ions historically used for identification26 and tandem MS to determine the structure of these two lipid A molecules from their respective signature ions. To our knowledge, this is the first successful demonstration of structural analysis of lipid A from a single bacterial colony.
Figure 1 illustrates FLATn results from a single colony of P. aeruginosa used for lipid A structure derivation. Figure 1A (arrow) shows the exact colony chosen for analysis (>2 mm). Figure 1B shows a mass spectrum obtained from the colony after FLAT processing (MS1). Using the precursor ion at m/z 1445.8673 (Figure 1C), we carried out tandem MS (MS2) and used that resultant data to confirm the structure of this lipid A molecule. Figure 1D indicates key bond cleavages by CID from FLATn. As expected, many neutral losses are observed, such as acyl chains from ester bonds, phosphoric acid (H3PO4), and cross-ring cleavages. The list of key fragment ions generated by FLATn is shown in Figure 1D. For instance, the loss of H3PO4 from P. aeruginosa lipid A corresponds to an ion at m/z 1347.8804, which is labeled as B2 according to accepted nomenclature.13,27 Multiple losses of acyl chains from ester bonds, such as m/z 1159.7391 (B2 with 3’α), 943.5666 (B2 with 3’α and 3ε), and 743.3889 (B2 with 3’α, 3ε, and 2’ ε) are shown in the Figure 1E. The cross-ring cleavage produces ions at m/z 722.3886 and 662.3675 corresponded to 0,2A2 and 0,4A2, respectively. These selective dissociations of acyl chains from ester bonds and crossring cleavage of the backbone were used to deduce the location of additional acylations and modifications of acyl chains.
Figure 1.
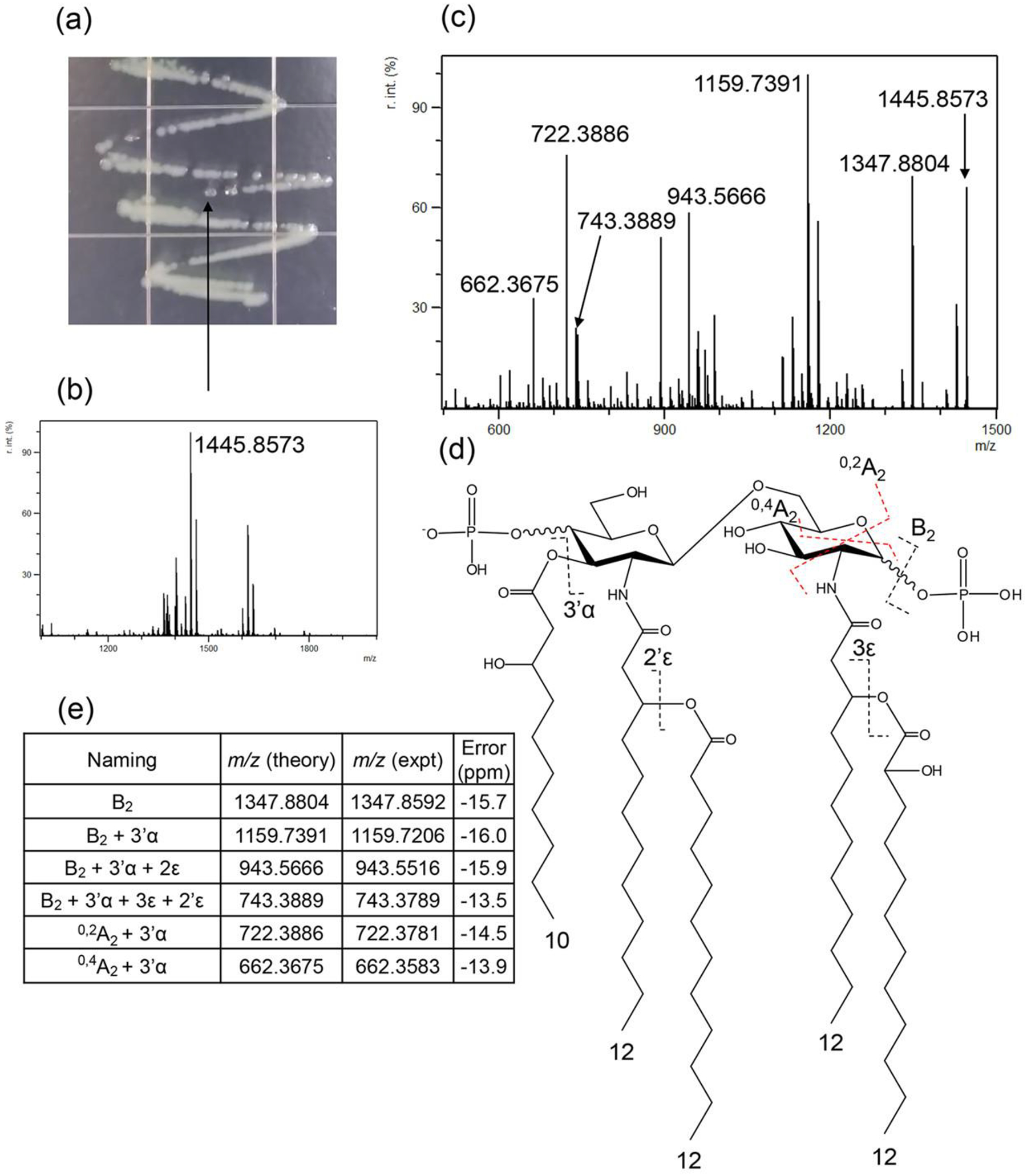
(a) An image of Pa single colonies on agar plate. (b) FLAT mass spectrum obtained from the colony (c) FLATn mass spectrum from precursor ion at m/z 1445.8573 (d) Structure of lipid A from FLATn, and (e) list of assigned ions.
Since the m/z error in these experiments resulting from the difference between theoretical and experimental value is approximately 20 ppm, fragment ions are readily identified using the high mass accuracy data. We note that FLATn of E. coli lipid A was conducted with similar results (see Figures S2 and S3). In order to test reproducibility of FLATn, three different individual colonies were selected and subjected to FLATn. These results suggest that FLATn provides highly reproducible data (see Figure S4). Interestingly, the acyl chain in the amide bond was not dissociated and the cross-ring cleavage was only observed in the reducing-end of the glucosamine backbone; however sufficient fragment ion detail was present to solve the structure represented by the ion at m/z 1347.8804 (B2), 1159.7391 (B2 + 3’α), 943.5666 (B2 + 3’α + 3ε), 743.3889 (B2 + 3’α + 3ε + 2’ε), 722.3886 (0,2A2 + 3’α), and 662.3675 (0,4A2 + 3’α).These type fragmentation phenomena of lipid A have been previously observed.28,29
Determination of lipid A structures that correlate with antibiotic resistance from a single bacterial colony
Electrostatic interaction between positively charged antibiotics, such as colistin and negatively charged LPS is a critical mechanism underlying the mechanism through which antibiotics target bacterial membranes.30 As colistin is often used as the ‘last resort’ antibiotic to treat multi-drug resistant Gram-negative bacterial infections, the emergence of colistin resistance is a high risk factor for patients.7 Colistin resistance in Gram-negative bacteria can be encoded chromosomally31–32 or be plasmid-based33,34. For this work, we focused on the recently identified Mobilized Colistin Resistance (mcr) genes that mediate bacterial colistin resistance via a bacterial plasmid.35 Previously, our group and others have shown mcr-1 gene products function as a lipid A phosphoethanolamine transferase resulting in the modification of the terminal phosphate groups of lipid A by addition of phosphoethanolamine (PEtN).33,34 However, it is not possible to assess which phosphate group position is modified without MS2 data. Here, we demonstrate that FLATn can be used to determine the exact location of the phosphate group modified by PEtN addition. All FLATn tandem mass spectra for each mcr-1 expressing bacterial (mcr-1 A. baumannii, mcr-1 K. pneumoniae, and mcr-1 P. aeruginosa) and their assigned structures are shown in Figures S5 to S8. Moreover, using the FLATn data, we were also able to identify acyl chain modifications from colistin-resistant bacteria, Acinetobacter baumannii (mcr-1 A. baumannii) and Klebsiella pneumoniae (mcr-1 K. pneumoniae). These data are shown in Figures S9 to S12. As shown for E. coli and P. aeruginosa above, FLATn tandem mass spectra used for structure elucidation could be obtained from analysis of a single colony. Data in all Supplemental Figures were produced from a single colony for each bacterial background. We were able to structurally analyze multiple lipid A structures from a single colony using FLATn (Figure S7 – 10).
Furthermore, we demonstrate the dependence on growth conditions of the location of PEtN modification on phosphate in E. coli lipid A. Figure 2A shows FLATn tandem mass spectra from solid media (Top) and mcr-1 E. coli obtained from a human urine sample (Bottom). Although the two mass spectra share the same precursor ion at m/z 1919.2206, each has a different tandem MS fragmentation pattern. This result indicates that the PEtN modification site is variable depending on growth conditions. The characteristic ion at m/z 1267.8095, representing the 4’ phosphate group modified with PEtN was also observed from single colony experiments (Figure 2A, Top) or when grown in the presence of gentamicin (liquid culture) as shown in Figure 2B (Top). Conversely, the characteristic ion at m/z 833.4335 representing the 1-phosphate group modification with PEtN, generated from Y1, was the predominant ion in the human urine sample (Figure 2A, Bottom) and after liquid culture without gentamicin (Figure 2B, Bottom). The structure of mcr-1 E. coli lipid A modification with PEtN on the 1 or 4’-phosphate group are shown in Figure 2C and D, respectively. These figures show the base peak and characteristic ions of each mcr-1 E. coli lipid A at m/z 1919.2206. When we expand any of these mass spectra, each one contains both the characteristic ion at m/z 1267.8095 and 833.4335. A bar graph in Figure 2E shows the ratio of intensities of two ions (m/z 1267.8095 and 833.4335), which represents the 4’-and 1-phosphate group modification, respectively. Individual data sets, except for the human urine sample, which was a single collection, were obtained from three independent biological replicates. The error bars in Figure 2E indicate the standard deviation that arise from biological replicates. This result indicates FLATn unambiguously distinguished structure isomers of lipid A from various growth conditions. Interestingly, other mcr-1 A. baumannii, mcr-1 K. pneumoniae, and mcr-1 P. aeruginosa did not show any alteration of the site of PEtN modification when growth conditions were changed. All data used to determine PEtN modification are provided in Table S1.
Figure 2.
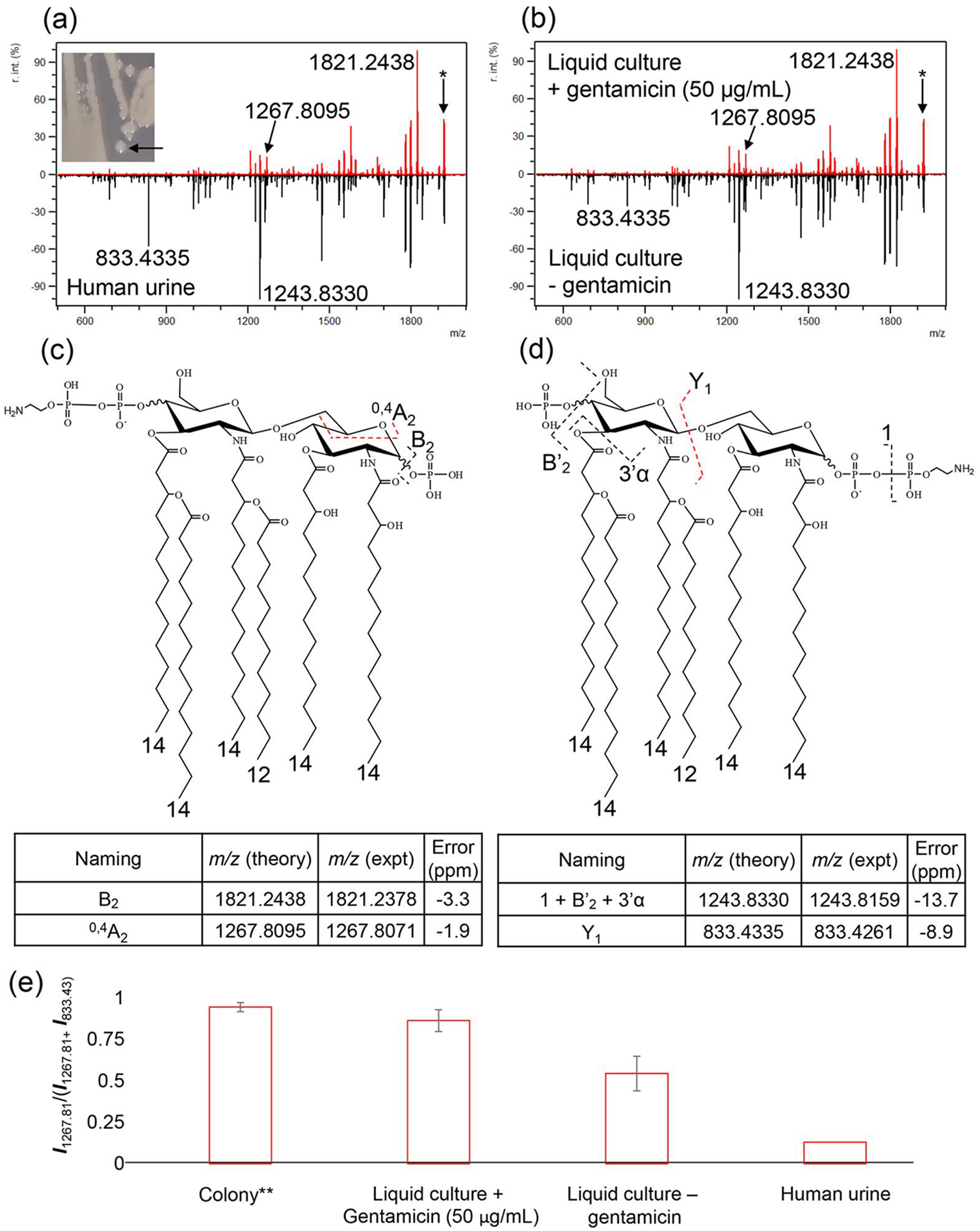
FLATn mass spectra of m/z 1919.2206, where the samples were obtained from (a) Single colony (Top) and human urine sample (Bottom), (b) liquid culture with gentamicin (50 μg/mL, Top) and without gentamicin (Bottom). Asterisk mark (*) in the mass spectra indicates precursor ion at m/z 1919.2206. The structure of 4’ (c) and 1-phosphate group (d) modification with PEtN (e) a bar graph of ratio of intensities of two ions between 1267.8095 and 833.4335, which represents 4’- and 1-phosphate group modification by PEtN addition, respectively. **Agar plate with gentamicin (50 μg/mL)
Conclusion
We developed FLATn, a method that provides highly reliable and robust tandem MS data for lipid A structural analysis without prior extraction. FLATn provided sufficient fragment ions from a single colony to confirm the structures of several Gram-negative bacteria and to discern structural changes induced by differences in growth conditions. Like FLAT, the FLATn method is conducted entirely after on-surface extraction of lipid A in a time that is significantly shorter compared to conventional methods (e.g., 48 hours vs 1 hr). Given the rapid rate that lipid A tandem MS data may now be generated we expect FLATn to play an important role in our quest to develop a better understanding of the lipid A SAR.
Supplementary Material
Acknowledgements
Professors Goodlett and Ernst thank the National Institutes of Health for funding from R01GM111066 and 1R01AI147314-01A1. DRG thanks the International Centre for Cancer Vaccine Science project carried out within the International Research Agendas program of the Foundation for Polish Science co-financed by the European Union under the European Regional Development Fund (MAB/2017/03) for support. DRG is grateful for funding for technology development and platform support for The Metabolomics Innovation Centre (TMIC), from Genome Canada, and Genome British Columbia through the Genomics Technology Platform (GTP) program for operations and technology development (265MET and MC4), as well as funding from the Canadian Foundation for Innovation’s Major Science Initiative program (35456). This research was supported in part by the Translational Analytical Core of the Intramural Research Program of the National Institute on Drug Abuse, NIH.
References
- (1).Peleg AY; Hooper DC N Engl J Med 2010, 362, 1804–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bertani B; Ruiz N EcoSal Plus 2018, 8. DOI: 10.1128/ecosalplus.ESP-0001-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Raetz CR; Reynolds CM; Trent MS; Bishop RE Annu Rev Biochem 2007, 76, 295–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Raetz CR; Whitfield C Annu Rev Biochem 2002, 71, 635–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Kabanov DS; Prokhorenko IR Biochemistry (Mosc) 2010, 75, 383–404. [DOI] [PubMed] [Google Scholar]
- (6).El-Sayed Ahmed MAE; Zhong LL; Shen C; Yang Y; Doi Y; Tian GB Emerg Microbes Infect 2020, 9, 868–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Aghapour Z; Gholizadeh P; Ganbarov K; Bialvaei AZ; Mahmood SS; Tanomand A; Yousefi M; Asgharzadeh M; Yousefi B; Kafil HS Infection and Drug Resistance 2019, 12, 965–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Molinaro A; Holst O; Di Lorenzo F; Callaghan M; Nurisso A; D’Errico G; Zamyatina A; Peri F; Berisio R; Jerala R; Jimenez-Barbero J; Silipo A; Martin-Santamaria S Chemistry 2015, 21, 500–519. [DOI] [PubMed] [Google Scholar]
- (9).Corsaro MM; Piaz FD; Lanzetta R; Parrilli M J Mass Spectrom 2002, 37, 481–488. [DOI] [PubMed] [Google Scholar]
- (10).Bedoux G; Vallee-Rehel K; Kooistra O; Zahringer U; Haras D J Mass Spectrom 2004, 39, 505–513. [DOI] [PubMed] [Google Scholar]
- (11).Shaffer SA; Harvey MD; Goodlett DR; Ernst RK J Am Soc Mass Spectrom 2007, 18, 1080–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Jones JW; Shaffer SA; Ernst RK; Goodlett DR; Turecek F Proc Natl Acad Sci U S A 2008, 105, 12742–12747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Jones JW; Cohen IE; Turecek F; Goodlett DR; Ernst RK J Am Soc Mass Spectrom 2010, 21, 785–799. [DOI] [PubMed] [Google Scholar]
- (14).Crittenden CM; Akin LD; Morrison LJ; Trent MS; Brodbelt JS J Am Soc Mass Spectrom 2017, 28, 1118–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Morrison LJ; Parker WR; Holden DD; Henderson JC; Boll JM; Trent MS; Brodbelt JS Anal Chem 2016, 88, 1812–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Crittenden CM; Herrera CM; Williams PE; Ricci DP; Swem LR; Trent MS; Brodbelt JS Analyst 2018, 143, 3091–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Henderson JC; O’Brien JP; Brodbelt JS; Trent MS J Vis Exp 2013,e50623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sidorczyk Z; Zahringer U; Rietschel ET European Journal of Biochemistry 1983, 137, 15–22. [DOI] [PubMed] [Google Scholar]
- (19).Sorensen M; Chandler CE; Gardner FM; Ramadan S; Khot PD; Leung LM; Farrance CE; Goodlett DR; Ernst RK; Nilsson E Sci Rep 2020, 10, 21536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Yang H; Chandler CE; Jackson SN; Woods AS; Goodlett DR; Ernst RK; Scott AJ Anal Chem 2020, 92, 13667–13671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Gauthier AE; Chandler CE; Poli V; Gardner FM; Tekiau A; Smith R; Bonham KS; Cordes EE; Shank TM; Zanoni I; Goodlett DR; Biller SJ; Ernst RK; Rotjan RD; Kagan JC Sci Immunol 2021, 6. DOI: 10.1126/sciimmunol.abe0531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Chandler CE; Harberts EM; Pelletier MR; Thaipisuttikul I; Jones JW; Hajjar AM; Sahl JW; Goodlett DR; Pride AC; Rasko DA; Trent MS; Bishop RE; Ernst RK Proc Natl Acad Sci U S A 2020, 117, 22984–22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Richard K; Perkins DJ; Harberts EM; Song Y; Gopalakrishnan A; Shirey KA; Lai W; Vlk A; Mahurkar A; Nallar S; Hawkins LD; Ernst RK; Vogel SN Vaccine 2020, 38, 4298–4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Gregg KA; Harberts E; Gardner FM; Pelletier MR; Cayatte C; Yu L; McCarthy MP; Marshall JD; Ernst RK mBio 2017, 8. DOI: 10.1128/mBio.00492-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Smith RD; Izac JR; Ha M; Yang H; Johnson JK; Ernst RK Access Microbiol 2021, 3, 000309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Leung LM; Fondrie WE; Doi Y; Johnson JK; Strickland DK; Ernst RK; Goodlett DR Sci Rep 2017, 7, 6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Costello CE; Vath JE Methods Enzymol 1990, 193, 738–768. [DOI] [PubMed] [Google Scholar]
- (28).Sandor V; Kilar A; Kilar F; Kocsis B; Dornyei A J Mass Spectrom 2016, 51, 615–628. [DOI] [PubMed] [Google Scholar]
- (29).Sandor V; Dornyei A; Makszin L; Kilar F; Peterfi Z; Kocsis B; Kilar A J Mass Spectrom 2016, 51, 1043–1063. [DOI] [PubMed] [Google Scholar]
- (30).Velkov T; Thompson PE; Nation RL; Li J J Med Chem 2010, 53, 1898–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Olaitan AO; Morand S; Rolain JM Front Microbiol 2014, 5, 643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Groisman EA J Bacteriol 2001, 183, 1835–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Liu YY; Wang Y; Walsh TR; Yi LX; Zhang R; Spencer J; Doi Y; Tian G; Dong B; Huang X; Yu LF; Gu D; Ren H; Chen X; Lv L; He D; Zhou H; Liang Z; Liu JH; Shen J Lancet Infect Dis 2016, 16, 161–168. [DOI] [PubMed] [Google Scholar]
- (34).Liu YY; Chandler CE; Leung LM; McElheny CL; Mettus RT; Shanks RMQ; Liu JH; Goodlett DR; Ernst RK; Doi Y Antimicrob Agents Chemother 2017, 61. DOI: 10.1128/AAC.00580-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Gharaibeh MH; Shatnawi SQ Vet World 2019, 12, 1735–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.