

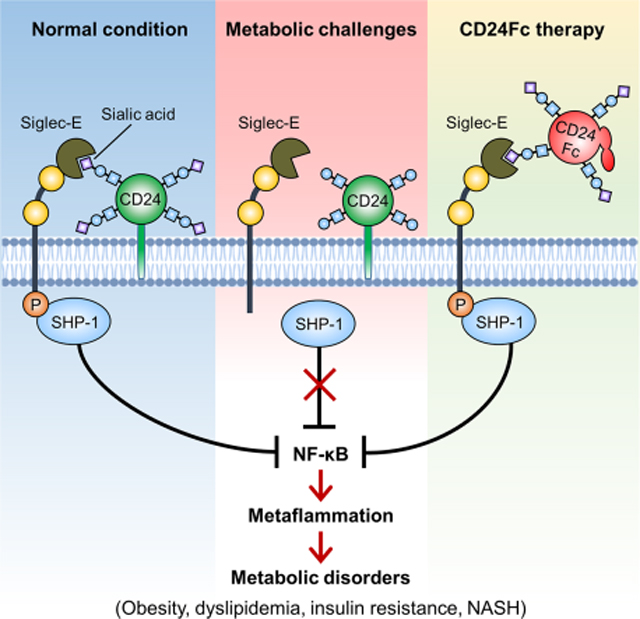
Summary
The molecular interactions that regulate chronic inflammation underlying metabolic disease remain largely unknown. Since the CD24-Siglec interaction regulates inflammatory response to danger-associated molecular patterns (DAMPs), we have generated multiple mouse strains with single or combined mutations of Cd24 or Siglec genes to explore the role of the CD24-Siglec interaction in metaflammation and metabolic disorder. Here we report that the CD24-Siglec-E axis, but not other Siglecs, is a key suppressor of obesity-related metabolic dysfunction. Inactivation of the CD24-Siglec-E pathway exacerbates, while CD24Fc treatment alleviates, diet-induced metabolic disorder, including obesity, dyslipidemia, insulin resistance and nonalcoholic steatohepatitis (NASH). Mechanistically, sialylation-dependent recognition of CD24 by Siglec-E induces SHP-1 recruitment and represses metaflammation to protect against metabolic syndrome. A first-in-human study of CD24Fc (NCT02650895) supports the significance of this pathway in human lipid metabolism and inflammation. These findings identify the CD24-Siglec-E axis as an innate immune checkpoint against metaflammation and metabolic disorder, and suggest a promising therapeutic target for metabolic disease.
Keywords: CD24, sialic acid-binding immunoglobulin-like lectins (Siglecs), Siglec-E, sialylation, metaflammation, metabolic syndrome, obesity, insulin resistance, NASH
Graphical Abstract
eTOC blurb
Metaflammation, chronic inflammation driven by nutrient and energy excess, lies at the center of metabolic disease. Here, Wang et al. show that inactivation of the CD24-Siglec-E axis exacerbates, while CD24Fc therapy ameliorates obesity-related metabolic disorder. Sialylated CD24 induces recruitment of SHP-1 to Siglec-E to dampen metaflammation. In a first-in-human phase I clinical trial, a single dose of CD24Fc was safe and well-tolerated in healthy subjects.
Introduction
Metabolic disorder, including obesity, dyslipidemia, diabetes, nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH) among others, have emerged as a major global health problem (Christ and Latz, 2019; Malik et al., 2013). Metaflammation, a chronic low-grade inflammatory state in metabolic tissues, is a hallmark of metabolic disease (Hotamisligil, 2006, 2017). Chronic tissue inflammation, accompanied by infiltration and activation of immune cells and increased inflammatory cytokines, impairs insulin signaling and disrupts systemic metabolic homeostasis (Hotamisligil, 2006; Lumeng and Saltiel, 2011; Schenk et al., 2008; Shoelson et al., 2006). It has been reported that several inflammatory signaling pathways (e.g., JNK and IKK) and inflammatory cytokines (e.g., TNF-α and IL-1β) are implicated in the pathogenesis of metabolic disease (Hirosumi et al., 2002; Hotamisligil et al., 1993; Maedler et al., 2002; Yuan et al., 2001). Despite strong evidence indicating a link between chronic inflammation and obesity, the initiation and regulation of metaflammation during obesity remains poorly understood.
Siglec proteins are immunoglobulin superfamily members that recognize sialic acid-containing structures (Crocker et al., 2007; Duan and Paulson, 2020). Most Siglecs have intracellular immunoreceptor tyrosine-based inhibitory motifs (ITIMs) and function as inhibitory receptors via the recruitment of tyrosine phosphatases SHP-1 and/or SHP-2 (Crocker et al., 2007; Macauley et al., 2014). Since Siglecs are predominately expressed on innate immune cells, it has been suspected that Siglecs confer negative regulation for innate immunity (Crocker et al., 2007; Pillai et al., 2012). Identification of natural Siglec ligands from host cells and bacterial components helps to establish the central role for Siglecs in the regulation of innate immune responses to PAMPs and DAMPs. Thus, our previous studies identified CD24, a small glycosylphosphatidylinositol (GPI)-anchored cell surface glycoprotein (Fang et al., 2010; Pirruccello and LeBien, 1986), as the first natural ligand of Siglec-G/10 and that the CD24-Siglec-G/10 interaction selectively suppresses inflammatory response to DAMPs in tissue injuries (Chen et al., 2011; Chen et al., 2009). More recent studies confirmed the significance of this regulatory pathway in autoimmune diseases (Bai et al., 2000; Zheng et al., 2013), graft versus host disease (Toubai et al., 2014; Toubai et al., 2017), macrophage phagocytosis of cancer cells (Barkal et al., 2019) and COVID-19 (Song et al., 2022; Welker et al., 2022). Most importantly, in a randomized, double-blind, placebo-controlled, phase III study, statistically significant and clinically meaningful therapeutic effect of CD24Fc was observed in patients with COVID-19 (Welker et al., 2022). The broad spectrum of interactions between Siglecs and TLRs further support the notion that Siglecs may emerge as central regulators of innate immune response (Chen et al., 2014).
CD24-Siglec interaction has emerged as an innate immune checkpoint regulating host response to tissue injuries (Chen et al., 2011; Chen et al., 2009; Liu et al., 2009) and cancer immunotherapy (Barkal et al., 2019). However, a role for the CD24-Siglec axis in modulating metabolic disease and metaflammation has not yet been addressed. Here we show that disruption of the CD24-Siglec-E interaction (but not other Siglecs) aggravates, whereas CD24Fc therapy improves obesity-related metabolic disorder. Sialoside-based recognition of CD24 by Siglec-E negatively regulates metaflammation to protect against metabolic syndrome. The clinical study of CD24Fc further supports the significance of this pathway in lipid metabolism and inflammation in humans. These findings reveal a key inhibitory role of the CD24-Siglec-E axis in metabolic dysfunction and metaflammation, and provide a potential immunotherapy for obesity, dyslipidemia, insulin resistance and NASH.
Results
CD24 deficiency aggravates metabolic disorder in mice
CD24-Siglec pathway has not been investigated in obesity-related metabolic disorder. To bridge this major gap, we evaluated the development of metabolic syndrome in CD24-deficient mice fed a high-fat diet (HFD). Relative to WT littermates, Cd24−/− mice displayed higher body weight gain (Figure 1A). Both inguinal and epididymal depots were larger in Cd24−/− mice than those of WT mice (Figure 1B). Dual energy X-ray absorption (DEXA) analysis also showed a selective increase in total body fat content in Cd24−/− mice (Figure 1C). Body weight indicates a balance of energy intake and expenditure. Food intake did not differ between the genotypes (Figure S1A), but Cd24−/− mice exhibited larger brown adipose tissues (BAT), increased lipid content in BAT and decreased body temperature (Figures S1B–S1D). These phenotypic changes were accompanied by reduced expression of genes associated with thermogenesis and mitochondrial biogenesis (Figure S1E), suggesting that reduced energy expenditure may be a potential mechanism for the increased body weight gain.
Figure 1. CD24 deficiency aggravates diet-induced metabolic disorder in mice.
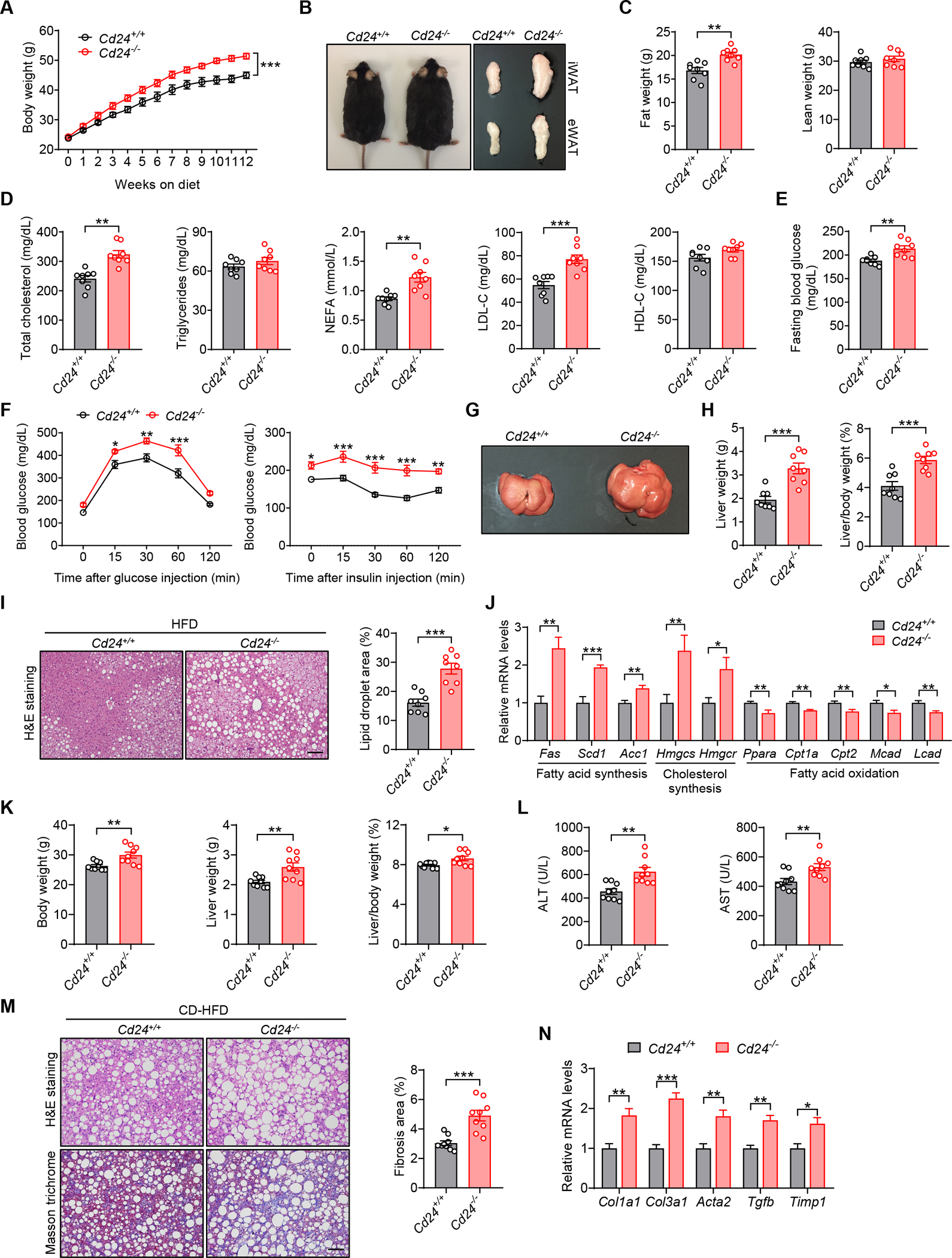
(A-J) Cd24−/− mice and WT littermates were fed a HFD for 12 weeks.
(A) Body weight of mice in the indicated groups. n = 8 per group.
(B) Representative photographs, inguinal and epididymal fat pads of mice in the indicated groups.
(C) Body composition of mice was detected by DEXA. n = 8 per group.
(D) TC, TG, FFA, LDL-C and HDL-C levels of mice in the indicated groups. n = 8 per group.
(E) Fasting blood glucose levels of mice in the indicated groups. n = 8 per group.
(F) GTT and ITT results of mice in the indicated groups. n = 8 per group.
(G) Representative photographs of livers from mice in the indicated groups.
(H) Liver weight and liver/body weight ratio of mice in the indicated groups. n = 8 per group.
(I) Representative images of H&E staining of liver sections. Scale bar, 100 μm. Graph shows the quantitation of lipid droplet area. n = 8 per group.
(J) Relative mRNA levels of key metabolic genes in the livers of mice in the indicated groups. n = 6 per group.
(K-N) Cd24−/− mice and littermate controls were fed a CD-HFD for 12 weeks.
(K) Body weight, liver weight and liver/body weight ratio of mice in the indicated groups. n = 9 per group.
(L) Serum levels of ALT and AST of mice in the indicated groups. n = 9 per group.
(M) H&E and Masson’s trichrome staining of liver sections. Scale bar, 100 μm. Graph shows the quantitation of liver fibrosis area. n = 9 per group.
(N) Relative mRNA levels of profibrotic genes in the livers of mice in the indicated groups. n = 6 per group.
Data are mean ± SEM and representative of two or three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, unpaired Student’s t-test (C-E, H-N), two-way analysis of variance (ANOVA) (A, F). See also Figure S1.
Since CD24 deficiency aggravated HFD-induced obesity, we examined whether this gene could play a role in lipid and glucose homeostasis. The lipid profile study showed lipid metabolic dysfunction in Cd24−/− mice, as evidenced by increased serum levels of total cholesterol (TC), free fatty acid (FFA) and low-density lipoprotein cholesterol (LDL-C), while triglycerides (TG) and high-density lipoprotein cholesterol (HDL-C) levels were unaffected (Figure 1D). In addition, Cd24−/− mice showed higher fasting blood glucose levels, exacerbated glucose intolerance and insulin resistance, as revealed by glucose tolerance tests (GTTs) and insulin tolerance tests (ITTs) (Figures 1E and 1F).
Hepatic steatosis is an important manifestation of metabolic disorder. We found that Cd24−/− mice developed more severe hepatomegaly than WT littermates after HFD feeding, as indicated by increased liver weight and liver-to-body weight ratio (Figures 1G and 1H). Consistently, hepatic lipid accumulation was markedly increased in Cd24−/− mice, as shown by H&E staining (Figure 1I). Excessive fat accumulation in the liver may be due to increased fat synthesis or reduced oxidation. Cd24−/− mice exhibited increased expression of genes related to fatty acid synthesis (Fas, Acc1 and Scd1) and cholesterol synthesis (Hmgcs and Hmgcr). In contrast, the expression of genes responsible for fatty acid oxidation (Ppara, Cpt1a, Cpt2, Mcad and Lcad) were significantly decreased in the livers of HFD-fed Cd24−/− mice (Figure 1J).
To examine the role of CD24 in NASH pathogenesis, we fed Cd24−/− mice and WT littermates with a choline deficient HFD (CD-HFD) to induce NASH with features that include hepatic steatosis, advanced inflammation and fibrosis (Matsumoto et al., 2013). In line with the response to HFD challenge, Cd24−/− mice exhibited increased body weight and liver weight compared to WT littermates after CD-HFD feeding (Figure 1K). A significant increase of serum ALT and AST activities suggested exacerbated liver damage in CD-HFD-fed Cd24−/− mice (Figure 1L). Furthermore, hepatic lipid accumulation and fibrosis were significantly higher in Cd24−/− mice, as indicated by H&E and Masson staining (Figure 1M). These were accompanied by increased expression of NASH-related profibrotic genes (Col1a1, Col3a1, Acta2, Tgfb and Timp1) in the livers of Cd24−/− mice (Figure 1N). Taken together, these results demonstrate that CD24 deficiency aggravates diet-induced obesity, insulin resistance and NASH.
We also assessed the development of metabolic syndrome in Cd24−/− mice fed a normal chow diet (NCD). Although metabolic profiling was equally unremarkable in the young mice (data not shown), aged Cd24−/− mice developed more severe metabolic disorder than control mice, as evidenced by increased body weight gain, impaired lipid profiles, glucose tolerance and insulin sensitivity, elevated liver weight, lipid accumulation and steatosis (Figures S1F–S1K). These data indicate an essential role for CD24 in preventing development of age-related metabolic dysfunction.
CD24Fc alleviates obesity-related metabolic dysfunction in mice
CD24Fc is a recombinant fusion protein consisting of the extracellular domain of mature human CD24 linked to the human immunoglobulin G1 (IgG1) Fc domain. To substantiate the role of CD24 signaling in the pathogenesis of metabolic syndrome, we evaluated the effects of CD24Fc treatment in diet-induced obese (DIO) mice. Preventive treatment with CD24Fc significantly reduced body weight gain and fat content (Figures 2A and 2B). We also observed improved serum lipid profiles in CD24Fc-treated DIO mice, as evidenced by decreased TC, TG, FFA and LDL-C levels (Figure 2C). In addition, CD24Fc treatment significantly reduced fasting blood glucose levels and alleviated glucose intolerance and insulin resistance in DIO mice (Figures 2D and 2E). Improved insulin sensitivity was further supported by greater activation of insulin signaling in CD24Fc-treated mice after insulin challenge (Figure 2F). Furthermore, we observed significant improvements in HFD-induced hepatic steatosis in CD24Fc-treated mice, as indicated by reduced liver weight, liver-to-body weight ratio and lipid accumulation (Figures 2G and 2H). The improvements were confirmed by metabolic gene profiles. Specifically, the expression of genes related to fatty acid synthesis and cholesterol synthesis were significantly reduced, whereas the expression of genes involved in fatty acid oxidation were much higher in the livers of CD24Fc-treated mice than those of control mice (Figure 2I).
Figure 2. CD24Fc alleviates obesity-related metabolic dysfunction in mice.
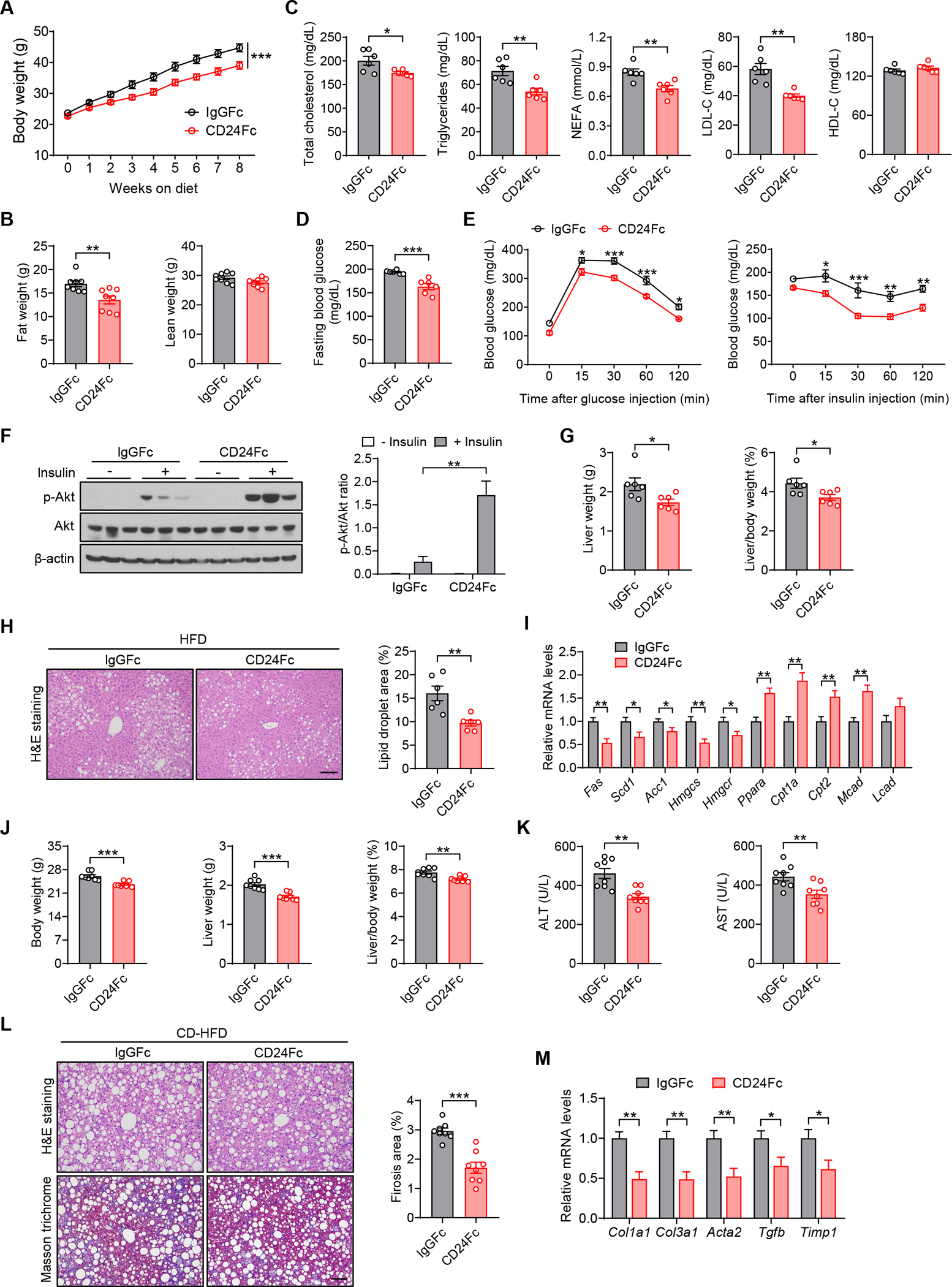
(A-I) WT mice were fed a HFD and concurrently treated with CD24Fc or IgGFc control twice a week for 8 weeks.
(A) Body weight of mice in the indicated groups. n = 8 per group.
(B) Body composition of mice was detected by DEXA. n = 8 per group.
(C) TC, TG, FFA, LDL-C and HDL-C levels of mice in the indicated groups. n = 6 per group.
(D) Fasting blood glucose levels of mice in the indicated groups. n = 6 per group. (E) GTT and ITT results of mice in the indicated groups. n = 8 per group.
(F) Insulin-stimulated phosphorylation of Akt in the livers of mice. Graph shows the quantitation of p-Akt relative to total Akt. n = 3 per group.
(G) Liver weight and liver/body weight ratio of mice in the indicated groups. n = 6 per group.
(H) Representative images of H&E staining of liver sections. Scale bar, 100 μm. Graph shows the quantitation of lipid droplet area. n = 6 per group.
(I) Relative mRNA levels of key metabolic genes in the livers of mice in the indicated groups. n = 6 per group.
(J-M) WT mice were fed a CD-HFD and simultaneously treated with CD24Fc or IgGFc control twice a week for 8 weeks. </p/>(J) Body weight, liver weight and liver/body weight ratio of mice in the indicated groups. n = 8 per group.
(K) Serum levels of ALT and AST of mice in the indicated groups. n = 8 per group.
(L) H&E and Masson’s trichrome staining of liver sections. Scale bar, 100 μm. Graph shows the quantitation of liver fibrosis area. n = 8 per group.
(M) Relative mRNA levels of profibrotic genes in the livers from mice in the indicated groups. n = 6 per group.
Data are mean ± SEM and representative of two or three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, unpaired Student’s t-test (B-D, G-M), two-way analysis of variance (ANOVA) (A, E, F). See also Figure S2.
We next explored whether CD24Fc can ameliorate NASH phenotypes triggered by CD-HFD. Similar to the results in HFD model, CD24Fc-treated mice had lower body weight and liver weight than control mice after CD-HFD consumption (Figure 2J). The degree of liver damage, as detected by serum levels of ALT and AST, was significantly improved in CD24Fc-treated mice (Figure 2K). Moreover, CD24Fc treatment significantly decreased hepatic lipid accumulation and collagen deposition induced by CD-HFD (Figure 2L). Consistent with decreased liver fibrosis, CD24Fc treatment markedly downregulated hepatic expression of profibrotic genes (Figure 2M).
We also evaluated the therapeutic effects of CD24Fc in mice with established obesity. Although there was no impact on body weight gain (Figure S2A), the short-term CD24Fc therapy significantly attenuated HFD-induced metabolic dysfunction, as evidenced by improved lipid profiles, glucose tolerance and insulin sensitivity (Figures S2B–S2D), as well as decreased liver weight, hepatic steatosis and inflammation (Figures S2E–S2I). Altogether, these findings clearly demonstrate that CD24Fc therapy ameliorates obesity-related metabolic disorder.
Siglec-E is the CD24 receptor that protects against metabolic syndrome
Since CD24 is a heavily sialylated cell surface protein with no intracellular domain for signal transduction (Fang et al., 2010), and since Siglecs recognize sialic acid-containing proteins (Crocker et al., 2007), we hypothesized that Siglec might act as the CD24 receptor to regulate metabolic syndrome. To address this, we evaluated the specificity of CD24-Siglec interactions using capture ELISA. We compared mouse Siglec fusion proteins for their binding to CD24 on spleen cells. We observed significant associations of endogenous CD24 with Siglec-E, Siglec-F and Siglec-G, but not Siglec-1 and Siglec-2 (Figure 3A). To test if CD24 directly interacts with Siglecs, we used recombinant CD24Fc and observed a similar pattern of interaction between CD24Fc and recombinant Siglecs (Figure 3B).
Figure 3. Siglec-E is the CD24 receptor that protects against metabolic syndrome.
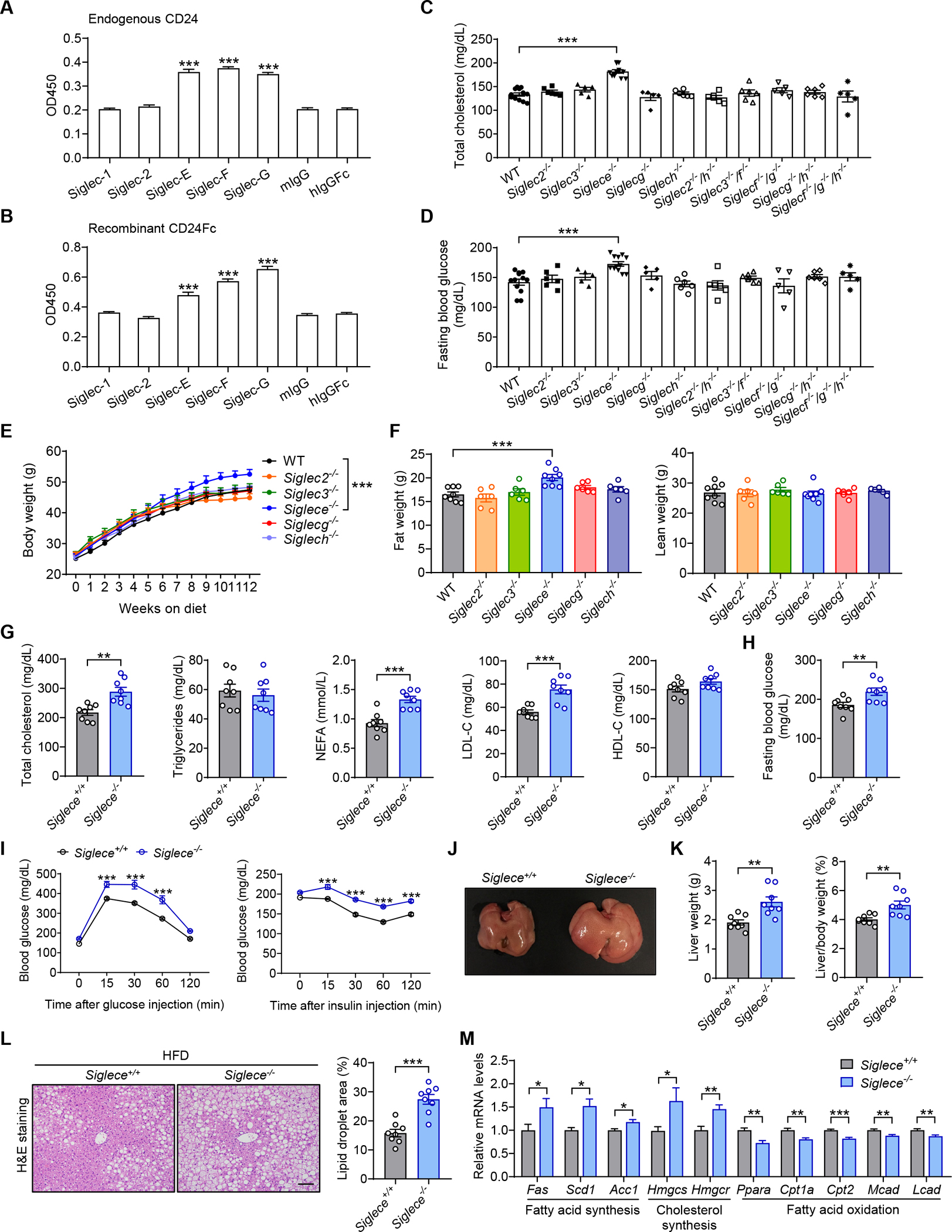
(A) Interactions between endogenous CD24 and recombinant Siglecs. n = 3 per group.
(B) Direct interactions between CD24Fc and recombinant Siglecs. n = 3 per group. </p/>(C and D) Siglec-deficient mice were maintained on a normal diet. Serum levels of total cholesterol (C) and fasting blood glucose (D) were detected at 8 months of age. n = 5–12 per group. </p/>(E and F) WT and Siglec-deficient mice were fed a HFD for 12 weeks. Body weight (E) and body composition (F) were detected. n = 6–8 per group. </p/>(G-M) Siglece−/− mice and WT littermates were fed a HFD for 12 weeks.
(G) TC, TG, FFA, LDL-C and HDL-C levels of mice in the indicated groups. n = 8 per group.
(H) Fasting blood glucose levels of mice in the indicated groups. n = 8 per group.
(I) GTT and ITT results of mice in the indicated groups. n = 6 per group.
(J) Photographs of representative livers from mice in the indicated groups.
(K) Liver weight and liver/body weight ratio of mice in the indicated groups. n = 8 per group.
(L) Representative images of H&E staining of liver sections. Scale bar, 100 μm. Graph shows the quantitation of lipid droplet area. n = 8 per group.
(M) Relative mRNA levels of key metabolic genes in the livers from mice in the indicated groups. n = 8 per group.
Data are mean ± SEM and representative of two or three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, unpaired Student’s t-test (G, H, K-M), one-way analysis of variance (ANOVA) (A-D, F) or two-way analysis of variance (ANOVA) (E, I). See also Figures S3 and S4.
To identify the CD24 receptor responsible for metabolic regulation, we took a genetic approach to determine whether the mutation in any Siglec gene may recapitulate the metabolic phenotypes of CD24 ablation. In addition to mice with mutation of either Siglecg or Siglece gene that we have previously reported (Ding et al., 2007; Flores et al., 2019), we produced mice with mutations of one or more of the additional Siglec genes using CRISPR/Cas9 system (Figure S3 and Table S1). All mutant strains were viable and fertile, with no obvious developmental or behavioral phenotypes. Among these Siglec-deficient mice, we observed that, single mutations in Siglec2, Siglec3, Siglecg or Siglech had no effect on blood glucose and lipid levels, and neither did combinatorial mutations of Siglec2 and Siglech, Siglec3 and Siglecf, Siglecf/g, Siglecg/h or Siglecf/g/h. In contrast, Siglece−/− mice showed impaired glucose and lipid homeostasis, as indicated by significantly increased fasting blood glucose and TC levels (Figures 3C and 3D), suggesting that Siglec-E might be the CD24 receptor regulating glucose and lipid metabolism in mice.
To substantiate the phenocopying of CD24 ablation by Siglec-E deficiency, we evaluated additional metabolic phenotypes in Siglece−/− mice after HFD feeding. Relative to control mice, Siglece−/− mice displayed significantly increased body weight gain and fat content, while mice with mutations in other Siglec genes did not (Figures 3E and 3F). Although there was no significant difference in food intake (Figure S4A), Siglece−/− mice showed dysregulation of energy homeostasis (Figures S4B–S4E). In addition, Siglece−/− mice exhibited disorders in lipid and glucose metabolism, with increased serum levels of TC, LDL-C and FFA, elevated fasting blood glucose levels, exacerbated glucose intolerance and insulin resistance compared to control mice (Figures 3G–3I). Moreover, Siglec-E deficiency aggravated HFD-induced hepatomegaly and hepatic steatosis (Figures 3J–3L), accompanied by increased expression of lipogenic genes and reduced expression of genes related to fatty acid oxidation in the livers of Siglece−/− mice (Figure 3M). We next explored the role of Siglec-E in NASH pathogenesis. In line with the response to HFD treatment, Siglec-E deficiency exacerbated CD-HFD-induced NASH phenotypes, as evidenced by increased hepatic steatosis, liver damage and fibrosis (Figures S4F–S4I), comparable to the phenotypes observed in Cd24−/− mice.
We also determined the development of metabolic syndrome in Siglece−/− mice fed a normal chow diet. As NCD-fed mice age, we observed more severe obesity, insulin resistance and hepatic steatosis in aged Siglece−/− mice (Figures S4J–S4O). In summary, all the findings illustrate that Siglec-E deficiency fully recapitulates the metabolic phenotypes of CD24 deletion in mice, and that Siglec-E is the functional CD24 receptor responsible for metabolic regulation.
Siglec-E signaling is required for CD24-mediated protection against metabolic disorder
To further evaluate that Siglec-E signaling mediates the protective effect of CD24 against metabolic disorder, we compared WT and Siglece−/− mice with established obesity for their response to the therapeutic effect of CD24Fc. CD24Fc therapy improved lipid profiles and fasting blood glucose in WT but not Siglece−/− mice (Figures 4A and 4B). Treatment of WT mice with CD24Fc showed ameliorated glucose intolerance and insulin resistance. However, the therapeutic effects were absent in Siglece−/− mice (Figure 4C). Although there was no impact on body weight gain (Figure 4D), CD24Fc therapy produced significant improvement in hepatomegaly in WT mice after HFD feeding, but not in Siglece−/− mice (Figure 4E). In addition, CD24Fc treatment of WT mice caused a marked decrease in hepatic lipid accumulation, as shown by H&E staining. However, the therapeutic effects were abolished in Siglece−/− mice (Figure 4F). Overall, these data demonstrate that Siglec-E signaling is required for CD24-mediated protection against metabolic disorder, and reveal a key negative role of the CD24-Siglec-E axis in metabolic disease.
Figure 4. Siglec-E signaling is required for CD24-mediated protection against metabolic disorder.
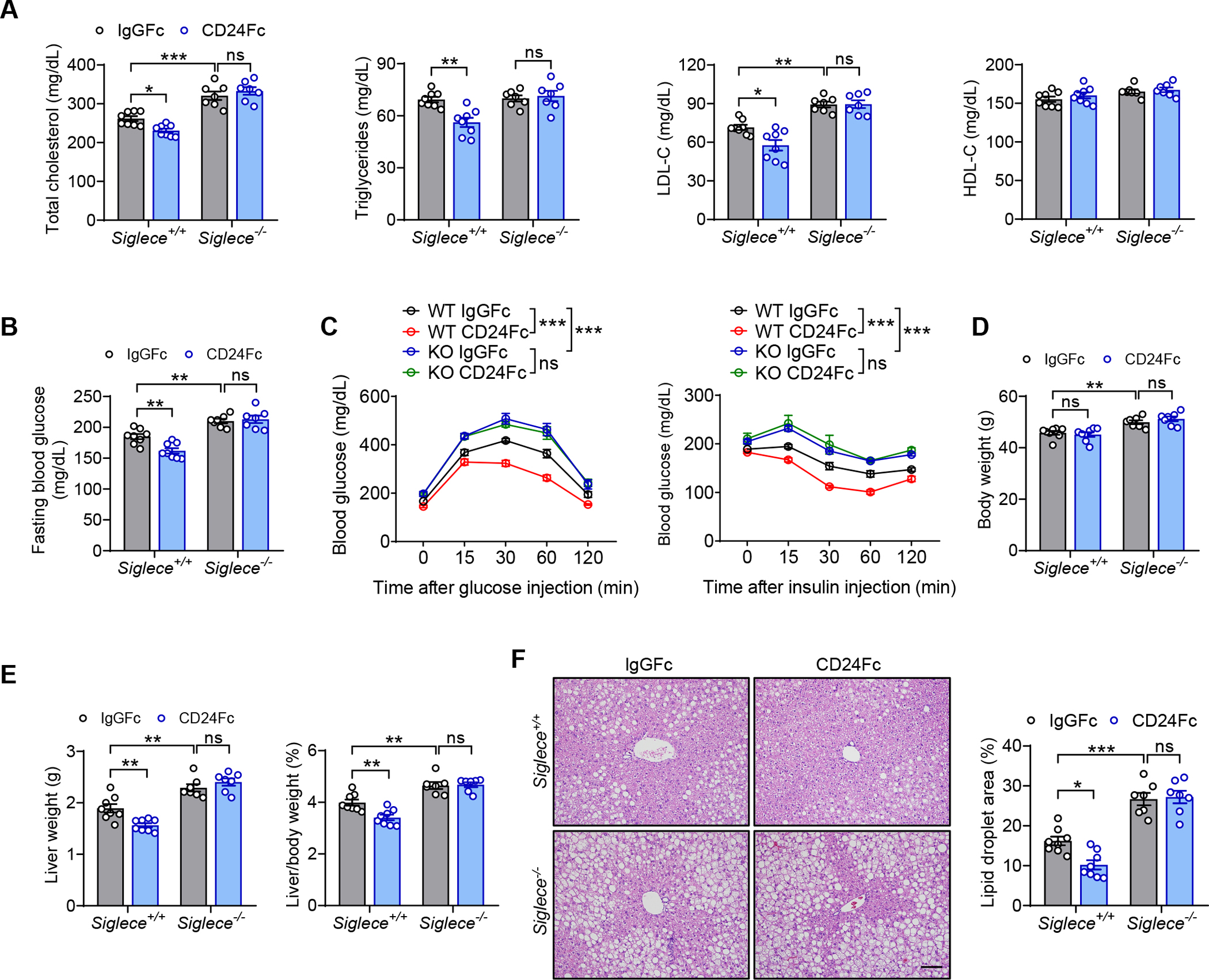
(A-F) Siglece−/− mice and WT littermates were fed a HFD for 8 weeks, followed by injection of CD24Fc or IgGFc control twice a week for 4 weeks while continuing HFD.
(A) TC, TG, LDL-C and HDL-C levels of mice in the indicated groups. n = 7–8 per group.
(B) Fasting blood glucose levels of mice in the indicated groups. n = 7–8 per group.
(C) GTT and ITT results of mice in the indicated groups. n = 6 per group.
(D) Body weight of mice in the indicated groups. n = 7–8 per group.
(E) Liver weight and liver/body weight ratio of mice in the indicated groups. n = 7–8 per group.
(F) Representative images of H&E staining of liver sections. Scale bar, 100 μm. Graph shows the quantitation of lipid droplet area. n = 7–8 per group.
Data are mean ± SEM and representative of two independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, two-way analysis of variance (ANOVA).
CD24-Siglec-E axis represses metaflammation to ameliorate metabolic disorder
Metaflammation is a major underlying cause for metabolic disease. We previously identified an essential role of the CD24-Siglec interaction in suppressing immune response to danger signals (Chen et al., 2011; Chen et al., 2009). This led us to examine whether the CD24-Siglec-E axis can negatively regulate metabolic inflammation in obesity. After challenge with HFD, both CD24 and Siglec-E deficient mice showed higher serum levels of inflammatory cytokines (TNF-α and IL-6) (Figure 5A). The infiltration of inflammatory cells was markedly increased in the liver and adipose tissues from either Cd24−/− or Siglece−/− mice compared to WT controls, as assessed by H&E staining (Figure 5B). These were accompanied by a significant increase in the expression of key inflammatory genes (Tnfa, Il6, Il1b, Ccl2, Ccl3 and Ccl5) in the liver and adipose tissues (Figure 5C). Since the NF-κB pathway plays a central role in metabolic inflammation (Baker et al., 2011), we examined the NF-κB signaling in either Cd24−/− or Siglece−/− mice after HFD feeding. Immunoblotting analysis indicated an enhanced activation of NF-κB signaling in either Cd24−/− or Siglece−/− livers compared to WT controls, as evidenced by increased phosphorylation level of the p65 subunit of NF-κB and reduced IκBα protein levels (Figure 5D). Furthermore, CD24Fc treatment alleviated HFD-induced metaflammation in WT mice. However, the therapeutic effects were abolished in Siglece−/− mice (Figures 5E and 5F). Thus, CD24-Siglec-E pathway attenuates systemic and local inflammation in response to nutrient excess.
Figure 5. CD24-Siglec-E axis represses metaflammation to ameliorate metabolic disorder.
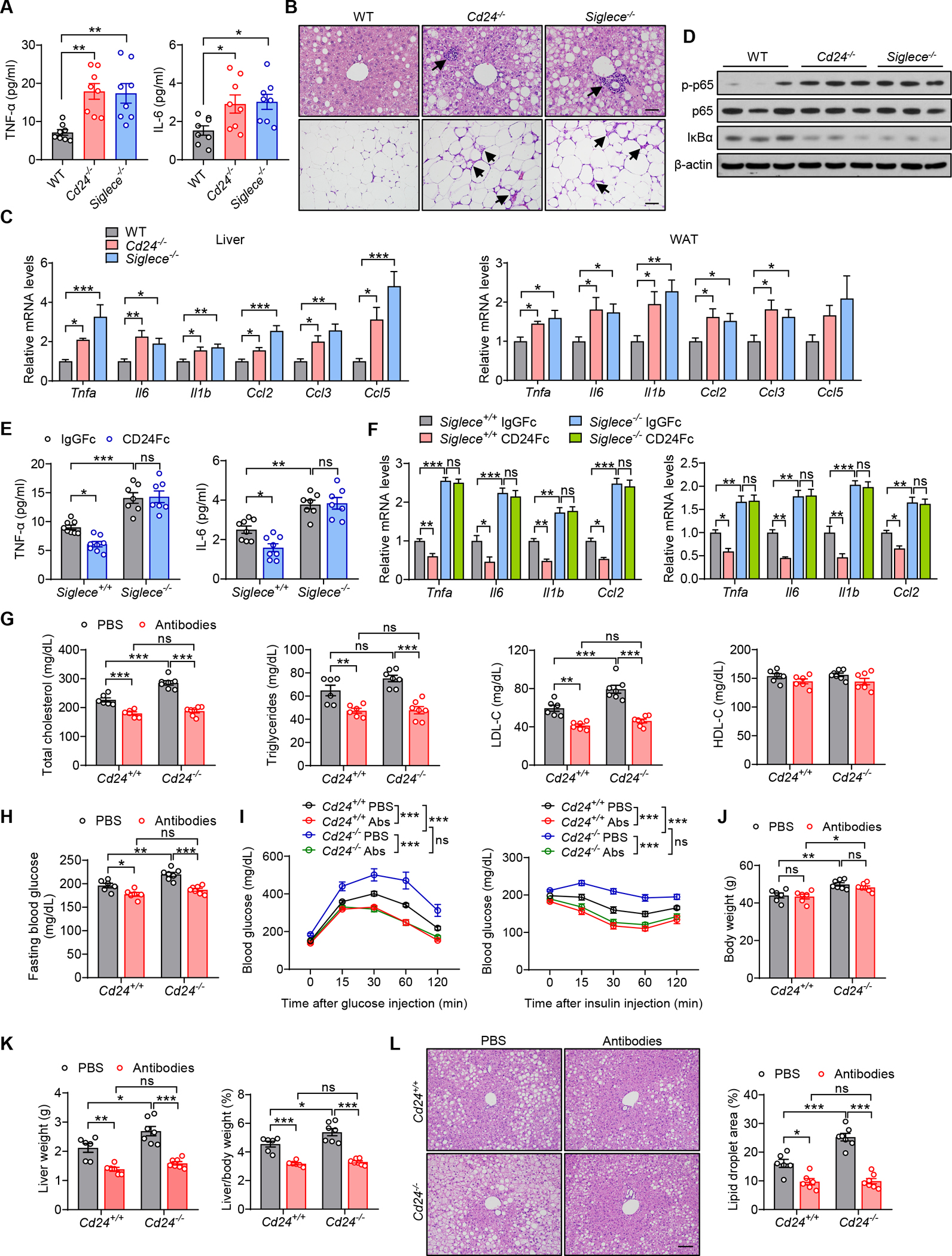
(A-D) WT, Cd24−/− and Siglece−/− mice were fed a HFD for 12 weeks.
(A) Serum concentrations of inflammatory cytokines of mice in the indicated groups. n = 8 per group.
(B) Representative images of H&E staining of liver and eWAT sections. Arrows indicate immune cells infiltrates. Scale bar, 50 μm.
(C) Relative mRNA levels of inflammatory genes in liver and eWAT from mice in the indicated groups. n = 6 per group.
(D) Immunoblotting analysis of NF-κB signaling pathway in the livers. β-actin was used as internal loading control. n = 3 per group.
(E and F) Siglece−/− mice and WT littermates were fed a HFD for 8 weeks, followed by injection of CD24Fc or IgGFc control twice a week for 4 weeks.
(E) Serum levels of inflammatory cytokines of mice in the indicated groups of mice. n = 7–8 per group.
(F) Relative mRNA levels of inflammatory genes in liver (left) and eWAT (right) from mice in the indicated groups. n = 6 per group.
(G-L) Cd24−/− mice and WT littermates were fed a HFD for 8 weeks, followed by injection of neutralizing antibodies (anti-TNFα, anti-IL-6 and anti-IL-1β monoclonal antibodies) twice a week for 4 weeks while continuing HFD.
(G) TC, TG, LDL-C and HDL-C levels of mice in the indicated groups. n = 6–7 per group.
(H) Fasting blood glucose levels of mice in the indicated groups. n = 6–7 per group.
(I) GTT and ITT results of mice in the indicated groups. n = 5–6 per group.
(J) Body weight of mice in the indicated groups. n = 6–7 per group.
(K) Liver weight and liver/body weight ratio of mice in the indicated groups. n = 6–7 per group.
(L) Representative images of H&E staining of liver sections. Scale bar, 100 μm. Graph shows the quantitation of lipid droplet area. n = 6–7 per group.
Data are mean ± SEM and representative of two or three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, one-way analysis of variance (ANOVA) (A, C), two-way analysis of variance (ANOVA) (E-L). See also Figure S5.
To explore the involvement of metaflammation in metabolic disorder associated with defective CD24-Siglec-E pathway, we blocked inflammatory cytokines in HFD-fed Cd24−/−, Siglece−/− and WT littermates by using a neutralizing antibody cocktail (anti-TNF-α, anti-IL-6 and anti-IL-1β monoclonal antibodies). Notably, blockade of inflammatory cytokines significantly improved serum lipid profiles, fasting blood glucose and insulin sensitivity in both WT and Cd24−/− mice (Figures 5G–5I). Although there was no significant difference in body weight gain (Figure 5J), systemic cytokines neutralization was accompanied by reduced liver weight, hepatic lipid accumulation and steatosis in both WT and Cd24−/− mice (Figures 5K and 5L). Thus, all of the metabolic phenotypes exacerbated by CD24 ablation were abolished by inflammatory cytokines neutralization. In line with the results of CD24-deficient mice, blockade of inflammatory cytokines reversed the Siglec-E deficiency-induced exacerbation of dyslipidemia, insulin resistance and hepatic steatosis (Figures S5A–S5F). Collectively, these findings indicate that the CD24-Siglec-E axis ameliorates HFD-induced metabolic disorder via dampening metaflammation.
Sialoside-based recognition of CD24 by Siglec-E modulates metabolic inflammation
To gain mechanistic insight into the aggravated metaflammatory phenotype, we evaluated whether the CD24-Siglec-E axis directly affects metaflammation. Macrophages are critical in orchestrating metabolic inflammation (McNelis and Olefsky, 2014) and express both CD24 (Fang et al., 2010) and Siglec-E (Macauley et al., 2014). We therefore examined the role of the CD24-Siglec-E pathway in macrophages under metabolic stress. We used peritoneal macrophages from Cd24−/−, Siglece−/− and WT mice and stimulated with palmitate, which is elevated in obesity and recapitulates lipid exposure in vitro. Both CD24 and Siglec-E deficiency aggravated lipid-induced mRNA expression and protein production of inflammatory cytokines in macrophages (Figures 6A and S6A). We further observed greater activation of NF-κB signaling in either Cd24−/− or Siglece−/− macrophages (Figure 6B). Consistently, there were greater increases in nuclear translocation of p65 in either Cd24−/− or Siglece−/− macrophages compared to WT controls under nutrient stress (Figures S6B and S6C). Moreover, treatment with CD24Fc significantly abolished lipid-induced metaflammation in WT but not Siglece−/− macrophages (Figures 6C, 6D and S6D). Thus, CD24-Siglec-E axis attenuates inflammatory response to metabolic stress in macrophages.
Figure 6. Sialoside-based recognition of CD24 by Siglec-E modulates metabolic inflammation.
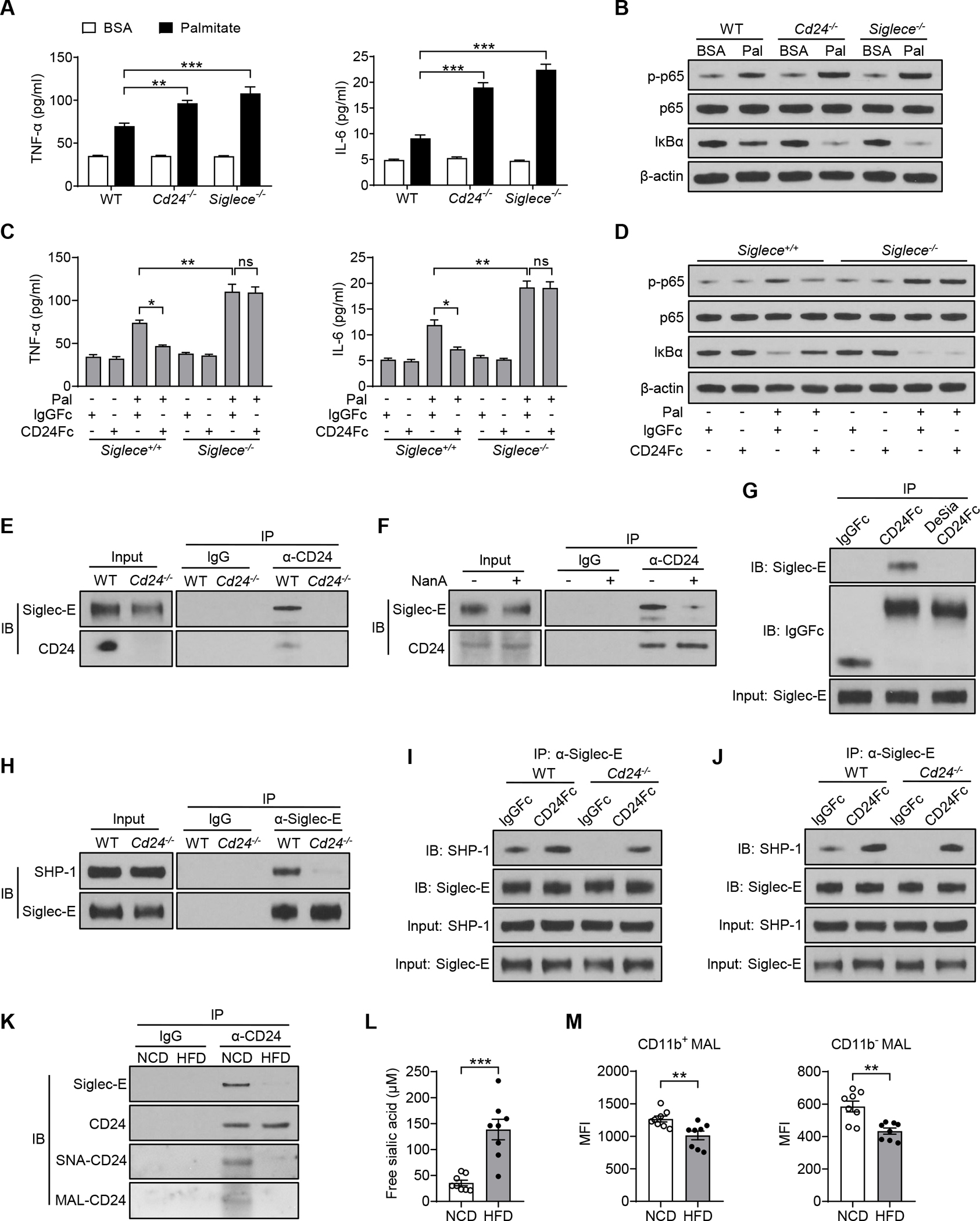
(A-B) Peritoneal macrophages from WT, Cd24−/− and Siglece−/− mice were stimulated with palmitate or BSA control for 16 hours before collection.
(A) TNF-α and IL-6 cytokines production in macrophages treated with palmitate or BSA control. n = 3 per group.
(B) Immunoblotting analysis of NF-κB signaling pathway in macrophages treated with palmitate or BSA control. n = 3 per group.
(C and D) Peritoneal macrophages from WT and Siglece−/− mice were stimulated with palmitate or BSA control, concurrently treated with CD24Fc or control IgGFc for 16 hours before collection.
(C) TNF-α and IL-6 cytokines production in macrophages with or without palmitate or CD24Fc treatment. n = 3 per group.
(D) Immunoblotting analysis of NF-κB signaling in macrophages with or without palmitate or CD24Fc treatment. n = 3 per group.
(E) Co-immunoprecipitation of CD24 and Siglec-E in WT and Cd24−/− spleen cells. (F) Co-immunoprecipitation of CD24 and Siglec-E in spleen cells with or without NanA treatment.
(G) The associations of CD24Fc or desialylated CD24Fc with Siglec-E in spleen cells. Protein A/G beads were used to pull down Fc.
(H) Co-immunoprecipitation of Siglec-E and SHP-1 in WT and Cd24−/− spleen cells.
(I) Co-immunoprecipitation of Siglec-E and SHP-1 in WT and Cd24−/− spleen cells treated with CD24Fc or IgGFc control.
(J) Co-immunoprecipitation of Siglec-E and SHP-1 in WT and Cd24−/− peritoneal macrophages treated with CD24Fc or IgGFc control.
(K) Co-immunoprecipitation of CD24 and Siglec-E in spleen cells from WT mice fed a NCD or HFD. Sialylation level of CD24 was detected by SNA and MAL II lectin blotting.
(L) Serum levels of free sialic acid in NCD or HFD-fed mice. n = 8 per group.
(M) Sialylation levels on PBMCs from NCD or HFD-fed mice were detected by flow cytometry with MAL II lectins. n = 8 per group.
Data are mean ± SEM and representative of two or three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, unpaired Student’s t-test (L, M), two-way analysis of variance (ANOVA) (A, C). See also Figures S6 and S7.
Since CD24 is most abundant in hematopoietic cells (Fang et al., 2010), and Siglec-E is only known to be expressed by cells of hematopoietic origin, including macrophages, dendritic cells and neutrophils (Macauley et al., 2014), we specifically evaluated the contribution of CD24 on immune cells to the observed metabolic phenotypes. We conducted bone marrow transplantation (BMT) experiments to generate chimeric mice in which irradiated WT or Cd24−/− mice received bone marrow cells from either WT or Cd24−/− mice. Upon HFD feeding, WT or Cd24−/− mice receiving Cd24−/− bone marrow cells showed exacerbated obesity, dyslipidemia, insulin resistance, hepatic steatosis and inflammation than those receiving WT bone marrow cells (Figures S6E–S6L), comparable to the phenotypes observed in Cd24−/− mice. Since CD24 ablation in hematopoietic cells alone recapitulated the phenotypes of germline deletion, CD24 expressed in the immune system confers the protective effects against metabolic disorder.
Next, we investigated how the CD24-Siglec-E axis negatively regulates metaflammation. We confirmed the endogenous CD24-Siglec-E interaction by co-immunoprecipitation (Figure 6E). Since sialoside-based recognition is a cardinal feature of Siglecs, we tested whether the CD24-Siglec-E interaction depends on CD24 sialylation. The WT spleen cells were treated with sialidase NanA. We observed that NanA treatment abolished the association of CD24 with Siglec-E (Figure 6F). Consistently, exogenous CD24Fc was able to immunoprecipitate Siglec-E, whereas the desialylation of CD24Fc abrogated the interaction (Figure 6G and S7A). These data demonstrate a sialoside-based recognition of CD24 by Siglec-E.
We further evaluated whether CD24 signals Siglec-E on the same cells (cis-interaction) or other cells (trans-interaction). We incubated equal numbers of Cd24−/− and Siglece−/− spleen cells together, and then detected Siglec-E signaling by measuring the recruitment of SHP-1 to Siglec-E by co-immunoprecipitation. We observed robust activation of Siglec-E signaling by CD24 from cells lacking Siglec-E (Figure S7B), indicating a trans-activation of Siglec-E by CD24. To address whether cis signaling of Siglec-E by endogenous CD24 is sufficient to activate Siglec-E, we abrogated the trans signaling by dilution to minimize cell contacts. While the dilution decreased recruitment of SHP-1 to Siglec-E in Cd24−/−/Siglece−/− co-culture, it did not affect the Siglec-E-SHP-1 interaction in WT cells cultured at the same density (Figure S7B). These data suggest that both cis and trans-interaction between CD24 and Siglec-E can activate Siglec-E signaling.
Since Siglec-E associates with the tyrosine phosphatase SHP-1, a known negative regulator of inflammation (Abram and Lowell, 2017), we hypothesized that CD24 might suppress inflammation via modulating the Siglec-E-SHP-1 signaling. Notably, Siglec-E was able to immunoprecipitate SHP-1 from WT but not Cd24−/− spleen cells (Figure 6H), suggesting that Silgec-E-SHP-1 interaction strictly depended on endogenous CD24 signaling. Since CD24Fc attenuated metabolic inflammation in obesity, we next evaluated whether CD24Fc affects the recruitment of SHP-1 to Siglec-E. As shown in Figure 6I, CD24Fc treatment enhanced Siglec-E-SHP-1 interaction in WT cells, and further restored the association in Cd24−/− cells. The effect was reflected in all known Siglec-E-expressing cells, especially macrophages (Figures 6J and S7C). These results indicate that CD24 directly modulates the Siglec-E-SHP-1 signaling to dampen inflammatory response.
The key inhibitory role of the CD24-Siglec-E axis in metabolic syndrome further raises the intriguing possibility that this pathway might be blunted during obesity. Interestingly, the endogenous CD24-Siglec-E interaction was substantially abolished in spleen cells from HFD-fed mice compared to NCD-fed mice (Figure 6K). Correspondingly, a dramatically decreased sialylation level of CD24 was observed after HFD challenge, as indicated by SNA and MAL II lectin blotting (Figure 6K). Consistent with clinical observations in obese patients (Browning et al., 2004; Rajappa et al., 2013; Yerlikaya et al., 2015), serum concentrations of free sialic acid significantly increased in HFD-fed mice compared to NCD-fed mice (Figure 6L). We also observed a reduction of global sialylation on immune cells from HFD-fed mice (Figure 6M), although the causality between increased free sialic acid and decreased sialylation on cells remains unknown. These results are consistent with the notion that defect in sialoside-based pattern recognition is a potential underlying cause of metabolic disorder.
CD24Fc regulates lipid metabolism and inflammation in humans
Preclinical studies have demonstrated that CD24Fc provides therapeutic effects for autoimmune diseases (Bai et al., 2000; Zheng et al., 2013), viral colitis and pneumonia (Tian et al., 2020; Tian et al., 2018) and graft versus host diseases (Toubai et al., 2014; Toubai et al., 2017). These studies prompted clinical development of CD24Fc for immunological diseases (NCT02663622; NCT04095858) and COVID-19 (NCT04317040). As a first-in-human study, CD24Fc was tested in a phase I, randomized, double-blind, placebo-controlled, single ascending dose study for safety, tolerability, and pharmacokinetics. We found that the single dose of CD24Fc up to 240 mg was safe and well tolerated in healthy subjects (NCT02650895; Figure S7D).
As exploratory analyses, we evaluated the effect of CD24Fc on blood biomarkers. Blood biochemical parameters were measured at pre-dosing baseline, and at 7, 14 and 42 days after dosing. Interestingly, there was a dose-dependent reduction of LDL-C on days 7 and 14 (Figure 7A). In particular, when compared with placebo control, a significant reduction of LDL-C was observed in subjects receiving 240 mg of CD24Fc on day 14 (Figure 7A). These data imply a potential biological activity of CD24Fc in human lipid metabolism, although the effect was modest because of limited dosing.
Figure 7. CD24Fc regulates lipid metabolism and inflammation in humans.
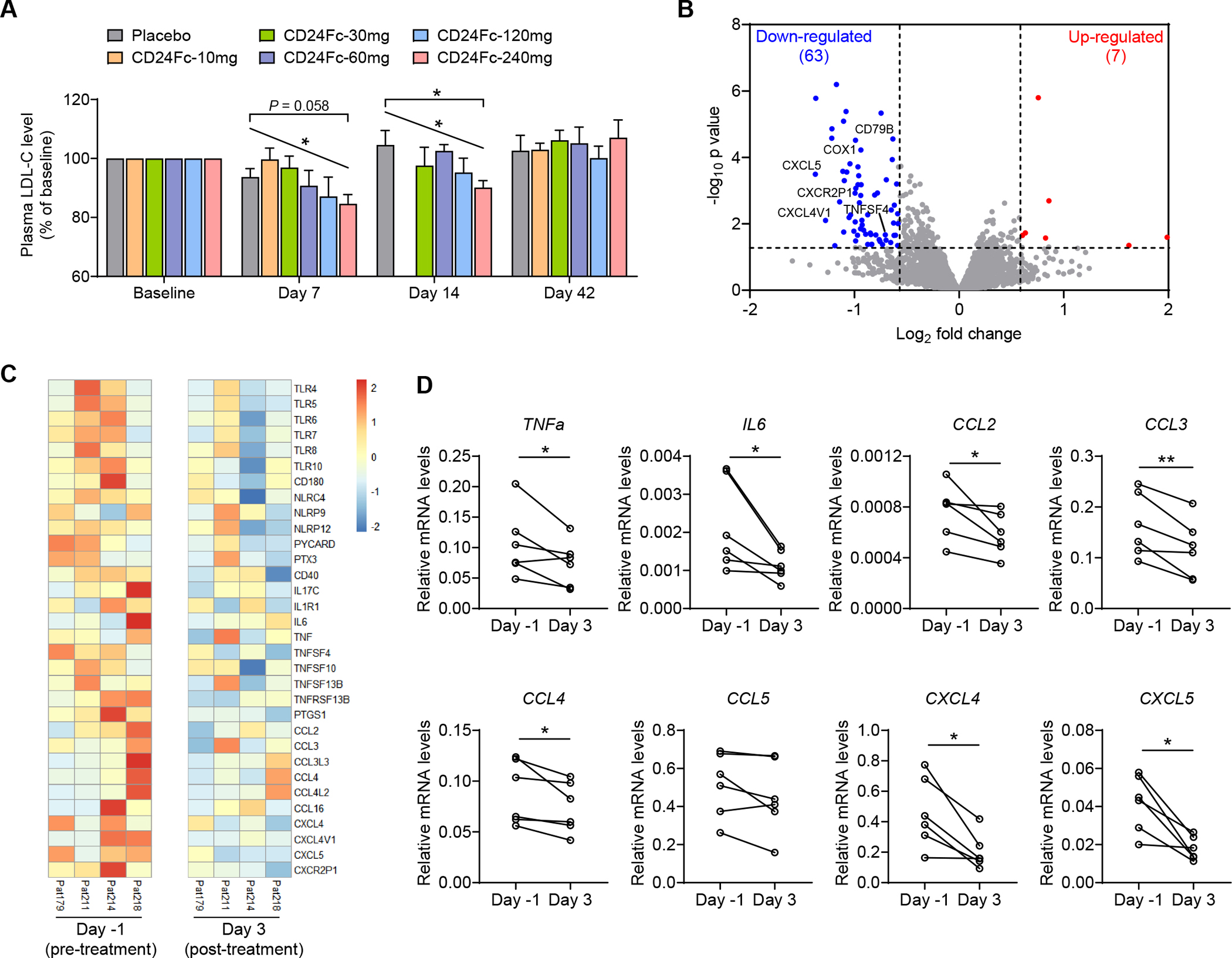
(A) The human subjects were treated with a single dose of placebo or CD24Fc at different doses (10, 30, 60, 120 and 240 mg per injection). Plasma LDL-C levels were detected at pre-dosing baseline, and at 7, 14 and 42 days after dosing. Relative LDL-C levels were normalized to the baseline. LDL-C of Day 14 samples in 10 mg group were not measured. n = 5–10 per group.
(B and C) RNA-sequencing was performed on PBMC samples obtained on Day −1 (pre-treatment) and Day 3 (post-treatment) from subjects receiving 240 mg of CD24Fc. All samples that pass quality control were used. </p/>(B) Volcano plot analysis. Significantly differentially expressed genes (P < 0.05 and fold change > 1.5) were highlighted in red (up-regulated) and blue (down-regulated). Selected genes related to inflammation were indicated.
(C) Heat map of alterations in expression of inflammatory genes.
(D) Expression of inflammatory genes in PBMC samples was validated by real-time PCR. Gene expression levels were calculated after normalization to the housekeeping gene GAPDH.
Data are mean ± SEM. *p < 0.05, **p < 0.01, one-way analysis of variance (ANOVA) for multiple comparisons (A), linear regression analysis for dose-dependency (A) or paired t-test (D). See also Figure S7.
For global assessment of CD24Fc activity on host immune system, we performed RNA-sequencing of peripheral blood mononuclear cells (PBMCs) obtained on Day −1 (pre-treatment) and Day 3 (post-treatment) from subjects receiving 240 mg of CD24Fc. We identified 7 up-regulated genes and 63 down-regulated genes (Figure 7B). The strong bias towards gene down-regulation is consistent with CD24Fc as an inhibitory immune modulator. More importantly, when levels of inflammatory gene transcripts were systematically compared, we observed reductions among the transcripts of genes encoding pattern recognition receptors (TLR, NLR, PTX, ASC), as well as inflammatory cytokines, chemokines and their receptors (Figures 7B and 7C). We further validated the expression of inflammatory genes by real-time PCR. In particular, CD24Fc treatment significantly reduced the mRNA levels of inflammatory cytokines (TNFa, IL6) and chemokines (CCL2, CCL3, CCL4, CXCL4 and CXCL5) (Figure 7D). Taken together, our first-in-human study of CD24Fc supports the relevance of CD24 in regulation of lipid metabolism and inflammation.
To evaluate the spectrum of CD24Fc receptors among human Siglecs, we compared human Siglec fusion proteins for their binding to recombinant CD24Fc. We observed significant associations of CD24Fc with Siglec-3, Siglec-6, Siglec-9 and Siglec-10 (Figure S7E).
Discussion
Metaflammation, a chronic inflammation driven by nutrient and energy excess, lies at the center of metabolic disease (Hotamisligil, 2006; Lumeng and Saltiel, 2011; Schenk et al., 2008; Shoelson et al., 2006). Therefore, identification of molecules and mechanisms directly controlling metaflammation is of great importance to develop treatments for metabolic disease. Here we identified the CD24-Siglec pathway as a missing link between chronic inflammation and metabolic disorder, and provided a novel therapeutic approach for treatment of obesity, insulin resistance and NASH by fortifying sialoside-based pattern recognition using CD24Fc.
Major advances have been made to control lipid and glucose metabolism. However, the disorders in lipid and glucose metabolism are treated by drugs specific for either lipid or glucose metabolism. CD24Fc therapy differs from the dominant approach that specifically reduces systemic lipid or glucose levels. By lowering inflammatory responses in metabolic tissues, CD24Fc may have helped to clear out a major root cause of metabolic disorder. It has been reported that therapeutics targeting of individual inflammatory cytokines, such as IL-1β and TNF-α, have so far yielded limited success in clinical studies (Donath, 2014; Larsen et al., 2007; Stanley et al., 2011), which calls for the development of broader and more efficacious strategies to address the unmet medical needs for metabolic disease associated with metaflammation. Here we have shown that CD24Fc affects the expression of multiple inflammatory cytokines in both mouse and human, the most likely explanation for the broad biological effects in metabolic disorder could be based on its general impact of a host of inflammatory cytokines, each of which may affect different aspects of metabolic dysfunction.
The therapeutic effect of CD24Fc corresponds with endogenous CD24 function in guarding the host against metabolic syndrome. We hereby demonstrated that targeted deletion of CD24 in mice exacerbated metabolic disorder associated with obesity and aging. It was previously reported that, after a short-term HFD feeding, CD24-deficient mice exhibited modestly increased body weight and fasting glucose levels, while no differences in WAT mass, lipid profiles or insulin sensitivity. Paradoxically, under normal diet, CD24 deficiency decreased WAT mass in comparison with non-littermate WT mice (Fairbridge et al., 2015). Yet another group recently reported that Cd24−/− male mice had increased body weight and adipose mass but paradoxically increased insulin sensitivity under normal diet (Shapira et al., 2021). Thus, these earlier studies suggest a limited and inconsistent role of CD24 in metabolic regulation. Here we have performed more extensive studies with proper littermate controls, and used genetic and pharmaceutical intervention-based approaches to establish a broad function of CD24 in metabolic disorder associated with both obesity and aging.
CD24 suppresses host response to DAMPs by interacting with Siglec (Chen et al., 2011; Chen et al., 2009; Liu et al., 2009, 2011). We generated mouse strains with mutations of one or more Siglec genes in order to identify the Siglec that mediates CD24 function in metabolic disease. Of all Siglec mutations that we have studied, including Siglec2, Siglec3, Siglece, Siglecf, Siglecg and Siglech, either alone or in combination, we found that only Siglece mutation fully recapitulates the metabolic phenotypes of CD24 deficiency. The essentially identical phenotypes, and the physical interaction between CD24 and Siglec-E, made it highly likely that Siglec-E is the functional CD24 receptor responsible for metabolic regulation. Consistent with this notion, we showed that CD24Fc alleviated metabolic disorder in a Siglec-E-dependent manner.
Siglec-E has been implicated in the pathogenesis of inflammatory lung disease, autoimmune disease, atherosclerosis and cancer (Flores et al., 2019; Hsu et al., 2021; Laubli et al., 2014; McMillan et al., 2013). Gagneux and co-workers found that Siglec-E-deficient mice exhibited reduced longevity and accelerated signs of aging, and that accelerated aging was related to increased ROS production and oxidative damage (Schwarz et al., 2015). However, the role of Siglec-E in obesity-related metabolic disorder has not yet been explored. Here our genetic and pharmacological studies suggest the CD24-Siglec-E axis as a physiological guard against metabolic syndrome associated with obesity and aging.
It has been suggested that the role of different Siglecs in a disease depends on their expression patterns and the importance of different cell populations to the disease (Duan and Paulson, 2020; Macauley et al., 2014). Siglec-E is a major Siglec on macrophages, which are critical in orchestrating metabolic inflammation in obesity. Therefore, it is tempting to suggest that the CD24-Siglec-E axis in macrophage is responsible for its suppression of metaflammation and metabolic disorder, although additional studies are needed to test this hypothesis. Furthermore, while our data demonstrated that CD24Fc induced association between Siglec-E and SHP-1, the molecular mechanism of this induction remains to be elucidated. In particular, how CD24 activates requisite tyrosine kinases for Siglec-E phosphorylation to prime Siglec-E for SHP-1 binding, remains to be identified.
Finally, it is of interest to consider whether and how the CD24-Siglec axis goes awry to allow the development of metabolic syndrome. One potential mechanism is desialylation of CD24 and/or other cell surface ligands for Siglecs. Clinical studies indicate that increased serum level of sialic acid is significantly associated with obesity and metabolic syndrome (Browning et al., 2004; Rajappa et al., 2013; Yerlikaya et al., 2015). Emerging evidence also demonstrates that sialyltransferases or sialidases, that catalyze the transfer or removal of sialic acid residues of glycoproteins respectively, are implicated in the pathogenesis of obesity (Kaburagi et al., 2017; Natori et al., 2013). These studies are consistent with the notion of crucial roles of sialic acid and sialylation in obesity and metabolic homeostasis. Here, we reported a remarkable increase of serum free sialic acid, and a global reduction of sialylation on immune cells in obese mice, although the causality between these remains unknown. We further observed dramatically decreased sialylation of CD24 on immune cells in obese mice. It is of note that the reduction of total sialylation appeared less than that of CD24, suggesting that obesity may have preferentially affected CD24 sialylation, although the difference may also be partially due to different methodologies. Overall, these findings suggest a potential role of desialylation or hyposialylation in obesity-related metabolic disorder.
Taken together, our work defines the CD24-Siglec-E interaction as an innate immune checkpoint against metaflammation and metabolic disorder. Since CD24Fc is under clinical development for a variety of diseases in human, our work provides a promising therapeutic approach for metabolic disease by fortifying sialoside-based pattern recognition.
Limitations of Study
Although our studies based on bone marrow transplantation established the role for CD24 in hematopoietic cells in suppression of metabolic disorder, the specific hematopoietic cell-type by which the CD24-Siglec-E axis achieves the protective effect remains to be identified. Since CD24Fc binds to multiple human Siglecs, additional studies are needed to evaluate these receptors are functionally relevant in CD24Fc mediated suppression of metaflammation in human.
STAR Methods
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yang Liu (yangl@oncoc4.com).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The RNA-seq raw data files have been deposited in the GEO repository with GEO accession number GSE140724.
This paper does not report original code.
Data S1 represents an Excel file containing the values that were used to create all the graphs in the paper, as well as a PDF file containing the full-length, unprocessed western blots. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental Model and Subject Details
CD24Fc phase I clinical study
CD24Fc was tested for safety, tolerability and pharmacokinetics in a phase I, randomized, double-blind, placebo-controlled, single ascending dose study in healthy subjects (NCT02650895). The population for this study was healthy males and females between the ages of 18 and 55 years, inclusive, with a body mass index between 18 and 30 kg/m2, inclusive. A total of 40 subjects were enrolled (17 females and 23 males, age 34.8 ± 9.3 years), in 5 cohorts of 8 subjects each. Six of the 8 subjects in each cohort received CD24Fc and 2 subjects received placebo. The first cohort was dosed with 10 mg of CD24Fc. Succeeding cohorts received 30, 60, 120, and 240 mg of CD24Fc or matching placebo. Administration of the next higher dose to a new cohort was permitted only if adequate safety and tolerability had been demonstrated in each prior cohort. One subject in the CD24Fc 10 mg group withdrew early from the study due to other reasons. The sample size for this study was based on clinical rather than statistical rationale. The sample size was considered adequate to address the study goals. A CONSORT diagram for the trial is provided in Figure S7D.
The primary objective of the study was to evaluate the safety and tolerability of single ascending intravenous doses of CD24Fc in healthy subjects. The secondary objective of the study was to characterize the single-dose pharmacokinetics of CD24Fc in healthy subjects. As exploratory analyses, we assessed the effect of CD24Fc on blood biomarkers and gene expression in PBMCs. Blood biochemical parameters were determined from plasma samples collected on Day −1, 7, 14 and 42. PBMC samples were collected from subjects receiving 240 mg of CD24Fc on Day −1 and Day 3 for RNA-sequencing and real-time PCR. Of the samples collected from 6 subjects, 4 pairs of RNA samples passed quality control and were subject to RNA-seq. The study protocol was approved by the duly constituted institutional review board. The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. All participants provided written informed consent before enrolment.
Animals
WT C57BL/6 mice (8–10 weeks of age) were purchased from Charles River. After necessary acclimatization, the imported WT mice were used in experiments. Cd24−/−, Siglece−/− and Siglecg−/− mice have been described (Ding et al., 2007; Flores et al., 2019; Nielsen et al., 1997). Siglece−/− mice were backcrossed with C57BL/6 mice for 8 generations. Cd24+/+ and Siglece+/+ littermates were used as the control of Cd24−/− and Siglece−/− mice respectively. Male C57BL/6 mice were used throughout the study as male mice are more susceptible to HFD-induced metabolic dysfunction than female mice (Hwang et al., 2010). All the mice were housed in a temperature-controlled environment (23 ± 2°C) under a 12-hour light/dark cycle with free access to water and normal chow diet (Teklad Diets, 2018SX). The mice were checked for health status two times a week during the experiment.
To establish an obesity model, male mice were fed a HFD consisting of 60% of calories from fat (Research Diets, D12492) starting at 8–10 weeks of age for 12 weeks. To establish a NASH model, male mice were fed a CD-HFD consisting of 60% fat, 0.1% methionine and no added choline (Research Diets, A06071302) starting at 8–10 weeks of age for 12 weeks. Mouse body weight and food intake were measured every week. All protocols were approved by the Institutional Animal Care and Use Committee of Children’s National Medical Center or University of Maryland School of Medicine.
Macrophages culture and stimulation
Peritoneal macrophages from male WT, Cd24−/− and Siglece−/− mice were isolated 3 days after intraperitoneal injection of 3% thioglycollate (Sigma). The cells were plated in 6-well plates at a density of 1.2 × 106 cells/well and cultured in RPMI-1640 medium containing 10% fetal bovine serum (FBS) at 37°C in a 5% CO2 incubator. The cells were then stimulated with palmitate-BSA (500μM) or unmodified BSA control for 16 hours. For CD24Fc treatment studies, peritoneal macrophages from WT and Siglece−/− mice were challenged with palmitate-BSA (500 μM) or BSA control and concurrently treated with CD24Fc (10 μg/ml) or IgG control for 16 hours. Supernatant and cell lysates were collected for ELISA, immunoblotting and gene expression analysis. Palmitate (Sigma-Aldrich, P9767) was conjugated with BSA before treatment. Palmitate was dissolved in 95% ethanol at 60°C and prepared as 50 mM solution. The palmitate solution was then diluted with RPMI-1640 medium containing 1% BSA to obtain the 500 μM palmitate concentration.
Method Details
Generation of genetically modified mice
The CRISPR/Cas9 system was used to induce Siglec genes mutations in C57BL/6 mice as previously described (Wang et al., 2013). Briefly, we injected Cas9 and guide RNAs for all 6 Siglec genes, including 5 Siglecs with intracellular ITIM domains (Siglec2, Siglec3, Siglece, Siglecf and Siglecg) and the DAP12-associated Siglech into C57BL/6 zygotes to generate mice with single and different combinations of Siglec deletions. The final mutant alleles for each locus were listed in Table S1.
CD24Fc therapeutic studies in DIO mice
For the therapeutic studies, WT or Siglece−/− mice were fed a HFD for 8 weeks, followed by intraperitoneal injection of CD24Fc (100 μg per mouse, OncoImmune, Inc.) or IgGFc control twice a week for 4 more weeks while continuing HFD. For the prophylactic studies, mice were fed a HFD and concurrently treated with CD24Fc or IgGFc control twice a week for 8 weeks. Metabolic phenotypes were detected after CD24Fc or IgGFc treatment.
Blockade of inflammatory cytokines in DIO mice
Cd24−/−, Siglece−/− and WT littermates were fed a HFD for 8 weeks to establish obesity, followed by intraperitoneal injection of neutralizing antibodies, including anti-TNFα antibody (200 μg per mouse, BioXCell, BE0058), anti-IL-6 antibody (200 μg per mouse, BioXCell, BE0046) and anti-IL-1β antibody (200 μg per mouse, BioXCell, BE0246), twice a week for 4 weeks while continuing HFD.
Tissue processing and histological analysis
After HFD feeding, DIO mice were anesthetized with isoflurane. Representative images of physical appearance were taken and body composition was detected by dual energy X-ray absorptiometry (DEXA). Mice were then euthanized, livers, white adipose and brown adipose tissues were immediately harvested, photographed and weighed. Tissues for histology were fixed in 10% formalin. Liver and eWAT sections were embedded in paraffin and then stained with H&E (performed by Histoserv, Inc, Germantown) to visualize the lipid accumulation and inflammatory status. Liver fibrosis was assessed by Masson’s trichrome staining (performed by Histoserv, Inc, Germantown). The histological features were observed and imaged using Olympus BX53 light microscope. The corresponding positive-staining area was quantified with Image-Pro Plus software, and the results are expressed as a percentage of the total area of a HPF.
Mouse metabolic studies
For the glucose tolerance tests (GTTs), mice were injected intraperitoneally with 1 g/kg glucose after 16 hours of fasting. Blood glucose levels were measured at 0, 15, 30, 60 and 120 min from tail blood using the One Touch Ultra glucometer (Lifescan). For the insulin tolerance tests (ITTs), an intraperitoneal injection of 1 U/kg insulin was given to mice after 6 hours of fasting. Blood glucose levels were determined as described above.
Mouse serum lipid and cytokine assays
The serum TC, TG and NEFA levels were measured with commercial kits (Randox), according to the manufacturer’s instruction. Serum LDL and HDL levels were analyzed using HPLC system (Skylight Biotech, Akita, Japan) as previously described (Usui et al., 2002). Serum cytokines were determined using mouse cytokine bead array designed for inflammatory cytokines (BD Biosciences). Serum free sialic acid levels were detected using sialic acid assay kit (Abnova, KA1655).
Liver function assay
Liver functions were evaluated in mice by determining the serum ALT and AST activity using ALT activity assay kit (Biovision, K752) and AST activity assay kit (Biovision, K753), according to the manufacturer’s instruction.
Rectal temperature measurement
Mouse body temperature was determined using thermocouple meter and rectal probe for mouse (Kent Scientific Corporation).
Immunofluorescence
For immunofluorescence staining, cells were seeded on chamber slides (Thermo Fisher Scientific). The slides were washed in PBS, fixed in 4% fresh paraformaldehyde for 15 min, permeabilized with 0.5% Triton X-100 in PBS for 5 min and blocked with 3% BSA in PBS for 60 min at room temperature. The slides were then stained with rabbit anti-mouse p65 antibody (Cell Signaling Technology, 8242) in PBS overnight at 4 °C. After washed with PBST for 3 times, the slides were incubated with Alexa Fluor 594-conjugated goat anti-rabbit IgG (Invitrogen) for 60 min at room temperature. Nuclei were stained with DAPI for 5 min. Fluorescent images were obtained using Olympus BX53 fluorescent microscope.
RNA extraction and Real-time PCR analysis
Total RNA was isolated from tissues and cells using TRIzol reagent (Invitrogen). For reverse transcription, cDNA was synthesized from RNA samples with a Superscript First-Strand Synthesis System (Invitrogen). Quantitative real-time PCR was performed with SYBR Green PCR Master Mix (Applied Biosystems) using the Applied Biosystems 7500 Real-time PCR System according to the manufacturer’s instructions. Gene expression levels were calculated after normalization to the housekeeping gene β-actin or GAPDH. The primer sequences are provided in Table S2.
Immunoprecipitation
The spleens of the indicated mice were collected, minced and filtered through 100 μm cell strainer to get single cells. The red blood cells were removed using the ACK buffer (Gibco). Then the splenocyte lysates were prepared in the lysis buffer (1% Triton X-100, 150 mM NaCl, 3 mM MnCl2, 1 mM CaCl2, 1 mM MgCl2, 25 mM Tris-HCl, pH 7.6) with protease inhibitor cocktail (Sigma-Aldrich). For immunoprecipitation, cell lysates were pre-cleared with Protein A/G-conjugated agarose beads (Thermo Fisher Scientific) at 4°C for 2 hours with rotation, then incubated with corresponding antibodies or control IgG overnight at 4°C. The cell lysates were then incubated with Protein A/G beads for an additional 2 hours. The beads were washed four times with lysis buffer and re-suspended in SDS sample buffer for western blot analysis.
For immunoprecipitation of CD24Fc receptors, the spleen cells were incubated with control IgGFc, CD24Fc or desialylated CD24Fc (10 μg/ml) for 1 hour. After washing away the unbound CD24Fc with PBS, the spleen cells were lysed in lysis buffer. Protein A/G beads were used to pull down Fc. The amounts of Siglec-E associated with CD24Fc were determined by immunoblotting.
Western blot analysis
Tissues and cells were lysed with RIPA lysis buffer (Thermo Fisher Scientific) containing protease inhibitor (Sigma-Aldrich) and phosphatase inhibitor (Sigma-Aldrich). Total protein was quantified with BCA assay (Thermo Fisher Scientific). Equal amounts of each protein sample were separated by NuPAGE 4–12% Bis-Tris Protein Gels (Invitrogen) and transferred to PVDF membranes (Millipore). Individual proteins were determined with the specific antibodies and β-actin was used as internal loading control. Blots were semi-quantified using Image-Pro Plus software. All the antibodies are listed in key resource table.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Akt | Cell Signaling Technology | Cat# 2920; RRID:AB_1147620 |
| Rabbit monoclonal anti-phospho-Akt | Cell Signaling Technology | Cat# 4060; RRID:AB_2315049 |
| Rabbit monoclonal anti-NF-κB p65 | Cell Signaling Technology | Cat# 8242; RRID:AB_10859369 |
| Rabbit monoclonal anti-phospho-NF-κB p65 | Cell Signaling Technology | Cat# 3033; RRID:AB_331284 |
| Mouse monoclonal anti-IκBα | Cell Signaling Technology | Cat# 4814; RRID:AB_390781 |
| Goat polyclonal anti-Siglec-E | R&D Systems | Cat# AF5806; RRID:AB_10971778 |
| Rat monoclonal anti-Siglec-E | BioLegend | Cat# 677102; RRID:AB_2565899 |
| Rat monoclonal anti-mouse CD24 (M1/69) | BioLegend | Cat# 101802; RRID:AB_312835 |
| Mouse monoclonal anti-human CD24 (ML5) | BD Biosciences | Cat# 555426; RRID:AB_395820 |
| Mouse monoclonal anti-human CD24 (SN3) | Santa Cruz | Cat# sc-19585; RRID:AB_626989 |
| Rabbit monoclonal anti-SHP1 | Abcam | Cat# ab32559; RRID:AB_777912 |
| Mouse monoclonal anti-β-actin | Sigma-Aldrich | Cat# A2228; RRID:AB_476697 |
| Rabbit monoclonal anti-Histone H3 | Cell Signaling Technology | Cat# 4499; RRID:AB_10544537 |
| Mouse monoclonal anti-α-tubulin | Cell Signaling Technology | Cat# 3873; RRID:AB_1904178 |
| Goat polyclonal anti-human IgGFc, HRP | Abcam | Cat# ab98624; RRID:AB_10673832 |
| Sambucus Nigra Lectin (SNA), Biotinylated | Vector Laboratories | Cat# B-1305; RRID:AB_2336718 |
| Maackia Amurensis Lectin II (MAL II), Biotinylated | Vector Laboratories | Cat# B-1265; RRID:AB_2336569 |
| Anti-mouse Siglec-E, BV421 | BD Biosciences | Cat# 748154; RRID:AB_2872615 |
| Anti-mouse Siglec-G, BV421 | BD Biosciences | Cat# 744875; RRID:AB_2742552 |
| Anti-mouse CD22, APC | BioLegend | Cat# 126110; RRID:AB_2561630 |
| Anti-mouse CD33, PE | Thermo Fisher Scientific | Cat# 12-0331-82; RRID:AB_2637179 |
| Anti-mouse Siglec-H, PerCP/Cyanine5.5 | BioLegend | Cat# 129614; RRID:AB_10643995 |
| Anti-mouse Siglec-F, PerCP-eFluor 710 | Thermo Fisher Scientific | Cat# 46-1702-80; RRID:AB_2573723 |
| Anti-mouse/human CD45R/B220, FITC | BioLegend | Cat#103206; RRID:AB_312991 |
| Anti-mouse CD193 (CCR3), APC | BioLegend | Cat# 144512; RRID:AB_2565738 |
| Anti-mouse CD11b, APC-eFluor 780 | Thermo Fisher Scientific | Cat# 47-0112-82; RRID:AB_1603193 |
| Anti-mouse CD45.2, PE | Thermo Fisher Scientific | Cat# 12-0454-82; RRID:AB_465678 |
| Anti-mouse CD45.2, PerCP-Cyanine5.5 | Thermo Fisher Scientific | Cat# 45-0454-82; RRID:AB_953590 |
| Anti-mouse CD16/CD32(2.4G2) | BioXCell | Cat# BE0307; RRID:AB_2736987 |
| InVivoMAb anti-mouse TNFα | BioXCell | Cat# BE0058; RRID:AB_1107764 |
| InVivoMAb anti-mouse IL-6 | BioXCell | Cat# BE0046; RRID:AB_1107709 |
| InVivoMAb anti-mouse IL-1β | BioXCell | Cat# BE0246; RRID:AB_2687727 |
| Biological samples | ||
| Human plasma | CD24Fc phase I clinical study | NCT02650895 |
| Human PBMCs | CD24Fc phase I clinical study | NCT02650895 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant CD24Fc | This paper, OncoImmune, Inc. | Lot: CT0639 |
| IgGFc | Athens Research and Technology | Cat# 16-16-090707; RRID:AB_575815 |
| Recombinant mouse Siglec-1 | R&D Systems | Cat# 5610-SL |
| Recombinant mouse Siglec-2 | R&D Systems | Cat# 2296-SL |
| Recombinant mouse Siglec-E | R&D Systems | Cat# 5806-SL |
| Recombinant mouse Siglec-F | R&D Systems | Cat# 1706-SF |
| Recombinant mouse Siglec-G | R&D Systems | Cat# 10103-SL |
| Recombinant human Siglec-1 | R&D Systems | Cat# 5197-SL |
| Recombinant human Siglec-2 | R&D Systems | Cat# 1968-SL |
| Recombinant human Siglec-3 | R&D Systems | Cat# 1137-SL |
| Recombinant human Siglec-5 | R&D Systems | Cat# 1072-SL |
| Recombinant human Siglec-6 | R&D Systems | Cat# 2859-SL |
| Recombinant human Siglec-7 | R&D Systems | Cat# 1138-SL |
| Recombinant human Siglec-9 | R&D Systems | Cat# 1139-SL |
| Recombinant human Siglec-10 | R&D Systems | Cat# 2130-SL |
| Recombinant human Siglec-11 | R&D Systems | Cat# 3258-SL |
| Human insulin solution | Sigma-Aldrich | Cat# I9278 |
| Glucose | Sigma-Aldrich | Cat# G8270 |
| Palmitate | Sigma-Aldrich | Cat# P9767 |
| BSA, fatty acid free | Sigma-Aldrich | Cat# A8806 |
| Thioglycollate | Sigma-Aldrich | Cat# B2551 |
| TRIzol | Invitrogen | Cat# 15596018 |
| SuperScript First-Strand Synthesis System | Thermo Fisher Scientific | Cat# 11904018 |
| Power SYBR Green PCR Master Mix | Applied Biosystems | Cat# 4367659 |
| ACK Lysing Buffer | Gibco | A1049201 |
| RIPA buffer | Thermo Fisher Scientific | Cat# 89900 |
| Protein A/G-conjugated agarose beads | Thermo Fisher Scientific | Cat# 20421 |
| Carbo-Free Blocking Solution | Vector Laboratories | Cat# SP-5040 |
| Neuraminidase A (NanA) | This paper | N/A |
| Critical commercial assays | ||
| H&E staining | Histoserv, Inc. | Cat# ST001A |
| Masson’s trichrome staining | Histoserv, Inc. | Cat# ST001A |
| BD Cytometric Bead Array (CBA) Mouse Enhanced Sensitivity Master Buffer Kit | BD Biosciences | Cat# 562246 |
| Mouse TNF Enhanced Sensitivity Flex Set | BD Biosciences | Cat# 562336 |
| Mouse IL-6 Enhanced Sensitivity Flex Set | BD Biosciences | Cat# 562236 |
| Total Cholesterol | Randox Laboratories | Cat# CH200 |
| Triglycerides | Randox Laboratories | Cat# TR210 |
| HDL Cholesterol | Randox Laboratories | Cat# CH2652 |
| LDL Cholesterol | Randox Laboratories | Cat# CH2657 |
| NEFA (Non-Esterified Fatty Acids) | Randox Laboratories | Cat# FA115 |
| ALT activity assay kit | BioVision | Cat# K752 |
| AST activity assay kit | BioVision | Cat# K753 |
| BCA protein assay kit | Thermo Fisher Scientific | Cat# 23227 |
| Deposited data | ||
| CD24Fc phase I clinical trial | https://clinicaltrials.gov/ | NCT02650895 |
| RNA-seq raw data | This paper | GSE140724 |
| Raw data | This paper | Data S1 |
| Experimental models: Cell lines | ||
| Peritoneal macrophages (WT, Cd24−/− and Siglece−/−) | This paper | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6NCr | NCI | Cat# 556 |
| Mouse: Cd24−/− | Nielsen et al., 1997 | N/A |
| Mouse: Siglece−/− | Flores et al., 2019 | N/A |
| Mouse: Siglecg−/− | Ding et al., 2007 | N/A |
| Mouse: Siglec2−/− | This paper | N/A |
| Mouse: Siglec3−/− | This paper | N/A |
| Mouse: Siglech−/− | This paper | N/A |
| Mouse: Siglec2−/−/h−/− | This paper | N/A |
| Mouse: Siglec3−/−/f−/− | This paper | N/A |
| Mouse: Siglecf−/−/g−/− | This paper | N/A |
| Mouse: Siglecg−/−/h−/− | This paper | N/A |
| Mouse: Siglecf−/−/g−/−/h−/− | This paper | N/A |
| Oligonucleotides | ||
| Primers for qPCR, see Table S2 | This paper | N/A |
| Software and algorithms | ||
| Graphpad Prism 8 | Graphpad | https://www.graphpad.com |
| Flowjo_V10 | BD Biosciences | https://www.flowjo.com |
| Image-Pro Plus 6 | Media Cybernetics | http://www.mediacy.com/imageproplus |
| Other | ||
| High-fat diet | Research Diets | Cat# D12492 |
| Choline-deficient HFD | Research Diets | Cat# A06071302 |
Lectin blot analysis
CD24 sialylation was evaluated by lectin blotting using SNA or MAL II (Vector Laboratories), which respectively recognizes either α2–6- or α2–3-linked sialic acids. Protein samples were separated by NuPAGE 4–12% Bis-Tris Protein Gels (Invitrogen) and transferred to PVDF membranes (Millipore). After blocking with Carbo-Free blocking solution (Vector Laboratories), the membranes were incubated with biotinylated SNA or MAL II (diluted with the blocking buffer), followed by HRP-conjugated streptavidin (Thermo Fisher Scientific) and chemiluminescence methods.
CD24-Siglecs binding assay
96-Well plates were coated with either Siglecs or IgGFc in 50 mM carbonate/bicarbonate buffer, pH 9.5, overnight at 4 °C. Wells were blocked with binding buffer (20 mM HEPES, 2% BSA, 150 mM NaCl, 3 mM MnCl2, 1 mM CaCl2, 1 mM MgCl2, pH 7.6) for 1 hour. For endogenous CD24 binding, mouse splenocyte lysates were prepared in the lysis buffer (1% Triton X-100, 150 mM NaCl, 3 mM MnCl2, 1 mM CaCl2, 1 mM MgCl2, 25 mM Tris-HCl, pH 7.6). Cell lysates were added to the plate and incubated for 2 hours. After washing, biotinylated anti-mouse CD24 antibody (BioLegend) was used to detect bound CD24. The plate-associated biotinylated proteins were detected by HRP-conjugated streptavidin. For recombinant CD24Fc binding, cell lysates were replaced with biotinylated CD24Fc in order to measure direct interaction between Siglecs.
Sialidase treatment of spleen cells
The spleen cells were treated with recombinant NanA (40 μg/ml) or vehicle control for 1 h at 37 °C. After washing away the sialidase with PBS, the spleen cells were lysed in the lysis buffer (1% Triton X-100, 150 mM NaCl, 3 mM MnCl2, 1 mM CaCl2, 1 mM MgCl2, 25 mM Tris-HCl, pH 7.6) for immunoprecipitation.
Sialidase treatment of CD24
CD24Fc was incubated with recombinant NanA (20 μg/ml) produced in the laboratory overnight at 37 °C. The desialylation of CD24Fc was confirmed by immunoblotting with anti-CD24 antibodies that recognize total CD24 (BD Biosciences, 555426, clone ML5) or sialylated CD24 (Santa Cruz, sc-19585, clone SN3), respectively.
Bone marrow transplantation
WT or Cd24−/− male recipient mice (8 weeks old) received a lethal dose of two 5 Gy radiation (total 10 Gy) at a 4 hours interval. The recipient mice were intravenously injected with 5 × 106 bone marrow cells from either WT or Cd24−/− mice via tail vein. After 4 weeks of BMT, CD24 expression on PBMCs was detected by FACS to analyze reconstitution efficiency. After a 6-week recovery on a normal diet, all mice were placed on a HFD for an additional 12 weeks.
Quantification and Statistical Analysis
RNA-seq data analysis and quantification
Raw fastq files were analyzed using FastQC v0.11.5 and trimming was performed on raw reads using Trimmomatic v0.35 (Bolger et al., 2014): standard parameters for phred33 encoding were used. Reads mapping to the reference genome (GRCh38) was performed on quality-checked and trimmed reads using HISAT2 (Kim et al., 2015). The reference annotation is Ensembl v87. The overlap of reads with annotation features found in the reference.gtf was calculated using HT-seq v0.6.1 (Anders et al., 2015). The output computed for each sample (raw read counts) was then used as input for DESeq2 analysis (Love et al., 2014). Raw counts were normalized using DESeq2’s function “fpkm” and the expression value of FPKM was used for differential gene expression analysis.
Statistics summary
All data are presented as means ± SEM. The specific tests used to analyze each set of experiments are indicated in the figure legends. Data were analyzed using an unpaired two-tailed Student’s t test to compare between two groups, one-way analysis of variance (ANOVA) for multiple comparisons, two-way ANOVA for studies with multiple parameters. For human studies, data were analyzed using a linear regression for dose-dependency, paired t-test for measurements taken at different times from the same individual. All statistical tests were performed using GraphPad Prism, and p < 0.05 was considered statistically significant. The sample size in each study was based on experience in previous studies in our lab. The data showed a normal distribution and they were all continuous. No methods were used to determine whether the data met assumptions of the statistical approach.
Supplementary Material
Data S1. Unprocessed data underlying the display items in the manuscript, related to Figures 1–7 and S1, S2, S4–S7.
Highlights.
The CD24-Siglec-E axis protects against metabolic disorder in mice
A CD24Fc therapy ameliorates obesity-related metabolic dysfunction in mice
Sialylated CD24 induces SHP-1 recruitment to Siglec-E and represses metaflammation.
In a phase I clinical trial, a single dose of CD24Fc was safe and well-tolerated in healthy subjects
Acknowledgements
We thank members of Liu/Zheng Laboratories for helpful discussion and support. This study is supported by grants from the National Institutes of Health (AI64350, NS080821 and CA227671) and OncoC4, Inc. Part of the studies were performed while some of the co-authors were at the Children’s National Medical Center in Washington DC.
Footnotes
Declaration of interests
P.Zheng and Y.L. are cofounders of and have significant equity interest in OncoC4, Inc. and OncoImmune, Inc. Y.L., P.Zheng, X.W., M.L. and M.D. are inventors on patent applications related to CD24Fc (WO/2020/163523 and WO/2020/163529). Other authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abram CL, and Lowell CA (2017). Shp1 function in myeloid cells. J Leukoc Biol 102, 657–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai XF, Liu JQ, Liu X, Guo Y, Cox K, Wen J, Zheng P, and Liu Y (2000). The heat-stable antigen determines pathogenicity of self-reactive T cells in experimental autoimmune encephalomyelitis. J Clin Invest 105, 1227–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RG, Hayden MS, and Ghosh S (2011). NF-kappaB, inflammation, and metabolic disease. Cell Metab 13, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, Krishnan V, Hatakeyama J, Dorigo O, Barkal LJ, et al. (2019). CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 572, 392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning LM, Jebb SA, Mishra GD, Cooke JH, O’Connell MA, Crook MA, and Krebs JD (2004). Elevated sialic acid, but not CRP, predicts features of the metabolic syndrome independently of BMI in women. Int J Obes Relat Metab Disord 28, 1004–1010. [DOI] [PubMed] [Google Scholar]
- Chen GY, Brown NK, Wu W, Khedri Z, Yu H, Chen X, van de Vlekkert D, D’Azzo A, Zheng P, and Liu Y (2014). Broad and direct interaction between TLR and Siglec families of pattern recognition receptors and its regulation by Neu1. Elife 3, e04066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GY, Chen X, King S, Cavassani KA, Cheng J, Zheng X, Cao H, Yu H, Qu J, Fang D, et al. (2011). Amelioration of sepsis by inhibiting sialidase-mediated disruption of the CD24-SiglecG interaction. Nat Biotechnol 29, 428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GY, Tang J, Zheng P, and Liu Y (2009). CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science 323, 1722–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ A, and Latz E (2019). The Western lifestyle has lasting effects on metaflammation. Nat Rev Immunol 19, 267–268. [DOI] [PubMed] [Google Scholar]
- Crocker PR, Paulson JC, and Varki A (2007). Siglecs and their roles in the immune system. Nat Rev Immunol 7, 255–266. [DOI] [PubMed] [Google Scholar]
- Ding C, Liu Y, Wang Y, Park BK, Wang CY, Zheng P, and Liu Y (2007). Siglecg limits the size of B1a B cell lineage by down-regulating NFkappaB activation. PLoS One 2, e997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donath MY (2014). Targeting inflammation in the treatment of type 2 diabetes: time to start. Nat Rev Drug Discov 13, 465–476. [DOI] [PubMed] [Google Scholar]
- Duan S, and Paulson JC (2020). Siglecs as Immune Cell Checkpoints in Disease. Annu Rev Immunol 38, 365–395. [DOI] [PubMed] [Google Scholar]
- Fairbridge NA, Southall TM, Ayre DC, Komatsu Y, Raquet PI, Brown RJ, Randell E, Kovacs CS, and Christian SL (2015). Loss of CD24 in Mice Leads to Metabolic Dysfunctions and a Reduction in White Adipocyte Tissue. PLoS One 10, e0141966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Zheng P, Tang J, and Liu Y (2010). CD24: from A to Z. Cell Mol Immunol 7, 100–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores R, Zhang P, Wu W, Wang X, Ye P, Zheng P, and Liu Y (2019). Siglec genes confer resistance to systemic lupus erythematosus in humans and mice. Cell Mol Immunol 16, 154–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, and Hotamisligil GS (2002). A central role for JNK in obesity and insulin resistance. Nature 420, 333–336. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS (2006). Inflammation and metabolic disorders. Nature 444, 860–867. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS (2017). Inflammation, metaflammation and immunometabolic disorders. Nature 542, 177–185. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS, and Spiegelman BM (1993). Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259, 87–91. [DOI] [PubMed] [Google Scholar]
- Hsu YW, Hsu FF, Chiang MT, Tsai DL, Li FA, Angata T, Crocker PR, and Chau LY (2021). Siglec-E retards atherosclerosis by inhibiting CD36-mediated foam cell formation. J Biomed Sci 28, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang LL, Wang CH, Li TL, Chang SD, Lin LC, Chen CP, Chen CT, Liang KC, Ho IK, Yang WS, et al. (2010). Sex differences in high-fat diet-induced obesity, metabolic alterations and learning, and synaptic plasticity deficits in mice. Obesity (Silver Spring) 18, 463–469. [DOI] [PubMed] [Google Scholar]
- Kaburagi T, Kizuka Y, Kitazume S, and Taniguchi N (2017). The Inhibitory Role of alpha2,6-Sialylation in Adipogenesis. J Biol Chem 292, 2278–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, Mandrup-Poulsen T, and Donath MY (2007). Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 356, 1517–1526. [DOI] [PubMed] [Google Scholar]
- Laubli H, Pearce OM, Schwarz F, Siddiqui SS, Deng L, Stanczak MA, Deng L, Verhagen A, Secrest P, Lusk C, et al. (2014). Engagement of myelomonocytic Siglecs by tumor-associated ligands modulates the innate immune response to cancer. Proc Natl Acad Sci U S A 111, 14211–14216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Chen GY, and Zheng P (2009). CD24-Siglec G/10 discriminates danger- from pathogen-associated molecular patterns. Trends Immunol 30, 557–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Chen GY, and Zheng P (2011). Sialoside-based pattern recognitions discriminating infections from tissue injuries. Curr Opin Immunol 23, 41–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng CN, and Saltiel AR (2011). Inflammatory links between obesity and metabolic disease. J Clin Invest 121, 2111–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macauley MS, Crocker PR, and Paulson JC (2014). Siglec-mediated regulation of immune cell function in disease. Nat Rev Immunol 14, 653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, Kaiser N, Halban PA, and Donath MY (2002). Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest 110, 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik VS, Willett WC, and Hu FB (2013). Global obesity: trends, risk factors and policy implications. Nat Rev Endocrinol 9, 13–27. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Hada N, Sakamaki Y, Uno A, Shiga T, Tanaka C, Ito T, Katsume A, and Sudoh M (2013). An improved mouse model that rapidly develops fibrosis in non-alcoholic steatohepatitis. Int J Exp Pathol 94, 93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan SJ, Sharma RS, McKenzie EJ, Richards HE, Zhang J, Prescott A, and Crocker PR (2013). Siglec-E is a negative regulator of acute pulmonary neutrophil inflammation and suppresses CD11b beta2-integrin-dependent signaling. Blood 121, 2084–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNelis JC, and Olefsky JM (2014). Macrophages, immunity, and metabolic disease. Immunity 41, 36–48. [DOI] [PubMed] [Google Scholar]
- Natori Y, Ohkura N, Nasui M, Atsumi G, and Kihara-Negishi F (2013). Acidic sialidase activity is aberrant in obese and diabetic mice. Biol Pharm Bull 36, 1027–1031. [DOI] [PubMed] [Google Scholar]
- Nielsen PJ, Lorenz B, Muller AM, Wenger RH, Brombacher F, Simon M, von der Weid T, Langhorne WJ, Mossmann H, and Kohler G (1997). Altered erythrocytes and a leaky block in B-cell development in CD24/HSA-deficient mice. Blood 89, 1058–1067. [PubMed] [Google Scholar]
- Pillai S, Netravali IA, Cariappa A, and Mattoo H (2012). Siglecs and immune regulation. Annu Rev Immunol 30, 357–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirruccello SJ, and LeBien TW (1986). The human B cell-associated antigen CD24 is a single chain sialoglycoprotein. J Immunol 136, 3779–3784. [PubMed] [Google Scholar]
- Rajappa M, Ikkruthi S, Nandeesha H, Satheesh S, Sundar I, Ananthanarayanan PH, and Harichandrakumar KT (2013). Relationship of raised serum total and protein bound sialic acid levels with hyperinsulinemia and indices of insulin sensitivity and insulin resistance in non-diabetic normotensive obese subjects. Diabetes Metab Syndr 7, 17–19. [DOI] [PubMed] [Google Scholar]
- Schenk S, Saberi M, and Olefsky JM (2008). Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest 118, 2992–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz F, Pearce OM, Wang X, Samraj AN, Laubli H, Garcia JO, Lin H, Fu X, Garcia-Bingman A, Secrest P, et al. (2015). Siglec receptors impact mammalian lifespan by modulating oxidative stress. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira S, Kazanov D, Dankner R, Fishman S, Stern N, and Arber N (2021). High Expression Level of PPARgamma in CD24 Knockout Mice and Gender-Specific Metabolic Changes: A Model of Insulin-Sensitive Obesity. J Pers Med 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoelson SE, Lee J, and Goldfine AB (2006). Inflammation and insulin resistance. J Clin Invest 116, 1793–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song NJ, Allen C, Vilgelm AE, Riesenberg BP, Weller KP, Reynolds K, Chakravarthy KB, Kumar A, Khatiwada A, Sun Z, et al. (2022). Treatment with soluble CD24 attenuates COVID-19-associated systemic immunopathology. J Hematol Oncol 15, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley TL, Zanni MV, Johnsen S, Rasheed S, Makimura H, Lee H, Khor VK, Ahima RS, and Grinspoon SK (2011). TNF-alpha antagonism with etanercept decreases glucose and increases the proportion of high molecular weight adiponectin in obese subjects with features of the metabolic syndrome. J Clin Endocrinol Metab 96, E146–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian RR, Zhang MX, Liu M, Fang X, Li D, Zhang L, Zheng P, Zheng YT, and Liu Y (2020). CD24Fc protects against viral pneumonia in simian immunodeficiency virus-infected Chinese rhesus monkeys. Cell Mol Immunol 17, 887–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian RR, Zhang MX, Zhang LT, Zhang P, Ma JP, Liu M, Devenport M, Zheng P, Zhang XL, Lian XD, et al. (2018). CD24 and Fc fusion protein protects SIVmac239-infected Chinese rhesus macaque against progression to AIDS. Antiviral Res 157, 9–17. [DOI] [PubMed] [Google Scholar]
- Toubai T, Hou G, Mathewson N, Liu C, Wang Y, Oravecz-Wilson K, Cummings E, Rossi C, Evers R, Sun Y, et al. (2014). Siglec-G-CD24 axis controls the severity of graft-versus-host disease in mice. Blood 123, 3512–3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toubai T, Rossi C, Oravecz-Wilson K, Zajac C, Liu C, Braun T, Fujiwara H, Wu J, Sun Y, Brabbs S, et al. (2017). Siglec-G represses DAMP-mediated effects on T cells. JCI Insight 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui S, Hara Y, Hosaki S, and Okazaki M (2002). A new on-line dual enzymatic method for simultaneous quantification of cholesterol and triglycerides in lipoproteins by HPLC. J Lipid Res 43, 805–814. [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, and Jaenisch R (2013). One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welker J, Pulido JD, Catanzaro AT, Malvestutto CD, Li Z, Cohen JB, Whitman ED, Byrne D, Giddings OK, Lake JE, et al. (2022). Efficacy and safety of CD24Fc in hospitalised patients with COVID-19: a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Infect Dis 22, 611–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yerlikaya FH, Toker A, Cicekler H, and Aribas A (2015). The association of total sialic acid and malondialdehyde levels with metabolic and anthropometric variables in obesity. Biotech Histochem 90, 31–37. [DOI] [PubMed] [Google Scholar]
- Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, and Shoelson SE (2001). Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science 293, 1673–1677. [DOI] [PubMed] [Google Scholar]
- Zheng X, Wu W, Liu Y, and Zheng P (2013). Methods of use of soluble cd24 for therapy of rheumatoid arthritis U. PTO, ed. (USA: OncoImmune, Inc; ). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Unprocessed data underlying the display items in the manuscript, related to Figures 1–7 and S1, S2, S4–S7.
Data Availability Statement
The RNA-seq raw data files have been deposited in the GEO repository with GEO accession number GSE140724.
This paper does not report original code.
Data S1 represents an Excel file containing the values that were used to create all the graphs in the paper, as well as a PDF file containing the full-length, unprocessed western blots. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.