

Abstract
We report on a child with prenatal findings of increased nuchal translucency, polydramnios, ascites, and overgrowth. At birth, she presented length >97° centile, minor facial anomalies, megalencephaly, and Wolff–Parkinson–White syndrome. Whole‐exome sequencing showed a pathogenic variant in the NRAS gene, but no mutations were found in PI3K/AKT/mTOR pathway genes.
Keywords: cardiovascular disorders, genetics, neurology, obstetrics and gynecology, pediatrics and adolescent medicine
This report expands the phenotypic spectrum of Noonan syndrome, because it is the first Italian case carrying a pathogenic variant in the NRAS gene associated with megalencephaly and overgrowth, without any anomaly in the PI3K/AKT/mTOR pathway.
1. INTRODUCTION
Noonan syndrome (NS, MIM # 163950) is a common genetic disorder. It is characterized by the presence of short stature, minor facial anomalies, congenital heart defects, variable degrees of developmental delay and intellectual disability, skeletal malformations, and coagulation defects. 1 , 2
The incidence of NS is estimated to be between 1:1000 and 1:2500 live births; there is no difference between sexes.
NS is caused by germline mutations in genes involved in the Ras‐mitogen‐activated protein kinase (RAS‐MAPK) pathway, while somatic mutations are frequently found in various types of tumors. This pathway is implicated in cell proliferation, differentiation, and survival.
PTPN11 (MIM * 176876) was the first gene to be associated with NS, and it is mutated in about 50% of individuals affected by Noonan syndrome.
Other genes that can be involved are SOS1 (MIM * 182530), RAF1 (MIM * 164760), RIT1 (MIM * 609591), KRAS (MIM * 190070), NRAS (MIM * 164790), BRAF (MIM * 64757) MAP2K1 (MIM * 176872), and LZTR1 (MIM * 600574).
Noonan syndrome has an autosomal dominant model of inheritance, with the exception of certain mutations of LZTR1 gene. 3 , 4
Congenital heart defects are a major problem in NS. The most frequent anomaly is pulmonary valve stenosis, whereas hypertrophic cardiomyopathy, atrial or ventricular septal defects, tetralogy of Fallot, and branch pulmonary artery stenosis are described in a minority of cases. 5
Megalencephaly is defined as a head circumference 2 Standard Deviations (SD) above the age‐ and sex‐related mean, caused by an increased growth of cerebral structures. This is due to disruptions of the normal brain development, especially during neuronal proliferation and migration. Megalencephaly can be evident at birth, or it can develop over time, during infancy or childhood. 6 , 7 To date, megalencephaly associated with NS has been reported only once in medical literature, in an individual affected by megalencephaly‐capillary malformation (MCAP) syndrome (OMIM #602501) carrying a 22% mosaic variant in PIK3CA (MIM * 171834) and a PTPN11 germline variant. 8
Relative megalencephaly has been associated with variants in SHOC2 gene (MIM * 602775), causing Noonan syndrome‐like disorder with loose anagen hair‐1 (MIM # 607721). 9
2. CASE HISTORY
We report on a child who came to our attention because of the presence of increased nuchal translucency (3.3 mm) in the first trimester of pregnancy. Second trimester morphology ultrasound revealed right pyelocaliectasis and single umbilical artery. Third trimester ultrasound showed the presence of polydramnios (Amniotic Fluid Index 22 cm) and fetal macrocephaly (head circumference >95° centile). Fetal brain MRI detected enlargement of periencephalic liquoral spaces and mild lateral ventricular asymmetry. Fetal echocardiography registered the presence of supraventricular extrasystoles. Further prenatal ultrasounds revealed ascites and hepatosplenomegaly, and confirmed the presence of abnormal fetal biometric parameters (head circumference >95° centile and abdominal circumference >95° centile). The patient was born to non‐consanguineous and healthy parents after a spontaneous pregnancy at 39 weeks of gestation by cesarean section. Family history was silent. At birth, she was appropriate for gestational age (AGA), with a birth weight of 3.63 kg, her head circumference was 37.3 cm (>97°), and her length was 54 cm (>97° centile). Apgar scores were 9 and 10 at 1 and 5 min after birth, respectively. At birth, she presented minor facial anomalies: hypertelorism, low‐set ears, posterior cleft palate, long fingers and toes, single umbilical artery, and axial hypotonia.
Subsequent abdominal ultrasound examinations showed the presence of splenomegaly without pyelocaliectasis. Brain MRI, performed at nearly 2 months of age, revealed moderately increased brain volume, mild lateral ventricular asymmetry, thinned dysmorphic corpus callosum, and dysmorphic aspects of brain stem (Figure 1).
FIGURE 1.
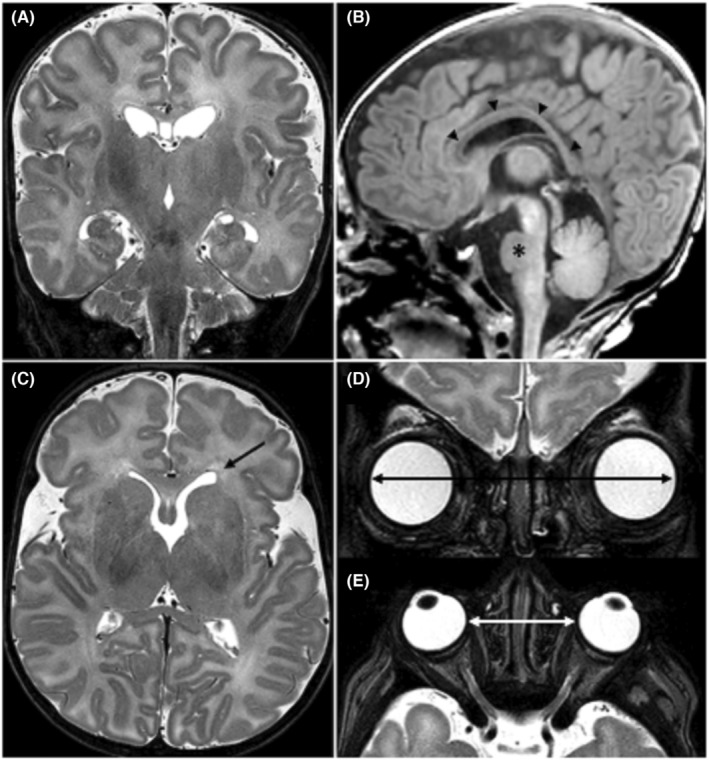
MR imaging examination performed at nearly 2 months of age. Coronals (A, D) and axials (C, E) TSE T2‐weighted images and sagittal (B) 3D‐TFE T1‐weighted image. MR showed (A) a moderately increased brain volume, (B) a thin and dysmorphic corpus callosum (arrow heads) and a small pons (asterisk). (C) Mild asymmetry of the lateral ventricles with squared off left ventricular frontal horn (arrow). Increased (D) binocular and (E) interocular distances. Abbreviations: TSE, turbo spin echo; 3D‐TFE, three‐dimensional turbo field echo
Electrocardiographic screening for long QT syndrome demonstrated the presence of Wolf–Parkinson–White Syndrome, treated with beta‐blockers and anti‐arrhythmic drugs.
Soon after birth, severe leukocytosis appeared (43,950/mm3), although further blood examinations revealed normal leucocyte count at 9 months of age.
With regard to psychomotor development, at 10 months of age she was able to smile in the interaction with the adult, vocalize, turn her head to follow objects, reach out and grasp them, and sit with anterior support. However, she presented reduced eye movement coordination and lower extremities hypotonia.
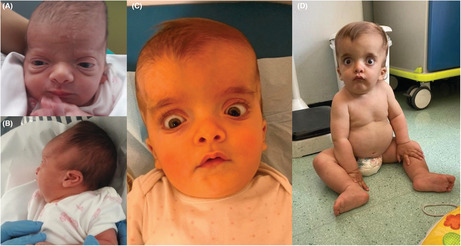
We evaluated again the child when she was 16 months old. Physical examination showed macrocephaly, frontal bossing, sparse eyebrows, hypertelorism, exophthalmos, depressed nasal root, low‐set and posteriorly rotated ears, ligamentous laxity, and hyposthenia (Figure 2). Her weight was 11.3 kg (75° centile), her length was 72.5 cm (5° centile), and her occipital‐frontal circumference was 53 cm (>97°centile). She was able to babble and to walk with adult support.
FIGURE 2.
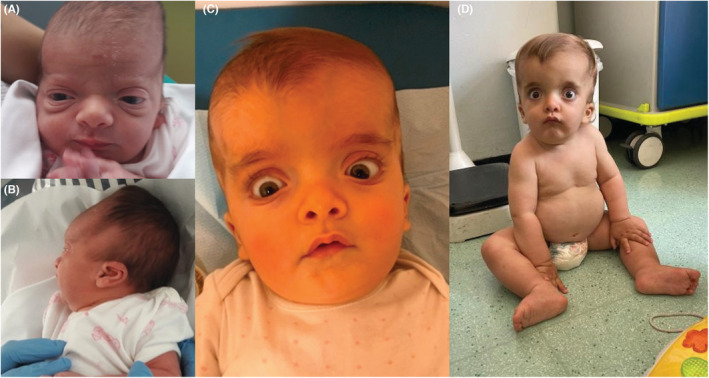
Photographs of the patient at birth (A, B), at the age of 10 months (C), and at the age of 16 months (D). Note, macrocephaly, frontal bossing, sparse eyebrows, hypertelorism, exophthalmos, depressed nasal root, and low‐set and posteriorly rotated ears
3. INVESTIGATIONS
3.1. WES analysis
Genomic DNA was extracted from peripheral blood samples of proband and parents using standard procedures. The exonic regions and flanking splice junctions of the genome were captured using the Clinical Research Exome v.2 kit (Agilent Technologies). Sequencing was done on a NextSeq500 Illumina system with 150 bp paired‐end reads. Reads were aligned to human genome build GRCh37/UCSC hg19 and analyzed for sequence variants using a custom‐developed analysis tool. 14 Additional sequencing technology and variant interpretation protocol have been previously described. 14 Coverage on target for the proband was ≥10× for 98% with a mean coverage of 203×.
3.2. Targeted sequencing
The Ion AmpliSeq Custom Panel of the 21 genes involved in the PI3K/AKT/mTOR pathway (i.e., PIK3R1, PIK3R2, PIK3CA, PTEN, PDK1, PDK2, KRAS, AKT1, AKT2, AKT3, RICTOR, MAPKAP1, MLST8, MTOR, IRS1, GAB1, GAB2, THEM4, MAPK8I1, PTPN11, and RAPTOR) was used according to our previous report. 15
3.3. Alignment
Data analysis was performed using the Torrent Suite Software v5.0.4 (Life Technologies). Reads were aligned to the hg19 human reference genome from the UCSC Genome Browser (http://genome.ucsc.edu/) and to the BED file designed using Ion AmpliSeq Designer. Alignments were visually verified with the software Alamut® v2.13 (Interactive Bio software).
3.4. Coverage analysis
The mean average read depth and the percentage of reads mapping on the ROI out of the total number of reads (reads on target) were calculated using the Coverage Analysis plugin (Torrent Suite v5.0.4 software, Life Technologies). For each sample, the percentage of ROI with a minimum coverage of 100× was calculated using the amplicon coverage matrix file.
3.5. Variant analysis
Variant calling was performed with the Variant Caller plugin configured with somatic high stringency parameters. Variants were annotated using the Ion Reporter 5.14.1.0 software (https://ionreporter.lifetechnologies.com/ir/).
Common single nucleotide variants (minor allele frequency [MAF] > 5%), exonic synonymous variants, and intronic variants were removed from the analysis, while exonic nonsynonymous, splice site, and loss‐of‐function variants were analyzed.
The sequence analysis software Alamut® v2.13 (Interactive Bio software) was used to interpret variants.
Online databases, including dbSNP (database the single nucleotide polymorphism database), 1000 Genomes, ClinVar, EXAC (exome aggregation consortium), COSMIC (catalog of somatic mutations in cancer), and ESP (exome sequencing project), were used. The pathogenicity prediction programs such as PolyPhen2, SIFT, Mutation Taster, and splice prediction programs were used to evaluate variants not previously described.
4. RESULTS
The karyotype and the array‐CGH analysis from chorionic villus sampling (CVS) showed a normal female profile.
Fast trio‐based whole exome sequencing (WES) analysis, performed when the patient was in very critical condition, detected a previously reported de novo heterozygous NRAS missense variant (NM_002524.4:c.34G > A; p.Gly12Ser) in 9 days (REF: Cirstea et al [2010] Nat Genet; ClinVar; Variation ID 177778).
We performed targeted deep sequencing of 21 selected genes belonging to the PI3K/AKT/ mTOR pathway on saliva sample. No pathogenic variants were found.
5. DISCUSSION
To date, only few studies regarding the perinatal findings in Noonan syndrome have been described in medical literature. In the prenatal period, the most common clinical characteristics that have been reported are lymphatic anomalies, such as increased nuchal translucency, pleural effusions, cystic hygroma, and hydrops fetalis. Long bones <5° centile have been described in almost half of cases. Other common prenatal findings in NS are as follows: polyhydramnios, macrosomia (defined as abdominal circumference >90th centile), occipital‐frontal circumference >90th centile, and congenital heart defects. Arrhythmias have been reported only once in medical literature.
Regarding neonatal findings, cardiovascular anomalies, such as structural heart defects, valvular dysplasia, hypertrophic cardiomyopathy are the most common features. Lymphatic dysplasia, birth weight and occipital‐frontal circumference >90th centile, renal defects, and hypotonia are anomalies frequently described in infants affected by NS. 10
During the prenatal period, our patient showed specific findings of NS: increased nuchal translucency, right caliectasis, polyhydramnios, abdominal circumference, and occipital‐frontal circumference >90th centile, hydrops fetalis.
At birth, other anomalies became evident, such as length >97th centile, long fingers and toes, megalencephaly, and Wolff–Parkinson–White syndrome (WPW). Those characteristics are not typical of Noonan syndrome. Wolff–Parkinson–White syndrome has never been associated with Noonan syndrome.
WPW syndrome is characterized by the presence of an accessory atrioventricular pathway, which bypasses the normal atrioventricular conduction. This pathway leads to a ventricular pre‐excitation, causing palpitations, syncope, ventricular fibrillation, and even sudden cardiac death. 11 , 12
Recently, variants in the PI3K‐AKT pathway have been described to be associated with overgrowth syndromes (PIK3CA‐Related Overgrowth Spectrum, PROS), such as Megalencephaly‐capillary malformation‐polymicrogyria syndrome (MCAP, MIM # 602501) and Megalencephaly‐polymicrogyria‐polydactyly‐hydrocephalus syndrome‐1 (MPPH, # MIM 603387), but also Congenital lipomatous asymmetric overgrowth, epidermal naevi, skeletal and spinal anomalies syndrome (CLOVES, # MIM 612918), as well as hemimegalencephaly and isolated macrodactyly. 13 For this reason, the analysis of a panel of genes involved in the PI3K/AKT/mTOR pathway was performed from a saliva sample of our patient and the gene panel testing came back negative. Since PROS shows phenotypic variability and it is mostly caused by somatic PI3KCA mutations, it could be important to follow the child over time, in order to monitor the onset of other signs related to PROS. It could be interesting to perform the analysis of a panel of genes involved in the PI3K‐AKT pathway in other tissues as well. Indeed, it cannot be excluded that PIK3CA‐mutant cells might exert growth‐promoting effects on adjacent or distant cells by signal propagation of secreted biomolecules or exosomes.
AUTHOR CONTRIBUTIONS
All the authors have contributed equally.
CONFLICT OF INTEREST
All authors declare no competing interests.
CONSENT
Written informed consent was obtained from the parents of the patient in order to publish this report in accordance with the journal's patient consent policy.
ACKNOWLEDGMENTS
We thank the patients' parents to participate to this study. This work has been generated within the European Reference Network on Rare Congenital Malformations and Rare Intellectual Disability (ERN‐ITHACA) (EU Framework Partnership Agreement ID: 3HP‐HP‐FPA ERN‐01‐2016/739516).
Beltrami B, Cerasani J, Consales A, et al. Prenatal overgrowth and polydramnios: Would you think about Noonan syndrome? Clin Case Rep. 2022;10:e06256. doi: 10.1002/ccr3.6256
DATA AVAILABILITY STATEMENT
None.
REFERENCES
- 1. Roelofs RL, Janssen N, Wingbermühle E, Kessels RP, Egger JI. Intellectual development in Noonan syndrome: a longitudinal study. Brain Behav. 2016;6(7):e00479. doi: 10.1002/brb3.479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yart A, Edouard T. Noonan syndrome: an update on growth and development. Curr Opin Endocrinol Diabetes Obes. 2018;25(1):67‐73. doi: 10.1097/MED.0000000000000380 [DOI] [PubMed] [Google Scholar]
- 3. Altmüller F, Lissewski C, Bertola D, et al. Genotype and phenotype spectrum of NRAS germline variants. Eur J Hum Genet. 2017;25(7):823‐831. doi: 10.1038/ejhg.2017.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. El Bouchikhi I, Belhassan K, Moufid FZ, et al. Noonan syndrome‐causing genes: molecular update and an assessment of the mutation rate. Int J Pediatr Adolesc Med. 2016;3(4):133‐142. doi: 10.1016/j.ijpam.2016.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shaw AC, Kalidas K, Crosby AH, Jeffery S, Patton MA. The natural history of Noonan syndrome: a long‐term follow‐up study. Arch Dis Child. 2007;92(2):128‐132. doi: 10.1136/adc.2006.104547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pavone P, Praticò AD, Rizzo R, et al. A clinical review on megalencephaly: a large brain as a possible sign of cerebral impairment. Medicine (Baltimore). 2017;96(26):e6814. doi: 10.1097/MD.0000000000006814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pirozzi F, Nelson B, Mirzaa G. From microcephaly to megalencephaly: determinants of brain size. Dialogues Clin Neurosci. 2018;20(4):267‐282. doi: 10.31887/DCNS.2018.20.4/gmirzaa [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Döcker D, Schubach M, Menzel M, et al. Germline PTPN11 and somatic PIK3CA variant in a boy with megalencephaly‐capillary malformation syndrome (MCAP) pure coincidence? Eur J Med Genet. 2015;23(3):409‐412. doi: 10.1038/ejhg.2014.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gripp KW, Zand DJ, Demmer L, et al. Expanding the SHOC2 mutation associated phenotype of Noonan syndrome with loose anagen hair: structural brain anomalies and myelofibrosis. Am J Med Genet A. 2013;161A(10):2420‐2430. doi: 10.1002/ajmg.a.36098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Myers A, Bernstein JA, Brennan ML, et al. Perinatal features of the RASopathies: Noonan syndrome, cardiofaciocutaneous syndrome and Costello syndrome. Am J Med Genet A. 2014;164A(11):2814‐2821. doi: 10.1002/ajmg.a.36737 [DOI] [PubMed] [Google Scholar]
- 11. De Castro RL Jr, de Alcantara Lima N, da Costa Lino DO, Bannon SF. Concealed Wolff‐Parkinson‐White Syndrome revealed by acute coronary syndrome. Ann Noninvasive Electrocardiol. 2019;25(5):e12735. doi: 10.1111/anec.12735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miyamoto L. Molecular pathogenesis of familial Wolff‐Parkinson‐White syndrome. J Med Invest. 2018;65(1.2):1‐8. doi: 10.2152/jmi.65.1 [DOI] [PubMed] [Google Scholar]
- 13. Keppler‐Noreuil KM, Rios JJ, Parker VE, et al. PIK3CA‐related overgrowth spectrum (PROS): diagnostic and testing eligibility criteria, differential diagnosis, and evaluation. Am J Med Genet A. 2015;167A(2):287‐295. doi: 10.1002/ajmg.a.36836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pezzani L, Marchetti D, Cereda A, et al. Atypical presentation of pediatric BRAF RASopathy with acute encephalopathy. Am J Med Genet A. 2018;176(12):2867‐2871. [DOI] [PubMed] [Google Scholar]
- 15. Loconte DC, Grossi V, Bozzao C, et al. Molecular and Functional Characterization of Three Different Postzygotic Mutations in PIK3CA‐Related Overgrowth Spectrum (PROS) Patients: Effects on PI3K/AKT/mTOR Signaling and Sensitivity to PIK3 Inhibitors. PLoS ONE. 2015;10(4):e0123092. doi: 110.1371/journal.pone.0123092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
None.