

To the editor:
Screening all pregnancies for sickle cell disease (SCD), as recommended by the American College of Gynecologists,1 is essential to enable informed decisions about diagnostic testing, clinical care, and expand available gene therapy treatment options.2–4 However, traditional prenatal screening, in which both maternal and paternal DNA are required for carrier screening, has a low sensitivity of 42% due to insufficient paternal screening uptake in the U.S.5 Even when paternal screening is performed and the carrier result is positive, the maximum fetal disease risk is 1 in 4.
Single-gene noninvasive prenatal testing (sgNIPT) is a promising new technology, able to achieve accurate prenatal results without requiring paternal screening. We previously reported the proof-of-principle development of a sequencing-based sgNIPT test for five conditions, including SCD, in 2019.6 sgNIPT analyzes cell-free DNA (cfDNA) from maternal plasma to provide a personalized fetal residual disease risk ranging from >9 in 10 to <1 in 20,000. The aim of this study was to build upon our previous work to validate the sgNIPT in clinical samples and identify high-risk SCD fetuses in a cohort of at-risk pregnancies.
This retrospective clinical study collected 77 maternal blood samples between October 2018 and December 2019 from pregnant patients at the Baylor College of Medicine or the University of Alabama at Birmingham who were known to have at least one pathogenic HBB allele. Newborn HBB genotype was determined by newborn screening chart review or genotyping of umbilical cord blood.
For sgNIPT processing, genomic DNA (gDNA) and cfDNA were extracted from the maternal blood sample. The SCD maternal carrier status was determined by next-generation sequencing of the gDNA. The cfDNA fraction was then sequenced to determine (1) fetal fraction, (2) molecular counts of cfDNA, (3) maternal variant fraction, and (4) variants that are not present in the maternal genotype (paternally inherited variants).6 All patients start with an a priori risk calculated from the pregnant patient’s carrier status, the highest subpopulation HBB carrier frequency in the U.S. (1 in 8),7 and the likelihood a fetus inherits two SCD alleles. A likelihood ratio is calculated through relative dosage analysis of the most abundant allele found in cfDNA, comparing the likelihood of inheriting one copy or two copies of the most abundant allele.6 The likelihood ratio was used (1) to calculate the genotype score and predict fetal HBB genotype (Figure 1A) and (2) to adjust the a priori risk and calculate a residual risk that the fetus is affected with SCD (Figure 1B). In a clinical report, the residual and associated risk categories (Low Risk, Decreased Risk, or High Risk) would be provided.
Figure 1.
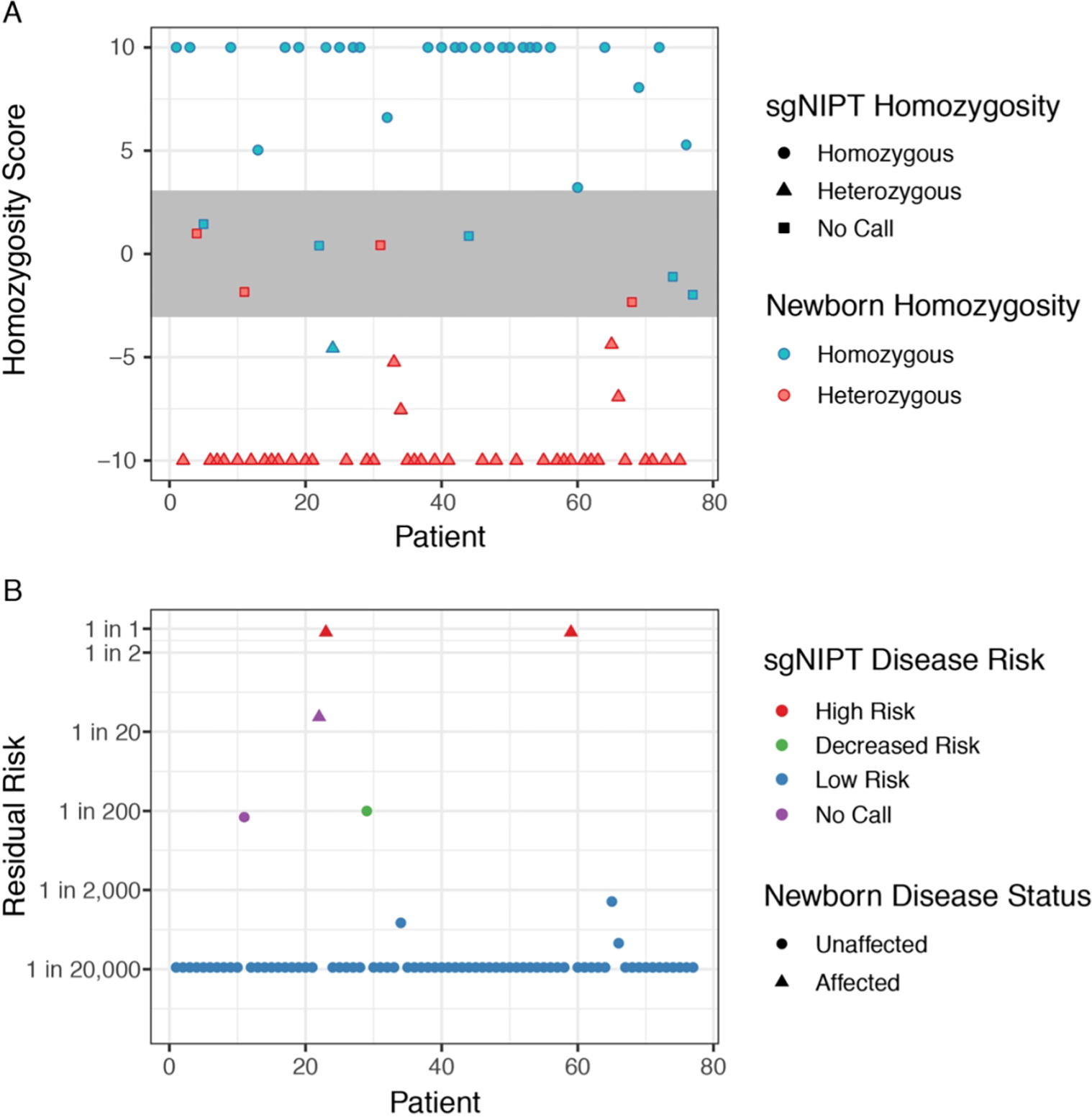
sgNIPT uses HBB genotype prediction to accurately determine fetal SCD risk. (A) Prediction of fetal homozygosity. sgNIPT classified a sample as homozygous (HbAA or HbSS) when the homozygosity score was above 3, and as heterozygous (HbAC, HbAS, or HbSC) when the homozygosity score was below −3. Samples with scores more extreme than 10 (−10) were set to 10 (−10). Samples with scores between −3 and 3 had an undetermined genotype. Once homozygosity is determined, the genotype is determined by inspection of sequencing data. (B) Residual risk of fetal sickle cell disease after sgNIPT. The residual risk is calculated from the a priori risk derived from population carrier frequency (for African-Americans: 1 in 32 for all pregnancies when mother is heterozygous) and the likelihood ratio derived from sgNIPT.
Out of the 77 pregnancies in the cohort, maternal HBB genotypes included HbAS (n = 59), HbAC (n = 13), HbSS (n = 4), and HbCC (n = 1). The median fetal fraction was 9.3% (IQR = 5.8–13.6%) for gestational ages ranging from 16.4 weeks to collection at delivery (Supplementary Table 1). The fetal fraction was similar to a large clinical study that reported a median fetal fraction of 10.0% (IQR = 7.8–13.0%) for gestational ages 11–13 weeks.8 Therefore, this cohort is likely representative of the expected fetal fraction in the first trimester, when prenatal screening is most common.
sgNIPT returned a fetal HBB genotype prediction for 68 of the 77 pregnancies, with 9 undetermined. sgNIPT accurately distinguished heterozygous from homozygous fetuses (Figure 1A) with 100% sensitivity (90.8% - 100%, 95% CI) and 96.5% specificity (82.2% - 99.9%, 95% CI). The fetal genotype predictions were concordant with newborn genotypes in 67 out of 68 pregnancies (98.5%) (Supplementary Tables 1–2). In the single discordant result, sgNIPT returned a fetal genotype prediction of HbAS when the newborn genotype was HbAA, both of which lead to low-risk results for sickle cell disease.
To calculate the residual risk, the data from sgNIPT assay and HBB genotype prediction are used to adjust the a priori risk (the a priori risk for an affected fetus for an African American patient is 1 in 32 when mother is heterozygous) to classify the fetus as “Low Risk,” “Decreased Risk,” or “High Risk.” Both the residual risk and risk classification would be included on the clinical report. sgNIPT returned a result for 75 of the 77 pregnancies with 2 no calls (2.6% no call rate). From these 75 pregnancies, sgNIPT correctly identified the two newborns affected with SCD (Figure 1B). Both high-risk sgNIPT results had a greater than 9 in 10 residual disease risk (Figure 1B). One case was a newborn affected with sickle cell anemia (genotype HbSS) whose mother who also had sickle cell anemia. The other case was a newborn affected with Hemoglobin SC disease (genotype HbSC) whose mother had an HbAC genotype. The fetal fraction of cfDNA used to determine the risk call for these two samples were 3.2% and 1.9%, respectively, highlighting the ability of sgNIPT to make informative calls even with low (< 5%) fetal fraction.
sgNIPT also correctly identified all unaffected fetuses (73 out of 75 pregnancies). Most low-risk calls had a fetal residual risk of 1 in 20,000; a few of the low-risk calls had a residual risk as high as 1 in 2,000 (Figure 1B). One sample was designated as “Decreased Risk” because carrier screening indicated that the mother was HbAS, and the paternal allele assay detected that the fetus inherited an HbC allele from the father. Dosage analysis on the maternal HbS allele determined that the fetus was likely an unaffected carrier with genotype HbAC. Therefore, the residual risk was lowered from 1 in 2 to 1 in 200, which is the lower limit of risk reduction when a paternal pathogenic allele is detected.
An undetermined fetal HBB genotype or a risk classification no call occurs when the likelihood ratio falls between the internal high and low risk thresholds. This typically happens when there are an inadequate number of fetal molecules in cfDNA sample. In our study, 9 genotypes were undetermined, but only 2 led to risk classification no calls. In the other 7 samples, the data was consistent with the fetus inheriting at least one wild-type allele (genotypes HbAA or HbAS). Therefore, the sgNIPT could still accurately classify the fetus as low risk since neither genotype is associated with SCD. In a clinical setting, a risk classification no call would trigger a request for a second blood sample to re-run sgNIPT. The additional molecules can be used for data analysis to increase the likelihood of obtaining a reportable result.
The personalized fetal risk assessment reported by sgNIPT is more sensitive and precise than the current standard of care and allow parents and genetic counselors to make better informed decisions regarding family and clinical planning. Furthermore, advanced detection of SCD allows the family and clinicians to prepare for cord blood collection to be used for gene-based therapies for an affected child, or an allogenic transplant for an affected sibling. There are multiple gene therapy strategies underway as part of Phase I, II, and III clinical trials that use gene insertion, fetal hemoglobin induction or editing of the sickle mutation to treat sickle cell disease.2 These SCD-related gene therapies are expected to be in widespread clinical use in the near future, increasing the importance of widespread and informative prenatal screening for SCD.
Together, we have validated that SCD sgNIPT predicts fetal genotype with high sensitivity and specificity, which leads to an accurate determination of fetal SCD risk (Supplemental Figure). Crucially, sgNIPT returned an informative fetal disease risk for 97.4% (75 out of 77) pregnancies compared to only 42% with traditional screening.5 Furthermore, sgNIPT determined residual disease risks as high as >9 in 10, therefore providing a more personalized risk assessment compared to traditional carrier screening. These results, combined with the unique workflow of reflex sgNIPT for carrier mothers without the need for a paternal sample, highlight that this screen should be considered for broad clinical adoption to promote efficient and accurate fetal risk assessment for SCD in pregnant patients.
Supplementary Material
Acknowledgements
Research reported in this publication was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under award number R43HL144322. In the study, 10% was funded by federal sources, and 90% was funded by BillionToOne. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Dr. Rong Mao and Dr. Jacqueline Carozza for providing editorial support.
Footnotes
Conflict of Interest Disclosures
DST, OA, BL, PPY, DCB, BA and JH are employees of BillionToOne and hold stock or options to hold stock in the company. VAS received a grant from BillionToOne directly and as a subcontractor from NIH. ERW, AS, KMP, SGJ, FDG, and TMT report no conflict of interest.
References
- 1.The American College of Obstetricians and Gynecologists. Committee Opinion No. 691: Carrier Screening for Genetic Conditions. Obstet. Gynecol 129, e41–e55 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Dever DP & Porteus MH The changing landscape of gene editing in hematopoietic stem cells: a step towards Cas9 clinical translation. Curr. Opin. Hematol 24, 481–488 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fitzhugh C, Hsieh MM, Bolan CD, Saenz C & Tisdale JF Granulocyte colony-stimulating factor (G-CSF) administration in individuals with sickle cell disease: time for a moratorium? Cytotherapy 11, 464–471 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ribeil J-A et al. Gene Therapy in a Patient with Sickle Cell Disease. N. Engl. J. Med. 376, 848–855 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Giles Choates M et al. It takes two: uptake of carrier screening among male reproductive partners. Prenat. Diagn. 40, 311–316 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Tsao DS et al. A novel high-throughput molecular counting method with single base-pair resolution enables accurate single-gene NIPT. Sci. Rep. 9, 1–14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.1000 Genomes Project. Population Genetics for Variant rs334. Ensembl GRCh37 Release 104 (2021). Available at: https://grch37.ensembl.org/Homo_sapiens/Variation/Population?db=core;r=11:5247732-5248732;v=rs334;vdb=variation;vf=340756472#population_freq_AFR. (Accessed: 4th August 2021) [Google Scholar]
- 8.Ashoor G, Syngelaki A, Poon LCY, Rezende JC & Nicolaides KH Fetal fraction in maternal plasma cell-free DNA at 11–13 weeks’ gestation: relation to maternal and fetal characteristics. Ultrasound Obstet. Gynecol. 41, 26–32 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.