

Abstract

In this article, we describe a combined experimental and theoretical mechanistic investigation of the C(sp2)–F bond formation from neutral and cationic high-valent organobismuth(V) fluorides, featuring a dianionic bis-aryl sulfoximine ligand. An exhaustive assessment of the substitution pattern in the ligand, the sulfoximine, and the reactive aryl on neutral triarylbismuth(V) difluorides revealed that formation of dimeric structures in solution promotes facile Ar–F bond formation. Noteworthy, theoretical modeling of reductive elimination from neutral bismuth(V) difluorides agrees with the experimentally determined kinetic and thermodynamic parameters. Moreover, the addition of external fluoride sources leads to inactive octahedral anionic Bi(V) trifluoride salts, which decelerate reductive elimination. On the other hand, a parallel analysis for cationic bismuthonium fluorides revealed the crucial role of tetrafluoroborate anion as fluoride source. Both experimental and theoretical analyses conclude that C–F bond formation occurs through a low-energy five-membered transition-state pathway, where the F anion is delivered to a C(sp2) center, from a BF4 anion, reminiscent of the Balz–Schiemann reaction. The knowledge gathered throughout the investigation permitted a rational assessment of the key parameters of several ligands, identifying the simple sulfone-based ligand family as an improved system for the stoichiometric and catalytic fluorination of arylboronic acid derivatives.
Introduction
The development of methodologies to forge C(sp2)–F bonds is of capital importance, as fluorine-containing molecules find applications as drugs,1 agrochemical products,2 organic materials,3 or [18F]Fluoride-labeled radiotracers for positron emission tomography (PET).4 In addition to the traditional nucleophilic aromatic substitution,5 fluorine has typically been anchored to aromatic substrates via the Balz–Schiemman reaction6 or through the Halex process.7 Despite numerous applications, these methods still suffer from harsh reaction conditions and a limited substrate scope. Attractive alternatives have recently emerged that facilitate C(sp2)–F bond formation under much milder reaction conditions which broaden the spectrum of compatible functional groups during C–F formation. These methods rely on the use of d-block elements, which have demonstrated to be excellent candidates for this purpose.8 Yet, metal-catalyzed C(sp2)–F bond-forming reactions are still arduous, due to the challenging reductive elimination from the small and highly electronegative fluoride anion. Therefore, the handful of examples reported successfully forged the C(sp2)–F bond mainly via high-valent metal centers or crafted ligands.
Early mechanistic studies by Grushin and Yandulov on late-transition-metal fluorides identified the main challenges to promote reductive elimination from Pd(II) centers.9 These seminal studies served as the stepping stone for the development of a groundbreaking nucleophilic fluorination process based on the Pd(0)/Pd(II) redox couple by Buchwald.10 In order to understand the challenges associated with the C(sp2)–F bond-forming step, the isolation and study of high-valent intermediates has been shown to serve as a valid strategy. In this context, Ritter reported the C(sp2)–F reductive elimination from well-defined σ-aryl Pd(IV)–F species.11 This work, together with subsequent studies from Sanford,12 revealed key structural features to guide the development of Pd-based fluorination methods, including relevant works on C(sp2)–H and C(sp2)–B functionalization.13 In addition to Pd, mechanistic investigations of σ-aryl Pt(IV)–F compounds by Gagné,14 Vigalok and Vedernikov,15 and Haghighi16 identified pathways to achieve smooth C(sp2)–F formation, overcoming unproductive side reactions. With the focus on more earth-abundant elements, aryl–F reductive elimination from Ni centers has been recently established. Ritter17 and Sanford,18 who independently identified C(sp2)–F reductive elimination pathways occurring from σ-aryl Ni(III)–F and Ni(IV)–F species, respectively, showed that aryl fluoride formation is feasible from different oxidation states. Along with group 10 metals, coinage metals have also shown promising results for both nucleophilic and electrophilic fluorination.19 Mechanistic studies on σ-aryl Cu–F complexes by Ribas demonstrated the intermediacy of Cu(III)–F species, which were further suggested by Hartwig.19a,19d Silver has also received significant attention in electrophilic fluorination.20 Clues about its mode of action where reported by Ribas, who described σ-aryl Ag(III) species endowed with the ability to engage in C(sp2)–F bond-forming events via a putative aryl–Ag(III)–F intermediate.20f Nevertheless, bimetallic Ag(II)–Ag(II) species have also been proposed to mediate aryl–F bond formation via one-electron participation of two Ag atoms, both in stoichiometric and catalytic fashion.20b,20c
Beyond the need to find alternative solutions to the imminent threat posed by the availability of noble metals, it is desirable to explore unchartered territories beyond the d-block to seek new reactivity. Accordingly, certain main group elements have recently been identified as potential candidates to mimic organometallic transformations.21 In the context of aryl–F bond formation, however, only a privileged selection has been demonstrated to satisfactorily forge C–F bonds. In the early 1980s, Van Der Puy unveiled σ-aryl hypervalent iodine(III) compounds as powerful reagents to readily obtain fluoroarenes,22 and subsequent mechanistic studies were key to introduce [18F]Fluoride into organic molecules.23 Recent examples of aryl–F bond formation have also been reported among chalcogens by the use of sulfonium salts,24 which are proposed to undergo reductive elimination from hypervalent sulfurane intermediates.25 Thermal decomposition of certain organolead26 and organothallium27 compounds has also been shown to forge the corresponding C–F bond. Comparatively, a heavy element that received much less attention is bismuth (Bi).28 While simple halogen-containing Ph3Bi(V)X2 (X = Cl, Br, I) compounds can thermally decompose to forge aryl–X bonds, analogous studies using F as anion resulted in traces of Ar–F.29 In an isolated example, Akiba reported that aryl–F bond formation is feasible from octahedral Bi(V) difluorides;30 yet, no additional information on this particular step was reported. Inspired by these promising precedents, together with the well-known benign properties associated with Bi,31 we started a research program capitalizing on the organometallic properties of high- and low-valent Bi complexes, both in redox and nonredox catalysis.32 Inspired by the sulfone-based bismacyclic scaffolds by Suzuki,33 our group reported sulfoximine-based Bi compounds capable of forging C(sp2)–F bond formation.32b Specifically, we provided conditions for the stoichiometric and catalytic oxidative fluorination of arylboronic acid derivatives in a redox process. In the former, aryl fluoride is formed upon oxidation of 1 with XeF2 and subsequent thermal decomposition at 90 °C (Figure 1). In the latter, 1-fluoro-2,6-dichloropyridinium tetrafluoroborate (2) acts as the sole oxidant to access a Bi(V) intermediate, which rapidly delivers fluorobenzene (3). Preliminary stoichiometric investigations led us to propose cationic σ-aryl Bi(V)–F intermediates; however, the genuine structure of the species promoting reductive elimination, together with the effect of exogenous additives such as fluoride, remained mysterious and needed further evaluation.
Figure 1.

Fluorination protocol from 1 via an oxidation/C(sp2)–F bond formation sequence.
Herein, we report a mechanistic study aimed at providing a detailed analysis of the aryl–F reductive elimination event from σ-aryl Bi(V) fluoride species. To do so, we assess the role of electronic and geometric perturbations on the ligand scaffold in 1, as well as on the pendant aryl moiety, with the aim of identifying the steric and electronic factors that govern the aryl–F bond formation. Theoretical investigations, kinetic studies, and an in-depth scrutiny of solvent effects and additives allowed us to fully identify the species involved in the aryl–F bond-forming event from neutral and cationic complexes. The outcome of this analysis led us to design a second generation of bismuth complexes that permit both stoichiometric and catalytic aryl–F bond formation with a wider substrate scope and milder reaction conditions.
Results
Solid-State Analysis of Bi(V) Difluoride 4
At the onset of our investigations, we focused on the structural characterization of pentavalent σ-aryl Bi(V) difluoride 4 (Figure 2), which served as a model complex during this study. When 1 is oxidized with 1.0 equiv of XeF2 in CHCl3 at 0 °C, a white solid corresponding to 4 is obtained after evaporation of the volatiles. Cooling a concentrated solution in MeCN at 4 °C led to suitable single-crystals to be analyzed by X-ray diffraction (XRD). As shown in Figure 2, 4 presents a quasi-symmetric dimeric structure with both Bi centers in oxidation state +5. Each Bi atom in 4 adopts a distorted octahedral geometry, with two fluorine atoms positioned trans to each other (F1–Bi1–F2, 157.40(13) ° and F3–Bi2–F4, 157.32(13) °). Interestingly, the pendant phenyl substituents are located syn to each other in close proximity (centroid–centroid distance of 3.766 Å), suggesting π–π attractive interactions. Additionally, one of the F atoms is shared with the other monomer, thus forming a four-membered ring with a μ-difluoro diamond-like core. The shared F atoms do not have equal distances to both Bi atoms (Bi1–F3, 2.585(3) Å and Bi2–F2, 2.582(3) Å are much longer compared to Bi1–F2, 2.185(3) Å and Bi2–F3, 2.178(3) Å). Importantly, the N atoms of the NCF3 moiety are in close proximity to the Bi centers (Bi1–N1, 3.535(6) Å and Bi2–N2, 3.665(4) Å). Overall, the solid-state structure of 4 resembles the dimer previously reported featuring a SO2 motif in place of the S(O)NCF3 in the ligand backbone;32b yet, the distance between Bi–N is ca. 0.3 Å longer than Bi-O distance of the sulfone.
Figure 2.

Synthesis of pentavalent complex 4 and XRD structure analysis. Hydrogen atoms, disordered parts, and solvent molecules omitted for clarity.
Solution-State Analysis of Bi(V) Difluoride 4
In our previous study, 4 was postulated to be a monomer in solution. To obtain more information, variable-temperature (VT) NMR was performed in CD2Cl2 (Figure 3A). At 298 K, 1H NMR measurements reveal 4 as a symmetric compound: both aryl groups of the sulfoximine are equivalent.34 Cooling down the sample to 183 K results in a significant broadening of all signals, which complicated interpretation. Similar results were obtained using other solvents, such as CD3CN at 233 K.34 The 19F NMR spectrum at 298 K (Figure 3A) showed a broad singlet at −112 ppm corresponding to the Bi–F unit. Interestingly, cooling the solution to 183 K caused the appearance of several broad signals at the region of δ = −70 to – 140 ppm as well as a new poorly defined peak around δ = −42.5 ppm, the region corresponding to the NCF3 unit. In order to discard the possibility of solubility issues when cooling down a solution of 4, an analogous Bi(V) compound bearing tBu groups in the ligand scaffold (5) was synthesized and analyzed by VT-NMR.34 Indeed a parallel behavior was observed for 5, which at 183 K also shows multiple species in solution. In addition, dilution experiments of 4 and 5 show peak broadening and movement at room temperature, pointing towards aggregation in solution. Additionally, 19F–19F COSY and EXSY NMR experiments of a solution of 4 in CD2Cl2 at 183 K (Figure 3B) unambiguously point at a chemical exchange between all F atoms (except CF3). These results manifest a complex dynamic behavior, showing a variety of species in solution undergoing rapid F exchange even at very low temperatures. Unfortunately, characterization of complex 4 at low temperature proved extremely difficult due to broad bands, partial precipitation, and low concentration of different species. However, warming the mixture to 298 K allowed the measurement of 1H–19F through-space interactions via HOESY NMR (Figure 3C). 1H–19F contacts between the NCF3 moiety with the ortho-H in the pendant aryl (H(a), Figure 3C) and the meta-H in the ligand scaffold (in respect to Bi, H(b), Figure 3C) were observed. This latter result is consistent with a dimeric species in solution such as the crystal structure of 4 in Figure 2. Yet, a monomeric cis-difluoride complex (int-cis) is predicted to possess similar spectroscopic features. As a result of such fast dynamic configurational processes, dimeric and monomeric species are proposed to coexist at higher temperatures, averaging the signals in 1H and 19F NMR and subsequently posing a severe challenge to identify the species responsible for aryl–F bond formation.
Figure 3.
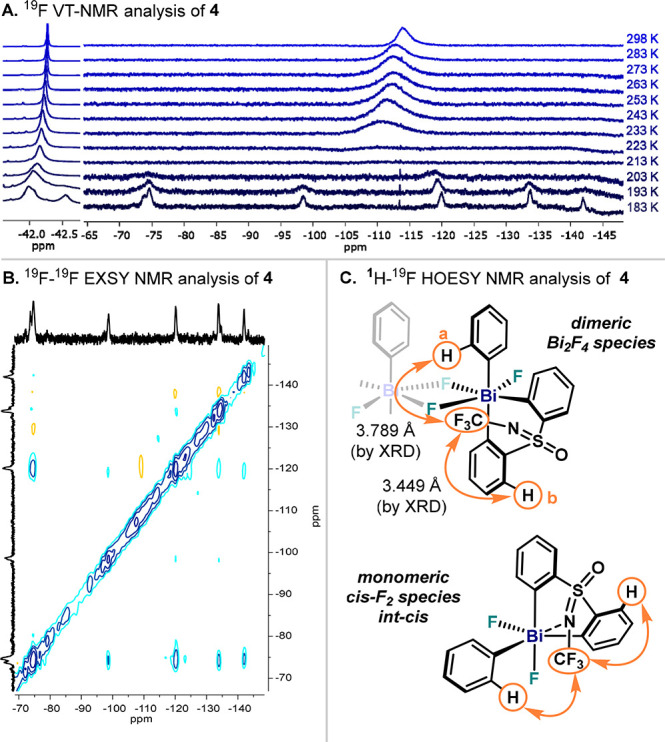
(A) VT 19F NMR measurements of 4 in CD2Cl2. (B) 19F–19F EXSY NMR spectrum at 183 K in CD2Cl2 showing chemical exchange. (C) 1H–19F HOESY cross peaks of 4 in CD2Cl2 at 298 K.
Reductive Elimination from Pentavalent Bi(V) Difluoride 4
First, thermal decomposition at 70 °C of 4 was attempted in solvents with diverse dielectric constants (ε), and reactions were monitored by 19F and 1H NMR spectroscopy. Interestingly, similar rates were observed in CDCl3 (ε = 4.8, kobs = 3.09 ± 0.02 × 10–5 s–1), CD2Cl2 (ε = 8.9, kobs = 3.05 ± 0.05 × 10–5 s–1), and CD3CN (ε = 37.5, kobs = 3.33 ± 0.02 × 10–5 s–1, pointing to a nonionic pathway. Further mechanistic information was obtained from the study of thermal decay from species 4, which was previously shown to follow first order kinetics.32b This result was further validated when reactions over a range of concentrations showed an unchanged rate constant (kobs ≈ 1.3 × 10–4 s–1, Figure 4A), indicating a unimolecular phenyl–F bond-forming event that is first order in 4. Collectively, these data suggest that C–F bond formation proceeds from 4, after rapid pre-equilibrium with monomeric species (cis and trans). Further information was obtained when the reductive elimination was monitored by 19F NMR (Figure 4B). Strikingly, the broad singlet at δ = −118 ppm corresponding to the Bi(V)–F2 unit in 4 did not fade away simultaneously with the appearance of a peak at δ = −182 ppm, which corresponds to Bi(III)–F byproduct 6. Instead, the Bi(V)–F2 NMR signal gradually shifts toward the Bi(III)–F unit (6), indicating a fast exchange between fluorides from Bi(V) and Bi(III) species. Similar results were obtained when mixtures of 4 and 6 were analyzed by NMR, showing unchanged 1H NMR spectra but different 19F signals depending on the concentration of the components.34 Thus, reductive elimination from dimeric species 4 produces fluorobenzene together with the corresponding Bi(III)–F complex 6 and a monomeric Bi(V) species (Figure 4C). These compounds, which are released in close proximity, presumably exchange fluoride ligands in a mixed Bi(V)–Bi(III) complex such as 7. This mixed-valence bimetallic compound is proposed to undergo complex downstream equilibria that will eventually lead back to 4.
Figure 4.

(A) Left, reaction profile of reductive elimination from 4 over a range of concentrations. Right, plot of kobs vs [4]0. (B) 19F NMR reaction monitoring of reductive elimination from 4 (red) with 1-fluoro-4-nitrobenzene (purple) as internal standard, showing formation of fluorobenzene (3, green) and 6 (blue). t = time. (C) Putative mixed-valent Bi(V)–Bi(III) species after C–F formation from 4.
Effect of Substitution on the Pendant Aryl Ring in the C–F Bond Formation from 4
In our previous study, we showed that the presence of electron-withdrawing groups at the para-position of the σ-aryl Bi(V) difluorides accelerates C–F bond formation.32b This tendency was explored further by including additional para-substituted complexes (Figure 5). Deviation of linearity (R2 = 0.88) with standard σp parameters is mainly caused by p-OMe substituted complex 10 (Figure 5A), indicating the great influence of strong π-donating groups. As shown in Figure 5B, better linearity (R2 = 0.97) is obtained when including resonance effects using σp+ values. These results indicate that there exists a buildup of negative charge around the Cipso atom in the transition state (TS) compared to the ground state, consistent with the nucleophilic attack by a fluoride in the rate-determining step. These data can be interpreted as the Cipso acting as an electrophile in the TS. When electronic effects at the meta position were evaluated, linearity became nonobvious when analyzed with various methods and using several Hammett parameters.34 However, a trend could be observed when comparing sterical bulkiness: Large substituents accelerate the reductive elimination. However, a model that accommodates all the observations could not be established and is currently under investigation.
Figure 5.

Electronic analysis of reductive elimination from 4–13. (A) Hammett plot vs σp.values. (B) Hammett plot vs σp+ values.
It is important to mention that Bi(V) difluoride compounds bearing an ortho substituent were shown to undergo extremely fast reductive elimination. For example, a Bi(V) complex with a pending o-tolyl group (20) underwent fluorination ca. 22 times faster than model complex 4.34 Larger groups at ortho-position such as −Et (21) or −iPr (22) resulted in instantaneous reductive elimination at 90 °C, and formation of the corresponding fluoroarene was observed even at 25 °C. Although 20–22 could be characterized at low temperature by NMR, structural information through XRD or solution-state NMR was prevented by their intrinsic high instability. However, we believe that in 20–22, the Bi–Caryl distance in pentavalent Bi species bearing ortho substitution becomes larger compared to 4, thus leading to a more electrophilic C center, a weaker C–Bi bond and a subsequently faster reductive elimination.
Solid-State Analysis of Sterically Hindered Bi(V) Difluorides 25 and 26
After an exhaustive assessment of several parameters influencing reductive elimination from model species 4, our efforts focused on the study of σ-aryl Bi(V) complexes presenting steric congestion on the ligand backbone. As we demonstrated earlier,32b the introduction of Me groups at the ortho position with respect to the Bi center allowed the synthesis of distorted trigonal bipyramidal (TBP) monomeric Bi(V) difluoride complex 25 upon oxidation of 23 with XeF2 at 0 °C, which was possible to characterize by XRD after it was crystallized from CHCl3/pentane mixture (Figure 6A, left). To provide additional evidence on the influence of the Me groups in the structure of the Bi(V) center, complex 26 bearing two additional Me groups was also synthesized and characterized by XRD (Figure 6A, right). This complex also presents a distorted trigonal bipyramidal geometry with similar structural features to 25; in this case, however, the Bi center is flanked by two Me groups in both sides of the sulfoximine ligand.
Figure 6.

(A) Synthesis of monomeric pentavalent bismine fluoride complex 25 and 26 and XRD structure analysis. Hydrogen atoms and solvent molecules omitted for clarity. (B) VT 19F NMR measurements of 26 in CD2Cl2. (C) 19F–19F and 13C–19F J-coupling constants of 26 in CD2Cl2 and CDCl3 respectively, together with JFF constants at different temperatures. (D) 1H–19F HOESY measurements of 26 in CD2Cl2. For simplicity, orange and blue arrows are not used to show all C–F and H–F interactions, but only to represent the distinct ones.
Solution-State Analysis of Sterically Hindered Bi(V) Difluorides 25 and 26
Due to its highly symmetric structure and simplified spectroscopic features, complex 26 was analyzed in solution. VT 19F NMR of 26 in CD2Cl2 revealed a broad singlet with a chemical shift of δ = −120.1 ppm at 343 K, corresponding to the Bi–F2 unit. However, measurements at 183 K resulted in a separate set of two doublets with a chemical shift of δ = −125.6 ppm and a JFF = 114.4 Hz, with a coalescence temperature of Tc = 323 K (Figure 6B). The appearance of these doublets with a JFF = 114.4 Hz indicates two nonequivalent fluoride ligands, indicating a trans-difluoride Bi(V) configuration in solution. Indeed, comparable chemical shift values and coupling constants were described for previously reported triaryl-Sb(V) and triaryl-Bi(V) trans-difluoride complexes.30,35 Analogous splitting was observed for the previously reported complex 25, with a JFF = 115.1 Hz; in this case, the coalescence temperature of the doublets was significantly lower, with a value of Tc = 233 K.34 The reduced JFF value of 26 at higher temperatures (JFF = 112.1 Hz at 253 K; JFF = 102.1 Hz at 273 K; JFF = 83.5 Hz at 283 K) suggests a higher contribution of the cis-conformer to the NMR signal, where the time-averaged F–F angle is reduced. The coalescence of the Bi–F signals at temperatures above 323 K is a result of a chemical exchange, probably due to fast F–F interconversion through rotation processes such as Berry pseudorotation and turnstile rotation with the intermediacy of cis-difluoride species.36 Analysis by 19F–19F EXSY NMR of 26 in CD2Cl2 showed significant exchange between both F atoms even at 223 K (Figure 6C), similarly to complex 4. Further confirmation of the trans-difluoride disposition was obtained by 1H–19F HOESY measurements in CD2Cl2 at 223 K (Figure 6D), which showed through-space H–F contacts consistent with this configuration in solution.34 Interestingly, dilution experiments of 25 and 26 showed no significant change in chemical shifts and peak broadening at room temperature, suggesting no aggregation in solution. Altogether, these results indicate that complexes 25 and 26 preserve the TBP geometry in solution with a trans-difluoride configuration. Installation of steric hindrance in the ligand certainly avoids dimerization and favors trans-difluoride monomers; yet fast F–F exchange still occurs even at low temperatures, thus highlighting the talent of pentavalent Bi complexes to undergo a collection of dynamic processes.
Reductive Elimination from Sterically Hindered Pentavalent Bi(V) Difluorides 25 and 26
Complexes 25 and 26 were also subjected to thermal decomposition at 90 °C in CDCl3, and their kinetic profiles were measured (Figure 7). C(sp2)–F bond formation from 25 resulted in nearly identical kinetic profiles compared to 4 (kobs = 1.54 ± 0.02 × 10–4 s–1), while sterically more crowded complex 26 showed a slower decay (kobs = 3.81 ± 0.02 × 10–5 s–1) and formation of fluorobenzene (kobs = 2.93 ± 0.02 × 10–5 s–1).34 Contrarily to 4, 19F NMR revealed a clean conversion of 25 and 26 to the corresponding Bi(III)–F (27 and 28, respectively) without a gradual shift of the Bi–F signal.34 This behavior suggests no F–F exchange processes during aryl–F reductive elimination, presumably proceeding from the monomeric Bi(V).
Figure 7.
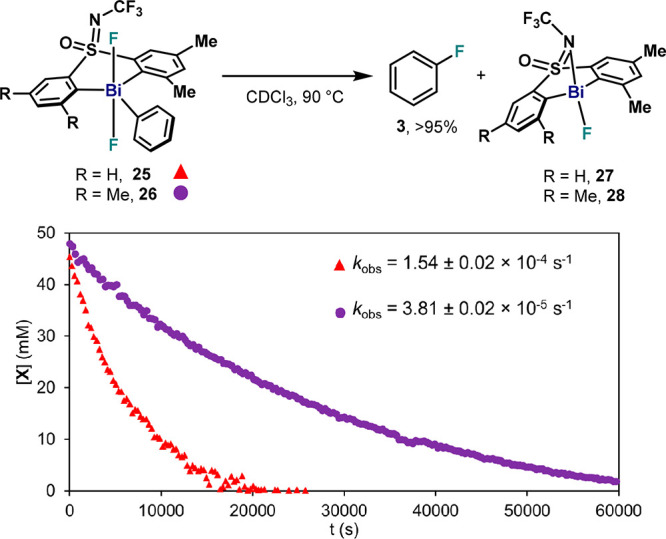
Reaction profile of reductive elimination from 25 (red) and 26 (purple) measured by 19F NMR with 1-fluoro-4-nitrobenzene as internal standard.
Effect of the Substitution on the Pendant Aryl Ring in the C(sp2)– – F Bond Formation from 25
Electronic modulation of the pendant aryl ring was also assessed in monomeric Bi(V) difluoride complexes. Due to synthetic simplicity, we focused on the reductive elimination from 25, containing a sole ortho-Me in the ligand scaffold. Thus, we synthesized several para-substituted σ-aryl Bi(V) difluoride complexes (29–31, Figure 8), and their thermal decomposition was evaluated in CDCl3 at 90 °C.34 Similarly to model complex 4, the Hammett plot using σp parameters resulted in poor linearity (R2 = 0.77); yet, when plotting log(kX/kH) vs σp+, a R2 = 0.9545 was obtained (Figure 8A). The ρ = 1.15 indicates a faster reductive elimination when electron-withdrawing groups (EWG) are present in the pendant aryl ring. In addition, this reaction presents higher sensitivity to para substitution compared to model complex 4 (ρ = 0.43). Evaluation of the thermodynamic parameters through Eyring analysis of 25 in CDCl3 (Figure 8B) revealed a ΔH⧧ = 25.7 ± 1.6 kcal·mol–1 and a ΔS⧧ = −5.7 ± 4.4 cal·mol–1·K–1, similar to values obtained for sterically crowded 26 (ΔH⧧ = 26.5 ± 1.5 kcal·mol–1 and ΔS⧧ = −6.1 ± 4.2 cal·mol–1·K–1).34 The rather small values on the entropic contribution are in stark contrast with that obtained for model complex 4 (ΔS⧧ = −34.7 ± 1.9 cal·mol–1·K–1). This latter value, combined with the structural analysis of 4, suggests that dimerization processes prior to C(sp2)–F bond formation could play an important role in the reductive elimination from model complex 4. Large entropic contributions have also been observed in other dimerization equilibrium in Bi(II) species.37 Hence, the small ΔS⧧ obtained for sterically congested 25 and 26 points to a reductive elimination from monomeric species, without prior dimerization.
Figure 8.

Electronic and thermodynamic analysis of reductive elimination from 25 and 29–31. (A) Hammett plot for the reductive elimination of para-substituted σ-aryl-Bi(V) difluorides 25 and 29–31. (B) Eyring analysis for 25.
Effect of the Substitution on the Sulfoximine Scaffold in the C–F Reductive Elimination
Various σ-aryl Bi(V) difluoride complexes bearing ligands with substituents in meta-position with respect to the Bi center (4, 5, 32–34, Figure 9) were thermally decomposed at 90 °C, and the decay was monitored by 19F NMR together with formation of 3 and the corresponding Bi(III)–F. Arguably, a study employing ligands with para-substituents to the Bi center would have been more appropriate. However, due to synthetic limitations on the synthesis of the parent Bi(III)–Ph complexes, symmetric diphenyl sulfoximine scaffolds were utilized. As shown in Figure 9A, a Hammett analysis resulted in a value of ρ = 1.72 ± 0.03 when σm was used in the x-axis, excluding complex 33 bearing −OMe moieties, which followed a differing trend. Introducing resonance effects via the Swain–Lupton equation,38 a similar slope of ρ = 1.47 ± 0.08 was obtained (Figure 9B), now including 33.34 These results indicate an important role of resonance contributions from strong π-donor substituents as well as a faster reductive elimination with ligands bearing m-EWG. We hypothesize that the Bi center is mainly affected by field, while the S(O)NCF3 unit is strongly affected by resonance, and for these reasons, pure σm and σp values could not be used.
Figure 9.

Electronic analysis of reductive elimination from 4, 5, and 32–34. (A) Hammett plot vs σm values. (B) Hammett plot vs σS–L values.
Evaluation of Fluoride Inhibition in the C(sp2)–F Bond Formation from 4
In our previous study, we noted that formation of fluorobenzene was prevented when 1.0 equiv of tetrabutylammonium fluoride (TBAF) was added, which led us to propose a cationic intermediate in equilibrium with neutral pentavalent Bi(V) species 4.32b Together with a slower rate, we also observed significant shifts in 1H NMR and partial decomposition of the initial Bi(V) difluoride, which we attributed to possible interactions with THF or even H2O, which is present in 5 wt % in commercial 1.0 M TBAF solutions. With the goal of elucidating the effect of fluoride anions in the aryl–F reductive elimination step, fluoride sources that present high solubility in common organic solvents were selected and mixed with complex 4 (Figure 10A). Addition of 1.0 equiv of tetrabutylammonium difluorotriphenylsilicate (TBAT) or tris(dimethylamino)sulfonium difluorotrimethylsilicate (TAS-F) to 4 resulted in the smooth formation of a new species together with concomitant formation of the corresponding R3Si–F species. These results encouraged us to re-evaluate our previous experiment using 1.0 equiv of TBAF from a 1.0 M solution in THF; indeed, the same species obtained with TBAT and TAS-F were observed. These experiments collectively suggest the formation of an anionic Bi(V) compound consisting of three fluorine atoms directly bound to the Bi center. Indeed, HRMS analysis of these samples confirmed the presence of anionic species 35 (experimental m/z = 626.0438; theoretical m/z = 626.0431). Although it was not possible to obtain a suitable single-crystal for XRD analysis, 35 (from reaction with TAS-F) was fully characterized spectroscopically by NMR in CD2Cl2.34 Initial proof of the presence of three F atoms directly bonded to the Bi center was collected measuring 13C NMR at 223 K, which revealed two reasonably resolved Bi–13C signals as quadruplets with a coupling constant of JCF ≈ 20 Hz, which compares to the values obtained for monomeric complex 26. This result points to three equivalent fluorine atoms directly bound to the Bi center experiencing fast dynamic processes. VT 19F NMR provided additional insight on the dynamics of this anionic Bi(V). Whereas at 298 K, a broad Bi–F signal appears at −88.9, at 183 K, the signal splits into three independent signals at −69.9, −96.7, and −107.7 ppm, with a coupling constant within the range of F–Bi(V)–F (JFF ≈ 85 Hz). Noteworthy, 19F–19F COSY measurements confirmed F–Bi–F through-bond interactions,34 while 19F–19F EXSY studies (inset Figure 10A) revealed fast exchange between F atoms even at 183 K. Unfortunately, no 19F–19F through-space coupling was observed between Bi–F and the CF3 moiety, impeding a full assignment of the F signals. However, the presence of three 19F signals, two of them being doublets with JFF = 85 Hz and similar chemical shift, suggests that 35 could adopt a pseudo-octahedral structure such as the one depicted in Figure 10A.
Figure 10.
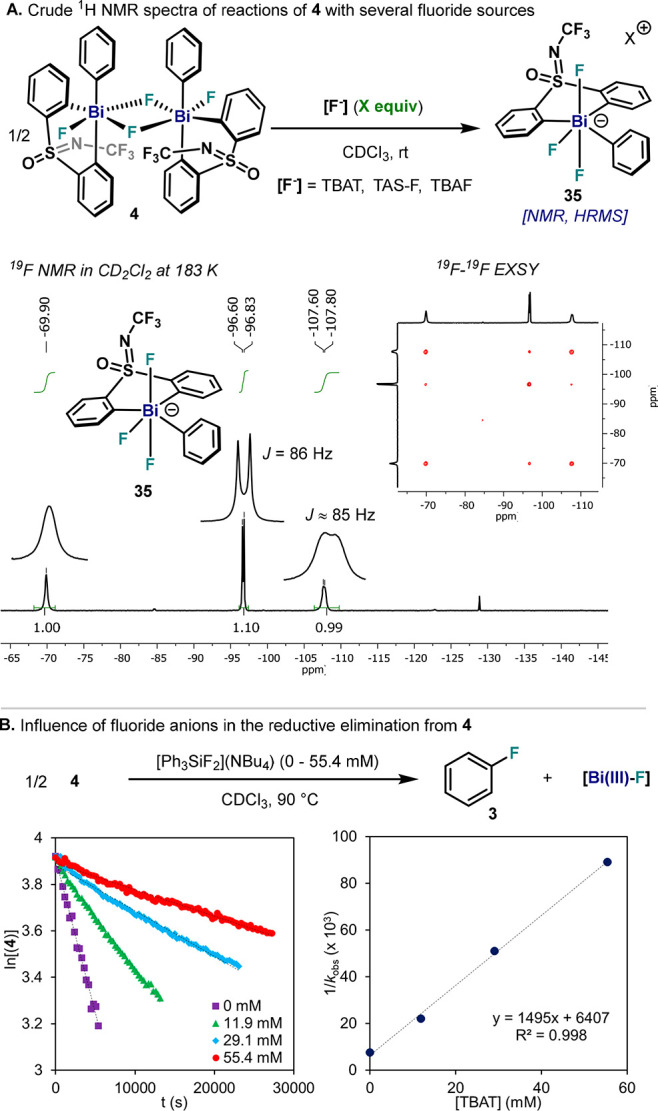
(A) Synthesis of anionic species 35 from 4 in the presence of several fluoride sources. (B) Left, reductive elimination of fluorobenzene from 4 in the presence of different amounts of TBAT. Right, decay of 4 dependence on TBAT concentration.
At this point, 4 was subjected to thermal decomposition in the presence of different amounts of TBAT, and the rate of reductive elimination was monitored. As depicted in Figure 10B (left), slower reaction rates were obtained with higher concentrations of TBAT, which indicate an inverse rate dependence as a function of TBAT concentration. Indeed, a positive, linear dependence of the reciprocal of the rate constant of Bi(V) decay (1/kobs) vs TBAT concentration was observed together with a nonzero intercept (Figure 10B, right). Furthermore, while the yield of fluorination was 94% for 4 without additional fluorides, it decreased to 71% in the presence of 1.1 equiv of TBAT. Altogether, these results unambiguously indicate that the slower rate in the presence of fluoride anions is a consequence of the formation of hexacoordinated anionic species 35. This species is proposed to engage in aryl–F reductive elimination events via neutral pentavalent Bi(V) intermediates, a process that appears to be more feasible compared to the previously proposed cationic species.
Theoretical Analysis of the Reductive Elimination Step from Neutral Bi(V) Difluoride 4
Intrigued by the experimental results obtained with neutral pentavalent σ-aryl Bi(V) difluoride complexes, we performed a collection of DFT calculations to support and fully understand the aryl–F bond-forming step. After a brief method evaluation,34 geometry optimizations and frequency calculations were carried out at the B3LYP-D3BJ level of theory39 with the def2-TZVP(-f) basis sets and matching auxiliary basis set (def2/J).40 The default small-core effective core potential was used for Bi,41 and solvent effects (chloroform) were incorporated using a conductor-like polarizable continuum model. Geometry optimization, normal-mode analysis, and single-point calculations were carried out with a development version of the ORCA 4.2 suite of programs.42 Natural bond orbital (NBO) analysis was performed at the same level of theory.43 Initially, the reductive elimination of fluorobenzene from species 4 was evaluated at 363 K (Figure 11A). Due to the symmetric nature of 4, three different TSs for the reductive elimination from one Bi center (Bi2, see Figure 2) were identified, which involved fluoride ligands in pendant (F4, equivalent to F1) and μ-bridged positions (F3 and F2). Reductive elimination from the pendant fluoride ligand was highly energetic, with an activation barrier of ΔG⧧ = 29.9 kcal·mol–1 (TSF4), similar to the C–F bond formation from the shared fluoride ligand within the μ-bridge (ΔG⧧ = 27.3 kcal·mol–1, TSF2). Interestingly, a value of ΔG⧧ = 25.5 kcal·mol–1 was obtained when reductive elimination was computed from F3, releasing fluorobenzene and leading to int-cis, which is the most stable monomeric 4 isomer (vide infra) together with 4. This value is in agreement with the activation barrier obtained experimentally by Eyring analysis, ΔG⧧ = 25.9 ± 0.9 kcal·mol–1, which suggests the reductive elimination of fluorobenzene could proceed through TSF3. In addition, the monometallic aryl–F bond-forming event described in Figure 11 constitutes a rare example of μ-difluoride-bridged species, leading to fluorobenzene in synthetically relevant yields, as μ-difluoride-bridged dimers in transition-metal chemistry tend to inhibit further reactivity due to their high stability.9e Characterization of structural and electronic parameters of TSF3 through NBO analysis revealed a positive charge on C6 in the TS with a value of qC6 = +0.21 (Figure 11B). Meanwhile, the fluoride anion remained nucleophilic (qF3 = −0.66), in agreement with the Hammett plot obtained with para-substituted 4 and 8–13 (Figure 5). Interestingly, Bi2 presents a smaller positive charge (qBi1 = +1.95) compared to Bi1 (qBi1 = +2.25), which denotes its partial reduction to Bi(III). Furthermore, the Wiberg bond index (WBI) and bond distance analysis clearly show the cleavage of the Bi2–C6 (WBI(Bi2–C6) = 0.49, d = 2.536 Å) and Bi2–F3 (WBI(Bi2–F3) = 0.07, d = 2.784 Å) bonds occurs simultaneously with C6–F3 formation (WBI(C6–F3) = 0.14, d = 2.050 Å), suggesting a concerted reductive elimination of fluorobenzene. It is important to note that dimeric species 4 is calculated to be ≥1.1 kcal·mol–1 more stable than two individual int-cis or int-trans monomers, indicating 4 as the lowest-energy species.34 Nonetheless, the aryl–F bond formation from monomeric species was also evaluated (Figure 11A). Results obtained for the reductive elimination of monomeric int-cis proceed through a concerted mechanism (TSD) and are higher in energy than TSF3 (ΔΔG⧧(TSF3–TSD) = −1.0 kcal·mol–1). In this case, however, additional 1.1 kcal·mol–1 would be required in the TSD to overcome the dissociation of 4, resulting in an overall 27.6 kcal·mol–1. Pathways from int-trans were located >28 kcal·mol–1 and, hence, not considered.34 These results suggest that reductive elimination for model complex 4 takes place preferentially from a bimetallic Bi(V) species in solution, albeit the reductive elimination from int-cis is also feasible. This is consistent with the large negative ΔS⧧ value obtained experimentally for 4, which can be explained by a highly ordered TS in bimetallic TSF3 (Figure 11) or the possible monomer–dimer equilibriums previous to the aryl–F reductive elimination step.
Figure 11.

(A) Gibbs energy profile of the reductive elimination of fluorobenzene from species 4 at 363 K. (B) Selected structural and electronic parameters for TSF3. Relative Gibbs energy values are given in kcal·mol–1.
Theoretical Analysis of the Reductive Elimination Step from Neutral Bi(V) Difluoride 26
As shown in Figure 6, sterically bulky 26 is characterized as a monomer, and no dimers were formed in solution or in the solid state. Yet, C–F bond formation is also possible from this complex, leading to good yields of 3. To investigate the differences between complexes such as 26 and 4, reductive elimination from symmetric monomeric compound 26 was evaluated at 363 K, and its possible pathways for fluorobenzene formation are depicted in Figure 12. Similarly to monomeric configurations of model complex 4 (Figure 11A), trans and cis isomers of 26 were studied. In this case, trans-26 resulted to be more stable than cis-26, which is consistent with the characterization of this compound in solution as well as in solid state (Figure 6). Fluorobenzene from monomeric trans-26 stems from a concerted C–F bond-forming event involving the bottom (TSA′) or the top (TSB′) fluoride ligand, with an activation energy of 34.1 kcal·mol–1 and 29.2 kcal·mol–1, respectively. Equatorial C–F bond formation occurs from cis-26 through a highly energetic TSC′, with a value of ΔG⧧ = 41.2 kcal·mol–1. On the other hand, equatorial C and axial F in TSD′ results in a more favorable pathway (27.3 kcal·mol–1) for C–F bond formation, in agreement with the experimental activation barrier obtained from the Eyring analysis for 26, ΔG⧧ = 28.3 kcal·mol–1 ± 0.9 kcal·mol–1. Results depicted in Figure 12 show the feasibility of the aryl–F bond-forming event for monomeric σ-aryl Bi(V) difluoride species in solution. The activation energy for 26, however, is still higher than that obtained for the lowest-energy pathway for model complex 4, which is consistent with the slower kinetic profile obtained for sterically crowded σ-aryl Bi(V) difluoride 26 (Figure 7).
Figure 12.

Gibbs energy profile of the reductive elimination of fluorobenzene from sterically congested monomeric species 26 at 363 K. Relative Gibbs energy values are given in kcal·mol–1.
Solid-State Analysis of a Fluorobismuthonium Bi(V)–F
The addition of Lewis acids such as BF3 to 4 could lead to the formation of fluorobismuthonium species in solution bearing BF4– as counteranions.32b While the compound 36 obtained from model complex 4 was originally characterized by NMR and HRMS, solid-state characterization was precluded due to the poor thermal stability and high hygroscopic properties of these complexes. Surprisingly, during crystallization attempts of neutral difluoride species 33, we isolated hexafluorosilicate salt 37 instead (Figure 13A). It was speculated that the interaction of difluoride 33 with glass generated SiF4 in situ, leading to fluoride abstraction from the neutral difluoride 33. To reproducibly obtain this species, a mixture of difluoride 33 in dry CH2Cl2 was bubbled with in situ generated SiF4 gas, leading to 37 in modest yields.34 Compound 37 crystallizes as a symmetric salt, with two monocationic Bi moieties sharing one SiF62– anion with a Bi···FSiF5 contact of 2.6607(13) Å (Figure 13B). The SiF62– anion shows elongated Si–F distances (d = 1.7215(13) Å) for the coordinating F atoms compared to noncoordinating Si–F bonds (d = 1.6701(14)–1.6809(14) Å). The cationic Bi fragment in 37 exhibits a distorted TBP geometry, with a single Bi–F bond (d = 2.0414(16) Å) and a short Bi–N interaction (d = 2.836(2) Å). Species 37 shows similar Bi–C1 (d = 2.224(2) Å) and Bi–C2 (d = 2.222(2) Å) distances and a slightly shorter Bi–C3 (d = 2.207(2) Å) bond, slightly bent toward C2 (C3–Bi–C2, 139.03(8) °; C1–Bi–C3, 110.95(8) °).
Figure 13.

(A) Synthesis of fluorobismuthonium species 37 and (B) ORTEP representation of XRD structure of 37. Hydrogen atoms and solvent molecules omitted for clarity.
Effect of the Substitution on the Sulfoximine Scaffold in the C–F Reductive Elimination from Fluorobismuthonium Bi(V)–F
Intriguingly, reductive elimination from 37 did not occur at 25 °C in CDCl3, and it was sluggish at 60 °C (31% of 3). The slow reactivity for cation 37 was ascribed to the strong electron-releasing properties of −OMe groups in the ligand scaffold and prompted us to conduct an assessment on the effects of the substituents in the ligand on the reductive elimination. To this end, a variety of complexes (4, 5, and 32–34) were thermally decomposed at 25 °C in the presence of BF3 and the kinetics monitored by NMR (Figure 14). As shown in Figure 14A, a Hammett analysis of the reaction kinetics resulted in a value of ρ = 3.84 ± 0.78 when σm was plotted in the x-axis, excluding complex 33 bearing m-OMe moieties. Introducing resonance effects via the Swain–Lupton equation (Figure 14B),38 a similar ρ value was obtained (ρ = 3.43 ± 0.31) with improved linearity (R2 = 0.9756), now including −OMe groups (33). The trend obtained for this Hammett plot is similar to the results obtained for neutral σ-aryl Bi(V) difluorides (Figure 9), highlighting the increased nucleofuge character of the Bi center when EWGs are installed in the ligand backbone. Interestingly, thermal decomposition of 33 bearing m-OMe substituents produces a significant 92% yield of 3. This result is in stark contrast with species 37 bearing a SiF62– counteranion, which produced fluorobenzene in <5% yield at 298 K after 48 h. Indeed, high yields of fluorobenzene when mixing species 33 with BF3 suggest a possible involvement of the BF4 anion in the C–F bond-forming step. Use of other Lewis acids to abstract a fluoride ligand, such as B(C5H6)3 and SbF5, resulted in similar outcomes to SiF4,34 presenting slower reaction rates or decomposition of starting material.
Figure 14.

Electronic analysis of reductive elimination from 4, 5, and 32–34 in the presence of 1.0 equiv of BF3·OEt2. (A) Hammett plot vs σm values. (B) Hammett plot vs σS–L values.
Effect of Substitution on the Pendant Aryl Ring in the C(sp2)–F Bond Formation from Fluorobismuthonium Bi(V)–F
p-Substituted aryl Bi(V) difluorides were dissolved in CDCl3 at 25 °C together with 1.0 equiv of BF3·OEt2, and the decay of the in situ generated cationic complex was monitored by NMR spectroscopy (Figure 15). Despite the poor linearity observed in Figure 15A, a positive slope (ρ = 6.31 ± 2.96) was obtained for 4 and 8–12 (R2 = 0.695). An increased correlation (R2 = 0.976) was obtained when Hammett analysis was done using σp+ values obtaining a ρ = 2.64 ± 0.29 (Figure 15B). Interestingly, this value corresponds to a 5-fold increase compared to neutral difluorides (Figure 5), pointing to a much larger change in electron density in the TS for these fluorobismuthonium species. Strikingly, no reaction was observed with p-EWG (p-CF3, 11 and p-Cl, 12). Although formation of fluorobismuthonium complexes was confirmed by NMR and HRMS studies, fluoroarenes 17 and 18 were only obtained after warming the reaction mixture to 90 °C over 2 h, leading to large amounts of decomposition.34 This result is in bold contrast to the Hammett plot for neutral difluorides, which showed rapid reaction kinetics in the presence of p-CF3 or p-CN. We speculated that the complete inhibition of reductive elimination from fluorobismuthonium species bearing electron-deficient arenes could be connected to the need of elongation of the Bi–Cipso bond, thus becoming a highly energetic rate-determining step prior to the nucleophilic attack of the BF4– anion in the TS.34 In the Hammett plot, this change in the rate-determining step would be represented in a very sharp break with a large negative ρ value for EWGs.
Figure 15.

Electronic analysis of reductive elimination from 4 and 8–12 in the presence of 1.0 equiv of BF3·OEt2. (A) Hammett plot vs σp. (B) Hammett plot vs σp+ taking into account resonance contributions.
Theoretical Analysis of the Reductive Elimination Step from Fluorobismuthonium 36
XRD studies of 37 and previously conducted NMR studies of 36(32b) point to species cis-36 being the most stable isomer; hence, different C–F bond formation pathways from cis-36 were evaluated at 298 K (Figure 16A). Although reductive elimination pathways from thermodynamically less stable cis2–36 species (ΔG = 9.6 kcal·mol–1) were also studied, energetic barriers resulted in prohibitive values (ΔG(TSA′′)⧧ = 46.1 kcal mol–1, ΔG(TSB′′)⧧ = 42.3 kcal·mol–1, and ΔG(TSC′′)⧧ = 32.8 kcal·mol–1). Therefore, reductive elimination from the most stable isomer, cis-36, was studied in more detail. Three pathways for C–F bond formation with significantly distinct energy barriers were identified. On one hand, the direct C–F bond formation through a three-membered TS involving the Bi–F bond resulted in a barrier of ΔG(TSD′′)⧧ = 25.8 kcal·mol–1. On the other hand, lower values of ΔG⧧ were obtained when BF4 was used as the fluoride source. Indeed, a three-membered TS involving B–F cleavage delivered an activation energy of ΔG(TSE′′)⧧ = 24.3 kcal·mol–1, while a five-membered TS resulted in the energetically lowest pathway in Figure 16A, with a theoretical value of ΔG(TSF′′)⧧ = 22.8 kcal·mol–1, which is in agreement with the experimental value obtained (ΔG⧧ = 22.4 ± 2.2 kcal·mol–1).32b Structural and electronic analyses of TSF′′ by NBO analysis show a dramatic buildup of positive charge at C3 during the TS, with a value of qC3 = +0.38, which represents a 2-fold increase compared to neutral difluorides (see Figure 16B). The fluoride F2 in the BF4 unit remains nucleophilic (qF2 = −0.49), and the Bi center presents a smaller positive charge Bi (qBi = +1.79) compared to the neutral difluoride TSs previously analyzed. These results, together with the Hammett plot presented in Figure 15, suggest that the Bi center in TSF′′ presents more Bi(III) character, and it can be regarded as a highly polarized, late TS. Indeed, the WBI and bond distance analysis clearly show an almost cleaved Bi–C3 bond (WBI(Bi–C3) = 0.30, d = 2.825 Å) together with a partially formed C3–F2 bond (WBI(C3–F2) = 0.12, d = 2.134 Å). This concerted-asynchronous ligand coupling event involves an initial elongation of the Bi–C3 bond, leading to a highly polarized TS for the final C3–F2 bond formation. The required elongation of the Bi–C3 in TSF′′ results in an energetic penalty in the TS for fluorobismuthonium complexes with pendant aryl moieties bearing para-EWG, consistent with the absence of C–F bond formation from 11 and 12 at 25 °C.34 Indeed, activation barriers following TSF′′ for fluorobismuthonium derivative of 11 resulted in a ΔG⧧ = 25.3 kcal·mol–1, significantly higher compared to 36.34
Figure 16.

(A) Gibbs energy profile of the reductive elimination of fluorobenzene from fluorobismuthonium species 36 at 298 K. (B) Selected structural and electronic parameters for TSF′′. Relative Gibbs energy values are given in kcal·mol–1.
Collectively, these results point to a preferred five-membered TS using BF4– as a fluoride source, similar to previous reports with heavy main group metals such as Pb and Tl26,27 or recent examples using Bi and OTf, ONf32c or phenols44 as ligands. Collectively, the data presented herein show that aryl–F bond formation from fluorobismuthonium σ-aryl Bi(V) fluoride species 36 proceeds through a different mechanism when compared to neutral σ-aryl Bi(V) difluorides.
Identification of Cationic Species in the Oxidation of 1 with 1-Fluoro-2,6-dichloropyridinium Tetrafluoroborate (2)
Due to the faster reaction rates for C–F bond formation from fluorobismuthonium species, 2 has been identified as a suitable electrophilic fluorinating agent for Bi(III). Despite the excellent yield of fluorobenzene after thermal decomposition, evidence of the intermediacy of similar fluorobismuthonium complexes such as 36 was eluded by the poor solubility of 2 salt in CDCl3. Hence, the eaction of 1 with 1.0 equiv of 2 was performed in MeCN-d3 (Figure 17, reaction 1) at 0 °C, and the reaction crude was analyzed by 1H and 19F NMR after 10 min. In parallel, with the aim of furnishing fluorobismuthonium intermediate 36, pentavalent difluoride 4 was reacted with BF3·OEt2 in the presence of 1.0 equiv of 2,6-dichloropyridine in MeCN-d3 (Figure 17, reaction 2). Similar 1H and 19F NMR spectra were obtained in both cases, thus coinciding with the characterization data obtained for cationic species 36 in reaction 2 (Figure 17). Furthermore, HRMS crude analysis of reaction 1 showed a peak with m/z = 588.0456 corresponding to the [36-BF4]+ ion (theoretical m/z = 588.0456).34
Figure 17.
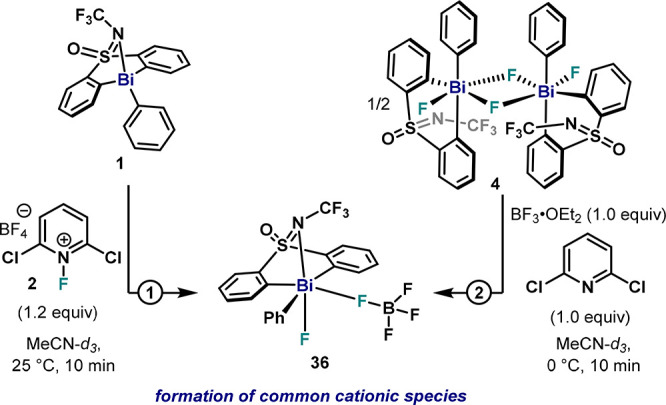
Reactivity of 1 with 1-fluoro-2,6-dichloropyridinium tetrafluoroborate 2 in MeCN-d3 (reaction 1) and 4 with BF3·OEt2 complex in the presence of 2,6-dichloropyridine in MeCN-d3 (reaction 2).
Evaluation of the Ligand Scaffold in the C–F Bond Formation from Bi(III)–Ph and 1-Fluoro-2,6-dichloropyridinium Tetrafluoroborate (2)
In order to gain insight on the features required to promote and inhibit formation of fluorobenzene, oxidation of various sulfone- and sulfoximine-based Bi complexes was examined using 2 (Table 1). Similar to neutral difluorides,32b,34 complexes with sulfone-based backbones without substituents (39) resulted in poor yields of 3. Introduction of EWG in the flanking aryl rings resulted in a dramatic increase of yield (40 and 41), similar to the effect observed for fluorobismuthonium species 34 in Figure 9. Sulfoximine-based complexes varying the substituent on the N atom were also evaluated. Species bearing N–CF3 (1) and N–CF2CF3 (42) groups were demonstrated to be excellent platforms for the synthesis of 3, while the installation of a N–Me group (43) failed to provide the desired product. Complexes bearing N–Ar units (44–46) were also tested, identifying N–Ar moieties with p-EWG as superior ligands. The results obtained in Table 1 are therefore in agreement with all the data collected up to now on the C–F bond formation: Electron-deficient ligand scaffolds promote aryl–F reductive elimination, making the Bi center a better nucleofuge toward the incoming F nucleophile.
Table 1. Substitution Effects on the Diphenyl Sulfone Scaffold and the Sulfoximine Moiety on the Oxidation/Reductive Elimination Sequence from Phenyl Bismine Speciesa.


Yields determined by 19F NMR using 1-fluoro-4-nitrobenzene as internal standard.
Reaction performed using CD3CN as solvent.
Improved Methods for the Fluorination of Boronic Acids
The mechanistic considerations inferred from data presented in Table 1 provide relevant information toward a more practical protocol for fluorination. Indeed, the excellent yield obtained with complex 40 evades the use of compounds bearing −S(O)NCF3– moieties (1), which are synthetically tedious, low yielding, and expensive compared to sulfone-based ligands. Thus, with the aim of developing an improved and easily accessible method, we evaluated compounds 40 and 41, which can be easily synthesized and furnished fluorobenzene in excellent yields. First, we assessed the stoichiometric fluorination of boronic acids through a two-step method involving a transmetalation and a one-pot oxidation/reductive elimination. For the first step, we employed the Bi–OTs complex 47 (Table 2).34 Transmetalation with a variety of arylboronic acids was assessed using Ball’s conditions,44c furnishing a variety of Bi–aryl compounds in excellent yields independently of the substitution pattern of the arylboronic acid (Table 2).34 It is important to note that this transmetalation protocol employs 1.0 equiv of arylboronic acid, while in our previous report, we were restricted to an excess of transmetallating reagent.32b After transmetalation, oxidation of Bi–aryl compounds with 1.0 equiv of 2 in CDCl3 at 90 °C furnished the corresponding arylfluorides. It is worth mentioning that due to decomposition of 2 in the presence of water,45 it was not possible to perform a one-pot reaction without previous isolation of the corresponding Bi–aryl compounds. Interestingly, this system bodes well with a variety of para-substituents (3, 15–17, 48–53), including CF3 (17, 55%), halogens (R = Cl, 18, 51%; R = F, 49, 52%), TMS (50, 93%), and alkynyl (52, 22%) moieties. Despite the broader functional group tolerance, trace amounts of arylfluoride were obtained when ether substituents were evaluated (16 and 53, <5%), highlighting some limitations of the methodology. Alkyl chains (54–56) and silyl groups (57) in meta-position resulted in good yields, including compounds with large substituents (58 and 63), while the installation of meta-EWG (59–62) slightly decreased the efficiency of the oxidation/reductive elimination. The electrophilic fluorination could also be performed on Bi–aryl compounds bearing ortho-substituents, such as Me (64, 92%) and Br (65, 33%) groups, albeit with lower yields for the latter. Furthermore, vinyl groups (66, 63%) and polyaromatic arenes (67, 63%) could also be accommodated. Overall, the Bi-mediated fluorination of arylboronic acids developed herein shows a broader scope and uses a readily available Bi–OTs species (47), in contrast to our previous methodology, which is based on the use of ligand scaffolds incorporating the −S(O)NCF3– unit.
Table 2. Bismuth-Mediated Two-Step Method for the Fluorination of Arylboronic Acids with 47a.
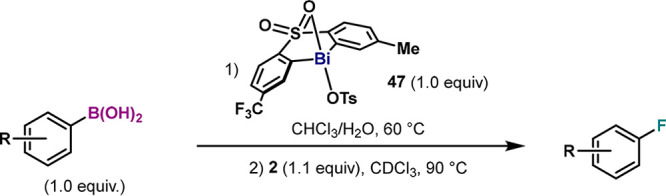

Yields are given for step 1 (isolated) and step 2 (determined by 19F NMR using 1-fluoro-4-nitrobenzene as internal standard).
Step 2 performed in the presence of 5.0 equiv of NaF at 110 °C.
Isolated yield by preparative TLC. Products contain trace amounts (<5%) of protodeboronation byproducts.
After providing an improved stoichiometric method based on complex 47 for the fluorination of arylboronic acids, we focused our attention on transferring the benefits of sulfone-based complexes to high-valent Bi-catalyzed fluorination reactions. A major limitation of our previously reported method was the need of 3.0 equiv of arylboronic ester, making this transformation impractical for valuable substrates. Optimal conditions for catalytic fluorination from aryl boronic esters (76% of 3) were found using sulfone-based catalyst 68 bearing two meta-CF3 groups, in combination with oxidant 2 (1.1 equiv) in CDCl3 (Table 3). Noteworthy, arylboronic esters could be utilized as limiting reagents, and most importantly, the reaction worked smoothly in the absence of base. Indeed, analysis of the reaction crude after fluorobenzene formation revealed the presence of BF3 in solution, as well as in the headspace,34 which suggests that the BF4– anion also acts as a fluoride source during the catalytic transformation. Under the optimized conditions, a variety of para-substituents could be accommodated in good yields, including alkyl groups (14, 85%; 15, 65%), Ph (51, 73%), and TMS (50, 77%). In addition, arylboronic esters bearing para-EWG groups such as halogen atoms required the use of a base (5.0 equiv of NaF) and higher reaction temperatures to yield arylfluorides in moderate yields (18, 69, and 70). Arylboronic esters containing meta-substituents were also tolerated (54, 59, 62, and 71), although strong meta-EWG inhibited the formation of fluoroarenes (61 and 72) even in the presence of NaF. ortho-Substitution was also well accommodated (64 and 73), albeit low yields were obtained with o-Br (65) and o-CO2Me (74) groups. Interestingly, this catalytic system allowed the introduction of several strong para-EWG groups such as CF3 (17, 56%), CO2Me (75, 56%), Br (76, 69%), and SO2Me (77, 35%), which were previously shown to inhibit reactivity when using sulfoximine-based Bi-catalysts.32b In fact, the use of sulfone-based catalyst 68 also boded well with sterically hindered substrates (78, 64%) and alkynyl groups (79, 49%), showing a wide scope and practicality. Although these protocols produce high yields of fluorinated arenes, isolation of these compounds in pure form without traces of Ar–H becomes tedious, requiring HPLC separations which, in some cases, result in low yields (14 and 51, Table 3).
Table 3. Bismuth-Catalyzed Fluorination of Arylboronic Esters with Sulfone-Based Catalyst 68a.
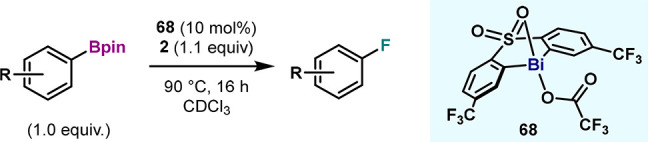
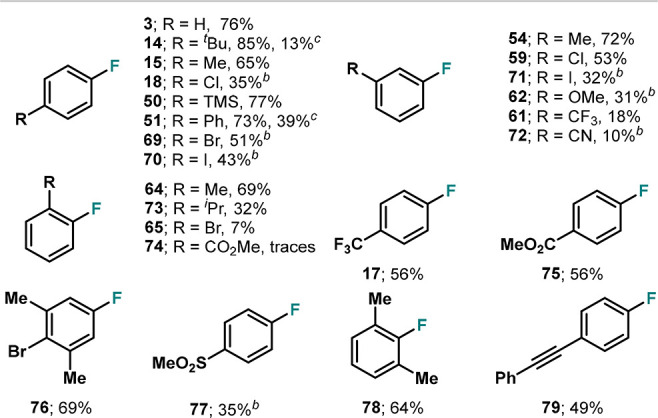
Yields determined by 19F NMR using 1-fluoro-4-nitrobenzene as internal standard.
Reaction performed in the presence of 5.0 equiv of NaF.
Isolated yield by preparative HPLC.
Conclusion
We provide herein a mechanism of the reductive elimination of aryl–F bonds from neutral triarylbismuth difluorides as well as cationic fluorobismuthonium species (Figure 18). Solid-state (XRD) and spectroscopic characterization in solution (1D and 2D NMR) suggests that 4 presents a dimeric structure and undergoes fast dynamic processes in solution. Evaluation of the electronic and steric effects on the pendant aryl ligand revealed that para-EWG enhances the rate of fluorobenzene (3) formation. Installation of Me groups—ortho with respect to the Bi center—in the sulfoximine ligand scaffold permitted the synthesis and characterization of monomeric TBP aryl Bi(V) difluorides 25 and 26. In contrast to model complex 4, these compounds have been characterized as trans-difluoride monomers in solid state and in solution. Evaluation of electronic effects affecting the reductive elimination of fluorobenzene revealed analogous effects on the reactive aryl compared to 4. Yet, the Eyring plot revealed a ΔS⧧ ≈ −6 cal·mol–1 K–1 for monomeric 25 and 26, which is in stark contrast to the ΔS⧧ ≈ −34 cal·mol–1 K–1 for complex 4. Evaluation of the effect of external fluoride anions in the reductive elimination of 4 revealed the formation of anionic species 35, which has a detrimental effect on fluorobenzene formation. Theoretical studies of the C–F bond formation from dimeric and monomeric neutral difluorides showed kinetic barriers in agreement with experimentally determined parameters. Indeed, for species 4, the aryl–F bond formation from neutral Bi(V) difluoride centers is postulated to proceed through a dimeric TS, albeit a reductive elimination event from monomeric species cannot be disregarded.
Figure 18.

Overview of the C(sp2)–F reductive elimination from Bi(V).
Evaluation of the C–F bond formation from cationic fluorobismuthonium species was also assessed. Isolation of hexafluorosilicate compound 37 allowed solid-state characterization of the fluorobismuhtonium species. Electronic modulations on the pendant aryl and the ligand scaffold suggested a highly polarized TS, consistent with the cationic nature of this complex. DFT studies of cationic species 36 unveiled the BF4– anion as the true fluoride source, forging the C–F bond through a five-membered TS. Reaction of 1 with the milder 1-fluoro-2,6-pyridinium tetrafluoroborate (2) also delivered a high-valent Bi(V) species 36, further supporting the involvement of fluorobismuthonium intermediates. With this mechanistic picture, re-evaluation of the ligand features led to the development of improved stoichiometric and catalytic fluorination reactions of arylboronic acid derivatives, using a simpler and easy to handle Bi catalyst. This second-generation fluorination has been successfully applied to >40 substrates, thus improving the yields over our previously reported methodologies. Overall, the detailed mechanistic investigation provided herein enabled the identification of the key parameters and limitations of the C–F bond-forming step from Bi(V) centers, revealing different pathways between neutral and cationic species. Finally, this article illustrates that by means of a mechanistic understanding, a rational design for an improved methodology for the fluorination of organic compounds based on Bi could be established.
Acknowledgments
Financial support for this work was provided by Max-Planck-Gesellschaft, Max-Planck-Institut für Kohlenforschung and Fonds der Chemischen Industrie (FCI-VCI). This project has received funding from European Union’s Horizon 2020 research and innovation program under the agreement nos. 850496 (ERC Starting Grant, J.C.) and 833361 (Marie Skłodowska Curie Fellowship, O.P.). We thank Prof. Dr. A. Fürstner for insightful discussions and generous support. N.N. especially thanks Dr. Richard Goddard for assistance in the XRD analysis. We also thank the analytical department at the MPI-Kohlenforschung for support in the characterization of compounds. We thank the reviewers of this article for their insights, which helped to improve the manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c01072.
Complete experimental details, NMR, computational details and crystallographic data (PDF)
Open access funded by Max Planck Society.
The authors declare no competing financial interest.
Supplementary Material
References
- a Purser S.; Moore P. R.; Swallow S.; Gouverneur V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. 10.1039/B610213C. [DOI] [PubMed] [Google Scholar]; b O’Hagan D. Fluorine in health care: Organofluorine containing blockbuster drugs. J. Fluor. Chem. 2010, 131, 1071–1081. 10.1016/j.jfluchem.2010.03.003. [DOI] [Google Scholar]; c Zhou Y.; Wang J.; Gu Z.; Wang S.; Zhu W.; Acena J. L.; Soloshonok V. A.; Izawa K.; Liu H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II-III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. 10.1021/acs.chemrev.5b00392. [DOI] [PubMed] [Google Scholar]
- a Jeschke P. The unique role of fluorine in the design of active ingredients for modern crop protection. Chembiochem 2004, 5, 570–589. 10.1002/cbic.200300833. [DOI] [PubMed] [Google Scholar]; b Ogawa Y.; Tokunaga E.; Kobayashi O.; Hirai K.; Shibata N. Current Contributions of Organofluorine Compounds to the Agrochemical Industry. iScience 2020, 23, 101467. 10.1016/j.isci.2020.101467. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Jeschke P. Current Trends in the Design of Fluorine-Containing Agrochemicals. Organofluorine Chemistry 2021, 363–395. 10.1002/9783527825158.ch11. [DOI] [Google Scholar]
- a Johns K.; Stead G. Fluoroproducts — the extremophiles. J. Fluor. Chem. 2000, 104, 5–18. 10.1016/S0022-1139(00)00251-7. [DOI] [Google Scholar]; b Kirsch P.; Bremer M. Nematic Liquid Crystals for Active Matrix Displays: Molecular Design and Synthesis. Angew. Chem., Int. Ed. 2000, 39, 4216–4235. . [DOI] [PubMed] [Google Scholar]; c Pagliaro M.; Ciriminna R. New fluorinated functional materials. J. Mater. Chem. 2005, 15, 4981–4991. 10.1039/b507583c. [DOI] [Google Scholar]; d Babudri F.; Farinola G. M.; Naso F.; Ragni R. Fluorinated organic materials for electronic and optoelectronic applications: the role of the fluorine atom. Chem. Commun. 2007, 1003–1022. 10.1039/B611336B. [DOI] [PubMed] [Google Scholar]
- a Fowler J. S.; Wolf A. P. Working against Time: Rapid Radiotracer Synthesis and Imaging the Human Brain. Acc. Chem. Res. 1997, 30, 181–188. 10.1021/ar960068c. [DOI] [Google Scholar]; b Ametamey S. M.; Honer M.; Schubiger P. A. Molecular imaging with PET. Chem. Rev. 2008, 108, 1501–1516. 10.1021/cr0782426. [DOI] [PubMed] [Google Scholar]; c Preshlock S.; Tredwell M.; Gouverneur V. (18)F-Labeling of Arenes and Heteroarenes for Applications in Positron Emission Tomography. Chem. Rev. 2016, 116, 719–766. 10.1021/acs.chemrev.5b00493. [DOI] [PubMed] [Google Scholar]
- a Neumann C. N.; Ritter T. Facile C-F Bond Formation through a Concerted Nucleophilic Aromatic Substitution Mediated by the PhenoFluor Reagent. Acc. Chem. Res. 2017, 50, 2822–2833. 10.1021/acs.accounts.7b00413. [DOI] [PubMed] [Google Scholar]; b Terrier F.Modern Nucleophilic Aromatic Substitution; Wiley-VCH: Weinheim, 2013. [Google Scholar]
- a Balz G.; Schiemann G. Über aromatische Fluorverbindungen, I.: Ein neues Verfahren zu ihrer Darstellung. Ber. Dtsch. Chem. Ges. B 1927, 60, 1186–1190. 10.1002/cber.19270600539. [DOI] [Google Scholar]; b Laali K. K.; Gettwert V. J. Fluorodediazoniation in ionic liquid solvents: new life for the Balz–Schiemann reaction. J. Fluor. Chem. 2001, 107, 31–34. 10.1016/S0022-1139(00)00337-7. [DOI] [Google Scholar]; c Cresswell A. J.; Davies S. G.; Roberts P. M.; Thomson J. E. Beyond the Balz-Schiemann reaction: the utility of tetrafluoroborates and boron trifluoride as nucleophilic fluoride sources. Chem. Rev. 2015, 115, 566–611. 10.1021/cr5001805. [DOI] [PubMed] [Google Scholar]
- Adams D. J.; Clark J. H. Nucleophilic routes to selectively fluorinated aromatics. Chem. Soc. Rev. 1999, 28, 225–231. 10.1039/a808707e. [DOI] [Google Scholar]
- a Campbell M. G.; Ritter T. Modern carbon-fluorine bond forming reactions for aryl fluoride synthesis. Chem. Rev. 2015, 115, 612–633. 10.1021/cr500366b. [DOI] [PubMed] [Google Scholar]; b Campbell M. G.; Ritter T. Late-Stage Fluorination: From Fundamentals to Application. Org. Process Res. Dev. 2014, 18, 474–480. 10.1021/op400349g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Fraser S. L.; Antipin M. Y.; Khroustalyov V. N.; Grushin V. V. Molecular Fluoro Palladium Complexes. J. Am. Chem. Soc. 1997, 119, 4769–4770. 10.1021/ja963984u. [DOI] [Google Scholar]; b Grushin V. V.; Marshall W. J. Ar–F Reductive Elimination from Palladium(II) Revisited. Organometallics 2007, 26, 4997–5002. 10.1021/om700469k. [DOI] [Google Scholar]; c Grushin V. V. Palladium fluoride complexes: one more step toward metal-mediated C-F bond formation. Chem.—Eur. J. 2002, 8, 1006–1014. . [DOI] [PubMed] [Google Scholar]; d Yandulov D. V.; Tran N. T. Aryl-fluoride reductive elimination from Pd(II): feasibility assessment from theory and experiment. J. Am. Chem. Soc. 2007, 129, 1342–1358. 10.1021/ja066930l. [DOI] [PubMed] [Google Scholar]; e Grushin V. V. The organometallic fluorine chemistry of palladium and rhodium: studies toward aromatic fluorination. Acc. Chem. Res. 2010, 43, 160–171. 10.1021/ar9001763. [DOI] [PubMed] [Google Scholar]
- a Watson D. A.; Su M.; Teverovskiy G.; Zhang Y.; Garcia-Fortanet J.; Kinzel T.; Buchwald S. L. Formation of ArF from LPdAr(F): catalytic conversion of aryl triflates to aryl fluorides. Science 2009, 325, 1661–1664. 10.1126/science.1178239. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Maimone T. J.; Milner P. J.; Kinzel T.; Zhang Y.; Takase M. K.; Buchwald S. L. Evidence for in situ catalyst modification during the Pd-catalyzed conversion of aryl triflates to aryl fluorides. J. Am. Chem. Soc. 2011, 133, 18106–18109. 10.1021/ja208461k. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Sather A. C.; Lee H. G.; De La Rosa V. Y.; Yang Y.; Muller P.; Buchwald S. L. A Fluorinated Ligand Enables Room-Temperature and Regioselective Pd-Catalyzed Fluorination of Aryl Triflates and Bromides. J. Am. Chem. Soc. 2015, 137, 13433–13438. 10.1021/jacs.5b09308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Furuya T.; Ritter T. Carbon-fluorine reductive elimination from a high-valent palladium fluoride. J. Am. Chem. Soc. 2008, 130, 10060–10061. 10.1021/ja803187x. [DOI] [PubMed] [Google Scholar]; b Furuya T.; Benitez D.; Tkatchouk E.; Strom A. E.; Tang P.; Goddard W. A. 3rd; Ritter T. Mechanism of C-F reductive elimination from palladium(IV) fluorides. J. Am. Chem. Soc. 2010, 132, 3793–3807. 10.1021/ja909371t. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Brandt J. R.; Lee E.; Boursalian G. B.; Ritter T. Mechanism of Electrophilic Fluorination with Pd(IV): Fluoride Capture and Subsequent Oxidative Fluoride Transfer. Chem. Sci. 2014, 5, 169–179. 10.1039/C3SC52367E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball N. D.; Sanford M. S. Synthesis and reactivity of a mono-sigma-aryl palladium(IV) fluoride complex. J. Am. Chem. Soc. 2009, 131, 3796–3797. 10.1021/ja8054595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hull K. L.; Anani W. Q.; Sanford M. S. Palladium-catalyzed fluorination of carbon-hydrogen bonds. J. Am. Chem. Soc. 2006, 128, 7134–7135. 10.1021/ja061943k. [DOI] [PubMed] [Google Scholar]; b Wang X.; Mei T. S.; Yu J. Q. Versatile Pd(OTf)2 × 2 H2O-catalyzed ortho-fluorination using NMP as a promoter. J. Am. Chem. Soc. 2009, 131, 7520–7521. 10.1021/ja901352k. [DOI] [PubMed] [Google Scholar]; c Lou S.-J.; Chen Q.; Wang Y.-F.; Xu D.-Q.; Du X.-H.; He J.-Q.; Mao Y.-J.; Xu Z.-Y. Selective C–H Bond Fluorination of Phenols with a Removable Directing Group: Late-Stage Fluorination of 2-Phenoxyl Nicotinate Derivatives. ACS Catal. 2015, 5, 2846–2849. 10.1021/acscatal.5b00306. [DOI] [Google Scholar]; d Mao Y. J.; Lou S. J.; Hao H. Y.; Xu D. Q. Selective C(sp(3))-H and C(sp(2))-H Fluorination of Alcohols Using Practical Auxiliaries. Angew. Chem., Int. Ed. 2018, 57, 14085–14089. 10.1002/anie.201808021. [DOI] [PubMed] [Google Scholar]; e Chen X. Y.; Sorensen E. J. Pd-Catalyzed, ortho C-H Methylation and Fluorination of Benzaldehydes Using Orthanilic Acids as Transient Directing Groups. J. Am. Chem. Soc. 2018, 140, 2789–2792. 10.1021/jacs.8b00048. [DOI] [PubMed] [Google Scholar]
- Zhao S. B.; Wang R. Y.; Nguyen H.; Becker J. J.; Gagne M. R. Electrophilic fluorination of cationic Pt-aryl complexes. Chem. Commun. 2012, 48, 443–445. 10.1039/C1CC15006E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kaspi A. W.; Goldberg I.; Vigalok A. Reagent-dependent formation of C-C and C-F bonds in Pt complexes: an unexpected twist in the electrophilic fluorination chemistry. J. Am. Chem. Soc. 2010, 132, 10626–10627. 10.1021/ja101436w. [DOI] [PubMed] [Google Scholar]; b Dubinsky-Davidchik I.; Goldberg I.; Vigalok A.; Vedernikov A. N. Selective aryl-fluoride reductive elimination from a platinum(IV) complex. Angew. Chem., Int. Ed. 2015, 54, 12447–12451. 10.1002/anie.201503116. [DOI] [PubMed] [Google Scholar]
- Sarkissian E.; Golbon Haghighi M. Strategy for Selective Csp2-F and Csp2-Csp2 Formations from Organoplatinum Complexes. Inorg. Chem. 2021, 60, 1016–1020. 10.1021/acs.inorgchem.0c03122. [DOI] [PubMed] [Google Scholar]
- a Lee E.; Hooker J. M.; Ritter T. Nickel-mediated oxidative fluorination for PET with aqueous [18F] fluoride. J. Am. Chem. Soc. 2012, 134, 17456–17458. 10.1021/ja3084797. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hoover A. J.; Lazari M.; Ren H.; Narayanam M. K.; Murphy J. M.; van Dam R. M.; Hooker J. M.; Ritter T. A Transmetalation Reaction Enables the Synthesis of [(18)F]5-Fluorouracil from [(18)F]Fluoride for Human PET Imaging. Organometallics 2016, 35, 1008–1014. 10.1021/acs.organomet.6b00059. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lee H.; Borgel J.; Ritter T. Carbon-Fluorine Reductive Elimination from Nickel(III) Complexes. Angew. Chem., Int. Ed. 2017, 56, 6966–6969. 10.1002/anie.201701552. [DOI] [PubMed] [Google Scholar]
- Meucci E. A.; Ariafard A.; Canty A. J.; Kampf J. W.; Sanford M. S. Aryl-Fluoride Bond-Forming Reductive Elimination from Nickel(IV) Centers. J. Am. Chem. Soc. 2019, 141, 13261–13267. 10.1021/jacs.9b06896. [DOI] [PubMed] [Google Scholar]
- a Casitas A.; Canta M.; Sola M.; Costas M.; Ribas X. Nucleophilic aryl fluorination and aryl halide exchange mediated by a Cu(I)/Cu(III) catalytic cycle. J. Am. Chem. Soc. 2011, 133, 19386–19392. 10.1021/ja2058567. [DOI] [PubMed] [Google Scholar]; b Fier P. S.; Hartwig J. F. Copper-mediated fluorination of aryl iodides. J. Am. Chem. Soc. 2012, 134, 10795–10798. 10.1021/ja304410x. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ye Y.; Sanford M. S. Mild copper-mediated fluorination of aryl stannanes and aryl trifluoroborates. J. Am. Chem. Soc. 2013, 135, 4648–4651. 10.1021/ja400300g. [DOI] [PubMed] [Google Scholar]; d Fier P. S.; Luo J.; Hartwig J. F. Copper-mediated fluorination of arylboronate esters. Identification of a copper(III) fluoride complex. J. Am. Chem. Soc. 2013, 135, 2552–2559. 10.1021/ja310909q. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Truong T.; Klimovica K.; Daugulis O. Copper-catalyzed, directing group-assisted fluorination of arene and heteroarene C-H bonds. J. Am. Chem. Soc. 2013, 135, 9342–9345. 10.1021/ja4047125. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Tredwell M.; Preshlock S. M.; Taylor N. J.; Gruber S.; Huiban M.; Passchier J.; Mercier J.; Genicot C.; Gouverneur V. A general copper-mediated nucleophilic 18F fluorination of arenes. Angew. Chem., Int. Ed. 2014, 53, 7751–7755. 10.1002/anie.201404436. [DOI] [PubMed] [Google Scholar]; g Mu X.; Zhang H.; Chen P.; Liu G. Copper-catalyzed fluorination of 2-pyridyl aryl bromides. Chem. Sci. 2014, 5, 275–280. 10.1039/C3SC51876K. [DOI] [Google Scholar]
- a Furuya T.; Ritter T. Fluorination of boronic acids mediated by silver(I) triflate. Org. Lett. 2009, 11, 2860–2863. 10.1021/ol901113t. [DOI] [PubMed] [Google Scholar]; b Furuya T.; Strom A. E.; Ritter T. Silver-mediated fluorination of functionalized aryl stannanes. J. Am. Chem. Soc. 2009, 131, 1662–1663. 10.1021/ja8086664. [DOI] [PubMed] [Google Scholar]; c Tang P.; Furuya T.; Ritter T. Silver-catalyzed late-stage fluorination. J. Am. Chem. Soc. 2010, 132, 12150–12154. 10.1021/ja105834t. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Tang P.; Ritter T. Silver-mediated fluorination of aryl silanes. Tetrahedron 2011, 67, 4449–4454. 10.1016/j.tet.2011.02.077. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Dubbaka S. R.; Narreddula V. R.; Gadde S.; Mathew T. Silver-mediated fluorination of potassium aryltrifluoroborates with Selectfluor®. Tetrahedron 2014, 70, 9676–9681. 10.1016/j.tet.2014.10.055. [DOI] [Google Scholar]; f Font M.; Acuna-Pares F.; Parella T.; Serra J.; Luis J. M.; Lloret-Fillol J.; Costas M.; Ribas X. Direct observation of two-electron Ag(I)/Ag(III) redox cycles in coupling catalysis. Nat. Commun. 2014, 5, 4373. 10.1038/ncomms5373. [DOI] [PubMed] [Google Scholar]
- a Power P. P. Main-group elements as transition metals. Nature 2010, 463, 171–177. 10.1038/nature08634. [DOI] [PubMed] [Google Scholar]; b Weetman C.; Inoue S. The Road Travelled: After Main-Group Elements as Transition Metals. ChemCatChem. 2018, 10, 4213–4228. 10.1002/cctc.201800963. [DOI] [Google Scholar]; c Melen R. L. Frontiers in molecular p-block chemistry: From structure to reactivity. Science 2019, 363, 479–484. 10.1126/science.aau5105. [DOI] [PubMed] [Google Scholar]
- Van Der Puy M. Conversion of diaryliodonium salts to aryl fluorides. J. Fluor. Chem. 1982, 21, 385–392. 10.1016/S0022-1139(00)81524-9. [DOI] [Google Scholar]
- a Pike V. W.; Aigbirhio F. I. Reactions of cyclotron-produced [18F]fluoride with diaryliodonium salts—a novel single-step route to no-carrier-added [18]fluoroarenes. J. Chem. Soc., Chem. Commun. 1995, 2215–2216. 10.1039/C39950002215. [DOI] [Google Scholar]; b Shah A.; Pike V. W.; Widdowson D. A. The synthesis of [18F]fluoroarenes from the reaction of cyclotron-produced [18F]fluoride ion with diaryliodonium salts. J. Chem. Soc., Perkin Trans. 1 1998, 2043–2046. 10.1039/a802349b. [DOI] [Google Scholar]; c Wang B.; Qin L.; Neumann K. D.; Uppaluri S.; Cerny R. L.; DiMagno S. G. Improved arene fluorination methodology for I(III) salts. Org. Lett. 2010, 12, 3352–3355. 10.1021/ol101154h. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Graskemper J. W.; Wang B.; Qin L.; Neumann K. D.; DiMagno S. G. Unprecedented directing group ability of cyclophanes in arene fluorinations with diaryliodonium salts. Org. Lett. 2011, 13, 3158–3161. 10.1021/ol201080c. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Rotstein B. H.; Stephenson N. A.; Vasdev N.; Liang S. H. Spirocyclic hypervalent iodine(III)-mediated radiofluorination of non-activated and hindered aromatics. Nat. Commun. 2014, 5, 4365. 10.1038/ncomms5365. [DOI] [PubMed] [Google Scholar]
- Xu P.; Zhao D.; Berger F.; Hamad A.; Rickmeier J.; Petzold R.; Kondratiuk M.; Bohdan K.; Ritter T. Site-Selective Late-Stage Aromatic [(18) F]Fluorination via Aryl Sulfonium Salts. Angew. Chem., Int. Ed. 2020, 59, 1956–1960. 10.1002/anie.201912567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron T.; Sander K.; Cybulska K.; Benhamou L.; Sin P. K. B.; Khan A.; Wood M.; Porter M. J.; Arstad E. Ring-Closing Synthesis of Dibenzothiophene Sulfonium Salts and Their Use as Leaving Groups for Aromatic (18)F-Fluorination. J. Am. Chem. Soc. 2018, 140, 11125–11132. 10.1021/jacs.8b06730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a De Meio G. V.; Pinhey J. T. Aryl fluorides from the reaction of boron trifluoride with aryl-lead(IV) triacetates, which may be generated in situ from aryltrimethylsilanes, triarylboroxines, and arenes.. J. Chem. Soc., Chem. Commun. 1990, 1065–1066. 10.1039/c39900001065. [DOI] [Google Scholar]; b De Meio G.; Morgan J.; Pinhey J. T. Aryl fluoride syntheses involving reaction of aryllead triacetates with boron trifluoride-diethyl ether complex. Tetrahedron 1993, 49, 8129–8138. 10.1016/S0040-4020(01)88032-7. [DOI] [Google Scholar]
- Taylor E. C.; Bigham E. C.; Johnson D. K.; McKillop A. Thallium in organic synthesis. 45. Synthesis of aromatic fluorides. J. Org. Chem. 1977, 42, 362–363. 10.1021/jo00422a048. [DOI] [Google Scholar]
- a Ligand coupling involving organobismuth compounds. In Ligand Coupling Reactions with Heteroatomic Compounds, Finet J.-P., Ed.; Elsevier: Amsterdam, 1998; Vol. 18, pp 159–204. [Google Scholar]; b Abramovitch R. A.; Barton D. H. R.; Finet J.-P. Newer methods of arylation. Tetrahedron 1988, 44, 3039–3071. 10.1016/S0040-4020(01)85938-X. [DOI] [Google Scholar]; c Ollevier T. New trends in bismuth-catalyzed synthetic transformations. Org. Biomol. Chem. 2013, 11, 2740–2755. 10.1039/c3ob26537d. [DOI] [PubMed] [Google Scholar]
- Ooi T.; Goto R.; Maruoka K. Fluorotetraphenylbismuth: a new reagent for efficient regioselective alpha-phenylation of carbonyl compounds. J. Am. Chem. Soc. 2003, 125, 10494–10495. 10.1021/ja030150k. [DOI] [PubMed] [Google Scholar]
- Minoura M.; Kanamori Y.; Miyake A.; Akiba K.-y. Structure of Azabismocines, Hexacoordinate Pentavalent Organobismuth Compounds. Chem. Lett. 1999, 28, 861–862. 10.1246/cl.1999.861. [DOI] [Google Scholar]
- a Mohan R. Green bismuth. Nat. Chem. 2010, 2, 336. 10.1038/nchem.609. [DOI] [PubMed] [Google Scholar]; b Krabbe S. W.; Mohan R. S.. Environmentally Friendly Organic Synthesis Using Bismuth(III) Compounds. In Bismuth-Mediated Organic Reactions, Ollevier T., Ed.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2012; pp 45–68. [DOI] [PubMed] [Google Scholar]
- a Wang F.; Planas O.; Cornella J. Bi(I)-Catalyzed Transfer-Hydrogenation with Ammonia-Borane. J. Am. Chem. Soc. 2019, 141, 4235–4240. 10.1021/jacs.9b00594. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Planas O.; Wang F.; Leutzsch M.; Cornella J. Fluorination of arylboronic esters enabled by bismuth redox catalysis. Science 2020, 367, 313–317. 10.1126/science.aaz2258. [DOI] [PubMed] [Google Scholar]; c Planas O.; Peciukenas V.; Cornella J. Bismuth-Catalyzed Oxidative Coupling of Arylboronic Acids with Triflate and Nonaflate Salts. J. Am. Chem. Soc. 2020, 142, 11382–11387. 10.1021/jacs.0c05343. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Pang Y.; Leutzsch M.; Nöthling N.; Cornella J. Catalytic Activation of N2O at a Low-Valent Bismuth Redox Platform. J. Am. Chem. Soc. 2020, 142, 19473–19479. 10.1021/jacs.0c10092. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Magre M.; Kuziola J.; Nöthling N.; Cornella J. Dibismuthanes in catalysis: from synthesis and characterization to redox behavior towards oxidative cleavage of 1,2-diols. Org. Biomol. Chem. 2021, 19, 4922–4929. 10.1039/D1OB00367D. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Pang Y.; Leutzsch M.; Nöthling N.; Katzenburg F.; Cornella J. Catalytic Hydrodefluorination via Oxidative Addition, Ligand Metathesis, and Reductive Elimination at Bi(I)/Bi(III) Centers. J. Am. Chem. Soc. 2021, 143, 12487–12493. 10.1021/jacs.1c06735. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Planas O.; Cornella J. High-valent bismuth redox catalysis. Nachr. Chem. 2021, 69 (10), 79–83. 10.1002/nadc.20214116551. [DOI] [Google Scholar]; h Planas O. Catálisis Redox con Bismuto. Ann. Quím. 2021, 117, 266–273. [Google Scholar]; i Moon H. W.; Cornella J. Bismuth Redox Catalysis: An Emerging Main-Group Platform for Organic Synthesis. ACS Catal. 2022, 12, 1382–1393. 10.1021/acscatal.1c04897. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Magre M.; Cornella J. Redox-Neutral Organometallic Elementary Steps at Bismuth: Catalytic Synthesis of Aryl Sulfonyl Fluorides. J. Am. Chem. Soc. 2021, 143, 21497–21502. 10.1021/jacs.1c11463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H.; Murafuji T.; Azuma N. Synthesis and reactions of some new heterocyclic bismuth-(III) and -(V) compounds. 5,10-Dihydrodibenzo[b,e]bismine and related systems. J. Chem. Soc., Perkin Trans. 1 1992, 1593–1600. 10.1039/p19920001593. [DOI] [Google Scholar]
- See Supporting Information for further details.
- a Sen S.; Ke I.-S.; Gabbaï F. P. Anion-Controlled Positional Switching of a Phenyl Group about the Dinuclear Core of a AuSb Complex. Inorg. Chem. 2016, 55, 9162–9172. 10.1021/acs.inorgchem.6b01290. [DOI] [PubMed] [Google Scholar]; b Kuziola J.; Magre M.; Nöthling N.; Cornella J. Synthesis and Structure of Mono-, Di-, and Trinuclear Fluorotriarylbismuthonium Cations. Organometallics 2022, 41 (14), 1754–1762. 10.1021/acs.organomet.2c00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ugi I.; Marquarding D.; Klusacek H.; Gillespie P.; Ramirez F. Berry pseudorotation and turnstile rotation. Acc. Chem. Res. 1971, 4, 288–296. 10.1021/ar50044a004. [DOI] [Google Scholar]; b Couzijn E. P.; Slootweg J. C.; Ehlers A. W.; Lammertsma K. Stereomutation of pentavalent compounds: validating the Berry pseudorotation, redressing Ugi’s turnstile rotation, and revealing the two- and three-arm turnstiles. J. Am. Chem. Soc. 2010, 132, 18127–18140. 10.1021/ja105306s. [DOI] [PubMed] [Google Scholar]
- Ishida S.; Hirakawa F.; Furukawa K.; Yoza K.; Iwamoto T. Persistent Antimony- and Bismuth-Centered Radicals in Solution. Angew. Chem., Int. Ed. 2014, 53, 11172–11176. 10.1002/anie.201405509. [DOI] [PubMed] [Google Scholar]
- Swain C. G.; Unger S. H.; Rosenquist N. R.; Swain M. S. Substituent effects on chemical reactivity. Improved evaluation of field and resonance components. J. Am. Chem. Soc. 1983, 105, 492–502. 10.1021/ja00341a032. [DOI] [Google Scholar]
- a Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]; b Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]; c Grimme S.; Ehrlich S.; Goerigk L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- a Weigend F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. 10.1039/b515623h. [DOI] [PubMed] [Google Scholar]; b Weigend F.; Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Metz B.; Stoll H.; Dolg M. Small-core multiconfiguration-Dirac–Hartree–Fock-adjusted pseudopotentials for post-d main group elements: Application to PbH and PbO. J. Chem. Phys. 2000, 113, 2563–2569. 10.1063/1.1305880. [DOI] [Google Scholar]
- a Neese F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. 10.1002/wcms.81. [DOI] [Google Scholar]; b Neese F. Software update: the ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 8, e1327 10.1002/wcms.1327. [DOI] [Google Scholar]
- Reed A. E.; Curtiss L. A.; Weinhold F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. 10.1021/cr00088a005. [DOI] [Google Scholar]
- a Barton D. H. R.; Blazejewski J.-C.; Charpiot B.; Lester D. J.; Motherwell W. B.; Papoula M. T. B. Comparative arylation reactions with pentaphenylbismuth and with triphenylbismuth carbonate. J. Chem. Soc., Chem. Commun. 1980, 827–829. 10.1039/c39800000827. [DOI] [Google Scholar]; b Barton D. H. R.; Lester D. J.; Motherwell W. B.; Papoula M. T. B. Observations on the cleavage of the bismuth–carbon bond in BiVcompounds: a new arylation reaction. J. Chem. Soc., Chem. Commun. 1980, 246–247. 10.1039/C39800000246. [DOI] [Google Scholar]; c Jurrat M.; Maggi L.; Lewis W.; Ball L. T. Modular bismacycles for the selective C-H arylation of phenols and naphthols. Nat. Chem. 2020, 12, 260–269. 10.1038/s41557-020-0425-4. [DOI] [PubMed] [Google Scholar]
- Rozatian N.; Hodgson D. R. W. Reactivities of electrophilic N–F fluorinating reagents. Chem. Commun. 2021, 57, 683–712. 10.1039/D0CC06339H. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.