

Abstract
Hypervalent iodine(V) reagents are a powerful class of organic oxidants. While the use of I(V) compounds Dess–Martin periodinane and IBX is widespread, this reagent class has long been plagued by issues of solubility and stability. Extensive effort has been made for derivatizing these scaffolds to modulate reactivity and physical properties but considerable room for innovation still exists. Herein, we describe the preparation, thermal stability, optimized geometries, and synthetic utility of an emerging class of I(V) reagents, Bi(N)-HVIs, possessing datively bound bidentate nitrogen ligands on the iodine center. Bi(N)-HVIs display favorable safety profiles, improved solubility, and comparable to superior oxidative reactivity relative to common I(V) reagents. The highly modular synthesis and in situ generation of Bi(N)-HVIs provides a novel and convenient screening platform for I(V) reagent and reaction development.
Graphical Abstract

■ INTRODUCTION
Hypervalent iodine reagents (HVIs) have received tremendous attention from the synthetic community in recent years. HVIs, commonly seen in both the I(III) and I(V) oxidation state, are environmentally benign, inexpensive, and versatile oxidants, providing an attractive alternative to metal-based reagents across a wide range of transformations.1 I(V) species are effective reagents for heteroatom oxidation,1e arene dearomatization,1f and ketone desaturation;1g however, their applications have long been plagued by practical and safety limitations.1,2 The simplest I(V) reagents, the acyclic iodylbenzene (1, PhIO2) or cyclic IBX2c,d (5), are polymeric in nature due to intermolecular I···X interactions, resulting in generally poor solubility in most common organic solvents.2b,3 Furthermore, the low thermal and shock stability of PhIO2 and IBX has been well-documented, posing a significant safety hazard and limiting their scalability (Figure 1A).2,4 In an effort to address these limitations, an array of pseudocyclic (2–4) and cyclic (6–8) I(V) derivatives have been developed that possess intramolecular I···X bonding interactions that disrupt the polymeric nature, or arene, X-ligand, or benziodoxolone variations to tune reactivity, leading to more soluble, practical, and efficient oxidants.5–7 In particular, the venerable Dess–Martin periodinane (DMP, 6), derived upon acylation of IBX with Ac2O,8 has seen broad application in natural product synthesis due to its superior solubility, selective reactivity, and commercial availability; however, the high cost and moisture sensitivity pose challenges for its large-scale preparation and storage. While it has been demonstrated that these strategies are effective in modulating reactivity and stability of I(V) scaffolds, the derivatives often still possess safety concerns and can require linear and sometimes lengthy synthetic sequences, making individual reaction optimization using these handles impractical.7a Therefore, the continued development of new scaffolds and, in particular, those capable of generating soluble, stable, low-cost, and readily tunable I(V) reagents remains of considerable interest.
Figure 1.
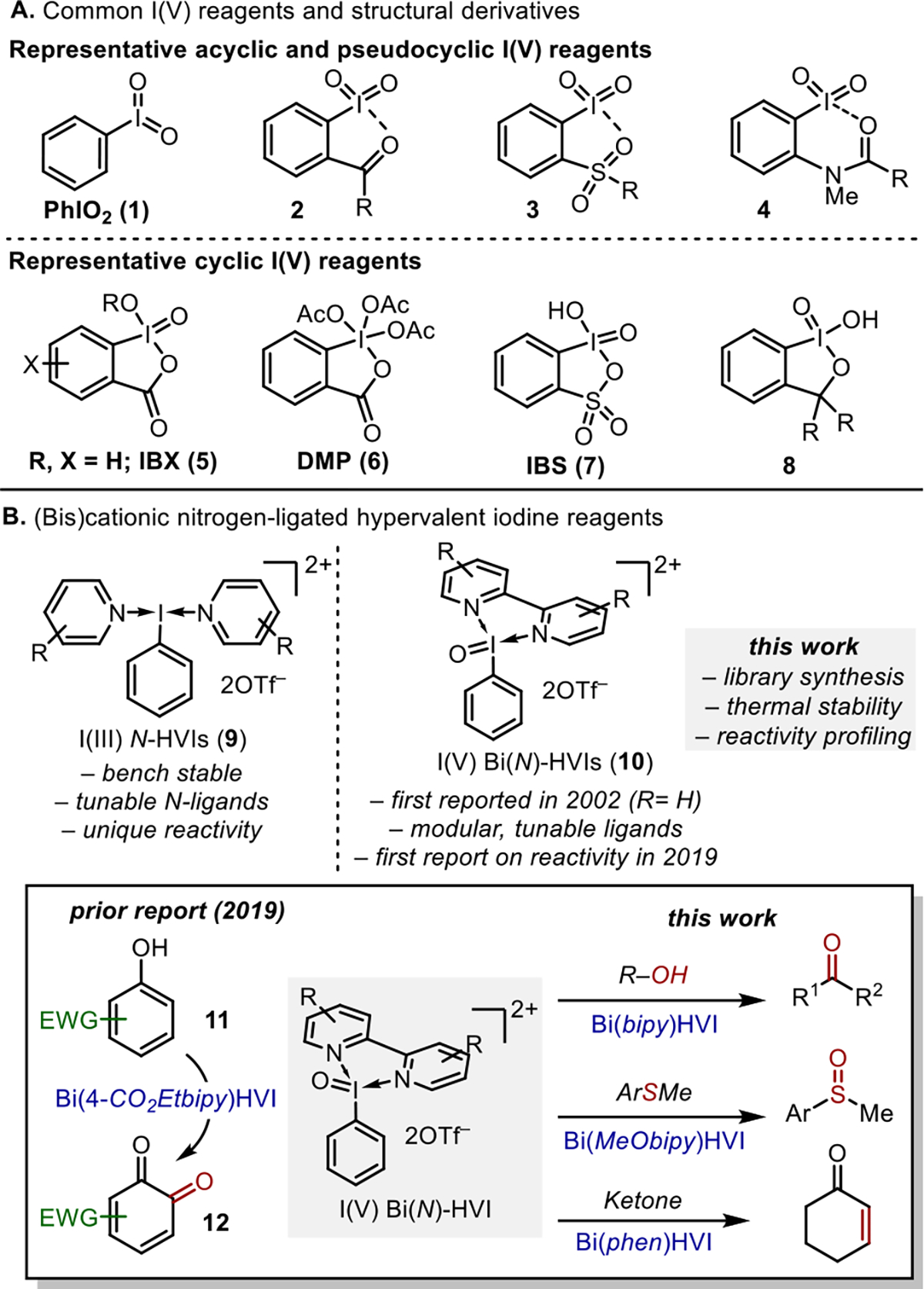
Common I(V) reagents and Bi(N)-HVI synthesis and reactivity.
In 2002, Zhdankin reported a bidentate nitrogen-ligated I(V) species with a 2,2′-bipyridine ligand (Figure 1B, 10, R = H);9 however, no further studies on its reactivity or potential synthetic utility were described. Based on the work of our laboratory and others regarding (bis)cationic nitrogen-ligated I(III) reagents, or N-HVIs (9), which display enhanced and unique reactivity relative to typical ArIX2 reagents,10 we hypothesized that the analogous nitrogen-ligated I(V) compounds, which we have termed Bi(N)-HVIs (10), warranted further study. In addition to their potential novel reactivity, our prior work indicated that the bidentate nitrogen ligands could provide a convenient and modular handle for tuning reactivity and thus offer a practical advantage over current I(V) scaffolds.
We recently reported the first synthetic application of Bi(N)-HVIs in the oxidative dearomatization of electron-deficient phenols to o-quinones (11 to 12, Figure 1B).11 These substrates have proven resistant to oxidation with almost all known oxidants, demonstrating the novel reactivity of Bi(N)-HVIs.12 The Bi(N)-HVIs could be isolated or prepared in situ from iodylbenzene, and commercially available bidentate nitrogen ligands were found to be soluble in common organic solvents and, as we had postulated, modulating the nitrogen ligand had a marked effect on reactivity. Given these promising preliminary findings, we conducted a more detailed study on the synthesis and reactivity of Bi(N)-HVI to probe their general utility as synthetic oxidants.
Herein, we report on the synthesis, isolation, and stability of Bi(N)-HVI derivatives as well as their reactivity in a panel of representative oxidative transformations. The Bi(N)-HVIs can be isolated or used in situ, display good solubility, have favorable safety profiles, and exhibit robust reactivity. Thermogravimetric analysis (TGA) indicates that Bi(N)-HVIs possess superior thermal stability to common I(V) reagents, addressing a long-standing liability of this reagent class. Computationally derived structures for isolable derivatives are reported, which provide insights into the coordination environment and geometry around the iodine center to aide in further reagent design. Their reactivity in alcohol and sulfide oxidation as well as ketone desaturation is found to be equivalent or superior to current I(V) reagents. Across these transformations, a different N-ligand was found to be the most effective in each case, further demonstrating the role of the N-ligands in tuning and modulating reactivity and demonstrating the potential of Bi(N)-HVIs as a convenient, operationally simple I(V) platform for reaction screening and development.
■ RESULTS AND DISCUSSION
Synthesis of Bi(N)-HVIs.
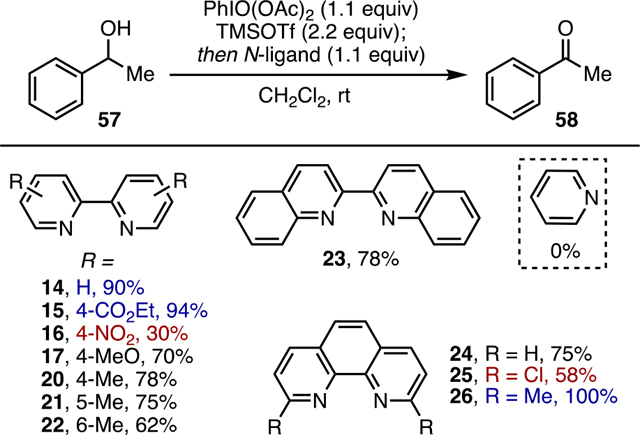
Bi(N)-HVIs can be synthesized in accordance with Zhdankin’s originally reported procedure, starting from commercial iodylbenzene (1).9 Due to its insolubility, iodylbenzene must first be converted to the bis(acetoxy)iodylbenzene (PhI(O)(OAc)2, 13) by stirring in neat acetic anhydride followed by concentration (Scheme 1). This has been performed in our laboratory on up to 1.5 g scale to give 13 in quantitative yield, which can then be stored at −20 °C under argon for up to 1 month; however, it is prone to loss of −OAc with reversion back to PhIO2, so purity should be checked by 1H NMR prior to use. Treatment of 13 with TMSOTf, presumed to give a highly active triflate intermediate,13 is followed by the addition of nitrogen ligand to give the desired Bi(N)-HVI. To date, we have investigated a range of bidentate nitrogen ligands (14–26) in this Bi(N)-HVI synthesis. In our previous report, Bi(N)-HVIs incorporating ligands 14–26 were all synthesized and used in situ and their presence was inferred by the successful subsequent reaction or by 1H NMR without isolation. In this study, the focus was on identifying and fully characterizing isolable derivatives. Of the compounds previously synthesized, only four Bi(N)-HVIs proved consistently isolable; two bipyridine (27 and 28) and two phenanthroline (29 and 30) derivatives were isolated in good-to-high yields after crystallization (Scheme 1). Even within the scope of the isolable compounds, identity of the nitrogen ligand did have a notable effect on stability; Bi(bipy)-HVI 27 and Bi(4-MeO-bipy)-HVI 28 can be stored at −20 °C for up to 1 month, whereas Bi(phen)-HVI 29 and Bi(5-Mephen)-HVI 30 have to be used immediately or isolated and stored in a glovebox, due to significant moisture sensitivity. Several practical considerations could be taken from our studies: (1) the Bi(N)-HVIs display good solubility in a range of common organic solvents including CH3CN, dimethyl sulfoxide (DMSO), MeOH, CH2Cl2, and CHCl3, a broader and more practical selection than many common I(V) reagents. (2) Decomposition back to PhIO2 and free ligand was the primary method of degradation, occurring during attempts at crystallization, trituration, or upon prolonged exposure to moisture and with the propensity affected by the identity of the N-ligand.
Scheme 1.

Synthesis and Isolation of I(V) Bi(N)-HVIs
Thermal Stability of Bi(N)-HVIs.
A common concern when using hypervalent iodine reagents, and in particular I(V) compounds, is their thermal instability. PhIO2 and IBX have both been reported to undergo violent exothermic decomposition at temperatures around 200 °C, leading to significant concerns when working on scale.1a–d,4 As the stability of Bi(N)-HVIs had yet to be studied, we sought to establish their thermal safety profiles. For these studies, only Bi(bipy)-HVI (27) and Bi(4-MeObipy)-HVI (28) were bench-stable for long enough periods of time to perform reliable analyses. The thermal decomposition profiles of 27 and 28, as well as PhI(O)(OAc)2 (13) and IBX (5), were examined using TGA (Figure 2). IBX (5) experienced two-stage degradation with initial onset temperature at 103 °C and a second and steeper decomposition at 230 °C, which is consistent with the reported decomposition temperature of 233 °C.4 PhI(O)(OAc)2 (13) underwent a steep and single stage decomposition at an onset temperature of 227 °C with an abrupt decomposition at 240 °C. In contrast, we were pleased to see that both Bi(N)-HVI reagents displayed gradual decomposition profiles; the onset points for Bi(bipy)-HVI 27 and Bi(4-MeO-bipy)-HVI 28 were 164 °C and 179 °C, respectively, with relatively gradual degradation curves in the range 200–600 °C. These results indicate that while the PhI(O)(OAc)2 (13) precursor should be handled with caution, Bi(N)-HVI reagents seem to offer improved thermal stability relative to IBX (5), potentially providing safer I(V) alternatives. A complete description of the samples’ thermal properties can be found in the Supporting Information.
Figure 2.
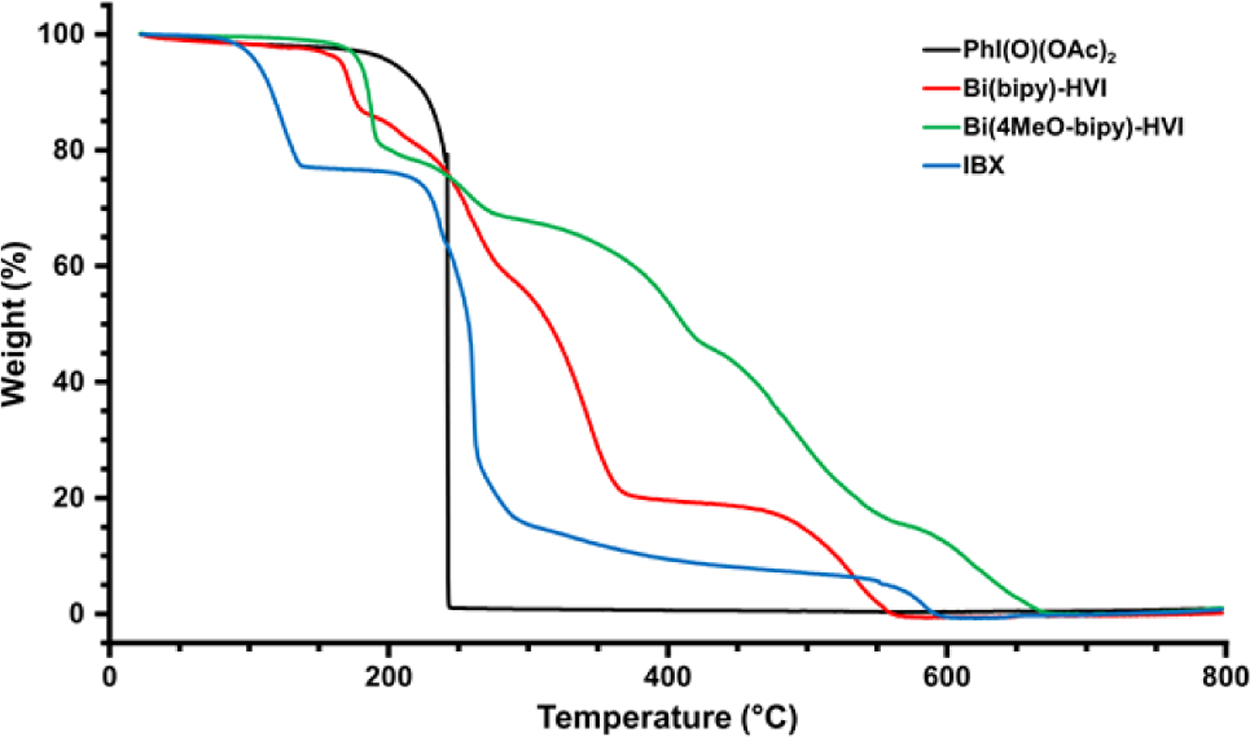
TGA for decomposition of I(V) reagents: Bi(bipy)-HVI 27 (red), Bi(4-MeObipy)-HVI 28 (green), precursor PhI(O)(OAc)2 13 (black), and IBX 5 (blue).
Optimized Geometries of Bi(N)-HVIs.
Due to the potential implications on reactivity and subsequent reagent design, we were interested in gaining insights into the geometry and coordination environment at the I(V) center of Bi(N)-HVIs. Despite extensive efforts, all attempts to obtain crystals for X-ray structural analysis were unsuccessful. In lieu of crystallographic data, we turned to the computational study of the reagent scaffolds. We have undertaken the density functional theory calculations at the SMD/M06-2X/def2-TZVP//SMD/M06-2X/LANL2DZ(d),6-31G(d) level of theory for obtaining structure and energy of various isomers of bipyridine-ligated Bi(bipy)-HVI (27).14
The four lowest energy isomers for Bi(bipy)-HVI (27-min1–27-min4) are shown in Figure 3, with selected bond distances and angles for the lowest energy isomer 27-min1, presented in Table 1. 27-min1 adopts a distorted octahedral geometry around the iodine(V) center, with the oxo (O1), bidentate nitrogen ligand (N1 and N2), and a triflate anion (O2) occupying equatorial positions.15 The aryl ring occupies one axial position while another, more weakly bound, triflate anion is bound in a pseudoaxial position, with a distortion toward the heterocyclic ligand. All of the equatorial ligands form slightly acute bond angles with an I1–C1 iodine–carbon bond with bond angles ranging from 82.8166 to 86.2108°. This basic ligand geometry, with equatorial positioning of X-ligands and an axial carbon substituent, is in accordance with previously reported X-ray structures of I(V) reagents.3a,14–16 Closer examination of 27-min1 reveals that the binding of the bipyridine ligand is unsymmetrical, with significantly different bond lengths of I1–N1 2.235 Å and I1–N2 2.567 Å for those trans to the triflate and oxo, respectively. The two heterocycles of the bipyridine ligand are also not completely coplanar, with a dihedral angle of 10.8° between the two nitrogens. The other isomers, 27-min2–27-min4, range from 3.3 to 10.6 kcal mol−1 less stable than 27-min1. 27-min2 results from an axial-equatorial isomerization of a triflate (O3) and one of the bipyridine nitrogens (N2). 27-min3 shows dissociation of the axial nitrogen in 27-min2, and 27-min4 has only one bound triflate anion (unbound −OTf has been omitted for clarity), but otherwise the coordination geometry most closely resembles that of 27-min1. It should be noted that recent computational studies on IBX-mediated oxidations have indicated that isomerization to a less stable isomer precedes either ligand exchange or substrate oxidation, and thus, it should not be presumed that 27-min1 would be the predominant reactive isomer in solution.14
Figure 3.
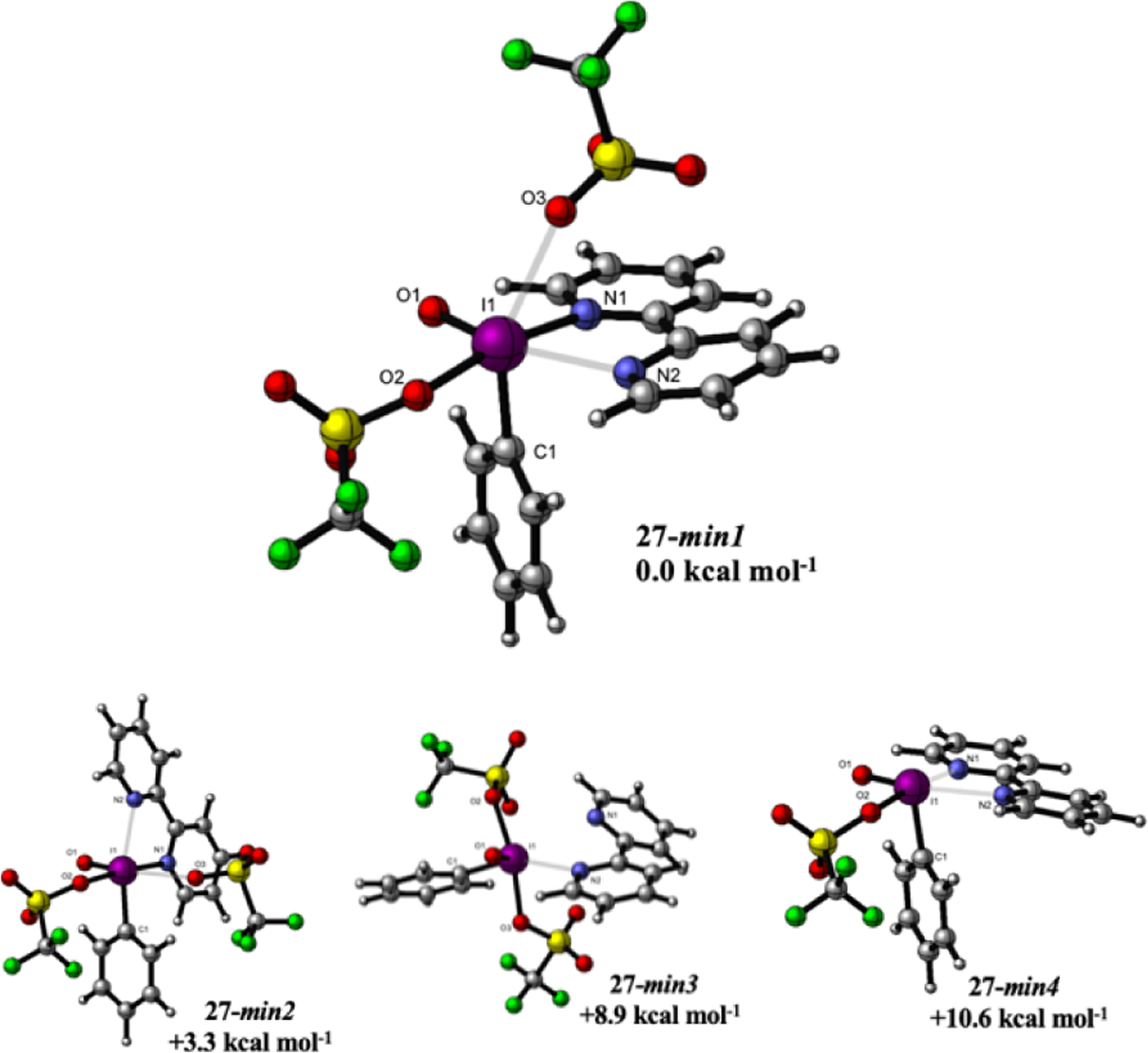
Optimized geometries for Bi(bipy)HVI (27-min, 1–4).
Table 1.
Selected Bond Distances and Angles in 27-min1
| bond lengths | distance [Å] | bond angles | degree (deg) |
|---|---|---|---|
| I1–C1 | 2.106 | C1–I1–N1 | 86.2 |
| I1–O1 | 1.763 | C1–I1–N2 | 82.8 |
| I1–O2 | 2.231 | C1–I1–O2 | 84.5 |
| I1–O3 | 2.732 | C1–I1–O3 | 151.3 |
| I1–N1 | 2.235 | O1–I1–N2 | 157.3 |
| I1–N2 | 2.567 | O2–I1–N1 | 170.1 |
| N1–I1–N2 | 67.7 |
Reactivity Profiling.
With reliable syntheses and encouraging safety data in hand, attention turned for establishing the utility of Bi(N)-HVIs as practical synthetic oxidants. As previously mentioned, the reactivity of Bi(N)-HVIs in the dearomatization of electron-deficient phenols to o-quinones has already been reported by our laboratory and the results are briefly summarized in Scheme 2. I(V) reagents have emerged as leading oxidants for the regioselective dearomatization of phenols to o-quinones and o-quinols; however, phenols lacking electron-donating groups are typically unreactive.1f Through the use of Bi(N)-HVIs, we described the first general method for the dearomatization of electron-deficient phenols to o-quinones. In situ Bi(4-CO2Etbipy)-HVI 31 was found to be the most effective and general reagent; however, other derivatives also showed significant activity against these challenging substrates.11 Computational studies to further understand the mechanism and unique reactivity of Bi(N)-HVIs in these oxidations are currently underway and will be reported in due course. Building on these findings, we now examine their reactivity in alcohol oxidation, sulfide oxidation, and ketone desaturation, all common transformations mediated by I(V) reagents.
Scheme 2.
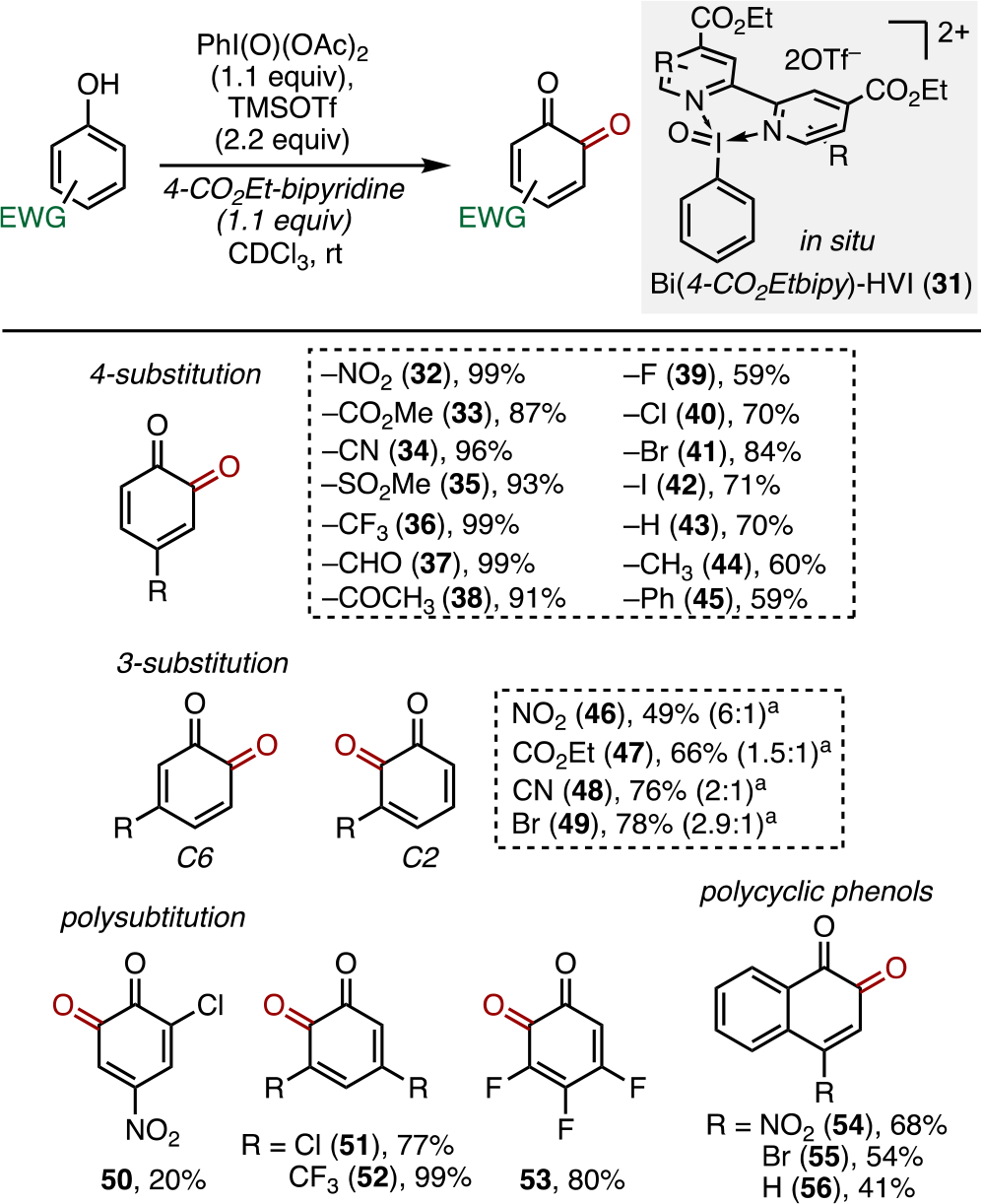
Dearomatization of Electron-Poor Phenols to ortho-Quinones with Bi(4-CO2Etbipy)-HVI11
aYields reported are of combined C2/C6 oxidation products as they were inseparable. The C2/C6 ratio is reported in parentheses.
Oxidation of Alcohols.
We began by investigating the reactivity of Bi(N)-HVIs in the oxidation of alcohols. While the use of DMP for this transformation is ubiquitous due to its commercial availability, solubility, and mild reaction conditions, it is also expensive and prone to hydrolysis, and its reactivity is not tunable, leaving room for potential improvement.1e,2 Initial screening consisted of using both the isolated and in situ-generated Bi(bipy)-HVI 27 alongside several common I(V) reagents in the oxidation of 1-phenylethanol (57) to acetophenone (58) (Table 2). As expected, DMP (6) gave the desired product with excellent yield after just 30 min (entry 1). Both IBX (5) and PhIO2 (1) performed poorly in CH2Cl2 due to insolubility (entries 2, 3), which could be alleviated in the case of IBX by running the reaction in DMSO to give 82% yield. Oxidation with pseudocyclic iodylarene o-tBuSO2PhIO2 (59) gave good yields but only when performed in NO2Me, in accordance with the literature.5 We were pleased to see that both the isolated (entry 5) and in situ (entry 6) Bi(bipy)-HVI (27) performed comparably to DMP, giving ketone 58 in 90% yield after 2 h. Low conversions obtained from control experiments with Bi(N)-HVI precursors, PhIO(OAc)2 (13) (entry 7), and the highly active intermediate generated upon treatment with TMSOTf (entry 8) supported that the Bi(bipy)-HVI complex was the active oxidant under the in situ conditions.
Table 2.
Oxidation of 1-Phenylethanola
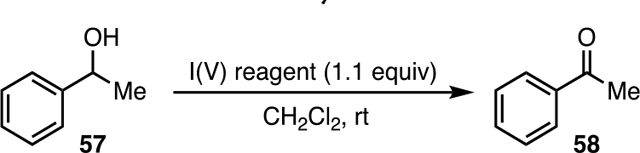
| |||
|---|---|---|---|
| entry | reagents | time | yield (%)b |
| 1 | DMP | 30 min | 97 |
| 2 | IBX | 24 h | 33 (82)c |
| 3 | PhIO2 | 24 h | trace |
| 4 | o-tBuSO2PhIO2 (59) | 24 h | 87d,e |
| 5 | 27 (isolated) | 2 h | 90 |
| 6 | 27 (in situ) | 2 h | 90 (88)e |
| 7 | PhIO(OAc)2 (13) | 24 h | 0 |
| 8 | PhIO(OAc)2, TMSOTf | 24 h | 10f |
Reactions were carried out on the 0.2 mmol scale in CH2Cl2 (0.2 M).
NMR yields, determined with 1,2-dibromoethane as the internal standard.
DMSO was used as a solvent. Low yield in CH2Cl2 due to limited solubility.
Nitromethane was used as a solvent.
Isolated yield.
Complex mixture of products.
To identify the most efficient Bi(N)-HVI, a ligand screen was then conducted under in situ conditions (Table 3). In almost all cases, good-to-high yields were obtained, with electron-deficient 4-NO2-bipy (16) and 5,5′-Clphen (25) ligands giving the lowest yields, at least in part due to poor solubility of the resulting Bi(N)-HVIs. Along with simple bipyridine (14), 4-CO2Etbipy (15) and 5,5′-Mephen (26) gave the best yields at greater than 90% in all cases. The use of monodentate pyridine did not afford any desired product, as seen in our prior dearomatization report,11 supporting the importance of a bidentate ligand. Due to the low cost and commercial availability, bipyridine was selected as the optimal ligand for further studies of Bi(N)-HVI alcohol oxidation.
Table 3.
Ligand Screening for In Situ Bi(N)-HVI Oxidation of 1-Phenylethanola
|
All the reactions were carried out with the 0.2 mmol scale in CH2Cl2 (0.2 M) and reported yields were determined by 1HNMR with 1,2-dibromoethane as the internal standard.
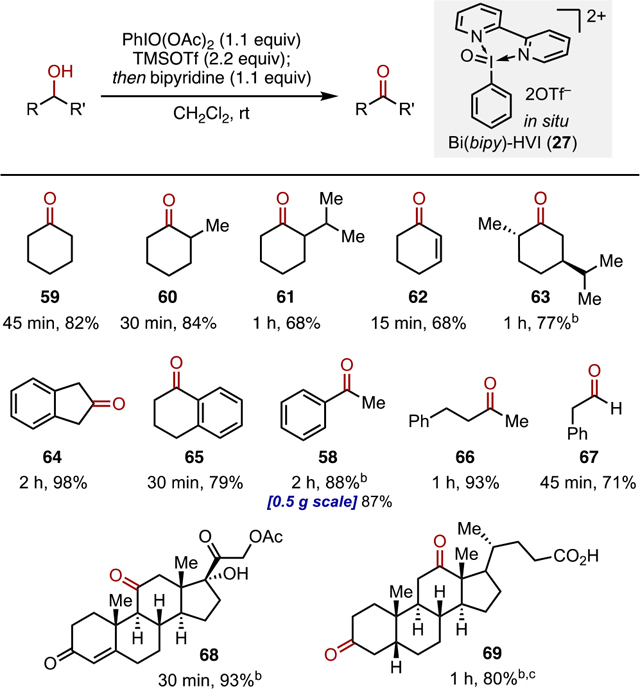
The substrate scope was then examined using in situ Bi(bipy)-HVI 27 (Table 4). Efficient oxidation of cyclic, acyclic, aliphatic, and aromatic secondary alcohols proceeded in good-to-excellent yields to give 58–66. Controlled oxidation of 2-phenyl-ethanol to aldehyde 67 could also be achieved in 71% yield. Steroid derivatives 68 and 69 possessing a tertiary alcohol, enone, and free carboxylic acid between them were also oxidized in 93 and 80%, respectively, including a double oxidation in the case of 69. This study demonstrates the utility of in situ Bi(bipy)-HVI 27 as a cost-effective, practical alternative to DMP or IBX for alcohol oxidation.
Table 4.
Alcohol Scope with In Situ Bi(bipy)-HVI Oxidationa
|
All the reactions were carried out on the 0.2 mmol scale in CH2Cl2 (0.2 M) and reported yields were determined by 1HNMR with 1,2-dibromoethane as the internal standard unless otherwise noted.
Isolated yield.
2.2 equiv in situ Bi(bipy)-HVI was used.
Sulfide oxidation.
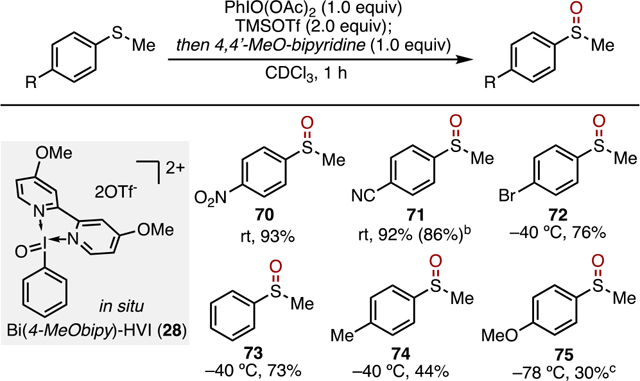
I(V) reagents have also found broad application in the controlled oxidation of sulfides to sulfoxides.1,6a,f,17 Screening of in situ Bi(N)-HVIs in the oxidation of thioanisole revealed that the more electron-rich Bi(4-MeObipy)-HVI 28 provided the best and most consistent yields of sulfoxide products (Table 5).18 Applying the optimized conditions to a panel of electronically diverse aryl sulfides, a similar trend emerged to that of Bi(N)-HVI-mediated phenol dearomatization; the more electron-deficient aryl sulfides 70–72 were oxidized to sulfoxides in high yields, whereas the more reactive, electron-rich derivatives 73–75 gave low-to-moderate yields along with unidentified byproducts. The electronic trend was counteracted to some extent by lowering the reaction temperatures; however, yields remained low. This trend is opposite to that of other I(V)-mediated sulfide oxidations, wherein more electron-rich substrates are more readily and cleanly oxidized.1,6a,f,17
Table 5.
Sulfide Oxidation with In Situ Bi(4-MeObipy)-HVIa
|
All the reactions were carried out with the 0.1 mmol scale in CDCl3 (0.05 M) and yields were determined by 1HNMR with 1,2-dibromoethane as the internal standard.
Isolated yield.
−78 °C in CH2Cl2.
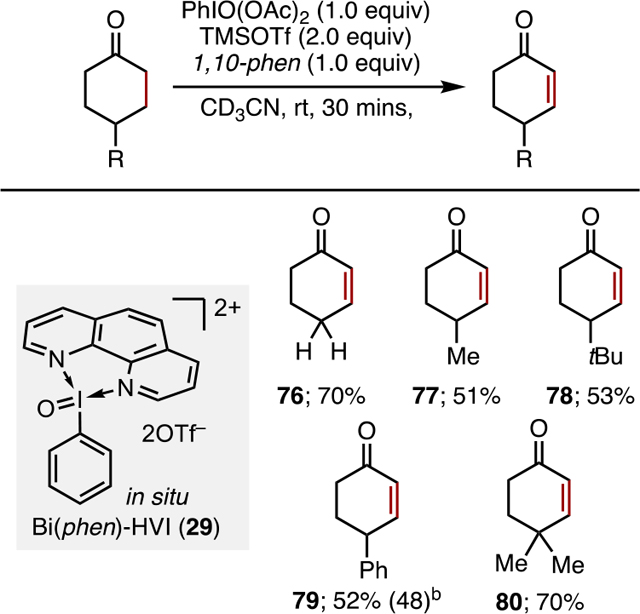
Ketone Desaturation.
Since Nicolaou and Baran’s pioneering reports on the use of IBX/DMSO,1g,19a,b the use of this and other I(V) reagents for ketone desaturation has become a staple of organic synthesis.1,19c Upon screening in situ Bi(N)-HVIs in this transformation, it was found that in situ 1,10-phenanthroline-ligated Bi(phen)-HVI 29 was effective in the desaturation of cyclohexanone, while other Bi(N)-HVIs showed markedly lower activity (Table 6).18 Several other notable reaction features included the use of acetonitrile as a solvent, which was previously ineffective, as well as the need for very short reaction times to achieve high yields. The small scope study shown in Table 6 demonstrates that while Bi(phen)-HVI 29 is capable of achieving desaturation, its utility is somewhat limited relative to other I(V) reagents such as IBX. The reason for the inferior reactivity in this case remains unclear; however, based on Nicolaou’s proposed mechanism,1g we propose two possible explanations: (1) Bi(N)-HVIs are less-efficient single-electron oxidants than other I(V) species or (2) the lack of dissociating hydroxyl ligands to act as a base hinders the desaturation process.
Table 6.
Ketone Desaturation with In Situ Bi(phen)-HVIsa
|
All the reactions were carried out on the 0.1 mmol scale in CD3CN (0.05 M) and yields were determined by 1H NMR with 1,2-dibromoethane as the internal standard.
Isolated yield.
■ CONCLUSIONS
In conclusion, we have reported on the synthesis, stability, and reactivity profiling of novel derivatives of bidentate nitrogen-ligated I(V) reagents, or Bi(N)-HVIs. These reagents can be synthesized and used in situ, or we have now identified four isolable derivatives some of which can be stored for extended periods. Bi(N)-HVIs have improved solubility in common organic solvents relative to traditional I(V) reagents and TGA analysis revealed favorable thermal stability profiles for Bi(bipy)-HVI 27 and Bi(4-MeObipy)-HVI 28 relative to PhI(O)(OAc)2 or IBX, providing a practical and potentially safer alternative to long-standing I(V) reagents. Computational study of Bi(bipy)-HVI 27 revealed the four lowest energy geometries and provided the first insights into the coordination environment and geometry at the iodine center. In addition to our prior report on phenol dearomatization, Bi(N)-HVI reactivity has now been demonstrated in alcohol and sulfide oxidation, as well as ketone desaturation, with each process optimized to use a different bidentate nitrogen ligand. With the exception of ketone desaturation, Bi(N)-HVIs performed equivalently or superior to I(V) oxidants such as IBX, DMP, or pseudocyclic analogues, while often enabling the use of a more convenient solvent or milder temperature. As a result of their facile synthesis and breadth of available bidentate heterocyclic scaffolds, Bi(N)-HVIs provide an I(V) reagent design platform with unprecedented modularity and tunability. Studies to further elucidate the structure, mechanism, and synthetic applications of these reagents are ongoing in our laboratory.
■ EXPERIMENTAL SECTION
General Information.
1H and 13C NMR spectra were recorded at 500 MHz and 125 MHz on a Bruker ADVANCE 500, 500 MHz and 125 MHz on a Bruker ADVANCE III HD, or 400 and 100 MHz on a Bruker ADVANCE 400. 1H NMR chemical shifts were reported in parts per million (ppm) from the solvent resonance (CDCl3 7.26 ppm, CD3CN 1.94 ppm, DMSO-d6 2.50 ppm). The data were reported as follows: chemical shift number, multiplicity (s = singlet, d = doublet, t = triplet, sept = septet, dd = doublet of doublets, dt = doublet of triplets, td = triplet of doublets, m = multiplet, and br = broad signal). Proton decoupled attached proton test 13C NMR shifts were reported in ppm from the solvent resonance (CDCl3 77.16 ppm, CD3CN 1.32 ppm, 118.26 ppm, DMSO-d6 39.52 ppm). The reaction solvent CDCl3 was neutralized through basic alumina and CD3CN was used from commercial sources without further purification. All the other reaction solvents were anhydrous (HPLC-grade solvent passed through an activated-alumina column). TMSOTf and pyridine were freshly distilled over CaH2 and stored over molecular sieves under Ar. All other reagents were used without further purification unless otherwise noted. Flash chromatography was carried out using Sorbent Technologies silica gel 60 Å (40–63 μm) in the solvent system listed in the individual experiments. The reactions were monitored by 1H NMR or analytical thin-layer chromatography (TLC) on Merck silica gel (60 F2s4) plates. Accurate masses for derivatized products were conducted on an Agilent 6520 Accurate-Mass Q-TOF LC/MS. Samples were taken up in a suitable solvent for analysis. The signals were mass measured against an internal lock mass reference of perfluorotributylamine for EI-GCMS and leucine enkephalin for ESI-LCMS. Waters software calibrates the instruments and reports measurements, by the use of neutral atomic masses. The mass of the electron is not included. Infrared spectra were obtained using a Thermo Nicolet iS5 FTIR spectrometer with an iD5 ATR accessory. Melting points were obtained on a Standford Research Systems MPA100 OptiMelt automated melting point system and are uncorrected.
Hypervalent iodine reagents PhIO2,20 2-t-BuSO2PhIO2,6e and ligand 1621 and 2522 were synthesized according to the corresponding literature procedures.
The dearomatization of electron-poor phenols with Bi(N)-HVIs (Scheme 2) was previously reported by our group and a summary of the key findings was included here for the sake of completion. Detailed experimental procedures and full characterization for compounds 32–56 can be found in our prior publication.11
Synthesis of Bis(acetoxy)iodylbenzene (13).
Procedure is in accordance with that reported by Yagupolski.23 To a 100 mL roundbottom flask containing iodylbenzene (0.5 g, 2.1 mmol) was added acetic anhydride (60 mL), and the mixture was stirred at room temperature for 3 h. Acetic anhydride was then removed in vacuo to give PhIO(OAc)2 (13) as white powder in quantitative yield. Note: PhI(O)(OAc)2 can be reliably stored for up to ~1 month at −20 °C under argon. 1H NMR (500 MHz, CDCl3/TFA): δ 8.33–8.03 (m, 2H), 7.93–7.60 (m, 3H), 2.17(s, 6H); 13C{1H} NMR (125 MHz, DMSO-d6): δ 172.0, 150.9, 131.4, 129.0, 126.5, 21.1.
General Procedure A: Synthesis of Isolable Bi(N)-HVI Reagents (27–30).
Procedure is in accordance with that reported by Zhdankin.9 To a flame-dried flask under argon were added bis(acetoxy)iodylbenzene (13) (0.388 g, 1.0 mmol) and dry CH2Cl2 (20 mL) to generate a white suspension. Distilled TMSOTf (0.36 mL, 2.0 mmol) in 10 mL of dry CH2Cl2 was then added, to generate a homogenous, clear solution. A solution of nitrogen ligand (1.0 mmol) in 20 mL of dry CH2Cl2 was then added slowly and the reaction mixture was stirred for 30 min followed by the removal of solvent via a rotary evaporator. The residue was then treated with dry CH2Cl2 (~30 mL) with gentle heating to 38 °C. The solution was carefully decanted to a new flask to avoid any insoluble byproducts. This solution was then treated with hexanes (~20 mL) and placed in a freezer, affording the desired Bi(N)-HVI as a white solid.
2,2′-Bipyridyl λ5-Iodane Bistriflate; Bi(bipy)-HVI (27).
Prepared according to general procedure A to give 27 as a white solid (0.505 g, 75% yield). The spectroscopic data for Bi(bipy)-HVI (27) are consistent with that previously reported.9,11 1H NMR (500 MHz, 1:20 TFA/CDCl3): δ 9.15 (d, J = 5.6 Hz, 2H), 8.93 (t, J = 8.0 Hz, 2H), 8.70 (d, J = 8.0 Hz, 2H), 8.37 (dd, J = 7.8, 5.9 Hz, 2H), 8.10–8.04 (m, 2H), 7.81–7.70 (m, 3H); 1H NMR (500 MHz, DMSO-d6): δ 8.82 (d, J = 5.0 Hz, 2H), 8.56 (d, J = 8.0 Hz, 2H), 8.26 (dt, J = 8.0, 1.5 Hz, 2H), 8.37 (dt, J = 8.0, 1.5 Hz, 2H), 7.75–7.71 (m, 2H), 7.70–7.63 (m, 3H); 13C{1H} NMR (125 MHz, DMSO-d6): δ 149.7, 148.8, 147.1, 142.0, 132.8, 129.8, 127.0, 126.7, 123.2; Anal. Calcd for C18H13F6IN2O7S2·2H2O: C, 30.44; H, 2.41; N, 3.94. Found: C, 30.94; H, 2.10; N, 3.44.
2,2′-(4,4′-MeO)Bipyridyl λ5-Iodane Bistriflate; Bi(4-MeObipy)-HVI (28).
Prepared according to general procedure A to give 28 as a white solid (0.410 g, 56% yield). 1H NMR (500 MHz, DMSO-d6): δ 8.69 (d, J = 6.5 Hz, 2H), 8.24 (d, J = 2.5 Hz, 2H), 8.07 (dd, J = 8.5, 1.5 Hz, 2H), 7.71–7.63 (m, 3H), 7.44 (dd, J = 6.0, 2.5 Hz, 2H), 4.09 (s, 6H); Anal. 13C{1H} NMR (126 MHz, DMSO): δ 169.8, 149.8, 148.8, 147.6, 137.2 132.7, 130.7, 129.8, 127.8, 127.0, 120.7 (q, J = 320.3 Hz), 112.6, 110.0, 57.3; Calcd for C20H17F6IN2O9S2·2H2O: C, 31.18; H, 2.75; N, 3.64. Found: C, 31.48; H, 2.39; N, 3.31.
1,10-Phenathrolyl λ5-Iodane Bistriflate; Bi(phen)-HVI (29).
Prepared according to general procedure A. After removing the reaction solvent in vacuo, the resulting solids were washed with CH2Cl2 (1 × 20 mL, 2 × 5 mL) to give 29 as a white solid (0.518 g, 71% yield). 1H NMR (500 MHz, DMSO-d6): δ 9.32 (dd, J = 5.0, 1.5 Hz, 2H), 9.10 (dd, J = 8.5, 1.5 Hz, 2H), 8.38 (s, 2H), 8.25 (dd, J = 8.5, 4.5 Hz, 2H), 8.08–18.05 (m, 2H), 7.73–7.63 (m, 3H); 13C{1H} NMR (125 MHz, DMSO-d6): δ 149.7, 147.7, 142.1, 137.4, 133.1, 130.1, 129.7, 127.7, 127.1, 125.9, 120.8 (q, J = 320.3 Hz); Anal. Calcd for C20H15F6IN2O7S2·2H2O: C, 32.62; H, 2.60; N, 3.80. Found: C, 32.17; H, 2.86; N, 3.83.
1,10-(5,5′-Me)Phenathrolyl λ5-Iodane Bistriflate; Bi(Mephen)-HVI (30).
Prepared according to general procedure A. After removing the reaction solvent in vacuo, the resulting solids were washed with CH2Cl2 (1 × 20 mL, 2 × 5 mL) to give 30 as a white solid (0.463 g, 61% yield). 1H NMR (500 MHz, DMSO-d6): δ 8.92 (d, J = 8.5 Hz, 2H), 8.25 (s, 2H), 8.09 (d, J = 8.0 Hz, 2H), 8.09–8.07 (m, 2H), 7.72–7.64 (m, 3H), 3.04 (s, 6H); 13C{1H} NMR (125 MHz, DMSO-d6): δ 159.1, 149.7, 141.5, 137.2, 136.6, 132.9, 130.7, 129.9, 127.8, 127.7, 127.0, 126.8, 126.6, 120.7 (q, J = 320.1 Hz), 22.9; Anal. Calcd for C22H19F6IN2O7S2·2H2O: C, 34.57; H, 2.60; N, 3.66. Found: C, 34.29; H, 2.30; N, 3.17.
General Procedure B: Oxidation of Alcohols with Bi(N)-HVI.
To a flame-dried flask with a stir bar were added bis(acetoxy)iodylbenzene (13) (74.4 mg, 0.22 mmol) in dry CH2Cl2 (0.1 M, 2 mL) followed by dry TMSOTf (80.0 μL, 0.44 mmol), generating a clear solution which was stirred for 5 min. 2,2′-Bipyridine (34.4 mg, 0.22 mmol) was then added, followed by desired alcohol (0.2 mmol). Reactions were monitored by TLC. In cases where yield was determined by 1H NMR yields, the crude reaction mixtures were filtered to remove a white solid (bipy·OTf), concentrated in vacuo, and dissolved in CDCl3, and 1,2-dibromoethane (8.7 μL, 0.1 mmol, internal standard) was added. For isolated yields, the crude reaction mixtures were concentrated in vacuo and purified by flash chromatography.
Acetophenone (58).
Prepared according to general procedure B from 1-phenyl-1-ethanol. Purified via flash chromatography (5% EtOAc/Hex) and isolated as colorless oil (21.1 mg, 88% yield). A large-scale reaction was performed on 3.95 mmol 1-phenyl-1-ethanol to give 58 (0.411 g, 87% yield). The spectroscopic data for 58 are consistent with that previously reported.24 1H NMR (500 MHz, CDCl3): δ 7.97–7.95 (m, 2H), 7.57 (d, J = 7.5 Hz, 1H), 7.49–7.45 (m, 2H), 2.61 (t, 3H).
Cyclohexanone (59).
Prepared according to general procedure B from cyclohexanol (82% NMR yield). The spectroscopic data for 59 are consistent with that previously reported.25 1H NMR (500 MHz, CDCl3): δ 2.34 (t, J = 6.5 Hz, 4H), 1.89–1.84 (m, 4H), 1.75–1.69 (m, 2H).
2-Methyl Cyclohexanone (60).
Prepared according to general procedure B from 2-methyl cyclohexanol (84% NMR yield). The spectroscopic data for 60 are consistent with that previously reported.26 1H NMR(500 MHz, CDCl3): δ 2.43–2.34(m, 2H), 2.33–2.25 (m, 1H), 2.12–2.08 (m, 2H), 1.89–1.80 (m, 1H), 1.72–1.61 (m, 2H), 1.43–1.33 (m, 1H), 1.02 (d, J = 6.5 Hz, 3H).
2-Isopropyl Cyclohexanone (61).
Prepared according to general procedure B from cis-2-isopropyl cyclohexanol (68% NMR yield). The spectroscopic data for 61 are consistent with that previously 1H reported.27 1H NMR (500 MHz, CDCl3): δ 2.44–2.31 (m, 2H), 2.29–2.23 (m, 1H), 2.11 (sep, J = 6.5 Hz, 1H), 2.00–1.90 (m, 2H), 1.89–1.81 (m, 1H), 1.77–1.68 (m, 1H), 1.65–1.51 (m, 2H), 0.89 (d, J = 4.4 Hz, 3H), 0.87 (d, J = 4.3 Hz, 3H).
Cyclohexenone (62).
Prepared according to general procedure B from cyclohexanol (68% NMR yield). The spectroscopic data for 62 are consistent with that previously reported.28 1H NMR (500 MHz, CDCl3): δ 7.00 (dt, J = 10.5, 4.0 Hz, 1H), 6.03 (dt, J = 10.5, 2.0 Hz, 1H), 2.44 (t, J = 6.5 Hz, 2H), 2.38–2.34 (m, 2H), 2.05–1.99 (m, 2H).
(2R,5R)-5-Isopropyl-2-methylcyclohexan-1-one (63).
Prepared according to general procedure B from (2R,5R)-2-methyl-5-(isopropyl)cyclohexan-1-ol. Purified via flash chromatography (5% EtOAc/Hex) and isolated as colorless oil (23.7 mg, 77% yield). The spectroscopic data for 63 are consistent with that previously reported.29 1H NMR (500 MHz, CDCl3): δ 2.35 (ddd, J = 13.0, 4.0, 2.0 Hz, 1H), 2.17–2.10 (m, 1H), 2.08–1.95 (m, 3H), 1.93–1.81 (m, 2H), 1.42–1.29 (m, 2H), 1.00 (d, J = 6.0 Hz, 3H), 0.91 (d, J = 6.5 Hz, 3H), 0.85 (d, J = 7.0 Hz, 3H).
1,3-Dihydro-2H-inden-2-one (64).
Prepared according to general procedure B from 2,3-dihydro-1H-inden-2-ol (98% NMR yield). The spectroscopic data for 64 are consistent with that previously reported.30 1H NMR (500 MHz, CDCl3): δ 7.32–7.27 (m, 5H), 3.58 (s, 4H).
3,4-Dihydronaphthalen-1(2H)-one (65).
Prepared according to general procedure B from 1,2,3,4-tetrahydronaphthalen-1-ol (79% NMR yield). The spectroscopic data for 65 are consistent with that previously reported.31 1H NMR (500 MHz, CDCl3): δ 7.04 (dd, J = 8.5, 1.5 Hz, 1H), 7.47 (td, J = 6.5, 1.5 Hz, 1H), 7.33–7.29 (m, 1H), 7.26–7.23 (m, 1H), 2.97 (t, J = 6.5 Hz, 2H), 2.67 (t, J = 6.5 Hz, 2H), 2.17–2.11 (m, 2H).
4-Phenylbutan-2-one (66).
Prepared according to general procedure B from 4-phenylbutan-2-ol (93% NMR yield). The spectroscopic data for 66 are consistent with that previously reported.32 1H NMR (500 MHz, CDCl3): δ 7.29–7.26 (m, 2H), 7.21–7.17 (m, 3H), 2.89 (d, J = 7.5 Hz, 2H), 2.76 (d, J = 8.0 Hz, 2H), 2.14 (s, 3H).
2-Phenylacetaldehyde (67).
Prepared according to general procedure B from 2-phenylethan-1-ol (71% NMR yield). The spectroscopic data for 67 are consistent with that previously reported.33 1H NMR (500 MHz, CDCl3): δ 9.75 (t, J = 2.5 Hz, 1H), 7.37 (t, J = 7.0 Hz, 2H), 7.31 (t, J = 7.5 Hz, 1H), 7.22 (d, J = 8.0 Hz, 2H), 3.69 (d, J = 2.5 Hz, 2H).
Cortisone acetate (68).
Prepared according to general procedure B from hydrocortisone acetate. Purified via flash chromatography (70% EtOAc/Hex) and isolated as a gray solid (74.8 mg, 93% yield). The spectroscopic data for 68 are consistent with that previously reported.34 1H NMR (500 MHz, CDCl3): δ 5.73 (s, 1H), 5.09 (d, J = 17.5 Hz, 1H), 4.66 (d, J = 17.5 Hz, 2H), 2.76 (d, J = 8.0 Hz, 2H), 2.14 (s, 3H), 2.87 (d, J = 12.5 Hz, 1H), 2.85–2.74 (m, 2H), 2.68–2.57 (m, 1H), 2.52–2.23 (m, 6H), 2.16 (s, 3H), 2.02–1.89 (m, 4H), 1.10–1.60 (m, 2H), 1.50–1.44 (m, 1H), 1.40 (s, 3H), 0.68 (s, 3H).
(4R)-4-((5R,8R,9S,10S,13R,14S)-10,13-Dimethyl-3,12 Dioxohexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)pentanoic acid (69).
Prepared according to general procedure B from (4R)-4((5R,8R,9S,10S,13R,14S)-3,12-dihydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)pentanoic acid, using 2.2 equiv of in situ 27. Purified via flash chromatography (50% EtOAc/Hex) and isolated as a white solid (62.2 mg, 80% yield). The spectroscopic data for 69 are consistent with that previously reported.35 1H NMR (500 MHz, CDCl3): δ 2.67–2.54 (m, 2H), 2.50–2.27 (m, 3H), 2.21–2.03 (m, 4H), 1.98–1.74 (m, 8H), 1.67–1.59 (m, 1H), 1.50–1.25 (m, 8H), 1.11 (s, 3H), 1.06 (s, 3H), 0.88 (d, J = 8.0 Hz, 3H).
General Procedure C: Oxidation of Sulfides with Bi(N)-HVI.
To a flame-dried flask with a stir bar was added bis(acetoxy)iodylbenzene (13) (33.8 mg, 0.1 mmol). With stirring, CDCl3 (2 mL) was added to form a cloudy white suspension. To this suspension was added TMSOTf (36.2 μL, 0.2 mmol) resulting in a colorless solution. After 5 min, 4,4′-dimethoxybipyridine (21.6 mg, 0.1 mmol) was added to the mixture which was then stirred at temperature indicated for oxidation (23 °C, −40 °C, or −78 °C) for 5 min before sulfide was added (0.1 mmol). After 1 h, the reaction vessel was removed from the cold bath (unless at 23 °C) and immediately quenched with water and rapidly stirred while coming to room temperature. Water was removed and the organic layer was dried with sodium sulfate, filtered, and concentrated in vacuo. 1,2-Dibromoethane (4.35 μL, 0.05 mmol) was then added as an internal standard and yields were determined by 1H NMR.
4-Nitrophenyl Methyl Sulfoxide (70).
Prepared according to general procedure C from 4-nitrothioanisole. The reaction was carried out at room temperature to give 70 (93% NMR yield). The spectroscopic data for 70 are consistent with that previously reported.36 1H NMR (500 MHz, CDCl3): δ 8.39 (dt, J = 9.0, 2.0 Hz, 2H), 7.84 (dt, J = 9.0, 2.0 Hz, 2H), 2.79 (s, 3H).
4-Cyanophenyl Methyl Sulfoxide (71).
Prepared according to general procedure C from 4-cyano-thioanisole. The reaction was carried out at room temperature to give 71 (92% NMR yield). The reaction mixture was concentrated and purified by flash chromatography (10% EtOAc/Hex) to give 71 as a white solid (14.2 mg, 86% yield). The spectroscopic data for 71 are consistent with that previously reported.37 1H NMR (500 MHz, CDCl3): δ 7.84 (dt, J = 8.5, 2.0 Hz, 2H), 7.77 (dt, J = 8.5, 1.5 Hz, 2H), 2.77 (s, 3H).
4-Bromophenyl Methyl Sulfoxide (72).
Prepared according to general procedure C from 4-bromo-thioanisole. The reaction was carried out at −40 °C to give 72 (76% NMR yield). The spectroscopic data for 72 are consistent with that previously reported.38 1H NMR (500 MHz, CDCl3): δ 7.67 (dt, J = 8.5, 2.0 Hz, 2H), 7.52 (dt, J = 8.5, 2.0 Hz, 2H), 2.72 (s, 3H).
Methyl Phenyl Sulfoxide (73).
Prepared according to general procedure C from thioanisole. The reaction was carried out at −40 °C to give 73 (73% NMR yield). The spectroscopic data for 73 are consistent with that previously reported.37 1H NMR (500 MHz, CDCl3): δ 7.56–7.51 (m, 3H), 7.18 (dd, J = 6.5, 2.0 Hz, 2H), 2.82 (s, 3H).
4-Methylphenyl Methyl Sulfoxide (74).
Prepared according to general procedure C from 4-methyl-thioanisole. The reaction was carried out at −40 °C to give 74 (44% NMR yield). The spectroscopic data for 74 are consistent with that previously reported.37 1H NMR (500 MHz, CDCl3): δ 7.54 (dt, J = 8.5, 2.0 Hz, 2H), 7.33 (d, J = 9.0 Hz, 2H), 2.70 (s, 3H), 2.41 (s, 3H).
4-Methoxyphenyl Methyl Sulfoxide (75).
Prepared according to general procedure C from 4-methoxy-thioanisole. The reaction was carried out at −78 °C in CH2Cl2 (2 mL) to give 75 (30% NMR yield). The spectroscopic data for 75 are consistent with that previously reported.37 1H NMR: δ 7.59 (dd, J = 9.0, 1.0 Hz, 2H), 7.02 (dd, J = 9.0, 1.0 Hz, 2H), 3.85 (s, 3H), 2.70 (s, 3H).
General Procedure D: Ketone Desaturation with Bi(N)-HVI.
To a flame-dried flask with PhIO(OAc)2 (13) (33.8 mg, 0.1 mmol) and 1.0 mL of CD3CN was added TMSOTf (36.2 μL, 0.2 mmol) dropwise under argon, generating a clear solution which was stirred for 5 min. Phenanthroline (18.0 mg, 0.1 mmol) was added in one portion and stirred for another 5 min. The resulting solution of Bi(phen)-HVI (29) was then transferred via a syringe into a solution of ketone (0.1 mmol) in 1.0 mL of CD3CN dropwise over 30 min. The reaction mixture was stirred for another 30 min, 1,2-dibromoethane (4.35 μL, 0.05 mmol) was added as the internal standard and yields were determined by 1H NMR.
Cyclohex-2-en-1-one (76).
Prepared according to general procedure D from cyclohexanone to give 76 (70% NMR yield). The spectroscopic data for 76 are consistent with that previously reported.39 1H NMR (400 MHz, CD3CN): δ 7.09 (dt, J = 10.0, 4.0 Hz, 1H), 5.94 (dt, J = 10.4, 2.0 Hz, 1H), 2.37 (t, J = 8.0 Hz, 2H), 2.36–2.32 (m, 2H), 2.00–1.96 (m, 2H).
4-Methylcyclohex-2-en-1-one (77).
Prepared according to general procedure D from 4-methylcyclohexanone to give 77 (51% NMR yield). The spectroscopic data for 77 are consistent with that previously reported.19c 1H NMR (500 MHz, CD3CN): δ 6.89 (dq, J = 10.0, 1.5 Hz, 1H), 5.87 (ddd, J = 10.0, 2.5, 1.0 Hz, 1H), 2.58–2.52 (m, 1H), 2.39–2.33 (m, 2H), 2.13–2.07 (m, 1H), 1.66–1.58 (m, 1H), 1.12 (d, J = 7.5 Hz, 3H).
4-(tert-Butyl)-cyclohex-2-en-1-one (78).
Prepared according to general procedure D from 4-(tert-butyl)-cyclohexanone to give 78 (53% NMR yield). The spectroscopic data for 78 are consistent with that previously reported.19c 1H NMR (500 MHz, CD3CN): δ 7.11 (dt, J = 10.5, 2.0 Hz, 1H), 5.95 (ddd, J = 10.5, 3.0, 1.0 Hz, 1H), 2.39–2.30 (m, 2H), 2.27–2.22 (m, 1H), 2.10–2.07 (m, 1H), 1.75–1.68 (m, 1H), 0.96 (s, 9H).
4-Phenylcyclohex-2-en-1-one (79).
Prepared according to general procedure D from 4-phenylcyclohexanone (52% NMR yield). The reaction mixture was concentrated and directly purified via flash chromatography (10% EtOAc/Hex) to give 79 as a white solid (8.3 mg, 48% yield). The spectroscopic data for 79 are consistent with that previously reported.19c 1H NMR (400 MHz, CDCl3): δ 7.38–7.35 (m, 2H), 7.30–7.27 (m, 1H), 7.24–7.21 (m, 2H), 7.00 (ddd, J = 10.0, 2.5, 1.5 Hz, 1H), 6.17 (dd, J = 10.0, 2.5, Hz, 1H), 3.75–3.71 (m, 1H), 2.59–2.45 (m, 2H), 2.40–2.34 (m, 1H), 2.09–2.01 (m, 1H).
4,4-Dimethylcyclohex-2-en-1-one (80).
Prepared according to general procedure D from 4,4-dimethylcyclohexanone to give 80 (70% NMR yield). The spectroscopic data for 80 are consistent with that previously reported.19c 1H NMR (500 MHz, CD3CN): δ 6.74 (dt, J = 10.0, 1.0 Hz, 1H), 5.76 (d, J = 10.0 Hz, 1H), 2.40 (t, J = 7.0 Hz, 1H), 1.85 (td, J = 6.5, 1.0 Hz, 1H),1.14 (s, 6H).
TGA Characterization.
TGA was performed on samples with a Hi-Res TGA 2950 (TA Instruments), taking care to keep exposure to air to a minimum (<120 s). Measurements were conducted under nitrogen gas, at a heating rate of 10 °C/min, from room temperature to 800 °C. For full description of thermal decomposition of Bi(N)-HVIs, see associated Supporting Information.
Supplementary Material
■ ACKNOWLEDGMENTS
The authors are grateful to the National Institutes of Health (NIH R01 GM123098) for financial support of this work, and the authors would like to acknowledge the Donors of the American Chemical Society Petroleum Research Fund for partial support of this research (DNI-56603). We are thankful to Dr. Stephanie Wunder for the use of her instrumentation for TGA studies. We thank Bilal Hoblos for running a large-scale oxidation during revision. We thank Dr. Charles DeBrosse (Temple University) for NMR spectroscopic assistance and Dr. Charles W. Ross III, Director: Automated Synthesis and Characterization at University of Pennsylvania Chemistry for providing high-resolution mass spectral data. A.A. and M.J. also thank the Australian Research Council (ARC) for project funding (DP180100904) and the Australian National Computational Infrastructure and the University of Tasmania for the generous allocation of the computing time.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.1c00375.
Computational details for structural elucidation, details on thermal decomposition studies, optimization of alcohol oxidation, sulfide oxidation, and ketone desaturation, and 1H NMR and 13C{1H} NMR spectra (PDF)
The authors declare no competing financial interest.
Contributor Information
Xiao Xiao, Department of Chemistry, Temple University, Philadelphia, Pennsylvania 19122, United States.
Jessica M. Roth, Department of Chemistry, Temple University, Philadelphia, Pennsylvania 19122, United States
Nathaniel S. Greenwood, Department of Chemistry, Temple University, Philadelphia, Pennsylvania 19122, United States
Maria K. Velopolcek, Department of Chemistry, Temple University, Philadelphia, Pennsylvania 19122, United States
Jordan Aguirre, Department of Chemistry, Temple University, Philadelphia, Pennsylvania 19122, United States.
Mona Jalali, School of Natural Sciences—Chemistry, University of Tasmania, Hobart TAS 7001, Australia.
Alireza Ariafard, School of Natural Sciences—Chemistry, University of Tasmania, Hobart TAS 7001, Australia.
Sarah E. Wengryniuk, Department of Chemistry, Temple University, Philadelphia, Pennsylvania 19122, United States.
■ REFERENCES
- (1).(a) The Chemistry of Hypervalent Halogen Compounds; Olofsson B, Marek I, Rappoport Z, Eds.; John Wiley & Sons: Chichester, 2019. [Google Scholar]; (b) Hypervalent Iodine Chemistry; Wirth T, Ed.; Springer: Switzerland, 2016. [Google Scholar]; (c) Zhdankin VV; Stang PJ Chemistry of Polyvalent Iodine. Chem. Rev. 2008, 108, 5299. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yoshimura A; Zhdankin VV Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev. 2016, 116, 3328. [DOI] [PubMed] [Google Scholar]; (e) Tohma H; Kita Y Hypervalent Iodine Reagents for the Oxidation of Alcohols and Their Application to Complex Molecule Synthesis. Adv. Synth. Catal. 2004, 346, 111. [Google Scholar]; (f) Xiao X; Wengryniuk SE Recent Advances in the Selective Oxidative Dearomatization of Phenols to o-Quinones and o-Quinols with Hypervalent Iodine Reagents. Synlett 2021, 32. DOI: 10.1055/s-0037-1610760. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Nicolaou KC; Montagnon T; Baran PS; Zhong Y-L Iodine (V) Reagents in Organic Synthesis. Part 4. O-Iodoxybenxoic Acid as a Chemospecific Tool for Single Electron Transfer-Based Oxidation Processes. J. Am. Chem. Soc. 2002, 124, 2245. [DOI] [PubMed] [Google Scholar]
- (2).(a) For reviews on I(V) reagents, see: Zhdankin VV Organoiodine (V) Reagents in Organic Synthesis. J. Org. Chem. 2011, 76, 1185. [DOI] [PubMed] [Google Scholar]; (b) Ladziata U; Zhdankin VV Hypervalent Iodine (V) Reagents in Organic Synthesis. Arkivoc 2006, 2006, 26. [Google Scholar]; (c) Satam V; Harad A; Rajule R; Pati H 2-Iodoxybenzoic acid (IBX): an efficient hypervalent iodine reagent. Tetrahedron 2010, 66, 7659. [Google Scholar]; (d) Duschek A; Kirsch SF 2-Iodoxybenzoic Acid– A Simple Oxidant with a Dazzling Array of Potential Applications. Angew. Chem., Int. Ed. 2011, 50, 1524. [DOI] [PubMed] [Google Scholar]
- (3).(a) Stevenson PJ; Treacy AB; Nieuwenhuyzen M Preparation of Dess–Martin periodinane– the role of the morphology of 1-hydroxy-1,2-benziodoxol-3(1H)-one 1-oxide precursor. J. Chem. Soc., Perkin Trans. 2 1997, 589. [Google Scholar]; (b) Alcock NW; Sawyer JF Secondary Bonding. Part 6. Distorted Octohedral Geometry in Seleninyl Dichloride–Dioxane (1/1) and Iodylbenzene. J. Chem. Soc., Dalton Trans. 1980, 115. [Google Scholar]
- (4).(a) Plumb JB; Harper DJ 2-Iodoxybenzoic acid. Chem. Eng. News 1990, 68, 2. [Google Scholar]; (b) Frigerio M; Santagostino M; Sputore S A User-Friendly Entry to 2-Iodoxybenzoic Acid (IBX). J. Org. Chem. 1999, 64, 4537. [Google Scholar]
- (5).For a review on pseudocyclic I(V) reagents, see: Yoshimura A; Yusubov MS; Zhdankin VV Synthetic applications of pseudocyclic hypervalent iodine compounds. Org. Biomol. Chem. 2016, 14, 4771. [DOI] [PubMed] [Google Scholar]
- (6).(a) For examples of acyclic and pseudocyclic I(V) derivatives, see: Koposov AY; Karimov RR; Geraskin IM; Nemykin VN; Zhdankin VV 2-Iodylphenyl Ethers: Preparation, X-Ray Crystal Structure, and Reactivity of New Hypervalent Iodine(V) Oxidizing Reagents. J. Org. Chem. 2006, 71, 8452. [DOI] [PubMed] [Google Scholar]; (b) Zhdankin VV; Koposov AY; Netzel BC; Yashin NV; Rempel BP; Ferguson MJ; Tykwinski RR IBX Amides: A New Family of Hypervalent Iodine Reagents. Angew. Chem., Int. Ed. 2003, 42, 2194. [DOI] [PubMed] [Google Scholar]; (c) Ladziata U; Koposov AY; Lo KY; Willging J; Nemykin VN; Zhdankin VV Synthesis, Structure, and Chemoselective Reactivity of N-(2-Iodylphenyl)acylamides: Hypervalent Iodine Reagents Bearing a Pseudo-Six-Membered Ring Scaffold. Angew. Chem., Int. Ed. 2005, 44, 7127. [DOI] [PubMed] [Google Scholar]; (d) Zhdankin VV; Koposov AY; Litvinov DN; Ferguson MJ; McDonald R; Luu T; Tykwinski RR Esters of 2-Iodoxybenzoic Acid: Hypervalent Iodine Oxidizing Reagents with a Pseudobenziodoxole Structure. J. Org. Chem. 2005, 70, 6484. [DOI] [PubMed] [Google Scholar]; (e) Mailyan AK; Geraskin IM; Nemykin VN; Zhdankin VV Preparation, X-Ray Structure, and Oxidative Reactivity of N-(2-Iodylphenyl)tosylamides and 2-Iodylphenyl Tosylate: Iodylarenes Stabilized by Ortho-Substitution with a Sulfonyl Group. J. Org. Chem. 2009, 74, 8444. [DOI] [PubMed] [Google Scholar]; (f) Moorthy JN; Senapati K; Parida KN 6-Membered Pseudocyclic IBX Acids: Syntheses, X-ray Structural Characteristics, and Oxidation Reactions in Common Organic Solvents. J. Org. Chem. 2010, 75, 8416. [DOI] [PubMed] [Google Scholar]; (g) Meprathu BV; Justik MW; Protasiewicz JD ortho-Phosphoryl stabilized hypervalent iodosyl- and iodyl-benzene reagents. Tetrahedron Lett. 2005, 46, 5187. [Google Scholar]
- (7).(a) For examples of cyclic I(V) derivatives see: Koposov AY; Litvinov DN; Zhdankin VV; Ferguson MJ; McDonald R; Tykwinski RR Preparation and Reductive Decomposition of 2-Iodoxybenzenesulfonic Acid. X-ray Crystal Structure of 1-Hydroxy-1H-1,2,3-benziodoxathiole 3,3-Dioxide. Eur. J. Org. Chem. 2006, 2006, 4791. [Google Scholar]; (b) Richardson RD; Zayed JM; Altermann S; Smith D; Wirth T Tetrafluoro-IBA and -IBX: Hypervalent Iodine Reagents. Angew. Chem., Int. Ed. 2007, 46, 6529. [DOI] [PubMed] [Google Scholar]; (c) Zhdankin VV; Nemykin VN; Karimov RR; Kazhkenov Z-G Preparation and X-ray crystal structure of 2-iodyl-N,N-dialkylanilineoxides: first entry into the heterocyclic system of benziodoxazole. Chem. Commun. 2008, 6131. [DOI] [PubMed] [Google Scholar]; (d) Moorthy JN; Senapati K; Parida KN; Jhulki S; Sooraj K; Nair NN Twist Does a Twist to the Reactivity: Stoichiometric and Catalytic Oxidations with Twisted Tetramethyl-IBX. J. Org. Chem. 2011, 76, 9593. [DOI] [PubMed] [Google Scholar]; (e) Cui L-Q; Dong Z-L; Liu K; Zhang C Design, Synthesis, Structure, and Dehydrogenation Reactivity of a Water-Soluble o-Iodoxybenzoic Acid Derivative Bearing a Trimethylammonium Group. Org. Lett. 2011, 13, 6488. [DOI] [PubMed] [Google Scholar]; (f) Yusubov MS; Svitich DY; Yoshimura A; Nemykin VN; Zhdankin VV 2-Iodoxybenzoic acid organosulfonates: preparation, X-ray structure and reactivity of new, powerful hypervalent iodine(V) oxidants. Chem. Commun. 2013, 49, 11269. [DOI] [PubMed] [Google Scholar]; (g) Yusubov MS; Postnikov PS; Yusubova RY; Yoshimura A; Jurjens G;Kirschning A; Zhdankin VV 2-Iodoxybenzoic Acid Tosylates: the Alternative to Dess-Martin Periodinane Oxidizing Reagents. Adv. Synth. Catal. 2017, 359, 3207. [Google Scholar]; (h) Yusubov MS; Soldatova NS; Postnikov PS; Valiev RR; Yoshimura A; Wirth T; Nemykin VN; Zhdankin VV 2-Iodoxybenzoic acid ditriflate: the most powerful hypervalent iodine(V) oxidant. Chem. Commun. 2019, 55, 7760. [DOI] [PubMed] [Google Scholar]
- (8).Dess DB; Martin JC Readily Accessible 12-I-51 Oxidant for the Conversion of Primary and Secondary Alcohols to Aldehydes and Ketones. J. Org. Chem. 1983, 48, 4155. [Google Scholar]
- (9).Zhdankin VV; Koposov AY; Yashin NV Complexes of hypervalent iodine compounds with nitrogen ligands. Tetrahedron Lett. 2002, 43, 5735. [Google Scholar]
- (10).(a) For the seminal report on N-HVIs see: Weiss R; Seubert J Electrostatic Activation of Hypervalent Organo-Iodine Compounds: Bis(onio)-Substituted Aryliodine(III) Salts. Angew. Chem., Int. Ed. 1994, 33, 891. [Google Scholar]; (b) For a review see: Corbo R; Dutton JL Weiss’ Reagents: A synthetically useful class of iodine(III) coordination compounds. Coord. Chem. Rev. 2018, 375, 69. [Google Scholar]; (c) For structural and computational studies see: Pell TP; Couchman SA; Ibrahim S; Wilson DJD; Smith BJ; Barnard PJ; Dutton JL Diverse Reactions of PhI(OTf)2 with Common 2-Electron Ligands: Complex 2 Formation, Oxidation, and Oxidative Coupling. Inorg. Chem. 2012, 51, 13034. [DOI] [PubMed] [Google Scholar]; (d) Pell TP; Couchman SA; Ibrahim S; Wilson DJD; Smith BJ; Barnard PJ; Dutton JL Diverse Reactions of PhI(OTf)2 with Common 2-Electron Ligands: Complex Formation, 2 Oxidation, and Oxidative Coupling. Inorg. Chem. 2015, 51, 13034. [DOI] [PubMed] [Google Scholar]; (e) For synthetic applications see: De Mico A; Margarita R; Piancatelli G Gazz. Chim. Ital. 1995, 215, 325. [Google Scholar]; (f) Kniep F; Walter SM; Herdtweck E; Huber SM 4,4’-Azobis(halopyridinium) Derivatives: Strong Multidentate Halogen-Bond Donors with a Redox-Active Core. Chem.—Eur. J. 2012, 18, 1306–1310. [DOI] [PubMed] [Google Scholar]; (g) Yuan Z; Cheng R; Chen P; Liu G; Liang SH Efficient Pathway for the Preparation of Aryl(isoquinoline)iodonium-(III) Salts and Synthesis of Radiofluorinated Isoquinolines. Angew. Chem., Int. Ed. 2016, 55, 11882. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Kelley BT; Walters JC; Wengryniuk SE Access to Diverse Oxygen Heterocycles via Oxidative Rearrangement of Benzylic Tertiary Alcohols. Org. Lett. 2016, 18, 1896. [DOI] [PubMed] [Google Scholar]; (i) Walters JC; Tierno AF; Dubin AH; Wengryniuk SE (Poly)cationicλ3-Iodane Mediated Oxidative Ring Expansion of Secondary Alcohols. Eur. J. Org. Chem. 2018, 2018, 1460. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Mikhael M; Adler SA; Wengryniuk SE Chemoselective Oxidation of Equatorial Alcohols with N-Ligated λ3-Iodanes. Org. Lett. 2019, 21, 5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Xiao X; Greenwood NS; Wengryniuk SE Dearomatization of Electron-Deficient Phenols to ortho-Quinones: Bidentate Nitrogen-Ligated Iodine(V) Reagents. Angew. Chem., Int. Ed. 2019, 131, 16327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).For a review on the oxidative dearomatization of phenols with I(V) reagents, see (ref 1f).
- (13).Tania; Houston SD; Sharp-Bucknall L; Poynder TB; Albayer M; Dutton JL PhI(OTf)2 Does Not Exist (Yet). Chem.—Eur. J 2020, 26, 15863. [DOI] [PubMed] [Google Scholar]
- (14).(a) Su JT; Goddard WA Enhancing 2-Iodoxybenzoic Acid Reactivity by Exploiting a Hypervalent Twist. J. Am. Chem. Soc. 2005, 127, 14146. [DOI] [PubMed] [Google Scholar]; (b) Jiang H; Sun T-Y; Wang X; Xie Y; Zhang X; Wu Y-D; Schaefer HF A Twist of the Twist Mechanism, 2-Iodoxybenzoic Acid (IBX)- Mediated Oxidation of Alcohol Revisited: Theory and Experiment. Org. Lett. 2017, 19, 6502. [DOI] [PubMed] [Google Scholar]; (c) Kaur A; Ariafard A Mechanistic investigation into phenol oxidation by IBX elucidated by DFT calculations. Org. Biomol. Chem. 2020, 18, 1117. [DOI] [PubMed] [Google Scholar]; (d) Chipman A; Farshadfar K; Smith JA; Yates BF; Ariafard A DFT-Based Comparison between Mechanistic Aspects of Amine and Alcohol Oxidation Mediated by IBX. J. Org. Chem. 2020, 85, 515. [DOI] [PubMed] [Google Scholar]
- (15).Schröckeneder A; Stichnoth D; Mayer P; Trauner D The crystal structure of the Dess–Martin periodinane. Belstein J. Org. Chem. 2012, 8, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhdankin VV; Litvinov DN; Koposov AY; Luu T; Ferguson MJ; McDonald R; Tywinski RR Preparation and structure of 2-iodoxybenzoate esters: soluble and stable periodinane oxidizing reagents. Chem. Commun. 2004, 106. [DOI] [PubMed] [Google Scholar]
- (17).Yoshimura A; Banek CT; Yusubov MS; Nemykin VN; Zhdankin VV Preparation, X-ray Structure, and Reactivity of 2-Iodylpyridines: Recyclable Hypervalent Iodine(V) Reagents. J. Org. Chem. 2011, 76, 3812. [DOI] [PubMed] [Google Scholar]
- (18).See Supporting Information for full details on screening and reaction optimization.
- (19).(a) Nicolaou KC; Zhong Y-L; Baran PS A New Method for the One-Step Synthesis of α,β-Unsaturated Carbonyl Systems from Saturated Alcohols and Carbonyl Compounds. J. Am. Chem. Soc. 2000, 122, 7596. [Google Scholar]; (b) Nicolau KC; Montagnon T; Baran PS Modulation of the Reactivity Profile of IBX by Ligand Complexation: Ambient Temperature Dehydrogenation of Aldehydes and Ketones to α,β-Unsaturated Carbonyl Compounds. Angew. Chem., Int. Ed. 2002, 41, 993. [DOI] [PubMed] [Google Scholar]; (c) Uyanik M; Akakura M; Ishihara K 2-Iodoxybenze-nesulfonic Acid as an Extremely Active Catalyst for the Selective Oxidation of Alcohols to Aldehydes, Ketones, Carboxylic Acids, and Enones with Oxone. J. Am. Chem. Soc. 2009, 131, 251. [DOI] [PubMed] [Google Scholar]
- (20).Ranganathan S; Ranganathan D; Ramachandran PV Iodoxybenzene: A Remarkably Close Ozone Equivalent. Tetrahedron 1984, 40, 3145. [Google Scholar]
- (21).Ganesan V; Sivanesan D; Yoon S Correlation between the Structure and Catalytic Activity of [Cp*Rh(Substituted Bipyridine)] Complexes for NADH Regeneration. Inorg. Chem. 2017, 56, 1366. [DOI] [PubMed] [Google Scholar]
- (22).Frey J; Kraus T; Heitz V; Sauvage J-P Synthesis of a Bismacrocycle Containing Two Back-to-Back Rigidly Connected 1,10-Phenanthroline Units as a Central Core and its Incorporation in a Handcuff-Like Catenane. Chem.—Eur. J. 2007, 13, 7584. [DOI] [PubMed] [Google Scholar]
- (23).Yagupolski LM; Maletina II; Orda VV Tetrakis-[perfluoroacyloxy]iodoarenes and Bis[acetoxy]iodylarenes; New Pentavalent Iodine Derivatives. Synthesis 1977, 1977, 574. [Google Scholar]
- (24).Moriyama K; Takemura M; Togo H Direct and Selective Benzylic Oxidation of Alkylarenes via C–H Abstraction Using Alkali Metal Bromides. Org. Lett. 2012, 14, 2414. [DOI] [PubMed] [Google Scholar]
- (25).Wang P; Cai J; Yang J; Sun C; Li L; Hu H; Ji M Zinc(II)-catalyzed oxidation of alcohols to carbonyl compounds with chloramine-T. Tetrahedron Lett. 2013, 54, 533. [Google Scholar]
- (26).Fu H; Hu C; Huang Z; Zhou J; Peng X Ammonium Tungstate as an Effective Catalyst for Selective Oxidation of Alcohols to Aldehydes or Ketones with Hydrogen Peroxide under Water – A Synergy of Graphene Oxide. Synlett 2018, 29, 447. [Google Scholar]
- (27).Malosh CF; Ready JM Catalytic Cross-Coupling of Alkylzinc Halides with α-Chloroketones. J. Am. Chem. Soc. 2004, 126, 10240. [DOI] [PubMed] [Google Scholar]
- (28).Cosner CC; Cabrera PJ; Byrd KM; Thomas AMA; Helquist P Selective Oxidation of Benzylic and Allylic Alcohols Using Mn(OAc)3/Catalytic 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone. Org. Lett. 2011, 13, 2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).LeGay CM; Gorobets E; Iftinca M; Ramachandran R; Altier C; Derksen DJ Natural-Product-Derived Transient Receptor Potential Melastatin 8 (TRPM8) Channel Modulators. Org. Lett. 2016, 18, 2746. [DOI] [PubMed] [Google Scholar]
- (30).Zhang G; Hu X; Chiang C-W; Yi H; Pei P; Singh AK; Lei A Anti-Markovnikov Oxidation of β-Alkyl Styrenes with H2O as the Terminal Oxidant. J. Am. Chem. Soc. 2016, 138, 12037. [DOI] [PubMed] [Google Scholar]
- (31).Saisaha P; Buettner L; van der Meer M; Hage R; Feringa BL; Browne WR; de Boer JW Selective Catalytic Oxidation of Alcohols, Aldehydes, Alkanes and Alkenes Employing Manganese Catalysts and Hydrogen Peroxide. Adv. Synth. Catal. 2013, 355, 2591. [Google Scholar]
- (32).Kantam ML; Kishore R; Yadav J; Sudhakar M; Venugopal A Chemoselective Hydrogenation of the Olefinic Bonds Using a Palladium/Magnesium-Lanthanum Mixed Oxide Catalyst. Adv. Synth. Catal. 2012, 354, 663. [Google Scholar]
- (33).Modak A; Deb A; Patra T; Rana S; Maity S; Maiti D A general and efficient aldehyde decarbonylation reaction by using a palladium catalyst. Chem. Commun. 2012, 48, 4253. [DOI] [PubMed] [Google Scholar]
- (34).Commercial available, CAS NO. 50-04-4.
- (35).Han YT; Yun H A Practical and Eco-friendly Synthesis of Oxo-bile Acids. Org. Prep. Proced. Int. 2016, 48, 55. [Google Scholar]
- (36).Shen H-M; Zhou W-J; Wu H-K; Yu W-B; Ai N; Ji HB; Shi H-X; She Y-B Metal-free chemoselective oxidation of sulfides to sulfoxides catalyzed by immobilized taurine and homotaurine in aqueous phase at room temperature. Tetrahedron Lett. 2015, 56, 4494. [Google Scholar]
- (37).Yu B; Guo C-X; Zhong C-L; Diao Z-F; He L-N Metal-free chemoselective oxidation of sulfides by in situ generated Koser’s reagent in aqueous media. Tetrahedron Lett. 2014, 55, 1818. [Google Scholar]
- (38).Secci F; Frongia A; Piras PP Ammonium salt catalyzed oxidation of organosulfides to organosulfoxydes. Tetrahedron Lett. 2014, 55, 603. [Google Scholar]
- (39).Lee Y-M; Hong S; Morimoto Y; Shin W; Fukuzumi S; Nam W Dioxygen Activation by a Non-Heme Iron(II) Complex: Formation of an Iron(IV)–Oxo Complex via C–H Activation by a Putative Iron(III)–Superoxo Species. J. Am. Chem. Soc. 2010, 132, 10668. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.