

Keywords: autophagy, cisplatin, fibrosis, kidney, repeated dosing
Abstract
The nephrotoxicity of cisplatin remains a major hurdle in the field of oncology. Thirty percent of patients treated with cisplatin develop acute kidney injury, and all patients are at risk for long-term impacts on kidney function. There are currently no Federal Drug Administration-approved agents to prevent or treat cisplatin-induced kidney injury. The dosing regimen used in preclinical models of nephrotoxicity may impact the success of therapeutic candidates in clinical trials. Here, we demonstrated that pharmacological inhibitors of autophagy have opposite effects when used as interventions in two different models of cisplatin-induced kidney injury. Eight-week-old male C57BL/6 mice were treated with either one dose of 20 mg/kg cisplatin or weekly doses of 9 mg/kg cisplatin for 4 wk or until body weight loss exceeded 30%. Concurrently, mice were administered multiple doses of 60 mg/kg chloroquine or 15 mg/kg 3-methyladenine attempting to globally inhibit autophagy. Mice that received a single high dose of cisplatin had worsened kidney function, inflammation, and cell death with the addition of chloroquine. 3-Methlyadenine did not impact the development of acute kidney injury in this model. In contrast, mice that received repeated low doses of cisplatin showed improved kidney function, reduced inflammation, and reduced fibrosis when treated with either chloroquine or 3-methyladenine. This study highlights how therapeutic candidates can have drastically different effects on the development of cisplatin-induced kidney injury depending on the dosing model used. This emphasizes the importance of choosing the appropriate model of injury for preclinical studies.
NEW & NOTEWORTHY This study examined how inhibition of autophagy has opposite effects on the development of acute and chronic kidney injury. Autophagy inhibition exacerbated the development of acute kidney injury following a single high dose of cisplatin but prevented the development of injury and fibrosis following repeated low doses of cisplatin.
INTRODUCTION
Despite major advances in cancer care, nephrotoxicity remains a major obstacle in the treatment of patients with cancer (1–3). The commonly used chemotherapeutic cisplatin is a prime example of this, with its success in treating cancer being largely hindered by its damaging effects on the kidney (4–6). An estimated 30% of patients treated with cisplatin develop acute kidney injury (AKI) (4, 7). Patients that do not develop AKI by clinical standards are still at risk for the long-term decline in renal function, development of fibrosis, and chronic kidney disease (CKD) (8–12). Although intravenous fluids are often given to try and reduce these side effects, there are no Federal Drug Administration-approved agents to treat or prevent the development of cisplatin-induced kidney injury. The lack of success in developing nephroprotective drugs necessitates a reevaluation of the models used for preclinical studies (13, 14).
In this study, we used two different models of cisplatin-induced kidney injury. We demonstrated that the dose and frequency of cisplatin treatment induce fundamentally different biologies, as evidenced by the different effects produced with the addition of commonly used autophagy inhibitors. Autophagy is a complex process of cellular recycling that is important in maintaining kidney homeostasis and mounting stress responses (15, 16). Autophagy has been shown to be protective in acute models of cisplatin nephrotoxicity, with inhibition leading to worsened renal function, increased DNA damage, and increased cell death (17–20). In contrast, autophagy inhibition in the unilateral ureteral obstruction (UUO) model of renal fibrosis has led to mixed results. Proximal tubule-specific knockout of autophagy-related 7 (Atg7) and pharmacological global inhibition of autophagy reduced the development of renal fibrosis (21); however, distal tubule-specific knockout of Atg7 led to increased renal fibrosis (22).
The results of this work support the hypothesis that autophagy plays different roles in the context of AKI and fibrosis development. More importantly, we demonstrated that intervention with the autophagy inhibitors chloroquine (CQ) and 3-methyladenine (3-MA) led to opposite outcomes in the acute model of cisplatin-induced kidney injury compared with the repeated low-dose model. We believe that these results highlight the importance of using multiple models of kidney injury in preclinical studies, as even the same damaging agent can cause vastly different pathologies depending on the dosing schedule.
MATERIALS AND METHODS
Animal Experiments
Seven-week-old male C57BL/6 mice were purchased from The Jackson Laboratory (Stock No. 000664) and allowed 1 wk to acclimate before all experiments began. All mice were maintained on a 12:12-h light-dark cycle and provided food and water ad libitum. Animals were maintained under standard laboratory conditions. All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Louisville and followed the guidelines of the American Veterinary Medical Association. For acute experiments, mice were intraperitoneally injected with either 0.9% N saline vehicle or cisplatin at 20 mg/kg at 8:00 AM EST. Mice were euthanized 72 h later. Mice were intraperitoneally injected with either 60 mg/kg CQ (C6628, Sigma), 15 mg/kg 3-MA (M9281, Sigma), or 0.9% N saline vehicle 1 h before cisplatin treatment and every day after until euthanasia. All mice received equal volumes of fluid. For chronic experiments, mice were intraperitoneally injected with either 0.9% N saline vehicle or 9 mg/kg cisplatin at 8:00 AM EST once a week for either 4 wk or until cisplatin-treated animals lost >30% of their body weight. Mice were intraperitoneally injected with either 60 mg/kg CQ, 15 mg/kg 3-MA, or 0.9% N saline vehicle three times a week, beginning the day before the first cisplatin treatment. All mice received equal volumes of fluid. Pharmaceutical grade cisplatin purchased from the University of Louisville hospital pharmacy (1 mg/mL in 0.9% N saline from Intas Pharmaceuticals) was used for all experiments. Mice were monitored for weight loss or evidence of high levels of discomfort/stress. Upon euthanasia, plasma was prepared and stored at −80°C. One kidney was divided into sections to be flash frozen in liquid nitrogen or fixed in 10% neutral buffered formalin. The other kidney was taken for immune cell analysis by flow cytometry.
Blood Urea Nitrogen and Neutrophil Gelatinase-Associated Lipocalin Determination
Blood urea nitrogen (BUN) was measured in the plasma of mice using a kit from AMS Diagnostics (No. 80146, AMS Diagnostics), per the manufacturer’s instructions. ELISA for neutrophil gelatinase-associated lipocalin (NGAL; DY1857, R&D Systems) was performed on mouse urine per the manufacturer’s protocol.
Gene Expression
Total RNA was isolated from the kidney cortex, and cDNA was made as previously published (23). The following predesigned TAQman primers (Life Technologies) were used: tumor necrosis factor-α (Tnfα; Mm00443258_m1), chemokine (C-X-C motif) ligand 1 (Cxcl1; Mm04207460_m1), monocyte chemoattractant protein-1 (Mcp-1; Mm00441242_m1), and β2-microglobulin (B2m; Mm00437762_m1). The following self-designed primers were used: kidney injury molecule (Kim-1) forward 5′- AGATCCACACATGTACCAACATCAA-3′ and reverse 5′- CAGTGCCATTCCAGTCTGGTTT-3′. Quantitative real-time RT-PCR was performed using iTaq Universal Probes Supermix (No. 172-5134, Bio-Rad) or iTaq Universal SYBR Green Supermix (No. 172-5124, Bio-Rad). B2m was used as the reference gene for expression analysis. Data are expressed as fold changes in the relative expression of the tested gene from vehicle-treated mice.
Protein Quantification and Western Blot Analysis
Homogenates were made from the kidney cortex using cell extraction buffer (FNN0011, Thermo Fisher Scientific), containing a cOmplete protease inhibitor cocktail tablet (No. 4693159001, Roche) and PhosSTOP phosphatase inhibitor cocktail tablet (No. 4906837001, Roche). Protein concentrations were determined using a Pierce BCA Protein Assay Kit (No. 23225, Thermo Fisher Scientific). Kidney homogenate (40 μg) was separated on 4–12% gradient Tris-glycine-SDS polyacrylamide gels and transferred to PVDF membranes, and proteins were detected by chemiluminescence substrate. The following antibodies were purchased from Cell Signaling unless otherwise noted: cleaved caspase-3 (No. 9664), C/EBP homologous protein (CHOP; No. 2895), sequestosome 1/p62 (p62; No. 5114), microtubule-associated protein light chain 3 (LC3B; No. 3868), α-smooth muscle actin (α-SMA; No. 14968), beclin-1 (No. 3495), glyceraldehyde-3-phosphate dehydrogenase (GAPDH; No. 5174), fibronectin (A3648; Sigma), and α-tubulin (sc-5286, Santa Cruz Biotechnology). Primary antibodies were used at 1:5,000 dilution and secondary antibodies were used at 1:20,000 dilution. Densitometry of Western blots was performed using ImageJ. Protein expression was graphed as the sample band density relative to its own control gene and normalized to the first vehicle sample on the gel.
Immunohistochemistry, Sirius Red/Fast Green, and Masson’s Trichrome Staining
α-SMA immunohistochemistry (ab5694, Abcam) for myofibroblasts and sirius red/fast green (SR/FG) staining for total collagen deposition were performed on paraffin-embedded kidney sections, as previously published (24). The α-SMA primary antibody was used at a dilution of 1:400. Masson’s trichrome staining was performed using a Trichrome Stain (Masson) Kit (HT15, Sigma-Aldrich), as previously described (25). Quantification of optical density for SR/FG and Masson’s trichrome staining was done as previously described (26).
Flow Cytometry
Whole kidneys were homogenized into single-cell suspensions and prepared for flow cytometry assessment, as previously described (26). Cells were blocked with CD16/32 (No. 101321, BioLegend) and extracellularly stained with CD45-PerCP (No. 103130, BioLegend). Flow cytometry was done using a BD LSRFortessa, collecting one million events per sample. Data are presented as the percentage of positively stained cells from the total number of single cells observed in that sample.
Statistical Analysis
Data are expressed as means ± SE for all experiments. Comparisons of normally distributed data sets were analyzed by one-way ANOVA, and group means were compared using Tukey’s post tests. The criteria for statistical differences were P < 0.05.
RESULTS
CQ Exacerbates and 3-MA Has No Effect on the Development of AKI Following Acute Cisplatin Treatment
Eight-week-old male C57BL/6 mice were intraperitoneally injected with 20 mg/kg cisplatin or 0.9% N saline vehicle to model cisplatin-induced AKI. CQ (60 mg/kg), 3-MA (15 mg/kg), or saline (0.9% N) were intraperitoneally administered 1 h before cisplatin injections and every day after until euthanasia on day 3. All mice received equal volumes of fluid. Following treatment, we observed that mice treated with CQ and cisplatin had significantly higher BUN than mice treated with cisplatin alone (Fig. 1A). In addition, mice treated with CQ and cisplatin had increased urinary NGAL (Fig. 1B) and significantly increased expression of Kim-1 (Fig. 1C) compared with cisplatin-only-treated mice. Mice treated with 3-MA and cisplatin had elevated BUN, urinary NGAL, and Kim-1 expression compared with vehicle-treated mice, but these elevations were not statistically different from the cisplatin-only-treated mice (Fig. 1, D–F). These results suggest that the autophagy inhibitors CQ and 3-MA exacerbate or have no effect on the development of AKI, respectively.
Figure 1.

Chloroquine (CQ) exacerbates and 3-methyladenine (3-MA) has no effect on acute kidney injury development following high-dose cisplatin (CIS) treatment. The development of acute kidney injury in the presence of CQ or 3-MA autophagy inhibition was assessed 72 h after 20 mg/kg CIS or vehicle (VEH) treatment via blood urea nitrogen (BUN; A and D), urinary neutrophil gelatinase-associated lipocalin (NGAL; B and E), and kidney injury molecule-1 (Kim-1) mRNA expression in the kidney cortex normalized to β2-microglobulin (C and F). Statistical differences were determined by one-way ANOVA followed by a Tukey’s post test. *Statistically different than VEH-treated mice. #Statistically different than CIS-treated mice. ^Statistically different than CQ- or 3-MA-treated mice. Data expressed as means ± SE; n = 2–10.
CQ Increases and 3-MA Has No Effect on High-Dose Cisplatin-Induced Kidney Inflammation
Following a single high dose of cisplatin, mice treated concurrently with CQ and cisplatin had significantly increased CD45+ immune cells in the kidney compared with mice treated with cisplatin alone (Fig. 2 and Fig. 3A). In contrast, mice that received 3-MA and cisplatin had similarly elevated levels of CD45+ immune cells in the kidney compared with mice treated with cisplatin alone (Fig. 2 and Fig. 3B). Induction of inflammatory cytokine and chemokine mRNA expression in the renal cortex followed a similar pattern. CQ treatment with cisplatin increased the expression of Mcp-1, Tnfα, and Cxcl1 compared with cisplatin treatment alone (Fig. 3, C–E). The addition of 3-MA to cisplatin had no effect on the elevation of Mcp-1, Tnfα, and Cxcl1 expression (Fig. 3, F–H). These data suggest that CQ exacerbates cisplatin-induced kidney inflammation in the high-dose model of injury, whereas 3-MA has no effect.
Figure 2.

CD45+ gating strategy. Representative images of flow cytometry gating performed on all samples are shown. Whole kidneys were homogenized before sample staining. One million events were recorded on a BD LSRFortessa. Gating was designed to eliminate dead cells and doublets before the CD45+ population was assessed. CD45+ gates were drawn based on unstained samples fun in each experiment.
Figure 3.

Chloroquine (CQ) increases high-dose cisplatin (CIS)-induced kidney inflammation, whereas 3-methyladenine (3-MA) has no effect. CD45+ total immune cell infiltration in the kidney was measured via flow cytometry of whole kidney homogenates from mice treated with 20 mg/kg CIS and either CQ (A) or 3-MA (B). Data are presented as percentages of positively labeled cells from the total number of single cells counted for each sample. mRNA expression of monocyte chemoattractant protein-1 (Mcp-1; C and F), tumor necrosis factor-α (Tnfα; D and G), and chemokine (C-X-C motif) ligand 1 (Cxcl1; E and H) were measured from renal cortex tissue. Expression was normalized to β2-microglobulin. Statistical differences were determined by one-way ANOVA followed by a Tukey’s post test. *Statistically different than vehicle (VEH)-treated mice. #Statistically different than CIS-treated mice. ^Statistically different than CQ- or 3-MA-treated mice. Data are expressed as means ± SE; n = 2–10.
CQ Amplifies High-Dose Cisplatin-Induced Endoplasmic Reticulum Stress and Cell Death, Whereas 3-MA Has No Effect
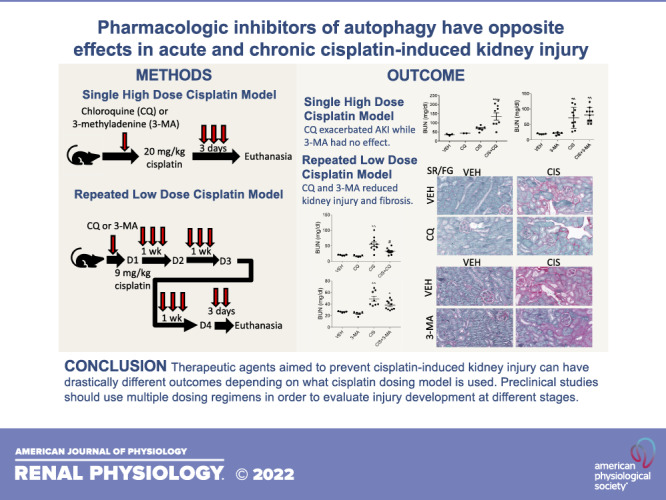
Following high-dose cisplatin treatment, levels of CHOP and cleaved caspase-3 were examined to assess endoplasmic reticulum stress and apoptosis, respectively. Mice treated with CQ and cisplatin had increased expression of both CHOP and cleaved caspase-3 compared with mice treated with cisplatin alone (Fig. 4, A–C). 3-MA had no effect on the elevation of CHOP or cleaved caspase-3 with cisplatin treatment (Fig. 4, E – G). Furthermore, we examined the expression of LC3 to assess the level of autophagy occurring in the renal cortex following treatment. Mice treated with CQ and cisplatin appeared to have increased expression of LC3-II compared with cisplatin-only-treated mice (Fig. 4, A and D). This is expected as CQ blocks the degradation of autophagosomes. 3-MA treatment with cisplatin appeared to slightly decrease the overall expression of LC3 compared with cisplatin treatment alone (Fig. 4, A and H). This is expected as 3-MA blocks the initiation of autophagy. These results suggest that CQ exacerbates high-dose cisplatin-induced endoplasmic reticulum stress and cell death, whereas 3-MA has no effect. LC3 expression suggests that both CQ and 3-MA are inhibiting autophagy, as is expected, but both drugs may also be acting through off-target effects.
Figure 4.
Chloroquine (CQ) amplifies high-dose cisplatin (CIS)-induced endoplasmic reticulum stress and cell death, whereas 3-methyladenine (3-MA) has no effect. Western blot analysis was performed to assess relative protein levels of the indicated proteins in the renal cortex of mice treated with 20 mg/kg CIS and either CQ (A) or 3-MA (E). Densitometry was performed and relative protein expression was graphed (B–D and F–H). Each sample’s value is expressed relative to its control gene and normalized to the first vehicle (VEH) sample on the gel. CC3, cleaved caspase-3; CHOP, C/EBP homologous protein; LC3, microtubule-associated protein light chain 3. n = 3–4.
CQ and 3-MA Improve Kidney Function and Reduce Injury Following Repeated Low-Dose Cisplatin Treatment
Eight-week-old male C57BL/6 mice were intraperitoneally injected with 9 mg/kg cisplatin or 0.9% N saline vehicle once a week for either 4 wk or until body weight loss reached 30%. CQ (60 mg/kg), 3-MA (15 mg/kg), or saline (0.9% N) were intraperitoneally administered three times a week beginning the day before the first cisplatin treatment. All mice received equal volumes of fluid. Mice in the CQ experiment were able to receive all four doses of cisplatin. Mice in the 3-MA experiment were only able to receive three doses of cisplatin, as the cisplatin-only-treated mice had excessive body weight loss. This variability has been previously reported (26). Nevertheless, following cisplatin treatment, we observed that both CQ and 3-MA significantly reduced BUN and urinary NGAL compared with cisplatin-only-treated mice (Fig. 5, A, B, D, and E). Interestingly, the cisplatin-induced elevation of Kim-1 expression was unchanged by the addition of either CQ or 3-MA (Fig. 5, C and F). Taken together, these results suggest that CQ and 3-MA improve kidney function and reduce injury when given concurrently with repeated low doses of cisplatin.
Figure 5.

Chloroquine (CQ) and 3-methyladenine (3-MA) prevent repeated low-dose cisplatin (CIS)-induced kidney injury. The development of CIS-induced kidney injury in the presence of CQ or 3-MA autophagy inhibition was assessed after three or four weekly 9 mg/kg CIS or vehicle (VEH) treatments. Kidney injury was evaluated via blood urea nitrogen (BUN; A and D), urinary neutrophil gelatinase-associated lipocalin (NGAL; B and E), and kidney injury molecule-1 (Kim-1) mRNA expression in the kidney cortex normalized to β2-microglobulin (C and F). Statistical differences were determined by one-way ANOVA followed by a Tukey’s post test. *Statistically different than VEH-treated mice. #Statistically different than CIS-treated mice. ^Statistically different than CQ- or 3-MA-treated mice. Data are expressed as means ± SE; n = 5–10.
CQ and 3-MA Alter Levels of Repeated Low-Dose Cisplatin-Induced Kidney Inflammation
Following repeated low-dose cisplatin treatment, mice that received CQ with cisplatin had a reduction in CD45+ immune cells in the kidney compared with cisplatin-only-treated mice, albeit not statistically significant (Fig. 6A). 3-MA treatment significantly reduced CD45+ immune cells in the kidney of cisplatin-treated mice (Fig. 6B). Interestingly, inflammatory cytokine and chemokine expression appeared to be more influenced by the addition of CQ treatment compared with 3-MA. Mcp-1 expression was significantly decreased in mice treated with CQ and cisplatin compared with mice treated with cisplatin alone (Fig. 6C). CQ treatment also nonsignificantly decreased expression of Tnfα and Cxcl1 in cisplatin-treated mice (Fig. 6, D and E). In contrast, 3-MA treatment had no effect on the cisplatin-induced elevation of Mcp-1 or Tnfα expression (Fig. 6, F and G) but did decrease the expression of Cxcl1 (Fig. 6H). Taken together, these data indicate that both CQ and 3-MA blunt inflammatory responses induced by repeated low-dose cisplatin treatment.
Figure 6.

Chloroquine (CQ) and 3-methyladenine (3-MA) reduce signs of kidney inflammation following repeated low-dose cisplatin (CIS) treatment. CD45+ total immune cell infiltration in the kidney was measured via flow cytometry of whole kidney homogenates from mice treated with three or four weekly doses of 9 mg/kg CIS and either CQ (A) or 3-MA (B). Data are presented as percentages of positively labeled cells from the total number of single cells counted for each sample. mRNA expression of monocyte chemoattractant protein-1 (Mcp-1; C and F), tumor necrosis factor-α (Tnfα; D and G), and chemokine (C-X-C motif) ligand 1 (Cxcl1; E and H) were measured from renal cortex tissue. Expression was normalized to β2-microglobulin. Statistical differences were determined by one-way ANOVA followed by a Tukey’s post test. *Statistically different than vehicle (VEH)-treated mice. #Statistically different than CIS-treated mice. ^Statistically different than CQ- or 3-MA-treated mice. Data are expressed as means ± SE; n = 5–10.
CQ and 3-MA Reduce Fibrosis Development Following Repeated Low-Dose Cisplatin Treatment
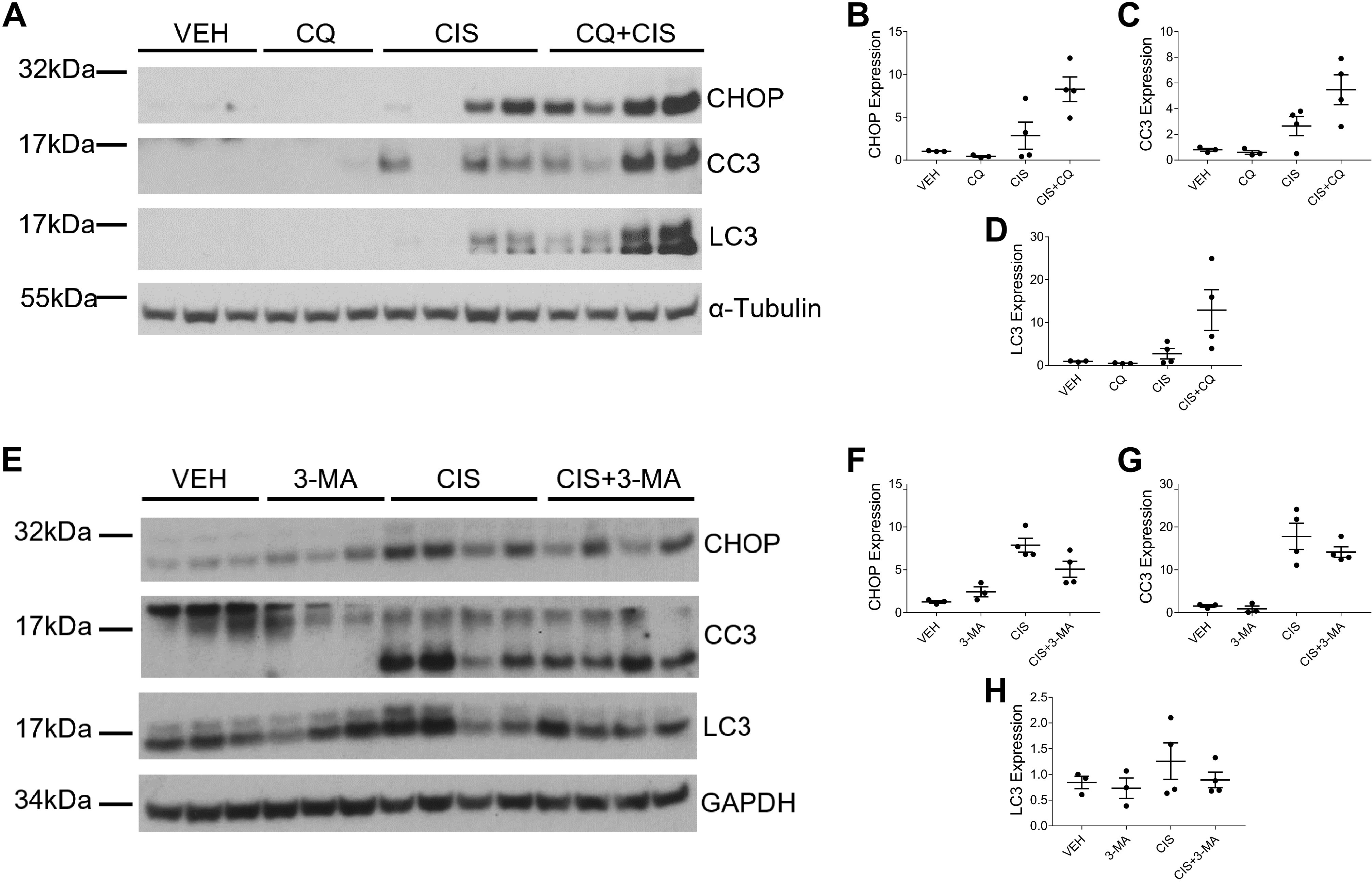
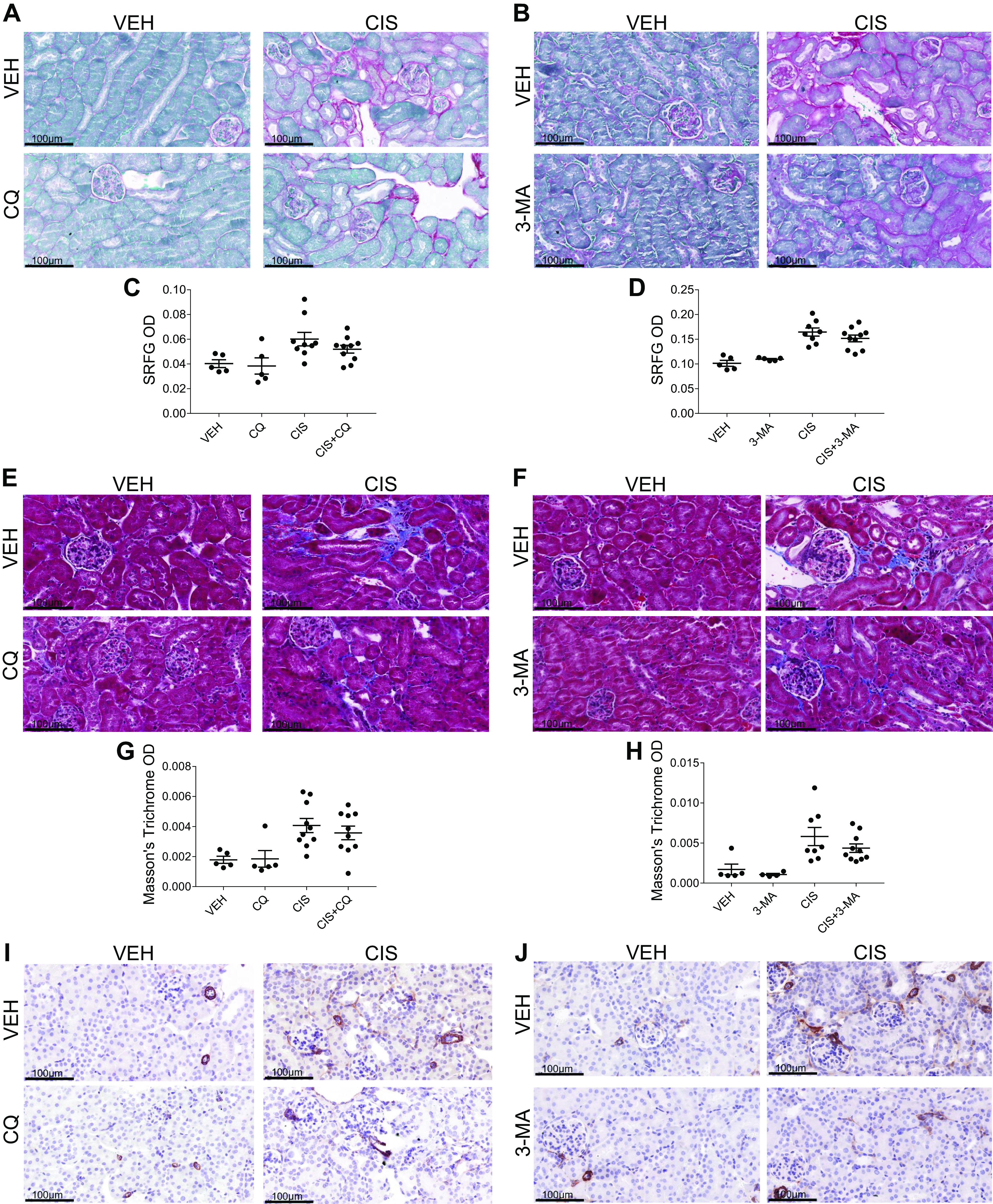
We assessed the development of fibrosis by examining levels of collagen deposition with SR/FG and Masson’s trichrome staining. We also examined the accumulation of myofibroblasts with immunohistochemistry staining for α-SMA. Repeated low-dose cisplatin treatment induces both collagen deposition and myofibroblast accumulation. SR/FG and Masson’s trichrome staining revealed that both CQ and 3-MA reduced interstitial collagen deposition when given in combination with cisplatin (Fig. 7, A–H). Likewise, CQ and 3-MA decreased the positive staining of α-SMA when given with cisplatin (Fig. 7, I and J). Furthermore, we assessed protein expression of fibronectin and α-SMA in the kidney cortex via Western blot analysis. Mice treated with CQ and cisplatin had decreased expression of both fibronectin and α-SMA (Fig. 8, A–C) compared with mice treated with cisplatin alone. Similarly, 3-MA decreased the expression of α-SMA but had less of an effect on fibronectin expression in cisplatin-treated mice (Fig. 8, D–F). These results suggest that both CQ and 3-MA reduce the development of fibrosis following repeated low-dose cisplatin treatment.
Figure 7.
Chloroquine (CQ) and 3-methyladenine (3-MA) decrease cisplatin (CIS)-induced kidney fibrosis. The development of cisplatin-induced kidney fibrosis in the presence of CQ or 3-MA autophagy inhibition was assessed after three or four weekly 9 mg/kg CIS or vehicle (VEH) treatments. Sirius red/fast green (A–D), Masson’s trichrome (E–H), and α-smooth muscle actin (α-SMA; I and J) immunohistochemistry staining of paraffin-embedded kidney sections were used to assess collagen deposition and myofibroblast accumulation. Quantification (C, D, G, and H) was calculated based on the optical density (OD) of positive staining as assessed with ImageJ. n = 5–10.
Figure 8.
Chloroquine (CQ) and 3-methyladenine (3-MA) decrease cisplatin (CIS)-induced fibronectin and α-smooth muscle actin (α-SMA) expression. Western blot analysis was performed to assess relative protein levels of the indicated proteins in the renal cortex of mice treated with three or four weekly doses of 9 mg/kg CIS or vehicle (VEH) and either CQ (A) or 3-MA (D). Densitometry was performed, and relative protein expression was graphed (B, C, E, and F). Each sample’s value is expressed relative to its control gene and normalized to the first VEH sample on the gel. n = 2–4.
CQ and 3-MA Alter Expression of Autophagy Markers
Both CQ and 3-MA are commonly used in research as pharmacological inhibitors of autophagy. We observed a decrease in LC3 expression and a slight increase in p62 expression in mice treated with CQ and cisplatin compared with cisplatin-only-treated mice (Fig. 9, A–C). 3-MA treatment decreased LC3, p62, and beclin-1 expression in cisplatin-treated mice (Fig. 9, D–G). As autophagy is a dynamic process, it is difficult to assess with static markers. We acknowledge that both CQ and 3-MA have off-target effects that may contribute to the phenotypes we observed.
Figure 9.
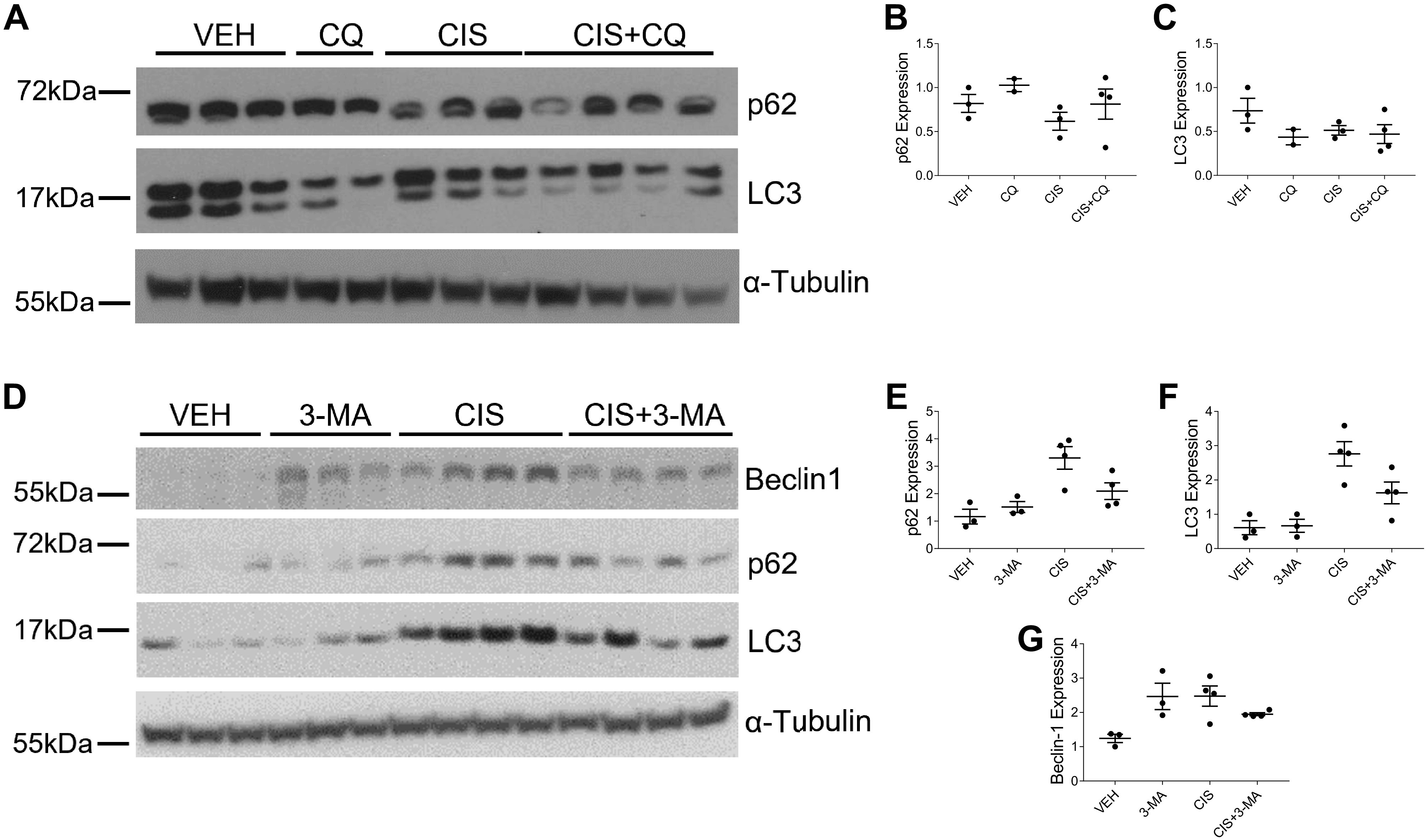
Chloroquine (CQ) and 3-methyladenine (3-MA) alter expression of autophagy markers in repeated low-dose cisplatin (CIS) treatment. Western blot analysis was performed to assess relative protein levels of the indicated proteins in the renal cortex of mice treated with three or four weekly doses of 9 mg/kg CIS or vehicle (VEH) and either CQ (A) or 3-MA (D). Protein expression was assessed on the same gel run as shown in Fig. 8; therefore, the loading control images are the same. Densitometry was performed, and relative protein expression was graphed (B, C, and E–G). Each sample’s value is expressed relative to its control gene and normalized to the first VEH sample on the gel. n = 2–4. LC3, microtubule-associated protein light chain 3; p62, sequestosome 1/p62.
DISCUSSION
In this study, we examined the effects of the commonly used autophagy inhibitors CQ and 3-MA on the development of cisplatin-induced kidney injury in two different cisplatin dosing models. We administered a single high dose of cisplatin (20 mg/kg) to mice to model nephrotoxic AKI. In this injury model, the addition of CQ exacerbated the development of AKI, increasing BUN, urinary NGAL, Kim-1 expression, inflammation, and cell death among cisplatin-treated mice. 3-MA treatment appeared to have no effect on these markers when given alongside cisplatin. In contrast, both CQ and 3-MA reduced the development of cisplatin-induced kidney injury when administered alongside weekly doses of 9 mg/kg cisplatin. Mice that received CQ or 3-MA in addition to cisplatin had reduced BUN, urinary NGAL, inflammation, and interstitial fibrosis compared with mice that received cisplatin alone. These results emphasize that different pathological processes are induced by cisplatin depending on the dosing regimen used.
The role of autophagy in cisplatin-induced AKI has been studied by several groups. Common findings suggest that the activation of autophagy is a vital stress response to high levels of cisplatin treatment. Inhibition of autophagy using either genetic models, CQ, or 3-MA has been shown to protect from cisplatin-induced DNA damage, cell death, and AKI development (17, 18, 27, 28). In addition, strategies used to induce autophagy have been demonstrated to protect mice from cisplatin-induced AKI (29–32). Our results agree that inhibition of autophagy protects mice from high levels of cisplatin nephrotoxicity.
In models of renal fibrosis, autophagy appears to play a more complex and cell-type-specific role. In the UUO model, Atg7 knockout in proximal tubules reduced the development of interstitial fibrosis (21). In contrast, Atg7 knockout in distal tubules increased the development of interstitial fibrosis in this model (22). Global inhibition of autophagy with CQ and 3-MA reduced the development of interstitial fibrosis following UUO (21). However, global genetic deficiency of LC3 and beclin-1 resulted in increased collagen deposition in this model (33).
Beclin-1 has recently gained attention as a regulator of kidney injury and fibrosis development. Ischemia-reperfusion-induced AKI was reduced in mice with knockin gain-of-function mutant beclin-1. Likewise, mice with reduced beclin-1 expression developed worse AKI following ischemia-reperfusion. The protective effects of beclin-1 in these experiments were attributed to the activation of autophagy (34). To assess the effect of beclin-1 on fibrosis development, this group also evaluated kidney injury in these mice following repeated low-dose cisplatin treatment, UUO, and recovery from ischemia-reperfusion. In each of these models, mice with decreased beclin-1 expression had worsened renal fibrosis, whereas increased beclin-1 expression reduced fibrosis development compared with wild-type mice. They also found that administration of exogenous beclin-1 after the development of ischemia-reperfusion-induced AKI suppressed subsequent fibrosis. However, the effects of beclin-1 in these studies were not determined to be autophagy dependent or independent (35).
In the ischemia-reperfusion model of injury, inhibition of autophagy with 3-MA exacerbated injury at early time points by increasing cell death and worsening renal function (36, 37). In contrast, deletion of Atg5 in proximal tubules reduced levels of renal fibrosis 30 days after ischemia-reperfusion, despite signs of increased cell death 2 h after injury (38). These results suggest that autophagy may be a key regulator of AKI-to-CKD transition processes. Our findings support this hypothesis, as we found that CQ and 3-MA treatment worsened the development of cisplatin-induced AKI but reduced the development of cisplatin-induced renal fibrosis. Understanding the role of autophagy at different time points in kidney injury is vital to developing therapeutics to improve kidney health (39).
In this study, we used CQ and 3-MA because they are commonly used in research to inhibit autophagy. However, both drugs have also been shown to have off-target effects. The dosing models of 60 mg/kg CQ and 15 mg/kg 3-MA were chosen conservatively, based on previous in vivo studies (17, 21, 40). CQ acts as a weak base, accumulating in any acidic compartment. This action inhibits autophagy by preventing autophagosome-lysosome fusion but also affects lysosome morphology, Golgi organization, and endosomal trafficking (41). In addition, CQ is known to have immunosuppressive effects, altering Toll-like receptor signaling and cytokine production (42). 3-MA is thought to act primarily as an inhibitor of phosphatidylinositol 3-kinases (PI3K). Inhibition of class III PI3K blocks autophagosome formation, preventing autophagy. However, inhibition of class I PI3K can induce autophagy in certain contexts (43, 44). Furthermore, 3-MA has been shown to stimulate lipolysis and alter immune responses (45, 46). It is imperative to consider that these alternative mechanisms of action may also be playing a role in the biologies we observed in this study. Regardless of the mechanism of action, the importance of this work lies in the fact that we observed opposite effects when the same two pharmaceuticals were given in different cisplatin dosing regimens.
To better treat and prevent cisplatin-induced kidney injury in humans, we need to have a better understanding of what pathological processes are occurring at different stages of injury. Knowing where patients are in the progression from AKI to CKD is instrumental in determining which therapeutic strategies will be successful. This study highlights just one example of this, whereby autophagy inhibition worsens AKI but prevents the development of fibrosis. The dosing regimen used in the study of nephrotoxic agents such as cisplatin is very important. The same drug can have different outcomes depending on the dosing model used. Therefore, choosing a dosing regimen that is most relevant to the clinical situation being modeled is essential. Furthermore, analyzing both acute and chronic outcomes of nephrotoxic kidney injury is essential to developing effective therapeutics.
Perspectives and Significance
This study examined the effect of classically used autophagy inhibitors, CQ and 3-MA, on the development of kidney injury and fibrosis in two different models of cisplatin-induced kidney injury. We found that these pharmacological agents exacerbated the development of AKI following a single high dose of cisplatin but reduced the development of kidney injury and fibrosis when administered alongside repeated low doses of cisplatin. These findings highlight the importance of using multiple models of cisplatin-induced kidney injury in preclinical studies.
GRANTS
This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases Grants R01DK124112 and 3R01DK124112-01S1 (to L.J.S.) and F31DK126400 (to S.M.S.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.M.S., L.J.B., and L.J.S. conceived and designed research; S.M.S., J.L.F., A.A.V., A.M.K., and M.A.D. performed experiments; S.M.S., J.L.F., A.O., P.P.S., and M.A.D. analyzed data; S.M.S. and J.L.F. interpreted results of experiments; S.M.S. prepared figures; S.M.S. drafted manuscript; S.M.S. and L.J.S. edited and revised manuscript; S.M.S., J.L.F., A.O., A.A.V., A.M.K., P.P.S., M.A.D., L.J.B., and L.J.S. approved final version of manuscript.
REFERENCES
- 1. Santos MLC, de Brito BB, da Silva FAF, Botelho A, de Melo FF. Nephrotoxicity in cancer treatment: an overview. World J Clin Oncol 11: 190–204, 2020. doi: 10.5306/wjco.v11.i4.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jagieła J, Bartnicki P, Rysz J. Nephrotoxicity as a complication of chemotherapy and immunotherapy in the treatment of colorectal cancer, melanoma and non-small cell lung cancer. IJMS 22: 4618, 2021. doi: 10.3390/ijms22094618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Perazella MA. Onco-nephrology: renal toxicities of chemotherapeutic agents. Clin J Am Soc Nephrol 7: 1713–1721, 2012. doi: 10.2215/CJN.02780312. [DOI] [PubMed] [Google Scholar]
- 4. Miller RP, Tadagavadi RK, Ramesh G, Reeves WB. Mechanisms of cisplatin nephrotoxicity. Toxins (Basel) 2: 2490–2518, 2010. doi: 10.3390/toxins2112490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int 73: 994–1007, 2008. doi: 10.1038/sj.ki.5002786. [DOI] [PubMed] [Google Scholar]
- 6. Ozkok A, Edelstein CL. Pathophysiology of cisplatin-induced acute kidney injury. Biomed Res Int 2014: 967826, 2014. doi: 10.1155/2014/967826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McMahon KR, Rassekh SR, Schultz KR, Blydt-Hansen T, Cuvelier GDE, Mammen C, Pinsk M, Carleton BC, Tsuyuki RT, Ross CJD, Palijan A, Huynh L, Yordanova M, Crépeau-Hubert F, Wang S, Boyko D, Zappitelli M; Applying Biomarkers to Minimize Long-term Effects of Childhood/Adolescent Cancer Treatment (ABLE) Research Study Group. epidemiologic characteristics of acute kidney injury during cisplatin infusions in children treated for cancer. JAMA Netw Open 3: e203639, 2020. doi: 10.1001/jamanetworkopen.2020.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chawla LS, Amdur RL, Amodeo S, Kimmel PL, Palant CE. The severity of acute kidney injury predicts progression to chronic kidney disease. Kidney Int 79: 1361–1369, 2011. doi: 10.1038/ki.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chawla LS, Eggers PW, Star RA, Kimmel PL. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med 371: 58–66, 2014. doi: 10.1056/NEJMra1214243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Coca SG, Singanamala S, Parikh CR. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int 81: 442–448, 2012. doi: 10.1038/ki.2011.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McMahon KR, Harel-Sterling M, Pizzi M, Huynh L, Hessey E, Zappitelli M. Long-term renal follow-up of children treated with cisplatin, carboplatin, or ifosfamide: a pilot study. Pediatric Nephrology 33: 2311–2320, 2018. doi: 10.1007/s00467-018-3976-5. [DOI] [PubMed] [Google Scholar]
- 12. Latcha S, Jaimes EA, Patil S, Glezerman IG, Mehta S, Flombaum CD. Long-term renal outcomes after cisplatin treatment. Clin J Am Soc Nephrol 11: 1173–1179, 2016. doi: 10.2215/CJN.08070715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hukriede NA, Soranno DE, Sander V, Perreau T, Starr MC, Yuen PST, Siskind LJ, Hutchens MP, Davidson AJ, Burmeister DM, Faubel S, de Caestecker MP. Experimental models of acute kidney injury for translational research. Nat Rev Nephrol 18: 277–293, 2022. doi: 10.1038/s41581-022-00539-2. [DOI] [PubMed] [Google Scholar]
- 14. Sears S, Siskind L. Potential therapeutic targets for cisplatin-induced kidney injury: lessons from other models of AKI and fibrosis. J Am Soc Nephrol 32: 1559–1567, 2021. doi: 10.1681/ASN.2020101455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tang C, Livingston MJ, Liu Z, Dong Z. Autophagy in kidney homeostasis and disease. Nat Rev Nephrol 16: 489–508, 2020. doi: 10.1038/s41581-020-0309-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lin TA, Wu VC, Wang CY. Autophagy in chronic kidney diseases. Cells 8: 61, 2019. doi: 10.3390/cells8010061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jiang M, Wei Q, Dong G, Komatsu M, Su Y, Dong Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int 82: 1271–1283, 2012. doi: 10.1038/ki.2012.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Takahashi A, Kimura T, Takabatake Y, Namba T, Kaimori J, Kitamura H, Matsui I, Niimura F, Matsusaka T, Fujita N, Yoshimori T, Isaka Y, Rakugi H. Autophagy guards against cisplatin-induced acute kidney injury. Am J Pathol 180: 517–525, 2012. doi: 10.1016/j.ajpath.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 19. Liao W, Wang Z, Fu Z, Ma H, Jiang M, Xu A, Zhang W. p62/SQSTM1 protects against cisplatin-induced oxidative stress in kidneys by mediating the cross talk between autophagy and the Keap1-Nrf2 signalling pathway. Free Radic Res 53: 800–814, 2019. doi: 10.1080/10715762.2019.1635251. [DOI] [PubMed] [Google Scholar]
- 20. Sears SM, Dupre TV, Shah PP, Davis DL, Doll MA, Sharp CN, Vega AA, Megyesi J, Beverly LJ, Snider AJ, Obeid LM, Hannun YA, Siskind LJ. Neutral ceramidase deficiency protects against cisplatin-induced acute kidney injury. J Lipid Res 63: 100179, 2022. doi: 10.1016/j.jlr.2022.100179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Livingston MJ, Ding HF, Huang S, Hill JA, Yin XM, Dong Z. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy 12: 976–998, 2016. doi: 10.1080/15548627.2016.1166317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nam SA, Kim W-Y, Kim JW, Park SH, Kim HL, Lee M-S, Komatsu M, Ha H, Lim JH, Park CW, Yang CW, Kim J, Kim YK. Autophagy attenuates tubulointerstital fibrosis through regulating transforming growth factor-β and NLRP3 inflammasome signaling pathway. Cell Death Dis 10: 78, 2019. doi: 10.1038/s41419-019-1356-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sharp CN, Doll MA, Megyesi J, Oropilla GB, Beverly LJ, Siskind LJ. Subclinical kidney injury induced by repeated cisplatin administration results in progressive chronic kidney disease. Am J Physiol Renal Physiol 315: F161–F172, 2018. doi: 10.1152/ajprenal.00636.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sharp CN, Doll MA, Dupre TV, Shah PP, Subathra M, Siow D, Arteel GE, Megyesi J, Beverly LJ, Siskind LJ. Repeated administration of low-dose cisplatin in mice induces fibrosis. Am J Physiol Renal Physiol 310: F560–568, 2016. doi: 10.1152/ajprenal.00512.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sears SM, Vega AA, Kurlawala Z, Oropilla GB, Krueger A, Shah PP, Doll MA, Miller R, Beverly LJ, Siskind LJ. F4/80hi resident macrophages contribute to cisplatin-induced renal fibrosis. Kidney360 3: 818–833, 2022. doi: 10.34067/KID.0006442021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sears SM, Sharp CN, Krueger A, Oropilla GB, Saforo D, Doll MA, Megyesi J, Beverly LJ, Siskind LJ. C57BL/6 mice require a higher dose of cisplatin to induce renal fibrosis and CCL2 correlates with cisplatin-induced kidney injury. Am J Physiol Renal Physiol 319: F674–F685, 2020. doi: 10.1152/ajprenal.00196.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Periyasamy-Thandavan S, Jiang M, Wei Q, Smith R, Yin X-M, Dong Z. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int 74: 631–640, 2008. doi: 10.1038/ki.2008.214. [DOI] [PubMed] [Google Scholar]
- 28. Wang S, Zhuang S, Dong Z. IFT88 deficiency in proximal tubular cells exaggerates cisplatin-induced injury by suppressing autophagy. Am J Physiol Renal Physiol 321: F269–F277, 2021. doi: 10.1152/ajprenal.00672.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu J, Livingston MJ, Dong G, Tang C, Su Y, Wu G, Yin X-M, Dong Z. Histone deacetylase inhibitors protect against cisplatin-induced acute kidney injury by activating autophagy in proximal tubular cells. Cell Death Dis 9: 322, 2018. doi: 10.1038/s41419-018-0374-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li J, Gui Y, Ren J, Liu X, Feng Y, Zeng Z, He W, Yang J, Dai C. Metformin protects against cisplatin-induced tubular cell apoptosis and acute kidney injury via AMPKα-regulated autophagy induction. Sci Rep 6: 23975, 2016. doi: 10.1038/srep23975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Park CH, Lee B, Han M, Rhee WJ, Kwak MS, Yoo T-H, Shin J-S. Canagliflozin protects against cisplatin-induced acute kidney injury by AMPK-mediated autophagy in renal proximal tubular cells. Cell death Discov 8: 12–12, 2022. doi: 10.1038/s41420-021-00801-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Song Z, Zhu J, Wei Q, Dong G, Dong Z. Canagliflozin reduces cisplatin uptake and activates Akt to protect against cisplatin-induced nephrotoxicity. Am J Physiol Renal Physiol 318: F1041–F1052, 2020. doi: 10.1152/ajprenal.00512.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ding Y, Kim S, Lee S-Y, Koo JK, Wang Z, Choi ME. Autophagy regulates TGF-β expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J Am Soc Nephrol 25: 2835–2846, 2014. doi: 10.1681/ASN.2013101068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li P, Shi M, Maique J, Shaffer J, Yan S, Moe OW, Hu MC. Beclin 1/Bcl-2 complex-dependent autophagy activity modulates renal susceptibility to ischemia-reperfusion injury and mediates renoprotection by Klotho. Am J Physiol Renal Physiol 318: F772–F792, 2020. doi: 10.1152/ajprenal.00504.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shi M, Maique J, Shepard S, Li P, Seli O, Moe OW, Hu MC. In vivo evidence for therapeutic applications of beclin 1 to promote recovery and inhibit fibrosis after acute kidney injury. Kidney Int 101: 63–78, 2022. doi: 10.1016/j.kint.2021.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang Y-L, Zhang J, Cui L-Y, Yang S. Autophagy activation attenuates renal ischemia-reperfusion injury in rats. Exp Biol Med (Maywood) 240: 1590–1598, 2015. doi: 10.1177/1535370215581306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Guan X, Qian Y, Shen Y, Zhang L, Du Y, Dai H, Qian J, Yan Y. autophagy protects renal tubular cells against ischemia/reperfusion injury in a time-dependent manner. Cell Physiol Biochem 36: 285–298, 2015. doi: 10.1159/000374071. [DOI] [PubMed] [Google Scholar]
- 38. Baisantry A, Bhayana S, Rong S, Ermeling E, Wrede C, Hegermann J, Pennekamp P, Sörensen-Zender I, Haller H, Melk A, Schmitt R. Autophagy induces prosenescent changes in proximal tubular S3 segments. J Am Soc Nephrol 27: 1609–1616, 2016. doi: 10.1681/ASN.2014111059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hu X, Ma Z, Wen L, Li S, Dong Z. Autophagy in cisplatin nephrotoxicity during cancer therapy. Cancers 13: 5618, 2021. doi: 10.3390/cancers13225618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li Q, Li L, Fei X, Zhang Y, Qi C, Hua S, Gong F, Fang M. Inhibition of autophagy with 3-methyladenine is protective in a lethal model of murine endotoxemia and polymicrobial sepsis. Innate Immun 24: 231–239, 2018. doi: 10.1177/1753425918771170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema K-J, Coppes RP, Engedal N, Mari M, Reggiori F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 14: 1435–1455, 2018. doi: 10.1080/15548627.2018.1474314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schrezenmeier E, Dörner T. Mechanisms of action of hydroxychloroquine and chloroquine: implications for rheumatology. Nat Rev Rheumatol 16: 155–166, 2020. doi: 10.1038/s41584-020-0372-x. [DOI] [PubMed] [Google Scholar]
- 43. Wu Y-T, Tan H-L, Shui G, Bauvy C, Huang Q, Wenk MR, Ong C-N, Codogno P, Shen H-M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem 285: 10850–10861, 2010. doi: 10.1074/jbc.M109.080796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lin Y-C, Kuo H-C, Wang J-S, Lin W-W. Regulation of inflammatory response by 3-methyladenine involves the coordinative actions on akt and glycogen synthase kinase 3β rather than autophagy. J Immunol 189: 4154–4164, 2012. doi: 10.4049/jimmunol.1102739. [DOI] [PubMed] [Google Scholar]
- 45. Heckmann BL, Yang X, Zhang X, Liu J. The autophagic inhibitor 3-methyladenine potently stimulates PKA-dependent lipolysis in adipocytes. Br J Pharmacol 168: 163–171, 2013. doi: 10.1111/j.1476-5381.2012.02110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dai S, Wang B, Li W, Wang L, Song X, Guo C, Li Y, Liu F, Zhu F, Wang Q, Wang X, Shi Y, Wang J, Zhao W, Zhang L. Systemic application of 3-methyladenine markedly inhibited atherosclerotic lesion in ApoE−/− mice by modulating autophagy, foam cell formation and immune-negative molecules. Cell Death Dis 7: e2498, 2016. doi: 10.1038/cddis.2016.376. [DOI] [PMC free article] [PubMed] [Google Scholar]