

Abstract
Background
The mortality of castration‐resistant prostate cancer (CRPC) is high due to lack of an effective treatment. Chimeric antigen receptor (CAR)‐based therapy is a promising immunotherapeutic strategy. Here, we aimed to design a novel CAR‐natural killer (NK) cells with a clinically significant tumoricidal effect on CRPC.
Methods
We constructed novel CAR‐NK92MI cells with a CD244‐based recombinant lentiviral vector. Different intracellular segments (CD244, NKG2D, or CD3ζ) were screened to identify the best candidate according to cell lysis assay and CD107a expression levels. To enhance the affinity of the CAR to the tumor antigen, we compared an antibody specific for prostate‐specific membrane antigen (anti‐PSMA) with PSMA‐targeted polypeptide (p‐PSMA), which was screened by phage display combinatorial library. Then, CAR‐NK92MI cells with both a high affinity for PSMA and a strong tumoricidal capacity were generated. In addition, we verified their tumor‐killing effect in vitro and in vivo. The release of cytokine by NK92MI cells was compared with that by CAR‐NK92MI cells through flow cytometry and enzyme‐linked immunosorbent assay. Moreover, ferroptosis‐related cell death was explored as a possible underlying mechanism.
Results
Three different CAR intracellular regions CAR1 (CD244), CAR2 (CD244, NKG2D) and CAR3 (CD244, NKG2D, and CD3ζ) were constructed. CAR2 was chosen to confer a stronger tumoricidal ability on CAR‐NK92MI cells. Compared with anti‐PSMA, p‐PSMA exhibited enhanced affinity for the tumor antigen. Thus, p‐PSMA‐CAR‐NK92MI cells, which expressed CAR with a polypeptide‐based antigen‐binding region, an intracellular CD244 and a NKG2D costimulatory domain, were generated. They could selectively and successfully kill PSMA+ target cells and exhibited specific lysis rate of 73.19% for PSMA‐positive C4‐2 cells and 33.04% for PSMA‐negative PC3 cells. Additionally, p‐PSMA‐CAR‐NK92MI cells had significantly higher concentrations of IFN‐γ, TNF‐α and granzyme B than NK92MI cells. In a CRPC cancer xenograft model, p‐PSMA‐CAR‐NK92MI cells significantly inhibited tumor growth and exerted a more consistent killing effect than NK92MI cells. Moreover, ferroptosis is a potential mechanism through which CAR‐NK92MI cells attack cancer cells, and is triggered by IFN‐γ.
Conclusions
p‐PSMA‐CAR‐NK92MI cells can effectively kill CRPCPSMA+ cells in vitro and in vivo. This strategy may provide additional treatment options for patients with CRPC.
Keywords: CAR‐NK92MI, chimeric antigen receptors, immunotherapy, prostate‐specific membrane antigen ferroptosis
List of abbreviations
- anti‐PSMA
PSMA antibody
- CAR
Chimeric antigen receptor
- CD244
lymphocyte activation molecule‐related receptor 2B4
- CRPC
Castration‐resistant prostate cancer
- ELISA
Enzyme‐linked immunosorbent assay
- GSH
Glutathione
- IFN‐γ
Interferon gamma
- ITAM
Immunoreceptor tyrosine‐based activation motif
- MHC
Major histocompatibility complex
- NK
Natural killer
- NKG2D
Natural killer group 2 member D
- PCa
Prostate cancer
- PI
Propidium iodide
- p‐PSMA
PSMA‐targeting polypeptide
- PSMA
Prostate‐specific membrane antigen
- scFv
Single‐chain fragment variable
- TCGA
The Cancer Genome Atlas
- TNF‐α
Tumor necrosis factor‐α
- WB
Western blotting
1. BACKGROUND
Prostate cancer (PCa) is one of the most common malignancies in males throughout the world [1]. At early stages, PCa can be surgically removed or effectively treated with radiation. However, in most patients, PCa has progressed due to lack of recognition of early symptoms [2]. Androgen deprivation therapy is commonly used for advanced PCa. However, almost all patients become less sensitive to the treatment, and the disease evolves into castration‐resistant prostate cancer (CRPC) after 18 to 24 months of hormone deprivation treatment [3]. Given the lack of effective treatment options, clinical statistics show that the median survival of patients with CRPC is less than 20 months and that CRPC can easily metastasize throughout the whole body and has a 5‐year survival rate of 28% [4]. Hence, the exploration of new treatment options is imperative for prolonging patient survival.
Due to improvements in technology and a better understanding of immunoregulatory mechanisms [5], one of the innovative therapies being investigated for use against cancers refractory to current therapies is chimeric antigen receptor (CAR)‐T cells. Studies conducted in the past few years have demonstrated that CAR‐T‐cell immunotherapy is a safe and effective treatment due to its promising results in treating adult and pediatric relapsed and/or refractory acute lymphoblastic leukemia [6, 7]. Although trials have demonstrated that the USA Food and Drug Administration (FDA)‐approved CAR‐T‐cell therapy is helpful against CD19‐expressing tumors, its efficacy remains uncertain [8, 9]. A previous study has also shown that CAR‐T cells may lead to an on‐target off‐tumor toxicity [10]. In comparison, natural killer (NK) cells could be a better alternative as a tumoricidal agent. NK cells are natural immune cells without major histocompatibility complex (MHC) restriction and do not require antigen stimulation to recognize and kill tumor cells. The early secretion of a variety of cytokines and chemokines by NK cells can regulate the acquired immune response. Moreover, these cells can be mass‐produced in vitro, and allogenic transplantation is viable, resulting in reduced costs and better treatment outcomes. In addition, NK cells have shorter life cycles than T cells and are thus safer for clinical application.
Currently, many studies have attempted to perform genetic modifications of fresh NK cells or the NK92MI cell lines with the aim of driving the expression of cytokines, Fc receptors and/or CARs specific for tumor antigens [11]. However, little progress in solid tumors has been achieved due to the complexity of the tumor microenvironment and negative immune regulation. Thus, the development of an effective CAR construct composed of an antigen‐binding region (single‐chain fragment variable, scFv), a transmembrane region and a signal transduction region (immunoreceptor tyrosine‐based activation motif, ITAM) for NK cells is urgently needed. The lymphocyte activation molecule‐related receptor 2B4 (CD244) and NKG2‐D type II integral membrane protein (NKG2D) are important immune regulators that mediate potent stimulatory and costimulatory signaling in NK cells [12]. Prostate‐specific membrane antigen (PSMA) is relatively specifically expressed in PCa. The highest expression of PSMA is observed in metastatic PCa and CRPC, which shows a PSMA‐positive frequency of 98% [13].
Herein, we established CAR‐NK cells with both a high affinity for PSMA and a clinically significant tumoricidal effect on CRPC, and these features were verified both in vitro and in vivo. Additionally, we explored the possible mechanisms underlying the CAR‐NK cell‐mediated killing of CRPC cells.
2. MATERIALS AND METHODS
2.1. Cell lines and cell culture
The cell lines C4‐2, PC‐3, LNCaP, SKVO3, NK92MI and 293T were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA). The NK92MI cell line is an interleukin‐2 (IL‐2)‐independent NK‐cell line derived from the NK92 cell line via transfection with retrovirus MFG‐hIL‐2 vector. The cancer cell lines (C4‐2, PC‐3, LNCaP, and SKVO3) were grown in RPMI 1640 medium (Gibco, Grand Island, NY, USA) supplemented with 1% penicillin/streptomycin (P/P; HyClone, Logan, UT, USA), and 10% fetal bovine serum (FBS; Bioind, Kibbutz Beit Haemek, Israel) at 37°C in a humidified atmosphere with 5% CO2. 293T cells were maintained in dulbecco's modified eagle medium (DMEM, HyClone) supplemented with 10% FBS and 1% P/P. NK92MI cells were cultured with minimum essential medium α (MEMα) (Invitrogen, Carlsbad, CA, USA) supplemented with 12.5% horse serum (Gibco), 12.5% FBS, 0.1 mmol/L β‐mercaptoethanol, 0.2 mmol/L inositol, and 1% P/P.
2.2. Bioinformatic analysis and PCa specimens
PSMA (FOHL1 gene) expression and patient survival data were obtained and analyzed from The Cancer Genome Atlas database (TCGA; https://portal.gdc.cancer.gov/) and Gene Set Cancer Analysis website (GSCA; http://bioinfo.life.hust.edu.cn/GSCA/) [14] by entering the gene name “FOLH1”, selecting the tumor type “PRAD” and the analysis content “Differential expression” and “Expression Survival”.
Tissue samples for immunohistochemistry were collected from patients who were diagnosed with prostate adenocarcinoma and treated with radical prostatectomy in Cancer Hospital, Chinese Academy of Medical Sciences (Beijing, China). Patients who received additional treatment, such as radiotherapy or chemotherapy, were not included. Included patients underwent radical prostatectomy with or without pelvic lymph node dissection.
2.3. Generation of CAR‐NK cells
The full‐length human CD244 sequence was used as the main skeleton of the CAR. The segment of the immunoglobulin‐like domain of the CD244 sequence was substituted by an anti‐mesothelin (meso) antibody scFv (SS1)‐PE38 [15], an anti‐PSMA antibody J591 scFv [16], or a PSMA‐targeting polypeptide (p‐PSMA). Either the NKG2D or the CD3ζ costimulatory domain was added to simultaneously activate NK92MI cells. The retroviral vector pLenti‐CMV/TO‐eGFP‐Puro (Addgene, Watertown, MA, USA) was lected as the backbone, and the eGFP segment was substituted by CAR to construct pLenti‐CMV/TO‐CAR‐Puro. Subsequently, 293T cells (3 × 106) were transfected with pLenti‐CMV/TO‐CAR‐Puro, psPAX2 (Addgene) and pMD2. G (Addgene) plasmid at a mass ratio of 5 μg: 3.2 μg: 1.8 μg in basic DMEM. After 8 hours, the culture medium was replaced with fresh DMEM. Every 24 hours over a 72‐hour period, the cell culture medium was collected. The collected culture medium (containing lentivirus) was passed through a 0.45‐μm filter, and centrifuged for 1 hour at 50,000 ×g. The vectors were then suspended in 1 mL of complete medium and then transfected into 2 × 105 NK92MI cells. The selection antibiotic puromycin was added to the cell culture medium. The monoclonal cells were then harvested. To confirm the expression of the CAR on NK92MI cells, CAR‐NK92MI cells were stained for PSMA and analyzed by flow cytometry.
2.4. Selection of PSMA‐binding phages and construction of polypeptide PSMA
PhD™‐12 Phage Display Peptide Library were purchased from New England Biolabs (Ipswich, MA, USA). The phage libraries were amplified and titrated according to the manufacturer's instructions. A total of 20 plaques with high affinity for recombinant PSMA protein were selected for sequencing using the PhD peptide phage libraries. Additionally, we found three other peptide carriers that efficiently targeted PSMA antigens from previous studies (GTI: GTIQPYPFSWGY; YVN: YVNSHSILGYTG; WQP: WQPDTAHHWATL) [17, 18]. We then evaluated these peptide carriers for their respective individual affinity for PSMA antigens, including peptides from PhD peptide phage libraries and three other peptides from earlier investigations. These respective binding forces for PCa cells were assessed through an immunofluorescence assay. Finally, we selected three peptides with the highest affinity to build a PSMA‐targeting polypeptide (p‐PSMA). Two replicates of each peptide were intersected with linker for linkage.
2.5. Cell lysis assay
Human cancer target cells were incubated with carboxyfluorescein succinimidyl ester (CFSE, Invitrogen) for 10 minutes at 37°C, washed three times, and cocultured with NK92MI cells at the indicated effector‐to‐target (E:T) ratios. After the incubation period, the cells were harvested and incubated with propidium iodide (PI, Sigma‐Aldrich, Louis, MI, USA) for 15 minutes in the dark. The cells were then washed with staining buffer. The samples were subjected to flow cytometric analysis using a Guava EasyCyte flow cytometer (Luminex, Shanghai, China). The specific lysis percentage was determined using the following equation: percentage of specific lysis = 100% × PI‐positive cells/CFSE‐positive cells.
2.6. Assay of CD107a expression
CAR‐NK92MI cells were incubated with or without CRPC target cells at the specified E:T ratios (1:2). After incubation for 30 minutes at 4°C, the cells were washed with fluorescence‐activated cell sorting buffer and stained with an anti‐CD56 antibody (BioLegend, San Diego, CA, USA) and anti‐CD107a antibody (BioLegend). The expression of CD107a was analyzed by flow cytometry.
2.7. Flow cytometry
The Guava@ EasyCyte system was used in this study. Approximately 5 × 105 CAR‐NK92MI cells were collected and centrifuged to obtain 200 μL of a dulbecco phosphate‐buffered saline (DPBS) suspension for evaluation. Before incubation with C4‐2 cells, CAR‐NK92MI cells were labelled with CFSE. CAR‐NK92MI cells were incubated with or without target cancer cells at an E:T ratio of 2:1. After 4 hours of incubation, we stained the cells with phycoerythrin (PE)‐labeled antibodies against granzyme B (Invitrogen), and APC‐labeled anti‐IFN‐γ antibodies (BioLegend). The binding affinity of the CAR for PSMA antigens was assessed with FITC‐labeled human PSMA (ACROBiosystems, Beijing, China).
2.8. Assays of cytokine release by Western blotting (WB) and enzyme‐linked immunosorbent assay (ELISA)
WB was performed as described previously [12]. Briefly, total cells were collected and lysed in cell lysis buffer. sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis was used for the separation of total proteins with varying molecular weights. The protein was then transferred to a solid‐phase membrane and detected by a specific antigen‐antibody binding reaction. Anti‐tag‐HRP‐DirecT (1:1000, PM020‐7, Medical Biological Laboratories Seoul, South Korea) and anti‐PSMA antibodies (1:50,000, ab133579, Abcam, Shanghai, China) were used for the detection of CAR‐NK92MI and PCa cells by WB analysis.
A total of 2 × 105 p‐PSMA‐CAR‐NK92MI cells were incubated with C4‐2 cells in 500 μL of MEMα. Twenty‐four hours later, the culture medium was collected for ELISA. The levels of IFN‐γ and TNF‐α secreted by CAR‐NK cells were measured with a commercial ELISA kit (Invitrogen) according to the manufacturer's protocol.
2.9. Immunohistochemistry (IHC) and immunofluorescence assays
Immunohistochemical staining was performed to detect the expression of proteins in tissues and cells. Anti‐PSMA antibody (1:250; ab133579), anti‐CD56 antibody (1:2000; ab220360), anti‐solute carrier family 3‐member 2 (SLC3A2) antibody (1:2500; ab244356) and anti‐solute carrier family 7‐member 11 (SLC7A11) antibody (1:500; ab37185) were purchased from Abcam. The PSMA, CD56, SLC3A2, and SLC7A11 protein expression levels were assessed by IHC according to the recommended protocol [19]. Briefly, the protocol included tissue or cell fixation, serum blocking, primary antibody incubation (4°C, 12 hours), marked second antibody incubation (25°C, 30 minutes), staining, judgment of the results and imaging. The DNA dye 4',6‐diamidino‐2‐phenylindole (DAPI) was obtained from Molecular Probes (San Francisco, CA, USA). The secondary antibody used was Cy3‐conjugated donkey anti‐rabbit IgG (Jackson ImmunoResearch Laboratory, West Grove, PA, USA). All stained cells were examined and photographed with a Leica SP5 confocal fluorescence microscope (Wetzlar, Germany).
2.10. Assessment of lipid peroxidation
To investigate the potential synergistic mechanism underlying the antitumor effect of CAR‐NK92MI cells, the induction of ferroptosis in C4‐2 cells by NK‐cell immunotherapy was studied. C4‐2 cells (20,000 cells per well) were seeded in 24‐well plates. The next day, the cells were treated with CAR‐NK92MI cells at a ratio of 1:2 for 24 hours. The cells were washed gently with PBS buffer to remove suspended NK92MI cells. The cells were stained with PerCP‐labeled anti‐CD56 antibody (BioLegend). The cells were then washed and resuspended in 1 ml of Hanks’ balanced salt solution containing 10 μmol/L BODIPY 581/591 C11 (Invitrogen) and incubated for 15 minutes at 37°C in a tissue culture incubator. The cells were washed with 200 μL of fresh PBS and then analyzed immediately by immunofluorescence staining and flow cytometry.
2.11. Transmission electron microscopy
C4‐2 cells were cultured with or without p‐PSMA‐CAR‐NK92MI cells at a ratio of 1:1 for 2 hours. The culture medium and suspended p‐PSMA‐CAR‐NK92MI cells were then removed, which left only C4‐2 cells. The cells were washed with 0.1 mol/L PBS treated with 0.1% Millipore‐filtered cacodylate‐buffered tannic acid, post‐fixed with 1% buffered osmium, and stained with 1% Millipore‐filtered uranyl acetate. After dehydration and embedding, the samples were incubated in an oven at 60 °C for 24 hours and observed with a transmission electron microscope (Field Electron and Ion Company, Hillsborough, OR, USA).
2.12. Iron assay
The intracellular ferrous iron levels were determined using an iron assay kit (Abcam) according to the manufacturer's instructions. In the iron assay protocol, ferric carrier protein dissociates ferric into solution in the presence of the acid assay buffer. Free ferrous iron (Fe2+) reacts with Iron Probe to produce a stable colored complex with absorbance at 593 nm.
2.13. Glutathione (GSH) quantification
C4‐2 cells (1,000 cells per well) were seeded in a clear‐bottom 96‐well plate. The next day, C4‐2 cells were cultured with p‐PSMA‐CAR NK92MI cells for 2 hours, and the GSH level was then measured using a GSH‐Glo Glutathione Assay kit (Promega, Madison, WI, USA). In brief, 100 μL of 1× GSH‐Glo reagent was added to each well for 30 minutes after removing the culture medium, and p‐PSMA‐CAR‐NK92MI cells were suspended. Subsequently, 100 μL of reconstituted luciferin detection reagent was added for 15 minutes. The luminescence intensity was measured using a luminometer (Tecan, Myron, Canton Zurich, Switzerland).
2.14. Animal experiments
All experimental procedures using animals were approved by the Committee on Animal Experimentation from Cancer Hospital, Chinese Academy of Medical Sciences. NOD/SCID mice were purchased from Beijing Vital River Laboratory (Beijing, China) and fed under standard laboratory conditions. After one week of adaptation, the mice were randomly divided into 3 groups: 4 mice in the blank control group, 4 mice in the NK92MI cell control group, and 4 mice in the CAR‐NK92MI cell group. For in vivo tumor establishment and bioluminescence imaging, C4‐2 cells were transduced with a lentiviral vector encoding the GFP gene. C4‐23‐eGFP cells (2 × 106) were injected into the prothorax of nude mice at 8 weeks of age (approximately 20 g). When the tumor volume reached approximately 100 mm3 (day 0), in vivo fluorescence imaging was performed. PBS, NK92MI cells or p‐PSMA‐CAR‐NK92MI cells (1×107 per mouse in 150 μL of PBS) were injected via the lateral tail vein on the following day and then every 7 days. Fluorescence imaging (Caliper Life Science, Hopkinton, MA, USA) was performed to measure the in vivo tumor volume before the injections. Once the tumor burden reached >1000 mm3 at 30 days, the mice were euthanized with carbon dioxide narcosis according to the Rutgers IACUC guidelines.
2.15. Statistical analysis
All the data were analyzed with SPSS (17.0) (International Business Machines Corporation, Armonk, NY, USA) and GraphPad Prism 8 (GraphPad Software, San Diego, CA, USA) and are reported as the mean ± standard deviation (SD). Student's t test was used to test for statistical significance. Differences among groups were compared by one‐way analysis of variance (ANOVA). The P values from pairwise comparisons were conservatively adjusted for multiple comparisons. A two‐tailed P value less than 0.05 was considered to indicate statistical significance. All the data from three independent experiments are presented.
3. RESULTS
3.1. PSMA expression in PCa
The highest expression of the FOLH1 gene (the gene encodes PSMA protein) was found in prostate adenocarcinoma (PRAD) according to pan‐cancer analysis using the GSCA website (Figure 1A). The expression of FOLH1 mRNA in PRAD tissue was significantly higher than that in normal prostate tissue (P <0.001) from the GSCA website (Figure 1B). A comparison of the survival of the two subsets (the higher expression subset and the lower expression subset) showed that higher expression of PSMA was associated with lower probability of progression‐free survival (PFS) (P = 0.040) (Figure 1C). As shown in Figure 1D, the LNCaP and C4‐2 cell lines expressed high levels of PSMA, as verified by flow cytometry. Similar results were obtained by WB (Figure 1E). Compared with normal prostate tissues, the PCa tissues of the patients showed strong positive staining for PSMA (Figure 1F). These findings suggested that PSMA molecules are specific surface antigens in PCa and could be a valid target for PCa treatment.
FIGURE 1.
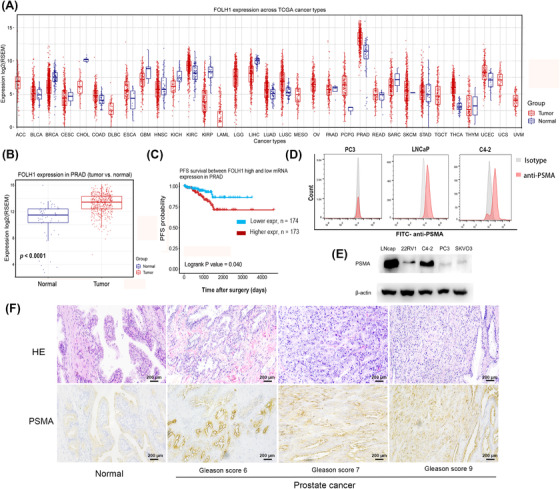
Expression of PSMA in prostate cancer. (A) FOLH1 gene expression was significantly increased in prostate cancer according to pan‐cancer analysis using the GSCA website. (B) An analysis of the GSCA website revealed significant differences in FOLH1 gene expression between normal tissues (n = 52) and prostate cancer tissues (n = 52). (C) An analysis of TCGA data revealed that the high FOLH1 expression group (n = 173) had a shorter PFS than the low expression group (n = 174) of patients with prostate cancer. (D) Flow cytometry showed higher surface expression of PSMA in LNCaP and C4‐2 cell lines than in PC3 cell lines. (E) Western blotting showed high PSMA expression in LNCaP and C4‐2 cell lines. (F) Immunohistochemical staining of PSMA in patients with prostate cancer and different Gleason scores. The PCa tissue of patients showed stronger positive staining for PSMA than normal prostate tissue. Scale bar: 200 μm. Abbreviations: PSMA, prostate‐specific membrane antigen; FOLH1, folate hydrolase 1; GSCA, Gene Set Cancer Analysis; TCGA, The Cancer Genome Atlas Program; PFS, progression‐free survival
3.2. Screening the intracellular segment of the CAR constructs
Three different optimized ITAM constructs are shown in Figure 2A. All experiments were performed with CAR‐NK constructs against the tumor‐associated antigen meso because antibodies against meso are well developed. In addition, in this scenario, it was better to design the signal transduction domain during CAR‐NK construction without interference from the other part yet to be developed. As shown in Figure 2A, CAR1 was the simplest construct based on the CD244 main skeleton. CAR2 was constructed by inserting a structure of the NKG2D intracellular domain, as a costimulatory domain, based on CAR1. CAR3 added the CD3ζ signaling intracellular domain based on CAR2. Cells from a single colony expressing one of these three constructs were tested for their ability to kill SKOV3 cells (meso+). NK92MI cells expressing NK‐cell specific CAR2 or CAR3 showed greater increases in antitumor activity when stimulated by mesohigh target cells compared with those expressing CAR1 (Figure 2B). Cells expressing these CARs also exhibited specifically enhanced CD107a expression (indicative of cytotoxic granule release) after meso+ target cell stimulation (P = 0.014, Figure 2C). CAR2‐NK92MI also had a shorter costimulatory domain than CAR3‐NK92MI. Therefore, the CAR2‐NK92MI ITAM construct was elected to confer CAR‐NK92MI cells with an enhanced tumoricidal ability.
FIGURE 2.
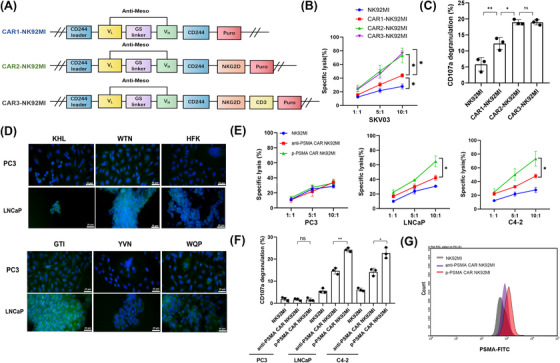
Screening of CAR constructs that enhanced NK92MI cell activity. (A) Schematic diagram of the structure of three different intracellular segments for screening CAR‐NK92MI cells with the highest antitumor activity. (B) A cell lysis assay showed that NK92MI cells expressing CAR2 or CAR3 exhibited the greatest increases in antitumor activity after coculture with mesohigh SKOV3 target cells at the indicated effector‐to‐target ratios for 4 hours. (C) The CD107a expression assay demonstrated that CAR2‐NK92MI and CAR3‐NK92MI cells showed stronger degranulation than CAR1‐NK92MI. (D) The peptides KHL, WTN and GTI exhibited stronger affinity for PSMA antigen, as demonstrated by immunofluorescence assays. The GTI, KHL, and WTN peptides showed higher affinities for their respective PSMA antigens compared with the HFK, YVN, and WQP peptides. The peptides were edited for green fluorescence. Prostate cancer cells were edited for blue fluorescence. Scale bar: 20 μm. (E) p‐PSMA‐CAR‐NK92MI cells exerted more potent antitumor effects than anti‐PSMA‐CAR‐NK92MI cells according to the cell lysis assay. (F) The CD107a expression assay indicated that p‐PSMA‐CAR‐NK92MI cells exhibited more degranulation after activation by PSMA antigen than anti‐PSMA‐CARNK92MI cells. (G) Flow cytometric results showed that the binding affinity for PSMA antigens of p‐PSMA‐CAR was superior to that of anti‐PSMA scFv. The data are presented as the means ± SEMs from three independent experiments. *, P <0.05; **, P <0.01; ns, not significant; ns, not significant. Abbreviations: CAR, chimeric antigen receptor; KHL, KHLHYHSSVRYG; WTN, WTNHHQHSKVRE; GTI, GTIQPYPFSWGY; PSMA, prostate‐specific membrane antigen; scFv, single‐chain fragment variable; Meso, mesothelin; SEM, standard error of the mean
3.3. Choice of the extracellular segment for the CAR‐NK construct
First, we searched for high‐affinity binding peptides for PSMA. Three peptides (KHL: KHLHYHSSVRYG; WTN: WTNHHQHSKVRE; and HFK: HFKAPHTVRLKS) with high repetition rates were obtained after 4 rounds of screening using the PhD™‐12 Phage Display Peptide Library (Supplementary Figure S1A). Additionally, we also found three other peptide (GTI, YVN and WQP) carriers from previous studies [17, 18]. We then evaluated all six peptide (KHL, WTN, HFK, GTI, YVN and WQP) carriers for their respective affinities for PSMA antigens. The immunofluorescence results shown in Figure 2D demonstrated that the GTI, KHL, and WTN peptides outperformed HFK, YVN, and WQP. Therefore, GTI, KHL, and WTN were selected to build p‐PSMA. We subsequently compared p‐PSMA‐CAR‐NK92MI with anti‐PSMA‐CAR‐NK92MI (J591 scFv) to find the most actively targeted structure for the extracellular segment of CAR‐NK92MI. A cytotoxicity assay of LNCaP and C4‐2 showed that p‐PSMA‐CAR‐NK92MI cells exhibited greater antitumor activity than anti‐PSMA‐CAR‐NK92MI cells (Figure 2E). The expression of these CARs also specifically enhanced CD107a expression after PSMA+ target cell stimulation (Figure 2F). The flow cytometric results showed that the binding affinity of p‐PSMA‐CAR for PSMA antigens was superior to that of anti‐PSMA scFv (Figure 2G). Therefore, the p‐PSMA‐CAR extracellular segment was elected to confer CAR‐NK93MI cells with an enhanced tumoricidal ability and affinity.
3.4. Expression of p‐PSMA‐CAR‐NK92MI and specific activation by PSMA+ cell lines
The final construct p‐PSMA‐CAR‐NK92MI included an extracellular antigen‐binding region (GTI, KHL and WTN), a transmembrane region (CD244) and an intracellular ITAM (CD244 and the NKG2D costimulatory domain) (Figure 3A). A WB analysis of modified NK92MI cells using an anti‐tag HRP‐direct antibody demonstrated the expression of p‐PSMA‐CAR (Figure 3B). Similar results were obtained by flow cytometric analysis (Figure 3C). To further validate PSMA as a specific target in PCa, cytotoxicity assays were performed. PSMA+ PCa cell lines (including LNCaP and C4‐2) and PSMA– cell lines (including PC‐3 and SKVO3) were killed by NK92MI, p‐PSMA‐CAR‐NK92MI, and anti‐meso‐CAR‐NK92MI cells. The cytotoxicity of p‐PSMA‐CAR‐NK92MI was specifically enhanced against PSMA+ PCa cell lines, whereas that of anti‐meso‐CAR‐NK92MI was specifically enhanced against meso+ SKVO3 cell lines (Figure 3D). An analysis of CD107a expression after PSMA+ target cell stimulation showed similar results (Figure 3E). Together, the data indicated that p‐PSMA‐CAR‐modified NK92MI cells could selectively and specifically kill PSMA+ target cells.
FIGURE 3.
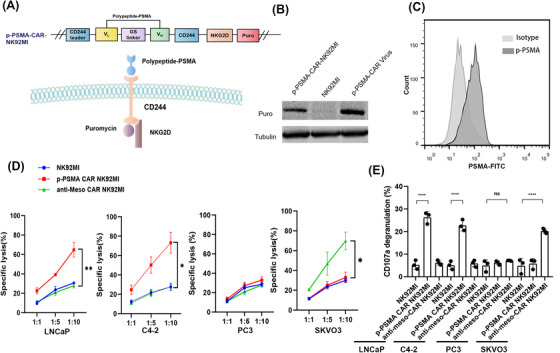
Expression of p‐PSMA‐CAR‐NK92MI and PSMA+ cell line‐specific activation of p‐PSMA‐CAR‐NK92MI cells. (A) Schematic diagram of p‐PSMA‐CAR‐NK92MI with an extracellular antigen‐binding polypeptide region (p‐PSMA), a transmembrane region (CD244), an intracellular ITAM (CD244 and the NKG2D costimulatory domain) and a puromycin‐tag. (B) Western blotting analysis indicated positive expression of CAR structures by the lentivirus and p‐PSMA‐CAR‐NK92MI cells. (C) Assessment of expression of p‐PSMA and surface expression of CAR in NK92MI cells by flow cytometry. (D) CAR‐NK92MI cells could be specifically activated to perform tumor killing as exemplified by a cell lysis assay. NK92MI, p‐PSMA‐CAR‐NK92MI and anti‐meso‐CAR‐NK92MI cells were co‐cultured with C4‐2 cells (PSMA+/meso–), LNCaP cells (PSMA+/meso–), PC3 cells (PSMA–/meso–) and SKVO3 (PSMA–/meso+) cells at the indicated effector to target ratios for 4 hours. p‐PSMA‐CAR‐modified NK92MI cells could selectively and specifically kill PSMA+ target cells. (E) Quantitative data for the percentage of surface CD107a expression after different stimulations. The data are presented as the means ± SEMs from three independent experiments. *, P <0.05; **, P <0.01; ***, P <0.001; ****, P < 0.0001; ns, not significant. Abbreviations: CD244, lymphocyte activation molecule‐related receptor 2B4; ITAM, immunoreceptor tyrosine‐based activation motif; NKG2D, natural killer group 2 member D; CAR, chimeric antigen receptor; PSMA, prostate‐specific membrane antigen; meso, mesothelin; SEM, standard error of the mean.
3.5. p‐PSMA‐CAR‐NK92MI cells kill CRPC cells in vitro
The C4‐2 cell line is a CRPC cell line derived from LNCaP cells that grow in the absence of androgens. As shown in Figure 4A, p‐PSMA‐CAR‐NK92MI cells were cocultured with C4‐2 cells at different E:T ratios for 4 hours. The cell lysis rate reached 77.87% when the ratio of p‐PSMA‐CAR‐NK92MI cells to C4‐2 cells was 15:1. p‐PSMA‐CAR‐NK92MI cells were cocultured with C4‐2 cells at an E:T ratio of 1:1 for different periods of time (Figure 4B). The killing activity of p‐PSMA‐CAR‐NK92MI cells was dose‐ and time‐dependent. The process of C4‐2 cell lysis by p‐PSMA‐CAR‐NK92MI cells was monitored (Figure 4C, Supplementary Video S1). During incubation with C4‐2 cells, p‐PSMA‐CAR‐NK92MI cells exhibited not only greater cytotoxicity but also higher release of IFN‐γ than NK92MI cells (Figure 4D). Treatment with p‐PSMA‐CAR‐NK92MI cells induced a significant increase in the concentration of TNF‐α compared with that obtained with treatment with NK92MI cells (Figure 4E). During the incubation of p‐PSMA‐CAR‐NK92MI cells with C4‐2 cells, granzyme B release was significantly dominant (Figure 4F). In addition, tumor cells were observed to undergo apoptosis once granzyme B entered the cytoplasm (Figure 4G).
FIGURE 4.
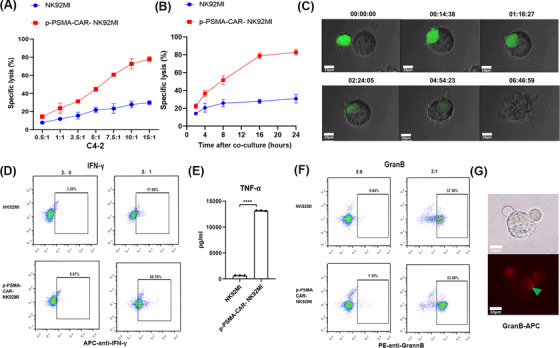
p‐PSMA‐CAR‐NK92MI cells killed a CRPC cell line in vitro. (A) p‐PSMA‐CAR‐NK92MI cells were cocultured with C4‐2 cells at different effector‐to‐target ratios for 4 hours. The specific lysis rate was dose‐dependent. (B) p‐PSMA‐CAR‐NK92MI cells were cocultured with C4‐2 cells at an effector‐to‐target ratio of 1:1 for different time periods. The killing activity of p‐PSMA‐CAR‐NK92MI cells was time‐dependent. (C) The process through which p‐PSMA‐CAR‐NK92MI cells labeled with green fluorescence lysed C4‐2 cells. p‐PSMA‐CAR‐NK92MI cancer cells were edited for green fluorescence. Scale bar: 10 μm. (D) Flow cytometry showed that p‐PSMA‐CAR enhanced IFN‐γ release by NK92MI cells. (E) ELISA revealed that p‐PSMA‐CAR‐NK92MI cells exhibited a significant higher concentration of TNF‐α than NK92MI cells. (F) NK92MI cells engineered with CAR‐CD244‐p‐PSMA secreted more granzyme B than the control after incubation with C4‐2 target cancer cells, as determined by flow cytometry. (G) Immunofluorescence results showed that granzyme B labeled with APC was released into C4‐2 cells. Activated CAR‐NK cells, stimulated by target cell binding, produced granzyme B to lyse target cells. Scale bar: 10 μm. The data are presented as the means ± SEMs from three independent experiments. ****, P <0.0001. Abbreviations: CRPC, castration‐resistant prostate cancer. ELISA, enzyme linked immunosorbent assay; IFN‐γ, interferon gamma; TNF‐α, tumor necrosis factor‐α; APC, allophycocyanin; SEM, standard error of the mean; GranB, granzyme B.
3.6. p‐PSMA‐CAR‐NK92MI cells significantly repressed CRPC tumor proliferation in vivo
C4‐2‐GPF cells (2 × 106 cells) were injected into the prothorax of NOD/SCID mice, and starting 10 days later, the mice were injected with PBS, NK92MI cells or p‐PSMA‐CAR‐NK92MI cells (1×107 per mouse in 150 μL PBS) via the lateral tail vein every 7 days (Figure 5A). The C4‐2‐GFP tumor was monitored by bioluminescent imaging (Figure 5B). p‐PSMA‐CAR‐NK92MI cells produced a significantly greater reduction in the tumor burden at 27 days compared to NK92MI cells (P = 0.005, Figure 5C). Disease progression was halted in mice treated with p‐PSMA‐CAR‐NK92MI cells, and these mice exhibited a reduction in the CRPC volume; comparable body weights were detected across all the groups (Figure 5D‐E). Immunohistochemical staining for CD56 antigen was then performed with tumor samples collected at 4, 8, or 16 hours after injection (Figure 5F). The results showed that the p‐PSMA‐CAR‐NK92MI group exerted a more consistent and effective killing effect than the NK92MI group. In addition, the level of IFN‐γ was also significantly increased (P = 0.002, Figure 5G), which suggested that p‐PSMA‐CAR‐NK92MI cells could mediate an immune response by producing IFN‐γ, and thereby boosting the antitumor effect. High levels of IL‐6 can activate clotting pathways and vascular endothelial cells and thereby cause “cytokine storms” that produce severe acute systemic inflammatory responses. We determined the safety of p‐PSMA‐CAR‐NK92MI in vivo by detecting IL‐6. The results showed no significant difference compared with the PBS group (P = 0.387, Figure 5H).
FIGURE 5.
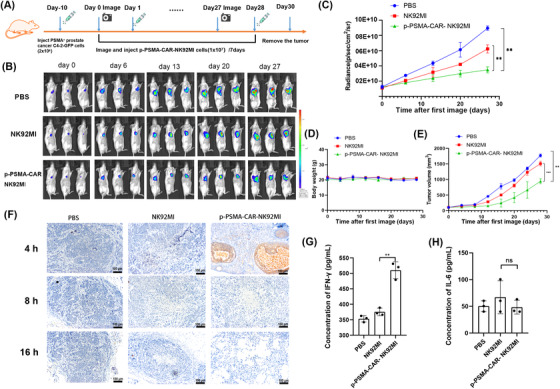
p‐PSMA‐CAR‐NK92MI cells significantly repressed CRPC tumor proliferation in vivo. (A) Diagram of the experimental design of the CRPC xenograft model. On day 0, the tumor burden (∼100 mm3) was measured, and the mice were randomly grouped. Of note, day 0 was defined as the day before CAR‐NK cell treatment. PBS, NK92MI cells or p‐PSMA‐CAR‐NK92MI cells (1×107 per mouse in 150 μL of PBS) were injected via the lateral tail vein on the following day and then every 7 days. (B) The C4‐2‐GFP tumor was monitored weekly by bioluminescent imaging. The tumor burdens of CRPC xenograft‐bearing mice treated with p‐PSMA‐CAR‐NK92MI cells, parental NK‐92MI cells, or PBS are shown. (C) The tumor burden was evaluated by bioluminescence imaging. p‐PSMA‐CAR‐NK92MI cells resulted in a significant reduction in the tumor burden at 27 days compared with that observed with either PBS or NK92MI cells. (D) The body weights did not significantly differ among the groups from B. (E) Disease progression was halted in mice treated with p‐PSMA‐CAR‐NK92MI cells from B, and a reduction in CRPC volume was observed at 27 days. (F) Immunohistochemical staining for CD56 was performed with tumor samples at 4, 8, or 16 hours after injection of PBS, NK92MI cells or p‐PSMA‐CAR‐NK92MI cells. p‐PSMA‐CAR‐NK92MI cells exhibited a more consistent tumoricidal performance than NK92MI cells. Scale bar: 100 μm. (G) The IFN‐γ levels in the tail vein blood of mice collected at 24th hour after PBS, NK92MI cells or p‐PSMA‐CAR‐NK92MI cell infusion were detected by ELISA. (F) The TNF‐a levels at 24th hour after NK92MI cell PBS, NK92MI cells or p‐PSMA‐CAR‐NK92MI cell infusion were detected by ELISA. The data are presented as the means ± SEMs from three independent experiments. **, P <0.01; ***, P <0.001; ****, ns, not significant. Abbreviations: CRPC, castration‐resistant prostate cancer. ELISA, enzyme linked immunosorbent assay; IFN‐γ, interferon gamma; TNF‐α, tumor necrosis factor‐α; GFP, green fluorescent protein; SEM, standard error of the mean
3.7. Involvement of ferroptosis in the mechanisms of p‐PSMA‐CAR‐NK92MI cell‐mediated killing
Cells that died by ferroptosis primarily harbored shrunken and damaged mitochondria, as visualized by electron microscopy, and a few other evident morphological changes prior to the point of cell death (Figure 6A). In the coculture of C4‐2 and p‐PSMA‐CAR‐NK92MI cells, activated p‐PSMA‐CAR‐NK92MI cells generated more lipid reactive oxygen species (ROS) (Figure 6B and Supplementary Figure S1B‐C). Iron exists in two oxidation states (Fe2+ or Fe3+), whereas Fe2+ accumulation is an early signal for the initiation of ferroptosis. Our biochemical analyses revealed that C4‐2 cells treated with CAR‐NK92MI cells for 2 hours contained higher intracellular Fe2+ levels than untreated C4‐2 (P = 0.004, Figure 6C). As shown in Figure 6D, both p‐PSMA‐CAR‐NK92MI cells and RSL3 increased the lipid ROS levels in C4‐2 cells; these increases were inhibited by ferrostatin‐1. Similarly, RSL3‐induced cell death was augmented in C4‐2 cells, and these effects were reversed by ferrostatin‐1 (Figure 6E). p‐PSMA‐CAR‐NK92MI cell therapy resulted in significantly increased lipid ROS levels in C4‐2 cells at different times (Figure 6F). A recent study conducted by Wang et al. [20] demonstrated that IFN‐γ produced by T cells could kill cancer cells via ferroptosis. IFN‐γ downregulated both system xc− subunits (SLC3A2 and SLC7A11) and thereby caused depletion of the intracellular GSH pool and triggered ferroptosis in cancer cells. Similarly, we hypothesized that p‐PSMA‐CAR‐NK92MI cells induced ferroptosis via this mechanism. ELISA results revealed evident increases in IFN‐γ production over time after the introduction of p‐PSMA‐CAR‐NK92MI cells, similar to the ROS results (Figure 6G). The increases in lipid ROS in C4‐2 cells induced by the p‐PSMA‐CAR‐NK92MI cell supernatant could be reversed by anti‐IFN‐γ antibodies but not by anti‐TNF antibodies (Figure 6H). As shown in Figure 6I, the expression of SLC3A2 and SLC7A11 in tumor sections was significantly reduced in the p‐PSMA‐CAR‐NK92MI cell group. Together, the results from the two experiments suggested that p‐PSMA‐CAR‐NK92MI cells reliably downregulated SLC3A2 and SLC7A11 expression by releasing more IFN‐γ, which resulted in depletion of the intracellular GSH pool and the triggering of ferroptosis in cancer cells (Figure 6J).
FIGURE 6.
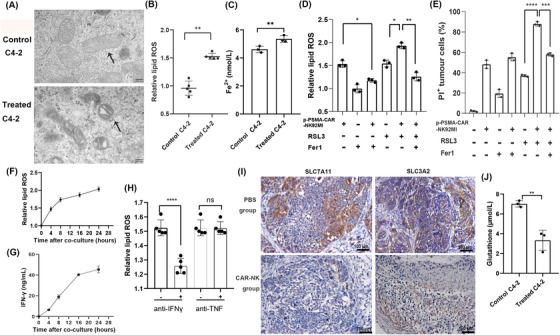
Involvement of ferroptosis in p‐PSMA‐CAR‐NK92MI cell‐mediated killing. (A) C4‐2 cell morphology was observed by transmission electron microscopy after 2 hours of culture alone or coculture with p‐PSMA‐CAR‐NK92MI cells. The cells that died by ferroptosis primarily exhibited shrunken and damaged mitochondria. Scale bar: 200 nm. (B) Flow cytometry revealed a significant difference in the relative lipid ROS levels in C4‐2 cells after coculturing with p‐PSMA‐CAR‐NK92MI cells (E:T ratio of 0.5:1) for 24 hours. (C) Our biochemical analyses revealed that C4‐2 cells treated with CAR‐NK92MI cells for 4 hours contained higher intracellular Fe2+ levels than untreated C4‐2 cells. (D) C4‐2 cells cocultured with p‐PSMA‐CAR‐NK92MI cells (E:T ratio of 0.5:1) for 24 hours were treated with RSL3 (2 μmol/L) or ferrostatin‐1 (10 μmol/L). p‐PSMA‐CAR‐NK92MI cells increased the lipid ROS levels in C4‐2 cells, and RSL3 increased the lipid ROS levels, these increases were inhibited by ferrostatin‐1. (E) C4‐2 cells cocultured with p‐PSMA‐CAR‐NK92MI cells (E:T ratio of 0.5:1) for 24 hours were treated with RSL3 (2 μmol/L) or ferrostatin‐1 (10 μmol/L). And augmented RSL3‐induced cell death in C4‐2 cells, and these effects were reversed by ferrostatin‐1. (F) The concentration of lipid ROS in C4‐2 cells after p‐PSMA‐CAR‐NK92MI cell therapy increased over time. (G) ELISA revealed evident increases in IFN‐γ production over time after p‐PSMA‐CAR‐NK92MI cell introduction. (H) The increase in lipid ROS in C4‐2 cells induced by the p‐PSMA‐CAR‐NK92MI cell supernatant could be reversed by anti‐IFN‐γ antibodies but not by anti‐TNF antibodies. (I) Immunohistochemistry revealed a significant reduction in SLC7A11 and SLC3A2 expression in tumor sections from the p‐PSMA‐CAR‐NK92MI cell treated mice. Scale bar: 100 μm. (G) The intracellular GSH levels were significantly higher in C4‐2 cells treated with CAR‐NK92MI cells (E:T ratio of 1:1) for 2 hours than in untreated C4‐2 cells. The data are presented as the means ± SEMs from three independent experiments. *, P <0.05; **, P <0.01; ***, P <0.001; ****, P <0.0001; ns, not significant. Abbreviations: ROS, reactive oxygen species; E:T, effector‐to‐target; ELISA, enzyme linked immunosorbent assay; IFN‐γ, interferon gamma; TNF, tumor necrosis factor; SLC7A11, solute carrier family 7 member 11; SLC3A2, solute carrier family 3 member 2; GSH, glutathione; SEM, standard error of the mean.
4. DISCUSSION
Genetically modified immune cells, such as T cells and NK cells, have shed light on cancer immunotherapy [21]. For hematological tumors, CAR‐T‐cell immunotherapy has been identified as a strategy with great impact [22, 23, 24]. However, CAR‐T therapy still has a few limitations. The long‐term survival of CAR‐T cells may lead to on‐target off‐tumor toxicity, such as B‐cell aplasia observed with anti‐CD19 CAR‐T cells [25, 26]. Additionally, this approach has a significant difficulty in the isolation and application of autologous cells. Furthermore, a common complication of CAR‐T cells is cytokine release syndrome (CRS) which refers to the overproduction of several proinflammatory cytokines, such as IFN‐γ, TNFα, and IL‐6. The symptoms of CRS include fever, nausea, headache, skin rash, tachycardia, hypotension, and shortness of breath. Mild reactions to CAR‐T cells can be common, but CRS can be serious and even life‐threatening.
The selection of an alternative tumoricidal agent might be the solution to the aforementioned drawbacks. Mature NK cells are short lived and expected to disappear rapidly after exerting their tumor cell‐killing function [27]; thus, these cells might be superior to T cells. Several studies on the use of CAR‐NK cells for the treatment of solid tumors have been published [28, 29]. In 2020, Montagne et al. [30] reported anti‐PSMA CAR‐NK92 cells for the treatment of PCa. These researchers believed that CAR‐modified NK‐92 cells could be an effective and safe immunotherapy treatment for solid malignancies. A phase I trial is also underway to assess the safety and feasibility of anti‐PSMA CAR NK cells (NCT03692663). In this study, we evaluated the partial safety by detecting the amount of IL‐6 released by CAR‐NK92MI cells in the tail vein blood of NOD/SCID mice, and observed no overproduction of IL‐6. Hence, no problem related to CRS was detected. In addition, because NK cells do not express specific antigen recognition receptors and are different from T and B lymphocytes, the allogeneic transplantation of CAR‐NK cells can be performed in the clinic [31, 32] which enables the development of an off‐the‐shelf product for clinical use. Moreover, engineered NK cells also retain their native receptors and exhibit cytotoxicity [33] mediated through mechanisms other than those dictated by the specificity of CAR to perform their function without requiring human leukocyte antigen (HLA) matching [34]. Therefore, NK cells act as allogeneic effectors and do not need a specific HLA‐matched donor. Additionally, NK92 cells have already been demonstrated to be a safe and effective antitumor therapy and have received FDA approval [35, 36].
Some trials have NK cells modified with a CAR designed for T cells that was not optimized for NK‐cell signaling [37]. Montagner et al. [30] constructed a CAR by inserting the CD28 and CD3ζ intracellular domains downstream of the leader‐SCFV‐MyC sequence. In this study, we screened different ITAM constructs (CD244, NKG2D and CD3ζ) to identify the best candidate. Interestingly, CAR2‐NK92MI cell‐mediated killing was almost equal to CAR3‐NK92MI cell‐mediated killing. Thus, the importance of CD3ζ as a costimulatory domain was less pronounced in CAR‐NK cells based on the structure of CD244‐CAR. Additionally, p‐PSMA could activate CRPC cells more effectively than anti‐PSMA. It is known that the length of the inserted exogenous gene affects its copy number and expression levels in host cells. High‐affinity binding peptides can reduce the size of the extracellular binding domain and exhibit enhanced affinity for the tumor antigen. Therefore, we constructed p‐PSMA‐CAR‐NK92MI based on the main CD244 skeletal structure and achieved enhanced tumoricidal activity against CRPC. Most of the CD244 regions, extracellular hinge region, transmembrane region and an intracellular ITAM costimulatory domain, were retained. The immunoglobulin‐like domain of CD244 in CAR‐NK92MI cells was replaced by an antigen‐binding extracellular region (p‐PSMA). Intracellular division with a puromycin‐tag and the NKG2D costimulatory domain was investigated.
In NK cells, granzyme B is packaged in cytotoxic granules with perforin, which mediates the delivery of granzymes into endosomes in the target cell. IFN‐γ can inhibit tumor growth by inducing degeneration of the microvasculature produced during tumor angiogenesis, resulting in a relatively hypoxic environment and tumor regression [38]. In this study, we found that p‐PSMA‐CAR‐NK92MI cells could more efficiently lyse PSMA+ cells by releasing increased amounts of IFN‐γ and granzyme B. After the incubation of CAR‐NK cells with tumor cells for 4 hours, we labeled the cells with an anti‐granzyme B monoclonal antibody. CAR‐NK cells conjugated with tumor cells were observed. The results found that p‐PSMA‐CAR‐NK92MI cells killed CRPCPSMA+ cells mainly via the perforin/granzyme system. This result is similar to that reported by Shimasaki et al. [39], who revealed that the killing process of NK cells occurs mainly through the insertion of perforin into the plasma membrane of target cells, which results in the formation of pores. These effects lead to target cell osmotic lysis and granzyme delivery through the pore and thereby results in apoptosis.
Ferroptosis is an atypical modality of cell death following failure of a GSH‐dependent antioxidant system. Cancer cells can be sensitized to ferroptosis following a reduction in the expression of SLC7A11/SLC3A2 caused by IFN‐γ, and H2O2 derived from the Fenton reaction can be generated by NADPH oxidase 2 in T cells [20]. Extensive studies have covered the topic of NK‐cell modulation in the tumor immune microenvironment and the more potent effects of increased NK‐cell infiltration. NK cells can utilize different mechanisms to elicit cancer cell death, and these mechanisms include delivery of cytotoxic granules via the perforin/granzyme system, secretion of death receptor ligands, and production of IFN‐γ. It is crucial to weigh the relative importance of each of these cytotoxic mechanisms in a systematic manner. Moreover, whether IFN‐γ can exert effects on other elements remains to be determined, and whether NK cells kill cancer cells by inducing ferroptosis has not been studied. In our studies, we observed that CAR‐NK92MI cells could kill cancer cells through the induction of ferroptosis. IFN‐γ downregulated both system xc− subunits (SLC3A2 and SLC7A11), which resulted in depletion of the intracellular GSH pool and the triggering of ferroptosis in cancer cells. RSL3‐induced cell ferroptosis worked synergistically with CAR‐NK cells to cause C4‐2 cell death in a coculture of C4‐2 and p‐PSMA‐CAR‐NK92MI cells. Hence, it is possible to combine immunotherapy with the existing treatments to increase the propensity of tumor cells to succumb to ferroptosis.
Despite the robust findings of this study, there are some limitations that need to be discussed. The mechanism by which CAR NK cells induce ferroptosis requires deeper exploration. Although the p‐PSMA‐CAR‐NK92MI cell therapy has been proved in our in vivo mouse model, the safety and efficacy still require further confirmation in more prospective clinical investigations. Future studies are guaranteed to enrich the research field.
5. CONCLUSIONS
p‐PSMA‐CAR‐NK92MI cells can effectively kill CRPCPSMA+ cells in vitro and in vivo. This strategy may provide additional treatment options for patients with CRPC.
DECLARATIONS
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
All animal experiments were approved by the National Cancer Center's Institutional Animal Care and Use Committee (IACUC) and were executed according to the ACUC guidelines (NCC2019‐A018).
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
COMPETING INTERESTS
The authors declare that they have no competing interests.
FUNDING
This study was financially supported by the Capital Science and Technology Leading Talent Project (Z181100006318007), the National Natural Science Foundation of China (81972400 and 32100631), the Beijing Excellent Talents Program‐Youth Backbone Project (2018000032600G393), the Young Elite Scientists Sponsorship Program by China Association for Science and Technology (YESS20210056) and the Beijing Hope Run Special Fund of Cancer Foundation of China (LC2019B02).
AUTHOR CONTRIBUTIONS
Yuchun Gu and Nianzeng Xing designed the entire project and supervised all the experiments. Liyuan Wu, Fei Liu, Fangming Wang, and Le Yin performed the experiments. Liyuan Wu, Fei Liu, Qinxin Zhao, and Hui Shi collected the data. Liyuan Wu, Fei Liu, Feiya Yang, Dong Chen, Yuchun Gu, Xiying Dong, and Nianzeng Xing analyzed the data and drafted and revised the manuscript. All the authors read and approved the final manuscript.
Supporting information
Supporting information
Supporting information
ACKNOWLEDGEMENTS
Not applicable.
Wu L, Liu F, Yin L, Wang F, Shi H, Zhao Q, et al. The establishment of polypeptide PSMA‐targeted chimeric antigen receptor‐engineered natural killer cells for castration‐resistant prostate cancer and the induction of ferroptosis‐related cell death. Cancer Commun. 2022;42:768–783. 10.1002/cac2.12321
Contributor Information
Yuchun Gu, Email: y.gu1@aston.ac.uk, Email: Guyuchun@allifetech.com.
Nianzeng Xing, Email: xingnianzeng@126.com.
REFERENCES
- 1. Qiu H, Cao S, Xu R. Cancer incidence, mortality, and burden in China: a time‐trend analysis and comparison with the United States and United Kingdom based on the global epidemiological data released in 2020. Cancer Commun (Lond). 2021;41(10):1037–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hu Y, Zhao Q, Rao J, Deng H, Yuan H, Xu B. Longitudinal trends in prostate cancer incidence, mortality, and survival of patients from two Shanghai city districts: a retrospective population‐based cohort study, 2000‐2009. BMC Public Health. 2014;14:356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Reciprocal Network between Cancer Stem‐Like Cells and Macrophages Facilitates the Progression and Androgen Deprivation Therapy Resistance of Prostate Cancer. Clinical Cancer Research. 2018:clincanres.0461.2018. [DOI] [PubMed]
- 4. Crawford ED, Higano CS, Shore ND, Hussain M, Petrylak DP. Treating Patients with Metastatic Castration Resistant Prostate Cancer: A Comprehensive Review of Available Therapies. J Urol. 2015;194(6):1537–47. [DOI] [PubMed] [Google Scholar]
- 5. Liu F, Wang C, Huang H, Yang Y, Dai L, Han S, et al. SEMA3A‐mediated crosstalk between prostate cancer cells and tumor‐associated macrophages promotes androgen deprivation therapy resistance. Cell Mol Immunol. 2021;18(3):752–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kochenderfer JN, Rosenberg SA. Treating B‐cell cancer with T cells expressing anti‐CD19 chimeric antigen receptors. Nat Rev Clin Oncol. 2013;10(5):267–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Deng W, Chen P, Lei W, Xu Y, Xu N, Pu JJ, et al. CD70‐targeting CAR‐T cells have potential activity against CD19‐negative B‐cell Lymphoma. Cancer Commun (Lond). 2021;41(9):925–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor‐modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor‐modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, et al. Mesothelin‐specific chimeric antigen receptor mRNA‐engineered T cells induce anti‐tumor activity in solid malignancies. Cancer Immunol Res. 2014;2(2):112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chu J, Deng Y, Benson DM, He S, Hughes T, Zhang J, et al. CS1‐specific chimeric antigen receptor (CAR)‐engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28(4):917–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC‐Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti‐tumor Activity. Cell Stem Cell. 2018;23(2):181–92.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Marchal C, Redondo M, Padilla M, Caballero J, Rodrigo I, Garcia J, et al. Expression of prostate specific membrane antigen (PSMA) in prostatic adenocarcinoma and prostatic intraepithelial neoplasia. Histol Histopathol. 2004;19(3):715–8. [DOI] [PubMed] [Google Scholar]
- 14. Liu CJ, Hu FF, Xia MX, Han L, Zhang Q, Guo AY. GSCALite: a web server for gene set cancer analysis. Bioinformatics. 2018;34(21):3771–2. [DOI] [PubMed] [Google Scholar]
- 15. Chowdhury PS, Viner JL, Beers R, Pastan I. Isolation of a high‐affinity stable single‐chain Fv specific for mesothelin from DNA‐immunized mice by phage display and construction of a recombinant immunotoxin with anti‐tumor activity. Proc Natl Acad Sci U S A. 1998;95(2):669–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang Q, Helfand BT, Carneiro BA, Qin W, Yang XJ, Lee C, et al. Efficacy Against Human Prostate Cancer by Prostate‐specific Membrane Antigen‐specific, Transforming Growth Factor‐beta Insensitive Genetically Targeted CD8(+) T‐cells Derived from Patients with Metastatic Castrate‐resistant Disease. Eur Urol. 2018;73(5):648–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wei J, Qin B, Chen Z, Hao L, Cheng K. Discovery of PSMA‐Specific Peptide Ligands for Targeted Drug Delivery. Int J Pharmaceutics. 2016;513(1):138–47. [DOI] [PubMed] [Google Scholar]
- 18. Aggarwal, S . A dimeric peptide that binds selectively to prostate‐specific membrane antigen and inhibits its enzymatic activity. Cancer Res. 2006;66(18):9171–7. [DOI] [PubMed] [Google Scholar]
- 19. Gan L, Meng J, Xu M, Liu M, Qi Y, Tan C, et al. Extracellular matrix protein 1 promotes cell metastasis and glucose metabolism by inducing integrin beta4/FAK/SOX2/HIF‐1alpha signaling pathway in gastric cancer. Oncogene. 2018;37(6):744–55. [DOI] [PubMed] [Google Scholar]
- 20. Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569(7755):270–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Park JH, Geyer MB, Brentjens RJ. CD19‐targeted CAR T‐cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date. Blood. 2016;127(26):3312–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang X, Popplewell LL, Wagner JR, Naranjo A, Blanchard MS, Mott MR, et al. Phase 1 studies of central memory‐derived CD19 CAR T‐cell therapy following autologous HSCT in patients with B‐cell NHL. Blood. 2016;127(24):2980–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Singh N, Frey NV, Grupp SA, Maude SL. CAR T Cell Therapy in Acute Lymphoblastic Leukemia and Potential for Chronic Lymphocytic Leukemia. Curr Treat Options Oncol. 2016;17(6):28. [DOI] [PubMed] [Google Scholar]
- 24. Gardner R, Wu D, Cherian S, Fang M, Hanafi LA, Finney O, et al. Acquisition of a CD19‐negative myeloid phenotype allows immune escape of MLL‐rearranged B‐ALL from CD19 CAR‐T‐cell therapy. Blood. 2016;127(20):2406–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B‐cell depletion and remissions of malignancy along with cytokine‐associated toxicity in a clinical trial of anti‐CD19 chimeric‐antigen‐receptor‐transduced T cells. Blood. 2012;119(12):2709–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19‐targeted T cells in patients with relapsed or chemotherapy refractory B‐cell leukemias. Blood. 2011;118(18):4817–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Claudia B, Sabine H, Andrea Q, Ruth E, Melanie B, Stephan K, et al. IL‐2 Stimulated but Not Unstimulated NK Cells Induce Selective Disappearance of Peripheral Blood Cells: Concomitant Results to a Phase I/II Study. PLoS One. 2011;6(11):e27351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Han J, Chu J, Keung Chan W, Zhang J, Wang Y, Cohen JB, et al. CAR‐Engineered NK Cells Targeting Wild‐Type EGFR and EGFRvIII Enhance Killing of Glioblastoma and Patient‐Derived Glioblastoma Stem Cells. Sci Rep. 2015;5:11483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nowakowska P, Romanski A, Miller N, Odendahl M, Bonig H, Zhang C, et al. Clinical grade manufacturing of genetically modified, CAR‐expressing NK‐92 cells for the treatment of ErbB2‐positive malignancies. Cancer Immunol Immunother. 2018;67(1):25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Montagner IM, Penna A, Fracasso G, Carpanese D, Dalla Pieta A, Barbieri V, et al. Anti‐PSMA CAR‐engineered NK‐92 Cells: An Off‐the‐shelf Cell Therapy for Prostate Cancer. Cells. 2020;9(6):1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nicholson IC, Lenton KA, Little DJ, Decorso T, Lee FT, Scott AM, et al. Construction and characterisation of a functional CD19 specific single chain Fv fragment for immunotherapy of B lineage leukaemia and lymphoma. Mol Immunol. 1997;34(16‐17):1157–65. [DOI] [PubMed] [Google Scholar]
- 32. Tassev DV, Cheng M, Cheung NK. Retargeting NK92 cells using an HLA‐A2‐restricted, EBNA3C‐specific chimeric antigen receptor. Cancer Gene Ther. 2012;19(2):84–100. [DOI] [PubMed] [Google Scholar]
- 33. Caligiuri MA, Velardi A, Scheinberg DA, Borrello IM. Immunotherapeutic approaches for hematologic malignancies. Hematology Am Soc Hematol Educ Program. 2004:337–53. [DOI] [PubMed] [Google Scholar]
- 34. Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer. 2016;16(1):7–19. [DOI] [PubMed] [Google Scholar]
- 35. Ljunggren HG, Malmberg KJ. Prospects for the use of NK cells in immunotherapy of human cancer. Nat Rev Immunol. 2007;7(5):329–39. [DOI] [PubMed] [Google Scholar]
- 36. Malmberg KJ, Bryceson YT, Carlsten M, Andersson S, Bjorklund A, Bjorkstrom NK, et al. NK cell‐mediated targeting of human cancer and possibilities for new means of immunotherapy. Cancer Immunol Immunother. 2008;57(10):1541–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bollino D, Webb TJ. Chimeric antigen receptor‐engineered natural killer and natural killer T cells for cancer immunotherapy. Transl Res. 2017;187:32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kammertoens T, Friese C, Arina A, Idel C, Briesemeister D, Rothe M, et al. Tumour ischaemia by interferon‐gamma resembles physiological blood vessel regression. Nature. 2017;545(7652):98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shimasaki N, Jain A, Campana D. NK cells for cancer immunotherapy. Nat Rev Drug Discov. 2020;19(3):200–18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information
Data Availability Statement
The data supporting the findings of this study are available from the corresponding author upon reasonable request.