

Abstract
Lung-resident memory B cells (MBCs) provide localized protection against reinfection in respiratory airways. Currently, the biology of these cells remains largely unexplored. Here, we combined influenza and SARS-CoV-2 infection with fluorescent-reporter mice to identify MBCs regardless of antigen specificity. We found that two main transcriptionally distinct subsets of MBCs colonized the lung peribronchial niche after infection. These subsets arose from different progenitors and were both class switched, somatically mutated, and intrinsically biased in their differentiation fate toward plasma cells. Combined analysis of antigen specificity and B cell receptor repertoire segregated these subsets into “bona fide” virus-specific MBCs and “bystander” MBCs with no apparent specificity for eliciting viruses generated through an alternative permissive process. Thus, diverse transcriptional programs in MBCs are not linked to specific effector fates but rather to divergent strategies of the immune system to simultaneously provide rapid protection from reinfection while diversifying the initial B cell repertoire.
Keywords: lungs, memory B cells, bona fide, bystander, respiratory infection, influenza virus, SARS-CoV-2
Graphical abstract
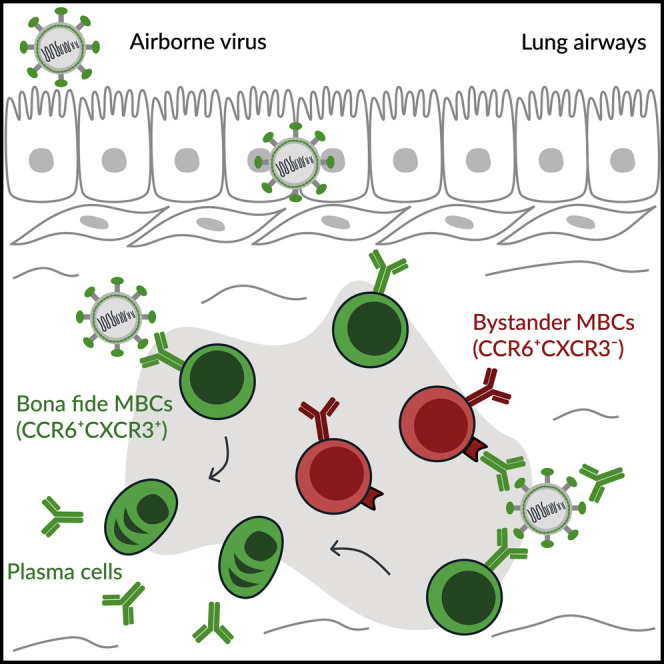
Lung-resident memory B cells (MBCs) provide rapid protection against reinfection with airborne pathogens. Gregoire et al. demonstrate that two MBC subsets with discrete transcriptional programs reside in the lung peribronchial niche: “bona fide” virus-specific MBCs, which dominate recall responses, and “bystander” non-specific MBCs, which locally retain antigens as immune complexes.
Introduction
The immune system mounts protective responses to counteract the threat posed by pathogens during infection. These responses leave immunological memory, a strategy that allows the body to remember previously encountered pathogens. Memory B cells (MBCs) and T cells are long-lived lymphocytes that constitute an essential component of this strategy (Akkaya et al., 2020). These cells take up residence in secondary lymphoid organs and remain in a quiescent state until a secondary antigen encounter. Upon re-challenge, they rapidly produce large numbers of effector cells that deliver fast and effective protection (Weisel and Shlomchik, 2017). In the case of MBCs, they can either differentiate into short-lived plasma cells (PCs), which produce high-affinity neutralizing antibodies, or re-enter germinal centers (GCs) and provide, up to a certain extent, an additional source of long-lasting protection with increased affinity and breadth (Kurosaki et al., 2015; McHeyzer-Williams et al., 2015; Mesin et al., 2020; Viant et al., 2020). MBC fate decision can be shaped by cell-intrinsic features, B cell receptor (BCR) isotype, antigen affinity, and the magnitude of CD40 signaling (Dogan et al., 2009; Koike et al., 2019; Pape et al., 2011; Viant et al., 2020, 2021; Zuccarino-Catania et al., 2014).
Memory lymphocytes do not exclusively reside in secondary lymphoid organs. For instance, a lineage of T cells can occupy the tissue barriers after infection without recirculating. These tissue-resident memory T cells are transcriptionally and functionally distinct from the recirculating ones and provide site-specific responses against infection (Szabo et al., 2019). Yet, if a B cell counterpart of these cells existed remained unknown. A recent study has shown, through the use of elegant parabiotic experiments, that a population of tissue-resident MBCs settles in the lungs after influenza virus infection (Allie et al., 2019). Lung MBCs not only ensure the first layer of protection directly at the tissue barrier but also display high cross-reactivity to viral escapes, highlighting the potential for targeting them to develop broadly protective vaccines (Adachi et al., 2015; Onodera et al., 2012). At present, it remains unknown if lung MBCs occupy specific tissue niches in the lung mucosa, if they bear special transcriptional programs that allow their survival in the lung airways, or if discrete MBC subsets coexist upon infection.
Here, we combined the use of Aicda-creERT2 Rosa26-EYFP reporter mice with influenza and SARS-CoV-2 infection models to track lung MBCs based on past Aicda expression at the time of viral infection. As this approach does not rely on the ability of MBCs to bind viral antigens, it gave us access to cells with little or no affinity for specific antigens, which can represent a large proportion of MBCs (Viant et al., 2020) and were disregarded in previous studies. We show that two main subsets of MBCs permanently colonize the lung peribronchial niche upon respiratory viral infection. These subsets arose from different progenitors that underwent class switching, somatic hypermutation (SHM) in GCs, and subsequently acquired divergent transcriptional programs and chemokine and Fc receptor patterns. Unexpectedly, both MBC subsets were intrinsically biased in their differentiation fate toward PCs, highlighting the fact that divergent transcriptional programs were not associated with specific effector fates. Instead, these subsets segregated “bona fide” MBCs, which dominated recall responses by producing high-affinity antibody-secreting cells (ASCs), from “bystander” MBCs, with no specificity for the immunogen, unable to produce protective antibodies, but with the ability to retain and display antigen in the form of immune complexes (ICs). These results challenge the notion that GCs only work as “machines” to produce high-affinity bona fide MBCs. Instead, a permissive mechanism simultaneously operates in these reactions, giving rise to bystander MBCs.
Results
Past Aicda expression allows the unbiased identification of lung-homing MBCs
To identify lung-homing MBCs without introducing a bias on antigen specificity, we used the Aicda-cre ERT2 Rosa26-EYFP mouse strain, from now on referred to as Aid-EYFP (Le Gallou et al., 2018). This model allows us to permanently label cells that have expressed Aid, the enzyme that initiates class switching and SHM, at the time of tamoxifen treatment. As a vast fraction of MBCs has gone through at least one of these two processes, we expect them to be labeled by tamoxifen. Accordingly, we intranasally infected Aid-EYFP mice with 5 plaque-forming units (PFUs) of influenza A virus H1N1 PR8 strain and gave tamoxifen on days 6, 8, and 10. We assessed the efficiency and specificity of this strategy at week 10 of infection by flow cytometry (Figure 1 A). We observed the presence of a discrete population of YFP+ cells in the lungs and secondary lymphoid organs, mostly corresponding to CD19+ B cells and a small population of CD19− PCs (Figures 1B and S1A). More than 90% of YFP+ lung cells showed a phenotype associated with MBCs (CD38+ GL-7−) (Figure 1C). By contrast, only 30% of YFP+ mediastinal lymph node cells showed an MBC phenotype, and a great majority of them were committed to GCs (CD38− GL-7+) that persisted long after viral clearance (Figure 1C; Yewdell et al., 2021).
Figure 1.

MBCs in influenza infection
(A) Experimental approach.
(B) Contour plots displaying YFP+ and YFP− B cells in Aid-EYFP animals treated as in (A).
(C) Contour plots showing MBCs (CD38+GL7−) and GC (CD38−GL-7+) cells gated from the CD19+YFP+ population in (B).
(D) Contour plots showing MBC binding to NP and HA.
(E) Histograms depicting IgD expression by YFP+ MBCs and YFP− B cells.
(F) Histograms showing the labeling of YFP+ MBCs and YFP− B cells with anti-CD45 antibody administered i.v. The quantification shows B cells protected from staining.
(G) Absolute numbers of YFP+ MBCs in mice treated as in (A) at indicated time points.
(H) Contour plots displaying YFP+ B cells in mice infected with 5 PFU of PR8 virus or challenged with 105 PFU of UV-inactivated PR8 virus. Unless indicated, all analyses were performed on day 70 of the infection. In all the panels, bar charts show the quantification of one representative experiment out of three, mean ± SEM. Each dot represents one mouse. t test: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
See also Figure S1.
To determine whether these YFP+ cells were genuine MBCs, we measured antigen specificity, class switching, accessibility to blood circulation, and lifespan. Regarding specificity, we detected an average of 30% of YFP+ B cells binding to influenza nucleoprotein (NP) and 5% that bound to hemagglutinin (HA).Less than 2% of YFP− B cells bound to these proteins, indicating that the YFP+ population was enriched in B cells specific for influenza antigens (Figures 1D and S1B). Regarding class switching, we observed that YFP+ cells lacked IgD expression, while YFP− cells were mainly IgD+, showing that YFP+ cells have undergone class switching (Figure 1E). To measure accessibility to blood, we administered i.v. anti-CD45 antibody. We found that more than 95% of lung YFP+ cells, in marked contrast to YFP− B cells, were protected from in vivo antibody labeling, indicating that these cells preferentially accumulated in the lung parenchyma rather than in the blood circulation (Figures 1F and S1C). We finally enumerated YFP+ cells at different times of infection to get insight into their lifespan. We found that the kinetics of YFP+ cells followed a similar trend across lungs and lymphoid organs: YFP+ cells were detected at low numbers on day 12, peaked between days 20 and 30, and slowly declined thereafter (Figure 1G). YFP+ B cells were detected in significant numbers even 1 year after infection, suggesting that these cells were long lived (Figure 1G). The accumulation of YFP+ B cells was largely reduced in mice challenged with UV-inactivated influenza virus, pointing out that infection, or the inflammation process associated with it, was required for MBC establishment (Figures 1H, S1D, and S1E). These results highlight the effectiveness of our strategy to track long-lived class-switched MBCs across organs without introducing a bias on specificity.
Transcriptionally distinct subsets of MBCs coexist in the lungs
To unveil if influenza infection engenders discrete subsets of MBCs, we performed 5′-end single-cell RNA sequencing(scRNA-seq) on YFP+ MBCs sorted from lungs, mediastinal lymph node, and spleen on day 70 of influenza infection using the system Chromium (10X Genomics). Through dimensionality reduction, we found that MBCs bifurcated into three separate clusters within the lungs and lymph node and into five clusters in the spleen (Figures 2A–2D). We initially observed a fourth cluster in the lungs with a high expression of heat shock protein and early response genes, which was removed for further analysis (Figure S2A). This artifactual cluster was induced by tissue dissociation with collagenase at 37°C, as it disappeared when we repeated the scRNA-seq at 4°C with mechanical dissociation (Figures S2B–S2F). By analyzing gene expression, we identified groups of marker genes associated with specific B cell clusters, reflecting divergent transcriptional programs among MBCs (Figure 2E). We calculated the Szymkiewicz-Simpson index to compare the extent of similarity among MBC clusters. We found that lung clusters Lg1, Lg2, and Lg3 shared a high amount of marker genes’ overlap with lymph node clusters Ln1, Ln2, and Ln3, respectively. By contrast, the spleen contained specific MBC clusters, such as Sp4 and Sp5, that were absent from lungs (Figure 2F). These data show that the MBC pool is not homogeneous but, instead, is constituted of distinct subsets that are tissue specific or shared across organs.
Figure 2.

Heterogeneity of MBCs
(A–C) UMAP (Uniform Manifold Approximation and Projection)projections of MBCs in (A) the lungs, (B) mediastinal lymph node, and (C) spleen. Feature plots display the expression of indicated marker genes in MBCs laid out in the UMAP representation. Scale: normalized UMI (Unique Molecular Identifiers) counts.
(D) Bar chart showing the proportions of MBC clusters.
(E) Heatmaps exhibiting marker gene expression for each tissue cluster. Color: scaled mean expression.
(F) Szymkiewicz-Simpson similarity matrix for pairwise comparisons among clusters from different tissues. Squares with black borders indicate values higher than 0.32.
(G and H) Dot plots showing the presence of CCR6−CXCR3−, CCR6+CXCR3−, and CCR6+CXCR3+ MBC subsets in (G) the lungs and (H) lymph node.
(I) Gating strategy for spleen MBC subsets according to the expression of CD11c, CCR6, CD21, CD24, and CD55.
In (G)–(I), bar charts show the quantification of one representative experiment out of three, mean ± SEM. Each dot represents one mouse. One-way ANOVA test: ∗∗p < 0.01 and ∗∗∗∗p < 0.0001.
See also Figure S2.
Among the MBCs residing in the lungs and lymph node, those belonging to the two major clusters (Lg1-Ln1 and Lg2-Ln2) expressed the chemokine receptor Ccr6, whereas cells from the minor cluster (Lg3-Ln3) did not (Figures 2A, 2B, and 2E). Out of the top differentially expressed genes, we found that cluster 1 (Lg1-Ln1) cells expressed the IgE Fc receptor Fcer2a (CD23), the calcium-binding protein Cnn3, and the cysteine-rich protein Lmo2; cluster 2 (Lg2-Ln2) cells expressed the chemokine receptor Cxcr3, the placenta-specific protein Plac8, the cystatin Cst3, and the integrin Itga4; and cluster 3 (Lg3-Ln3) cells expressed the membrane-spanning 4-domains Ms4a6c, the transcription factor Nfatc1, and the zinc-finger-containing protein Zbtb32 (Figures 2A, 2B, and 2E). Importantly, we were able to distinguish these three MBC subsets by flow cytometry using the cluster-identifying markers CCR6 and CXCR3 (Figures 2G, 2H, and S2G).
In the spleen, MBCs from the major cluster Sp1 expressed Ccr6, Lmo2, Fcer2a, and Cxcr3, a gene expression pattern associated with follicular MBCs (Figures 2C and 2E). Cells from cluster Sp2 were characterized by high amounts of the Krüppel-like factor Klf2, the complement factor Cd55, and the cytoskeletal protein Vimentin, resembling transitional MBCs (Figures 2C and 2E). Cells from cluster Sp3 expressed the tyrosine kinase Lck, Nfatc1, Ms4a6c, and Zbtb32, a similar gene pattern to B1 cells (Figures 2C and 2E). Cells from cluster Sp4 expressed high amounts of the complement receptor Cr2 (CD21) and the sphingosine 1-phosphate receptors S1pr1 and S1pr3, resembling marginal zone MBCs (Figures 2C and 2E). Lastly, cells from the smallest cluster Sp5 expressed the transcription factor Tbx21 (T-bet) and the integrin Itgax (Cd11c), resembling age-associated MBCs (Figures 2C and 2E) (Riedel et al., 2020). We could segregate these five MBC subsets by flow cytometry based on the expression of CD11c, CCR6, CD21, CD55, and CD24 (Figures 2I and S2H). These data indicate that splenic B cells with diverse origins and developmental stages participate in the memory response.
MBC subsets colonize the lung peribronchial niche upon influenza infection
Our scRNA-seq data revealed the co-existence of discrete subsets of MBCs in the lung mucosa. As these subsets expressed contrasting patterns of CCR6 and CXCR3 receptors, we wondered if they exhibit differential micro-anatomic distributions. To gain insight into their spatial positioning, we imaged lung sections at day 70 of influenza infection by confocal microscopy. We found that YFP+ MBCs assembled in discrete B220+ clusters composed of hundreds of cells. These clusters were found in areas surrounding EpCAM+ epithelial cells, indicating that MBCs preferentially locate in peribronchial areas (Figure 3 A). We further extended the analysis to lymphoid organs by tracking the YFP+ cells devoid of GC (GL-7) and PC (CD138) markers. In the lymph node, MBCs preferentially accumulated in follicles and interfollicular areas but rarely in the paracortex or medulla (Figure 3B). In the spleen, YFP+ MBCs were mostly found in follicles, in close association with the marginal zone (Figure 3C). These results show that MBCs remain positioned at sites of antigen entry, a strategy that facilitates a fast encounter of pathogens upon re-challenge.
Figure 3.

Spatial positioning of MBCs
(A–D) Confocal images of (A and D) the lung, (B) mediastinal lymph node, and (C) spleen sections from Aid-EYFP mice treated as in Figure 1A. Scale bars: left 50 μm and right 25 μm in (A), left 300 μm and right 30 μm in (B), left 250 μm and right 55 μm in (C), and 20 μm in (D). The quantification in (A) displays the minimal distance of B cell clusters to the EpCAM+ cells. Bar charts in (B) and (C) show the proportion of MBCs residing in the indicated zones. MZ, marginal zone; PALS, periarteriolar lymphoid sheaths.
(E) Dot plot showing CCR6 and CXCR3 fluorescence intensity for MBCs (left). X and Y positions of MBC subsets are laid out in the B220 representation (middle). The quantification displays the distance of each MBC to the cluster’s centroid (right).
(F) Lung images of wild-type and CCR6-deficient Aid-EYFP mice treated as in Figure 1A. Scale bars, 30 μm. The quantification displays the density of YFP+ B cells in B cell clusters. Dots represent individual B cell clusters.
(G) Strategy to generate mixed bone marrow chimeras.
(H and I) Histograms showing (H) CCR6 and (I) CXCR3 expression in the lung MBCs and non-B cells in chimeras.
(J and K) Quantification of MBC numbers after 70 days of primary influenza infection in (J) CCR6- or (K) CXCR3-deficient chimeras.
(L and M) Enumeration of ASCs after 4 days of re-challenge in (L) CCR6- or (M) CXCR3-deficient chimeras. The quantification of one representative experiment is shown in the bar charts; each dot represents one mouse. In all panels: mean ± SEM, t test: ∗∗p < 0.01, ns, not significant.
See also Figure S3.
To visualize individual MBC subsets in the lungs, we stained tissue sections with antibodies against YFP, B220, CCR6, and CXCR3 (Figure 3D). We then generated a mask on YFP+ cells and excluded PCs based on their size and B220 expression (Figure S3A), and measured CCR6 and CXCR3 fluorescence on individual YFP+ cells. By plotting CXCR3 versus CCR6 fluorescence intensity, we observed a cell distribution pattern that closely resembled the one obtained by flow cytometry (Figure 3E). When we tracked each subset back into space, we found that cells from the two main CCR6+ subsets interspersed with each other across the entire B cell cluster, whereas cells lacking CCR6 were largely excluded to the peripheral areas (Figure 3E). Still, CCR6 and CXCR3 were not required to enter the peribronchial niche, as we found similar numbers of MBCs within B cell clusters in the wild-type, CCR6, and CXCR3-deficient mice (Figures 3F, S3B, and S3C). Then it is plausible that other chemokine pathways, such as the CXCR5-CXCL13 axis, are driving this process (Denton et al., 2019)
To test if CCR6 and CXCR3 were required by MBCs to mount recall responses, we generated chimeric mice by irradiating μMT animals and injecting a mixture of 80% μMT bone marrow and 20% of either wild-type, CCR6-, or CXCR3-deficient bone marrow (Figures 3G–3I). We observed comparable numbers of MBCs among all groups after 70 days of infection with influenza PR8, indicating that CCR6 and CXCR3 were dispensable for the maintenance of the MBC pool (Figures 3J, 3K, and S3B). We then re-challenged chimeric mice with 5.104 PFU of influenza H3N2 X31 strain and measured the formation of NP-specific ASCs by ELISpot 4 days after. We observed a 10-fold decrease in the formation of IgA ASCs in CCR6-deficient chimeras compared with their wild-type counterparts, whereas the numbers of IgM and IgG ASCs were similar among both groups (Figure 3L). This defect reflected the reduced proportion of IgA+ cells within the initial MBC pool of CCR6-deficient mice (Figure S3D), a phenomenon that was also observed in Peyer’s patches in a recent study (Reboldi et al., 2016). These results point to a general mechanism for CCR6 in IgA responses against diverse microbial challenges across organs. By contrast, we observed comparable numbers of ASCs in wild-type and CXCR3-deficient chimeras, indicating that CXCR3 is dispensable for recall responses (Figure 3M).
Lung MBC subsets are class switched and somatically hypermutated
We investigated whether the transcriptionally distinct subsets of MBCs co-existing across organs were related in their origin. To this end, we produced scBCR-seq libraries from the same cells analyzed by scRNA-seq. The analysis of the constant heavy chain (IgH) revealed that lung and lymph node MBCs displayed extensive class switching toward IgG isotypes, whereas most splenic MBCs expressed IgM (Figure 4 A). At the cluster level, the two major lung and lymph node CCR6+ clusters (Lg1-Ln1 and Lg2-Ln2) were class switched toward IgGs, whereas the minor CCR6− cluster (Lg3-Ln3) was composed of IgM+ cells (Figure 4B). In the spleen, the major follicular MBC cluster (Sp1) exhibited class switching toward IgGs, whereas transitional (Sp2), B1-like (Sp3), marginal zone (Sp4), and ABC (Sp5) MBCs were IgM+ (Figure 4B). Concerning the constant light chain, we observed that Kappa (IgK) was the most commonly expressed among all MBCs. Yet, Lambda (IgL) was also expressed by some cells from the minor IgM+CCR6− clusters (Lg3-Ln3) and by transitional (Sp2) and B1-like (Sp3) MBCs (Figure 4C). Then, the MBC pool displays pronounced intra- and inter-organ differences in isotype-class usage.
Figure 4.

Origin of MBCs
(A and B) Quantification of IgH isotype-class usage by MBCs according to (A) the organ and (B) tissue cluster from the 1st scRNA-seq dataset.
(C) Quantification of IgK and IgL usage by MBC clusters across organs.
(D and E) Heatmap of (D) IgH and (E) IgK variable gene frequency in individual MBC clusters.
(F) Mean CDR3 length in IgH for indicated MBC clusters, two-way ANOVA test.
(G) Overlap similarity matrices in individual mice for pairwise comparisons of clonal overlaps among clusters and tissues. Similarity trees show hierarchical clustering analysis.
(H) Contour plots displaying YFP+ GC B cells in the lymph node of Aid-EYFP animals treated as in Figure 1A. One group of mice was treated with anti-CD40L on days 6, 8, and 10.
(I) Dot plots showing the lung MBC subsets in mice treated as in Figure 1A plus/minus administration of anti-CD40L. Each dot in the bar charts represents one mouse, mean ± SEM, t test.
(J and K) Quantification of mean SHM amounts in the variable gene of (J) heavy and (K) light chain for MBCs in clusters Lg1 and Lg2 in 1st and 2nd datasets, paired t test (left). Bar charts showing the distribution of Lg1 and Lg2 cluster cells according to the extent of SHM, mean ± SEM, one-way ANOVA test (right). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
We measured the magnitude of BCR diversification driven by the differential use of variable genes (IgHV, IgKV, and IgLV) and the length of the complementary-determining region 3 (CDR3) within each subset. We observed that the two major lung and lymph node CCR6+ clusters (Lg1-Ln1 and Lg2-Ln2) and the splenic follicular (Sp1), marginal zone (Sp4), and ABC (Sp5) clusters displayed high usage of IgHV1, 5, 9, and14 and IgKV5 and 6 (Figures 4D and 4E). By contrast, the minor lung and lymph node CCR6− clusters (Lg3-Ln3) and the splenic transitional (Sp2) and B1-like (Sp3) clusters showed a different expression pattern: high usage of IgHV1, 4, and 6; IgKV1, 2, 4, 8, and 14; and IgLV1. These clusters further exhibited a lower length of the IgH CDR3 (Figures 4D–4F). These results show that MBC subsets were skewed toward divergent families of variable genes, in line with the notion that certain MBC subsets may originate from different precursors. To test this idea, we defined clonotypes and measured overlaps in MBC clusters. We detected an overlap of clonal BCR repertoires among lung and lymph node clusters containing class-switched CCR6+ cells and the splenic follicular cluster, Lg1-Ln1-Sp1 on one side and Lg2-Ln2-Sp1 on the other side (Figure 4G). We further detected high amounts of clonal overlap among the lung and lymph node clusters enriched in IgM+CCR6− cells and splenic transitional and B1-like clusters (Lg3-Ln3-Sp2-Sp3) (Figure 4G). These results show that (1) certain memory populations were interconnected in their generation or maintenance across organs and that (2) the minor “innate-like” group of IgM+CCR6− cells represented a separate lineage from class-switched CCR6+ MBCs (Figure 4G).
The two major class-switched CCR6+ MBC populations identified in the lungs and lymph node could be segregated based on their CXCR3 expression (Figure 2). To evaluate whether these two subsets arise from GC reactions, we treated influenza-infected mice with anti-CD40L antibodies between days 6 and 10, which allowed early B-T cell contacts while disrupting subsequent B-T follicular helper (Tfh) cell interactions in GCs (Figure 4H). We found that both the CXCR3− and the CXCR3+ subsets were markedly reduced in mice receiving anti-CD40L treatment, suggesting that both MBC subsets were products of GCs (Figure 4I). We then measured the rate of SHM in IgHV and IgKV genes in our scRNA-seq datasets. We found that the Lg1 cluster (CXCR3lo) displayed significantly higher amounts of SHM in the IgHV and IgKV genes than the Lg2 cluster (CXCR3hi) (Figures 4J and 4K). These results indicate that the two major CCR6+ class-switched MBC subsets were derived from cells that underwent active SHM in GCs but either at dissimilar rates or for different periods of time.
Lung MBC subsets display contrasting specificities and protective functions
We wondered whether these two subsets of class-switched CCR6+ MBCs were intrinsically biased in their fate toward PCs or GCs upon re-activation. To evaluate this, we sorted CCR6+CXCR3− and CCR6+CXCR3+ MBC subsets from lungs and lymph node on day 70 of the influenza infection and cultured them ex vivo with 40LB feeder cells, sources of BAFF and CD40L, and IL-21 for 3 days (Figure 5 A; Nojima et al., 2011). Under these conditions, MBCs have been shown to differentiate into plasma or GC cells according to cell-intrinsic features (Koike et al., 2019). Flow cytometry analysis revealed that both CCR6+ MBC subsets mostly differentiated into CD93+ PCs and rarely into GL-7+ GC B cells, regardless of their CXCR3 expression or organ of origin (Figures 5B and S4A). These cells produced IgGs to the same extent (Figures 5C and S4B). Yet, the antibodies derived from CXCR3+ MBCs cultures were specific for influenza NP, HA, and Neuraminidase (NA), whereas those from CXCR3− MBCs cultures showed low to undetectable recognition of native or denatured forms of influenza antigens (Figures 5D and S4C–S4E). We then examined the specificity of MBC subsets and found that CXCR3+ MBCs bound extensively to NP, HA, and NA, whereas CXCR3− cells showed minimal binding (Figures 5E and S4F–S4H). Thus, the CXCR3+ MBC subset is indeed enriched in antigen-specific cells, whereas the CXCR3− subset comprises cells with no apparent specificity for influenza proteins.
Figure 5.

MBC fate and specificity
(A) Schematic overview of the 40LB-MBCs co-culture.
(B) Contour plots showing CD93+ PCs and GL-7+ GC-like cells derived from CCR6+CXCR3− or CCR6+CXCR3+ MBCs isolated from the lungs at day 70 after the influenza infection and co-cultured for 3 days with 40LB cells.
(C and D) Bar charts displaying (C) total IgG amounts and (D) NP- or HA-specific IgG titers measured in supernatants.
(E) Contour plots showing MBC binding to NP and HA.
(F) Overview of the strategy to evaluate homotypic immunity.
(G) Survival curve of mice treated as described in (F).
(H) Enumeration of NP-specific ASCs in the lungs of mice treated with PBS or influenza PR8 and challenged at day 70 with PBS or influenza X31.
(I) Contour plots displaying MBC subsets in the lungs of mice infected with influenza PR8 alone (left panel) or re-challenged with influenza X31 after 70 days (right panel).
(J) Overview of the index cell sorting and FB5P-seq experimental workflow.
(K) Heatmap showing the expression of marker genes from Lg1, Lg2, and Lg3 clusters (10× dataset) by CCR6+CXCR3−, CCR6+CXCR3+, and CCR6−CXCR3− subsets. Color: scaled mean expression.
(L) Venn diagrams showing the clonal overlap among non-binding cells from CCR6+CXCR3−, CCR6+CXCR3+, and CCR6−CXCR3− subsets and NP-binding (upper panel) or HA-binding (lower panel) cells.
(M) Trees showing phylogenetic relationships of IgH and IgK sequences from clones containing NP-binding (left) and HA-binding (right) cells. In all quantifications: mean ± SEM, each dot represents one mouse; t test: ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗∗p < 0.0001.
See also Figure S4.
These results led us to investigate the involvement of these two MBC subsets in homotypic and heterosubtypic immunity. For the former, we used a previously described in vivo setup (Onodera et al., 2012). We i.v. transferred naive splenocytes to μMT mice and treated them intranasally with CpG to generate bronchoalveolar lymphoid structures. After 10 days, we transferred CXCR3− or CXCR3+ MBCs sorted from the lungs of previously PR8-infected animals together with splenic CD4+ T cells isolated from the same donors. We infected these animals with influenza PR8, monitored their weight daily, and euthanized those exhibiting ≥20% loss of initial mass (Figure 5F). We observed that mice receiving CXCR3+ MBCs were highly protected from infection, whereas more than 50% of those receiving CXCR3− MBCs needed to be euthanized between days 6 and 8 (Figure 5G). Therefore, CXCR3+ and CXCR3− subsets differ in their ability to provide homotypic protection. To investigate the ability of these subsets to respond to a different influenza subtype, we infected Aid-EYFP mice with influenza PR8, re-challenged them with influenza X31 on day 70, and analyzed them 4 days after. Secondary infection with X31 led to a robust formation of flu-specific ASCs (Figure 5H). These cells were mainly derived from lung MBCs, as this response was not observed in mice receiving PR8 or X31 infection alone (Figure 5H). Taking this into account, if one subset of MBCs is preferentially engaged in secondary responses, we should observe a change in the subsets ratio before and after the re-challenge (Allie et al., 2019). We found that the CXCR3+/CXCR3− memory ratio is close to 2 after primary infection (PR8 alone) (Figure 5I). We observed a marked decrease in the CXCR3+/CXCR3− ratio upon secondary infection (PR8 + X31) (Figures 5I and S4I). This change was not due to a transition of MBCs from the CXCR3+ subset to the CXCR3− one, as antigen-specific MBCs remained in the CXCR3+ population (Figure S4J). Instead, our results favor the notion that CXCR3+ MBCs actively differentiated into ASCs when compared with the CXCR3− subset. These results show that influenza infection gives rise to two distinct subsets of CCR6+ MBCs: (1) a CXCR3+ subset enriched in MBCs with specificity for influenza antigens that robustly respond upon secondary challenge by secreting protective antibodies and (2) a CXCR3− subset enriched in MBCs with no evident specificity for influenza antigens and restricted effector capacity.
Lung CCR6+CXCR3− and CCR6+CXCR3+ MBCs arise from different progenitors
We were intrigued by the results showing that the CCR6+CXCR3− MBC subset was composed of cells that underwent class switching and SHM but did not display evident specificity for influenza antigens. We investigated whether these cells were (1) low-affinity MBCs with undetectable capture in our antigen-binding assay or (2) non-antigen-specific MBCs produced through an alternative mechanism. Although in the first scenario CXCR3− MBCs can share progenitors with antigen-binding cells, no clonal overlap is expected in the second one. To discern among these contrasting situations, we performed single-cell index sorting of lung YFP+ MBCs on day 70 of the influenza infection. For each cell, we recorded the information of NP and HA binding and CCR6 and CXCR3 expression. We then performed a flow cytometry-based 5′ end (FB5P) scRNA-seq for the integrative analysis of transcriptome and BCR repertoire (Figure 5J; Attaf et al., 2020).
Firstly, we measured the expression of cluster-identifying genes from our 10× dataset in the different MBC subsets (Figures 2E and 5K). We found that the CCR6+CXCR3− subset expresses high amounts of Lg1 cluster genes (Fcer2a, Cnn3, and Lmo2), the CCR6+CXCR3+ subset expresses high amounts of Lg2 cluster genes (Cst3 and Itga4), and the minor CCR6−CXCR3− subset is characterized by the expression of Lg3 cluster genes (Ms4a6c and Zbtb32) (Figure 5K). Then, the segregation of MBCs based on the expression of CCR6 and CXCR3 is an accurate representation of the transcriptionally divergent populations. Secondly, we identified common BCR genes used by antigen-binding cells across all mice. We found that a large fraction of NP-binding cells used IGHVs 9–3 or 9–4 and IGKVs 5–48, 5–37, or 5–43 genes, indicating that the NP-specific BCR repertoire is in part composed of “public” BCR combinations (Figures S4K and S4L). Notably, MBCs using VH 9-3/4 and VK 5-48/37/43 in our large 10× dataset were enriched in CXCR3-expressing cells when compared with MBCs using alternative BCR gene combinations, reinforcing the notion that influenza-specific cells are mainly found within the CXCR3+ compartment (Figure S4M).
We then examined to which extent MBCs were clonally expanded. Regardless of the subset, we found that more than 60% of MBCs were constituted by single-cell clones, in line with a recent report (Figure S4N; Viant et al., 2020). Despite the limited number of expanded clones, we moved on to evaluate the overlaps among NP- and HA-binding MBCs with non-binding cells from the different subsets. We found notable overlaps among NP- and HA-binding cells with non-binding cells from the CCR6+CXCR3+ subset, and non-significant overlaps were detected with CCR6+CXCR3− and CCR6−CXCR3− subsets (Figure 5L). Furthermore, phylogenetic trees based on heavy- and light-chain nucleotide sequences show that clones comprising NP- and HA-specific cells were either entirely composed of antigen-binding cells or contained several non-binding cells, and regardless of the situation, these cells exclusively corresponded to the CCR6+CXCR3+ subset (Figure 5M). Overall, the simultaneous analysis of antigen binding, surface marker expression, and B cell repertoire revealed that CCR6+CXCR3+ and CCR6+CXCR3− populations represent discrete subsets of MBCs with divergent origins, in line with scenario (2). The CXCR3+ subset is composed of antigen-specific bona fide MBCs that share common progenitors with low-affinity cells. By contrast, the CXCR3− subset is enriched in MBCs with no apparent specificity for influenza antigens and originating from an alternative mechanism.
CXCR3 expression segregates lung CCR6+ MBCs into bona fide and bystander subsets
As CCR6+CXCR3− MBCs did not show evident specificity for influenza antigens or clonal overlap with influenza-specific cells, we investigated their potential origin. CCR6+CXCR3− MBCs did not arise from previously ongoing GCs in the spleen or Peyer's patches at the time of infection, as these cells were present at similar amounts in splenectomized mice and did not recognize microbiota antigens (Figures S5A–S5C). Furthermore, CCR6+CXCR3− MBCs did not originate from “on-site” GCs triggered against endogenous retrovirus or autoantigens exposed upon tissue damage, as these cells still appeared in mice treated with antiretrovirals and did not recognize common autoantigens (Figures S5D and S5E; Genton et al., 2006).
We hypothesized that CCR6+CXCR3− MBCs may originate from precursors that made it through the GC reaction due to bystander help received from Tfh cells. Tfh cells provide cognate help to antigen-specific B cells through CD40, ICAM-1, and SLAM receptor engagement. However, they can also produce soluble cytokines, such as IL-4, that are secreted in a multidirectional way instead of being polarized to the B-T cell contact (Huse et al., 2006). Indeed, it has recently been shown that IL-4 not only acts on antigen-specific interactions but it further diffuses to its proximity, influencing adjacent B cells in a bystander way (Duan et al., 2021). Notably, the IL-4 inducible gene Fcer2a (Kikutani et al., 1986) is one of the highest differentially expressed genes in the CCR6+CXCR3− MBCs in our transcriptomic datasets (Figures 2A and 5K); we confirmed this finding at the protein level by flow cytometry (Figure 6 A). Then, we investigated the role of IL-4 in the generation of CCR6+CXCR3− MBCs. Firstly, we measured IL-4 production at the peak of GC reactions following influenza infection. We found that more than 90% of IL-4 producing cells are Tfh cells, indicating that IL-4 secretion is highly restricted to GC reactions during influenza infection (Figure 6B). Then, we determined whether decreasing or increasing the availability of IL-4 in vivo can affect the extent of CCR6+CXCR3− MBC formation. To achieve this, we infected mice with the influenza virus and treated them with either anti-IL-4 blocking antibody, PBS, or complexed IL-4 (Figure 6C). In line with previous reports, diminishing IL-4 availability resulted in the decreased expression of Fcεr2a by B cells, whereas increasing IL-4 amounts promoted Fcεr2a expression (Figure 6D). Total MBC numbers in the lungs and lymph node were not significantly affected by the treatments (Figures 6E and 6F). Yet, changing IL-4 accessibility altered the balance between CXCR3+ and CXCR3− MBC subsets. When IL-4 availability was restricted, most MBCs were from the CCR6+CXCR3+ antigen-specific bona fide subset (Figures 6G and 6H). Instead, when IL-4 was largely available, the MBC pool was over populated by non-antigen-specific CCR6+CXCR3− MBCs (Figures 6G and 6H). Taken together, our data favor a scenario in which the CCR6+CXCR3− MBC subset is enriched in non-influenza-specific bystander MBCs generated as a result of a permissive mechanism within GC reactions.
Figure 6.

Bona fide and bystander MBCs
(A) Contour plots showing Fcεr2a expression by the lung MBC subsets after 70 days of influenza infection, paired t test.
(B) Contour plots showing the IL-4 GFP expression at day 9 of influenza infection in the mediastinal lymph node (left) and the proportion of GFP+ cells that express PD-1 and CXCR5 (right), one-way ANOVA.
(C) Experimental approach for the treatment of wild-type mice with anti-IL4 antibody, PBS, or complexed IL4 during infection.
(D) Histograms showing Fcεr2a expression in mice treated as in (C), one-way ANOVA.
(E and F) Contour plots of (E) lung and (F) lymph node CCR6+ MBCs in mice treated as in (C), one-way ANOVA.
(G and H) Contour plots of (G) the lung and (H) lymph node MBCs depicting the proportions of CCR6+CXCR3− and CCR6+CXCR3+ subsets in mice treated as in (C). Quantifications in the left panels show the CXCR3+/CXCR3− MBC ratio, one-way ANOVA. Quantifications in the middle panels show the number of NP+ MBCs, one-way ANOVA. Quantifications in the right panels show the proportion of NP+ MBCs within each subset, two-way ANOVA.
(I) Contour plots showing Fcmr expression by the lung MBCs at day 70 of the influenza infection, paired t test.
(J) Dot plots showing the binding of lung MBCs to labeled IgM immune complexes (ICs), paired t test.
(K and L) Enumeration of ASCs in the lungs of chimeric mice receiving (K) 80% μMT + 20% wild type versus 80% μMT + 20% Fcmr−/− bone marrow or (L) 50% CD45.1 + 50% wild-type CD45.2 versus 50% CD45.1 + 50% Fcmr−/− CD45.2 bone marrow. Mice were infected with influenza PR8, challenged with influenza X31 after 50 days, and sacrificed 4 days later. In (L), CD45.2 cells were sorted before ELISpot, two-way ANOVA test. In all the panels, each dot represents one mouse, mean ± SEM, ∗p < 0.01, ∗p < 0.05, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
See also Figure S5.
Bystander CCR6+CXCR3− MBCs express higher amounts of Fc receptors for IgE and IgM (Fcer2a and Fcmr) than CXCR3+ bona fide MBCs, as revealed by FB5P-seq transcriptomic and flow cytometry analysis (Figures 5K, 6A, 6I, and S5F). This led us to investigate if bystander MBCs could bind ICs despite their lack of specificity for influenza antigens, a phenomenon that would increase antigen density in the proximity of bona fide MBCs. We found that the incubation of lung MBCs with fluorescently labeled IgM ICs led to a preferential binding to bystander CCR6+CXCR3− MBCs (Figure 6J). To assess the contribution of this axis on the magnitude of memory responses, we generated mouse chimeras lacking Fcmr specifically on the B cell compartment. Chimeric mice were infected with influenza H1N1 PR8 and re-challenged with influenza H3N2 X31 at day 40. We found a profound reduction in the generation of influenza-specific IgA ASCs in Fcmr-deficient mice (Figure 6K). This defect was not intrinsic to bona fide MBCs, as the presence of congenic wild-type MBCs restored IgA recall responses (Figure 6L). Altogether, our results provide evidence for a role of the Fcmr axis on MBC responses to influenza infection.
SARS-CoV-2 infection generates bona fide and bystander MBC subsets
To find out whether the establishment of discrete subsets of MBCs with contrasting antigen specificities is a general feature of humoral responses to airborne pathogens, we intranasally infected K18-hACE2 mice with 2.103 PFU of SARS-CoV-2 (McCray et al., 2007). We detected foci of SARS-CoV-2 NP in peribronchial areas as early as day 2, and the infection rapidly spread toward deeper areas of the lung parenchyma by day 5 (Figures 7A and 7B). By day 70 of the infection, SARS-CoV-2 was effectively cleared, and we observed the accumulation of class-switched MBCs (CD19+CD45i.v.−IgD−CD38+GL-7−) in the lungs and mediastinal lymph node (Figures 7A–7D). We distinguished three discrete populations of MBCs based on their differential expression of CCR6 and CXCR3 (Figures 7E and 7F). The CCR6+CXCR3+ cells extensively bound SARS-CoV-2 Spike protein, indicating that this population represented the bona fide subset enriched in virus-specific cells (Figures 7G and 7H). By contrast, the CCR6+CXCR3− and CCR6−CXCR3− cells did not show substantial binding to Spike protein, indicating that these populations represented bystander and innate-like subsets, respectively. Lung-resident memory B and T cells were still observed in mice infected with low doses of SARS-CoV-2, even in those with no apparent weight loss, indicating that asymptomatic animals can still mount memory responses to SARS-CoV-2 (Figures S6A–S6E).
Figure 7.

MBC subsets in SARS-CoV-2 infection
(A and B) Confocal images of lung sections from (A and B) K18-hACE2 or (B) wild-type mice infected with 2.103 PFU of SARS-CoV-2. Scale bars, 130 μm.
(C and D) Contour plots showing (C) lung and (D) lymph node class-switched resident B cells (left) and MBCs (middle) from mice infected as in (A) and analyzed at day 70. The multicolor bar (right) shows IgH isotype distribution in MBCs.
(E and F) Dot plots showing the presence of MBC subsets in (E) lungs and (F) lymph node.
(G and H) Contour plots showing MBC binding to Spike protein in (G) lungs and (H) lymph node.
(I–K) Confocal images of lung sections at day 70 of the infection. Scale bars: 25 μm in (I), 18 μm in (J), and 20 μm in (K). The quantification in (I) displays the minimal distance of B cell clusters to EpCAM+ cells; dots represent B cell clusters. The quantification in (J) displays the minimal distance (nm) of IgM+ or IgG2b+ cells to the B cell cluster border divided by the cluster’s area (μm2); dots represent individual B cells.
(L) Dot plot showing the area and B220 fluorescence for the detected IgG2b+ surfaces in (K) (left). Dot plot showing CCR6 and CXCR3 fluorescence intensity for IgG2b+ MBCs (center). X and Y positions of MBC subsets are laid out in the B220 representation (right).
In (E)–(H), bar charts show the quantification of one experiment out of three, mean ± SEM. Each dot represents one mouse. (E and F) Two-way ANOVA test and (G and H) one-way ANOVA test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
See also Figure S6.
We finally analyzed the spatial distribution of MBCs in the lungs of SARS-CoV-2-infected animals by confocal imaging. As in this case, we could not rely on YFP expression, we tracked MBCs by using antibodies against B220, IgM, and IgG2b. We found that SARS-CoV-2 infection triggered the formation of B cell clusters that accumulated in peribronchial areas (Figure 7I). While IgM+ cells were restricted to the periphery of B cell clusters, IgG2b+ cells were largely confined in the center (Figure 7J). The analysis of CCR6 and CXCR3 expression in individual IgG2b+ cells revealed that both CCR6+CXCR3+ and CCR6+CXCR3− populations were equally distributed inside the B cell clusters (Figures 7K and 7L).
Our results show that SARS-CoV-2 infection, as observed with influenza virus, triggers the accumulation of bona fide, bystander, and innate-like MBCs in the respiratory niche with marked differences in antigen specificity and chemokine receptor expression. These results unveil common MBC mechanisms elicited during infection with diverse families of respiratory viruses.
Discussion
Several studies have shown that the MBC pool is composed of discrete cell subsets that coexist upon immunization and that bear a differential ability to seed GC reactions or form ASCs upon re-challenge (Weisel and Shlomchik, 2017). Some groups have demonstrated that MBC fate is associated with the BCR isotype, and while IgM+ MBCs are more predisposed to re-enter GC reactions, those expressing IgG are more prone to acquire an ASC phenotype upon re-challenge (Dogan et al., 2009; Kometani et al., 2013; Lutz et al., 2015; Pape et al., 2011). However, the BCR isotype does not always draw a clear-cut line, as class-switched cells were also shown to actively remodel their BCR within secondary GC reactions (McHeyzer-Williams et al., 2015). Work from Shlomchik and collaborators show that the subcategorization of MBCs according to the expression of CD80 and PD-L2, independently of BCR isotype, identifies cell subsets with distinct developmental kinetics and functions (Anderson et al., 2007; Weisel et al., 2016; Zuccarino-Catania et al., 2014). The differential ability to commit to the GC or PC program by CD80hi and CD80lo MBC subsets is associated with the unequal amount of CD40L help received (Koike et al., 2019). As most of these studies have been performed in the context of protein immunization, it remains unknown whether distinct subsets of MBCs coexist upon the resolution of infection. Here, we report that influenza infection gives rise to discrete subsets of MBCs with divergent origins, transcriptional programs, and functions.
In the last few years, it became evident that the presence of MBCs is not restricted to secondary lymphoid organs. Infection with the influenza virus leads to the development of lung-resident MBCs, which persist in the lung mucosa for long periods of time (Allie et al., 2019; MacLean et al., 2022; Oh et al., 2021; Tan et al., 2022). These studies identified MBCs based on their ability to bind fluorescently labeled flu proteins. Here, we took an unbiased fate-mapping approach in mice to track MBCs independently of their antigen specificity and based on their early expression of Aicda. By doing so, we were able to identify MBCs with a wide range of specificities, including those with no specificity for viral antigens that were disregarded in previous studies. Through unsupervised scRNA-seq analysis, we unveiled the presence of three discrete clusters of lung MBCs with unique expression patterns of CCR6 and CXCR3. The smallest subset consists of innate-like MBCs, which lack CCR6 and CXCR3 expression and exclusively express IgM. These cells do not seem to be part of the memory response to influenza, as a previous study identified a similar population of IgM+ MBCs with high IgHV6 usage in non-immunized Aid-EYFP mice (Le Gallou et al., 2018). Besides this minor subset, two large subsets of CCR6-expressing MBCs emerge after influenza infection, one of them lacking CXCR3 and the other one with high CXCR3 expression.
We found that both CXCR3− and CXCR3+ MBC subsets undergo extensive class switching toward IgG, share the lung peribronchial niche, and arise from GCs, based on their CD40L dependence and SHM rates. These subsets mainly differentiate into PCs, rather than into GC cells, when challenged ex vivo. Thus, these two transcriptionally distinct MBC subsets do not segregate cells regarding fate but, instead, cells generated through divergent mechanisms. The CXCR3+ subset constitutes bona fide MBCs, which are mostly pathogen specific and actively recruited during secondary immune responses, whereas the CXCR3− subset represents bystander MBCs with no evident specificity for pathogen-derived antigens. The following questions are inexorable: how do MBCs from the CXCR3− subset develop in GCs and what are they specific for? Work by Kelsoe and collaborators show that clonal selection is highly permissive in response to complex antigens, allowing the presence of B cells within GC reactions with a broad range of affinities and even cells with no detectable affinity for native immunogens (Kuraoka et al., 2016). Although the authors have shown that these “less-fit” GC B cells showed extensive SHM and clonal diversification, it remained unknown whether they could acquire a memory phenotype. Here, we show that these cells not only enter the long-lived memory compartment but also that they express a unique transcriptional program that distinguishes them from bona fide MBCs. Regarding antigen specificity, it seems unlikely that these cells are low affinity, as they do not display clonal overlap with antigen-specific cells. Furthermore, bystander MBCs do not recognize microbiota antigens, autoantigens, or non-native conformations of eliciting antigens, in line with previous reports (Horns et al., 2020; Kuraoka et al., 2016).
Then, how do these cells still make it to the memory compartment? Low-affinity B cells were shown to highly rely on ICAM-1 and CD40 surface expression to compensate for insufficient Tfh cell help (Nakagawa et al., 2021; Zaretsky et al., 2017). Furthermore, bystander B cells can survive in GCs due to the diffusion of soluble mediators, such as IL-4, from adjacent antigen-specific B-Tfh cell interactions (Duan et al., 2021; Wan et al., 2019). We found that IL-4 secretion was highly restricted to Tfh cells during the peak of GC responses triggered by influenza infection, in contrast to Th2 cell-oriented responses where IL-4 was available across the entire lymph node (Perona-Wright et al., 2010). Limiting the availability of IL-4 constrained the formation of bystander MBCs, whereas increasing IL-4 availability facilitated their appearance. As such, our data supports a model in which an alternative permissive mechanism operates within GCs regardless of antigen specificity, giving rise to bystander MBCs. This permissive mechanism seems to be a general strategy of the immune system, as we found that SARS-CoV-2 infection also engendered bona fide and bystander MBC subsets with contrasting antigen specificities.
Our findings raise the question of the potential benefit of generating bystander MBCs, as these cells can represent up to 50% of the MBC pool generated upon infection. We propose that the presence of high- and low-affinity MBCs within the bona fide subset helps the host to overcome future infections with the same pathogen, mutating variants, or even different viruses from the same family (Viant et al., 2020; Wong et al., 2020). By contrast, the prevalence of bystander MBCs with no detectable specificity for the infecting pathogen but evident SHM and high expression of Fc receptors could be beneficial to expand the diversity of the initial B cell repertoire and to retain ICs in close proximity to bona fide MBCs, enhancing the magnitude of the humoral memory response.
Limitations of the study
In this study, we identified bona fide and bystander lung MBCs. As specific depletion of MBC subsets was not possible, we performed adoptive transfer experiments to evaluate their protective functions. In the future, we will develop approaches to specifically deplete endogenous bona fide or bystander MBC subsets and evaluate the outcome of the humoral response after re-challenge. On top of this, we plan to generate strategies to specifically delete IL-4rα and Fcmr from the bystander MBC population to directly assess the impact of these axes on their development and function. On another note, we observed that mice lacking CCR6 or Fcmr show defective IgA, but not IgG, recall responses. Further work is required to unveil whether the activation of IgA+ and IgG+ bona fide MBCs follow different activation trajectories and whether bystander MBCs are activated in an antigen-independent manner upon re-challenge.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC/Cy7 Rat Anti-Mouse CD19, Clone 6D5 | Biolegend | Cat# 115530, RRID: AB_830707 |
| BUV395 Rat Anti-Mouse CD38, Clone 90/CD38 | BD | Cat# 740245, RRID: AB_2739992 |
| PE/Cy7 Rat Anti-Mouse CD38, Clone 90 | Biolegend | Cat# 102718, RRID: AB_2275531 |
| Alexa Fluor 647 Rat Anti-Mouse/Human GL7 Antigen, Clone GL7 | Biolegend | Cat# 144606, RRID: AB_2562185 |
| PerCP/Cy5.5 Rat Anti-Mouse/Human GL7 Antigen, Clone GL7 | Biolegend | Cat# 144610, RRID: AB_2562979 |
| PE Rat Anti-Mouse CD138 (Syndecan-1), Clone 281-2 | Biolegend | Cat# 142504, RRID: AB_10916119 |
| BV785 Rat Anti-Mouse CD138 (Syndecan-1), Clone 281-2 | Biolegend | Cat# 142534, RRID: AB_2814047 |
| BV650 Rat Anti-Mouse CD93, Clone AA4.1 | BD | Cat# 563807, RRID: AB_2738432 |
| BV605 Mouse Anti-Mouse CD95 (Fas), Clone SA367H8 | Biolegend | Cat# 152612, RRID: AB_2728202 |
| PE/Cy7 Rat Anti-Mouse IgD, Clone 29-2L17 | Biolegend | Cat# 405720, RRID: AB_2561876 |
| BV786 Rat Anti-Mouse IgD, Clone 11-26c.2a | BD | Cat# 563618, RRID: AB_2738322 |
| FITC Rat Anti-Mouse CD45, Clone 30-F11 | Biolegend | Cat# 103108, RRID: AB_312973 |
| PE Rat Anti-Mouse CD45, Clone 30-F11 | Biolegend | Cat# 103106, RRID: AB_312971 |
| PE/Dazzle 594 Hamster Anti-Mouse CD196 (CCR6), Clone 29-2L17 | Biolegend | Cat# 129822, RRID: AB_2687019 |
| BV421 Armenian Hamster Anti-Mouse CD183 (CXCR3), Clone CXCR3-173 | Biolegend | Cat# 126522, RRID: AB_2562205 |
| BV605 Armenian Hamster Anti-Mouse CD11c, Clone N418 | Biolegend | Cat# 117334, RRID: AB_2562415 |
| BV510 Rat Anti-Mouse CD21/CD35, Clone 7G6 | BD | Cat# 747764, RRID: AB_2872231 |
| PE/Cy7 Armenian Hamster Anti-Mouse CD55 (DAF), Clone RIKO-3 | Biolegend | Cat# 131814, RRID: AB_2800634 |
| PE/Cy5 Rat Anti-Mouse CD24, Clone M1/69 | Biolegend | Cat# 101812, RRID: AB_439714 |
| PerCP/Cy5.5 Rat Anti-Mouse IgM, Clone RMM-1 | Biolegend | Cat# 406512, RRID: AB_2075943 |
| V450 Rat Anti-Mouse IgG1, Clone A85-1 | BD | Cat# 562107, RRID: AB_10894002 |
| PE Rat Anti-Mouse IgG2b, Clone RMG2b-1 | Biolegend | Cat# 406708, RRID: AB_2563381 |
| Biotinylated Goat Anti-Mouse IgG2c, polyclonal | Southern BT | Cat# 1079-08, RRID: AB_2794467 |
| BV650 Rat Anti-Mouse IgG3, Clone R40-82 | BD | Cat# 744136, RRID: AB_2742025 |
| Alexa Fluor 647 Goat Anti-Mouse IgA, polyclonal | Southern BT | Cat# 1040-31, RRID: AB_2794377 |
| PerCP/Cy5.5 Armenian Hamster Anti-Mouse CD3e, clone 145-2C11 | Biolegend | Cat# 100328, RRID: AB_893318 |
| BV605 Rat Anti-Mouse CD4, clone RM4-5 | Biolegend | Cat# 100548, RRID: AB_2563054 |
| APC/Cy7 Rat Anti-Mouse CD8a, clone 53-6.7 | Biolegend | Cat# 100714, RRID: AB_312753 |
| PE Rat Anti-Mouse/Human CD44, clone IM7 | Biolegend | Cat# 103008, RRID: AB_312959 |
| APC Rat Anti-Mouse CD62L, clone MEL-14 | Biolegend | Cat# 104412, RRID: AB_313099 |
| PE/Cy7 Armenian Hamster Anti-Mouse CD69, clone H1.2F3 | Biolegend | Cat# 104512, RRID: AB_493564 |
| BV711 Armenian Hamster Anti-Mouse CD103, clone 2E7 | Biolegend | Cat# 121435, RRID: AB_2686970 |
| APC Rat Anti-Mouse CD23, clone B3B4 | Biolegend | Cat# 101620, RRID: AB_2563439 |
| Purified Mouse Anti-Mouse Fcmr, clone MM3 | Biolegend | Cat# 157802, RRID: AB_2814094 |
| PerCP/Cy5.5 Mouse anti-Mouse CD45.1, Clone A20 | Thermo Fisher Scientific | Cat# 45-0453-82, RRID: AB_1107003 |
| Pacific Blue Mouse anti-Mouse CD45.2, Clone 104 | Biolegend | Cat# 109820, RRID: AB_492872 |
| BV650 Armenian Hamster Anti-Mouse TCRβ, clone H57-597 | Biolegend | Cat# 109251, RRID: AB_2810348 |
| PE PBS57-loaded Mouse CD1d tetramer | NIH | N/A |
| PE/Cy7 Rat Anti-Mouse CD4, Clone GK1.5 | Biolegend | Cat# 116016, RRID: AB_2563111 |
| PerCP eFluor 710 Rat Anti-Mouse CD185 (CXCR5), Clone SPRCL5 | Thermo Fisher Scientific | Cat# 46-7185-82, RRID: AB_2573837 |
| Alexa Fluor 647 Rat Anti-Mouse CD279 (PD-1), Clone RMP1-30 | Biolegend | Cat# 109118, RRID: AB_2566550 |
| Pacific Blue Rat Anti-Mouse/Human CD45R (B220), Clone RA3-6B2 | Biolegend | Cat# 103227, RRID: AB_492876 |
| Alexa Fluor 488 Rat Anti-Mouse CD326 (Ep-CAM), Clone G8.8 | Biolegend | Cat# 118210, RRID: AB_1134099 |
| Alexa Fluor 647 Rat Anti-Mouse CD326 (Ep-CAM), Clone G8.8 | Biolegend | Cat# 118212, RRID: AB_1134101 |
| APC Rat Anti-Mouse CD3, clone 17A2 | Biolegend | Cat# 100236, RRID: AB_2561456 |
| Alexa Fluor 647 Rat Anti-Mouse CD169 (Siglec-1), Clone 3D6.112 | Biolegend | Cat# 142408, RRID: AB_2563621 |
| APC Armenian Hamster Anti-Mouse CD183 (CXCR3), Clone CXCR3-173 | Biolegend | Cat# 126512, RRID: AB_1088993 |
| eFluor 570 Rat Anti-Mouse IgM, Clone II/41 | eBioscience | Cat# 41579082, RRID: AB_2573617 |
| Alexa Fluor 488 Rat Anti-Mouse IgG2b, Clone RMG2b-1 | Biolegend | Cat# 406718, RRID: AB_2876732 |
| Chicken Anti-GFP, polyclonal | Abcam | Cat# ab13970, RRID: AB_300798 |
| Alexa Fluor 488 Goat Anti-Chicken IgY (H+L), polyclonal | Invitrogen | Cat# A11039, RRID: AB_142924 |
| Rabbit Anti-SARS-CoV-2 Nucleocapsid, polyclonal | GeneTex | Cat# GTX135357, RRID: AB_2868464 |
| Alexa Fluor 555 Goat Anti-Rabbit IgG (H+L), | Invitrogen | Cat# A21428, RRID: AB_2535849 |
| BV510 Mouse IgM anti-TNP (Clone G155-228) | Becton | Cat# 563082, RRID: AB_2869457 |
| Goat anti-mouse IgM-biot | Southern Biotech | Cat# 1020-08, RRID: AB_2737411 |
| Goat anti-mouse IgG-biot | Southern Biotech | Cat# 1010-08, RRID: AB_2794127 |
| Goat anti-mouse IgA-biot | Southern Biotech | Cat# 1040-08, RRID: AB_2794374 |
| Anti-mouse CD40L (clone MR1) | BioXCell | Cat# BE0017-1, RRID: AB_1107601 |
| Anti-mouse IL4 (11B11) | BioXCell | Cat# BE0045, RRID: AB_1107707 |
| Bacterial and virus strains | ||
| Influenza virus A/Puerto Rico/8/1934 (PR8) H1N1 | Eric Vivier’s lab | N/A |
| Influenza virus A/X-31 H3N2 | Ronan Le Goffic’s lab | N/A |
| BetaCoV/France/IDF0372/2020 (SARS-CoV-2) | Sylvie van der Werf’s lab | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| PE / R-Phycoerythrin Conjugation Kit - Lightning-Link® | Abcam | Cat# ab102918 |
| APC Conjugation Kit - Lightning-Link® | Abcam | Cat# ab201807 |
| Alexa Fluor® 700 Conjugation Kit (Fast) - Lightning-Link® | Abcam | Cat# ab269824 |
| Tamoxifen | Fisher Scientific | Cat# 11455161 |
| Corn oil, ACROS Organics™ | Fisher Scientific | Cat# 10616051 |
| Tenofovir disoproxil fumarate, 98%, Thermo Scientific™ | Fisher Scientific | Cat# 15668725 |
| Thermo Scientific™ Emtricitabine | Fisher Scientific | Cat# 15781789 |
| CpG ODN-1826 | Invivogen | Cat# tlrl-1826c |
| TNP-KLH biotin | Santa Cruz | Cat# sc-396497 |
| Streptavidin-PE | Biolegend | Cat# 405204 |
| Biotinylated anti-Ig | Southern Biotech | Cat# 1010-08 |
| Extravidin-Alkaline Phosphatase | Sigma | Cat# E2636 |
| IL-4 | Peprotech | Cat# 214-14 |
| IL-21 | Peprotech | Cat# 210-21 |
| Influenza A H1N1 (A/Puerto Rico/8/34) Hemagglutinin | Sino Biological | Cat# 11684-V08H |
| Influenza A H1N1 (A/Puerto Rico/8/34) Nucleoprotein | Sino Biological | Cat# 11675-V08B |
| Influenza A H1N1 (A/Puerto Rico/8/34) Neuraminidase | Sino Biological | Cat# 40196-VNAHC |
| SARS-CoV-2 spike protein | Sino Biological | Cat# 40589-V08B1 |
| Sm/RNP | Arotec Diagnostics | Cat# ATR01 |
| RNP 68 kD | Arotec Diagnostics | Cat# ATR04 |
| Critical commercial assays | ||
| Lung Dissociation Kit, mouse | Miltenyi Biotec | Cat# 130-095-927 |
| Chromium Single Cell 5’ Library & Gel Bead Kit | 10X Genomics | Cat# 1000014 |
| Chromium Single Cell A Chip Kit | 10X Genomics | Cat# 120236 |
| Silane Dynabeads™ MyOne™ | 10X Genomics | Cat# 2000048 |
| KAPA HiFi HotStart Ready Mix PCR Kit | Roche Diagnostics | Cat# 07958935001 |
| Agilent High Sensitivity DNA kit | Agilent | Cat# 5067-4626 |
| Nextera XT DNA sample Preparation kit | Illumina | Cat# FC-131-1096 |
| SPRIselect | Beckman Coulter | Cat# B23317 |
| Qubit™ dsDNA HS Assay kit | ThermoFisher Scientific | Cat# Q32851 |
| Deposited data | ||
| Single-Cell RNA-Seq data | This study | GEO: GSE174682 |
| Original Code | This study |
https://doi.org/10.5281/zenodo.5566675 https://github.com/CIML-bioinformatic/MGlab_PMlab_moFluMemB |
| Experimental models: Cell lines | ||
| Dog (female), MDCK cells | Marc Dalod’s lab | N/A |
| 40LB cells | Kitamura’s lab | N/A |
| Experimental models: Organisms/strains | ||
| Mouse, Wild type (C57BL/6J) | Janvier Labs | N/A |
| Mouse, B6.SJL-Ptprca Pepcb /BoyJ (CD45.1) | The Jackson Laboratory | Strain Code: 002014 |
| Mouse, B6.129P2-Aicdatm1.1(cre/ERT2)Crey/J (AID-Cre-ERT2) | The Jackson Laboratory | Strain Code: 033897 |
| Mouse, B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J (Rosa26-EYFP) | The Jackson Laboratory | Strain Code: 006148 |
| Mouse, B6.129S2-Ighmtm1Cgn/J (μMT) | The Jackson Laboratory | Strain Code: 002288 |
| Mouse, B6.129S2-Fcmrtm1Mak (Fcmr -/-) | (Lang et al., 2013) | N/A |
| Mouse, B6.129P2-Ccr6tm1Dgen/J (CCR6 -/-) | The Jackson Laboratory | Stain Code: 005793 |
| Mouse, B6.129P2-Cxcr3tm1Dgen/J (CXCR3 -/-) | The Jackson Laboratory | Stain Code: 005796 |
| Mouse, B6;129-Lattm2Mal (LAT Y136F) | (Aguado et al., 2002) | N/A |
| Mouse, C.129-Il4tm1Lky/J (IL-4 GFP) | The Jackson Laboratory | Strain code: 004190 |
| Mouse, B6.Cg-Tg(K18-ACE2)2Prlmn/J (K18-hACE2) | The Jackson Laboratory | Strain code: 034860 |
| Software and algorithms | ||
| DIVA (version 9) | BD Biosciences | https://www.bdbiosciences.com/en-eu/products/software/instrument-software/bd-facsdiva-software |
| FlowJO (version 10.8.1) | FlowJo, LLC | https://www.flowjo.com/ |
| Zen (version 3.5.093.00000) | Zeiss | https://www.zeiss.com/microscopy/int/downloads/zen.html |
| ImageJ (version 1.53q) | NIH Image | https://imagej.nih.gov/ij/ |
| Imaris (version 9.6) | Bitplane | http://www.bitplane.com/ |
| GraphPad Prism (version 9.3.1) | GraphPad Software LLC | https://www.graphpad.com/scientific-software/prism/ |
| SAPHIR | Mathieu Fallet | https://github.com/Imagimm-CIML/SAPHIR |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Mauro Gaya (gaya@ciml.univ-mrs.fr).
Materials availability
This study did not generate any unique reagents.
Experimental model and subject details
Mice
8-week old wild-type C57BL/6 mice were obtained from Janvier Labs. Aicda-CreERT2 mice were obtained from Claude-Agnès Reynaud and Jean Claude Weill, Institut Necker Enfants Malades, France. Rosa26-EYFP, CD45.1, CCR6-/- and K18-hACE2 mice were obtained from Jackson Laboratories, USA. μMT mice were obtained from Stéphane Mancini, Centre de Recherche en Cancérologie de Marseille, France. Lat Y136F mice were obtained from Bernard Malissen, Centre d’Immunologie de Marseille Luminy, France. IL-4 GFP mice were obtained from Mark Wilson, Genentech, US. CXCR3-/- and Fcmr-/- bone marrows were obtained from Jacqueline Marvel, Centre International de Recherche en Infectiologie, France and Tak Mak, University of Toronto, Canada, respectively. Aicda-CreERT2 mice were further crossed with Rosa26-EYFP and CCR6-/- mice.
For the generation of mixed bone marrow chimeras, μMT mice of 6-8 weeks of age were irradiated with 2 doses of 4.75 Gy, 4 hours apart. One day later, bone marrow cells were injected i.v. in recipient animals (1x106 cells in total). Experimental animals were kept on water with Bactrim for 3 days prior and 3 weeks post irradiation treatment. Chimeras were used after 8 weeks of reconstitution.
Mice were bred and maintained at the animal facilities of the Centre d’Immunologie de Marseille Luminy and Centre d'Immunophénomique. Up to five mice per cage were housed under a standard 12 hr light/dark cycle, with room temperature at 22°C (19°C-23oC change). They were fed with autoclaved standard pellet chow and reverse osmosis water. All cages contained 5 mm of aspen chip and tissue wipes for bedding and a mouse house for environmental enrichment. Mice were used at the age of 8 to 12 weeks and littermates (males or females) were randomly assigned to experimental groups. Generally, between 4 to 8 mice were used per experimental group. Experimental procedures were conducted in accordance with French and European guidelines for animal care under the permission number 16708-2018091116493528 following review and approval by the local animal ethics committee in Marseille.
Method details
Infections and injections
Mice were anesthetized i.p. with Ketamine/Xylazine (100 mg/kg body) and intranasally infected with 5 PFU of Influenza virus A/Puerto Rico/8/1934 (PR8) H1N1 strain or 104 PFU of Influenza virus A/X-31 H3N2 or 102-104 PFU of SARS-CoV-2 in 20 μl of PBS. In indicated experiments, Influenza PR8 was inactivated under 365 nm long-wave UV light for 10 minutes and administered intranasally at 105 PFU/mouse. Influenza virus was amplified on MDCK cells. Purification of viral particles was performed in a sucrose 30% cushion at 25,000RPM for 2 hours in an SW32Ti rotor. For Aid-EYFP mice, tamoxifen (Cayman chemical) was resuspended in corn oil (Sigma), sonicated and given by oral gavage at 5mg/100μl. For in vivo disruption of germinal centers, mice were i.p. injected with 300 μg of anti-CD40L (MR1, BioXCell) in 200 μl of PBS at different days of infection. For in vivo IL-4 blocking, mice were i.p. injected with 500 μg of anti-IL4 (11B11, BioXCell) in 200 μl of PBS at different days of infection. For the administration of IL-4 complex (IL-4c), 4 μg of recombinant mouse IL-4 (PrepoTech) was complexed to 20 μg of anti-mouse IL-4 (BioXCell, 11B11), diluted in 200 μl of PBS and administered i.p. on different days of infection. For antiretroviral treatment, mice were treated with Tenofovir disoproxil fumarate 100 mg/kg and Emtricitabine 60 mg/kg (both from ACROS Organics) every other day by oral gavage from one week before influenza infection and for two weeks after challenge. For in vivo labeling of immune cells in circulation, 3 μg of anti-CD45 antibody was administered i.v. 5 minutes before sacrifice. For adoptive transfers, bone marrow cells (1×106) were resuspended in 100 μl of PBS and i.v. injected. For the protection experiment, μMT mice received 5×106 C57BL/6 splenocytes i.v. and 15μg of CpG ODN-1826 (Invivogen) intranasally in PBS (Onodera et al., 2012). After 10 days, mice received 106 splenic CD4+ T cells and 3×103 CCR6+CXCR3- or CCR6+CXCR3+ MBCs sorted from lungs of B6 mice infected with Influenza PR8 70 days before. The day after, mice were challenged with PR8 virus, weight was measured daily and euthanized if exhibiting ≥20% loss of initial mass.
The strain BetaCoV/France/IDF0372/2020 (SARS-CoV-2) was supplied by the National Reference Centre for Respiratory Viruses hosted by Institut Pasteur (Paris, France) and headed by Pr. Sylvie van der Werf. The human sample from which strain BetaCoV/France/IDF0372/2020 was isolated has been provided by Dr. X. Lescure and Pr. Y. Yazdanpanah from the Bichat Hospital, Paris, France. Moreover, the strain BetaCoV/France/IDF0372/2020 was supplied through the European Virus Archive goes Global (Evag) platform, a project that has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No 653316. Infectious stocks were grown by inoculating Vero E6 cells and collecting supernatant upon observation of cytopathic effect; debris were removed by centrifugation and passage through a 0.22-μm filter. Supernatant was then aliquoted and stored at -80 °C. Vero E6 (CRL-1586; American Type Culture Collection) were cultured at 37°C in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES (pH 7.3), 1 mM sodium pyruvate, 1 x non-essential amino acids and 100 U/ml penicillin–streptomycin. Work with SARS-CoV-2 was performed in the biosafety level 3 laboratory of Center for Immunophenomics (CIPHE) by personnel equipped with powered air-purifying respirators. All the CIPHE BSL3 facility operations are overseen by a Biosecurity/Biosafety Officer and accredited by Agence Nationale de Sécurité du Médicament (ANSM).
Flow Cytometry
Single cell suspensions of mediastinal lymph nodes and spleens were obtained by pressing organs through a 70μm nylon mesh cell strainer with a plastic plunge in PBS 2%FCS 2mM EDTA. For lungs, single cell suspensions were obtained with the mouse lung dissociation kit (Miltenyi) according to manufacturer instructions. For spleen and lungs, cell suspensions were further incubated with red blood cell lysis buffer for 5 minutes. To block nonspecific antibody binding, cell suspensions were incubated with hybridoma supernatant 2.4G2, diluted 1/5 in PBS 2%FCS 2mM EDTA, for 20 minutes on ice. For labeling of surface markers, cells were stained for 20 minutes on ice with the indicated anti-mouse antibodies and fluorescently-labeled influenza hemagglutinin, nucleoprotein, neuraminidase or SARS-CoV-2 spike protein (Sino biological). Labeling of recombinant proteins was carried out using APC, PE or PE-Cy5 conjugation lightning-link kits (Abcam). When using biotinylated antibodies, cell suspensions were washed following antibody labeling and incubated for 20 minutes on ice with labeled streptavidin. Immune complexes were prepared by mixing 5 μg/ml mouse IgM-BV510 (Clone G155-228, BD), 10 μg/ml TNP-KLH biotin (Santa Cruz) and 1 μg/ml streptavidin-PE (Biolegend) and incubated with cell suspensions for 30 minutes. Finally, cells were either resuspended in 400μl of PBS 2%FCS 2mM EDTA and analyzed on Fortessa-X20/Symphony cytometers (BD Biosciences) or in RPMI media and used for cell sorting on a FACSAria II (BD, Biosciences). DAPI (0.1 μg/ml final concentration) was added right before passing samples on cytometers. In the case of samples from SARS-CoV-2 infected animals, cell suspensions were incubated with Zombie UV fixable viability kit (Biolegend) to exclude dead cells and samples were fixed 30 minutes with PFA 4%. Data was analyzed using FlowJo (TreeStar).
Immunohistochemistry
Lungs, lymph nodes and spleens were fixed in 4% PFA for 6 hours at 4°C, washed with PBS, incubated overnight in PBS 30% sucrose solution, immersed in OCT and snap-frozen in liquid nitrogen-cooled isopentane. Cryostat sections (10 to 30 μm thick) were dried in silica beads, permeabilized with PBS Saponin 0.5% for 30 minutes and blocked with PBS 0.5% saponin 2% BSA 1% goat serum 1% FCS for 30 minutes. Sections were then incubated with primary antibodies in PBS 0.5% saponin 2% BSA 1% goat serum 1% FCS for at least 1 hour, washed and incubated with secondary antibodies for a further hour. After a final wash, sections were mounted in Fluoromount-G mounting media. Imaging was carried out on a LSM 780 (Zeiss) inverted confocal microscope using a Plan-Apochromat 40x NA 1.3 oil immersion objective or a Plan-Apochromat 20X/0.8 M27 objective in the case of full organ section.
Analysis of confocal images
Image segmentation was performed using a specific ImageJ macro based on classical binary watershed to generate a mask on YFP+ cells. Selections were corrected manually by overlay masking. Mean fluorescence intensity for the B220, CXCR3 and CCR6 channels as well as the area in each COI (Cell Of Interest) was exported. The ROI (Region Of Interest) was defined manually at the beginning of the macro and exported in order to calculate cell density. The macro exports 4 files: the image in TIF, the COI in zip format (.roi), the mean fluorescence intensity and area for each cell in csv, and the channel legend. A specific software named SAPHIR (Shiny Analytical Plot of Histological Image Results) was developed using R, the code source can be downloaded here: https://github.com/elodiegermani/SAPHIR. This software uses the 4 files exported by the ImageJ macro and allows scatterplot gating (cross, polygon or lasso) to filter and define cell populations interactively with the COI overlaid on the image. The ROI used to select the mask on YFP+ B cells was further used to calculate the centroid of the clusters with ImageJ software. The distance of each MBC to the centroid was manually measured with ImageJ.
Ex vivo culture of MBCs on 40LB cells
40LB cells were cultured in DMEM supplemented with 10% FCS, penicillin 100u/ml, streptomycin 100μg/ml, sodium pyruvate 1mM and beta-mercaptoethanol 5x10-5M as previously described (Nojima et al., 2011). 40LB cells were irradiated (120Gy) and seeded in 24-well plates at a density of 0.25x106 cells/well. The following day, sorted MBCs were added on irradiated feeder cells in RPMI, 10% FCS, penicillin 100u/ml, streptomycin 100μg/ml, sodium pyruvate 1mM and beta-mercaptoethanol 5x10-5M. Mouse recombinant IL-21 (PeproTech) was added to co-cultures at 10ng/ml. At indicated time points, cells were analyzed by flow cytometry and supernatant collected for posterior antibody analysis.
ELISpot
To measure influenza-specific ASCs, enzyme-linked immunosorbent spot (ELISpot) multiscreen filtration plates (Millipore) were activated with absolute ethanol, washed with PBS and coated overnight at 4oC with 1μg/ml of nucleoprotein (Sino Biological) diluted in PBS. Plates were subsequently blocked for 1 hour with complete medium and incubated for 24 hours at 37°C with serial dilutions of lymph node, lung and spleen single cell suspensions. For lungs, the lymphocyte fraction was previously enriched using 40:80% Percoll (GE Healthcare). Plates were washed with PBS 0.01% Tween-20 and incubated for 1 hour with 1μg/ml of biotinylated anti-IgM, IgG or IgA (Southern Biotech) diluted in PBS 1% BSA. Then, plates were washed and incubated for 30 minutes with 1μg/ml Streptavidin-Alkaline Phosphatase (Sigma). Finally, plates were washed and developed with BCIP®/NBT (Sigma).
ELISA
To measure influenza-specific antibodies, ELISA (enzyme-linked immunosorbent assay) plates were coated overnight at 4oC with either 1μg/ml of nucleoprotein, hemagglutinin or neuraminidase (Sino Biological) diluted in PBS or with influenza PR8 particles that were previously inactivated for 30 mins with UV light, 1 minute at 95°C, 1 hour at pH 2 or 1 hour with Triton 1%. To detect autoantibodies, plates were coated with 5μg/ml of genomic DNA, RNA extracted from lungs of influenza infected mice, Sm/RNP or RNP 68 kD (Arotec). To measure antibodies against commensal bacteria, we homogenized 200 mg of fecal pellets from naive C57BL/6 SPF mice (8 weeks old) in sterile PBS, filtered through a 70 μm cell strainer, and separated from debris/mouse cells by removing the pellet after centrifugation at 1000 rpm for 5 minutes. Isolated fecal bacteria were washed twice, heat-killed at 85°C for 1 h, resuspended in 10 ml of PBS and used for coating plates. Plates were washed with PBS 0.01% Tween, blocked for 2 hours at room temperature with PBS 2.5% FCS and incubated overnight at 4oC with serial dilutions of co-culture/serum supernatants. The following day, plates were washed and probed at room temperature for 1 hour with 1μg/ml of biotinylated anti-Ig (Southern Biotech) in PBS 2.5%FCS. Plates were washed and incubated at room temperature for 30 minutes with 1μg/ml Streptavidin-Alkaline Phosphatase (Sigma). Plates were washed and developed with p-NitrophenylPhosphate (Sigma). 405nm absorbance was detected using a SPECTRAmax190 plate reader (Molecular Device).
10x 5’ scRNA-Seq library preparation
In a first scRNA-seq experiment (Figures 2 and 4), single-cell suspensions from spleen, lymph nodes and lungs were prepared as described above. Because enzymatic digestion of tissues at 37°C induced the expression of dissociation-induced genes in some lung MBCs (Figure S2), we performed a second experiment using only mechanical dissociation of lung tissue at 4°C. For both experiments, we used cell hashing with hashtag oligonucleotides (HTO) to multiplex 3 organ samples from 3 individual mice as previously described (Mimitou et al., 2019). After cell surface staining with the mix of antibodies used for gating MBCs, single-cell suspensions from each organ of each individual mice were independently stained with a distinct barcoded anti-mouse CD45 antibody (in-house conjugated) in PBS 2%FCS 2mM EDTA for 30 min on ice, then washed and resuspended in PBS. For each sample, live MBCs (DAPI-CD19+eYFP+CD38+GL7-) were bulk-sorted with BD FACS Aria II. Sorted cell samples (experiment 1: 13,126 lung cells, 3,414 lymph node cells, 12,838 spleen cells; experiment 2: 1,319 lung cells, 1,436 lymph node cells, 1,500 spleen cells) were pooled and loaded in a single capture well for subsequent 10x Genomics Single Cell 5’ v1 workflow.
10x 5’ scRNA-seq libraries were prepared according to the manufacturer’s instructions with modifications for generating the BCR-seq libraries. Following cDNA amplification, SPRI select beads were used to separate the large cDNA fraction derived from cellular mRNAs (retained on beads) from the HTO-containing fraction (in supernatant). For the cDNA fraction derived from mRNAs, 50ng were used to generate transcriptome library and around 5ng were used for BCR library construction. Gene expression libraries were prepared according to manufacturer’s instructions. For BCR libraries, heavy and light chain cDNA were amplified by two rounds of PCR (6 cycles + 8 cycles) using external primers recommended by 10x Genomics, and 800 pg of purified amplified cDNA was tagmented using Nextera XT DNA sample Preparation kit (Illumina) and amplified for 12 cycles using the SI-PCR forward primer (10x Genomics) and a Nextera i7 reverse primer (Illumina). For the HTO-containing fraction, 5ng was used to generate the HTO library. The resulting libraries were pooled and sequenced together on an Illumina NextSeq550 platform, using High Output 75-cycle flow cells, targeting 5×104 reads per cell for gene expression, 5x103 reads per cell for BCR, 2x103 reads per cell for hashtag, in paired-end single-index mode (Read 1: 26 cycles, Read i7: 8 cycles, Read 2: 57 cycles).
FB5P-seq library preparation
Single-cell suspensions from lungs of three previously infected Aid-EYFP mice were prepared as described above with enzymatic digestion, and stained with a panel of antibodies for identifying subsets of antigen-specific MBCs (GL7-PerCP-Cy5.5, HA-PE, CCR6-PE-Dazzle594, CD38-PE-Cy7, NP-APC, CD19-APC-Cy7, CXCR3-BV421, Live/Dead Aqua stain). FB5P-seq protocol was performed as previously described (Attaf et al., 2020). Briefly, single MBCs were FACS sorted on a BD Influx into 96-well PCR plates containing 2μl lysis mix per well. The index-sorting mode was activated to record the different fluorescence intensities of each sorted cell. Immediately after cell sorting, plates containing single cells in lysis mix were frozen on dry ice and stored at -80°C until further processing. For each plate, library preparation consisted in RT with template switching for incorporating well-specific barcodes and UMIs, cDNA amplification with 22 cycles of PCR, pooling of 96 wells into one tube, and 5’-end RNA-seq library preparation using tagmentation-based modified Nextera XT DNA sample Preparation kit (Illumina). Libraries were tagged with a plate-specific i7 index and were pooled for sequencing on an Illumina NextSeq2000 platform, with P2 flow cells, targeting 2.5×105 reads per cell in paired-end single-index mode (Read 1: 103 cycles, Read i7: 8 cycles, Read 2: 16 cycles).
scRNA-seq analysis
Preprocessing and analysis of data were done through the usage of standard tools and custom R and Python scripts. We used R version 3.5 and 3.6, Seurat package version 3 (Stuart et al., 2019), 10x Genomics CellRanger version 3, CITE-seq-count version 1.4 (Stoeckius et al., 2018). Docker and Singularity containers were used to ensure the reproducibility of analyses. All codes and data are available on Github and Zenodo.
Pre-processing of FB5P-seq dataset
We used a custom bioinformatics pipeline to process fastq files and generate single-cell gene expression matrices and BCR sequence files as previously described (Attaf et al., 2020). Detailed instructions for running the FB5P-seq bioinformatics pipeline can be found at https://github.com/MilpiedLab/FB5P-seq. Quality control was performed to remove poor quality cells. Cells with less than 500 genes detected and genes detected in fewer than 3 cells were removed. We further excluded bad quality cells expressing more than 3% of mitochondrial genes or less than 10% of ribosomal genes. For each cell, gene expression UMI count values were log-normalized with Seurat NormalizeData with a scale factor of 10,000 (Stuart et al., 2019).
Index-sorting FCS files were visualized in FlowJo software and compensated parameters values were exported in CSV tables for further processing. For visualization on linear scales in the R programming software, we applied the hyperbolic arcsine transformation on fluorescence parameters (Finak et al., 2010).
For BCR sequence reconstruction, the FB5P-seq pipeline used Trinity for de novo transcriptome assembly for each cell based on Read1 sequences, then MigMap for filtering the resulting contigs for productive BCR sequences and identifying germline V, D and J genes and CDR3 sequence for each contig. Filtered contigs were aligned to reference constant region sequences using Blastn. The FB5P-seq pipeline also ran the pseudoaligner Kallisto to map each cell’s FB5P-seq Read1 sequences on its reconstructed contigs and quantify contig expression. The outputs of the FB5P-seq pipeline were further processed and filtered with custom R scripts. For each cell, reconstructed contigs corresponding to the same V(D)J rearrangement were merged, keeping the largest sequence for further analysis. We discarded contigs with no constant region identified in-frame with the V(D)J rearrangement. In cases where several contigs corresponding to the same BCR chain had passed the above filters, we retained the contig with the highest expression. BCR metadata from the MigMap and Blastn annotations were appended to the gene expression and index sorting metadata for each cell. Finally, heavy and light chain contig sequences were trimmed to retain only sequences from FR1 to the first 36 nucleotides of FR4 regions, and were exported as fasta files for further analysis of clonotypes and BCR phylogenies.
Pre-processing of 10x 5’ datasets
Raw fastq files from gene expression libraries were processed using Cell Ranger software, with alignment on the mm10 reference genome. For each experiment, cells with less than 200 genes detected and genes detected in less than 3 cells were removed. We further excluded bad quality cells expressing less than 2,000 UMI, more than 40% mitochondrial genes or less than 10% ribosomal genes. HTO barcodes for sample demultiplexing after hashing were counted using CITE-seq-count and were normalized for each cell using a centered log ratio (CLR) transformation across cells implemented in the Seurat function NormalizeData. Cells were demultiplexed using Seurat MULTIseqDemux function and barcodes assigned as doublets or negative were excluded from further analysis. The resulting filtered UMI count matrices were log-normalized with Seurat NormalizeData with a scale factor of 10,000.
BCR-seq raw fastq files were processed with the FB5P-seq pipeline (Attaf et al., 2020) as described above for FB5P-seq datasets, omitting the part of the pipeline related to gene expression analysis, and using the list of cell-associated 10x barcodes from CellRanger analysis as inputs for splitting bam files upstream Trinity assembly of BCR contigs. BCR metadata from the MigMap and Blastn annotations were appended to the gene expression metadata for each cell. Heavy and light chain variable sequences, trimmed to retain only sequences from FR1 to the first 36 nucleotides of FR4 regions, were exported as fasta files for further analysis of clonotypes and BCR phylogenies.
BCR-seq based phylogenies
For selected large clones in the FB5P-seq dataset, we inferred the unmutated common ancestor (UCA) sequence by combining the IMGT-defined germline VL, JL and VH, DH, JH sequences with the observed VL-JL and VH-DH-JH junctional sequences from the least somatically mutated sequence observed in the clonotype. For phylogenetic analyses, concatenated variable (FR1 to FR4) IGH and IGL sequences of UCA and all clonally related FL cells were trimmed to equal length and aligned with the GCtree software (DeWitt et al., 2018). Phenotypic group metadata were used as labels to color the different nodes and leaves of the resulting BCR sequence phylogenetic trees.
Dataset analysis
Analysis of datasets were performed using custom R scripts. Variable genes (n=2000) were identified with Seurat FindVariableFeatures (vst method), BCR coding genes were excluded from the lists of variable genes. After centering with Seurat ScaleData, principal component analysis was performed on variable genes with Seurat RunPCA, and embedded in two-dimensional UMAP plots with Seurat RunUMAP on 20 principal components. UMAP embeddings colored by sample metadata or clusters were generated by Seurat DimPlot, those colored by single gene expression or module scores were generated by Seurat FeaturePlot, those colored by BCR sequence metadata were generated with ggplot2 ggplot.
Clustering was performed using the FindNeighbors and FindClusters methods of the Seurat package using 30 PC for SNN graph build and Leiden clustering method with sensitivity set to 0.5, 1.3, and 0.5 for the first and second 10x datasets, and the FB5P-seq dataset, respectively.
Marker genes between clusters were identified using the FindAllMarkers method of the Seurat package using the Wilcoxon Rank Sum test on genes expressed at least in 10% of the cells, a logFC threshold of 0.25 and a FDR threshold of 0.001.
Heatmap of gene expression along tissue clusters was done performing a mean of the expression of the genes of interest over the clusters, using the pheatmap package (version 1.0.12) for the plot. Heatmap of Szymkiewicz–Simpson coefficient were computed using the set of identified marker genes of each cluster and the ggplot2 ggplot function (version 3.3) for the plot. Szymkiewicz–Simpson coefficient of an intersection of two sets is the ratio between the size of the intersection between the two sets and the size of the smaller set of the two.
The Stress Index was computed at single-cell scale using the function AddModuleScore of the Seurat package, based on a list of genes from O’Flanagan et al. (2019) (10.1186/s13059-019-1830-0, Figure 3). This is a core gene set of 512 heat shock and stress response genes, including Fos and Jun, induced by collagenase (37°C), which are minimized by dissociation with a cold active protease (6°C). We selected the top 40 differentially expressed genes to create the geneset used in the AddModuleScore function. The individual scores were grouped by cluster in each datasets and displayed as violin plots. On dataset 1, a Kruskall-Wallis test followed by post-hoc pairwise wilcoxon tests were applied through clusters to determine if the excluded cluster contains a stress index distribution significantly higher (at 5% error) than other clusters.
Clonotype analysis
Clonotypes were defined using the composition of (i) associated V gene full name (ii) associated J gene full name (e.g. for heavy chain IGHV9-3∗01/IGHJ4∗01/42 or for light chain IGKV5-48∗01/IGKJ2∗01/33) of both heavy and light chain (e.g. Clonotype 1 = IGHV9-3∗01/IGHJ4∗01/42≪≫IGKV5-48∗01/IGKJ2∗01/33). We used Clustal Omega from msa R package (https://doi.org/10.1093/bioinformatics/btv494) to evaluate the sequence proximity of clonotypes and verify if full BCR sequences in clonotype were consistent. We observed that distances between sequences within clonotypes were much smaller than distances between clonotypes.
Only cell barcodes found in the transcriptomic data and associated with both light chains and heavy chain information were kept for further analysis. Analysis on shared V genes took into account only the main version of the genes (eg. IGHV9-3∗01 was identified to IGHV9-3). Clustal Omega was used to assess the clonotype sequence similarities (Madeira et al., 2019).
The clonotype overlap heatmap was produced considering the set of clonotypes in each cluster and computing the Szymkiewicz–Simpson coefficient between sets. Hierarchical Clustering was performed using the stats hclust function (version 3.6.3) with euclidean distance and average linkage. Plot was performed with the ggplot2 ggplot function (version 3.3).
Phenotype analysis
Index-sorting flow cytometry data were converted through hyperbolic arcsine transformation (asinh) on fluorescence parameters. A scale factor was applied to each transformation. This scale factor was 20, 10, 100 and 100 for CXCR3, CCR6, HA and NP parameters, respectively. CXCR3+, CCR6+, HA+ and NP+ subsets were defined according to the following respective threshold values on asinh transformed data: 0.5, 1.5, 0.8, 0.5.
Quantification and statistical analysis
Statistical parameters including the exact value of n with the description of what n represents, the mean, the SEM and the p value are reported in the Figures and the Figure Legends. Statistical analyses were performed using Prism (GraphPad Software): two-tailed t test, paired t test, one-way Anova, two-way Anova. Anova tests were followed by Tukey post-hoc tests. p values < 0.05 were considered significant. In figures, asterisks stand for: ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001; ∗∗∗∗, p < 0.0001.
Acknowledgments
We thank the flow cytometry, imaging, bioinformatics, and genomics platforms for their technical support and the biological resource unit for the breeding of animals (CIML). We thank C.A. Reynaud and J.C. Weill for the Aicda-CreERT2 mice. We thank S. Mancini for the μMT mice. We thank M. Wilson for the IL-4-GFP mice. We thank J. Marvel for the CXCR3−/− bone marrows. We thank T. Mak, D. Wakeham, and J. Haight for the Fcmr−/− bone marrows. We thank R. Le Goffic for the influenza X31. We thank D. Kitamura, M. Cogné, and E. Pinaud for the 40LB cells. We thank the NIH Tetramer Core Facility for the CD1d tetramers. We thank L. Alexopoulou for the autoantigens. We thank N. Mossadegh-Keller for the assistance with the in vivo experiments. We thank A. Sansoni, P. Hoest, L. Gelard, Q. Bardin, and M. Malissen for performing the studies with SARS-CoV-2 in the CIPHE biosafety level 3. We thank C. Lutz and The Jackson Laboratory for the K18-hACE2 mice. We thank S. van der Werf for the BetaCoV/France/IDF0372/2020 strain and B. La Scola for the advice on SARS-CoV-2 handling. We thank F. Batista and P. Golstein for the critical reading of the manuscript. We thank Centre de Calcul Intensif d’Aix-Marseille for granting access to its computing resources. This work was supported by the ATIP-AVENIR young group leader program (INSERM) and the Marie Curie reintegration fellowship (EU) to M.G.; the Fondation pour la Recherche Médicale (FRM) fellowship to S.V.-M.; ANR-17-CE15-0009-01 (MoDEx-GC) to P.M.; the COVIDHUMICE project (FRM-ANR Flash Covid-COVI-0066) to B.M.; ANR-10-INBS-04-01 France Bio Imaging; and the Centre d’Immunologie de Marseille Luminy, which receives its core funding from Aix Marseille University, CNRS, and INSERM.
Author contributions
C.G. designed and performed the experiments and analyzed the data. L.S. analyzed the scRNA-seq data. S.V.-M. performed the flow cytometry analysis. L.G. prepared the scRNA-seq libraries. M.P.H. performed the enzyme-linked immunosorbent assay (ELISA) experiments. M.M. performed the confocal imaging. C.D. performed the scRNA-seq data processing. J.-M.N. prepared the hashtag antibodies. M.F. analyzed the confocal images. A.Z. and B.M. provided the K18-hACE2 mice and performed the SARS-CoV-2 infections. P.M. conceived the project and supervised the scRNA-seq experiments and data analysis. M.G. conceived the project, supervised the work, and wrote the manuscript. All the authors were involved in the scientific discussions.
Declaration of interests
B.M. is part of the Immunity advisory board.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community.
Published: June 28, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.immuni.2022.06.002.
Supplemental information
Data and code availability
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession number is listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request. All original code has been deposited on GitHub and Zenodo, and they are publicly available as of the date of publication. DOIs are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Adachi Y., Onodera T., Yamada Y., Daio R., Tsuiji M., Inoue T., Kobayashi K., Kurosaki T., Ato M., Takahashi Y. Distinct germinal center selection at local sites shapes memory B cell response to viral escape. J. Exp. Med. 2015;212:1709–1723. doi: 10.1084/jem.20142284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguado E., Richelme S., Nuñez-Cruz S., Miazek A., Mura A.-M., Richelme M., Guo X.-J., Sainty D., He H.-T., Malissen B., et al. Induction of T helper type 2 immunity by a point mutation in the LAT adaptor. Science. 2002;296:2036–2040. doi: 10.1126/science.1069057. [DOI] [PubMed] [Google Scholar]
- Akkaya M., Kwak K., Pierce S.K. B cell memory: building two walls of protection against pathogens. Nat. Rev. Immunol. 2020;20:229–238. doi: 10.1038/s41577-019-0244-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allie S.R., Bradley J.E., Mudunuru U., Schultz M.D., Graf B.A., Lund F.E., Randall T.D. The establishment of resident memory B cells in the lung requires local antigen encounter. Nat. Immunol. 2019;20:97–108. doi: 10.1038/s41590-018-0260-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson S.M., Tomayko M.M., Ahuja A., Haberman A.M., Shlomchik M.J. New markers for murine memory B cells that define mutated and unmutated subsets. J. Exp. Med. 2007;204:2103–2114. doi: 10.1084/jem.20062571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attaf N., Cervera-Marzal I., Dong C., Gil L., Renand A., Spinelli L., Milpied P. FB5P-seq: FACS-based 5-prime end single-cell RNA-seq for integrative analysis of transcriptome and antigen receptor repertoire in B and T cells. Front. Immunol. 2020;11:216. doi: 10.3389/fimmu.2020.00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton A.E., Innocentin S., Carr E.J., Bradford B.M., Lafouresse F., Mabbott N.A., Mörbe U., Ludewig B., Groom J.R., Good-Jacobson K.L., et al. Type I interferon induces CXCL13 to support ectopic germinal center formation. J. Exp. Med. 2019;216:621–637. doi: 10.1084/jem.20181216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitt W.S., 3rd, Mesin L., Victora G.D., Minin V.N., Matsen F.A. 4th. Using genotype abundance to improve phylogenetic inference. Mol. Biol. Evol. 2018;35:1253–1265. doi: 10.1093/molbev/msy020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan I., Bertocci B., Vilmont V., Delbos F., Mégret J., Storck S., Reynaud C.-A., Weill J.-C. Multiple layers of B cell memory with different effector functions. Nat. Immunol. 2009;10:1292–1299. doi: 10.1038/ni.1814. [DOI] [PubMed] [Google Scholar]
- Duan L., Liu D., Chen H., Mintz M.A., Chou M.Y., Kotov D.I., Xu Y., An J., Laidlaw B.J., Cyster J.G. Follicular dendritic cells restrict interleukin-4 availability in germinal centers and foster memory B cell generation. Immunity. 2021;54:2256–2272.e6. doi: 10.1016/j.immuni.2021.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak G., Perez J.-M., Weng A., Gottardo R. Optimizing transformations for automated, high throughput analysis of flow cytometry data. BMC Bioinformatics. 2010;11:546. doi: 10.1186/1471-2105-11-546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genton C., Wang Y., Izui S., Malissen B., Delsol G., Fournié G.J., Malissen M., Acha-Orbea H. The Th2 lymphoproliferation developing in LatY136F mutant mice triggers polyclonal B cell activation and systemic autoimmunity. J. Immunol. 2006;177:2285–2293. doi: 10.4049/jimmunol.177.4.2285. [DOI] [PubMed] [Google Scholar]
- Horns F., Dekker C.L., Quake S.R. Memory B cell activation, broad anti-influenza antibodies, and bystander activation revealed by single-cell transcriptomics. Cell Rep. 2020;30:905–913.e6. doi: 10.1016/j.celrep.2019.12.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse M., Lillemeier B.F., Kuhns M.S., Chen D.S., Davis M.M. T cells use two directionally distinct pathways for cytokine secretion. Nat. Immunol. 2006;7:247–255. doi: 10.1038/ni1304. [DOI] [PubMed] [Google Scholar]
- Kikutani H., Inui S., Sato R., Barsumian E.L., Owaki H., Yamasaki K., Kaisho T., Uchibayashi N., Hardy R.R., Hirano T. Molecular structure of human lymphocyte receptor for immunoglobulin E. Cell. 1986;47:657–665. doi: 10.1016/0092-8674(86)90508-8. [DOI] [PubMed] [Google Scholar]
- Koike T., Harada K., Horiuchi S., Kitamura D. The quantity of CD40 signaling determines the differentiation of B cells into functionally distinct memory cell subsets. eLife. 2019;8 doi: 10.7554/eLife.44245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kometani K., Nakagawa R., Shinnakasu R., Kaji T., Rybouchkin A., Moriyama S., Furukawa K., Koseki H., Takemori T., Kurosaki T. Repression of the transcription factor Bach2 contributes to predisposition of IgG1 memory B cells toward plasma cell differentiation. Immunity. 2013;39:136–147. doi: 10.1016/j.immuni.2013.06.011. [DOI] [PubMed] [Google Scholar]
- Kuraoka M., Schmidt A.G., Nojima T., Feng F., Watanabe A., Kitamura D., Harrison S.C., Kepler T.B., Kelsoe G. Complex antigens drive permissive clonal selection in germinal centers. Immunity. 2016;44:542–552. doi: 10.1016/j.immuni.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaki T., Kometani K., Ise W. Memory B cells. Nat. Rev. Immunol. 2015;15:149–159. doi: 10.1038/nri3802. [DOI] [PubMed] [Google Scholar]
- Lang K.S., Lang P.A., Meryk A., Pandyra A.A., Boucher L.-M., Pozdeev V.I., Tusche M.W., Göthert J.R., Haight J., Wakeham A., et al. Involvement of Toso in activation of monocytes, macrophages, and granulocytes. Proc. Natl. Acad. Sci. USA. 2013;110:2593–2598. doi: 10.1073/pnas.1222264110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Gallou S., Zhou Z., Thai L.-H., Fritzen R., de Los Aires A.V., Mégret J., Yu P., Kitamura D., Bille E., Tros F., et al. A splenic IgM memory subset with antibacterial specificities is sustained from persistent mucosal responses. J. Exp. Med. 2018;215:2035–2053. doi: 10.1084/jem.20180977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz J., Dittmann K., Bösl M.R., Winkler T.H., Wienands J., Engels N. Reactivation of IgG-switched memory B cells by BCR-intrinsic signal amplification promotes IgG antibody production. Nat. Commun. 2015;6:8575. doi: 10.1038/ncomms9575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean A.J., Richmond N., Koneva L., Attar M., Medina C.A.P., Thornton E.E., Gomes A.C., El-Turabi A., Bachmann M.F., Rijal P., et al. Secondary influenza challenge triggers resident memory B cell migration and rapid relocation to boost antibody secretion at infected sites. Immunity. 2022;55:718–733.e8. doi: 10.1016/j.immuni.2022.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeira F., Park Y.M., Lee J., Buso N., Gur T., Madhusoodanan N., Basutkar P., Tivey A.R.N., Potter S.C., Finn R.D., et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019;47:W636–W641. doi: 10.1093/nar/gkz268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCray P.B., Jr., Pewe L., Wohlford-Lenane C., Hickey M., Manzel L., Shi L., Netland J., Jia H.P., Halabi C., Sigmund C.D., et al. Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus. J. Virol. 2007;81:813–821. doi: 10.1128/JVI.02012-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHeyzer-Williams L.J., Milpied P.J., Okitsu S.L., McHeyzer-Williams M.G. Class-switched memory B cells remodel BCRs within secondary germinal centers. Nat. Immunol. 2015;16:296–305. doi: 10.1038/ni.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesin L., Schiepers A., Ersching J., Barbulescu A., Cavazzoni C.B., Angelini A., Okada T., Kurosaki T., Victora G.D. Restricted clonality and limited germinal center reentry characterize memory B cell reactivation by boosting. Cell. 2020;180:92–106.e11. doi: 10.1016/j.cell.2019.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou E.P., Cheng A., Montalbano A., Hao S., Stoeckius M., Legut M., Roush T., Herrera A., Papalexi E., Ouyang Z., et al. Multiplexed detection of proteins, transcriptomes, clonotypes and CRISPR perturbations in single cells. Nat. Methods. 2019;16:409–412. doi: 10.1038/s41592-019-0392-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa R., Toboso-Navasa A., Schips M., Young G., Bhaw-Rosun L., Llorian-Sopena M., Chakravarty P., Sesay A.K., Kassiotis G., Meyer-Hermann M., et al. Permissive selection followed by affinity-based proliferation of GC light zone B cells dictates cell fate and ensures clonal breadth. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2016425118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nojima T., Haniuda K., Moutai T., Matsudaira M., Mizokawa S., Shiratori I., Azuma T., Kitamura D. In-vitro derived germinal centre B cells differentially generate memory B or plasma cells in vivo. Nat. Commun. 2011;2:465. doi: 10.1038/ncomms1475. [DOI] [PubMed] [Google Scholar]
- O'Flanagan, C.H., Campbell, K.R., Zhang, A.W., Kabeer, F., Lim, J.L.P., Biele, J., Eirew, P., Lai, D., McPherson, A., Kong, E., Bates, C., Borkowski, K., Wiens, M., Hewitson, B., Hopkins, J., Pham, J., Ceglia, N., Moore, R., Mungall, A.J., McAlpine, J.N.; CRUK IMAXT Grand Challenge Team, Shah, S.P., Aparicio, S. (2019). Dissociation of solid tumor tissues with cold active protease for single-cell RNA-seq minimizes conserved collagenase-associated stress responses. Genome Biol. 20, 210. 10.1186/s13059-019-1830-0. [DOI] [PMC free article] [PubMed]
- Oh J.E., Song E., Moriyama M., Wong P., Zhang S., Jiang R., Strohmeier S., Kleinstein S.H., Krammer F., Iwasaki A. Intranasal priming induces local lung-resident B cell populations that secrete protective mucosal antiviral IgA. Sci. Immunol. 2021;6 doi: 10.1126/sciimmunol.abj5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onodera T., Takahashi Y., Yokoi Y., Ato M., Kodama Y., Hachimura S., Kurosaki T., Kobayashi K. Memory B cells in the lung participate in protective humoral immune responses to pulmonary influenza virus reinfection. Proc. Natl. Acad. Sci. USA. 2012;109:2485–2490. doi: 10.1073/pnas.1115369109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape K.A., Taylor J.J., Maul R.W., Gearhart P.J., Jenkins M.K. Different B cell populations mediate early and late memory during an endogenous immune response. Science. 2011;331:1203–1207. doi: 10.1126/science.1201730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perona-Wright G., Mohrs K., Mohrs M. Sustained signaling by canonical helper T cell cytokines throughout the reactive lymph node. Nat. Immunol. 2010;11:520–526. doi: 10.1038/ni.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reboldi A., Arnon T.I., Rodda L.B., Atakilit A., Sheppard D., Cyster J.G. IgA production requires B cell interaction with subepithelial dendritic cells in Peyer’s patches. Science. 2016;352:aaf4822. doi: 10.1126/science.aaf4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel R., Addo R., Ferreira-Gomes M., Heinz G.A., Heinrich F., Kummer J., Greiff V., Schulz D., Klaeden C., Cornelis R., et al. Discrete populations of isotype-switched memory B lymphocytes are maintained in murine spleen and bone marrow. Nat. Commun. 2020;11:2570. doi: 10.1038/s41467-020-16464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckius M., Zheng S., Houck-Loomis B., Hao S., Yeung B.Z., Mauck W.M., 3rd, Smibert P., Satija R. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 2018;19:224. doi: 10.1186/s13059-018-1603-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo P.A., Miron M., Farber D.L. Location, location, location: tissue resident memory T cells in mice and humans. Sci. Immunol. 2019;4 doi: 10.1126/sciimmunol.aas9673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan H.-X., Juno J.A., Esterbauer R., Kelly H.G., Wragg K.M., Konstandopoulos P., Alcantara S., Alvarado C., Jones R., Starkey G., et al. Lung-resident memory B cells established after pulmonary influenza infection display distinct transcriptional and phenotypic profiles. Sci. Immunol. 2022;7:eabf5314. doi: 10.1126/sciimmunol.abf5314. [DOI] [PubMed] [Google Scholar]
- Viant C., Weymar G.H.J., Escolano A., Chen S., Hartweger H., Cipolla M., Gazumyan A., Nussenzweig M.C. Antibody affinity shapes the choice between memory and germinal center B cell fates. Cell. 2020;183:1298–1311.e11. doi: 10.1016/j.cell.2020.09.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viant C., Wirthmiller T., ElTanbouly M.A., Chen S.T., Cipolla M., Ramos V., Oliveira T.Y., Stamatatos L., Nussenzweig M.C. Germinal center-dependent and -independent memory B cells produced throughout the immune response. J. Exp. Med. 2021;218 doi: 10.1084/jem.20202489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Z., Lin Y., Zhao Y., Qi H. T cells in bystander and cognate interactions with B cells. Immunol. Rev. 2019;288:28–36. doi: 10.1111/imr.12747. [DOI] [PubMed] [Google Scholar]
- Weisel F., Shlomchik M. Memory B cells of mice and humans. Annu. Rev. Immunol. 2017;35:255–284. doi: 10.1146/annurev-immunol-041015-055531. [DOI] [PubMed] [Google Scholar]
- Weisel F.J., Zuccarino-Catania G.V., Chikina M., Shlomchik M.J. A temporal switch in the germinal center determines differential output of memory B and plasma cells. Immunity. 2016;44:116–130. doi: 10.1016/j.immuni.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong R., Belk J.A., Govero J., Uhrlaub J.L., Reinartz D., Zhao H., Errico J.M., D’Souza L., Ripperger T.J., Nikolich-Zugich J., et al. Affinity-restricted memory B cells dominate recall responses to heterologous flaviviruses. Immunity. 2020;53:1078–1094.e7. doi: 10.1016/j.immuni.2020.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yewdell W.T., Smolkin R.M., Belcheva K.T., Mendoza A., Michaels A.J., Cols M., Angeletti D., Yewdell J.W., Chaudhuri J. Temporal dynamics of persistent germinal centers and memory B cell differentiation following respiratory virus infection. Cell Rep. 2021;37 doi: 10.1016/j.celrep.2021.109961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaretsky I., Atrakchi O., Mazor R.D., Stoler-Barak L., Biram A., Feigelson S.W., Gitlin A.D., Engelhardt B., Shulman Z. ICAMs support B cell interactions with T follicular helper cells and promote clonal selection. J. Exp. Med. 2017;214:3435–3448. doi: 10.1084/jem.20171129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccarino-Catania G.V., Sadanand S., Weisel F.J., Tomayko M.M., Meng H., Kleinstein S.H., Good-Jacobson K.L., Shlomchik M.J. CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat. Immunol. 2014;15:631–637. doi: 10.1038/ni.2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession number is listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request. All original code has been deposited on GitHub and Zenodo, and they are publicly available as of the date of publication. DOIs are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.