

This cohort study investigates the timing at which β-amyloid–positron emission tomography (PET) binding starts showing associations with other markers of Alzheimer disease using the Alzheimer’s Disease Neuroimaging Initiative, the Harvard Aging Brain Study, and the Presymptomatic Evaluation of Experimental or Novel Treatments for Alzheimer Disease Research Group.
Key Points
Question
In 3 cohorts of cognitively unimpaired persons, does spatial distribution (regional vs widespread) of β-amyloid (Aβ) deposition modify associations with Alzheimer disease–related clinical and biological markers?
Findings
In this cohort study of 817 participants that contrasted Aβ-negative participants vs regional participants, those with regional Aβ binding showed proportionately more apolipoprotein E ε4 carriership, reduced cerebrospinal fluid Aβ1-42 levels, and greater longitudinal Aβ-PET binding accumulation. Participants with widespread amyloid binding further exhibited notable cognitive decline and greater cerebrospinal fluid phosphorylated tau181 and tau–positron emission tomography binding than others; using visual reads or each cohort’s specified dichotomous threshold for positivity almost all participants deemed Aβ-positive had widespread Aβ deposition.
Meaning
Regional Aβ binding appears to be biologically relevant among individuals without significant tau and related cognitive decline.
Abstract
Importance
Preventive trials of anti-amyloid agents might preferably recruit persons showing earliest biologically relevant β-amyloid (Aβ) binding on positron emission tomography (PET).
Objective
To investigate the timing at which Aβ-PET binding starts showing associations with other markers of Alzheimer disease.
Design, Setting, and Participants
This longitudinal multicentric cohort study included 3 independent cohorts: Presymptomatic Evaluation of Experimental or Novel Treatments for Alzheimer Disease (PREVENT-AD) (data collected from 2012-2020), Alzheimer Disease Neuroimaging Initiative (ADNI) (data collected from 2005-2019), and Harvard Aging Brain Study (HABS) (data collected from 2011-2019). In a 3-tiered categorization of Aβ-PET binding spatial extent, individuals were assigned as having widespread Aβ deposition if they showed positive signal throughout a designated set of brain regions prone to early Aβ accumulation. Those with binding in some but not all were categorized as having regional deposition, while those who failed to show any criterion Aβ signal were considered Aβ-negative. All participants who were cognitively unimpaired at their first Aβ PET scan.
Main Outcomes and Measures
Differences in cerebrospinal fluid (CSF), genetics, tau-PET burden, and cognitive decline.
Results
A total of 817 participants were included, including 129 from the PREVENT-AD cohort (mean [SD] age, 63.5 [4.7] years; 33 [26%] male; 126 [98%] White), 400 from ADNI (mean [SD] age, 73.6 [5.8] years; 190 [47%] male; 10 [5%] Hispanic, 338 [91%] White), and 288 from HABS (mean [SD] age, 73.7 [6.2] years; 117 [40%] male; 234 [81%] White). Compared with Aβ-negative persons, those with regional Aβ binding showed proportionately more APOE ε4 carriers (18 [64%] vs 22 [27%] in PREVENT-AD and 34 [31%] vs 38 [19%] in ADNI), reduced CSF Aβ1-42 levels (F = 24 and 71), and greater longitudinal Aβ-PET accumulation (significant β = 0.019 to 0.056). Participants with widespread amyloid binding further exhibited notable cognitive decline (significant β = −0.014 to −0.08), greater CSF phosphorylated tau181 (F = 5 and 27), and tau-PET binding (all F > 7.55). Using each cohort’s specified dichotomous threshold for Aβ positivity or a visual read classification, most participants (56% to 100%, depending on classification method and cohort) with regional Aβ would have been classified Aβ-negative.
Conclusions and Relevance
Regional Aβ binding appears to be biologically relevant and participants at this stage remain relatively free from CSF phosphorylated tau181, tau-PET binding, and related cognitive decline, making them ideal targets for anti-amyloid agents. Most of these individuals would be classified as negative based on classical thresholds of Aβ positivity.
Introduction
β-Amyloid (Aβ) and tau deposits are the pathological hallmarks of Alzheimer disease (AD). Deposition of these proteins is a continuous process that starts decades before the onset of AD symptoms.1,2 While tau deposition may occasionally precede Aβ accumulation,3 it is widely thought that Aβ pathology is required for tau to spread beyond the medial temporal lobe and begin the pathological cascade that leads to AD dementia. Therefore, early Aβ abnormality is often viewed as an ideal target for clinical trials.4,5,6,7 Several such trials have reduced brain Aβ without slowing AD clinical symptoms.8,9,10 These results have led to circumspection about the role of Aβ in the AD pathological cascade,11 but it also may be that Aβ should be targeted early on, before the spread of tau pathology.12 We therefore focused on cognitively unimpaired older adults and investigated the earliest timing when biologically relevant signal of Aβ–positron emission tomography (PET) pathology can be detected. To do so, we took advantage of the spatial distribution of Aβ deposition as a method for identification of individuals having different levels of Aβ burden (no burden, regional deposition, and widespread deposition). In 3 independent cohorts, we then investigated the association between spatial extent of Aβ burden and various AD markers including tau-PET and cognitive decline.
Methods
Participants and Study Design
Participants provided written informed consent, and research procedures were approved by the relevant ethics committees. Specific inclusion and exclusion criteria per cohort can be found in the eMethods in Supplement 1. Race and ethnicity were collected in all 3 cohorts by self-report.
Presymptomatic Evaluation of Experimental or Novel Treatments for Alzheimer Disease
The Presymptomatic Evaluation of Experimental or Novel Treatments for Alzheimer Disease (PREVENT-AD)13 is an ongoing longitudinal observational cohort study launched in 2011 including 385 individuals.14 Here we studied a subsample of 129 participants who underwent PET. Data were collected from 2012 to 2020.
Alzheimer Disease Neuroimaging Initiative
The Alzheimer Disease Neuroimaging Initiative (ADNI)15 was launched in 2003 as a public-private partnership. The primary goal of ADNI has been to test whether serial magnetic resonance imaging, PET, other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment and early AD. We studied data from 400 individuals from the ADNI-2 extension who underwent Aβ-PET with the 18F-AV-45 tracer. Data were collected from 2005 to 2019.
Harvard Aging Brain Study
The Harvard Aging Brain Study (HABS)16 was launched in 2010 and is funded by the National Institute on Aging.17 We included data from 288 persons from data release 2. Data were collected from 2011 to 2019.
Neuropsychological Evaluation
In all 3 cohorts, participants underwent cognitive testing annually. We analyzed both baseline (time of the first cognitive assessment visit) and longitudinal cognitive performance. In PREVENT-AD, the main neuropsychological measure was the Repeatable Battery for the Assessment of Neuropsychological Status.18 We used the total score and the 2 composite memory scores (immediate memory and delayed memory) as the composite scores of interest. Longitudinal cognitive assessment data were available for all participants, with a median (IQR) follow-up time of 7 (2-8) years.
In ADNI, we used the 2 composite scores reflective of memory and executive functions as previously described.19,20 Longitudinal cognitive assessment was available for 393 individuals (98%), with a median (IQR) follow-up time of 6 (1-14) years.
In HABS, we used the Preclinical Alzheimer’s Cognitive Composite, a composite score including memory, executive function, and semantic processing.21 All participants had a longitudinal cognitive assessment, with a median (IQR) follow-up time of 6 (1-9) years.
Cerebrospinal Fluid
Cerebrospinal fluid (CSF) Aβ1-42, phosphorylated tau181 (pTau181) were measured using enzyme-linked immunoassay (INNOTEST; Fujirebio) and available for 77 participants in PREVENT-AD and measured using immunoassays (Elecsys; Roche) and available for 276 participants in ADNI (eMethods in Supplement 1).
PET Tracers and Processing
In all cohorts, T1-weighted magnetic resonance imaging was processed using FreeSurfer (version 5.3 or 6) and parcellated according to the Desikan-Killiany atlas.22 The Aβ tracers differed between the cohorts: 18F-NAV-4694 was used in PREVENT-AD, florbetapir was used in ADNI, and 11C-Pittsburgh compound B was used in HABS. The tau PET tracer was flortaucipir in all cohorts. We used standardized uptake value ratios (SUVRs) in all cohorts; results were similar using distribution volume ratio in HABS.
Regional Thresholds of Aβ Positivity
For all cohorts, Aβ-PET values were extracted across 7 bilateral regions considered to be especially sensitive to early Aβ accumulation: medial orbitofrontal, rostral anterior cingulate, posterior cingulate, precuneus, rostral middle frontal, superior frontal, and inferior parietal cortices.23 Tracer uptake in the first 5 of these regions of interest (ROI) has been observed to be elevated in Aβ-negative individuals who subsequently had significant evidence of amyloid deposition.24 We used a Gaussian mixture modeling approach to quantify specific Aβ thresholds in the 7 specified bilateral ROIs. Because Aβ typically follows a bimodal distribution, we fitted 2 Gaussian distributions to categorize Aβ positivity.23,25,26 These 2 distributions acquired from Gaussian mixture modeling assigned to each participant a probability of belonging to either the lower or higher regional distributions and allowed identification of a threshold of positivity for each ROI (eTable 1 in Supplement 1). Individuals who were Aβ-positive in all 7 ROIs were classified as having widespread Aβ deposition; those who were positive in 1 to 6 ROIs were included in a regional Aβ deposition group; those who were negative in all the ROIs were termed Aβ-negative.
As expected, because of tracer differences, the SUVR regional distributions from various cohorts differed (eFigure 1 in Supplement 1). In PREVENT-AD and HABS (18F-NAV-4694 and 11C-Pittsburgh compound B tracer, respectively), Gaussian mixture modeling analyses provided a clear distinction between distributions using a threshold at the 90th percentile of the lower distribution. In ADNI (florbetapir tracer), regional positive and negative distinctions were less obvious; participants appeared to show a more continuous distribution of regional Aβ deposition, creating greater ambiguity in classification. Following approaches used by others,27,28,29 we therefore assigned the ROI cutoffs at 50% probability. In sensitivity analyses, we tested the effect of modifying the number of ROI for classification of spatial extent from 7 to 5 or 10 (eResults in Supplement 1).30
Comparison With Traditional Classification of Aβ Positivity
We also compared our 3-tiered spatial extent classification method to more conventional approaches: (1) binary classification based on cohort-specific global SUVR uptake,31,32,33 (2) binary classification based on visual read, and (3) a 3-tiered global quantification approach based on Centiloids (≤20, >20 to ≤40, and >40; eMethods in Supplement 1).
Tau-PET
For tau-PET, SUVR was calculated for 6 bilateral regions that characterize early tau-PET deposition: entorhinal cortex, amygdala, fusiform, parahippocampal, inferior temporal, and middle temporal cortex.34 Voxelwise analyses were also performed as secondary analyses (eResults in Supplement 1).
Statistical Analyses
In this cohort study, we analyze cross-sectional and longitudinal data from observations collected in 3 aging cohorts. We compared demographics, apolipoprotein E (APOE) ε4 status, CSF biomarkers, cross-sectional cognition, and tau-PET SUVR across the 3 Aβ groups in each cohort separately using analysis of covariance and χ2 tests for normally distributed continuous variables and categorical variables, respectively. We used the Tukey Honestly Significant Difference post hoc test and Bonferroni correction to help interpret differences between the 3 Aβ groups. Linear mixed-effects models investigated longitudinal Aβ accumulation (ADNI and HABS) and cognitive decline over 2 or more sequential measurements (all cohorts) across the 3 Aβ classes. For Aβ accumulation, age and sex were included as covariates in the models. For cognitive decline, education was further included as a covariate. The criterion for statistical significance was 2-sided P ≤ .05 after correction for multiple comparisons. Analysis took place between September 2019 and July 2021.
Results
Definition of Amyloid Groups Based on Aβ Spatial Extent
A total of 817 participants were included, including 129 from the PREVENT-AD cohort (mean [SD] age, 63.5 [4.7] years; 33 [26%] male; 126 [98%] White), 400 from ADNI (mean [SD] age, 73.6 [5.8] years; 190 [47%] male; 10 [5%] Hispanic, 338 [91%] White), and 288 from HABS (mean [SD] age, 73.7 [6.2] years; 117 [40%] male; 234 [81%] White). The distribution of participants in the 3 Aβ classes in the individual cohorts is shown in Figure 1A. Aβ-negative proportions ranged across cohorts from 48% (139 of 288) to 62% (81 of 129); the regional group from 22% (28 of 129) to 27% (108 of 400); and the widespread group from 16% (20 of 129) to 25% (73 of 288). Most regional participants would have been classified as negative using conventional quantitative or visual binary classifications (Figure 1B). Examples of Aβ uptake by cohort in the negative, regional, and widespread groups are shown in Figure 2. See eFigure 2 in Supplement 1 for the distribution of abnormal regions in the regional groups and eFigure 3A in Supplement 1 for voxelwise differences between groups.
Figure 1. Defining the β-Amyloid (Aβ) Groups.
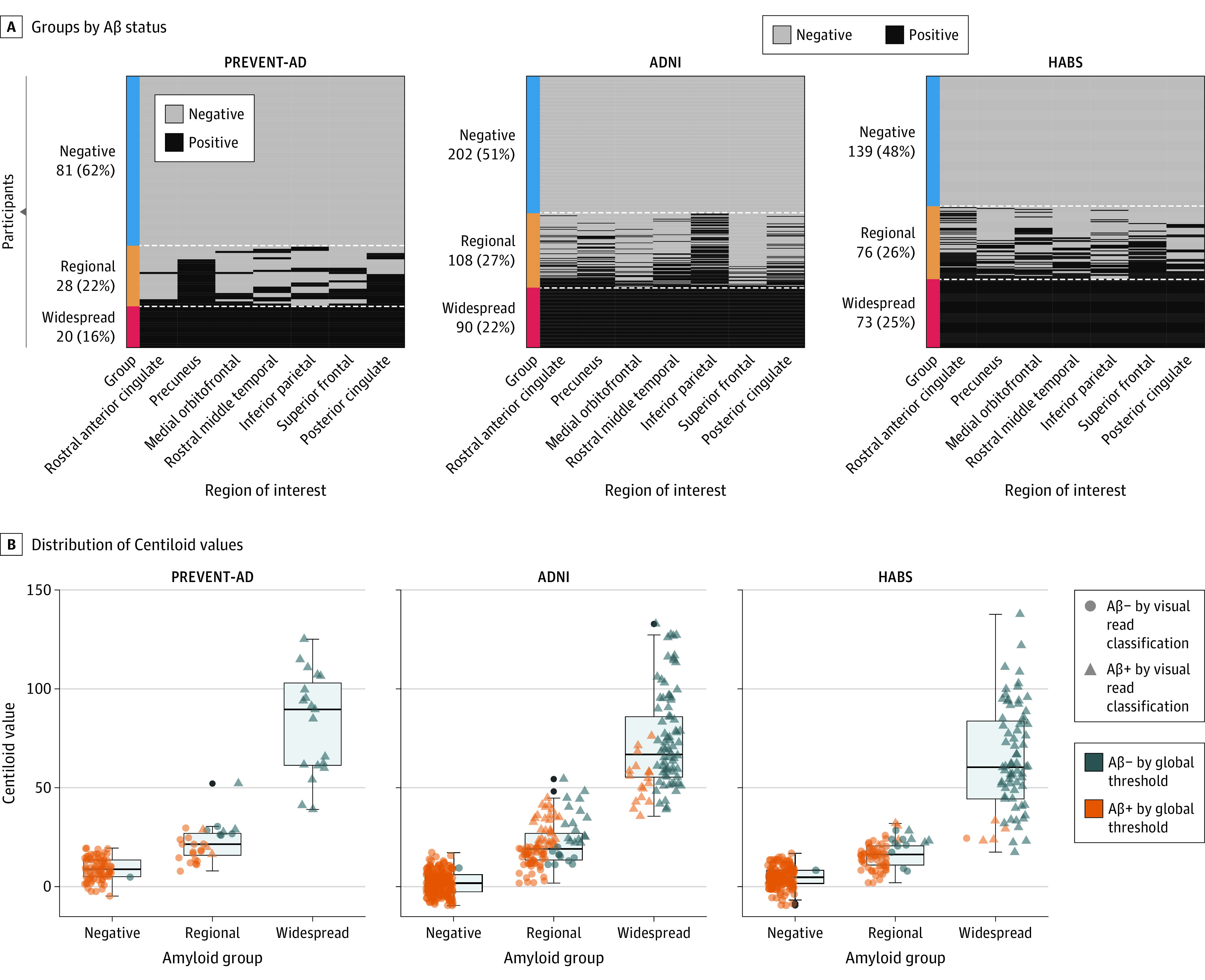
A, Individuals were separated into 3 groups based on their Aβ status in 7 cortical regions: rostral anterior cingulate, precuneus, medial orbitofrontal, rostral middle frontal, inferior parietal, superior frontal, and posterior cingulate. According to the region-specific positivity, individuals who were Aβ-positive in all 7 regions were classified as the widespread Aβ deposition group; those who were positive in 1 to 6 regions were included in the regional Aβ group; while those who fell below the cutoff in all the regions were considered as Aβ-negative. B, The boxplots represent the distribution of the Centiloid values of the 3 Aβ groups across the Aβ-negative, regional, and widespread groups in all cohorts. Different shapes for the data points indicate the visual read classification, and the color categorizes participants as Aβ+ or Aβ− based on quantitative binary amyloid index using previously established global thresholds for each cohort. ADNI indicates Alzheimer Disease Neuroimaging Initiative; HABS, Harvard Aging Brain Study; PREVENT-AD, Presymptomatic Evaluation of Experimental or Novel Treatments for Alzheimer Disease.
Figure 2. Examples of β-Amyloid (Aβ) Uptake From Participants in the Negative, Regional, and Widespread Groups.
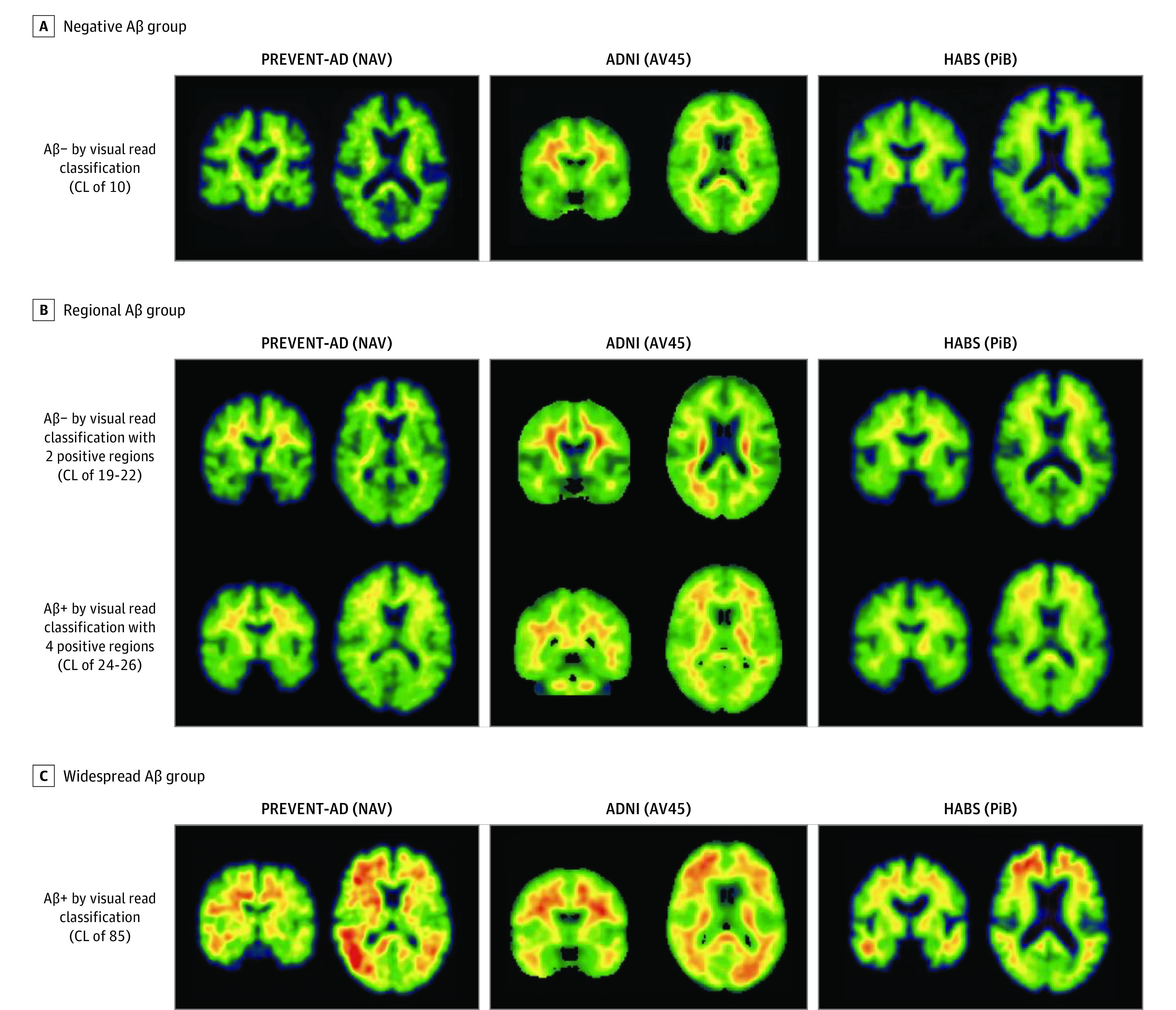
Illustrative examples of Aβ standardized uptake value ratios positron emission tomography images from participants in the negative (A), regional (B), and widespread (C) Aβ groups in the 3 cohorts. All shown images in the negative group were negative based on visual read and had a Centiloid (CL) value of 10. We show examples of participants in the regional group who were positive on 2 regions and negative based on visual read and participants who were positive on 4 regions and positive based on visual read. In the Presymptomatic Evaluation of Experimental or Novel Treatments for Alzheimer Disease (PREVENT-AD) study cohort, the 2 and 4 regions positive were precuneus and posterior cingulate (2), plus rostral anterior cingulate and medial orbitofrontal (4). In the Alzheimer Disease Neuroimaging Initiative (ADNI) study cohort, the regions were rostral middle frontal and inferior parietal (2) and the 4 were the inferior parietal, precuneus, posterior cingulate, and medial orbitofrontal. In the Harvard Aging Brain Study (HABS) cohort, the regions were rostral anterior cingulate and medial orbitofrontal (2), plus rostral middle frontal and superior frontal (4). All images shown in the Widespread group were positive based on visual read and had a CL value of 85. The standardized uptake value ratios scales were restricted to 0 to 4 in PREVENT-AD, 0 to 3 in ADNI, and 0 to 3.5 in HABS. AV45 indicates 18F-AV-45; NAV, 18F-NAV-4694; PiB, 11C-Pittsburgh compound B.
Biological and Clinical Markers of Interest
The widespread and regional Aβ groups had greater proportions of APOE ε4 carriers than the Aβ-negative group in PREVENT-AD (Table; eTable 2 in Supplement 1). In ADNI, the widespread group had a larger proportion of APOE ε4 carriers than the regional group. In HABS, only the widespread group had larger proportions of APOE ε4 carriers than both groups.
Table. Biological and Clinical Characteristics of Aβ Groups.
| Characteristic | Mean (SD) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| PREVENT-AD | ADNI | HABS | |||||||
| Negative (n = 81) | Regional (n = 28) | Widespread (n = 20) | Negative (n = 202) | Regional (n = 108) | Widespread (n = 90) | Negative (n = 139) | Regional (n = 76) | Widespread (n = 73) | |
| Age, y | 63 (4.61)a | 63 (3.83) | 66 (5.62)a | 73 (5.81)a | 73 (5.93)b | 76 (5.35)a,b | 73 (6.29)a | 74 (6.08) | 75 (5.93)a |
| Education, y | 16 (3.53) | 15 (2.75) | 14 (2.46) | 17 (2.59) | 17 (2.56) | 16 (2.70) | 16 (3.09) | 15 (3.20)b | 16 (2.81)b |
| Race and ethnicity | |||||||||
| Black/African American | 0 | 0 | <5 | 9 (5) | 8 (8) | <5 | 23 (17) | 14 (18) | 8 (11) |
| Hispanic | <5 | 0 | <5 | <5 | 6 (11) | 0 | <5 | <5 | 0 |
| White | 79 (97) | 28 (100) | 19 (95) | 174 (93) | 86 (86) | 78 (95) | 112 (81) | 58 (76) | 64 (88) |
| Otherc | 0 | 0 | 0 | 5 (2) | 6 (6) | <5 | <5 | <5 | <5 |
| Female, No. (%) | 60 (74) | 23 (82) | 13 (65) | 94 (47)a | 61 (57) | 55 (61)a | 75 (54)d | 55 (72)d | 41 (56) |
| Male, No. (%) | 21 (26) | 5 (18) | 7 (35) | 108 (53) | 47 (43) | 35 (39) | 64 (46) | 21 (28) | 32 (44) |
| APOE ε4 carriership, No. (%) | 22 (27)a,e | 18 (64)d | 13 (65)a | 38 (19)a,d | 34 (31)b,d | 45 (50)a,b | 20 (14)a | 18 (24)b | 41 (56)a,b |
| CSF Aβ1-42f | 1265 (37.78)a,d | 1043 (60.09)b,d | 718 (71.53)a,b | 1448 (30.13)a,d | 1158 (40.07)b,d | 802 (45.69)a,b | NA | NA | NA |
| CSF pTau181f | 46 (3.14)a | 55 (4.89) | 67 (6.15)a | 19 (0.72)a | 22 (0.96)b | 29 (1.10)a,b | NA | NA | NA |
Abbreviations: Aβ, β-amyloid; ADNI, Alzheimer Disease Neuroimaging Initiative; APOE, apolipoprotein; CSF, cerebrospinal fluid; HABS, Harvard Aging Brain Study; PREVENT-AD, Presymptomatic Evaluation of Experimental or Novel Treatments for Alzheimer Disease; pTau, phosphorylated tau.
P < .05 between Aβ-negative and widespread Aβ groups.
P < .05 between regional Aβ and widespread Aβ groups.
Other race included those who were American Indian or Alaskan Native, Asian, Native Hawaiian or Other Pacific Islander, and more than 1 race.
P < .05 between Aβ-negative and regional Aβ groups.
In PREVENT-AD, CSF samples were available for 46 Aβ-negative, 19 regional, and 12 widespread; in ADNI, CSF samples were available for 138 Aβ-negative, 78 regional, and 60 widespread.
CSF biomarker measures were available for 77 PREVENT-AD and 276 ADNI participants. In both cohorts, the regional and widespread groups had lower CSF Aβ1-42 levels than Aβ-negative persons, and the widespread group also had lower levels than the regional Aβ group (Table; eTable 2 in Supplement 1).
In PREVENT-AD, CSF pTtau181 was higher in the widespread Aβ group than the Aβ-negative group. In ADNI, CSF pTau levels were higher in the widespread group than in the 2 other groups.
Cross-Sectional and Longitudinal Cognition
There were negligible differences in cognitive performance at baseline between the 3 Aβ classes in each cohort. Only the PREVENT-AD widespread Aβ group had worse delayed memory scores than the Aβ-negative group (F2,126 = 6.53; eTable 3 in Supplement 1). Comparing cognitive decline over time, all cohorts consistently showed that the widespread Aβ groups experienced greater cognitive decline than the Aβ-negative or regional groups on all cognitive indices (all β < −0.06) (Figure 3; eTable 4 in Supplement 1). Further, in ADNI (with up to 14 years of follow-up), participants in the regional Aβ group experienced greater cognitive decline than the Aβ-negative group (all β < −0.03). These associations remained when restricting follow-up to 7 years (PREVENT-AD mean follow-up time), but the difference in the regional group was lost when restricting analysis to 6 years (HABS mean follow-up).
Figure 3. Change in Cognition Over Time Between the 3 β-Amyloid (Aβ) Groups.
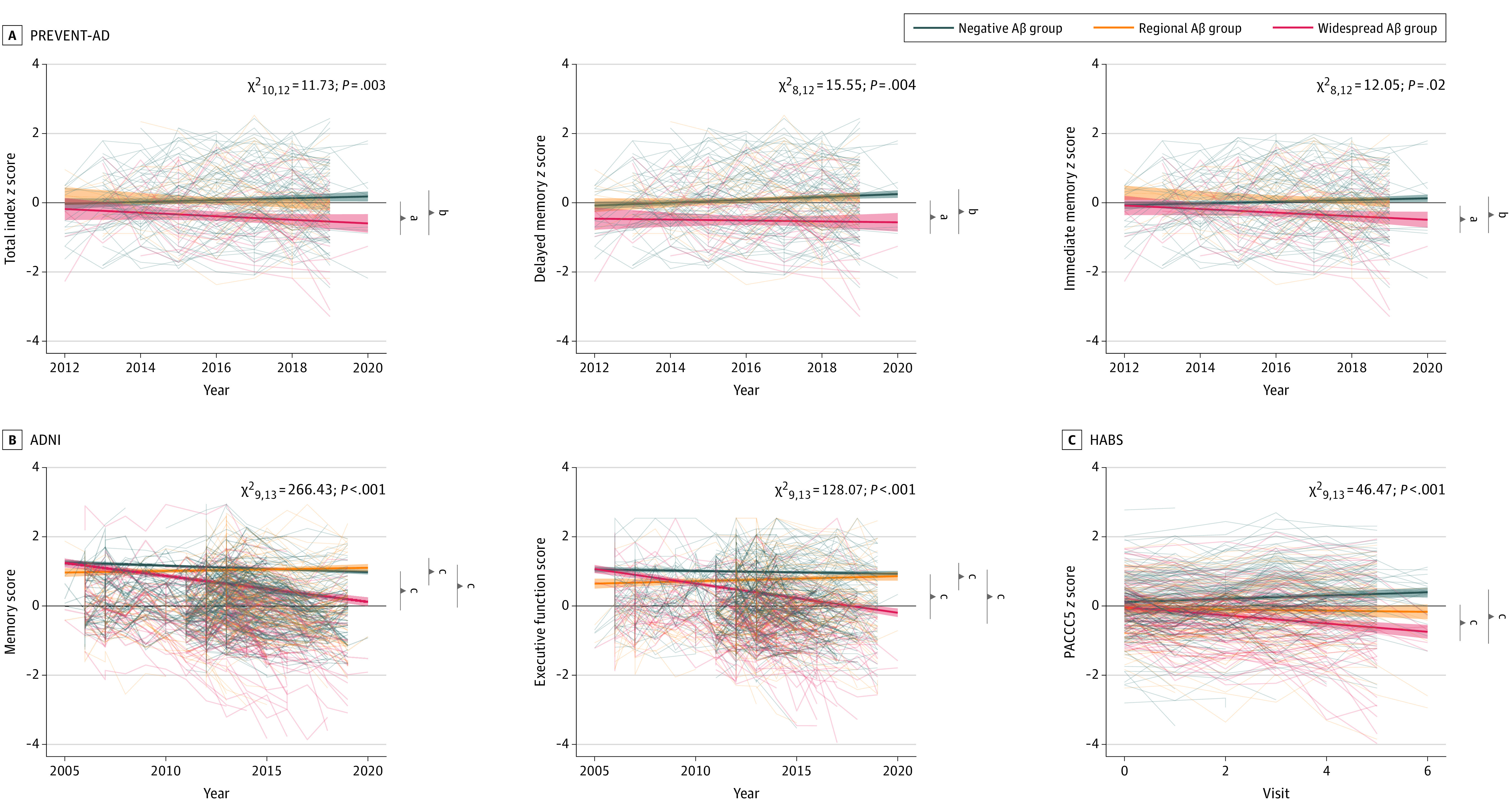
Linear mixed-effect models were used to assess the main association of Aβ groups with longitudinal cognition, corrected for age, sex, and education. The analyses were anchored at the participants’ baseline visit date. Cognitive test scores for the Presymptomatic Evaluation of Experimental or Novel Treatments for Alzheimer Disease (PREVENT-AD) (A), Alzheimer Disease Neuroimaging Initiative (ADNI) (B), and Harvard Aging Brain Study (HABS) (C) cohorts were represented over time in the 3 different groups. The widespread Aβ group showed a greater decline in their cognition scores when compared with the 2 other groups in all cohorts. In both ADNI and HABS, the regional group showed a greater cognitive decline compared to the Aβ-negative group. PACC5 indicates the 5-item Preclinical Alzheimer’s Cognitive Composite.
aP < .05.
bP < .01.
cP < .001.
Longitudinal Aβ Trajectories
Up to 4 years of longitudinal Aβ-PET data were available for all ADNI participants (median follow-up, 3 years) and for 222 HABS participants (median follow-up, 2 years). In these 2 cohorts, all 3 baseline Aβ groups showed Aβ accumulation rates significantly different from 0 and the rate of accumulation differed by group (eTable 5 in Supplement 1). In ADNI, the widespread and regional Aβ groups showed faster Aβ accumulation in all the 7 ROIs over time than the Aβ-negative group (all β > 0.03). Interestingly, no difference was found between the regional and widespread Aβ groups regarding Aβ accumulation over time in any of the ROIs (eFigure 4 in Supplement 1). In HABS, the widespread group accumulated Aβ faster than the Aβ-negative group in 6 of 7 ROIs (all β > 0.05), while the regional group showed greater Aβ accumulation than the Aβ-negative group in 5 of 7 ROIs (all β > 0.02). The widespread group had faster Aβ accumulation than the regional Aβ group in rostral anterior cingulate and precuneus (all β > 0.04; eFigure 4 and eTable 5 in Supplement 1).
Cross-Sectional Tau-PET
In PREVENT-AD, the widespread Aβ group had elevated tau-PET signal in 5 of 6 regions investigated when compared with Aβ-negative and regional Aβ groups (Figure 4; eTable 6 in Supplement 1). The regional Aβ group had greater tau-PET binding in the entorhinal cortex and middle temporal gyrus compared with the negative group. In both ADNI and HABS, the widespread Aβ group had elevated tau-PET signal compared with both Aβ-negative and regional groups across all regions investigated. Voxelwise analyses confirmed that the main group differences in tau-PET signal were found between the widespread and negative groups in the temporal lobe (eFigure 3B in Supplement 1).
Figure 4. Tau–Positron Emission Tomography (PET) Uptake Across the 3 β-Amyloid (Aβ) Groups.
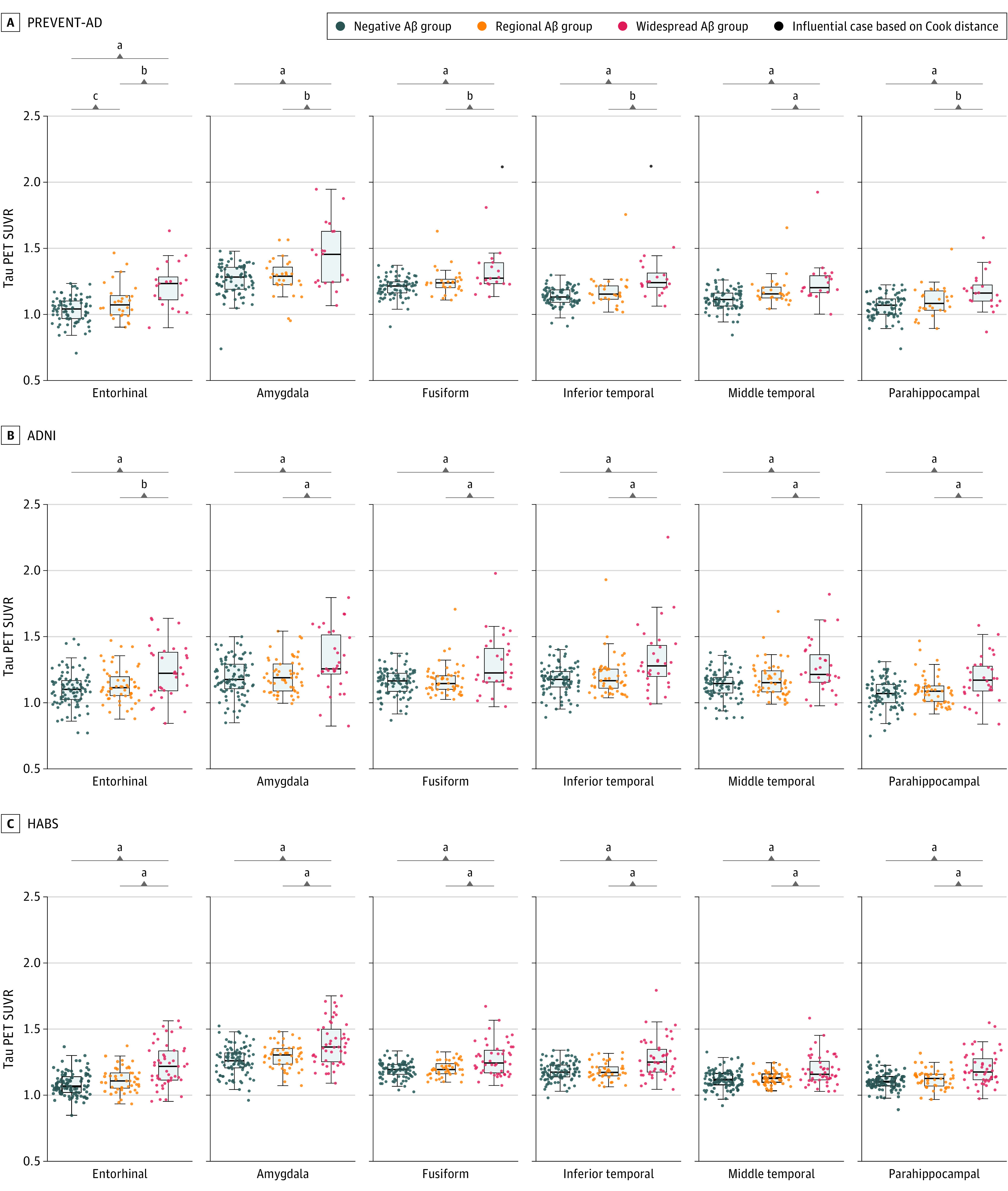
Six regions were chosen to represent areas of early tau-PET accumulation.35 Tau-PET scans were available for 129 Presymptomatic Evaluation of Experimental or Novel Treatments for Alzheimer Disease (PREVENT-AD) participants, 176 Alzheimer Disease Neuroimaging Initiative (ADNI) participants, and 195 Harvard Aging Brain Study (HABS) participants. A, In PREVENT-AD, the widespread Aβ group had elevated tau-PET signal when compared with Aβ-negative and regional Aβ groups across 6 regions. The regional Aβ group had elevated tau-PET binding only in the entorhinal cortex and middle temporal gyrus when compared with the Aβ-negative group. B and C, In both ADNI and HABS, the widespread Aβ group had elevated tau-PET signal compared with Aβ-negative and regional Aβ groups across all regions. Analyses were corrected for age and sex. SUVR indicates standardized uptake value ratios.
aP < .001.
bP < .01.
cP < .05.
Supplementary Analyses
Most of the regional participants would have been classified as Aβ-negative using conventional classifications (Figure 1B; eTable 7 for global binary classifications, eTable 8 for visual read, and eTable 9 for Centiloids in Supplement 1). The main results did not change when removing from the regional groups participants who would have been classified as Aβ-positive based on cohort specific global binary classifications or visual reads (eTables 10 and eTable 11 and eFigures 5-8 in Supplement 1). Removing individuals with high Centiloids (>40) from the regional group did not change the results. Removing individuals with high or intermediate Centiloids (>20) from the regional group had almost no association in ADNI but obscured most of the differences between negative and regional groups in PREVENT-AD and HABS (eTable 12 and eFigures 9 and 10 in Supplement 1). Changing the number of ROIs from 7 to either 5 or 10 yielded similar results across the main analyses (eTables 13 and 14 and eFigures 11 and 12 in Supplement 1).
Discussion
Most AD drugs are targeting single disease pathways. Removing Aβ when tau has already spread throughout the cortex might not be ideal given that tau is more closely related to cognitive decline than is Aβ.36,37 One way to identify individuals with Aβ, but with limited tau, could be to assess Aβ spatial extent severity. The hypothesis would be that individuals who have Aβ-PET binding restricted to a few brain regions might not yet have extensive tau and therefore be optimal candidates for anti-Aβ therapies.
The most common approach to analyze Aβ-PET is to classify individuals into Aβ-negative and Aβ-positive groups based on a global SUVR quantification or a visual read. However, this approach is not always optimal for detection of individuals with early Aβ levels,38 particularly if Aβ has started to accumulate regionally but is not yet globally widespread.23,35 We took advantage of the literature suggesting a spatiotemporal ordering of Aβ pathology to identify regions considered to be early Aβ accumulating regions23,24 and classified cognitively unimpaired participants into those with Aβ-PET signal that is widespread, regional, or negative. Our results (which did not vary when we tested 5 or 10 ROIs for Aβ deposition) suggest that by the time Aβ has spread extensively, tau has expanded beyond the entorhinal cortex and cognitive decline is prevalent.
Furthermore, our findings highlight the biological relevance of the regional Aβ group. These had intermediate CSF Aβ1-42 levels between the widespread (lower Aβ1-42) and Aβ-negative groups (higher Aβ1-42), indicating incipient cerebral accumulation of Aβ.39 In addition, the regional groups accumulated more Aβ fibrils (on PET) than the Aβ-negative group in ADNI and HABS (longitudinal PET data were not available in PREVENT-AD). Another crucial difference between groups in APOE ε4 carrier status: in contrast with the Aβ-negative group, both regional and widespread Aβ groups had higher percentages of APOE ε4 carriers (in PREVENT-AD and ADNI), suggesting increased risk of disease.40 Other recent studies have shown decreased CSF Aβ1-42 levels in participants with regional Aβ,26,41 and higher proportions of APOE ε4 carriers, as contrasted with Aβ-negative participants.42 APOE ε4 status is associated with increased Aβ load across all clinical diagnostic groups.43 Although the participants in this study with widespread Aβ did have detectable tau-PET signal in temporal brain regions, this tau PET binding was nearly absent in individuals in the regional Aβ-group using either region-based or voxelwise analyses. Regional participants also had similar levels of CSF pTau181 than Aβ-negative participants (data available for PREVENT-AD and ADNI). Therefore, as expected, cognitive decline was restricted mainly to widespread Aβ persons. These data suggest that most individuals with regional Aβ binding are in the earliest stages of the AD continuum, several years away from the onset of cognitive decline.
Importantly, regardless of cohort, most participants with regional Aβ binding had been classified as negative based using cohort-specific global Aβ thresholds or a visual read. Similar findings had been found in the Anti-Amyloid Treatment in Asymptomatic AD (A4) study,38 where quantitative methods suggested that only 50.1% of those classified as early accumulators had been identified visually as Aβ-positive. Therefore, unsurprisingly, sensitivity analyses removing participants previously classified as Aβ-positive using either quantitative threshold approaches or a visual read showed no important difference from our main results. When using a 3-tiered Centiloid approach, removing participants with high Centiloids (>40) in the regional group made almost no difference on the results, but removing participants with high and intermediate Centiloids (>20) obscured most PREVENT-AD and HABS cohorts’ findings in regional Aβ-binding participants.
Enrollment of regional Aβ-binding persons in clinical trials may nonetheless be challenging. A regional classification would be difficult to harmonize in multicenter trials, especially if these used different tracers. Our findings suggested that florbetapir, a US Food and Drug Administration–approved tracer used in ADNI, was less efficient at establishing clear categories of regional and widespread Aβ accumulation. In the present analyses, these categories were less distinct, probably owing to a lower signal-to-noise ratio (found in most 18F tracers when compared with 11C-Pittsburgh compound B and 18F-NAV-469444). Gaussian mixture modeling analysis, which we used to define the regional thresholds, was also tracer dependent, and further validation would be needed before applying current thresholds in other data sets. ROIs that first showed Aβ positivity also differed across individuals and cohorts, an observation that could result either from biological or interindividual pathological differences and tracer proprieties (or both). Despite these challenges, we found broadly consistent results across 3 independent cohorts, suggesting that a regional vs widespread binding approach is biologically meaningful and practicable. As a final caution, we nonetheless note that restriction of trial eligibility to regional participants would likely prevent the use of cognitive decline as the primary outcome of preventive trials as these individuals do not show decline over a window of approximately 7 years. Such trials might therefore require changes in primary outcomes, such as longitudinal change in AD biomarkers with the expectation that such changes will signal subsequent cognitive decline.
Limitations
We also note several limitations. The PREVENT-AD cohort presently lacks longitudinal PET scans. The HABS cohort lacks CSF data. Furthermore, across all cohorts, the number of cognitively unimpaired individuals with widespread and regional Aβ binding was relatively small, a difficulty that we attempted to mitigate in part through the inclusion of 3 independent cohorts.
Conclusions
We conclude that assessment of spatial Aβ burden may be a powerful method for identification of candidates well suited to clinical trials for prevention of AD progression. As Aβ-negative persons showed little Aβ accumulation over time or other evidence of advancing AD pathology, we suggest that anti-Aβ trials might advantageously enroll individuals limited to regional Aβ binding as they seek the earliest practical stage of amyloid signaling for AD pathogenesis.
eMethods.
eResults.
eTable 1. Regional Specific Thresholds
eTable 2. Comparison of Biological and Clinical Characteristics across the Aβ Groups
eTable 3. Baseline Cognition across the Aβ Groups
eTable 4. Change in Cognition Over Time Between the Aβ Groups
eTable 5. Aβ Accumulation Rate in ADNI and HABS
eTable 6. Tau-PET Uptake in Early Tau Regions
eTable 7. Global Binary Quantitative Classification by Spatial Extent groups
eTable 8. Visual Read Classification by Spatial Extent groups
eTable 9. Three-tiered Centiloid (CL) Classification by Spatial Extent groups
eTable 10. Biological and Clinical Characteristics excluding Regional Individuals that would have been classified as Positive based on Global Binary Classifications
eTable 11. Biological and Clinical Characteristics excluding Regional Individuals that would have been classified as Positive based on Visual Reads
eTable 12. Biological and Clinical Characteristics excluding Regional Individuals that would have been classified as having Intermediate and High Centiloid values (CL>20)
eTable 13. Biological and Clinical Characteristics across the Aβ Groups with 5 Regions
eTable 14. Biological and Clinical Characteristics across the Aβ Groups with 10 Regions
eFigure 1. Amyloid SUVR Distribution for the 7 Regions of Interest
eFigure 2. Distribution of Abnormal Regions in Regional Aβ Groups
eFigure 3. Group-level Voxel-wise Analysis of Differences in Aβ-PET and tau-PET signals between the three Aβ Groups
eFigure 4. Change in Aβ Uptake Over Time Between the three Aβ Groups in ADNI and HABS
eFigure 5. Tau-PET Uptake Across the 3 Aβ groups excluding Regional Individuals that would have been classified as Positive based on Global Binary Classifications
eFigure 6. Tau-PET Uptake Across the 3 Aβ groups excluding Regional Individuals that would have been Classified as Positive based on Visual Read
eFigure 7. Change in Cognition and Aβ Uptake over Time Between the Aβ Groups excluding Regional Individuals that would have been classified as Positive based on Global Binary Classifications
eFigure 8. Change in Cognition and Aβ Uptake over Time Between the Aβ Groups excluding Regional Individuals that would have been Classified as Positive based on Visual Read
eFigure 9. Tau-PET Uptake Across the 3 Aβ groups excluding Regional Individuals with CL>20
eFigure 10. Change in Cognition and Aβ Uptake Over Time Between the three Aβ Groups ex-cluding Regional Group Participants with CL>20
eFigure 11. Change in Cognition and Aβ Uptake Over Time Between the three Aβ Groups with 5 Regions
eFigure 12. Change in Cognition and Aβ Uptake Over Time Between the three Aβ Groups with 10 Regions
eReferences
Nonauthor Collaborators. Alzheimer’s Disease Neuroimaging Initiative, the Harvard Aging Brain Study, and the Presymptomatic Evaluation of Experimental or Novel Treatments for Alzheimer Disease Research Group
References
- 1.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239-259. doi: 10.1007/BF00308809 [DOI] [PubMed] [Google Scholar]
- 2.Thal DR, Rüb U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58(12):1791-1800. doi: 10.1212/WNL.58.12.1791 [DOI] [PubMed] [Google Scholar]
- 3.Braak H, Del Tredici K. The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol. 2011;121(2):171-181. doi: 10.1007/s00401-010-0789-4 [DOI] [PubMed] [Google Scholar]
- 4.Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991;12(10):383-388. doi: 10.1016/0165-6147(91)90609-V [DOI] [PubMed] [Google Scholar]
- 5.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353-356. [DOI] [PubMed] [Google Scholar]
- 6.Herrup K. The case for rejecting the amyloid cascade hypothesis. Nat Neurosci. 2015;18(6):794-799. doi: 10.1038/nn.4017 [DOI] [PubMed] [Google Scholar]
- 7.Selkoe DJ. Toward a comprehensive theory for Alzheimer’s disease: hypothesis: Alzheimer’s disease is caused by the cerebral accumulation and cytotoxicity of amyloid β-protein. Ann N Y Acad Sci. 2000;924(1):17-25. doi: 10.1111/j.1749-6632.2000.tb05554.x [DOI] [PubMed] [Google Scholar]
- 8.Huang YM, Shen J, Zhao H-L. Major clinical trials failed the amyloid hypothesis of Alzheimer’s disease. J Am Geriatr Soc. 2019;67(4):841-844. doi: 10.1111/jgs.15830 [DOI] [PubMed] [Google Scholar]
- 9.Mintun MA, Lo AC, Duggan Evans C, et al. Donanemab in early Alzheimer’s disease. N Engl J Med. 2021;384(18):1691-1704. doi: 10.1056/NEJMoa2100708 [DOI] [PubMed] [Google Scholar]
- 10.Uddin MS, Kabir MT, Rahman MS, et al. Revisiting the amyloid cascade hypothesis: from anti-Aβ therapeutics to auspicious new ways for Alzheimer’s disease. Int J Mol Sci. 2020;21(16):5858. doi: 10.3390/ijms21165858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Honig LS, Vellas B, Woodward M, et al. Trial of solanezumab for mild dementia due to Alzheimer’s disease. N Engl J Med. 2018;378(4):321-330. doi: 10.1056/NEJMoa1705971 [DOI] [PubMed] [Google Scholar]
- 12.Sperling RA, Jack CR Jr, Aisen PS. Testing the right target and right drug at the right stage. Sci Transl Med. 2011;3(111):111cm33. doi: 10.1126/scitranslmed.3002609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.StoP-AD Centre. Accessed July 18, 2022. https://prevent-alzheimer.net
- 14.Breitner JCS, Poirier J, Etienne PE, Leoutsakos JM. Rationale and structure for a new center for studies on prevention of Alzheimer’s disease (STOP-AD). J Prev Alzheimers Dis. 2016;3(4):236-242. doi: 10.14283/jpad.2016.121 [DOI] [PubMed] [Google Scholar]
- 15.Alzheimer’s Disease Neuroimaging Initiative. Accessed July 18, 2022. https://adni.loni.usc.edu/
- 16.Harvard Aging Brain Study. Accessed July 18, 2022. https://habs.mgh.harvard.edu/
- 17.Dagley A, LaPoint M, Huijbers W, et al. Harvard Aging Brain Study: dataset and accessibility. Neuroimage. 2017;144(pt B):255-258. doi: 10.1016/j.neuroimage.2015.03.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Randolph C, Tierney MC, Mohr E, Chase TN. The Repeatable Battery for the Assessment of Neuropsychological Status (RBANS): preliminary clinical validity. J Clin Exp Neuropsychol. 1998;20(3):310-319. doi: 10.1076/jcen.20.3.310.823 [DOI] [PubMed] [Google Scholar]
- 19.Crane PK, Carle A, Gibbons LE, et al. ; Alzheimer’s Disease Neuroimaging Initiative . Development and assessment of a composite score for memory in the Alzheimer's Disease Neuroimaging Initiative (ADNI). Brain Imaging Behav. 2012;6(4):502-516. doi: 10.1007/s11682-012-9186-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gibbons LE, Carle AC, Macking RS, et al. ; Alzheimer’s Disease Neuroimaging Initiative . A composite score for executive functioning, validated in Alzheimer's Disease Neuroimaging Initiative (ADNI) participants with baseline mild cognitive impairment. Brain Imaging Behav. 2012;6(4):517-527. doi: 10.1007/s11682-012-9176-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Papp KV, Rentz DM, Orlovsky I, Sperling RA, Mormino EC. Optimizing the preclinical Alzheimer’s cognitive composite with semantic processing: the PACC5. Alzheimers Dement (N Y). 2017;3(4):668-677. doi: 10.1016/j.trci.2017.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Desikan RS, Ségonne F, Fischl B, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31(3):968-980. doi: 10.1016/j.neuroimage.2006.01.021 [DOI] [PubMed] [Google Scholar]
- 23.Villeneuve S, Rabinovici GD, Cohn-Sheehy BI, et al. Existing Pittsburgh Compound-B positron emission tomography thresholds are too high: statistical and pathological evaluation. Brain. 2015;138(pt 7):2020-2033. doi: 10.1093/brain/awv112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jagust WJ, Landau SM; Alzheimer’s Disease Neuroimaging Initiative . Temporal dynamics of β-amyloid accumulation in aging and Alzheimer disease. Neurology. 2021;96(9):e1347-e1357. doi: 10.1212/WNL.0000000000011524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fantoni E, Collij L, Lopes Alves I, Buckley C, Farrar G; AMYPAD consortium . The spatial-temporal ordering of amyloid pathology and opportunities for PET imaging. J Nucl Med. 2020;61(2):166-171. doi: 10.2967/jnumed.119.235879 [DOI] [PubMed] [Google Scholar]
- 26.Grothe MJ, Barthel H, Sepulcre J, Dyrba M, Sabri O, Teipel SJ; Alzheimer’s Disease Neuroimaging Initiative . In vivo staging of regional amyloid deposition. Neurology. 2017;89(20):2031-2038. doi: 10.1212/WNL.0000000000004643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farrell ME, Jiang S, Schultz AP, et al. ; Alzheimer’s Disease Neuroimaging Initiative and the Harvard Aging Brain Study . Defining the lowest threshold for amyloid-pet to predict future cognitive decline and amyloid accumulation. Neurology. 2021;96(4):e619-e631. doi: 10.1212/WNL.0000000000011214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mormino EC, Betensky RA, Hedden T, et al. ; Alzheimer’s Disease Neuroimaging Initiative; Australian Imaging Biomarkers and Lifestyle Flagship Study of Ageing; Harvard Aging Brain Study . Amyloid and APOE ε4 interact to influence short-term decline in preclinical Alzheimer disease. Neurology. 2014;82(20):1760-1767. doi: 10.1212/WNL.0000000000000431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buckley RF, Sikkes S, Villemagne VL, et al. Using subjective cognitive decline to identify high global amyloid in community-based samples: a cross-cohort study. Alzheimers Dement (Amst). 2019;11(1):670-678. doi: 10.1016/j.dadm.2019.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fantoni E, Collij L, Lopes Alves I, Buckley C, Farrar G; AMYPAD consortium . The spatial-temporal ordering of amyloid pathology and opportunities for PET imaging. J Nucl Med. 2020;61(2):166-171. doi: 10.2967/jnumed.119.235879 [DOI] [PubMed] [Google Scholar]
- 31.Clark CM, Schneider JA, Bedell BJ, et al. ; AV45-A07 Study Group . Use of florbetapir-PET for imaging beta-amyloid pathology. JAMA. 2011;305(3):275-283. doi: 10.1001/jama.2010.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hedden T, Mormino EC, Amariglio RE, et al. Cognitive profile of amyloid burden and white matter hyperintensities in cognitively normal older adults. J Neurosci. 2012;32(46):16233-16242. doi: 10.1523/JNEUROSCI.2462-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Villeneuve Lab PET Pipeline. Github. Accessed July 18, 2022. https://github.com/villeneuvelab/vlpp
- 34.McSweeney M, Pichet Binette A, Meyer PF, et al. ; PREVENT-AD Research Group . Intermediate flortaucipir uptake is associated with Aβ-PET and CSF tau in asymptomatic adults. Neurology. 2020;94(11):e1190-e1200. doi: 10.1212/WNL.0000000000008905 [DOI] [PubMed] [Google Scholar]
- 35.Farrell ME, Chen X, Rundle MM, Chan MY, Wig GS, Park DC. Regional amyloid accumulation and cognitive decline in initially amyloid-negative adults. Neurology. 2018;91(19):e1809-e1821. doi: 10.1212/WNL.0000000000006469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bejanin A, Schonhaut DR, La Joie R, et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. Brain. 2017;140(12):3286-3300. doi: 10.1093/brain/awx243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hanseeuw BJ, Betensky RA, Jacobs HIL, et al. Association of amyloid and tau with cognition in preclinical Alzheimer disease: a longitudinal study. JAMA Neurol. 2019;76(8):915-924. doi: 10.1001/jamaneurol.2019.1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sperling RA, Donohue MC, Raman R, et al. ; A4 Study Team . Association of factors with elevated amyloid burden in clinically normal older individuals. JAMA Neurol. 2020;77(6):735-745. doi: 10.1001/jamaneurol.2020.0387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palmqvist S, Mattsson N, Hansson O; Alzheimer’s Disease Neuroimaging Initiative . Cerebrospinal fluid analysis detects cerebral amyloid-β accumulation earlier than positron emission tomography. Brain. 2016;139(Pt 4):1226-1236. doi: 10.1093/brain/aww015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mattsson N, Groot C, Jansen WJ, et al. Prevalence of the apolipoprotein E ε4 allele in amyloid β positive subjects across the spectrum of Alzheimer’s disease. Alzheimers Dement. 2018;14(7):913-924. doi: 10.1016/j.jalz.2018.02.009 [DOI] [PubMed] [Google Scholar]
- 41.Collij LE, Heeman F, Salvadó G, et al. ; ALFA Study, for the Alzheimer’s Disease Neuroimaging Initiative; AMYPAD Consortium . Multitracer model for staging cortical amyloid deposition using PET imaging. Neurology. 2020;95(11):e1538-e1553. doi: 10.1212/WNL.0000000000010256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sakr FA, Grothe MJ, Cavedo E, et al. ; INSIGHT-preAD study group; Alzheimer Precision Medicine Initiative (APMI) . Applicability of in vivo staging of regional amyloid burden in a cognitively normal cohort with subjective memory complaints: the INSIGHT-preAD study. Alzheimers Res Ther. 2019;11(1):15. doi: 10.1186/s13195-019-0466-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li C, Loewenstein DA, Duara R, Cabrerizo M, Barker W, Adjouadi M; Alzheimer’s Disease Neuroimaging Initiative . The relationship of brain amyloid load and APOE status to regional cortical thinning and cognition in the ADNI cohort. J Alzheimers Dis. 2017;59(4):1269-1282. doi: 10.3233/JAD-170286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Su Y, Flores S, Wang G, et al. Comparison of Pittsburgh compound B and florbetapir in cross-sectional and longitudinal studies. Alzheimers Dement (Amst). 2019;11(1):180-190. doi: 10.1016/j.dadm.2018.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eMethods.
eResults.
eTable 1. Regional Specific Thresholds
eTable 2. Comparison of Biological and Clinical Characteristics across the Aβ Groups
eTable 3. Baseline Cognition across the Aβ Groups
eTable 4. Change in Cognition Over Time Between the Aβ Groups
eTable 5. Aβ Accumulation Rate in ADNI and HABS
eTable 6. Tau-PET Uptake in Early Tau Regions
eTable 7. Global Binary Quantitative Classification by Spatial Extent groups
eTable 8. Visual Read Classification by Spatial Extent groups
eTable 9. Three-tiered Centiloid (CL) Classification by Spatial Extent groups
eTable 10. Biological and Clinical Characteristics excluding Regional Individuals that would have been classified as Positive based on Global Binary Classifications
eTable 11. Biological and Clinical Characteristics excluding Regional Individuals that would have been classified as Positive based on Visual Reads
eTable 12. Biological and Clinical Characteristics excluding Regional Individuals that would have been classified as having Intermediate and High Centiloid values (CL>20)
eTable 13. Biological and Clinical Characteristics across the Aβ Groups with 5 Regions
eTable 14. Biological and Clinical Characteristics across the Aβ Groups with 10 Regions
eFigure 1. Amyloid SUVR Distribution for the 7 Regions of Interest
eFigure 2. Distribution of Abnormal Regions in Regional Aβ Groups
eFigure 3. Group-level Voxel-wise Analysis of Differences in Aβ-PET and tau-PET signals between the three Aβ Groups
eFigure 4. Change in Aβ Uptake Over Time Between the three Aβ Groups in ADNI and HABS
eFigure 5. Tau-PET Uptake Across the 3 Aβ groups excluding Regional Individuals that would have been classified as Positive based on Global Binary Classifications
eFigure 6. Tau-PET Uptake Across the 3 Aβ groups excluding Regional Individuals that would have been Classified as Positive based on Visual Read
eFigure 7. Change in Cognition and Aβ Uptake over Time Between the Aβ Groups excluding Regional Individuals that would have been classified as Positive based on Global Binary Classifications
eFigure 8. Change in Cognition and Aβ Uptake over Time Between the Aβ Groups excluding Regional Individuals that would have been Classified as Positive based on Visual Read
eFigure 9. Tau-PET Uptake Across the 3 Aβ groups excluding Regional Individuals with CL>20
eFigure 10. Change in Cognition and Aβ Uptake Over Time Between the three Aβ Groups ex-cluding Regional Group Participants with CL>20
eFigure 11. Change in Cognition and Aβ Uptake Over Time Between the three Aβ Groups with 5 Regions
eFigure 12. Change in Cognition and Aβ Uptake Over Time Between the three Aβ Groups with 10 Regions
eReferences
Nonauthor Collaborators. Alzheimer’s Disease Neuroimaging Initiative, the Harvard Aging Brain Study, and the Presymptomatic Evaluation of Experimental or Novel Treatments for Alzheimer Disease Research Group