

The increase in the prevalence of individuals with Alzheimer’s disease (AD) combined with the lack of a cure calls for the development of novel therapies against AD (Canter et al., 2016). The key disease-defining pathological features of AD are the accumulation of extracellular amyloid-beta (Aβ) plaques (accompanied by increasing intracellular Aβ1–42) and higher intracellular neurofibrillary tangles, comprised mostly of hyperphosphorylated tau protein/pTau (Goedert, 2015; Hardy, 2017). It is evident that the elderly are more predisposed to develop AD, and thus aging is considered to be the primary risk factor for AD. By extrapolation, strategies that delay aging may also slow down (if not stop) AD.
The Nicotinamide adenine dinucleotide (NAD+)-mitophagy axis is compromised during aging. Working with Prof. Vilhelm Bohr at the National Institute on Ageing at the USA and colleagues, a series of our studies have shown that the key metabolite nicotinamide adenine dinucleotide (oxidized form NAD+) is reduced in common accelerated aging diseases, including xeroderma pigmentosum group A, ataxia telangiectasia, Cockayne syndrome (CS-A and CS-B), as well as in Werner syndrome. Importantly, NAD+ depletion is a key cause of pathological aging in these diseases (Fang et al., 2014, 2016, 2019b; Scheibye-Knudsen et al., 2014). While NAD+ participates in many cellular pathways that link to neuronal protection and healthy longevity, we show a pivotal role for NAD+-induced mitophagy in longevity and neuroprotection. More specifically, the deacetylase SIRT1 uses NAD+ to deacetylate FOXO3 and PGC1α, which are transcriptional regulators that upregulate different mitophagy genes, such as NIX/BNIP3L (detailed mechanisms are in Fang (2019)). NAD+ regulates mitophagy, a cellular pathway that specifically recognizes and degrades damaged mitochondria (Fang, 2019). Indeed, this NAD+-mitophagy axis is compromised unequivocally in the aforementioned diseases. Restoration of the NAD+-mitophagy axis alleviates disease pathologies and extends healthspan and lifespan in animal models, suggesting a causative role for the NAD+-mitophagy axis in longevity (Fang et al., 2014; Waltz et al., 2017).
In view of the fundamental role of the compromised NAD+-mitophagy axis in accelerating aging, we wondered whether the axis could also be relevant to the development of common age-predisposed diseases such as AD. The phenomenon of accumulated mitochondria in AD has been noted for decades but the molecular mechanism that leads to such accumulation was not fully understood, with oxidative stress cited as the most probable cause. In 2017, we hypothesized that the accumulation of damaged mitochondria in AD could be caused by defective mitophagy (Kerr et al., 2017). In fact, our subsequent experimental studies indicated that mitophagy was dramatically reduced in post-mortem hippocampal tissues from AD patients as well as in both Aβ and pTau animal models of AD (Fang et al., 2019a). In accelerated aging and Alzheimer’s disease, the NAD+-mitophagy axis is compromised likely due to increased NAD+ consumption (via Poly (adenosine diphosphate-ribose) polymerases and CD38) and reduced production. In line with these findings, NAD+ repletion repaired AD pathologies, and retained learning and memory in AD mice (Fang et al., 2019a; Figure 1). As NAD+ is reduced in normal aging brains, and age is the primary driver of AD, it is presumed that NAD+ reduction in AD could be at least significantly contributed to aging. Consequently, we hypothesize that NAD+ supplementation could be considered to treat early-onset AD with the clinical trial validation necessary.
Figure 1.
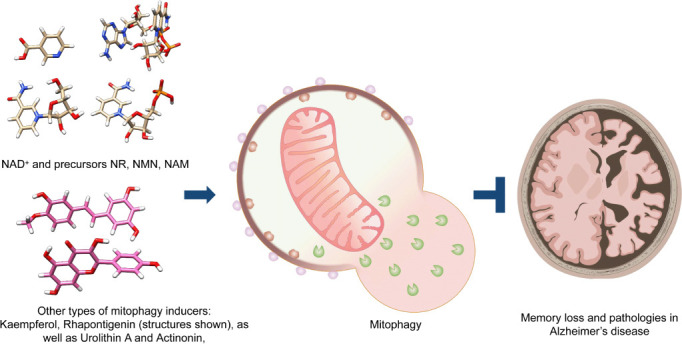
A schematic of turning up the NAD+-mitophagy axis to treat AD.
The structures of NAD+ and its precursors such as NR, NMN, and NAM are presented (upper left). In addition to NAD+ replenishment, novel mitophagy stimulators (lower left) could also upregulate neuronal mitophagy, leading to inhibition of AD pathologies and memory loss in animal models of AD. AD: Alzheimer’s disease; NAM: nicotinamide; NMN: nicotinamide mononucleotide; NR: nicotinamide riboside. Part of the figure was created with BioRender. com.
In addition to NAD+, other mitophagy inducers also show anti-AD potential. These compounds are actinonin (a naturally occurring antibacterial agent) and Urolithin A (Fang et al., 2019a). Very recently, we identified two robust mitophagy inducers, Kaempferol and Rhapontigenin, from a large natural compounds library through the combination of machine learning-based virtual screening and cross-species platform-supported wet lab validation (Xie et al., 2022; Figure 1). Kaempferol and Rhapontigenin inhibited memory loss and pathologies in both Aβ and pTau animal models of AD (Xie et al., 2022). The mechanisms behind how Kaempferol, Rhapontigenin, Urolithin A, and Actinonin inhibit AD pathology may work by directly turning up mitophagy, rather than through activation of the ‘NAD+-mitophagy axes’.
Our cross-species experimental models consistently support a causative role for a compromised NAD+-mitophagy axis in accelerated aging (as well as normal aging), likely forming a risk factor for AD. Strategies such as restoration of tissue NAD+ levels via augmentation of NAD+ precursors (such as nicotinamide riboside, nicotinamide mononucleotide, and nicotinamide) and mitophagy stimulation hold potential for clinical trials as the treatments against AD.
This work was supported by National Natural Science Foundation of China (No. 81971327), Akershus University Hospital (Nos. 269901, 261973), the Civitan Norges Forskningsfond for Alzheimers sykdom (No. 281931), the Czech Republic-Norway KAPPA programme (with Martin Vyhnálek, No. TO01000215), and the Rosa sløyfe/Norwegian Cancer Society & Norwegian Breast Cancer Society (No. 207819) to EFF.
Footnotes
Open peer reviewer: R Scott Turner, Georgetown University, USA
P-Reviewer: Turner RS; C-Editors: Zhao M, Liu WJ, Wang Lu; T-Editor: Jia Y
References
- 1.Canter RG, Penney J, Tsai LH. The road to restoring neural circuits for the treatment of Alzheimer's disease. Nature. 2016;539:187–196. doi: 10.1038/nature20412. [DOI] [PubMed] [Google Scholar]
- 2.Fang EF, Scheibye-Knudsen M, Brace LE, Kassahun H, SenGupta T, Nilsen H, Mitchell JR, Croteau DL, Bohr VA. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell. 2014;157:882–896. doi: 10.1016/j.cell.2014.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fang EF, Kassahun H, Croteau DL, Scheibye-Knudsen M, Marosi K, Lu H, Shamanna RA, Kalyanasundaram S, Bollineni RC, Wilson MA, Iser WB, Wollman BN, Morevati M, Li J, Kerr JS, Lu Q, Waltz TB, Tian J, Sinclair DA, Mattson MP, et al. NAD(+) Replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab. 2016;24:566–581. doi: 10.1016/j.cmet.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fang EF. Mitophagy and NAD(+) inhibit Alzheimer disease. Autophagy. 2019;15:1112–1114. doi: 10.1080/15548627.2019.1596497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, Lautrup S, Hasan-Olive MM, Caponio D, Dan X, Rocktaschel P, Croteau DL, Akbari M, Greig NH, Fladby T, Nilsen H, Cader MZ, Mattson MP, Tavernarakis N, Bohr VA. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer's disease. Nat Neurosci. 2019a;22:401–412. doi: 10.1038/s41593-018-0332-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fang EF, Hou Y, Lautrup S, Jensen MB, Yang B, SenGupta T, Caponio D, Khezri R, Demarest TG, Aman Y, Figueroa D, Morevati M, Lee HJ, Kato H, Kassahun H, Lee JH, Filippelli D, Okur MN, Mangerich A, Croteau DL, et al. NAD(+) augmentation restores mitophagy and limits accelerated aging in Werner syndrome. Nat Commun. 2019b;10:5284. doi: 10.1038/s41467-019-13172-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goedert M. NEURODEGENERATION. Alzheimer's and Parkinson's diseases:The prion concept in relation to assembled Abeta, tau, and alpha-synuclein. Science. 2015;349:1255555. doi: 10.1126/science.1255555. [DOI] [PubMed] [Google Scholar]
- 8.Hardy J. The discovery of Alzheimer-causing mutations in the APP gene and the formulation of the “amyloid cascade hypothesis”. FEBS J. 2017;284:1040–1044. doi: 10.1111/febs.14004. [DOI] [PubMed] [Google Scholar]
- 9.Kerr JS, Adriaanse BA, Greig NH, Mattson MP, Cader MZ, Bohr VA, Fang EF. Mitophagy and Alzheimer's disease:cellular and molecular mechanisms. Trends Neurosci. 2017;40:151–166. doi: 10.1016/j.tins.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scheibye-Knudsen M, Mitchell SJ, Fang EF, Iyama T, Ward T, Wang J, Dunn CA, Singh N, Veith S, Hasan-Olive MM, Mangerich A, Wilson MA, Mattson MP, Bergersen LH, Cogger VC, Warren A, Le Couteur DG, Moaddel R, Wilson DM, 3rd, Croteau DL, et al. A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 2014;20:840–855. doi: 10.1016/j.cmet.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Waltz TB, Fivenson EM, Morevati M, Li C, Becker KG, Bohr VA, Fang EF. Sarcopenia, aging and prospective interventional strategies. Curr Med Chem. 2017;25:5588–5596. doi: 10.2174/0929867324666170801095850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xie C, Zhuang XX, Niu Z, Ai R, Lautrup S, Zheng S, Jiang Y, Han R, Gupta TS, Cao S, Lagartos-Donate MJ, Cai CZ, Xie LM, Caponio D, Wang WW, Schmauck-Medina T, Zhang J, Wang HL, Lou G, Xiao X, et al. Amelioration of Alzheimer's disease pathology by mitophagy inducers identified via machine learning and a cross-species workflow. Nat Biomed Eng. 2022;6:76–93. doi: 10.1038/s41551-021-00819-5. [DOI] [PMC free article] [PubMed] [Google Scholar]