


Key Words: amyloid-beta 1–40, brain-derived neurotrophic factor, Fluoro-Gold, neuroprotection, retinal ganglion cells (RGC), retinal toxicity, tropomyosin receptor kinase B (TrkB)
Abstract
Amyloid-beta (Aβ)-related alterations, similar to those found in the brains of patients with Alzheimer’s disease, have been observed in the retina of patients with glaucoma. Decreased levels of brain-derived neurotrophic factor (BDNF) are believed to be associated with the neurotoxic effects of Aβ peptide. To investigate the mechanism underlying the neuroprotective effects of BDNF on Aβ1–40-induced retinal injury in Sprague-Dawley rats, we treated rats by intravitreal administration of phosphate-buffered saline (control), Aβ1–40 (5 nM), or Aβ1–40 (5 nM) combined with BDNF (1 µg/mL). We found that intravitreal administration of Aβ1–40 induced retinal ganglion cell apoptosis. Fluoro-Gold staining showed a significantly lower number of retinal ganglion cells in the Aβ1–40 group than in the control and BDNF groups. In the Aβ1–40 group, low number of RGCs was associated with increased caspase-3 expression and reduced TrkB and ERK1/2 expression. BDNF abolished Aβ1–40-induced increase in the expression of caspase-3 at the gene and protein levels in the retina and upregulated TrkB and ERK1/2 expression. These findings suggest that treatment with BDNF prevents RGC apoptosis induced by Aβ1–40 by activating the BDNF-TrkB signaling pathway in rats.
Introduction
Amyloid-beta (Aβ) deposition is a major neuropathological abnormality in Alzheimer’s disease (AD) (Murphy and LeVine, 2010; Sadigh-Eteghad et al., 2015; DeTure and Dickson, 2019). Aβ deposition also occurs in other neurodegenerative diseases, such as glaucoma, where its deposition in the retina is associated with a loss of retinal ganglion cells (RGCs) (Mancino et al., 2018; Wang and Mao, 2021). Multiple studies have shown a relationship between AD and glaucoma (Ratnayaka et al., 2015; Masuzzo et al., 2016). AD is one of the most widely investigated neurodegenerative diseases. It is characterized by the deposition of Aβ fibrils and formation of senile plaques in the brain. These pathological changes are associated with the formation of aggregates around cerebral blood vessels, oxidative stress, and glutamate excitotoxicity, leading to neuronal apoptosis (Dong et al., 2009; Uttara et al., 2009; Nita and Grzybowski, 2016). Interestingly, in patients with AD, similar neuropathological changes have been observed in association with RGC apoptosis and axonal loss, reduced thickness of the retinal nerve fibre layer, and increased cupping of the optic disc (Goldstein et al., 2003; Liu et al., 2009; Moncaster et al., 2010; Kesler et al., 2011; Frost et al., 2013; Javaid et al., 2016). In transgenic AD mouse models, RGC apoptosis and impaired visual functions have been found to be associated with increased Aβ deposition (Chiu et al., 2012; Parnell et al., 2012). These findings suggest that retinal neurodegeneration is associated with AD-like pathology (Normando et al., 2009). More recently, mechanisms underlying RGC loss in glaucoma have been shown to resemble those involved in AD-associated changes in the brain, suggesting that a common pathway could be involved in cell death in the two diseases (Vrabec and Levin, 2007; Caprioli, 2013; Ghiso et al., 2013; Pescosolido et al., 2014; Davis et al., 2016). Several studies have shown that glaucomatous RGC apoptosis is associated with increased retinal Aβ deposition (Guo et al., 2005, 2007; Guo and Cordeiro, 2008; Davis et al., 2016; Yan et al., 2017). For example, RGC apoptosis was significantly reduced in an experimental glaucoma model by targeting the endogenous Aβ synthesis pathway (Guo et al., 2007). Accumulating evidence has shown increased vitreous levels of Aβ in patients with glaucoma (Guo et al., 2007; Tsolaki et al., 2011; Mancino et al., 2018).
Hence, Aβ-associated molecular pathways are potential targets for the prevention of glaucomatous neurodegeneration. We previously showed that intravitreal administration of Aβ1–40-induced apoptosis of retinal cells in rats, leading to time- and dose-dependent changes in retinal and optic nerve morphology (Mohd Lazaldin et al., 2018). Aβ1–40-induced changes, marked by increased retinal oxidative stress and significantly reduced levels of retinal brain-derived neurotropic factor (BDNF), were more severe following 14 days of intravitreal administration (Mohd Lazaldin et al., 2018).
BDNF depletion is associated with both AD and glaucoma (Chitranshi et al., 2018; Mancino et al., 2018). BDNF, which exists as a precursor (proBDNF) and in a mature form, plays a critical role in neuronal functions and survival (Bathina and Das, 2015; Xue et al., 2022).
The sera and cerebrospinal fluids of patients with AD show low levels of BDNF (Diniz et al., 2014). Similarly, post-mortem brain tissues from patients with AD show decreased BDNF mRNA levels (Connor et al., 1997). Connor et al. (1997) also proposed that BDNF acts as a neuroprotective agent and delays the progression of neurodegenerative disorders, including AD (Connor et al., 1997). The differentiation and survival of neurons in the hippocampus and basal forebrain is promoted by BDNF (Tapia-Arancibia et al., 2008). In glaucoma, interruption of the retrograde transport of BDNF and the accumulation of its TrkB receptor at the optic nerve head in rats suggest that BDNF deprivation plays a role in the pathogenesis of RGC death (Pease et al., 2009). In contrast, overexpression of BDNF delays the progressive loss of RGCs and axons in the eyes of rats with elevated intraocular pressure (IOP) (Feng et al., 2016, 2017).
The neuroprotective effects of BDNF against retinal damage caused by Aβ1–40 have been reported previously (Mohd Lazaldin et al., 2020). Pre-treatment with BDNF significantly reduces Aβ1–40-induced increase in the number of TUNEL-positive cells. It also ameliorates Aβ1–40-induced changes in optic nerve morphology, axonal swelling, and the expression of retinal glutathione, superoxide dismutase, and catalase. Furthermore, pre-treatment with BDNF restores Aβ1–40-induced deterioration of the ability of a rat to recognize visual cues (Mohd Lazaldin et al., 2020). However, the mechanisms underlying the anti-apoptotic effects of BDNF against RGC loss remain unknown. Hence, in this study, we investigated whether the BDNF-TrkB signaling pathway acts by altering ERK1/2 activation and suppressing caspase-3 activation, thereby protecting RGCs against Aβ-induced apoptosis.
Methods
Animals
Forty-two male Sprague-Dawley rats (9 weeks old, 200–250 g body weight) free from any contamination under strict quarantine observation were housed one per cage under a 12-hour light/dark cycle before use. Normal pellet diet and water were provided ad libitum. The experimental protocols complied with the Association for Research in Vision and Ophthalmology’s Resolution on the Use of Animals in Research (ARVO, 2021). Ethical approval for this study was obtained from Committee on Animal Research & Ethics of Universiti Teknologi MARA, Puncak Alam, Selangor, Malaysia (UiTM Care: 117/2015, approval date: October 9, 2015).
Preparation and validation of aggregated Aβ1–40
Fresh Aβ1–40 solution was prepared as previously described (Watts et al., 2010; Li et al., 2011). Aβ1–40 peptides (AnaSpec, Fremont, CA, USA) were diluted with sterile 0.1 M PBS (pH 7.4; Affymetrix USB, Santa Clara, CA, USA). The solution was sonicated for 1 minute and incubated at 37°C for 1 week to aggregate the peptides. This stock solution of Aβ1–40 was stored at –20°C (Resende et al., 2008).
Scanning electron microscopy (SEM) images of amyloid fibrils were obtained using an ESEM system (Thermo Fisher Scientific, Waltham, MA, USA) with an accelerating voltage of 5 kV. The sample solution (1 nmol/µL) was diluted 50-fold in pure water, and 1 µL of the diluted solution was placed on silicone and air-dried overnight. To prevent electric charge build-up, the sample was coated with platinum using an Auto-Fine Coater (JEOL, Akishima, Tokyo, Japan) prior to imaging. Images were viewed at magnifications of 2000×, 4000×, and 10,000×. Fibril’s diameter data were collected from software build-up in scanning electron microscopy (Thermo Fisher Scientific, Waltham, MA, USA).
BDNF preparation
BDNF solution was prepared in accordance with manufacturer’s instructions (BioVision revision 11/14, Mountain View, CA, USA). Briefly, the BDNF-containing vial was centrifuged (6988 × g for 30 seconds at 4°C; Eppendorf 5430R, Germany) and then diluted in ddH2O to obtain a final 1.0 µg/µL concentration. BDNF aliquots were kept at –20°C for future use.
Study design
The rats were randomly divided into three groups using the random number table method, each independent selection containing 14 animals. Animals in group 1 received bilateral intravitreal injection of 3 μL PBS; those in group 2 received 3 μL of 5 nmol/µL Aβ1–40 in PBS, while those in group 3 received 2 μL of 5 nmol/µL Aβ1–40 in PBS, followed by 1 μL of 1 μg BDNF in PBS. In all cases, the PBS solution was sonicated and incubated in a manner similar to that of the Aβ1–40 solution (Figure 1).
Figure 1.

Study design.
Aβ: Amyloid-beta; BDNF: brain-derived neurotrophic factor; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; i.v.: intravitreal administration; PCR: polymerase chain reaction; RGC: retinal ganglion cell.
For intravitreal injections, ketamine (80 mg/kg, intraperitoneal) combined with xylazine (12 mg/kg, intraperitoneal) (Ilium Troy Laboratories, Glendenning, NSW, Australia) were administered to anesthetize the animals. A 30-gauge needle mounted on 10 μL Hamilton syringe was used. Using a microscope, the tip of the needle was used to puncture the sclera at the dorsal limbus of the eye, following which the Hamilton needle was inserted through the puncture site. The solution was injected slowly over at least 2 minutes to avoid retinal damage resulting from pressure caused by fluid injection. Neomycin-polymyxin ointment (Alcon, Geneva, Switzerland) was applied after the procedure.
For retrograde labeling, Fluoro-Gold (Fluorochrome, Denver, CO, USA) was injected very slowly into the superior colliculus of each hemisphere, and a stereotaxic instrument (Stoelting Co., Wood Dale, IL, USA) was used to locate the specified area. The fur was shaved, povidone-iodine was applied to the overlying skin, and a 2-cm-long incision was made in the midline of the scalp over the cranium to expose the skull. A micro dental drill was used to drill a hole 7.5 mm behind the bregma and 2.0 mm on each side from the midline at a depth of 4.0 to 4.5 mm to reach the superior colliculus area (Paxinos and Watson, 2007; Schlamp et al., 2013). Four microliters of 3% Fluoro-Gold (Fluorochrome) in sterile PBS was injected using a Hamilton syringe at a rate of 1 μL/min for 2 minutes on either side as previously described (Schlamp et al., 2013). The exposed skull was fully covered with dental cement, and the scalp was closed using a nylon surgical suture. The rats were allowed to fully recover from anesthesia before being transferred back to the animal house.
Intraperitoneal ketamine (> 80 mg/kg) conbined with xylazine (> 12 mg/kg) was used to sacrifice the animals. The eyes were enucleated at 24 hours, 3 days, and 7 days for western blotting (WB) and quantitative reverse transcription-polymerase chain reaction (qRT-PCR) analysis, and at 14 days for Congo red staining and counting of Fluoro-Gold-stained RGCs. For both WB and qRT-PCR, samples were derived from three rats at each time point in each group. Furthermore, for WB, each biological sample was subjected to assessment in two technical replicates whereas for qRT-PCR there were three technical replicates. Fluoro-Gold and Congo red staining was performed in four rats in each group on days 7 and 14, respectively. For histopathological examination, a 10% formaldehyde solution was used to fix the eyeballs for 24 hours at room temperature. Fluoro-Gold-stained RGCs were counted in freshly isolated retinas.
Congo red staining
To visualize fibrillar Aβ plaques in the retina, Congo red (MilliporeSigma, Burlington, MA, USA) staining was performed (Wang et al., 2010). Briefly, deparaffinized retinal sections were dehydrated using decreasing ethanol concentrations and stained with Congo red solution for 1 hour. Slides were rinsed, counterstained with hematoxylin, and examined under a light microscope (20× magnification; Nikon, Tokyo, Japan).
Retrograde labeling
To count Fluoro-Gold-stained RGCs, enucleated eyes were fixed with 4% formaldehyde solution for 2 hours at 4°C. The eyeballs were then washed in 5 mL of 1 M PBS at room temperature to remove excess formaldehyde. The anterior segment was removed, and the vitreous was carefully detached from the retina. The retinal cup was then isolated and four incisions were made at the retinal periphery to flatten it. Finally, the retinal flat mount was examined under a fluorescence microscope.
A fluorescence microscope (Olympus, Tokyo, Japan) was used to observe Fluoro-Gold-labeled RGCs using ultraviolet filters with maximal absorption at 560 nm (40× magnification). Fluoro-Gold-labeled RGCs were counted by three blinded investigators using the image analysis program, ImageJ (version 1.52, National Institutes of Health, Bethesda, MD, USA), based on 12 images representing 12 areas (120 × 160 μm2, each), three in each quadrant of each retina (Figure 2). The number of Fluoro-Gold-labeled RGCs in each image was then converted into RGC density per square millimetre (RGCs/mm2) of the retina. The average number of Fluoro-Gold-labeled RGCs in the 12 images was calculated to determine the cell density of each retina (Lafuente et al., 2002; Schlamp et al., 2013; Nadal-Nicolás et al., 2015; Nor Arfuzir et al., 2020; Lambuk et al., 2021).
Figure 2.

Outline of the flat mount of a four-quadrant (1–4) retina.
The optic nerve head has been excluded from the retina for retinal ganglion cell counting. The central section (C) was located 0.875 mm from the optic disc (OD). The mid (M) and peripheral (P) sections were located 1 mm apart from each other.
qRT-PCR
Extracted retinal tissues were rinsed with ice cold PBS (0.1 M) and stabilized in RNAlater Stabilization Solution (Ambion, Austin, TX, USA). AGeneJET RNA purification kit (Thermo Fisher Scientific) was used for RNA extraction and purification according to the manufacturer’s instructions. Contaminating genomic DNA was removed using the RapidOut DNA Removal Kit (Thermo Fisher Scientific). A NanoDrop spectrophotometer (Thermo Fisher Scientific) was used to measure the quantity and quality of RNA for qRT-PCR. Maxima First Strand cDNA Synthesis Kit (Thermo Fisher Scientific) was used to synthesize cDNA. Up to 1 µg of DNA- and DNase-free RNA was added to 5× reaction mix (containing reaction buffer, dNTPs, oligo (dT)18, and random hexamer primers), Maxima Enzyme Mix (containing Thermo Scientific Maxima™ Reverse Transcriptase and Thermo Scientific™ RiboLock™ RNase Inhibitor), and nuclease-free water in a sterile RNAse-free microcentrifuge tube. After gentle mixing and centrifugation for 1 minute, the mixture was incubated for 10 minutes at 25°C and then for 15 minutes at 50°C. The mixture was subjected to a final incubation at 85°C for 5 minutes to stop the reaction. Primer pair specificity against the rat genome was confirmed using the nucleotide Basic Local Alignment Search Tool, an online software tool maintained by the National Center for Biotechnology Information (U.S. National Library of Medicine). All primers (Table 1) were supplied by Macrogen Inc. (Seoul, Korea) as 100 µM stock solutions and diluted to 10 µM prior to use.
Table 1.
Primers used to amplify genes of interest and reference genes in the retina (18–24 bases)
| Gene symbol (primer source) | Sequence (5’–3’) | Annealing temperature (°C) |
|---|---|---|
| GAPDH | F: ATG ATT CTA CCC ACG GCA AG | 63 |
| R: CTG GAA GAT GGT GAT GGG TT | ||
| β-Actin | F: GAC ATC CGT AAA GAC CTC TAT GCC | 59 |
| R: ATA GAG CCA CCA ATC CAC ACA GAG | ||
| TrkB | F: TCT GTA CCA AAA TAC ACG TC | 59 |
| R: TTT GGG TTT GTC TCA TAG TC | ||
| Caspase-3 | F: TCC ATA AAA GCA CTG GAA TG | 59 |
| R: CTG TGA TCT TCC TTA GAA ACA C | ||
| ERK1/2 | F: CCA TTG ATA TTT GGT CTG TGG | 59 |
| R: ATC CAA GAA TAC CCA GGA TG |
F: Forword; R: reverse.
Real-time PCR was performed on the CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA, USA) using Luminaris Colour HiGreen qPCR Master Mix (Thermo Fisher Scientific). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and β-actin were used as reference genes, while Trk-B, ERK1/2, and caspase-3 were the genes of interest (GOI). All GOI data were normalized to those of the reference genes.
Western blotting
Five mL of PBS (0.1 M, pH 7.4) was used to rinse the retinal tissue, following which the wet weight was recorded. The retinal tissue was homogenized using radioimmunoprecipitation assay (RIPA) lysis buffer (150 mM NaCl, 1.0% IGEPAL CA-630, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), and 50 mM Tris; pH 8.0) containing 0.5 mL protease inhibitor. RIPA buffer was used at a ratio of 10 μL RIPA buffer to 1 mg of retinal tissue. The mixture was sonicated for 1 minute, centrifuged at 11,000 × g at 4°C for 13 minutes, and stored at -80°C until further use.
Retinal extract samples were incubated with 4× sample buffer (60 mM Tris-HCl, pH 7.4, 25% glycerol, 2% SDS, 14.4 mM 2-mercaptoethanol, and 0.1% bromophenol blue), in a 4:1 ratio, at 90°C for 5 minutes before being electrophoretically separated on a 12% sodium dodecyl sulphate polyacrylamide gel for 2.5 hours at 300 mA. The separated proteins were transferred to a nitrocellulose membrane (0.2 mm, Thermo Fisher Scientific), and protein bands were detected using a Ponceau-S solution (MilliporeSigma) after 5 minutes of gentle agitation. The membrane was incubated with 5% skim milk in TBST blocking buffer (20 mM Tris-HCl, pH 7.6, 137 mM NaCl, and 0.1% Tween-20) for 1 hour at room temperature (21–25°C) to reduce nonspecific binding of the primary antibody. Primary antibody diluted in TBST, at a ratio of 1:10,000, was added to the membrane and then incubated overnight at 4°C under gentle agitation. Next, the membrane was incubated with secondary antibodies diluted in TBST for 2 hours at room temperature under gentle agitation. Protein bands were detected using Amersham ECL prime; chemiluminescent substrate (Cytiva, Marlborough, MA, USA). The density of protein bands was measured using the image analysis software ImageJ. To test the constitutive activity of TrkB (1:1000, Santa Cruz Biotechnology, Santa Cruz, CA, USA, Cat# sc-377218, RRID: AB_2801499) and ERK1/2 (1:1000, Santa Cruz Biotechnology, Cat# sc-514302, RRID: AB_2571739), mouse antibodies against the aforementioned antigens were used. To detect pro-apoptotic marker caspase-3, anti-caspase3 anti-mouse antibody (1:500, Santa Cruz Biotechnology, Cat# sc-56053, RRID: AB_781826) was used. For all experiments, anti-β-actin mouse antibody (1:10,000, Santa Cruz Biotechnology, Cat# sc-47778, RRID: AB_2714189) was used to normalize the data. Horseradish peroxidase-coupled anti-mouse antibody (1:10000, Santa Cruz Biotechnology, Cat# sc-516102, RRID: AB_2687626) was used as the secondary antibody.
Statistical analysis
The sample size in this study was calculated according to the resource equation method (Arifin and Zahiruddin, 2017). In the present study, the sample size was calculated based on the “resource equation” proposed by Arifin and Zahiruddin. The sample size in each group should be a minimum of 10/k+1 and a maximum of 20/k+1 (k, the number of groups) in this equality. Thereby, we calculated the sample size of each group to be a minimum of four, and a maximum of six. The sample size for western blot analysis and RT-PCR were considered adequate to test the respective biological effects and our sample sizes are similar to those reported in previous publication (Lambuk et al., 2021). In addition, the sample size was also in accordance with recommendation of the university animal ethics research committee. In this study, investigators who collected the data were blinded to the experimental groups. Statistical analysis was performed using SPSS 26.0 (IBM Corp., Armonk, NY, USA). Fibril’s diameter data are presented as the mean ± SD. Statistical significance was analyzed using independent samples t-tests. Data from retrograde labelling, western blotting and real-time PCR analyses are expressed as the mean ± SEM. Significant differences between groups were determined using one-way analysis of variance with post hoc Bonferroni tests. P-values < 0.05 were considered statistically significant.
Results
Validation of aggregated Aβ1–40
The appearances of fresh Aβ1–40 and Aβ1–40 incubated for 7 days are shown in Figure 3A–C. Prolonged incubation led to an increase in fibrillar length (Figure 3D–F). Although generated from the same protein, fresh Aβ1–40 and Aβ1–40- incubated for 7 days had different structures. Fresh Aβ1–40 consisted of a non-branched straight fibril with a diameter of 631.5 ± 245.74 nm. In contrast, the diameter of the Aβ1–40 fibril after 7 days of incubation was 2.12-fold larger (P = 1.4 × 10–5), with a mean value of 1340.8 ± 291.66 nm (Figure 4).
Figure 3.

Scanning electron microscopy images of Aβ1–40.
(A, D) Fresh Aβ1–40 (A) and Aβ1–40 incubated for 7 days (D) at 2000× magnification; (B, E) fresh Aβ1–40 (B) and Aβ1–40 incubated for 7 days (E) at 4000× magnification; (C, F) fresh Aβ1–40 (C) and Aβ1–40 incubated for 7 days (F) at 10,000× magnification. Aβ1–40 sample solution (1 nmol/µL) was diluted 50-fold in pure water and coated with platinum. Arrows represent the amyloid fibrils. Aβ: Amyloid-beta.
Figure 4.

Fibril diameters of fresh Aβ1–40 and the Aβ1–40 for 7 days.
Measurements of fibril diameter was repeated ten times and the photograph captured swept across the surface on the film. Data are presented as the mean ± SD. P < 0.01 (independent samples t-test). Aβ: Amyloid-beta.
Retinal Aβ1–40 deposition in rats
Congo red staining is part of a set of histochemical techniques used to confirm the presence of amyloid deposits based on a characteristic deep-red or salmon color. In this study, Congo red staining clearly demonstrated the presence of amyloid deposits in both the Aβ1–40- and BDNF groups (Figure 5A–C).
Figure 5.

Representative light photomicrograph of Congo red-stained rat retinal sections showing the effect of intravitreal administration of BDNF on Aβ1–40 deposition in the rat retina.
(A) Control group; (B) Aβ1–40 group; and (C) BDNF group. Scale bars represent 100 µm at 20× magnification and arrows represent the amyloid deposition. Aβ: Amyloid-beta; BDNF: brain-derived neurotrophic factor; GCL: ganglion cell layer; IPL: inner plexiform layer; IR: inner retina.
Effects of BDNF on RGC survival in Aβ1–40-exposed retinas
Retrograde labeling of RGCs with Fluoro-Gold showed that RGC density was 3.99-fold lower in the Aβ1–40 group than in the control group (P = 4 × 10–6). Rats that received BDNF showed 3.98-fold higher number of Fluoro-Gold-labeled RGCs than those that received Aβ1–40 (P = 4 × 10–6). The number of Fluoro-Gold-positive cells was comparable between the control and BDNF groups (Figure 6A–D).
Figure 6.

Effect of BDNF on the density of Fluoro-Gold-positive cells in Aβ1–40 exposed retina.
(A) Control group, (B) Aβ1–40 group, (C) BDNF group, and (D) quantitative expression of the effect of BDNF on the density of Fluoro-Gold-positive cells in Aβ1–40 exposed retina. The image of live retinal ganglion cells was captured from four different rats in the same group by three blinded investigators. **P < 0.01, vs. control; ##P < 0.01, vs. amyloid (one-way analysis of variance with post hoc Bonferroni test). Scale bars represent 100 µm at 100× magnification and arrows represent the live retinal ganglion cells; n = 4 (mean ± SEM). Aβ: Amyloid-beta; BDNF: brain-derived neurotrophic factor.
Effects of BDNF on TrkB Expression in Aβ1–40-exposed retina
TrkB gene expression
TrkB gene expression in the retina following exposure to Aβ1–40 was significantly lower 24 hours (3.51-fold, P = 1 × 10–3), 3 days (2.45-fold, P = 1 × 10–3), and 7 days (2.00-fold, P = 1 × 10–3) after treatment compared with the control group (Figure 7). On day 7, TrkB expression was upregulated in the BDNF group when compared with the Aβ1–40 group (2.37-fold, P = 1 × 10–3). However, TrkB expression in the BDNF group was significantly reduced at 24 hours (3.23-fold, P = 1 × 10–3) and 3 days (1.6-fold, P = 7.9 × 10–3) after treatment. On day 7 after treatment, it was 1.20-fold higher in the BDNF group (P = 1.3 × 10–2) than that in the control group.
Figure 7.

Effect of BDNF on TrkB gene expression in Aβ1–40-exposed retina (qRT-PCR).
Total RNA was extracted after 24 hours, 3 days, and 7 days of intravitreal administration. TrkB gene expression was normalized against that of GAPDH and β-actin and compared to the PBS (control) group. Data represent three biological replicates with three technical replicates. n = 3 (mean ± SEM). *P < 0.05, **P < 0.01, ***P < 0.001, vs. control group (PBS); ###P < 0.001, vs. Aβ1–40 group (one-way analysis of variance with post hoc Bonferroni test). Aβ: Amyloid-beta; BDNF: brain-derived neurotrophic factor; PBS: phosphate-buffered saline; qRT-PCR: quantitative reverse transcription-polymerase chain reaction.
TrkB protein expression
Western blot analysis showed that at 24 hours, TrkB protein expression in the retina of rats in the Aβ1–40 group was significantly lower compared with the control group (2.48-fold, P = 1 × 10–3). In the BDNF group, protein expression was upregulated at 24 hours compared with the Aβ1–40 group (2.56-fold, P = 1 × 10–3). No significant differences were observed between the BDNF and control groups at any of the time points analyzed (Figure 8).
Figure 8.

Effect of BDNF on TrkB protein expression in Aβ1–40-exposed retinas after 24 hours, 3 days, and 7 days of intravitreal administration (western blotting).
Total TrkB protein expression was normalized against that of β-actin and compared with the PBS (control) group for statistical significance. Data represent three biological replicates with three technical replicates. n = 3 (mean ± SEM). *P < 0.05, **P < 0.01, ***P < 0.001, vs. control; ###P < 0.001, vs. Aβ1–40 group (one-way analysis of variance with post hoc Bonferroni test). Aβ: Amyloid-beta; BDNF: brain-derived neurotrophic factor; PBS: phosphate-buffered saline.
Effects of BDNF on ERK1/2 expression in Aβ1–40-exposed retina
ERK1/2 gene expression
ERK1/2 gene expression in the retina was significantly lower at 24 hours (2.26-fold, P = 1 × 10–3), 3 days (2.48-fold, P = 1 × 10–3), and 7 days (5.14-fold, P = 1 × 10–3) after Aβ1–40 exposure compared with the control group (Figure 9). Analysis at 24 hours and 3 days showed no significant differences in ERK1/2 gene expression in the retina of rats in the BDNF group compared with the Aβ1–40 group, but ERK1/2 gene expression at these two time points was significantly lower compared with the control group (2.4-fold, P = 1 × 10–3; 1.62-fold, P = 8 × 10–3, respectively). On day 7, ERK1/2 gene expression was significantly upregulated (4.33-fold, P = 1 × 10–3) in the BDNF group compared to the Aβ1–40 group. No significant differences in ERK1/2 gene expression were observed between the BDNF and control groups on day 7.
Figure 9.
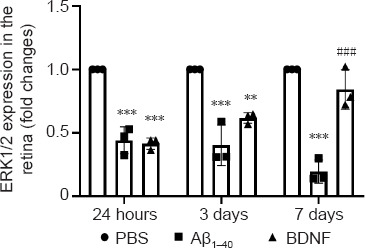
Effects of BDNF on ERK1/2 gene expression in Aβ1–40-exposed retina (qRT-PCR).
Total RNA was extracted after 24 hours, 3 days, and 7 days of intravitreal administration. ERK1/2 gene expression was normalized against that of GAPDH and β-actin and compared with the PBS (control) group for statistical significance. Data represent three biological replicates with three technical replicates. n = 3 (mean ± SEM): **P < 0.01, ***P < 0.001, vs. control group (PBS); ###P < 0.001, vs. Aβ1–40 group (one-way analysis of variance with post hoc Bonferroni test). Aβ: Amyloid-beta; BDNF: brain-derived neurotrophic factor; PBS: phosphate-buffered saline; qRT-PCR: quantitative reverse transcription-polymerase chain reaction.
ERK1/2 protein expression
ERK1/2 protein expression was significantly lower in the Aβ1–40 group than that in the control group at 24 hours (2.01-fold, P = 9 × 10–3) and 7 days (3.41-fold, P = 1.9 × 10–2) after exposure. In contrast, on day 7, ERK1/2 expression was significantly upregulated in the BDNF group compared with the Aβ1–40 group (3.03-fold, P = 4.1 × 10–2). No significant change in ERK1/2 expression in the retina was observed between BDNF and control groups at any of the time points analyzed (Figure 10).
Figure 10.

Effect of BDNF on ERK1/2 protein expression in Aβ1–40-exposed retina (western blotting).
Quantification was based on densitometry of three different biological samples. Total protein was extracted after 24 hours, 3 days, and 7 days of intravitreal administration. Total ERK1/2 protein expression was normalized against that of β-actin and compared to the PBS (control) group for statistical significance. Data represent three biological replicates with three technical replicates. n = 3 (mean ± SEM). *P < 0.05, **P < 0.01, vs. control group (PBS); #P < 0.05, vs. Aβ1–40 group (one-way analysis of variance with post hoc Bonferroni test). Aβ: Amyloid-beta; BDNF: brain-derived neurotrophic factor; PBS: phosphate-buffered saline; qRT-PCR: quantitative reverse transcription-polymerase chain reaction.
Effects of BDNF on Aβ1–40-induced retinal caspase-3 activation
Caspase-3 gene expression
Significant upregulation of caspase-3 gene expression in the retina was observed in the Aβ1–40 group at 24 hours (2.20-fold, P = 5 × 10–3) and 3 days (1.55-fold, P = 7 × 10–3) post-treatment (Figure 11). However, caspase-3 expression in this group was not significantly different from that in the control group at 7 days post-treatment. In contrast, caspase-3 gene expression in the BDNF group was significantly lower at 24 hours (1.53-fold, P = 3.9 × 10–2) and at 3 days post-treatment (2.42-fold, P = 1 × 10–3) compared with the Aβ1–40 group. Compared with the control group, caspase-3 gene expression in the BDNF group was significantly downregulated at 3 days post-treatment (1.56-fold, P = 5 × 10–2).
Figure 11.

Effects of BDNF on Aβ1–40-induced caspase-3 gene expression in the retina (qRT-PCR).
Total RNA was extracted after 24 hours, 3 days, and 7 days of intravitreal administration. Caspase-3 gene expression was normalized against that of GAPDH and β-Actin and compared with the PBS (control) group for statistical significance. Data represent three biological replicates with three technical replicates, n = 3 (mean ± SEM). Statistically significant difference was calculated using one-way analysis of variance followed by a multiple comparison test using Bonferroni method: 24 hours, F(2, 6) = 15.309, P = 4 × 10–3; 3 days, F(2, 6) = 35.103, P = 4 × 10–4; and 7 days, F(2, 6) = 3.207, P = 0.1. n = 3 (mean ± SEM): *P < 0.05, **P < 0.01, vs. control group (PBS); #P < 0.05, ###P < 0.001, vs. Aβ1–40 group. qRT-PCR: Quantitative reverse transcription-polymerase chain reaction.
Caspase-3 protein expression
Caspase-3 protein expression was 3.25-fold higher in the retina of rats in the Aβ1–40 group than that in the control group (P = 1 × 10–3) at 24 hours post-treatment. At 24 hours post-treatment, Aβ1–40-induced retinal caspase-3 upregulation was lower by 2.23-fold (P = 2 × 10–3) in the BDNF group than that in the Aβ1–40 group. Caspase-3 protein expression in the BDNF-treated retina was comparable to that in the control group at 24 hours post-treatment. There was no significant difference in caspase-3 protein expression in the retinas across all groups at 3 and 7 days post-treatment (Figure 12).
Figure 12.

Effect of BDNF on Aβ1–40-induced caspase-3 protein expression in the retina (western blotting).
Quantification was based on densitometry of three different biological samples. Total protein was extracted after 24 hours, 3 days, and 7 days of intravitreal administration. Total caspase-3 protein expression was normalized against that of β-actin and compared with the PBS (control) group for statistical significance. Data represent three biological replicates with three technical replicates, n = 3 (mean ± SEM). Statistically significant difference was calculated using one-way analysis of variance followed by a multiple comparison test using Bonferroni method: 24 hours, F(2, 6) = 33.798, P = 5 × 10–4; 3 days, F(2, 6) = 3.867, P = 8 × 10–2; and 7 days, F(2, 6) = 2.387, P = 0.1. n = 3 (mean ± SEM): ***P < 0.001, vs. control group (PBS); ##P < 0.01, vs. Aβ1–40 group.
Discussion
In the present study, the effects of intravitreally administered BDNF on Aβ1–40-induced RGC damage via the BDNF-TrkB signaling pathway was studied in rats. The retinoprotective effect of BDNF was correlated with the number of surviving RGCs following exposure to Aβ1–40. Retrograde labelling with Fluoro-Gold showed a significant increase in the RGC density in the group of animals treated with BDNF compared with the group that received Aβ1–40. Treatment with BDNF also abolished Aβ1–40-induced increase in retinal expression of caspase-3 and upregulated expression of TrkB and ERK1/2, as shown by western blot technique. These data were supported by the results of real-time PCR analysis, which demonstrated that BDNF suppressed Aβ1–40-induced transcriptional activity of caspase-3 and increased TrkB and ERK transcriptional activity involved in the regulation of apoptosis.
Formation of the polymerized fibrous form of Aβ1–40, which is potentially pathogenic, was confirmed using SEM. Aβ1–40 polymerisation and subsequent fibril formation were observed 7 days after incubation with parental seeds (fresh Aβ1–40). These data are in line with the stages of Aβ polymerisation described by Brännström et al. (2011, 2018).
We used Fluoro-Gold RGC labelling to determine the effect of BDNF on Aβ1–40-induced changes in RGC survival. Fluoro-Gold neuronal tracer travels retrogradely through axons when injected near the superior colliculi and labels approximately 98% of RGCs (Salinas-Navarro et al., 2009). Hence, RGC labelling by Fluoro-Gold injection near the superior colliculus using a stereotaxic apparatus is one of the best approaches for studying the population of live RGCs with intact axonal transport.
The number of Fluoro-Gold-labeled RGCs in the Aβ1–40 group was significantly lower than that in the control and BDNF groups. These data concur with those from a previous study, which showed significant RGC loss in response to Aβ1–40-induced damage (Mohd Lazaldin et al., 2020). In that study, the BDNF group showed significantly higher RGC count than the Aβ1–40-treated group, with the findings being positively correlated with differences in retinal morphometry between the two groups (Mohd Lazaldin et al., 2020). Furthermore, the results regarding RGC survival in both Aβ1–40- and BDNF groups in this study were in agreement with those from the earlier study, which showed similar changes in optic nerve morphology, retinal cell apoptosis, and retinal oxidative stress (Mohd Lazaldin et al., 2020).
Rats exposed to Aβ1–40 showed reduced TrkB expression at the gene and protein levels in the retina compared with rats in the control and BDNF groups. The expression of TrkB mRNA in the BDNF group was significantly reduced after 24 hours and 3 days of intravitreal injection of AB compared with the control group. However, TrkB protein expression in the same group was similar to the control at both time points. A mismatch between gene and protein expression has often been reported. Significant post-translational modification of proteins may cause detection of smaller quantities of proteins despite upregulation of corresponding genes. Similarly post-transcriptional modification of mRNA may result in detection of its lower quantities compared with protein or the half-life of mRNA and proteins may differ resulting in a mismatch (Greenbaum et al., 2003).
Similar to our observations, Jerónimo-Santos et al. (2015) previously showed that rats exposed to Aβ25–35 and Aβ1–42 had reduced TrkB-FL protein levels. In cultured cortical neurons, sub-lethal concentrations of Aβ1–42 have been shown to interfere with BDNF-induced activation of the ERK1/2 and PI3K/Akt pathways, resulting in the suppression of the activation of several transcription factors, such as CREB and Elk-1, leading to apoptosis. Since the site of interference with ERK1/2 and PI3-K/Akt signaling is downstream of the TrkB receptor, it is likely that the expression of TrkB itself is altered by Aβ1–42 (Tong et al., 2004), and the current study provides evidence supporting this phenomenon.
The loss of TrkB signaling reportedly plays a key role in the pathogenesis of AD, Huntington’s disease, and other neurodegenerative disorders (Gupta et al., 2013; Jin, 2020). Furthermore, we previously showed that intravitreal injections of Aβ1–40 induced retinal neurodegeneration and visual behavioural alterations (Mohd Lazaldin et al., 2020).
In the current study, intravitreal BDNF administration in rats exposed to Aβ1–40 caused an upregulation of TrkB expression at the gene and protein levels, indicating improved TrkB signaling. TrkB receptors are activated by binding to the cognate ligand BDNF and undergo dimerisation with the unliganded monomeric form, which is thought to be in equilibrium with its phosphorylated dimeric state (Massa et al., 2010). This dynamic monomer-dimer equilibrium may be of importance in the regulation of downstream intracellular signaling. Trk receptors possess an intracellular tyrosine kinase domain, which, upon phosphorylation, recruit intermediates in intracellular signaling cascades that affect gene transcription and final cellular responses to BDNF (Berra et al., 1998).
The current study clearly demonstrated that treatment with BDNF counteracts Aβ1–40-induced suppression of TrkB expression, indicating enhanced TrkB signaling. Earlier studies have shown that TrkB stimulation in the retina leads to activated PI3K/Akt and ERK1/2 signaling pathways in RGCs, and particularly, ERK1/2, appears to be of importance for promoting the survival of RGCs (Cheng et al., 2002). In fact, both BDNF and TrkB are involved in mediating RGC protection (Turner et al., 2006), and the loss of TrkB signaling, which is likely to be due to a blockade of axonal transport, leads to preferential RGCs degeneration (Pease et al., 2000; Marvanová et al., 2001; Vrabec and Levin, 2007; Johnson et al., 2009; Ly et al., 2011). Further evidence of this comes from a study that showed increased TrkB labeling behind the optic nerve head region and a decrease in RGC count under the conditions of elevated IOP (Pease et al., 2000). Other studies have also shown that the retinal toxicity induced by Aβ peptides can be prevented by upregulating TrkB signaling (Gupta et al., 2005, 2008). In the current study, higher TrkB expression following treatment with BDNF seemed to enhance RGC survival, as demonstrated using Fluoro-Gold-labeling. Similarly, previous studies have shown that TrkB gene therapy markedly promotes RGC survival after optic nerve axotomy, indicating that BDNF/TrkB axis activation in RGCs can be a useful therapeutic approach in the treatment of several optic neuropathies (Cheng et al., 2002).
Considering that mitogen-activated protein kinase (MAPK) cascades are fundamental downstream pathways of TrkB activation, we also evaluated the effects of BDNF on Aβ1–40-induced changes in ERK1/2 expression at the gene and protein levels in the retina of rats. Aberrant regulation of MAPK cascade contributes to neuronal cell degeneration. ERK1/2, a key MAPK, is commonly associated with cell survival through the activation of various transcription factors, including CREB and mTOR. In the current study, significantly reduced RGC survival was associated not only with reduced TrkB expression but also with the downregulation of ERK1/2 gene and protein expression at all three time points post-treatment in Aβ1–40-treated rats compared with the control and BDNF-treated rats. Since ERK1/2 activation occurs downstream of TrkB, it is logical to conclude that the sequential changes, Aβ1–40 → reduced TrkB expression → reduced ERK1/2, culminate in neurodegenerative changes in the retina. These results are also in agreement with those from a study conducted by Cruz et al. (2018), which demonstrated a significant reduction in ERK1/2 phosphorylation in primary rat hippocampal and cortical neurons with increased Aβ1–42 expression (Cruz et al., 2018).
Importantly, intravitreal BDNF administration in rats exposed to Aβ1–40 caused an upregulation of ERK1/2 gene expression at all three time points post-treatment compared with rats treated with Aβ1–40 only. The corresponding protein expression was upregulated at 3 and 7 days post-treatment, but not at 24 hours post-treatment. The relatively lower protein expression levels at the earlier time point post-treatment may be attributed to the time lag between gene and protein expression. Previously, ERK1/2 activation in RGCs was proposed to be primarily responsible for promoting the survival of RGCs (Gupta et al., 2013). Hence, it can be stated with reasonable certainty that the protective effect of BDNF against Aβ1–40-induced neurodegeneration, as observed in the current study, involves the activation of TrkB and ERK1/2. Although the current study did not evaluate whether ERK1/2 activation occurred following TrKB activation, evidence from literature indicates the likelihood of a sequential activation of BDNF → TrkB → ERK1/2. The translation of TrkB and ERK1/2 activation into the protective effects observed in the current study was clearly demonstrated by increased RGC survival in BDNF-treated retinas. Since TrkB signaling also leads to the activation of pathways other than ERK1/2, the contribution of other activated pathways to enhanced RGC survival could not be excluded in the current study. Nevertheless, it would be logical to conclude that increased ERK1/2 activation following increased TrkB expression at least partially contributes to enhanced RGC survival in response to BDNF treatment.
Since caspases are crucial mediators of programmed cell death (apoptosis), we further corroborated the observations made in relation to the effects of BDNF on TrkB and ERK1/2 expression by studying the expression of caspase-3 at the gene and protein levels. Caspase-3 was chosen because it is a typical hallmark of apoptosis and, as a final executioner caspase, it is indispensable for apoptotic chromatin condensation and DNA fragmentation (Porter and Jänicke, 1999). Rats exposed to Aβ1–40 showed upregulated expression of retinal caspase-3 at the gene and protein levels compared with the control and BDNF groups at all three time points post-treatment, indicating significant neuronal apoptosis. Hence, it can be proposed that in the current study, Aβ1–40-induced neuronal damage in the retina is likely the outcome of reduced TrkB expression → reduced ERK1/2 expression → increased caspase-3 expression. Previous studies have also shown that the suppression of ERK1/2 is associated with increased caspase expression and the resulting apoptosis (Berra et al., 1998). Intravitreal BDNF administration in rats exposed to Aβ1–40 caused downregulation of the expression of caspase-3 at the gene and protein levels. Based on the results of this study, it would be logical to propose that a BDNF-induced increase in TrkB and ERK1/2 expression culminates into reduced caspase-3 activation, leading to the pro-survival effect observed on RGCs. Earlier studies have shown that the activation of the MAPK/ERK and PI3-K/AKT pathways by TrkB/BDNF can block caspase-3 activation (Yuan et al., 2018). ERK1/2 activation is associated with reduced caspase activation and improved cell survival (Holmström et al., 2000; Means et al., 2006). Taken together, these findings demonstrate the potential of BDNF/TrkB axis activation as a therapeutic approach for enhancing neuronal survival and axon growth following injury or in degenerative diseases, such as glaucoma.
Conclusion
Treatment with BDNF prevents Aβ1–40-induced retinal injury by inhibiting Aβ1–40-induced neuronal apoptosis through the downregulation of caspase-3 expression and upregulation of TrkB and ERK1/2 expression. Further studies are needed to fully explore the enhanced efficacy of BDNF as a neuroprotective agent to enable its application in the treatment of AD as well as its ocular manifestation glaucoma. This study used intravitreal administration, which may not be appropriate for patients with glaucoma. Hence, developing appropriate non-invasive formulations of BDNF for future clinical applications is critically important. For example, nanoparticles can not only improve the passage of BDNF through the blood-retinal barrier but may also enhance cellular uptake and protect the drug from degradation. Based on these advantages, nanoparticle formulation may offer an enhanced therapeutic effect of BDNF in treating ocular manifestations of AD.
Acknowledgments:
We acknowledge the administrative and facility support from the Research Management Institute, the Institute of Medical Molecular Biotechnology (IMMB), the Laboratory Animal Care Unit, the Anatomy Department and the Centre for Neuroscience Research (NeuRon) at Universiti Teknologi MARA, Malaysia.
Footnotes
Funding: This study was supported by the Ministry of Higher Education, Government of Malaysia, No. FRGS/2/2014/SG03/UITM/02/2 [UiTM IRMI file No. 600-RMI/FRGS 5/3 (111/2014), to II] and Yayasan Penyelidikan Otak, Minda dan Neurosains Malaysia (YPOMNM), No. YPOMNM/2019-04(2) [UiTM IRMI No. 100-IRMI/PRI 16/6/2 (010/2019), to MAML].
Conflicts of interest: The authors state no conflict of interest.
Author statement: All authors have jointly decided to designate Prof. Igor Iezhitsa to be responsible for decision-making regarding the presence of authors and the order of their presence in the manuscript. Prof Dr Igor Iezhitsa has also been selected by all authors to be responsible for all future communication with the journal regarding this manuscript. We reported part of the data at the Alzheimer’s Association International Conference 2020 (AAIC 2020), Alzheimer’s Association, July 26-30, 2020.
C-Editor: Zhao M; S-Editor: Li CH; L-Editor: Song LP; T-Editor: Jia Y
References
- 1.Arifin WN, Zahiruddin WM. Sample size calculation in animal studies using resource equation approach. Malays J Med Sci. 2017;24:101–105. doi: 10.21315/mjms2017.24.5.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bathina S, Das UN. Brain-derived neurotrophic factor and its clinical implications. Arch Med Sci. 2015;11:1164–1178. doi: 10.5114/aoms.2015.56342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berra E, Diaz-Meco MT, Moscat J. The activation of p38 and apoptosis by the inhibition of Erk is antagonized by the phosphoinositide 3-kinase/Akt pathway. J Biol Chem. 1998;273:10792–10797. doi: 10.1074/jbc.273.17.10792. [DOI] [PubMed] [Google Scholar]
- 4.Brännström K, Ohman A, Olofsson A. Aβpeptide fibrillar architectures controlled by conformational constraints of the monomer. PLoS One. 2011;6:e25157. doi: 10.1371/journal.pone.0025157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brännström K, Islam T, Gharibyan AL, Iakovleva I, Nilsson L, Lee CC, Sandblad L, Pamrén A, Olofsson A. The properties of amyloid-βfibrils are determined by their path of formation. J Mol Biol. 2018;430:1940–1949. doi: 10.1016/j.jmb.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 6.Caprioli J. Glaucoma:a disease of early cellular senescence. Invest Ophthalmol Vis Sci. 2013;54 doi: 10.1167/iovs.13-12716. ORSF60-67. [DOI] [PubMed] [Google Scholar]
- 7.Cheng L, Sapieha P, Kittlerova P, Hauswirth WW, Di Polo A. TrkB gene transfer protects retinal ganglion cells from axotomy-induced death in vivo. J Neurosci. 2002;22:3977–3986. doi: 10.1523/JNEUROSCI.22-10-03977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chitranshi N, Dheer Y, Abbasi M, You Y, Graham SL, Gupta V. Glaucoma pathogenesis and neurotrophins:focus on the molecular and genetic basis for therapeutic prospects. Curr Neuropharmacol. 2018;16:1018–1035. doi: 10.2174/1570159X16666180419121247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiu K, Chan TF, Wu A, Leung IY, So KF, Chang RC. Neurodegeneration of the retina in mouse models of Alzheimer's disease:what can we learn from the retina? Age (Dordr) 2012;34:633–649. doi: 10.1007/s11357-011-9260-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Connor B, Young D, Yan Q, Faull RL, Synek B, Dragunow M. Brain-derived neurotrophic factor is reduced in Alzheimer's disease. Brain Res Mol Brain Res. 1997;49(1-2):71–81. doi: 10.1016/s0169-328x(97)00125-3. [DOI] [PubMed] [Google Scholar]
- 11.Cruz E, Kumar S, Yuan L, Arikkath J, Batra SK. Intracellular amyloid beta expression leads to dysregulation of the mitogen-activated protein kinase and bone morphogenetic protein-2 signaling axis. PLoS One. 2018;13:e0191696. doi: 10.1371/journal.pone.0191696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis BM, Crawley L, Pahlitzsch M, Javaid F, Cordeiro MF. Glaucoma:the retina and beyond. Acta Neuropathol. 2016;132:807–826. doi: 10.1007/s00401-016-1609-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer's disease. Mol Neurodegener. 2019;14:32. doi: 10.1186/s13024-019-0333-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diniz BS, Teixeira AL, Machado-Vieira R, Talib LL, Radanovic M, Gattaz WF, Forlenza OV. Reduced cerebrospinal fluid levels of brain-derived neurotrophic factor is associated with cognitive impairment in late-life major depression. J Gerontol B Psychol Sci Soc Sci. 2014;69:845–851. doi: 10.1093/geronb/gbu096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dong XX, Wang Y, Qin ZH. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin. 2009;30:379–387. doi: 10.1038/aps.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng L, Chen H, Yi J, Troy JB, Zhang HF, Liu X. Long-term protection of retinal ganglion cells and visual function by brain-derived neurotrophic factor in mice with ocular hypertension. Invest Ophthalmol Vis Sci. 2016;57:3793–3802. doi: 10.1167/iovs.16-19825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng L, Puyang Z, Chen H, Liang P, Troy JB, Liu X. Overexpression of brain-derived neurotrophic factor protects large retinal ganglion cells after optic nerve crush in mice. eNeuro. 2017;4 doi: 10.1523/ENEURO.0331-16.2016. ENEURO.0331-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frost SM, Kanagasingam Y, Sohrabi HR, Taddei K, Bateman R, Morris J, Benzinger T, Goate A, Masters CL, Martins RN. Pupil response biomarkers distinguish amyloid precursor protein mutation carriers from non-carriers. Curr Alzheimer Res. 2013;10:790–796. doi: 10.2174/15672050113109990154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghiso JA, Doudevski I, Ritch R, Rostagno AA. Alzheimer's disease and glaucoma:mechanistic similarities and differences. J Glaucoma. 2013;22(Suppl 5):S36–38. doi: 10.1097/IJG.0b013e3182934af6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldstein LE, Muffat JA, Cherny RA, Moir RD, Ericsson MH, Huang X, Mavros C, Coccia JA, Faget KY, Fitch KA, Masters CL, Tanzi RE, Chylack LT, Jr, Bush AI. Cytosolic beta-amyloid deposition and supranuclear cataracts in lenses from people with Alzheimer's disease. Lancet. 2003;361:1258–1265. doi: 10.1016/S0140-6736(03)12981-9. [DOI] [PubMed] [Google Scholar]
- 21.Greenbaum D, Colangelo C, Williams K, Gerstein M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 2003;4:117. doi: 10.1186/gb-2003-4-9-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo L, Moss SE, Alexander RA, Ali RR, Fitzke FW, Cordeiro MF. Retinal ganglion cell apoptosis in glaucoma is related to intraocular pressure and IOP-induced effects on extracellular matrix. Invest Ophthalmol Vis Sci. 2005;46:175–182. doi: 10.1167/iovs.04-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo L, Salt TE, Luong V, Wood N, Cheung W, Maass A, Ferrari G, Russo-Marie F, Sillito AM, Cheetham ME, Moss SE, Fitzke FW, Cordeiro MF. Targeting amyloid-beta in glaucoma treatment. Proc Natl Acad Sci U S A. 2007;104:13444–13449. doi: 10.1073/pnas.0703707104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo L, Cordeiro MF. Assessment of neuroprotection in the retina with DARC. Prog Brain Res. 2008;173:437–450. doi: 10.1016/S0079-6123(08)01130-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gupta VB, Anitha S, Hegde ML, Zecca L, Garruto RM, Ravid R, Shankar SK, Stein R, Shanmugavelu P, Jagannatha Rao KS. Aluminium in Alzheimer's disease:are we still at a crossroad? Cell Mol Life Sci. 2005;62:143–158. doi: 10.1007/s00018-004-4317-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gupta VB, Indi SS, Rao KS. Studies on the role of amino acid stereospecificity in amyloid beta aggregation. J Mol Neurosci. 2008;34:35–43. doi: 10.1007/s12031-007-0070-z. [DOI] [PubMed] [Google Scholar]
- 27.Gupta VK, You Y, Gupta VB, Klistorner A, Graham SL. TrkB receptor signalling:implications in neurodegenerative, psychiatric and proliferative disorders. Int J Mol Sci. 2013;14:10122–10142. doi: 10.3390/ijms140510122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holmström TH, Schmitz I, Söderström TS, Poukkula M, Johnson VL, Chow SC, Krammer PH, Eriksson JE. MAPK/ERK signaling in activated T cells inhibits CD95/Fas-mediated apoptosis downstream of DISC assembly. EMBO J. 2000;19:5418–5428. doi: 10.1093/emboj/19.20.5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Javaid FZ, Brenton J, Guo L, Cordeiro MF. Visual and ocular manifestations of Alzheimer's disease and their use as biomarkers for diagnosis and progression. Front Neurol. 2016;7:55. doi: 10.3389/fneur.2016.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jerónimo-Santos A, Vaz SH, Parreira S, Rapaz-Lérias S, Caetano AP, Buée-Scherrer V, Castrén E, Valente CA, Blum D, Sebastião AM, Diógenes MJ. Dysregulation of TrkB receptors and BDNF function by amyloid-βpeptide is mediated by calpain. Cereb Cortex. 2015;25:3107–3121. doi: 10.1093/cercor/bhu105. [DOI] [PubMed] [Google Scholar]
- 31.Jin W. Regulation of BDNF-TrkB signaling and potential therapeutic strategies for Parkinson's disease. J Clin Med. 2020;9:257. doi: 10.3390/jcm9010257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson EC, Guo Y, Cepurna WO, Morrison JC. Neurotrophin roles in retinal ganglion cell survival:lessons from rat glaucoma models. Exp Eye Res. 2009;88:808–815. doi: 10.1016/j.exer.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kesler A, Vakhapova V, Korczyn AD, Naftaliev E, Neudorfer M. Retinal thickness in patients with mild cognitive impairment and Alzheimer's disease. Clin Neurol Neurosurg. 2011;113:523–526. doi: 10.1016/j.clineuro.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 34.Lafuente MP, Villegas-Pérez MP, Sellés-Navarro I, Mayor-Torroglosa S, Miralles de Imperial J, Vidal-Sanz M. Retinal ganglion cell death after acute retinal ischemia is an ongoing process whose severity and duration depends on the duration of the insult. Neuroscience. 2002;109:157–168. doi: 10.1016/s0306-4522(01)00458-4. [DOI] [PubMed] [Google Scholar]
- 35.Lambuk L, Iezhitsa I, Agarwal R, Agarwal P, Peresypkina A, Pobeda A, Ismail NM. Magnesium acetyltaurate prevents retinal damage and visual impairment in rats through suppression of NMDA-induced upregulation of NF-κB, p53 and AP-1 (c-Jun/c-Fos) Neural Regen Res. 2021;16:2330–2344. doi: 10.4103/1673-5374.310691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li J, Wang YJ, Zhang M, Fang CQ, Zhou HD. Cerebral ischemia aggravates cognitive impairment in a rat model of Alzheimer's disease. Life Sci. 2011;89(3-4):86–92. doi: 10.1016/j.lfs.2011.04.024. [DOI] [PubMed] [Google Scholar]
- 37.Liu B, Rasool S, Yang Z, Glabe CG, Schreiber SS, Ge J, Tan Z. Amyloid-peptide vaccinations reduce {beta}-amyloid plaques but exacerbate vascular deposition and inflammation in the retina of Alzheimer's transgenic mice. Am J Pathol. 2009;175:2099–2110. doi: 10.2353/ajpath.2009.090159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ly T, Gupta N, Weinreb RN, Kaufman PL, Yücel YH. Dendrite plasticity in the lateral geniculate nucleus in primate glaucoma. Vision Res. 2011;51:243–250. doi: 10.1016/j.visres.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mancino R, Martucci A, Cesareo M, Giannini C, Corasaniti MT, Bagetta G, Nucci C. Glaucoma and Alzheimer disease:one age-related neurodegenerative disease of the brain. Curr Neuropharmacol. 2018;16:971–977. doi: 10.2174/1570159X16666171206144045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marvanová M, Lakso M, Pirhonen J, Nawa H, Wong G, Castrén E. The neuroprotective agent memantine induces brain-derived neurotrophic factor and trkB receptor expression in rat brain. Mol Cell Neurosci. 2001;18:247–258. doi: 10.1006/mcne.2001.1027. [DOI] [PubMed] [Google Scholar]
- 41.Massa SM, Yang T, Xie Y, Shi J, Bilgen M, Joyce JN, Nehama D, Rajadas J, Longo FM. Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J Clin Invest. 2010;120:1774–1785. doi: 10.1172/JCI41356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Masuzzo A, Dinet V, Cavanagh C, Mascarelli F, Krantic S. Amyloidosis in retinal neurodegenerative diseases. Front Neurol. 2016;7:127. doi: 10.3389/fneur.2016.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Means JC, Muro I, Clem RJ. Lack of involvement of mitochondrial factors in caspase activation in a Drosophila cell-free system. Cell Death Differ. 2006;13:1222–1234. doi: 10.1038/sj.cdd.4401821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mohd Lazaldin MA, Iezhitsa I, Agarwal R, Bakar NS, Agarwal P, Mohd Ismail N. Time- and dose-related effects of amyloid beta1-40 on retina and optic nerve morphology in rats. Int J Neurosci. 2018;128:952–965. doi: 10.1080/00207454.2018.1446953. [DOI] [PubMed] [Google Scholar]
- 45.Mohd Lazaldin MA, Iezhitsa I, Agarwal R, Bakar NS, Agarwal P, Mohd Ismail N. Neuroprotective effects of brain-derived neurotrophic factor against amyloid beta 1-40-induced retinal and optic nerve damage. Eur J Neurosci. 2020;51:2394–2411. doi: 10.1111/ejn.14662. [DOI] [PubMed] [Google Scholar]
- 46.Moncaster JA, Pineda R, Moir RD, Lu S, Burton MA, Ghosh JG, Ericsson M, Soscia SJ, Mocofanescu A, Folkerth RD, Robb RM, Kuszak JR, Clark JI, Tanzi RE, Hunter DG, Goldstein LE. Alzheimer's disease amyloid-beta links lens and brain pathology in Down syndrome. PLoS One. 2010;5:e10659. doi: 10.1371/journal.pone.0010659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murphy MP, LeVine H., 3rd Alzheimer's disease and the amyloid-beta peptide. J Alzheimers Dis. 2010;19:311–323. doi: 10.3233/JAD-2010-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nadal-Nicolás FM, Salinas-Navarro M, Vidal-Sanz M, Agudo-Barriuso M. Two methods to trace retinal ganglion cells with fluorogold:from the intact optic nerve or by stereotactic injection into the optic tract. Exp Eye Res. 2015;131:12–19. doi: 10.1016/j.exer.2014.12.005. [DOI] [PubMed] [Google Scholar]
- 49.Nita M, Grzybowski A. The role of the reactive oxygen species and oxidative stress in the pathomechanism of the age-related ocular diseases and other pathologies of the anterior and posterior eye segments in adults. Oxid Med Cell Longev 2016. 2016 doi: 10.1155/2016/3164734. 3164734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nor Arfuzir NN, Agarwal R, Iezhitsa I, Agarwal P, Ismail NM. Magnesium acetyltaurate protects against endothelin-1 induced RGC loss by reducing neuroinflammation in Sprague dawley rats. Exp Eye Res. 2020;194:107996. doi: 10.1016/j.exer.2020.107996. [DOI] [PubMed] [Google Scholar]
- 51.Normando EM, Coxon KM, Guo L, Cordeiro MF. Focus on:amyloid beta. Exp Eye Res. 2009;89:446–447. doi: 10.1016/j.exer.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 52.Parnell M, Guo L, Abdi M, Cordeiro MF. Ocular manifestations of Alzheimer's disease in animal models. Int J Alzheimers Dis 2012. 2012 doi: 10.1155/2012/786494. 786494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paxinos G, Watson C. 6th ed. Elsevier; 2007. The rat brain in stereotaxic coordinates: ebook version. [DOI] [PubMed] [Google Scholar]
- 54.Pease ME, McKinnon SJ, Quigley HA, Kerrigan-Baumrind LA, Zack DJ. Obstructed axonal transport of BDNF and its receptor TrkB in experimental glaucoma. Invest Ophthalmol Vis Sci. 2000;41:764–774. [PubMed] [Google Scholar]
- 55.Pease ME, Zack DJ, Berlinicke C, Bloom K, Cone F, Wang Y, Klein RL, Hauswirth WW, Quigley HA. Effect of CNTF on retinal ganglion cell survival in experimental glaucoma. Invest Ophthalmol Vis Sci. 2009;50:2194–2200. doi: 10.1167/iovs.08-3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pescosolido N, Pascarella A, Buomprisco G, Rusciano D. Critical review on the relationship between glaucoma and Alzheimer's disease. Adv Ophthalmol Vis Syst. 2014;1:112–123. [Google Scholar]
- 57.Porter AG, Jänicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- 58.Ratnayaka JA, Serpell LC, Lotery AJ. Dementia of the eye:the role of amyloid beta in retinal degeneration. Eye (Lond) 2015;29:1013–1026. doi: 10.1038/eye.2015.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Resende R, Ferreiro E, Pereira C, Resende de Oliveira C. Neurotoxic effect of oligomeric and fibrillar species of amyloid-beta peptide 1-42:involvement of endoplasmic reticulum calcium release in oligomer-induced cell death. Neuroscience. 2008;155:725–737. doi: 10.1016/j.neuroscience.2008.06.036. [DOI] [PubMed] [Google Scholar]
- 60.Sadigh-Eteghad S, Sabermarouf B, Majdi A, Talebi M, Farhoudi M, Mahmoudi J. Amyloid-beta:a crucial factor in Alzheimer's disease. Med Princ Pract. 2015;24:1–10. doi: 10.1159/000369101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Salinas-Navarro M, Jiménez-López M, Valiente-Soriano FJ, Alarcón-Martínez L, Avilés-Trigueros M, Mayor S, Holmes T, Lund RD, Villegas-Pérez MP, Vidal-Sanz M. Retinal ganglion cell population in adult albino and pigmented mice:a computerized analysis of the entire population and its spatial distribution. Vision Res. 2009;49:637–647. doi: 10.1016/j.visres.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 62.Schlamp CL, Montgomery AD, Mac Nair CE, Schuart C, Willmer DJ, Nickells RW. Evaluation of the percentage of ganglion cells in the ganglion cell layer of the rodent retina. Mol Vis. 2013;19:1387–1396. [PMC free article] [PubMed] [Google Scholar]
- 63.Tapia-Arancibia L, Aliaga E, Silhol M, Arancibia S. New insights into brain BDNF function in normal aging and Alzheimer disease. Brain Res Rev. 2008;59:201–220. doi: 10.1016/j.brainresrev.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 64.The Association for Research in Vision and Ophthalmology- Statement for the Use of Animals in Ophthalmic and Vision Research [Internet] Arvo.org. 2021. [cited 7 July 2021]. Available from: https://www.arvo.org/About/policies/statement-for-the-use-of-animals-in-ophthalmic-and-vision-research/#six .
- 65.Tong L, Balazs R, Thornton PL, Cotman CW. Beta-amyloid peptide at sublethal concentrations downregulates brain-derived neurotrophic factor functions in cultured cortical neurons. J Neurosci. 2004;24(30):6799–6809. doi: 10.1523/JNEUROSCI.5463-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tsolaki F, Gogaki E, Tiganita S, Skatharoudi C, Lopatatzidi C, Topouzis F, Tsolaki M. Alzheimer's disease and primary open-angle glaucoma:is there a connection? Clin Ophthalmol. 2011;5:887–90. doi: 10.2147/OPTH.S22485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Turner BA, Sparrow J, Cai B, Monroe J, Mikawa T, Hempstead BL. TrkB/BDNF signaling regulates photoreceptor progenitor cell fate decisions. Dev Biol. 2006;299:455–465. doi: 10.1016/j.ydbio.2006.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases:a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol. 2009;7:65–74. doi: 10.2174/157015909787602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vrabec JP, Levin LA. The neurobiology of cell death in glaucoma. Eye (Lond) 21 Suppl. 2007;1:S11–S14. doi: 10.1038/sj.eye.6702880. [DOI] [PubMed] [Google Scholar]
- 70.Wang L, Mao X. Role of retinal amyloid-βin neurodegenerative diseases:overlapping mechanisms and emerging clinical applications. Int J Mol Sci. 2021;22:2360. doi: 10.3390/ijms22052360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang YJ, Gao CY, Yang M, Liu XH, Sun Y, Pollard A, Dong XY, Wu XB, Zhong JH, Zhou HD, Zhou XF. Intramuscular delivery of a single chain antibody gene prevents brain Aβdeposition and cognitive impairment in a mouse model of Alzheimer's disease. Brain Behav Immun. 2010;24:1281–1293. doi: 10.1016/j.bbi.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 72.Watts HR, Anderson P, Ma D, Philpott KL, Jen SM, Croucher M, Jen LS, Gentleman SM. Differential effects of amyloid-βpeptide aggregation status on in vivo retinal neurotoxicity. Eye Brain. 2010;2:121–137. doi: 10.2147/eb.s9902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xue Z, Shui M, Lin X, Sun Y, Liu J, Wei C, Wu A, Li T. Role of BDNF/ProBDNF imbalance in postoperative cognitive dysfunction by modulating synaptic plasticity in aged mice. Front. Aging Neurosci. 2022;14:780972. doi: 10.3389/fnagi.2022.780972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yan Z, Liao H, Chen H, Deng S, Jia Y, Deng C, Lin J, Ge J, Zhuo Y. Elevated intraocular pressure induces amyloid-βdeposition and tauopathy in the lateral geniculate nucleus in a monkey model of glaucoma. Invest Ophthalmol Vis Sci. 2017;58:5434–5443. doi: 10.1167/iovs.17-22312. [DOI] [PubMed] [Google Scholar]
- 75.Yuan Y, Ye HQ, Ren QC. Proliferative role of BDNF/TrkB signaling is associated with anoikis resistance in cervical cancer. Oncol Rep. 2018;40:621–634. doi: 10.3892/or.2018.6515. [DOI] [PMC free article] [PubMed] [Google Scholar]