

Abstract
By controlling the proper folding of proteins imported into mitochondria and ensuring crosstalk between the reticulum and mitochondria to modulate intracellular calcium fluxes, Mortalin is a chaperone protein that plays crucial roles in neuronal homeostasis and activity. However, its expression and stability are strongly modified in response to cellular stresses, in particular upon altered oxidative conditions during neurodegeneration. Here, we report and discuss the abundant literature that has highlighted its contribution to the pathophysiology of Parkinson’s disease, as well as its therapeutic and prognostic potential in this still incurable pathology.
Key Words: chaperone, Hspa9, mitochondria, Mortalin, neurodegeneration, oxidative stress, Parkinson’s disease, prognostic and therapeutic potential
Introduction
Mortalin (also called Hspa9, mt-Hsp70, Grp75, Pbp74, Mot-2) is a heat-uninducible member of the 70-kDa heat shock protein (HSP70) family. Its expression has been shown to be regulated by different cellular stresses such as glucose deprivation, oxidative injury, ionizing radiation, and calorie restriction (Kaul et al., 2007). Despite being predominantly confined to the mitochondrial matrix, studies have shown multiple subcellular localizations of Mortalin including endoplasmic reticulum (ER), cytoplasmic vesicles, and cytosol for some immortalized cell lines only, evoking distinct roles and binding partners in each of these compartments (Bhattacharyya et al., 1995; Ran et al., 2000; Londono et al., 2012). The locations and functions of Mortalin are featured in Figure 1 and will be further detailed throughout this review article.
Figure 1.
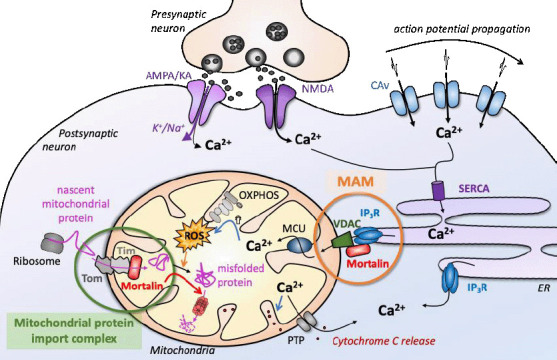
Mortalin functions in neurons under physiological conditions.
Mortalin functions depend on its subcellular localization and protein partners. Its two main functions rely on (1) tethering ER and mitochondrial membranes (MAM, in orange) that mostly enable intracellular calcium storage and fluxes, and (2) controlling the quality of proteins imported into the mitochondrial matrix, in association with the Tom/Tim mitochondrial import complexes (green). AMPA: α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; CAv: voltage-gated calcium channels; ER: endoplasmic reticulum; IP3R: inositol trisphosphate receptor; KA: kainate; MAM: mitochondria-associated endoplasmic reticulum membranes; MCU: mitochondrial calcium uniporter; NMDA: N-methyl-D-aspartate; OXPHOS: oxidative phosphorylation; PTP: permeability transition pore; ROS: reactive oxygen species; SERCA: sarco/endoplasmic reticulum Ca2+-ATPase; Tim: translocase of the inner membrane; Tom: translocase of the outer membrane; VDAC: voltage-dependent anion channel.
Within mitochondria, Mortalin interacts with inner mitochondrial lipid bilayers containing cardiolipin (Dores-Silva et al., 2020). Mortalin has been identified as the only ATPase component of the mitochondrial import complex and is required for translocation of most inner membrane or matrix proteins (Schneider et al., 1994; Brunner et al., 1995; Geissler et al., 2001). In association with Hsp60, Mortalin is especially involved in the detection of damaged or dysfunctional proteins to prevent their accumulation into mitochondria and proceed to either proper folding or degradation (Wadhwa et al., 2005; Rosenzweig et al., 2019). Indeed, non-physiological temperatures or chemical modifications such as oxidation by reactive oxygen species (ROS) can functionally and/or physically alter proteins integrity. Misfolded proteins are prone to form abnormal or irregular interactions due to the exposure of their normally buried hydrophobic parts. By establishing stable interaction with these hydrophobic surfaces, Mortalin can prevent protein concretion and thus toxic accumulation of insoluble aggregates (Havalová et al., 2021).
How Mortalin reaches ER rather than mitochondria is not yet understood. Nonetheless, extramitochondrial Mortalin was found to interact simultaneously with both the ER-membrane protein inositol trisphosphate receptor 1 (IP3R1) and the mitochondrial outer membrane protein voltage-dependent anion channels 1 (VDAC1), thereby functioning as a tethering factor between those 2 compartments (Szabadkai et al., 2006). ER-mitochondria membrane contacts, also called “mitochondria-associated endoplasmic reticulum membranes” (MAMs), are essential for the regulation of intracellular calcium flux, which is particularly important for neurons as it contributes to the spread of the depolarizing signals and to synaptic activity [for review: (Brini et al., 2014)]. Bringing together metabolic and signaling factors that potentiate glucose oxidation, mitochondrial respiration as well as citrate production, MAMs represent bioenergetic hubs that modulate the function of tricarboxylic acid cycle dehydrogenases and mitochondrial respiration (Theurey and Rieusset, 2017), which are systematically hampered in neurodegenerative disorders (Johri and Chandra, 2021).
Considering its different functions, Mortalin appears as a prime actor in cellular homeostasis, metabolic modulation, and oxidative stress sensing. Hence, deregulation of Mortalin activity in humans underlies the pathologies of several diseases including cancer, diabetes, cardiovascular diseases, atherosclerosis, stroke, inflammatory disorders, chronic fatigue syndrome, asthma, and neurodegenerative diseases [see Flachbartova and Kovacech (2013) for a general review]. Mutations in the gene encoding Mortalin are responsible for the development of EVEN-PLUS syndrome, a rare congenital pathology characterized by epiphyseal and vertebral dysplasia and abnormalities of the ears and nose (Royer-Bertrand et al., 2015). However, there is little evidence that genetic alterations of the Mortalin gene are involved in cancer, metabolic or neurodegenerative pathologies, for which we rather find variations of its post-translational modifications (Nitika et al., 2020; Havalová et al., 2021) and expression level (see Part Modulating Mortalin levels in neurons).
Search Strategy and Selection Criteria
Studies cited in this review were searched on PubMed (NCBI) and Google databases, using the following keywords: Hspa9, Mortalin, Grp75, Parkinson disease, synuclein, DJ1, mitochondria, MPTP, 6-OHDA, oxidative stress. We focused on publications from the last 5 years and only returned to older publications when necessary (first discovery or analysis). These searches were performed between September 2021 and April 2022.
Evidence of Mortalin Implication in Parkinson’s Disease Pathophysiology
Among other high-energy demanding cells, neurons are particularly sensitive to misfolded protein accumulation, ROS overproduction, and calcium fluxes impairment. Neurons are also poorly renewed in adult organisms. Since Mortalin is implicated in each of those hallmarks of archetypal neurodegeneration, it is not surprising that changes in Mortalin activity or expression level modify neuronal outcomes in several models of neurodegeneration, including glutamate or amyloid β mediated toxicity (Qu et al., 2011; Flachbartova and Kovacech, 2013; Park et al., 2014; Sharma and Kaur, 2018). Nevertheless, Mortalin plays a particular role in the specific case of Parkinson’s disease (PD), and this is the object of the present bibliographic analysis.
PD is a chronic neurodegenerative pathology that first affects dopaminergic (DA) neurons of the nigrostriatal pathway, before spreading to other regions of the central nervous system. The alteration of this neuronal pathway causes the typical motor symptoms of the disease: tremor, bradykinesia, and rigidity. PD occurs sporadically in over 90% of cases, resulting from a combination of genetic predispositions and environmental factors, including age and intoxication with mitochondrial toxins such as rotenone or paraquat (Elbaz and Moisan, 2008). However, some cases have an autosomal monogenic origin, with mutations mainly in the genes of α-synuclein, parkin, PTEN-induced kinase 1 (PINK1), deglycase J-1 (DJ-1), or leucine-rich repeat kinase 2 (LRRK2). Interestingly, these proteins are all directly or indirectly linked to mitochondria [for a broad review of monogenic cases of PD see (Jia et al., 2022); for a more in-depth review of PD genetics see (Lanore et al., 2022)]. Likewise, in both sporadic and monogenic cases of PD, DA neurons display high oxidative stress and mitochondrial dysfunction [for review about mitochondria dysfunctions in PD see (Borsche et al., 2021)] together with the neurotoxic agglomeration of α-synuclein in cellular inclusions of protein aggregates called Lewy bodies [for review about α-synuclein pathophysiology see (Ottolini et al., 2017)].
While rare Mortalin variants were only identified in a few PD patients for whom it has been considered the main contributor to the disease (Burbulla et al., 2010; Larsen et al., 2018), these variants were never recognized as monogenic familial origins of PD. Screening of additional cohorts of PD patients did not uncover any Mortalin mutation significantly associated with PD (Freimann et al., 2013; Chung et al., 2017). Hence, mutations in the HSPA9 gene are not usually considered to be among the major causes of familial PD. Nonetheless, several studies describe altered levels of Mortalin in the brain and blood of PD patients. Mortalin being a chaperone protein, which is crucial for proper mitochondrial function, and given the abundant literature on this topic, it is relevant to take an interest in its contribution in the context of PD.
Low Mortalin amounts were first revealed in post-mortem brains of PD patients and people suffering from Parkinsonism due to manganese exposure (Jin et al., 2006; Cook, 2014). Manganese and rotenone, which are known to be environmental factors to PD, indeed directly target mitochondria and cause a reduction of Mortalin amount in the mitochondrial matrix (Cook, 2014; Ferré et al., 2021). Hence, Mortalin depletion in the brain might be an aggravating factor in PD as a consequence of exposure to environmental toxicants.
As a matter of fact, it has also been shown that Mortalin interacts with proteins associated with familial forms of PD. In 2011, Rakovic et al. identified Mortalin (Grp75) as a binding partner of Pink1, using a tandem affinity purification assay. PINK1 is involved in mitophagy, the mitochondrial quality control system, which is strongly impaired in all forms of PD. Direct interactions between Mortalin and both α-synuclein and DJ-1 were initially described, albeit without any functional enlightenment (Jin et al., 2007). DJ-1, like Mortalin, is involved in the response to oxidative stress and is thought to participate in α-synuclein aggregation in Lewy bodies and DA neurons degeneration (Bonifati et al., 2003; Hijioka et al., 2017; Havalová et al., 2021). In line with this, agglomerated α-synuclein is able to bind to Mortalin (Jin et al., 2007), likely impeding its normal function and leading to its proteasomal degradation. This might somewhat explain α-synuclein toxicity towards mitochondria (Grassi et al., 2018). Taken together, these in vitro data suggest that Mortalin could be a relevant actor in PD pathophysiology, which was further established using animal models of PD (see Part Insights about Mortalin roles in PD using animal models).
Besides, it was shown that dopamine metabolites can form adducts with Mortalin (Van Laar et al. 2009). Dopamine metabolites in general are highly neurotoxic and can form adducts with many proteins, disrupting their functions (Segura-Aguilar et al. 2014). This is why dopamine metabolism has to be strictly regulated in order to avoid triggering a vicious cycle of oxidative stress, protein damage, and mitochondrial dysfunction. Although dopamine-Mortalin adducts toxicity was not further examined, this might explain both the specific vulnerability of DA neurons in PD and other forms of Parkinsonism, together with the strong implication of Mortalin in this particular neurodegenerative pathology.
Dissecting the Role of Mortalin in Parkinson’s Disease Animal Models
Various animal models have been designed in mammals (mostly rodents but also small monkeys and non-human primates), flies or zebrafish, in order to reproduce and study the pathologic hallmarks of PD [for review: (Chia et al., 2020)]. Some models are obtained using toxicants such as paraquat and rotenone, which disrupt the mitochondria and generate oxidative stress. Among them, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and 6- Hydroxydopamine (6-OHDA) are more widely used due to their specificity for the dopamine transporter, which ensures effective DA neurons targeting while sparing the other neurons (Simola et al., 2007; Mustapha and Mat Taib, 2021). These models have been used for decades to study the molecular and cellular mechanisms of PD, and some of them highlighted a major involvement of Mortalin.
Shen et al. (2005) used MPTP intoxication in mice to study the role of Mortalin in PD. MPTP is a lipophilic drug, which can cross the blood-brain barrier after systemic administration in mice. It is then metabolized by the MAO-B enzyme into MPP+, which is lipophobic and enters the DA neurons through the dopamine transporter DAT. MPP+ inhibits the mitochondrial complex I, which causes oxidative stress, the release of cytochrome C, and apoptosis, similarly to oligomerized α-synuclein (Grassi et al., 2018). In their study, Shen et al. found that a pre-treatment with geldanamycin, which elevates Mortalin levels in the central nervous system (CNS), mitigates DA denervation in the striatum following MPTP intoxication. However, MPTP treatment does not induce changes in CNS Mortalin by itself, as opposed to what Chiasserini et al. (2011) observed using the 6-OHDA model in rats. This could be explained by the fact that MPTP causes a less severe phenotype than 6-OHDA. The 6-OHDA rat model consists of a stereotaxic injection of the drug 6-OHDA directly into one of the rat’s dorsal striata. Based on proteomics analyses, Chiasserini et al. indeed observed a 25% decrease in Mortalin expression in the 6-OHDA rats’ striata when compared to controls, which was recently confirmed in vitro by Magalingam et al. (2022). Furthermore, using the Mortalin inhibitor MKT-077 on ex vivo brain slices, Chiasserini et al. demonstrated that Mortalin inhibition decreases field potential amplitude in the striatum and depolarizes the medium spiny neurons of the striatum. Those effects were enhanced in 6-OHDA injected rats. Hence, Mortalin might be important for maintaining the intracellular homeostasis required for the normal electrophysiological activity of neurons. Indeed, mitochondria, being calcium storage compartments, help regulate calcium homeostasis (see Part The crucial role of Mortalin at the ER-mitochondria contact sites). Elevated intracellular calcium levels due to mitochondrial dysfunction (following mortalin inhibition) could result in depolarization and abnormal excitability in medium spiny neurons, as what Chiasserini et al. observed. This could be extrapolated to cause excitotoxicity in the already vulnerable DA neurons, in the context of PD.
Zhu et al. (2013) used Mortalin interfering RNA to study its role in Drosophila neurons. They found that silencing Mortalin expression in either all neurons or DA neurons specifically was lethal to larvae, whereas silencing in muscle cells only did not affect viability. A milder reduction of Mortalin levels was not lethal but reduced lifespan, and induced abnormal body posture and impaired locomotion, reminiscent of bradykinesia and postural defects observed in PD patients. At the cellular level, an increased proportion of small round mitochondria was observed, associated with increased autophagy and mitophagy. Thus, lowering Mortalin function in Drosophila is sufficient to motor symptoms reminiscent of PD.
Altogether, data obtained from different animal models of PD suggest that neuronal degeneration is concomitant with a drop in Mortalin expression, analogous to what is observed in patients. Conversely, Mortalin overexpression in animal models is protective, suggesting that Mortalin might hamper the molecular mechanisms of DA degeneration.
Mortalin and the Molecular Mechanisms of Dopaminergic Neurons Degeneration
Loss of Mortalin causes characteristic changes in mitochondria morphology in various model organisms highlighting the importance of Mortalin in cellular homeostasis. Mortalin expression variations are associated with the preservation of normal cellular functioning, through anti-apoptotic and pro-proliferative properties as well as modulation of mitochondrial dynamics and functions. Tight control of Mortalin expression seems to be critical overall for the cellular fate and more particularly to mitochondrial homeostasis. Figure 2 recapitulates the following descriptions.
Figure 2.

Implication of Mortalin in the degenerative DA neuron during PD.
Intrinsic dopamine metabolites and synuclein aggregation generate a ROS overload in the DA neurons of PD patients, which is further augmented with the inhibition of the mitochondrial electron transport chain (OXPHOS). Dopamine metabolites and synuclein aggregates also trigger Mortalin degradation, which amplifies the impact of oxidative stress in neuronal homeostasis. C.I: Mitochondrial respiratory complex I; DAT: dopamine transporter; L-Dopa: levodopa; MAM: mitochondria-associated endoplasmic reticulum membranes; OXPHOS: oxidative phosphorylation; ROS: reactive oxygen species; Tyr: tyrosine.
Mortalin controls mitochondrial respiration and hampers reactive oxygen species overload
Dysfunctions of the mitochondrial respiratory chain are observed in all forms of PD, in particular the mitochondrial complex I, which is inhibited by both α-synuclein oligomers and environmental toxins (Haelterman et al., 2014). Interestingly, Mortalin plays a key role in maintaining the functional integrity of mitochondria respiratory chain by limiting electron leakage from complex I, probably through both its interactions with Tid1 (Hsp40) and Mouse Double Minute 2 (Arena et al., 2018) and its role in DJ-1 translocation to mitochondria (Zhou et al., 2020). Overexpression of Mortalin in catecholamine producing pheochromocytoma-derived (PC12) cells was found to be cytoprotective from metabolic stress such as glucose deprivation (Liu et al., 2005). Actually, a high Mortalin level was shown to stabilize respiratory chain complexes and subsequently preserve mitochondrial bioenergetic function without inducing excessive production of ROS. High Mortalin levels were linked with DJ-1 translocation to mitochondria, where DJ-1 interacts with proteins from the respiratory complex I (ND1 and NDUFA4) and stabilizes its activity while lowering ROS production (Zhou et al., 2020). Liu et al. (2005) also suggest that Mortalin interaction with antioxidant defenses may participate in the regulation of oxidative stress upon hypoglycemic condition. Two studies confirmed this hypothesis after ischemic brain injury or ischemia/reperfusion in neonatal rat cardiac myocytes (Williamson et al., 2008; Xu et al., 2009). In these models, Mortalin overexpression induced a reduction in infarct volume, with decreased ROS levels associated with a preserved function of the respiratory chain and increased ATP production and cell viability. The authors also observed a specific increase of manganese superoxide dismutase expression, the main mitochondrial antioxidant factor, hence comforting better management of ROS.
In DA neurons, dopamine degradation is a highly toxic process, which generates toxic metabolites and ROS (Segura-Aguilar et al., 2014). Under physiological conditions, dopamine catabolism mostly occurs inside vesicles; however, the basal level of oxidative stress in DA neurons is always quite high. In the PD context, oligomeric aggregates of α-synuclein can create pores in intracellular membranes, notably causing dopamine leakage (Ottolini et al., 2017). Conversely, high oxidative stress promotes α-synuclein aggregation (Ischiropoulos and Beckman, 2003), creating a vicious circle of accumulative oxidative stress in degenerative DA neurons, which further affects nucleic acid, lipids, and protein stability, and thus neuronal activity and defenses (Zhao et al., 2017). As a matter of fact, Mortalin was identified as a key oxidation-sensitive cellular chaperone (Choi et al., 2004). Upon induced ROS overproduction or H2O2 treatment, Mortalin functional knockdown leads to mitochondrial membrane potential collapse and ROS accumulation, which is partially rescued by Parkin overexpression (Burbulla et al., 2010). Indeed, ROS overload promotes the translocation of Parkin to mitochondria where it interacts with Mortalin, suggesting a common role in the quality control of these organelles (Yang et al., 2011). This hypothesis was confirmed a few years later with the demonstration that Pink1 and Parkin both lose their ability to rescue the Mortalin deficiency phenotype in the presence of inhibitors of autophagic machinery (Burbulla et al., 2014). Using a model of PD disease in Drosophila, Zhu et al. (2013) also reported reduced ATP levels and accumulation of fragmented mitochondria upon Mortalin homolog (Hsc70-5) knockdown in DA neurons. Altogether, those studies suggest a crucial role of Mortalin in sensing ROS accumulation and engagement of the Pink1/Parkin pathway of mitochondria quality control.
Mortalin modulates mitochondrial dynamics impairment during neurodegeneration
In PD, such as in most degenerative diseases, mitochondria functional damage comes along with impairment of mitochondrial morphology and biogenesis. Mitochondria are not isolated organelles but rather form a dynamic network whose interconnectedness relies on their ability to fuse and split to respond rapidly to fluctuations in metabolic demand, based on cellular physiological state. These continuous mitochondrial morphology changes, called mitochondrial dynamics, control the cellular management of stresses, allow quick adjustments to energy needs and direct the survival/death decision through the recruitment of the Bcl-2 pro-apoptotic factors at the outer membrane and the release of cytochrome C from cristae intermembrane space [see Adebayo et al. (2021) for a recent review about mitochondrial dynamics implication in cellular homeostasis]. Alterations of fission/fusion balance have major deleterious effects on neurons, and neuronal degeneration is associated with mitochondrial dynamics impairments for the majority of neurodegenerative diseases [see Bertholet et al. (2016) for a review on the mitochondrial dynamics and neurodegenerative diseases].
The implication of Mortalin on mitochondrial dynamics was first highlighted with the Mortalin homolog in yeast, SSC1, in which loss of function was associated with mitochondria aggregation (Kawai et al., 2001). However, ultrastructural features of cristae and tubular thread-like structure of the mitochondria were still maintained in yeast. Contrariwise, we and others demonstrated that modifications of Mortalin expression level have direct effects on the tubulation of the mitochondrial network, through a regulation of mitochondrial fission (Yang et al., 2011; Zhu et al., 2013; Park et al., 2014; Liu et al., 2015; Ferré et al., 2021). In Drosophila, Mortalin downregulation was shown to decrease mitochondria density and ATP supply at the synapses (Zhu et al., 2013). Using murine primary neuronal cultures, we also observed that interference with Mortalin expression induced a fragmented mitochondrial network and poor ability to manage oxidative stress (Ferré et al., 2021). Oppositely, overexpression of Mortalin induced over-filamentation of mitochondria and conferred protection against axonal degeneration mediated by rotenone exposure. By which molecular mechanisms Mortalin affects mitochondrial morphology remains unclear, but modulation of Mortalin expression comes along with modification of Drp1 (dynamin-related protein 1) activity, the main actor of mitochondrial fission (Park et al., 2014; Ferré et al., 2021). However, the fusogenic functions of OPA1, which also shapes cristae, might also rely on Mortalin. An imbalanced stoichiometry of mtHsp40 and Mortalin in the mitochondrial matrix was indeed shown to provoke Opa1 cleavage, which both impedes proper Opa1 functioning and stimulates the fission activity of Drp1, leading to mitochondrial fragmentation (Lee et al., 2015). In line with this, both Drp1 and Opa1 might participate in Mortalin depletion-mediated mitochondria fragmentation in Drosophila since either overexpression of Opa1 or down-regulation of Drp1 was able to rescue mitochondrial phenotype (Zhu et al., 2013). Hence, Mortalin is not only involved in the quality control of mitochondrial proteins but also represents a major administrator of mitochondrial morphology and proper functioning.
Mortalin is required for endoplasmic reticulum and mitochondria membrane tethering
Neuronal activity, DA neurons tonic activity, is coupled to a large Ca2+ entry, which is amplified upon oxidative stress. Induced overload of intracellular Ca2+ is firstly stored in the ER and then partially transferred to mitochondria through the MAMs junctions (Marchi et al., 2014). MAMs formation and mitochondrial Ca2+ uptake strongly rely on mitochondrial dynamics (Friedman et al., 2011; Marchi et al., 2014) and are altered in most neurodegenerative pathologies, especially PD [see Johri and Chandra (2021) for a general review of MAMs and neurodegeneration]. This could both disturb intracellular calcium signaling pathways, including metabolism and transcription, and modulate mitochondrial bioenergetics capacities (Brini et al., 2014).
Interestingly, extramitochondrial Mortalin was shown to be mostly associated with ER, acting as a “bridge” through covalent interactions with proteins from both ER (IP3R) and outer mitochondrial (VDAC1) membranes (Szabadkai et al., 2006). Mortalin is crucial to maintain the association between ER and mitochondria since knockdown of the chaperone was associated with decreased contact sites numbers (Honrath et al., 2017). Conversely, Mortalin overexpression decreases steady-state ER Ca2+ concentration (Szabadkai et al., 2006), suggesting a role of Mortalin in CA2+ transfer flux from ER to mitochondria. However, Honrath et al. (2017) observed that overexpression of Mortalin sensitized cells to oxidative stress by increasing contacts between ER and mitochondria, while decreased expression of Mortalin was found to be protective against glutamate-induced cell death, by disconnecting both organelles. They suggest that Mortalin silencing might modulate energetic metabolism with decreased mitochondrial respiration rate, and thus limited mitochondrial ROS formation. Variation in Mortalin expression is here thought to modify ER-mitochondria coupling, thereby maintaining mitochondrial calcium homeostasis. In this context, mitochondrial dysfunctions were attributed to an upregulation of Mortalin expression, which appears quite inconsistent with the assumption of a protective role of Mortalin in degenerative neurons. Actually, Mortalin was recently shown to be locally translated at the axon injury site following axotomy, which increases ER-mitochondria tethering and promotes axon regeneration (Lee et al., 2019). Hence, the precise role of Mortalin expression and MAMs function in degenerative neurons remains partly ambiguous.
The implication of extramitochondrial Mortalin in the specific context of PD was supported by recent demonstrations of the involvement of DJ-1 and α-synuclein in MAMs stabilization and function (Guardia-Laguarta et al., 2015; Liu et al., 2019; Basso et al., 2020; Erustes et al., 2021). α-Synuclein was previously shown to be located at mitochondria from where it would modulate calcium homeostasis by enhancing ER-mitochondria contact (Calì et al., 2012). Nevertheless, wild-type α-synuclein is naturally and specifically located at MAMs, while mutant α-synuclein (familial forms of PD) and aggregates disconnected from the ER-mitochondria contact sites (Guardia-Laguarta et al., 2015). Moreover, the overexpression of PD-associated α-synuclein mutants was found to disturb the IP3R-Mortalin association thereby decreasing the number of MAMs and thus Ca2+ trafficking towards mitochondria (Erustes et al., 2021). Besides, Liu et al. (2019) established the association of DJ-1 with the IP3R-Mortalin-VDAC-1 complex. DJ-1 knockout impaired the interactions between both organelles, as well as IP3R homeostasis and mitochondrial Ca2+ uptake. The authors suggest that DJ-1 might stabilize the calcium channel formed by IP3R and VDAC1, thereby regulating mitochondria and neuronal homeostasis and function, which was confirmed shortly after by Basso et al. (2020). Lastly, DJ-1 translocation to mitochondria was found to be dependent on its association with Mortalin (Zhou et al., 2020).
DJ-1 translocation to mitochondria and α-synuclein aggregation being key features of PD, the involvement of Mortalin in ER-mitochondria tethering might be of crucial importance in the course of PD pathophysiology.
Targeting Mortalin as a Therapeutic Approach to Parkinson’s Disease
Undoubtedly, Mortalin is essential for mitochondrial function and neuronal homeostasis. At the same time, Mortalin seems to be gradually depleted in the CNS over the course of PD. Further studies will be needed to understand exactly how and why this happens, but preserving Mortalin function in the brain could be a relevant research axis in order to find a way to slow the progression of PD.
Modulating Mortalin levels in neurons
Although Mortalin is not a heat-inducible chaperone, its expression is upregulated in response to stress, especially ROS overproduction. Several studies (see above) pointed out the drop in Mortalin expression level in the neurons of patients experiencing neurodegenerative diseases. Hence, it is tempting to speculate that increasing Mortalin expression in the brains of PD patients would improve neuronal outcomes.
It has been shown over the past decades that manipulating Mortalin expression alters survival in various organisms. The deletion of Mortalin homolog in yeast (SSC1) was shown to be lethal, while the proliferation of immortal cells was halted (Craig et al., 1987). Inversely, overexpression of Mortalin is sufficient to extend the lifespan of C. Elegans and human fibroblasts (Yokoyama et al., 2002; Kaul et al., 2003). In neuronal and non-neuronal cells lines, modulation of Mortalin expression does not affect their morphology, division, or lethality, but overexpression protects them from toxin-mediated toxicity, while knock-down of Mortalin gene enhances cellular damage (Jin et al., 2006; Burbulla et al., 2010; Zhu et al., 2013; Liu et al., 2015). Likewise, using rodent primary neuronal cultures, we recently demonstrated the perfect correlation between Mortalin expression level and mitochondrial function/morphology, ROS accumulation, and neuronal sensitivity to PD-related toxins such as rotenone (Ferré et al., 2021).
Immunomodulation through Mortalin expression
As with most neurodegenerative diseases, PD occurs in a chronic inflammatory context. Neuroinflammation even participates in neuronal degeneration, with both activation of resident cells (microglia, astrocytes, tissue-resident macrophages) and a massive infiltration of peripheric innate and adaptive immune cells monocytes, macrophages, dendritic cells, lymphocytes; for review about the neuroinflammation in PD context (Weiss et al., 2022). Interestingly, both CNS-resident and infiltrated neuroinflammatory cells also exhibit mitochondrial and metabolic alterations that affect their relationships with neighboring neurons (Rose et al., 2017). The CNS being a confined environment with highly energetic demands, this results in competition for nutrients and oxygen that are essential for their metabolic activity and progressive nutrient starvation and oxidative stress (Szepesi et al., 2018). Hence, modulation of mitochondrial functions and ROS buffering in immune cells during PD might be beneficial to restore healthy reciprocal interactions with neurons and is currently under investigation for therapeutic purposes.
Targeting Mortalin expression in neuroimmune cells during PD might therefore be an interesting avenue. However, little literature relays the functions of Mortalin in immune cells. In a few papers, Mortalin has been shown to participate in the cellular elimination of complement C9 oligomers through the release of extracellular vesicles by ectocytosis (a variant of exocytosis) in response to complement-mediated cytotoxicity, which triggers a translocation of Mortalin from mitochondria towards the plasma membrane (Pilzer and Fishelson, 2005; Mazkereth et al., 2016). This is of particular interest, since α-synuclein aggregation was recently found to induce complement-mediated toxicity in a neuroblastoma cell line (Gregersen et al., 2021), and might require a specific and dedicated study. On another hand, Mortalin over-expression was shown to reduce the pro-inflammatory activation of astrocytes, microglia, and macrophages in different CNS pathologies, mitigating neuronal death (Voloboueva et al., 2013; Joseph et al., 2017; Priyanka et al., 2020). Importantly, different studies pointed out a crucial role of T-lymphocytes in the progression of PD reviewed in (Baird et al., 2019)]. Despite the absence of studies analyzing the specific role of Mortalin in those cells, there is growing evidence that metabolic adaptation of T cells infiltrating the CNS might be a key pathogenic event in most neurological and neurodegenerative disorders, in which ER-mitochondria tethering sites appear as immunometabolic hubs (Bantug et al., 2018; Fessler and Angiari, 2021). Overall, manipulation of Mortalin expression and function in neuroimmune cells could be of definite therapeutic interest for PD.
Mortalin level detection in circulating fluids: a prognostic marker for Parkinson’s disease?
High expression of Mortalin was associated with a broad spectrum of cancers, poor patient prognosis, and resistance to therapy (Cui et al., 2017; Krawczyk et al., 2018; Cheng et al., 2019; Li et al., 2019; Starenki et al., 2019; Xu et al., 2020; Kabakov and Gabai, 2021; Zhang et al., 2021; Meng et al., 2022). Interestingly, Mortalin itself or antibodies against Mortalin were also found to be elevated in the circulating fluids of patients with several types of tumors, in sera or inside plasmatic vesicles (Lu et al., 2015; Huang et al., 2019; Kabakov and Gabai, 2021). This led to the idea that Mortalin could be a serological biomarker of cancer prognosis and progression (Xu et al., 2019; Rai et al., 2021).
Cook (2014) detected a positive correlation between the presence of antibodies directed against Mortalin in PD patients’ blood and the severity of the disease. Interestingly, a pilot study highlighted the presence of Mortalin in the sera of PD patients (Singh et al., 2018). In this study, Singh et al. found a 40% decrease in Mortalin amount in serum samples of PD patients compared to those of control healthy individuals, together with a slight but significant increase in seric α-synuclein concentration. This study is quite preliminary and would require larger cohorts and a comparison of different progression stages of the disease, however, it suggests the potential use of seric Mortalin detection as a biomarker for PD diagnosis. Hence, more usefully than a postmortem observation, Mortalin expression and immunogenicity might represent potential biomarkers of PD progression. Given the recent development of the quick ELISA technic for Mortalin detection in body fluids (Garg et al., 2019), this approach might present a fascinating opportunity for further amelioration of PD patients’ medical care.
Conclusion
By comparing the clinical analyzes carried out on PD patients with the studies using animal and cellular models of the pathology, it appears that Mortalin is not a priori a susceptibility gene for PD but controls most of the cellular pathways involved in the degeneration of dopaminergic neurons. Accordingly, Mortalin is a hub for sensing mitochondrial stress, maintenance of calcium flux, dopamine catabolism, and synuclein aggregates toxicities. Moreover, its expression levels correlate with neuronal susceptibility since degenerative neurons display low Mortalin levels while overexpression of Mortalin would provide neuronal protection. Furthermore, the quantity of Mortalin found in body fluids (cerebrospinal fluid, serum) would coincide inversely with the pathological evolution, suggesting a prognostic and/or therapeutic potential. Mortalin is also a key actor and biomarker for several cancers. Therefore, it is difficult to speculate on a general enhancement of Mortalin as a therapeutical approach. However, various studies including ours, suggest that specific and controllable overexpression of Mortalin in neurons would overcome most of the neuronal dysfunctions at causing neurologic symptoms in this still incurable pathology.
Additional file: Open peer review report 1 (92.4KB, pdf) .
Acknowledgments:
Authors thank Dr. Anne S. Dejean for critical reading of the manuscript and insightful editorial comments.
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
Availability of data and materials: All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open peer reviewer: Patricia Gomez-Suaga, University of Extremadura, Spain.
P-Reviewer: Gomez-Suaga P; C-Editors: Zhao M, Liu WJ, Qiu Y; T-Editor: Jia Y
References
- 1.Adebayo M, Singh S, Singh AP, Dasgupta S. Mitochondrial fusion and fission:The fine-tune balance for cellular homeostasis. FASEB J. 2021;35:e21620. doi: 10.1096/fj.202100067R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arena G, Cissé MY, Pyrdziak S, Chatre L, Riscal R, Fuentes M, Arnold JJ, Kastner M, Gayte L, Bertrand-Gaday C, Nay K, Angebault-Prouteau C, Murray K, Chabi B, Koechlin-Ramonatxo C, Orsetti B, Vincent C, Casas F, Marine JC, Etienne-Manneville S, et al. Mitochondrial MDM2 regulates respiratory complex I activity independently of p53. Mol Cell. 2018;69:594–609. doi: 10.1016/j.molcel.2018.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baird JK, Bourdette D, Meshul CK, Quinn JF. The key role of T cells in Parkinson's disease pathogenesis and therapy. Parkinsonism Relat Disord. 2019;60:25–31. doi: 10.1016/j.parkreldis.2018.10.029. [DOI] [PubMed] [Google Scholar]
- 4.Bantug GR, Fischer M, Grählert J, Balmer ML, Unterstab G, Develioglu L, Steiner R, Zhang L, Costa ASH, Gubser PM, Burgener AV, Sauder U, Löliger J, Belle R, Dimeloe S, Lötscher J, Jauch A, Recher M, Hönger G, Hall MN, et al. Mitochondria-endoplasmic reticulum contact sites function as immunometabolic hubs that orchestrate the rapid recall response of memory CD8. Immunity. 2018;48:542–555. doi: 10.1016/j.immuni.2018.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basso V, Marchesan E, Ziviani E. A trio has turned into a quartet:DJ-1 interacts with the IP3R-Grp75-VDAC complex to control ER-mitochondria interaction. Cell Calcium. 2020;87:102186. doi: 10.1016/j.ceca.2020.102186. [DOI] [PubMed] [Google Scholar]
- 6.Bertholet AM, Delerue T, Millet AM, Moulis MF, David C, Daloyau M, Arnaune-Pelloquin L, Davezac N, Mils V, Miquel MC, Rojo M, Belenguer P. Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol Dis. 2016;90:3–19. doi: 10.1016/j.nbd.2015.10.011. [DOI] [PubMed] [Google Scholar]
- 7.Bhattacharyya T, Karnezis AN, Murphy SP, Hoang T, Freeman BC, Phillips B, Morimoto RI. Cloning and subcellular localization of human mitochondrial hsp70. J Biol Chem. 1995;270:1705–1710. doi: 10.1074/jbc.270.4.1705. [DOI] [PubMed] [Google Scholar]
- 8.Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 9.Borsche M, Pereira SL, Klein C, Grünewald A. Mitochondria and Parkinson's disease:clinical, molecular, and translational aspects. J Parkinsons Dis. 2021;11:45–60. doi: 10.3233/JPD-201981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brini M, Cali T, Ottolini D, Carafoli E. Neuronal calcium signaling:function and dysfunction. Cell Mol Life Sci. 2014;71:2787–2814. doi: 10.1007/s00018-013-1550-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brunner M, Schneider HC, Lill R, Neupert W. Dissection of protein translocation across the mitochondrial outer and inner membranes. Cold Spring Harb Symp Quant Biol. 1995;60:619–627. doi: 10.1101/sqb.1995.060.01.066. [DOI] [PubMed] [Google Scholar]
- 12.Burbulla LF, Schelling C, Kato H, Rapaport D, Woitalla D, Schiesling C, Schulte C, Sharma M, Illig T, Bauer P, Jung S, Nordheim A, Schöls L, Riess O, Krüger R. Dissecting the role of the mitochondrial chaperone mortalin in Parkinson's disease:functional impact of disease-related variants on mitochondrial homeostasis. Hum Mol Genet. 2010;19:4437–4452. doi: 10.1093/hmg/ddq370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burbulla LF, Fitzgerald JC, Stegen K, Westermeier J, Thost AK, Kato H, Mokranjac D, Sauerwald J, Martins LM, Woitalla D, Rapaport D, Riess O, Proikas-Cezanne T, Rasse TM, Krüger R. Mitochondrial proteolytic stress induced by loss of mortalin function is rescued by Parkin and PINK1. Cell Death Dis. 2014;5:e1180. doi: 10.1038/cddis.2014.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Calì T, Ottolini D, Negro A, Brini M. α-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J Biol Chem. 2012;287:17914–17929. doi: 10.1074/jbc.M111.302794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng W, Zhang B, Zikeliyar M, Wang J, Jian H, Wu K, Zhang Y, Ding J. Elevated Mortalin correlates with poor outcome in hepatocellular carcinoma. Ann Diagn Pathol. 2019;42:59–63. doi: 10.1016/j.anndiagpath.2019.06.011. [DOI] [PubMed] [Google Scholar]
- 16.Chia SJ, Tan EK, Chao YX. Historical perspective:models of Parkinson's disease. Int J Mol Sci. 2020;21:2464. doi: 10.3390/ijms21072464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chiasserini D, Tozzi A, de Iure A, Tantucci M, Susta F, Orvietani PL, Koya K, Binaglia L, Calabresi P. Mortalin inhibition in experimental Parkinson's disease. Mov Disord. 2011;26:1639–1647. doi: 10.1002/mds.23647. [DOI] [PubMed] [Google Scholar]
- 18.Choi J, Forster MJ, McDonald SR, Weintraub ST, Carroll CA, Gracy RW. Proteomic identification of specific oxidized proteins in ApoE-knockout mice:relevance to Alzheimer's disease. Free Radic Biol Med. 2004;36:1155–1162. doi: 10.1016/j.freeradbiomed.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 19.Chung SJ, Kim MJ, Ryu HS, Kim J, Kim YJ, Kim K, You S, Kim SY, Lee JH. Lack of association of mortalin (HSPA9) and other mitochondria-related genes with risk of Parkinson's and Alzheimer's diseases. Neurobiol Aging. 2017;49:215.e9–215. doi: 10.1016/j.neurobiolaging.2016.09.017. [DOI] [PubMed] [Google Scholar]
- 20.Cook TJ. Seattle: University of Washington; 2014. Pathogenesis of mortalin in manganese-induced parkinsonism. [Google Scholar]
- 21.Craig EA, Kramer J, Kosic-Smithers J. SSC1, a member of the 70-kDa heat shock protein multigene family of Saccharomyces cerevisiae, is essential for growth. Proc Natl Acad Sci U S A. 1987;84:4156–4160. doi: 10.1073/pnas.84.12.4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cui X, Li Z, Piao J, Li J, Li L, Lin Z, Jin A. Mortalin expression in pancreatic cancer and its clinical and prognostic significance. Hum Pathol. 2017;64:171–178. doi: 10.1016/j.humpath.2017.03.015. [DOI] [PubMed] [Google Scholar]
- 23.Dores-Silva PR, Cauvi DM, Kiraly VTR, Borges JC, De Maio A. Human HSPA9 (mtHsp70, mortalin) interacts with lipid bilayers containing cardiolipin, a major component of the inner mitochondrial membrane. Biochim Biophys Acta Biomembr. 2020;1862:183436. doi: 10.1016/j.bbamem.2020.183436. [DOI] [PubMed] [Google Scholar]
- 24.Elbaz A, Moisan F. Update in the epidemiology of Parkinson's disease. Curr Opin Neurol. 2008;21:454–460. doi: 10.1097/WCO.0b013e3283050461. [DOI] [PubMed] [Google Scholar]
- 25.Erustes AG, D'Eletto M, Guarache GC, Ureshino RP, Bincoletto C, da Silva Pereira GJ, Piacentini M, Smaili SS. Overexpression of α-synuclein inhibits mitochondrial Ca. J Neurosci Res. 2021;99:2932–2947. doi: 10.1002/jnr.24952. [DOI] [PubMed] [Google Scholar]
- 26.Ferré CA, Thouard A, Bétourné A, Le Dorze AL, Belenguer P, Miquel MC, Peyrin JM, Gonzalez-Dunia D, Szelechowski M. HSPA9/Mortalin mediates axo-protection and modulates mitochondrial dynamics in neurons. Sci Rep. 2021;11:17705. doi: 10.1038/s41598-021-97162-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fessler J, Angiari S. The role of T cell senescence in neurological diseases and its regulation by cellular metabolism. Front Immunol. 2021;12:706434. doi: 10.3389/fimmu.2021.706434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flachbartova Z, Kovacech B. Mortalin - a multipotent chaperone regulating cellular processes ranging from viral infection to neurodegeneration. Acta Virologica. 2013;57:3–15. doi: 10.4149/av_2013_01_3. [DOI] [PubMed] [Google Scholar]
- 29.Freimann K, Zschiedrich K, Brüggemann N, Grünewald A, Pawlack H, Hagenah J, Lohmann K, Klein C, Westenberger A. Mortalin mutations are not a frequent cause of early-onset Parkinson disease. Neurobiol Aging. 2013;34:2694. doi: 10.1016/j.neurobiolaging.2013.05.021. [DOI] [PubMed] [Google Scholar]
- 30.Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–362. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garg S, Wu C, Ohmiya Y, Kaul SC, Wadhwa R. Express ELISA for detection of mortalin. Biotechniques. 2019;67:166–171. doi: 10.2144/btn-2018-0158. [DOI] [PubMed] [Google Scholar]
- 32.Geissler A, Rassow J, Pfanner N, Voos W. Mitochondrial import driving forces:enhanced trapping by matrix Hsp70 stimulates translocation and reduces the membrane potential dependence of loosely folded preproteins. Mol Cell Biol. 2001;21:7097–7104. doi: 10.1128/MCB.21.20.7097-7104.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grassi D, Howard S, Zhou M, Diaz-Perez N, Urban NT, Guerrero-Given D, Kamasawa N, Volpicelli-Daley LA, LoGrasso P, Lasmézas CI. Identification of a highly neurotoxic α-synuclein species inducing mitochondrial damage and mitophagy in Parkinson's disease. Proc Natl Acad Sci U S A. 2018;115:E2634–E2643. doi: 10.1073/pnas.1713849115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gregersen E, Betzer C, Kim WS, Kovacs G, Reimer L, Halliday GM, Thiel S, Jensen PH. Alpha-synuclein activates the classical complement pathway and mediates complement-dependent cell toxicity. J Neuroinflammation. 2021;18:177. doi: 10.1186/s12974-021-02225-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guardia-Laguarta C, Area-Gomez E, Schon EA, Przedborski S. A new role for alpha- synuclein in Parkinson's disease:Alteration of ER-mitochondrial communication. Mov Disord. 2015;30:1026–1033. doi: 10.1002/mds.26239. [DOI] [PubMed] [Google Scholar]
- 36.Haelterman NA, Yoon WH, Sandoval H, Jaiswal M, Shulman JM, Bellen HJ. A mitocentric view of Parkinson's disease. Annu Rev Neurosci. 2014;37:137–159. doi: 10.1146/annurev-neuro-071013-014317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Havalová H, Ondrovičová G, Keresztesová B, Bauer JA, Pevala V, Kutejová E, Kunová N. Mitochondrial HSP70 chaperone system-the influence of post-translational modifications and involvement in human diseases. Int J Mol Sci. 2021;22:8077. doi: 10.3390/ijms22158077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hijioka M, Inden M, Yanagisawa D, Kitamura Y. DJ-1/PARK7:a new therapeutic target for neurodegenerative disorders. Biol Pharm Bull. 2017;40:548–552. doi: 10.1248/bpb.b16-01006. [DOI] [PubMed] [Google Scholar]
- 39.Honrath B, Metz I, Bendridi N, Rieusset J, Culmsee C, Dolga AM. Glucose-regulated protein 75 determines ER-mitochondrial coupling and sensitivity to oxidative stress in neuronal cells. Cell Death Discov. 2017;3:17076. doi: 10.1038/cddiscovery.2017.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang MB, Xia M, Gao Z, Zhou H, Liu M, Huang S, Zhen R, Wu JY, Roth WW, Bond VC, Xiao J, Leng J. Characterization of exosomes in plasma of patients with breast, ovarian, prostate, hepatic, gastric, colon, and pancreatic cancers. J Cancer Ther. 2019;10:382–399. doi: 10.4236/jct.2019.105032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ischiropoulos H, Beckman JS. Oxidative stress and nitration in neurodegeneration:cause, effect, or association? J Clin Invest. 2003;111:163–169. doi: 10.1172/JCI17638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jia F, Fellner A, Kumar KR. Monogenic Parkinson's disease:genotype, phenotype, pathophysiology, and genetic testing. Genes (Basel) 2022;13:471. doi: 10.3390/genes13030471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jin J, Hulette C, Wang Y, Zhang T, Pan C, Wadhwa R, Zhang J. Proteomic identification of a stress protein, mortalin/mthsp70/GRP75:relevance to Parkinson disease. Mol Cell Proteomics. 2006;5:1193–1204. doi: 10.1074/mcp.M500382-MCP200. [DOI] [PubMed] [Google Scholar]
- 44.Jin J, Li GJ, Davis J, Zhu D, Wang Y, Pan C, Zhang J. Identification of novel proteins associated with both alpha-synuclein and DJ-1. Mol Cell Proteomics. 2007;6:845–859. doi: 10.1074/mcp.M600182-MCP200. [DOI] [PubMed] [Google Scholar]
- 45.Johri A, Chandra A. Connection lost, MAM:errors in ER-mitochondria connections in neurodegenerative diseases. Brain Sci. 2021;11:1437. doi: 10.3390/brainsci11111437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Joseph S, Yuen A, Singh V, Hmama Z. Cpn60.2 (GroEL2) blocks macrophage apoptosis via interaction with mitochondrial mortalin. Biol Open. 2017;6:481–488. doi: 10.1242/bio.023119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kabakov AE, Gabai VL. HSP70s in breast cancer:promoters of tumorigenesis and potential targets/tools for therapy. Cells. 2021;10:3446. doi: 10.3390/cells10123446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaul SC, Yaguchi T, Taira K, Reddel RR, Wadhwa R. Overexpressed mortalin (mot- 2)/mthsp70/GRP75 and hTERT cooperate to extend the in vitro lifespan of human fibroblasts. Exp Cell Res. 2003;286:96–101. doi: 10.1016/s0014-4827(03)00101-0. [DOI] [PubMed] [Google Scholar]
- 49.Kaul SC, Deocaris CC, Wadhwa R. Three faces of mortalin:a housekeeper, guardian and killer. Exp Gerontol. 2007;42:263–274. doi: 10.1016/j.exger.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 50.Kawai A, Nishikawa S, Hirata A, Endo T. Loss of the mitochondrial Hsp70 functions causes aggregation of mitochondria in yeast cells. J Cell Sci. 2001;114:3565–3574. doi: 10.1242/jcs.114.19.3565. [DOI] [PubMed] [Google Scholar]
- 51.Krawczyk Z, Gogler-Pigłowska A, Sojka DR, Scieglinska D. The role of heat shock proteins in cisplatin resistance. Anticancer Agents Med Chem. 2018;18:2093–2109. doi: 10.2174/1871520618666180817114952. [DOI] [PubMed] [Google Scholar]
- 52.Lanore A, Lesage S, Mariani LL, Menon PJ, Ravassard P, Cheval H, Corti O, Brice A, Corvol JC. Does the expression and epigenetics of genes involved in monogenic forms of Parkinson's disease influence sporadic forms? Genes (Basel) 2022;13:479. doi: 10.3390/genes13030479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Larsen SB, Hanss Z, Krüger R. The genetic architecture of mitochondrial dysfunction in Parkinson's disease. Cell Tissue Res. 2018;373:21–37. doi: 10.1007/s00441-017-2768-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee B, Ahn Y, Kang SM, Park Y, Jeon YJ, Rho JM, Kim SW. Stoichiometric expression of mtHsp40 and mtHsp70 modulates mitochondrial morphology and cristae structure via Opa1L cleavage. Mol Biol Cell. 2015;26:2156–2167. doi: 10.1091/mbc.E14-02-0762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee S, Wang W, Hwang J, Namgung U, Min KT. Increased ER-mitochondria tethering promotes axon regeneration. Proc Natl Acad Sci U S A. 2019;116:16074–16079. doi: 10.1073/pnas.1818830116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li S, Lv M, Qiu S, Meng J, Liu W, Zuo J, Yang L. NF-κB p65 promotes ovarian cancer cell proliferation and migration via regulating mortalin. J Cell Mol Med. 2019;23:4338–4348. doi: 10.1111/jcmm.14325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu FT, Chen Y, Yang YJ, Yang L, Yu M, Zhao J, Wu JJ, Huang F, Liu W, Ding ZT, Wang J. Involvement of mortalin/GRP75/mthsp70 in the mitochondrial impairments induced by A53T mutant α-synuclein. Brain Res. 2015;1604:52–61. doi: 10.1016/j.brainres.2015.01.050. [DOI] [PubMed] [Google Scholar]
- 58.Liu Y, Liu W, Song XD, Zuo J. Effect of GRP75/mthsp70/PBP74/mortalin overexpression on intracellular ATP level, mitochondrial membrane potential and ROS accumulation following glucose deprivation in PC12 cells. Mol Cell Biochem. 2005;268:45–51. doi: 10.1007/s11010-005-2996-1. [DOI] [PubMed] [Google Scholar]
- 59.Liu Y, Ma X, Fujioka H, Liu J, Chen S, Zhu X. DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proc Natl Acad Sci U S A. 2019;116:25322–25328. doi: 10.1073/pnas.1906565116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Londono C, Osorio C, Gama V, Alzate O. Mortalin, apoptosis, and neurodegeneration. Biomolecules. 2012;2:143–164. doi: 10.3390/biom2010143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lu WJ, Saxena N, Luk JM, Kaul SC, Wadhwa R. Circulating mortalin autoantibody--a new serological marker of liver cirrhosis. Cell Stress Chaperones. 2015;20:715–719. doi: 10.1007/s12192-015-0592-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Magalingam KB, Somanath SD, Ramdas P, Haleagrahara N, Radhakrishnan AK. 6- Hydroxydopamine induces neurodegeneration in terminally differentiated SH-SY5Y neuroblastoma cells via enrichment of the nucleosomal degradation pathway:a global proteomics approach. J Mol Neurosci. 2022 doi: 10.1007/s12031-021-01962-z. doi:10.1007/s12031-021-01962-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marchi S, Patergnani S, Pinton P. The endoplasmic reticulum-mitochondria connection:one touch, multiple functions. Biochim Biophys Acta. 2014;1837:461–469. doi: 10.1016/j.bbabio.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 64.Mazkereth N, Rocca F, Schubert JR, Geisler C, Hillman Y, Egner A, Fishelson Z. Complement triggers relocation of Mortalin/GRP75 from mitochondria to the plasma membrane. Immunobiology. 2016;221:1395–1406. doi: 10.1016/j.imbio.2016.07.005. [DOI] [PubMed] [Google Scholar]
- 65.Meng Z, Zhang R, Wu X, Zhang M, Zhang S, Jin T. Prognostic value of Mortalin correlates with roles in epithelial-mesenchymal transition and angiogenesis in lung adenocarcinoma. Carcinogenesis. 2022;43:40–51. doi: 10.1093/carcin/bgab081. [DOI] [PubMed] [Google Scholar]
- 66.Mustapha M, Mat Taib CN. MPTP-induced mouse model of Parkinson's disease:a promising direction of therapeutic strategies. Bosn J Basic Med Sci. 2021;21:422–433. doi: 10.17305/bjbms.2020.5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nitika, Porter CM, Truman AW, Truttmann MC. Post-translational modifications of Hsp70 family proteins:Expanding the chaperone code. J Biol Chem. 2020;295:10689–10708. doi: 10.1074/jbc.REV120.011666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ottolini D, Calí T, Szabò I, Brini M. Alpha-synuclein at the intracellular and the extracellular side:functional and dysfunctional implications. Biol Chem. 2017;398:77–100. doi: 10.1515/hsz-2016-0201. [DOI] [PubMed] [Google Scholar]
- 69.Park SJ, Shin JH, Jeong JI, Song JH, Jo YK, Kim ES, Lee EH, Hwang JJ, Lee EK, Chung SJ, Koh JY, Jo DG, Cho DH. Down-regulation of Mortalin exacerbates Abeta-mediated mitochondrial fragmentation and dysfunction. J Biol Chem. 2014;289:2195–2204. doi: 10.1074/jbc.M113.492587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pilzer D, Fishelson Z. Mortalin/GRP75 promotes release of membrane vesicles from immune attacked cells and protection from complement-mediated lysis. Int Immunol. 2005;17:1239–1248. doi: 10.1093/intimm/dxh300. [DOI] [PubMed] [Google Scholar]
- 71.Priyanka, Wadhwa R, Chaudhuri R, Nag TC, Seth P. Novel role of mortalin in attenuating HIV-1 Tat-mediated astrogliosis. J Neuroinflammation. 2020;17:276. doi: 10.1186/s12974-020-01912-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Qu M, Zhou Z, Xu S, Chen C, Yu Z, Wang D. Mortalin overexpression attenuates beta-amyloid-induced neurotoxicity in SH-SY5Y cells. Brain Res. 2011;1368:336–345. doi: 10.1016/j.brainres.2010.10.068. [DOI] [PubMed] [Google Scholar]
- 73.Rai R, Kennedy AL, Isingizwe ZR, Javadian P, Benbrook DM. Similarities and differences of Hsp70, hsc70, Grp78 and Mortalin as cancer biomarkers and drug targets. Cells. 2021;10:2996. doi: 10.3390/cells10112996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rakovic A, Grünewald A, Voges L, Hofmann S, Orolicki S, Lohmann K, Klein C. PINK1-interacting proteins:proteomic analysis of overexpressed PINK1. Parkinsons Dis 2011. 2011 doi: 10.4061/2011/153979. 153979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ran Q, Wadhwa R, Kawai R, Kaul SC, Sifers RN, Bick RJ, Smith JR, Pereira-Smith OM. Extramitochondrial localization of mortalin/mthsp70/PBP74/GRP75. Biochem Biophys Res Commun. 2000;275:174–179. doi: 10.1006/bbrc.2000.3237. [DOI] [PubMed] [Google Scholar]
- 76.Rose J, Brian C, Woods J, Pappa A, Panayiotidis MI, Powers R, Franco R. Mitochondrial dysfunction in glial cells:Implications for neuronal homeostasis and survival. Toxicology. 2017;391:109–115. doi: 10.1016/j.tox.2017.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rosenzweig R, Nillegoda NB, Mayer MP, Bukau B. The Hsp70 chaperone network. Nat Rev Mol Cell Biol. 2019;20:665–680. doi: 10.1038/s41580-019-0133-3. [DOI] [PubMed] [Google Scholar]
- 78.Royer-Bertrand B, Castillo-Taucher S, Moreno-Salinas R, Cho TJ, Chae JH, Choi M, Kim OH, Dikoglu E, Campos-Xavier B, Girardi E, Superti-Furga G, Bonafé L, Rivolta C, Unger S, Superti-Furga A. Mutations in the heat-shock protein A9 (HSPA9) gene cause the EVEN-PLUS syndrome of congenital malformations and skeletal dysplasia. Sci Rep. 2015;5:17154. doi: 10.1038/srep17154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schneider HC, Berthold J, Bauer MF, Dietmeier K, Guiard B, Brunner M, Neupert W. Mitochondrial Hsp70/MIM44 complex facilitates protein import. Nature. 1994;371:768–774. doi: 10.1038/371768a0. [DOI] [PubMed] [Google Scholar]
- 80.Segura-Aguilar J, Paris I, Muñoz P, Ferrari E, Zecca L, Zucca FA. Protective and toxic roles of dopamine in Parkinson's disease. J Neurochem. 2014;129:898–915. doi: 10.1111/jnc.12686. [DOI] [PubMed] [Google Scholar]
- 81.Sharma A, Kaur G. Tinospora cordifolia as a potential neuroregenerative candidate against glutamate induced excitotoxicity:an in vitro perspective. BMC Complement Altern Med. 2018;18:268. doi: 10.1186/s12906-018-2330-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shen HY, He JC, Wang Y, Huang QY, Chen JF. Geldanamycin induces heat shock protein 70 and protects against MPTP-induced dopaminergic neurotoxicity in mice. J Biol Chem. 2005;280:39962–39969. doi: 10.1074/jbc.M505524200. [DOI] [PubMed] [Google Scholar]
- 83.Simola N, Morelli M, Carta AR. The 6-hydroxydopamine model of Parkinson's disease. Neurotox Res. 2007;11:151–167. doi: 10.1007/BF03033565. [DOI] [PubMed] [Google Scholar]
- 84.Singh AP, Bajaj T, Gupta D, Singh SB, Chakrawarty A, Goyal V, Dey AB, Dey S. Serum Mortalin correlated with α-Synuclein as serum markers in Parkinson's disease:a pilot study. Neuromolecular Med. 2018;20:83–89. doi: 10.1007/s12017-017-8475-5. [DOI] [PubMed] [Google Scholar]
- 85.Starenki D, Sosonkina N, Hong SK, Lloyd RV, Park JI. Mortalin (GRP75/HSPA9) promotes survival and proliferation of thyroid carcinoma cells. Int J Mol Sci. 2019;20:2069. doi: 10.3390/ijms20092069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+channels. J Cell Biol. 2006;175:901–911. doi: 10.1083/jcb.200608073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Szepesi Z, Manouchehrian O, Bachiller S, Deierborg T. Bidirectional microglia-neuron communication in health and disease. Front Cell Neurosci. 2018;12:323. doi: 10.3389/fncel.2018.00323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Theurey P, Rieusset J. Mitochondria-associated membranes response to nutrient availability and role in metabolic diseases. Trends Endocrinol Metab. 2017;28:32–45. doi: 10.1016/j.tem.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 89.Voloboueva LA, Emery JF, Sun X, Giffard RG. Inflammatory response of microglial BV-2 cells includes a glycolytic shift and is modulated by mitochondrial glucose-regulated protein 75/mortalin. FEBS Lett. 2013;587:756–762. doi: 10.1016/j.febslet.2013.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wadhwa R, Takano S, Kaur K, Aida S, Yaguchi T, Kaul Z, Hirano T, Taira K, Kaul SC. Identification and characterization of molecular interactions between mortalin/mtHsp70 and HSP60. Biochem J. 2005;391:185–190. doi: 10.1042/BJ20050861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Weiss F, Labrador-Garrido A, Dzamko N, Halliday G. Immune responses in the Parkinson's disease brain. Neurobiol Dis. 2022;168:105700. doi: 10.1016/j.nbd.2022.105700. [DOI] [PubMed] [Google Scholar]
- 92.Williamson CL, Dabkowski ER, Dillmann WH, Hollander JM. Mitochondria protection from hypoxia/reoxygenation injury with mitochondria heat shock protein 70 overexpression. Am J Physiol Heart Circ Physiol. 2008;294:H249–256. doi: 10.1152/ajpheart.00775.2007. [DOI] [PubMed] [Google Scholar]
- 93.Xu L, Voloboueva LA, Ouyang Y, Emery JF, Giffard RG. Overexpression of mitochondrial Hsp70/Hsp75 in rat brain protects mitochondria, reduces oxidative stress, and protects from focal ischemia. J Cereb Blood Flow Metab. 2009;29:365–374. doi: 10.1038/jcbfm.2008.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xu M, Jin T, Chen L, Zhang X, Zhu G, Wang Q, Lin Z. Mortalin is a distinct bio-marker and prognostic factor in serous ovarian carcinoma. Gene. 2019;696:63–71. doi: 10.1016/j.gene.2019.02.033. [DOI] [PubMed] [Google Scholar]
- 95.Xu M, Zhang Y, Cui M, Wang X, Lin Z. Mortalin contributes to colorectal cancer by promoting proliferation and epithelial-mesenchymal transition. IUBMB Life. 2020;72:771–781. doi: 10.1002/iub.2176. [DOI] [PubMed] [Google Scholar]
- 96.Yang H, Zhou X, Liu X, Yang L, Chen Q, Zhao D, Zuo J, Liu W. Mitochondrial dysfunction induced by knockdown of mortalin is rescued by Parkin. Biochem Biophys Res Commun. 2011;410:114–120. doi: 10.1016/j.bbrc.2011.05.116. [DOI] [PubMed] [Google Scholar]
- 97.Yokoyama K, Fukumoto K, Murakami T, Harada S, Hosono R, Wadhwa R, Mitsui Y, Ohkuma S. Extended longevity of Caenorhabditis elegans by knocking in extra copies of hsp70F, a homolog of mot-2 (mortalin)/mthsp70/Grp75. FEBS Lett. 2002;516:53–57. doi: 10.1016/s0014-5793(02)02470-5. [DOI] [PubMed] [Google Scholar]
- 98.Zhang R, Meng Z, Wu X, Zhang M, Zhang S, Jin T. Mortalin promotes breast cancer malignancy. Exp Mol Pathol. 2021;118:104593. doi: 10.1016/j.yexmp.2020.104593. [DOI] [PubMed] [Google Scholar]
- 99.Zhao J, Yu S, Zheng Y, Yang H, Zhang J. Oxidative modification and its implications for the neurodegeneration of Parkinson's disease. Mol Neurobiol. 2017;54:1404–1418. doi: 10.1007/s12035-016-9743-3. [DOI] [PubMed] [Google Scholar]
- 100.Zhou TT, Wang XY, Huang J, Deng YZ, Qiu LJ, Liu HY, Xu XW, Ma ZX, Tang L, Chen HP. Mitochondrial translocation of DJ-1 is mediated by Grp75:implication in cardioprotection of resveratrol against hypoxia/reoxygenation-induced oxidative stress. J Cardiovasc Pharmacol. 2020;75:305–313. doi: 10.1097/FJC.0000000000000805. [DOI] [PubMed] [Google Scholar]
- 101.Zhu JY, Vereshchagina N, Sreekumar V, Burbulla LF, Costa AC, Daub KJ, Woitalla D, Martins LM, Kruger R, Rasse TM. Knockdown of Hsc70-5/mortalin induces loss of synaptic mitochondria in a Drosophila Parkinson's disease model. PLoS One. 2013;8:e83714. doi: 10.1371/journal.pone.0083714. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.