

Summary
Despite its crucial role in initiation of cytotoxic immune responses, the molecular pathways underlying antigen cross-presentation remain incompletely understood. The mechanism of antigen exit from endocytic compartments into the cytosol is a long-standing matter of controversy, confronting two main models: transfer through specific channels/transporters or rupture of endocytic membranes and leakage of luminal content. By monitoring the occurrence of intracellular damage in conventional dendritic cells (cDCs), we show that cross-presenting cDC1s display more frequent endomembrane injuries and increased recruitment of endosomal sorting complex required for transport (ESCRT)-III, the main repair system for intracellular membranes, relative to cDC2s. Silencing of CHMP2a or CHMP4b, two effector subunits of ESCRT-III, enhances cytosolic antigen export and cross-presentation. This phenotype is partially reversed by chemical inhibition of RIPK3, suggesting that endocytic damage is related to basal activation of the necroptosis pathway. Membrane repair therefore proves crucial in containing antigen export to the cytosol and cross-presentation in cDCs.
Keywords: cross-presentation, dendritic cells, necroptosis, ESCRT-III, membrane repair, endomembrane disruption
Graphical abstract
Highlights
-
•
Cross-presenting dendritic cells (cDC1s) undergo endomembrane damage at steady state
-
•
Occurrence of endocytic injuries triggers ESCRT-III recruitment in cDC1s
-
•
ESCRT-III deficiency results in persistence of unrepaired endomembrane damage
-
•
Endomembrane repair defects enhance cytosolic antigen export and cross-presentation
Despite their rate-limiting nature, the molecular events leading to cytosolic export of antigens during cross-presentation remain incompletely understood. Gros et al. identify the ESCRT-III membrane repair complex as a negative regulator of antigen export to the cytosol and point to endomembrane rupture as a major route for antigens in cross-presentation.
Introduction
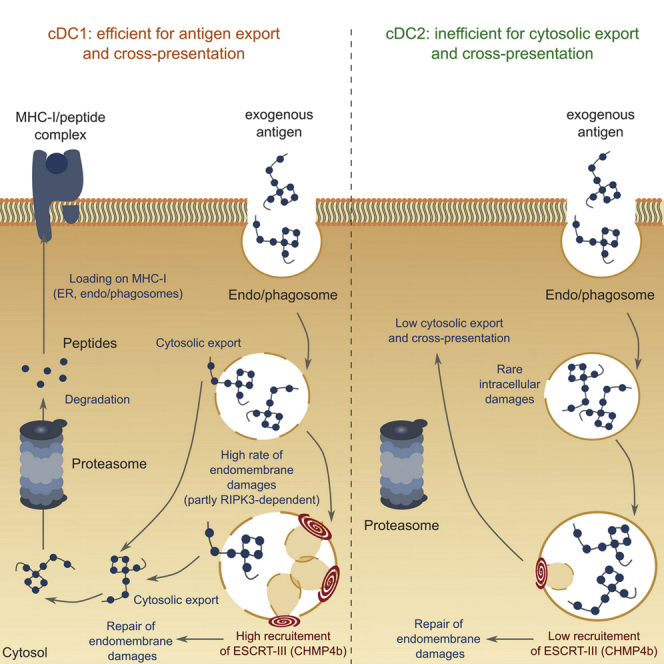
Conventional dendritic cells (cDCs) initiate immune responses against pathogens and tumors. To do so, they present exogenous antigens on major histocompatibility complex (MHC) class II and class I molecules. Presentation of internalized antigens by MHC class I, or “cross-presentation,” mediates priming of CD8+ naive T lymphocytes, or “cross-priming,” a process first described over 40 years ago (Bevan, 1976a, 1976b). At steady state, in mice, CD8+ CD11b− cDC1s display a superior ability to cross-present antigens compared with CD8− CD11b+ cDC2s due to specialized intracellular machineries delivering antigens to cross-presentation pathways (Blander, 2018). Upon uptake, antigens are exported from intracellular compartments to the cytosol, where they are degraded by the proteasome. The resulting peptides are then loaded on MHC class I (in the endoplasmic reticulum [ER] or in endocytic compartments) and subsequently presented at the cell surface (reviewed in Cruz et al., 2017). Whereas several steps of antigen cross-presentation are now well described, the molecular mechanisms underlying antigen export to the cytosol remain elusive (Gros and Amigorena, 2019).
Although the idea that antigens are exported to the cytosol has been proposed in the early 1990s (Kovacsovics-Bankowski and Rock, 1995), the first insights into the molecular mechanisms involved came from studies using Exotoxin A (ExoA), a bacterial inhibitor of SEC61 (Koopmann et al., 2000). ExoA blocked cytosolic export-dependent function of the ICP47 protein (which inhibits peptide import into the ER or phagosomes by interacting with the cytosolic face of TAP transporter), involving SEC61 in the process (Ackerman et al., 2006). This translocon mediates signal peptide-dependent protein import into the ER and could be involved in retro-translocation of misfolded proteins back into the cytosol (Römisch, 2017). SEC61-dependent export of antigens was further supported by results showing that small interfering RNA (siRNA)-mediated depletion of SEC61 as well as retention of SEC61 in the ER by an intrabody inhibited cytosolic antigen translocation and cross-presentation (Zehner et al., 2015). A recent study, however, showed that acute inhibition of SEC61α with a more specific inhibitor, mycolactone, had no direct effect on antigen export to the cytosol (Grotzke et al., 2017). Sustained SEC61 inhibition indirectly diminished antigen cross-presentation because of reduced expression of MHC class I molecules at the plasma membrane. This study argues against SEC61 acting as a retrotranslocon in antigen export to the cytosol.
Furthermore, the role of SEC61 in retro-translocation remains controversial, even in the ER. The pore size of SEC61, adapted for insertion of extended unfolded polypeptides (Voorhees et al., 2014), seems insufficient to export large folded, post-translationally modified proteins, known to be exported to the cytosol during antigen cross-presentation (Nalle et al., 2020; Singh and Cresswell, 2010), suggesting a contribution of additional export mechanisms.
Several recent studies have highlighted a role of reactive oxygen species (ROS)-dependent lipid peroxidation in triggering endosomal membrane destabilization and subsequent antigen escape into the cytosol (Canton et al., 2020; Dingjan et al., 2016; Nalle et al., 2020). However, if leakage from endocytic compartments occurs during antigen cross-presentation in cDCs, then why does it not result in cell death? Permeabilization of endolysosomal membranes leads to release of cathepsins and hydrolases that initiate cell death pathways (Boya and Kroemer, 2008). Therefore, if membrane leaks were involved in any physiological cell function, such as antigen cross-presentation, then damage would need to be strictly contained to avoid cell death.
A vast range of publications have identified the endosomal sorting complex required for transport (ESCRT) as a major player in repairing different types of cellular membranes following damage, including plasma (Jimenez et al., 2014), nuclear (Denais et al., 2016; Olmos et al., 2015; Raab et al., 2016; Vietri et al., 2015), and endolysosomal membranes (Radulovic et al., 2018; Skowyra et al., 2018). The highly conserved ESCRT machinery is composed of four different complexes (from ESCRT-0 to ESCRT-III), recruited sequentially to cellular membranes and displaying a wide range of functions, from cytokinetic abscission (Carlton et al., 2012) to virus budding (Garrus et al., 2001). During repair, ESCRT-III constitutes the core complex forming helical filaments around damaged portions of membranes. The latter are ultimately shed away from the cytosol in the form of buds, after filament constriction (Schöneberg et al., 2017).
Here we tested the hypothesis that ESCRT-III, by repairing membrane damage, controls antigen leakage into the cytosol during antigen cross-presentation in cDCs. We used short hairpin (sh) RNA to target CHMP4b, the main component of ESCRT-III helical filaments (Chiaruttini et al., 2015; Hanson et al., 2008; Shen et al., 2014), and CHMP2a, a subunit required for recruitment of the unfolding protein VPS4b (Adell et al., 2014; Fujita et al., 2004; Saksena et al., 2009). We show that depletion of ESCRT-III results in profound enhancement of antigen export to the cytosol and cross-presentation in a cell-intrinsic manner. ESCRT-III is recruited to damaged intracellular compartments, limiting cell death and antigen leakage into the cytosol, which, in turn, controls antigen cross-presentation. Our results thus support the model of membrane leakage for antigen export to the cytosol in cross-presenting cDCs and suggest that necroptotic pathway might modulate the occurrence and efficiency of this process.
Results
Increased damage in intracellular compartments of cross-presenting cDCs correlates with enhanced ESCRT-III recruitment to their endolysosomes
It has been shown previously that CD8+ cDC1s cross-present antigens more efficiently than CD11b+ cDC2s at steady state (Pooley et al., 2001; Schnorrer et al., 2006), partly owing to their higher capacity to export antigens into the cytosol (Imai et al., 2011; Lin et al., 2008). To confirm these findings, we used another experimental approach, the β-lactamase assay, to monitor antigen export to the cytosol. Briefly, splenic cDCs were fed with β-lactamase for 3 or 5 h and then loaded with CCF4, a cytosolic β-lactamase substrate emitting green fluorescence due to fluorescence resonance energy transfer (FRET) between its two constitutive subunits. When β-lactamase is efficiently transported from intracellular compartments to the cytosol, it cleaves CCF4, thereby disrupting the FRET signal and resulting in emission of blue fluorescence. The ratio between blue and green fluorescence is then measured by flow cytometry as a readout for antigen export to the cytosol. Similar to previous findings, this assay also shows that cDC1s export antigens to the cytosol more efficiently than cDC2s (Figure 1A). Antigen export to the cytosol in cDC1s can also be detected biochemically by cell fractionation and subsequent western blotting on cytosolic extracts, using the model antigen ovalbumin (OVA) (Figure 1B).
Figure 1.

ESCRT-III subunits are enriched in damage-prone cross-presenting CD8+ cDC1s
(A) Purified splenic cDCs were incubated with β-lactamase for various times before CCF4 loading, antibody staining, and analysis by flow cytometry (n = 3 experiments performed with technical duplicates or triplicates, two-way ANOVA, mean ± SEM, exact p values are indicated). The zero time point corresponds to incubation for 5 h without β-lactamase at 37°C.
(B) Splenic CD8+ cDC1s or CD11b+ cDC2s were purified from FLT3L-treated mice and pulsed for 30 min with OVA-biotin. After a 2 h chase, cells were subjected to cell fractionation, and western blotting was then performed on cytosolic extracts obtained by ultracentrifugation. Blots representative of 7 experiments are shown.
(C) Purified splenic cDCs were purified and treated for 30 min with 0.3 mM LLOME, endotoxin-containing (sOVA), or endotoxin-free (sEF-OVA) OVA for 4 h before fixation and galectin-3 intracellular staining. Images shown were acquired with a confocal microscope equipped with a 100× objective (scale bar, 5 μm). Frequencies (mean) in the right panel display the results of 3 (DMSO, LLOME, and sOVA) or 2 (sEF-OVA) independent experiments. Arrowheads indicate examples of galectin-3 focus location.
(D) Intracellular compartments of CD8+ cDC1s or CD11b+ cDC2s purified from B16-FLT3L-injected mice were isolated by subcellular fractionation and analyzed by mass spectrometry. Purity control (top panel), ESCRT-III components (center panel), or galectin-3 (bottom panel) found on the various compartments are shown. Values below 0.4 represent enrichment in endocytic compartments of cDC2s, and those above 0.4 show enrichment in endocytic compartments of cDC1s. ∗ indicates the references used for fraction purity controls. Ratio of spectral counts (RSC) was calculated as in Segura et al. (2010). n = 1. Raw peptide numbers and spectral counts found in cDC1 and cDC2 intracellular compartments are available in Tables S1, S2, and S3.
To investigate the mechanisms underlying this functional specialization, we monitored the possible occurrence of endomembrane rupture events by labeling purified splenic cDCs with galectin-3. This cytosolic lectin enters endocytic compartments after membrane damage, serving as a marker for membrane disruption (Aits et al., 2015). At steady state (DMSO control condition), cDC1s display significantly higher amounts of galectin-3 foci than their cDC2 counterparts (Figures 1C, S1A, and S1B), suggesting that cross-presenting cDC undergo more pronounced endolysosomal rupture events. These observations were further confirmed by endocytic compartment proteomics, which revealed enriched galectin-3 recruitment to cDC1 early endosomes and lysosomes (Figure 1D, bottom panel). Although cDC1s show increased expression of galectin-3 compared with cDC2s (Figure S1C; Luber et al., 2010), the absence of correlation between galectin-3 fluorescence intensity and the number of foci (Figure S1D) indicates that differences in numbers of galectin-3 foci between cDC1s and cDC2s are not due to variations in galectin-3 expression. Greater occurrence of endolysosomal damage in cDC1 was confirmed, and even exacerbated, after incubation of splenic cDCs with L-leucine methyl ester (LLOME) (Figures 1C and S1A), a drug causing endolysosomal damage after cleavage by cathepsin C in lysosomes (Aits et al., 2015; Maejima et al., 2013; Repnik et al., 2017). This observation cannot be explained by discrepant cathepsin C levels since cDC1s and cDC2s express equal levels of this protease (Luber et al., 2010). Additionally, in cDC1s, but not in cDC2s, endotoxin-containing OVA tends to increase the frequency of galectin-3 foci compared with treatment with endotoxin-free OVA (Figures 1C and S1A). In line with previous reports (Dingjan et al., 2016, 2017), endotoxin-containing OVA-mediated occurrence of endolysosomal damage is likely mediated by the NADPH-oxidase NOX2, as NOX2-deficient cDC1 display slightly fewer galectin-3 foci than their wild-type (WT) counterparts after incubation with this antigen (Figure S1E, bottom panel, and S1F, right panel). Although a trend toward reduction of endolysosomal rupture events is also observed in NOX2-deficient cDC1s at steady state (Figures S1E, top panel, and S1F, left panel), conclusive involvement of NOX2 cannot be firmly established in this context, most likely because of the narrow range and scarcity of events detected. In summary, the occurrence of endolysosomal damage, as detected by galectin-3 foci, mirrors the intrinsic capacity of antigens to be cross-presented efficiently as well as the enhanced ability of cDC1s to export and cross-present antigens.
We reasoned that, if endolysosomal membrane rupture underlies increased cytosolic antigen export in cDC1s, then molecular repair machineries, such as ESCRT-III, should be actively recruited to cDC1 intracellular compartments. We therefore searched for ESCRT-III family members in isolated endosomes from splenic cDC1s and cDC2s using semi-quantitative label-free mass spectrometry (Old et al., 2005). Typical markers of each cDC subset, such as LY75 (CD205) for cDC1s (Vremec and Shortman, 1997) and CD22 for cDC2s (Edwards et al., 2003), are present in the corresponding subsets (Figure 1D, top panel), confirming the relevance of our assay. At the sole exception of CHMP2a, which is found enriched in cDC1 late endosomes and lysosomes, most ESCRT-III subunits are enriched in cDC1 early endocytic compartments (Figure 1D, center panel) at steady state, supporting the notion that endocytic membrane damage occurs more frequently in this subset. To further confirm these results, we studied the recruitment dynamics of IST1, one of the terminal effectors of the ESCRT-III cascade, required for completion of membrane constriction and fission (Nguyen et al., 2020; Pfitzner et al., 2020). After short treatment with LLOME (15 min), cDC1s display significantly reduced recruitment of IST1 to endocytic compartments compared with cDC2, a situation inversely mirroring the pattern of galectin-3 focus appearance (Figures 2A and 2B). The occurrence of IST1 and galectin-3 foci negatively correlate in cDC1s (Figures S1G and S1H), suggesting that increased appearance of endolysosomal damage in this subset is at least partially related to reduced IST1-mediated endomembrane closure and repair.
Figure 2.

cDC1s fail to complete ESCRT-III-mediated repair
(A) Splenic cDCs were purified and left untreated (DMSO; top panel) or treated for 15 min (center panel) or 30 min (bottom panel) with 0.3 mM LLOME before fixation and galectin-3 and IST1 intracellular staining. Images shown were acquired with a confocal microscope equipped with a 40× objective (scale bar, 5 μm).
(B) Numbers and frequencies (mean) of GAL3/IST1 foci are the results of 2 independent experiments (Mann-Whitney t test, exact p-values are indicated). Each dot represents one cell. The numbers of cells analyzed are indicated in Table S4.
ESCRT-III is recruited to endocytic compartments to repair damage
To next investigate the recruitment of ESCRT-III to endocytic membranes (which requires a high number of cells), we used the cDC1 cell line MutuDC as a model system (Fuertes Marraco et al., 2012). MutuDCs are derived from splenic cDC1s and share phenotypic and functional properties with these cells (Fuertes Marraco et al., 2012; Kozik et al., 2020), including selective use of the cytosolic pathway for antigen cross-presentation (Canton et al., 2020). To confirm ESCRT-III recruitment to MutuDC endocytic compartments, we expressed recombinant human CHMP4b-mCherry (Figure S2A) and monitored its recruitment to phagosomes using fluorescence-activated cell sorting (FACS) on isolated phagosomes (PhagoFACS), as described previously (Hoffmann et al., 2016). CHMP4b-mCherry-expressing MutuDCs were pulsed with OVA-coated latex beads, as indicated (Figure 3A), and chased for various times in presence or absence of LLOME. As shown in Figure 3B, in the absence of LLOME, CHMP4b is recruited to approximately 13% of OVA-containing phagosomes. Recruitment is further increased to over 20% of mCherry-positive phagosomes by treatment with LLOME, a result concordant with previous reports (Radulovic et al., 2018; Skowyra et al., 2018). ESCRT-III recruitment to phagosomes may require the activity of ATP-dependent proteins, such as proton pumps (controlling phagosome maturation; Figure S2B) or kinases (which might trigger endocytic damage), as it is completely abolished upon ATP deprivation with 2-DG and NaN3 (Figure 3B). These results further connect ESCRT-III recruitment at sites of damage targeting endocytic compartments.
Figure 3.

ESCRT-III is recruited to damaged intracellular compartments and maintains their integrity
(A) Experiment timeline for PhagoFACS represented in (B).
(B) Recruitment of CHMP4b-mCherry on phagosomes was evaluated by flow cytometry. Plots and results representative of at least 2 independent experiments are shown in the left and right panels (two-way ANOVA, mean ± SEM, exact p values are indicated).
(C and D) CHMP4b-silenced, CHMP2a-silenced, or control cells were left untreated or incubated for 15 or 30 min with 0.5 mM LLOME before fixation and galectin-3 and IST1 staining. Images representative of 2 independent experiments are shown in (C) (scale bar, 5 μm). Ratio of area covered by IST1 foci on total cell area (left panel, mean, Student’s t test) and numbers of galectin-3 (right panel, Mann-Whitney t test) foci per cell are quantified in (D). Each dot represents one cell and exact p values are indicated. The numbers of cells analyzed are indicated in Table S4.
Silencing of ESCRT-III proteins increases endocytic damage
We hypothesized that the absence of functional ESCRT-III should cause persistence of endomembrane damage. To test this hypothesis, we silenced two ESCRT-III components required for membrane repair (Denais et al., 2016; Jimenez et al., 2014; Olmos et al., 2015; Vietri et al., 2015), CHMP4b and CHMP2a, in the cDC1 MutuDC line. For each target, we used two different shRNAs (sh#2 and sh#3 for CHMP4b and sh#1 and sh#2 for CHMP2a) that decreased mRNA (Figure S2C) and protein (Figure S2D) expression by day 3 after transduction. As described previously in other cell types, silencing of CHMP4b or CHMP2a induces cell death over time (Figure S5A), most likely due to unrepaired necroptotic MLKL-associated membrane damage (Gong et al., 2017). We therefore performed all functional experiments on day 3 after transduction, a timepoint that systematically yielded efficient silencing and maintained over 70% viability.
First, we confirmed that silencing of CHMP4b or CHMP2a impairs recruitment of downstream ESCRT-III proteins, such as IST1, to LLOME-triggered endolysosomal membrane disruptions (Figures 3C and 3D, left panel, and S2E). Then, to investigate the functional consequences of such perturbations in the sequence of events leading to ESCRT-III-mediated endomembrane repair, we incubated CHMP4b-silenced, CHMP2a-silenced, or control MutuDC in the presence or absence of LLOME and subsequently labeled them with a galectin-3-targeting antibody. While untreated control cells show diffuse, cytosolic galectin-3 distribution, untreated CHMP2a- and CHMP4b-deficient cells display intense galectin-3+ puncta (Figures 3C and 3D, right panel, and S2E), that resemble those observed after addition of LLOME (Figures 3C and 3D, right panel, and S2E). The appearance of galectin-3+ foci in ESCRT-III-silenced cells confirms that ESCRT-III is required to repair intracellular compartments and protects them from deleterious endosomal rupture. We conclude that ESCRT-III is recruited to repair damaged intracellular compartments and could therefore restrict antigen export to the cytosol.
Silencing of ESCRT-III proteins enhances antigen export to the cytosol
Before evaluating antigen access to the cytosol, we tested how silencing of ESCRT-III affects antigen uptake and degradation by MutuDCs. As shown in Figure S3A, left panel, ESCRT-III-silencing did not affect endocytosis of OVA (Figure S3A, right panel) or β-lactamase (Figure S3B). Previous studies have shown that depletion of CHMP3 (Bache et al., 2006) or CHMP4b (Mamińska et al., 2016) leads to delayed endolysosomal fusion after receptor-mediated endocytosis, whereas degradation of proteins endocytosed by fluid phase remains unaffected (Bache et al., 2006). To monitor degradation, we used red bovine serum albumin (BSA)-dye quenched (DQ), which emits increased fluorescence when degraded (Figure S3C). ESCRT-III-silenced cells display reduced degradation compared with control DCs (Figure S3D).
We then evaluated the capacity of ESCRT-III-silenced MutuDCs to export β-lactamase into the cytosol (Figure 4A). As shown in Figures 4B, top panel, and 4C, CHMP4b- and CHMP2a-silenced MutuDCs export β-lactamase into the cytosol more efficiently than DCs transduced with a control shRNA. The much higher efficiency of Chmp4b sh#2 relative to Chmp4b sh#3 may be explained by faster silencing with Chmp4b sh#2, as suggested by lower mRNA levels on days 1 and 2 (Figure S5B) but not on day 3 (Figures S2C and S5B) and lower CHMP4b protein expression on day 3 (Figure S2D). Additionally, ESCRT-III-deficient MutuDCs incubated with β-lactamase for 5 h on ice do not show any CCF4 conversion (Figure 4B, bottom panel), suggesting that internalization of β-lactamase is required. To obtain more direct evidence that β-lactamase enters the cytosol through endosomal perforations rather than through unrepaired holes in the plasma membrane, we incubated CHMP4b-silenced, CHMP2a-silenced, or control MutuDCs in the presence of a membrane-impermeant (DAPI) and a membrane-permeant dye (siR-DNA) to distinguish dead cells from live ones. After fixation and galectin-3 staining, we quantified the proportion of live (DAPI−) and dead (DAPI+) cells displaying galectin-3+ foci. Overall, we detected no correlation between DAPI fluorescence intensity and number of galectin-3+ foci (Figures S4A–S4C). We also confirm that, compared with control cells, ESCRT-III-deficient cDCs show increased occurrence of endolysosomal perforations, labeled by galectin-3 (Figures S4A–S4D). The vast majority of these foci are found in live cells (Figures S4A–S4D), whose unaltered plasma membrane is impermeable to DAPI, strengthening the idea that endomembrane damage precedes plasma membrane rupture (Vanden Berghe et al., 2010).
Figure 4.

ESCRT-III-silenced cDCs display increased antigen export to the cytosol
(A) Timeline for the β-lactamase assay in (B) and (C).
(B) Representative plots after 3 h of incubation with or without β-lactamase (gated on live cells).
(C) Quantification of 4 independent experiments (two-way ANOVA, mean ± SEM, exact p values are indicated). The zero time point corresponds to incubation for 5 h without β-lactamase at 37°C.
(D–F) ESCRT-III-deficient or control MutuDCs were pulsed with tetramethylrhodamine-labeled dextrans (3K [D], 10K [E], or 70K [F]) before extensive washes and subsequent chase. Representative images acquired with a confocal microscope equipped with a 40× objective are shown (scale bar, 5 μm). Quantification of the median of cytosolic fluorescence and of the ratio of dextran+ vesicle area on total cell area are shown (Student’s t test, mean, exact p values are indicated). Each dot represents one cell. One experiment representative of 2 independent experiments is shown. Arrowheads indicate cells showing cytosolic dextran fluorescence. Because the images shown in (D) originate from the same experiment as the one presented in Figure S3F, the control shRNA image and values are identical (and copied) in both figures.
(G) qRT-PCR expression analysis of endogenous Chmp4b (left panel) and shRNA-resistant Chmp4b (right panel) in MutuDCs expressing (resistant CHMP4b) or not expressing (empty vector) shRNA-resistant CHMP4b and the different shRNA. n = 3 (unpaired Student’s t test, exact p values are indicated).
(H) Representative flow cytometry plots after 3 h of incubation with β-lactamase (gated on live cells).
(I) Quantification of 2 independent experiments (two-way ANOVA, mean ± SEM, exact p values are indicated).
(J) Representative galectin-3 staining images obtained with a confocal microscope equipped with a 40× objective (scale bar, 5 μm). n = 2. Arrowheads indicate locations of galectin-3 foci.
Next, we sought to determine whether increased CCF4 conversion in ESCRT-III-depleted cells is due to slow cytosolic degradation of β-lactamase. Inhibition of the proteasome with MG-132 does not affect CCF4 conversion in control or ESCRT-III-silenced MutuDCs (Figure S3E). In the presence of MG-132, ESCRT-III silencing still induces increased export (Figure S3E), arguing that changes in β-lactamase degradation by the proteasome do not account for the observed phenotype. To confirm this result, we next tested whether protease-insensitive cargoes (e.g., dextrans), could also gain access to the cytosol after ESCRT-III depletion. For this purpose, ESCRT-III-silenced or control MutuDCs were pulsed for 1 h with tetramethylrhodamine-labeled dextrans of various molecular weights (ranging from 3K–70K). After a 3 h chase, cytosolic leakage of fluorescent dextrans was investigated by confocal microscopy. In control shRNA-transduced MutuDCs, 3K fluorescent dextrans remain mostly trapped in endocytic compartments (Figure 4D). Conversely, in ESCRT-III-silenced DCs, 3K dextrans leak into the cytosol, as reflected by the decreased area of dextran-containing vesicles, paralleled by increased median cytosolic fluorescence (Figure 4D). The aforementioned fluorescence pattern of ESCRT-III-silenced cells resembles the pattern observed in control MutuDCs treated with prazosin, a drug causing endocytic leakage (Kozik et al., 2020; Figure S3F). This result suggests that, similar to prazosin, ESCRT-III deficiency triggers endolysosomal damage, allowing export of endocytic content to the cytosol. In ESCRT-III-silenced DCs, cytosolic export of large 10K and 70K dextrans shows lower efficiency compared with 3K dextran (Figures 4D–4F), thereby confirming that antigen leakage to the cytosol is size selective (Rodriguez et al., 1999). Altogether, these results demonstrate that ESCRT-III deficiency causes leakage of large branched or folded antigens into the cytosol through unrepaired permeable endomembranes.
Finally, to validate the specificity of the shRNAs used in this study, we expressed a shRNA-resistant version of murine CHMP4b (Figure 4G, right panel) in MutuDCs that were simultaneously silenced for endogenous CHMP4b (Figure 4G, left panel). These MutuDCs expressing shRNA-resistant CHMP4b display rescued viability (Figure S3G) and are no longer activated (Figures S3H and S3I; see below) compared with CHMP4b-deficient cells (empty vector). Expression of shRNA-resistant CHMP4b not only reverses phenotypic characteristics of ESCRT-III silencing but also completely abolishes its functional features, such as enhanced antigen export into the cytosol (Figures 4H and 4I) and presence of galectin-3 foci (Figure 4J). These results show that ESCRT-III silencing in a cDC1 cell line specifically induces a strong increase in the export of soluble endocytic cargo to the cytosol.
ESCRT-III-mediated control of antigen export to the cytosol is cell intrinsic
As reported previously for other cells types (Mamińska et al., 2016), ESCRT-III silencing in DCs triggers nuclear factor κB (NF-κB) activation. CD86 and MHC class II surface expression (Figure S5C) as well as the production of Tnf, Il1b and Relb transcripts (Figure S5D) are increased in ESCRT-III-silenced MutuDCs. To examine whether enhanced antigen export to the cytosol in these cells is a consequence of silencing-mediated DC activation, we pre-activated ESCRT-III-silenced and control cells with CpG, a Toll-like receptor (TLR) ligand to which cDC1s (and MutuDCs) respond strongly (Fuertes Marraco et al., 2012). In comparison with their resting counterparts, CpG-treated control MutuDCs show slightly increased antigen export to the cytosol (Figures 5A and 5B, left panel) but retain low cytosol export activity compared with ESCRT-III-silenced cells (Figures 5A and 5B, right panel). As shown in Figures 5A and 5B, center and right panels, CpG treatment does not further enhance the robust export capacity of ESCRT-III-silenced cells. Incidentally, β-Lactamase is contaminated with bacterial products and induces DC activation per se in silenced and control cells (data not shown). Therefore, the increased export capacity of ESCRT-III-silenced cells is unlikely to be a direct consequence of NF-κB activation.
Figure 5.

ESCRT-III-mediated increased antigen export to the cytosol is cell intrinsic
(A) Representative flow cytometry plots after 3 h of incubation with β-lactamase (gated on live cells).
(B) Quantification of 3 independent experiments (two-way ANOVA, mean ± SEM, exact p values are indicated). The zero time point corresponds to incubation for 5 h, on ice, with β-lactamase.
(C) Timeline for the ESCRT-III intrinsic assay co-culture experiment.
(D and E) Representative 3 h plots of cells co-cultured or grown in separate wells (left panel) and quantified (right panel) (gated on live cells). n = 2 experiments (two-way ANOVA, mean ± SEM). Exact p values depicted in a solid-line box show a comparison between ESCRT-targeting and control shRNA-transduced MutuDCs cultured in separate wells for each time point. Exact p values depicted in a dotted-line box show a comparison between ESCRT-targeting and control shRNA-transduced MutuDCs co-cultured in the same well (pooled) for each time point.
If the effect of ESCRT-III silencing on export to the cytosol is due to reduced endosomal membrane repair, then it should be intrinsic to individually silenced DCs, and if it is related to DC activation, then it could be mediated by secreted factors. To address this question, we co-cultured MutuDCs silenced for CHMP4b or CHMP2a together with control cells, labeled or not labeled with a cytosolic dye (to distinguish them by flow cytometry). On day 3, we performed the β-lactamase assay on DCs pooled or cultured in separate wells for 2 days (Figure 5C). After co-culture with CHMP4b- or CHMP2a-deficient cells, control cells display increased surface expression of CD86 and MHC class II compared with control cells cultured in separate wells (data not shown), indicating that silenced MutuDCs secrete soluble factors that can activate control bystander cells. Despite exposure to ESCRT-III depletion-induced inflammatory cytokines, co-cultured control MutuDCs exhibit poor export capacity (Figures 5D and 5E). We conclude that enhanced export to the cytosol in ESCRT-III-silenced DCs is cell intrinsic. These results demonstrate that NF-κB activation in ESCRT-III-deficient cells is not sufficient to account for the strong enhancement of cytosolic antigen export and that the effect of ESCRT-III silencing on cytosolic export is cell intrinsic.
Inhibition of necroptosis impairs enhanced antigen export to the cytosol induced by ESCRT-III silencing
ESCRT-III has been shown previously to limit necroptotic cell death by excising portions of the plasma membrane damaged by insertion of MLKL oligomers (Gong et al., 2017). Consequently, reduced ESCRT-III expression results in unrepaired membrane lesions and may also lead to persistence of MLKL pores at the plasma membrane. In agreement with these results, ESCRT-III silencing in cDCs induces necroptosis, as reflected by increased cellular levels of active phospho-RIPK3 and phospho-MLKL (Figure 6A). In addition to necroptosis, cleavage of caspase-8, caspase-3, and Parp (Figure 6B) and caspase-1, caspase-11, and Gasdermin-D (Figure 6C) indicates parallel induction of apoptosis (Figure 6B) and pyroptosis (Figure 6C), respectively (Rühl et al., 2018). Apoptosis and pyroptosis are induced mostly on day 4 after transduction, whereas MLKL oligomers strongly appear on day 3 after transduction (Figure S6A, left panel, “stack” line) before obvious loss of cell viability (Figure S5A). These results show that the pore-forming function of MLKL precedes onset of cell death by at least 24 h, suggesting that necroptosis might be the primary pathway leading to cell death after ESCRT-III silencing.
Figure 6.

RIPK3 kinase inhibition restricts antigen export to the cytosol in ESCRT-III-deficient DCs
(A–C) ESCRT-III-silenced and control MutuDCs were lysed at different times after transduction, and induction of necroptotic (A), apoptotic (B), and pyroptotic (C) mediators was assessed by immunoblotting. Data are representative of 2 independent experiments.
(D) Representative flow cytometry plots after 3 h of incubation with β-lactamase in the presence or absence of 10 μM GSK′872 (gated on live cells).
(E) Quantification of 2 experiments (two-way ANOVA, mean ± SEM, exact p values are indicated). The zero time point corresponds to incubation for 5 h, on ice, with β-lactamase.
Considering that MLKL also assembles on endomembranes (Frank et al., 2019; Yoon et al., 2017), we hypothesized that ESCRT-III may also fulfill its damage-excising function there, limiting endomembrane rupture and subsequent antigen export to the cytosol. To test this possibility, we used GSK′872, a pharmacological inhibitor of RIPK3 (Mandal et al., 2014). By inducing a phosphorylation-dependent conformational change in MLKL (Hildebrand et al., 2014), the RIPK3 kinase governs acquisition of MLKL pore-forming activity and therefore regulates necroptosis. Treatment of CHMP2a-silenced MutuDCs with GSK′872 interferes with MLKL phosphorylation (Figure S6A, right panel) and decreases formation of MLKL oligomers (Figure S6A, left panel), revealing efficient and selective blockade of RIPK3 kinase function, while other cell death pathways are not affected (Figure S6B). GSK′872 also reduces the observed activation of CHMP2a-silenced MutuDCs (Figures S6C and S6D), confirming previous reports (Moriwaki et al., 2014). Although GSK′872 was not sufficient to rescue the viability of ESCRT-III-deficient MutuDCs (which could die by other cell death modalities; Figure S6E), it dramatically impairs their ability to export antigens into the cytosol relative to untreated cells (Figures 6D and 6E). Collectively, these results suggest that ESCRT-III-mediated repair of RIPK3-dependent necroptotic damage targeting endomembranes restrains antigen export to the cytosol.
Antigen cross-presentation is enhanced in ESCRT-III-silenced DCs
To explore whether the increased damage to endocytic membranes and cytosolic export observed after ESCRT-III silencing is associated with enhanced antigen cross-presentation, we first used the OVA-specific B3Z CD8+ T cell hybridoma (Karttunen et al., 1992). We incubated ESCRT-III-silenced or control MutuDCs with soluble (sEF-OVA) or bead-bound endotoxin-free OVA or with the OVA257–264 peptide as a control for MHC class I expression (Figure 7A). Although control MutuDCs fail to cross-present sEF-OVA or bead-bound EF-OVA efficiently, ESCRT-III-deficient DCs show strong enhancement of soluble or bead-coated EF-OVA cross-presentation (Figures 7B, left panel, and S7A). ESCRT-III silencing leads to increased sEF-OVA cross-presentation, mirroring the effects of the same shRNA on cytosolic export (compare with Figures 4B and 4C). This enhancement of cross-presentation was abolished in cells co-expressing shRNA-resistant CHMP4b (Figure S3J). Despite slight variations in MHC class I surface expression in cells silenced with Chmp4b sh#2 and Chmp2a sh#2 (Figures S7B and S7C), the OVA257–264 peptide, which does not require processing, is presented with equal efficiency by control or ESCRT-III-silenced DCs (Figure 7B, right panel). Additionally, MutuDC pre-activation with CpG failed to increase cross-presentation of sEF-OVA in control cells (Figure 7C), suggesting that NF-κB induction has little influence on cross-presentation in this model. We conclude that ESCRT-III is a negative regulator of antigen cross-presentation.
Figure 7.

ESCRT-III silencing enhances cross-presentation in vitro
(A) Timeline for the B3Z assay.
(B and C) Antigen cross-presentation assay with B3Z hybridoma in the presence (C) or absence (B) of CpG. n = at least 3 (B) or 2 (C) experiments (two-way ANOVA, mean ± SEM. Only exact p values of less than 0.05 are shown for clarity).
(D) Timeline for the OT-I and OT-II presentation assays.
(E and F) Antigen cross-presentation assay with OT-II (E) or OT-I (F) T cells. Pooled data are from 3 experiments (two-way ANOVA, mean ± SEM). Only exact p values of less than 0.05 are shown for clarity).
Next, to determine whether ESCRT-III silencing affects MHC class II antigen presentation, we used the OVA-specific CD4+ T cell receptor (TCR)-transgenic T cells OT-II (Figure 7D). As shown in Figure 7E, presentation of sEF-OVA to OT-II CD4+ T cells is unaffected by ESCRT-III depletion, whereas a tendency of increased presentation of OVA323–339 control peptide may reflect their higher MHC class II expression (Figure 7E). In contrast, cross-presentation of soluble EF-OVA to OT-I (OVA-specific CD8+ TCR-transgenic T cells) is increased in ESCRT-III-silenced cells relative to control cells (Figure 7F), again highlighting the crucial role of ESCRT-III in this process.
We next sought to investigate the capacity of ESCRT-III-silenced DCs to cross-prime CD8+ T cells in vivo. To circumvent the embryonic lethality and cell death observed in constitutional (Lee et al., 2007) and conditional (Zhou et al., 2019) knockouts of CHMP4b, respectively, we developed an alternative model: after adoptive transfer of OT-I T cells, CpG-activated ESCRT-III-silenced or control MutuDCs, loaded or not loaded with sEF-OVA or endotoxin-containing OVA, were injected into the footpad. The next day, popliteal draining lymph nodes (pLNs) and inguinal non-draining lymph nodes (iLNs) were harvested, and OT-I T cells were stained for CD69 and CD25 to monitor cross-priming in vivo (Figure S7D). CHMP4b-silenced and control MutuDCs display similar migratory capacities as the pLN (Figure S7E). No differences in frequencies of CD8+ OT-I T cells in the iLNs and pLN were detected on the day of sacrifice (Figure S7F). As an additional control, we also examined OT-I CD8+ T cells in the non-draining iLN and observed no CD8+ T cell activation (Figure S7G, left panel). In the pLN, CHMP4b-deficient cells show increased cross-priming ability compared with control shRNA-transduced cells (Figure S7G, right panel). We conclude that ESCRT-III is a critical regulator of cross-presentation in vitro and in vivo.
Discussion
Understanding how cDCs export antigens from intracellular compartments to the cytosol during cross-presentation has been a major challenge for immunologists since the discovery of this process over 20 years ago (Kovacsovics-Bankowski and Rock, 1995). The strongest evidence so far has suggested a role of the SEC61 translocon in the transport of extracellular antigen into the cytosol and, by extension, in cross-presentation (Ackerman et al., 2006; Koopmann et al., 2000; Zehner et al., 2015). Because SEC61 is required for signal-peptide-dependent import of membrane proteins into the ER, its inhibition targets numerous proteins for cytosolic degradation, including MHC class I. It is therefore difficult to exclude indirect effects of SEC61 blockade on antigen export to the cytosol and cross-presentation.
An alternative model for antigen export to the cytosol proposes local destabilization of endocytic membranes (Childs et al., 2021). In this study, we bring additional evidence supporting the alternative “membrane disruption” hypothesis by showing that ESCRT-III is recruited to repair disrupted intracellular membranes, regulating antigen export to the cytosol and cross-presentation. A body of literature suggests early endosomes to be the main site for antigen leakage into the cytosol (Burgdorf et al., 2008; Howland and Wittrup, 2008; Oura et al., 2011). Our results indicate that early and late endocytic compartments in cDC1s show signs of membrane damage and that this damage might facilitate antigen export during cross-presentation.
Central to understanding the biology of antigen cross-presentation is the mechanism that triggers membrane damage. In this regard, Ploegh (2007) suggested a lipid-based mechanism for antigen export from the ER to the cytosol. The model proposed that lipid body biogenesis involves transient formation of bicellar structures resulting from fusion of the cytoplasmic and luminal sides of the ER. Severing these lipid droplets from the ER could generate a transient pore in the ER membrane (reviewed in Saka and Valdivia, 2012). Considering the similar topology of the ER and intracellular compartments, one could hypothesize that analogous events could mediate cytosolic antigen export from endocytic structures. Along with others, we have shown that IGTP-dependent lipid body accumulation in cDCs correlates with high cross-presentation efficiency (Bougnères et al., 2009), possibly by mediating antigen export to the cytosol. However, pharmacological inhibition of lipid droplet formation proved this process to be dispensable for endosomal leakage of antigens (den Brok et al., 2016). Nevertheless, investigating the lipid composition of intracellular membranes may still be instrumental to decipher the nature of the damage-inducing signal. Recent studies have shown that NOX2-mediated (Canton et al., 2020; Dingjan et al., 2016) ROS production as well as AQP3-dependant endosomal import of mitochondrial ROS (Nalle et al., 2020) induce lipid peroxidation, causing local membrane disruption and endosomal antigen leakage. ROS-driven lipid peroxidation results in severe conformational changes, characterized by increased membrane curvature that destabilizes bilayers (Agmon et al., 2018) and triggers membrane permeability (Wong-Ekkabut et al., 2007), possibly contributing to induction of ferroptosis, a form of regulated cell death. Interestingly, ESCRT-III has been proposed to repair ferroptotic damage (Pedrera et al., 2020), reminiscent of its function in regulation of other cell death pathways: after MLKL activation and necroptosis induction, ESCRT-III ensures cellular integrity by shedding damaged portions of the plasma membrane (Gong et al., 2017). Consequently, silencing of CHMP2a or CHMP4b results in spontaneous necroptosis due to unrepaired MLKL-triggered plasma membrane damage. Given the fact that MLKL also oligomerizes on other biological membranes, it may also drive endomembrane damage and subsequent antigen export to the cytosol in ESCRT-III-silenced cells, as suggested by the present work. Additionally, necroptosis induction is often associated with MLKL-dependent release of mitochondrial ROS (Yang et al., 2018), which contributes to endomembrane rupture and ultimately results in antigen export to the cytosol (Nalle et al., 2020). Necroptosis could also provide insights into the molecular mechanisms triggering the cDC activation observed after ESCRT-III silencing. Indeed, oligomerization of RIPK3, a core protein in necroptosis induction, is responsible for RIPK1-dependent activation of NF-κB (Yatim et al., 2015). Therefore, genetic ablation of RIPK3 or MLKL may not only demonstrate their involvement in antigen export to the cytosol but also help dissociate silencing-mediated enhanced antigen export from cDC activation.
A role of ESCRT-I in antigen export to the cytosol and cross-presentation has been suggested by a previous study, as TSG101 depletion causes an increase in both processes (Zehner et al., 2011). This phenotype, similar to the one described in this study, has been attributed to TSG101’s high structural homology with E2-ubiquitin ligases, allowing its binding to ubiquitin residues. However, unlike E2 enzymes, TSG101 is unable to fulfill the same ubiquitination function and prevents poly-ubiquitination of the mannose receptor, which is required for OVA export to the cytosol in bone-marrow-derived dendritic cells (BMDCs). Because TSG101 silencing has been shown to delay but not abolish ESCRT-III recruitment to damaged endolysosomes (Skowyra et al., 2018), repair defects might also contribute to the previously shown phenotype. In contrast, and despite their requirement for ubiquitinated cargo sorting in multivesicular bodies (MVBs), ESCRT-0 and ESCRT-II seem to be mainly dispensable for biological membrane repair (with the exception of EAP30/VPS22, which is recruited to damaged lysosomes) (Jimenez et al., 2014; Radulovic et al., 2018). ESCRT-0 members were not detected in our proteomics dataset, and the only subunit of ESCRT-II we found (VPS25/EAP20) did not display clear cDC1/cDC2 segregation (enriched in cDC1 early endosomes and in cDC2 late endosomes), suggesting that the enhanced ESCRT-III recruitment in cDC1 endolysosomes may result from a higher response to damage rather than from intense cargo sorting in MVBs.
Our results show that ESCRT-III is a major negative regulator of antigen export to the cytosol and cross-presentation. These results confirm the importance of ESCRT-III as an ancestral and conserved membrane repair complex whose function might have been coopted to tightly and rapidly regulate a crucial pathway of immune responses.
Limitations of the study
Our approach uses a now well-characterized murine cDC1 line (MutuDCs) that allows in-depth characterization of ESCRT-III involvement in containing endomembrane damage and thereby restricting antigen export to the cytosol. Our conclusions require further validation in in vivo models that, for now, have proven inadequate for long cross-presentation experiments owing to the embryonic lethality and cell death observed in constitutional (Lee et al., 2007) and conditional (Zhou et al., 2019) knockouts of CHMP4b, respectively. Although our results suggest that ESCRT-III repairs RIPK3-dependant necroptotic damage targeting endomembranes, they do not exclude a more indirect link between the necroptotic and repair pathways. Endomembrane lipid peroxidation, responsible for local disruptions possibly repaired by ESCRT-III, could originate from RIPK3-dependant production of mitochondrial ROS (Nalle et al., 2020; Yang et al., 2018). Therefore, the precise contributions of RIPK3-dependant MLKL assembly to the mitochondrial or endolysosomal membranes in triggering damage repaired by ESCRT-III remain to be determined.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-CD86-PE (clone GL1) | BD Pharmingen | Cat#553692; RRID: AB_394994 |
| anti-CD69-PE (clone H1.2F3) | BD Pharmingen | Cat#553237; RRID: AB_394726 |
| anti-CD25-PerCPCy5.5 (clone PC61) | BD Pharmingen | Cat#551071; RRID: AB_394031 |
| anti-CD8α-APC (clone 53-6.7) | BD Pharmingen | Cat#553035; RRID: AB_398527 |
| anti-CD8α-Pacific Blue (clone 53-6.7) | BD Pharmingen | Cat#558106; RRID: AB_397029 |
| anti-TCR Vα2-PeCy7 (clone B20.1) | BD Pharmingen | Cat#560624; RRID: AB_1727584 |
| anti-CD8α-PerCPCy5.5 (clone 53-6.7) | BD Pharmingen | Cat#551162; RRID: AB_394081 |
| anti-TCR Vα2-PE (clone B20.1) | eBioScience | Cat#12-5812-82; RRID: AB_465949 |
| anti-MHC-II-APC eFluor780 (clone M5/114.15.2) | eBioScience | Cat#47-5321-82; RRID: AB_1548783 |
| anti-MHC-II-eFluor450 (clone M5/114.15.2) | eBioScience | Cat#48-5321-82; RRID: AB_1272204 |
| anti-MHC-II-PerCPCy5.5 (clone M5/114.15.2) | BD Pharmingen | Cat#562363; RRID: AB_11153297 |
| anti-CD11c-PeCy7 (clone N418) | eBioScience | Cat#25-0114-82; RRID: AB_469590 |
| anti-CD11c-PeCy7 (clone HL3) | BD Pharmingen | Cat#558079; RRID: AB_647251 |
| anti-H-2Kb-AF647 (clone AF6-88.5) | BD Pharmingen | Cat#562832; RRID: AB_2737824 |
| anti-H-2Kb-BV421 (clone AF6-88.5) | BD Pharmingen | Cat#562942; RRID: AB_2737908 |
| anti-CD44-FITC (clone IM7) | BD Pharmingen | Cat#553133; RRID: AB_2076224 |
| anti-CD62L-PeCy7 (clone MEL-14) | BD Pharmingen | Cat#560516; RRID: AB_1645257 |
| anti-CD4-APC (clone RM4-5) | BD Pharmingen | Cat#553051; RRID: AB_398528 |
| anti-CD11b-PerCPCy5.5 (clone M1/70) | eBioScience | Cat#45-0112-82; RRID: AB_953558 |
| anti-CD8α-FITC (clone 53-6.7) | BD Pharmingen | Cat#553031; RRID: AB_394569 |
| anti-TCRβ-PE (clone H57-597) | BD Pharmingen | Cat#553172; RRID: AB_394684 |
| anti-CD16/CD32 (mouse Fc block) | BD Pharmingen | Cat#553142; RRID: AB_394657 |
| anti-chicken egg albumin | Sigma Aldrich | Cat#C6534; RRID: AB_258953 |
| anti-Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 647 | ThermoFisher | Cat#A21245; RRID: AB_141775 |
| anti-LAMP-1-biotin (clone 1D4B) | eBioScience | Cat#13-1071-82; RRID: AB_657544 |
| anti-galectin-3-PE (clone M3/38) | eBioScience | Cat##12-5301-82; RRID: AB_842792 |
| anti-IST1 | ProteinTech | Cat##19842-1-AP; RRID: AB_2878612 |
| anti-Rabbit IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 405 | ThermoFisher | Cat#A-31556, RRID: AB_221605 |
| anti-CD8-Alexa Fluor 647 (clone 53-6.7) | BD Pharmingen | Cat#557682; RRID: AB_396792 |
| anti-CD11b-Alexa Fluor 488 (clone M1/70) | BD Pharmingen | Cat##557672; RRID: AB_396784 |
| anti-caspase-3 | Cell Signaling Technology | Cat#9662; RRID: AB_331439 |
| anti-cleaved caspase-8 | Cell Signaling Technology | Cat#9429; RRID: AB_2068300 |
| anti-caspase-1 | Schotte et al. (2004) | N/A |
| anti-caspase-11 | Novus Biologicals | Cat#120–10454; RRID: AB_788441 |
| anti-cleaved Parp | Cell Signaling Technology | Cat#9544; RRID: AB_2160724 |
| anti-Gasdermin-D | Genentech | N/A |
| anti-IL-1β | Genetex | Cat#GTX74034; RRID: AB_378141 |
| anti MLKL | Millipore | Cat#MABC604; RRID: AB_2820284 |
| anti MLKL | Sigma Aldrich | Cat#SAB1302339; RRID: AB_2687464 |
| anti-phosphoMLKL | Abcam | Cat#ab196436; RRID: AB_2687465 |
| anti-RIPK3 | Sigma Aldrich | Cat#R4277; RRID: AB_477454 |
| anti-RIPK1 | BD Biosciences | Cat#610459, RRID: AB_397832 |
| anti-Chmp4b | Cell Signaling Technology | Cat#42466S |
| anti-actin | Millipore | Cat#MAB1501; RRID: AB_2223041 |
| anti-Rabbit IgG (H + L) | Jackson ImmunoResearch Labs | Cat#111-035-144; RRID: AB_2307391 |
| anti-Mouse IgG (H + L) | Jackson ImmunoResearch Labs | Cat#115-035-146; RRID: AB_2307392 |
| Chemicals, peptides, and recombinant proteins | ||
| IMDM | Sigma Aldrich | Cat#I3390-500ML |
| CO2-independent medium | Gibco | Cat#18045088 |
| HEPES | Life Technologies | Cat#15630080 |
| Glutamax | Life Technologies | Cat#35050061 |
| Penicillin/Streptomycin | Life Technologies | Cat#15140122 |
| β-mercaptoethanol | Life Technologies | Cat#31350010 |
| RPMI-Glutamax | Gibco | Cat#61870010 |
| Sodium pyruvate | Life Technologies | Cat#11360070 |
| MEM non-essential amino acids | Life Technologies | Cat#11140035 |
| DMEM-Glutamax | Gibco | Cat#31966021 |
| Optimem | Life Technologies | Cat#31985062 |
| TransIT-LT1 | Mirus Bio | Cat#MIR2300-US |
| Bovine Serum Albumin (BSA) solution | Sigma Aldrich | Cat#A7979-50ML |
| Puromycin | Invivogen | Cat#ant-pr-1 |
| ODN 2395 | Invivogen | Cat#tlrl-2395-1 |
| Β-lactamase | Sigma Aldrich | Cat#P0389 |
| Probenecid | ThermoFisher | Cat#P36400 |
| MG-132 | Sigma Aldrich | Cat#C2211-5MG |
| Nitrocefin | Calbiochem | Cat#484400-5MG |
| Ovalbumin, Alexa Fluor™ 647 Conjugate | ThermoFisher | Cat#O34784 |
| Albumin from chicken egg white | Sigma Aldrich | Cat#A7641-250MG |
| H-2Kb (MHC-I)-restricted OVA(257–264) peptide | Invivogen | Cat#vac-sin |
| I-Ab (MHC-II)-restricted OVA(323–339) peptide | Invivogen | Cat#vac-isq |
| BSA-DQ | ThermoFisher | Cat#D12051 |
| BSA, Alexa Fluor™ 647 Conjugate | ThermoFisher | Cat#A34785 |
| Trizol | ThermoFisher | Cat#15596018 |
| MAXIMA Retrotranscriptase | ThermoFisher | Cat#EP0741 |
| RNasin® Ribonuclease Inhibitor | Promega | Cat#N2511 |
| CellTracker™ DeepRed Dye | ThermoFisher | Cat#C34565 |
| Glutaraldehyde | Euromedex | Cat#16200 |
| Glycine | Invitrogen | Cat#15527013 |
| Leu-Leu methyl ester hydrobromide | Sigma Aldrich | Cat#L7393-500MG |
| 2-deoxy-D-glucose | Sigma Aldrich | Cat#D8375 |
| Imidazole | Sigma Aldrich | Cat#I202 |
| DTT | ThermoFisher | Cat#P2325 |
| cOmplete™, EDTA-free Protease Inhibitor Cocktail | Sigma Aldrich | Cat#000000011873580001 |
| EndoGrade Ovalbumin | Hyglos | Cat#321001 |
| CPRG | Sigma Aldrich | Cat#000000010884308001 |
| Liberase™ TL Research Grade | Sigma Aldrich | Cat#05401020001 |
| DNAse I | Sigma Aldrich | Cat#11284932001 |
| 16% Paraformaldehyde | Electron Microscopy Science | Cat#15710 |
| Poly-L-Lysine 0,1% | Sigma Aldrich | Cat#P8920 |
| Dextran, Tetramethylrhodamine, 3000 MW, Anionic, Lysine Fixable | ThermoFisher | Cat#D3308 |
| Dextran, Tetramethylrhodamine, 10,000 MW, Lysine Fixable | ThermoFisher | Cat#D1817 |
| Dextran, Tetramethylrhodamine, 70,000 MW, Lysine Fixable | ThermoFisher | Cat#D1818 |
| Prazosin | Sigma Aldrich | Cat#P7791 |
| Glucose Solution | ThermoFisher | Cat#A2494001 |
| RIPK3 inhibitor, GSK′872 | Merck | Cat#5303890001 |
| SDS | Euromedex | Cat#EU0660 |
| Bromophenol Blue | Sigma Aldrich | Cat#B-5525 |
| Tris Base | Sigma Aldrich | Cat#10708976001 |
| Laemmli | BioRad | Cat#1610747 |
| 10X Tris-Buffered Saline (TBS) | BioRad | Cat#1706435 |
| Critical commercial assays | ||
| LIVE/DEAD™ fixable violet dead cell kit | ThermoFisher | Cat#L34955 |
| Fixable Viability Dye eFluor™ 780 | eBioScience | Cat#65-0865-14 |
| Nucleobond Xtra Midi EF kit | Macherey-Nagel | Cat#740420.10 |
| LiveBLAzer FRET-B/G Loading Kit | ThermoFisher | Cat#K1095 |
| RNeasy Micro Kit | Qiagen | Cat#74004 |
| Mouse Dendritic Cell Nucleofector™ Kit | Lonza | Cat#VPA-1011 |
| Naive CD8+ T cell Isolation Kit, mouse | Miltenyi | Cat#130-096-543 |
| Naive CD4+ T cell Isolation Kit, mouse | Miltenyi | Cat#130-104-453 |
| CellTrace™ Violet Cell Proliferation Kit | ThermoFisher | Cat#C34557 |
| SiR-DNA kit | Spirochrome | Cat#SC007 |
| Pan Dendritic Cell Isolation Kit, mouse | Miltenyi | Cat#130-100-875 |
| Pierce™ BCA Protein Assay Kit | ThermoFisher | Cat#23250 |
| Experimental models: Cell lines | ||
| MutuDC | Fuertes Marraco et al. (2012) | N/A |
| B3Z | Sanderson and Shastri (1994) | RRID: CVCL_6277 |
| HEK293T | ATCC | RRID: CVCL_0063 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J (wild type) | Charles River | Cat#632 |
| Mouse: B6.129S-Cybbtm1Din/J | Jackson Laboratory | Cat#002365; RRID: IMSR_JAC:002,365 |
| Oligonucleotides | ||
| Oligo(dT)15 primer | Promega | Cat#C1101 |
| Chmp4b Taqman Gene Expression Assay | ThermoFisher | Cat#Mm00551493_m1 |
| Chmp2a Taqman Gene Expression Assay | ThermoFisher | Cat#Mm00509883_m1 |
| Tnf Taqman Gene Expression Assay | ThermoFisher | Cat#Mm00443258_m1 |
| Il1b Taqman Gene Expression Assay | ThermoFisher | Cat#Mm00434228_m1 |
| Relb Taqman Gene Expression Assay | ThermoFisher | Cat#Mm00485664_m1 |
| Hprt Taqman Gene Expression Assay | ThermoFisher | Cat#Mm03024075_m1 |
| Gapdh Taqman Gene Expression Assay | ThermoFisher | Cat#Mm99999915_g1 |
| shRNA-resistant (exogenous) Chmp4b Taqman Gene Expression Assay (Custom) | ThermoFisher | Cat#APRWJAV |
| shRNA-sensitive (endogenous) Chmp4b Taqman Gene Expression Assay (Custom) | ThermoFisher | Cat#APPRPPX |
| Recombinant DNA | ||
| Plasmid: psPax2 | Addgene | Cat#12260; RRID:Addgene_12260 |
| Plasmid: pMD2.G | Addgene | Cat#12259; RRID:Addgene_12259 |
| Plasmid: Chmp4b shRNA (#SHCLNG-NM_029362) #2 | Sigma Aldrich | Cat#TRCN0000105502 |
| Plasmid: Chmp4b shRNA (#SHCLNG-NM_029362) #3 | Sigma Aldrich | Cat#TRCN0000105503 |
| Plasmid: Chmp2a shRNA (#SHCLNG-NM_026885) #1 | Sigma Aldrich | Cat#TRCN0000182144 |
| Plasmid: Chmp2a shRNA (#SHCLNG-NM_026885) #2 | Sigma Aldrich | Cat#TRCN0000198105 |
| Plasmid: pLKO.1-puro Non-Mammalian shRNA Control Plasmid DNA | Sigma Aldrich | Cat#SHC002 |
| Plasmid: pmCherry-N1-(human)Chmp4b | Jimenez et al. (2014) | N/A |
| Plasmid: pL-SFFV.Reporter.RFP657.PAC | Addgene | Cat#61395; RRID: Addgene_61395 |
| Plasmid: pL-SFFV.shRNA-resistant Chmp4b.RFP657.PAC | This paper | N/A |
| Software and algorithms | ||
| FlowJo version 9.3 | Tree Star | http://www.flowjo.com |
| GraphPadPrism version 9 | GraphPad Software | N/A |
| ImageJ/FIJI | Schindelin et al. (2012) | https://fiji.sc/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sebastian Amigorena (sebastian.amigorena@curie.fr).
Materials availability
Plasmid encoding a shRNA-resistant version of murine CHMP4b will be made available on request.
Experimental model and subject details
Animals
C57BL/6J mice were obtained from Charles River Laboratories. NOX2-deficient KO animals (B6.129S-Cybbtm1Din/J) were obtained from the Jackson Laboratory. C57BL/6N-recombination activating gene 1 deficient OT-I and OT-II TCR (Vα2, Vβ5.1) transgenic mice were bred at Institut Curie. Female mice were used, except for OT-I and OT-II isolation on Figures 7D, 7E and 7F (animals from both sexes were used). All mice used were less than six months old, except for one experiment with NOX2 KO mice in which animals were 9.5 months old (age-matched co-housed controls were used in that case). Animal care and use for this study were performed in accordance with the recommendations of the European Community (2010/63/UE) for the care and use of laboratory animals. Experimental procedures were specifically approved by the ethics committee of the Institut Curie CEEA-IC #118 (CEEA-IC, 2018-024) in compliance with the international guidelines.
Cell lines and cell culture
MutuDC 1940 (obtained from Hans Acha Orbea) cells were grown in IMDM (Sigma-Aldrich, #I3390-500ML), supplemented with 8% FCS (Biosera or Eurobio), 10 mM HEPES (#15630080), 2 mM glutamax (#35050061), 100 U/mL penicillin, 100 μg/mL streptomycin (#15140122) and 50 μM β-mercaptoethanol (#31350010) (all from Life Technologies). B3Z hybridomas were grown in RPMI-Glutamax (Gibco, #61870010), supplemented with 10% FCS (Biosera or Eurobio), 10 mM HEPES, 1 mM sodium pyruvate (#11360070), 1% MEM non-essential amino acids (#11140035), 100 U/mL penicillin, 100 μg/mL streptomycin and 50 μM β-mercaptoethanol (all from Life Technologies). HEK293T were grown in DMEM-Glutamax (Gibco, #31966021), supplemented with 10% FCS (Biosera or Eurobio).
All cell lines used tested negative from Mycoplasma by PCR.
Splenic dendritic cells were grown in RPMI-Glutamax (Gibco, #61870010), supplemented with 10% FCS (Biosera), 10 mM HEPES, 1 mM sodium pyruvate (#11360070), 1% MEM non-essential amino acids (#11140035), 100 U/mL penicillin, 100 μg/mL streptomycin and 50 μM β-mercaptoethanol (all from Life Technologies).
Method details
Flow cytometry
For flow cytometry, the following antibodies were used: anti-CD86-PE (clone GL1, BD Pharmingen #553692), anti-CD69-PE (clone H1.2F3, BD Pharmingen #553237), anti-CD25-PerCPCy5.5 (clone PC61, BD Pharmingen #551071), anti-CD8α-APC (clone 53-6.7, BD Pharmingen #553035), anti-CD8α-Pacific Blue (clone 53-6.7, BD Pharmingen #558106), anti-TCR Vα2-PeCy7 (clone B20.1, BD Pharmingen #560624), anti-CD8α-PerCPCy5.5 (clone 53-6.7, BD Pharmingen, #551162), anti-TCR Vα2-PE (clone B20.1, eBioscience #12-5812-82), anti-MHC-II-APC eFluor780 (clone M5/114.15.2, eBioscience #47-5321-82), anti-MHC-II-eFluor450 (clone M5/114.15.2, eBioscience #48-5321-82), anti-MHC-II-PerCPCy5.5 (clone M5/114.15.2, BD Pharmingen #562363), anti-CD11c-PeCy7 (clone N418, eBioscience #25-0114-82), anti-CD11c-PeCy7 (clone HL3, BD Pharmingen #558079), anti-H-2Kb-AF647 (clone AF6-88.5, BD Pharmingen #562832), anti-H-2Kb-BV421 (clone AF6-88.5, BD Pharmingen #562942), anti-CD44-FITC (clone IM7, BD Pharmingen #553133), anti-CD62L-PeCy7 (clone MEL-14, BD Pharmingen #560516), anti-CD4-APC (clone RM4-5, BD Pharmingen, #553051), anti-CD11b-PerCPCy5.5 (clone M1/70, eBioscience #45-0112-82), anti-CD8α-FITC (clone 53-6.7, BD Pharmingen #553031), anti-TCRβ-PE (clone H57-597, BD Pharmingen #553172).
Cells were transferred in conical-bottom well plates (Greiner Bio-One, #651201) and stained with Fc Block (BD Pharmingen, #553142) and fixable Live/Dead dye violet (ThermoFisher, #L34955) or fixable Live/Dead Dye eFluor780 (eBioscience, #65-0865-14) diluted in PBS, for 20 min, on ice. Cells were then washed in FACS buffer (PBS, 0.5% BSA, 2 mM EDTA) and labeled for surface staining with the aforementioned antibodies or corresponding isotype controls, for 25 min, on ice.
Lentiviral shRNA knockdown of CHMP4b and CHMP2a
Lentivirus production
Plasmids encoding lentiviruses expressing shRNAs were obtained from the library of The RNAi Consortium (TRC) (Moffat et al., 2006). Plasmids were purified with the Nucleobond Xtra Midi EF kit (Macherey-Nagel, #740420.10). Transfection was conducted as follows: HEK293T cells, plated the day before at a density of 3.5 × 106 cells/10cm Petri dish, were transfected with a three-plasmid system (psPax2 7 μg/dish, vsv-g 0.7 μg/dish and plasmid encoding shRNA 7 μg/dish), all diluted in 1 mL Optimem #31985062 from Life Technologies and supplemented with 45 μL TransIT-LT1 (#MIR2300-US, Mirus Bio) to produce lentivirus. After mixing by slow up-and-down pipetting, transfecting complexes were incubated for 30min at room temperature, before dropwise addition on cells, evenly over the surface of the dish.
Eighteen hours after transfection, medium was replaced by DMEM, 10% FCS and 2 mM glutamax supplemented with 1% BSA (Sigma-Aldrich, #A7979-50ML). Supernatants were harvested 48h after transfection and filtered on 0.22 μm filter units (#SCGP00525, Merck Millipore). Supernatants were frozen at −80°C and aliquots were only thawed once. Titration was performed on thawed virus supernatants: the dose used for transduction was the minimal virus dose that does not lead to mortality after puromycin selection (i. e. 100 μL per well in a 6-well plate in our system).
The following shRNAs were produced (all from Sigma-Aldrich): Chmp4b sh#2 (target sequence CCGGGCAGGCTCTGAAGCGCAAGAACTCGAGTTCTTGCGCTTCAGAGCCTGCTTTTTG, #SHCLNG-NM_029362 TRCN0000105502), Chmp4b sh#3 (target sequence CCGGAGATGACGACATGAAGGAATTCTCGAGAATTCCTTCATGTCGTCATCTTTTTTG, #SHCLNG-NM_029362 TRCN0000105503), Chmp2a sh#1 (target sequence CCGGCAAGCCATGAAGGGTGTTACTCTCGAGAGTAACACCCTTCATGGCTTGTTTTTTG, #SHCLNG-NM_026885, TRCN0000182144), Chmp2a sh#2 (target sequence CCGGCCTCAAGATACAGACTCTAAACTCGAGTTTAGAGTCTGTATCTTGAGGTTTTTTG, # SHCLNG-NM_026885 TRCN0000198105) and the non-targeting control (#SHC002, Sigma Aldrich).
Lentiviral transduction
MutuDC were plated in 6 well plates (TPP, #92006) at a concentration of 0.4 × 106 cells per well in 2 mL of aforementioned medium, 3h before virus transduction. Fifteen hours after transduction, viruses were removed; cells were washed once with medium and put back to incubation. The next day (= day 2 post transduction), cells were selected with 2 μg/mL of puromycin (#ant-pr-1, Invivogen). Cells were collected for analysis 24h after selection (= day 3 post transduction), a sufficient delay for puromycin-mediated cell killing.
Induction of DC maturation
Maturation of MutuDC was induced by a 16h treatment preceding the harvesting of ESCRT-III-deficient or control cells on day 3 post-transduction. For this purpose, CpG (ODN 2395, #tlrl-2395-1, Invivogen) at a concentration of 1 μg/mL was used. DC maturation was controlled both by surface staining for MHC-I, MHC-II and co-stimulatory (CD86) molecules and by RT-qPCR for NF-κB activation markers (TNF-α, IL-1β and RelB).
Β-lactamase export assay
MutuDC were seeded at 0.2 × 106 cells/well (or 0.15 × 106 cells/well for rescue and GSK′872 experiments) in round-bottom 96-well plates (Falcon, #353077) and incubated for various timepoints (5h, 3h or 1h) at 37°C with 2 mg/mL β-lactamase (#P0389, Sigma-Aldrich – it must be noted that some batches of this reagent are toxic for cells and each batch should therefore be tested for cell toxicity). As negative controls, MutuDC were incubated with β-lactamase for 5h at 4°C, or without β-lactamase for 5h at 37°C. Cells were then washed extensively in ice-cold PBS and loaded with 1 μM of the CCF4 probe during 1h at room temperature in EM buffer (120mM NaCl, 7mM KCl, 1.8mM CaCl2, 0.8mM MgCl2, 5mM glucose and 25mM HEPES at pH 7.3) (Cebrian et al., 2011) supplemented with 1:20 solution B (ThermoFisher, #K1095) and 1:100 (from a 250 mM stock concentration) probenecid (ThermoFisher, #P36400). In all steps following CCF4 loading, probenecid, a non-specific inhibitor of anion transport, was added in media and buffers to retain CCF4 in the cytosol. To increase the assay’s sensitivity, plates were then incubated for 16h, at room temperature in CO2-independent (ThermoFisher, #18045088) medium supplemented with 8% FCS, 2 mM glutamax and 1:100 probenecid. Immediately before flow cytometry analysis, cells were stained with eFluor-780 viability dye (#65-0865-14, eBioscience) diluted 1:2500 in PBS. Proportion of live cells with a high V450/V500 fluorescence ratio was used as readout for their exporting capacity.
Splenic dendritic cells were stained with live/dead dye and CD11c, CD8α and CD11b markers right after CCF4 loading, and analyzed by flow cytometry. Overnight incubation was not performed in that case to avoid high cell mortality.
Alternatively, cells were seeded at 0.2 × 106 cells/well in round-bottom 96-well plates and incubated with or without MG-132 10 μM (#C2211-5MG, Sigma-Aldrich) for 30 min at 37°C, before addition of 2 mg/mL β-lactamase. MG-132 was maintained at a concentration of 10 μM during the whole course of the assay (including β-lactamase incubation, CCF4 loading and overnight incubation steps). Similarly, in experiments using GSK′872, the inhibitor was also maintained during the whole course of the assay.
Samples were acquired on a FACS Verse (BD Biosciences) and analyzed using the FlowJo software (Tree Star).
Nitrocefin assay
Control or ESCRT-III-deficient MutuDC were seeded at 0.2 × 106 cells/well in flat-bottom 96-well plates and incubated for 20min or 2h with or without 2 mg/mL β-lactamase (#P0389, Sigma-Aldrich), at 37°C. Cells were then washed three times with ice-cold PBS, detached and resuspended in cold buffer containing 50 mM NaH2PO4, 300mM NaCl, 10mM imidazole and 5mM mercaptoethanol pH = 8 (Cebrian et al., 2011) at a concentration of 1 × 106 cells/100 μL buffer. Samples were then sonicated (5 rounds of sonication, 30s ON, 30s OFF each) on Bioruptor Pico (Diagenode) and centrifuged at 16 000g, 15 min, 4°C to remove insoluble material. Lysates were then incubated for 1h, at 37°C, with 100 μM nitrocefin (#484400-5MG, Calbiochem), a chromogenic lactamase substrate that undergoes distinctive color change from yellow to red (486 nm) upon lactamase hydrolysis. After 1h of incubation, optical density at 490 nm was measured by spectrophotometry. Soluble β-lactamase at 5 mg/mL was used as a positive saturation control.
Antigen uptake assay
ESCRT-III-deficient or control MutuDC were seeded at 0.2 × 106 cells/well in round-bottom 96-well plates and incubated at 37°C for various timepoints (15, 30, 60 or 180 min) with or without OVA-AF647 (#O34784, ThermoFisher) + unlabeled grade VII OVA (#A7641-250MG, Sigma-Aldrich) at the following concentration: 0.25 mg/mL each, for a total or 0.5 mg/mL OVA concentration. Cells were then washed extensively with ice-cold PBS before DAPI viability staining and subsequent flow cytometry analysis.
BSA-DQ assay
ESCRT-III-deficient or control MutuDC were pulsed with BSA-DQ (#D12051, ThermoFisher) and BSA-AF647 (#A34785, ThermoFisher), used as an internal uptake control, at a concentration of 0.25 mg/mL each for 20 min at 16°C (to avoid endolysosomal fusion), in CO2-independent medium. Cells were then washed extensively in ice-cold PBS, resuspended in MutuDC medium and distributed in round-bottom 96 well plates at 0.2 × 106 cells/well. At various timepoints (30, 45, 60, 120 or 240 min), cells were washed in ice-cold PBS, stained for viability with DAPI and analyzed by flow cytometry.
Samples were acquired on a MACSQuant VYB (Miltenyi) and analyzed using the FlowJo software (Tree Star).
RT-qPCR
Cells were harvested and lysed in 500 μL-1 mL Trizol buffer (#15596018, ThermoFisher). Chloroform was added to samples (200 μL per mL Trizol) that were subsequently mixed thoroughly before spinning on a tabletop centrifuge (10000g, 18min, 4°C). Aqueous phase containing RNA was collected, pooled to an equal volume of absolute ethanol and RNA extracted on RNAeasy micro kit columns (#74004, QIAGEN), according to manufacturer’s instructions (DNA digestion on column, using RNAse-free DNAse was included in the process). RNA concentrations were measured using Nanodrop 2000 (#ND-2000, ThermoFisher Scientific). 1 μg total RNA was retro-transcribed in a final volume of 20 μL using the MAXIMA retrotranscriptase (#EP0741, ThermoFisher) in combination with oligo dT (#C1101, Promega), RNase inhibitor (#N2511, Promega) and dNTPs (Promega), following manufacturer’s instructions. cDNA was diluted 10 times in nuclease-free ddH2O for further use. Transcripts were quantified by real time PCR on a 480 LightCycler instrument (Roche), using 384-well plates. Reactions were carried out in 10 μL, using master mix (#05-QP2X-03 + WOUN, Eurogentec) and the following Taqman Assays (ThermoFisher): Chmp4b (#Mm00551493_m1), Chmp2a (#Mm00509883_m1), Tnf (#Mm00443258_m1), Il1b (#Mm00434228_m1), Relb (#Mm00485664_m1), Hprt (#Mm03024075_m1), Gapdh (#Mm99999915_g1). For each well, 5 μL master mix, 0.5 μL 20X Taqman assay and 4.5 μL diluted cDNA were used. The relative expression of genes was calculated with the formula 2−ΔCt, where ΔCt = Ct target gene – Ct endogenous control gene (Livak and Schmittgen, 2001). The mean of Hprt and Gapdh housekeepting gene CTs was used in the aforementioned formula.
Co-culture assay
MutuDC were stained for 20 min, at 37°C, with 5 μM Deep Red Dye (#C34565, Invitrogen) or left unstained, before transduction with control shRNA or Chmp4b sh#2/Chmp2a sh#2 respectively. Fifteen hours after transduction, Chmp4b sh#2/Chmp2a sh#2 or control shRNA-transduced cells were extensively washed and pooled in equal amounts. As control, an activation staining (MHC-II, MHC-I and CD86) was performed to ensure that cells were not yet activated at the time of pooling (data not shown). At day 3 post-transduction, β-lactamase assay was performed on pooled ESCRT-III-silenced MutuDC and control cells (or on cells cultured in separate wells) as described previously.
Of note, Deep Red dye staining does not lead to MutuDC activation and still allows discrimination of labeled versus unlabeled cells until the β-lactamase assay readout by flow cytometry.
Transfection
Plasmid encoding the CHMP4b-mCherry (human CHMP4b sequence) was a kind gift of Franck Perez and described in (Jimenez et al., 2014). 3 × 106 MutuDC were washed and resuspended in 100 μL electroporation buffer (#VPA-1011, Lonza) containing 1 μg of the plasmid. Cells were then electroporated using Nucleofector device 2b (Lonza) (Y-001 program). Cells were recovered and diluted in warmed MutuDC medium for 6h before PhagoFACS assay.
PhagoFACS
Bead coating
Bead preparation was described elsewhere (Hoffmann et al., 2016). Briefly, 3 μm polybeads (#17145-5, Polysciences), were washed, resuspended in 1 mL PBS 8% glutaraldehyde (#16200, Euromedex) and incubated for 4h at room temperature, on a rotating wheel. Then, beads were washed again, resuspended in 0.5 mg/mL OVA (Worthington Biochemicals), and incubated overnight, on a rotating wheel, at 4°C. After overnight incubation, cells were washed, resuspended in 0.5M glycine (#15527013, Invitrogen) and incubated for 30min, at 4°C on a rotating wheel. After two more PBS washes, beads were resuspended in initial volume of CO2-independent medium supplemented with 4 mM glutamax.
Assay
MutuDC were incubated with 3 μm OVA-coated beads (with a bead:cell ratio of 10:1) for 25 min at 16°C, followed by 5 min of incubation at 37°C. Cells were immediately washed in ice-cold PBS, three times, and the remaining external beads were removed using an FCS gradient. Cells were then resuspended in pre-warmed PBS 8% FCS, 10 mM HEPES, containing or not 1 mM Leu-Leu methyl ester hydrobromide (#L7393-500MG, Sigma-Aldrich) in presence or absence of ATP. ATP deprivation was induced by addition of 10 mM NaN3 + 5 mM 2-deoxy-D-glucose (#D8375, Sigma-Aldrich) in glucose-free PBS 8% FCS, 10 mM HEPES. After various timepoints, cells were washed and outside beads were stained with anti-OVA (#C6534, Sigma-Aldrich) followed by anti-rabbit-AF647 staining (#A21245, ThermoFisher). Cells were then resuspended in homogenization buffer (PBS, 3 mM imidazole (#I202, Sigma-Aldrich), 8,6% sucrose, 2 mM DTT (#P2325, ThermoFisher), 2X protease inhibitors (#000000011873580001, Sigma-Aldrich), pH = 7.4) and mechanically disrupted with 2 mL syringes and 22-gauge needles (#050105B, Dutscher). Intact cells and nuclei were removed by a centrifugation at 150g for 4min at 4°C and postnuclear supernatant was then transferred to conical bottom-well plate. Non-specific binding was blocked by incubation in PBS 1% BSA, followed by labeling of phagosomes with anti-OVA (#C6534, Sigma-Aldrich) or anti-LAMP-1 (clone 1D4B, eBioscience #13-1071-82) antibodies (or isotype controls) and appropriate secondary antibodies. External AF647+ beads were excluded from the analysis. CHMP4b-mCherry expression was directly measured by flow cytometry. Samples were acquired on a MACSQuant VYB (Miltenyi) and analyzed using the FlowJo software (Tree Star).
Antigen presentation assay
Bead coating
3 μm beads (#17134, Polysciences) were coated with endotoxin-free OVA (#321001, Hyglos) by adsorption, as previously described (Alloatti et al., 2016). Briefly, 250 μL of beads were washed three times with PBS, before resuspension in final volume of 750 μL of various OVA:BSA concentration ratios (100% OVA = 10 mg/mL OVA, 50% OVA:50% BSA (Sigma-Aldrich, #A7979-50ML) = 5 mg/mL OVA and 5 mg/mL BSA, 25% OVA: 75% BSA = 2.5 mg/mL OVA and 7.5 mg/mL BSA, and 0% OVA:100% BSA = 10 mg/mL BSA). After overnight incubation, at 4°C, on a rotating wheel, beads were washed three times in PBS, and finally resuspended in 250 μL MutuDC culture medium.
Assay
ESCRT-III-deficient or control shRNA-transduced cells were plated at 0.1 × 106 cells/well in round-bottom 96-well plates and incubated for 5h with soluble or bead-coated (bead:cell ratio 170:1) endotoxin-free OVA (#321001, Hyglos), or with OVA257-264 peptide, as a control of DC′ ability to activate T cells. Then, DC were washed twice with PBS, fixed with 0.008% glutaraldehyde (#16200, Euromedex) for 3min at room temperature, washed twice with 0.4M glycine (#15527013, Invitrogen), once with B3Z medium and finally B3Z hybridoma cells were added (0.1 × 106 cells/well). After 16h of activation, cells were washed, and CPRG (#000000010884308001, Roche) β-galactosidase substrate was added for 4h, at 37°C before optical density measurement (590 nm) with a PerkinElmer Wallac 1420 Victor2 plate reader (#8381-30-1005). For experiments with beads, optical density was measured after incubation for 4h at 37°C, followed by 15h at room temperature.
Alternatively, DC′ ability to cross-present antigens via MHC-I or present antigens via MHC-II was evaluated using OT-I and OT-II T cells, respectively. OT-I and OT-II T cells were purified from spleen and lymph nodes using naive CD8+ T cells (#130-096-543, Miltenyi) or naive CD4+ T cells (#130-104-453, Miltenyi) isolation kits. Purity was routinely above 95%. MutuDC were plated at 0.1 × 105 cells/well in round-bottom well place and incubated for 5h with the aforementioned antigens. Cells were then washed twice with PBS, twice with medium, left unfixed, and 1.0 × 105 cells/well OT-I or 0.6 × 105 cells/well OT-II T cells were added per well. After 16h of activation, T cells were stained for CD25 and CD69 to monitor T cell activation. Samples were acquired on a FACS Verse (BD Biosciences) and analyzed using the FlowJo software (Tree Star).
In vivo cross-priming assay
Protocol was adapted from Pace et al., (2012). Briefly, mice were intravenously injected with 4 × 106 purified OT-I CD8+ T cells labeled with 5 μM CellTrace Violet (#C34557, ThermoFisher). The next day, CpG-activated (ODN 2395, #tlrl-2395-1, Invivogen, 1 μg/mL for 12-16h) CHMP4b-deficient or control MutuDC were left unloaded or loaded with 3 mg/mL endotoxin-free OVA or 3 mg/mL grade VII OVA for 1h, at 37°C. MutuDC were then extensively washed in PBS before injection (0.5 × 106 cells/footpad) in the footpads. 24h after MutuDC injection, inguinal and popliteal lymph nodes were harvested, mechanically disrupted and stained for CD25 and CD69 to monitor T cell activation. Samples were acquired on a MACS Quant Analyzer 10 (Miltenyi) or FACS Verse (BD Biosciences) and analyzed using the FlowJo software (Tree Star).
To evaluate migration of CHMP4b-deficient or control MutuDC in vivo, cells were activated with CpG (1 μg/mL, 16h) and stained with 5 μM Deep Red Dye (#C34565, Invitrogen) before injection in the footpad (0.5 × 106 cells/footpad). 24h after injection, popliteal lymph nodes were digested with liberase (0,3 mg/mL, #05401020001, Roche) in presence of DNAse (0,1 mg/mL, #11284932001, Sigma-Aldrich) for 30 min at 37°C. Following mashing of the residual lymph nodes pieces through a 40 μm strainer, cell suspensions were stained with DAPI and acquired on FACS Verse (BD Biosciences).
ESCRT-III recruitment and galectin-3 staining in MutuDC
LLOME kinetics
50,000 Ctrl, CHMP4b-deficient or CHMP2a-deficient MutuDC were plated on 0.01% poly-L-lysine-coated multiwell chambered coverslips (#C24779, ThermoFisher), left to adhere for 30min, and treated with 0.5 mM LLOME (L7393-500MG, Sigma-Aldrich) for 0, 15 or 30 min at 37°C. Cells were fixed with pre-warmed 2% PFA (#15710; Electron Microscopy Science) for 20 min at RT and washed with PBS. Cells were then permeabilized with PBS, 0.1% Triton X-100 for 2 min at RT, washed with PBS, and blocked for 1h in PBS 5% BSA for 1h at 37°C. Finally, intracellular staining was achieved with anti-galectin3-PE (1 μg/mL, clone M3/38, #12-5301-82, eBioscience) and when specified with anti-IST1 (1:100, #19842-1-AP, Proteintech), overnight at 4°C. The next day, cells were washed three times in PBS, stained with goat anti-rabbit Alexa Fluor 405 (1:100, Life Technologies, A-31556) for 1h at RT or with DAPI, mounted in PBS and observed at the microscope within the week.
Evaluation of plasma membrane integrity concomitant to galectin 3 staining
5 × 105 Ctrl or CHMP4b-deficient MutuDC were stained with SiR-DNA according to manufacturer’s instructions (Spirochrome #SC007, 1μM final), for 1h at 37°C. Cells were then plated on 0.01% poly-L-lysine-coated multiwell chambered coverslips (#C24779, ThermoFisher) and left to adhere for 30 min. Cells were washed in PBS and incubated with DAPI (Sigma Aldrich) in cold PBS, at 4°C, for 10 min. After extensive washes, cells were fixed in 2% pre-warmed PFA. Galectin-3 staining was performed as above.
Splenic cDC isolation
Splenic cDC isolation was performed as follows: spleens were collected from C57BL/J females. After injection with CO2-independant medium (#18045088, ThermoFisher) supplemented with 0.1 mg/mL DNase I (#11284932001, Sigma-Aldrich) and 0.3 mg/mL Liberase (#05401020001, Roche), spleens were first incubated for 15 min, before mechanical dissociation with a scalpel and further digestion for 15 additional minutes. Following mashing of the residual splenic pieces through a 40 μm strainer, cell suspension was then subjected to splenic cDC isolation with the Pan Dendritic Cell Isolation Kit (#130-100-875, Miltenyi). Purities over 80% of CD11c+ MHC-IIhi conventional DC were routinely obtained (the 20% non-conventional DC population contains about 10% CD11cint MHC-IIint plasmacytoid DC).
Splenic cDC treatments and microscopy staining
0,1 × 106 purified splenic DC were plated on multiwell chambered coverslips (#C24779, ThermoFisher) previously coated with poly-L-lysine (#P8920, Sigma-Aldrich) and left to adhere for 30 min, before addition of 3 mg/mL EF OVA (#321001, Hyglos), 3 mg/mL grade VII OVA (#A7641-250MG, Sigma-Aldrich) or 0.3 mM LLOME (L7393-500MG, Sigma-Aldrich) for respectively 4h (OVAs) or 30min (LLOME), at 37°C. After treatment, cells were immediately fixed with 2% pre-warmed paraformaldehyde (#15710; Electron Microscopy Science) for 20 min at room temperature. Following three PBS washes, non-specific antibody binding was prevented by 1h incubation with Fc block (BD Pharmingen, #553142) diluted at 10 μg/mL in PBS 5% BSA. After blocking, staining with anti-CD8-AF647 (clone 53-6.7, #557682, BD Pharmingen) and anti-CD11b-AF488 (clone M1/70, #557672, BD Pharmingen), both diluted at 1 μg/mL in PBS 5% BSA, was performed for 1h at room temperature. After three PBS washes, cells were permeabilized with PBS 0,1% Triton X-100 (5 min, room temperature), before 1h of blocking with PBS 5% BSA. Finally, intracellular staining was achieved with anti-galectin3-PE antibody (1 μg/mL, clone M3/38, #12-5301-82, eBioscience), and when specified with anti-IST1 (1:100, #19842-1-AP, Proteintech) overnight at 4°C. The next day, cells were washed three times in PBS, stained with goat anti-rabbit Alexa Fluor 405 (1:100, Life Technologies, A-31556) for 1h at room temperature or with DAPI, mounted in PBS and observed at the microscope within the week.
Dextran leakage experiment
0.1 × 106 MutuDC silenced for CHMP2a or CHMP4b (or transduced with control shRNA) were plated on 35 mm glass-bottom dishes (#P35G-1.5-10-C, Mattek LifeSciences) previously coated with poly-L-lysine (#P8920, Sigma-Aldrich), and left to adhere for 1h30. Cells were then pulsed with 1 mg/mL 3000 (#D3308, ThermoFisher), 10,000 (#D1817, ThermoFisher) or 70,000 MW (#D1818 ThermoFisher) tetramethylrhodamine (TRITC)-labeled dextrans for 1h, before extensive PBS washes (all dextrans suspensions were filtered on 0.22 μm before incubation with cells, to ensure sterility). After dextran pulse, ESCRT-III-silenced or control MutuDC were further incubated in medium containing or not 10 μM of Prazosin (#P7791, Sigma Aldrich), as a positive control, for 3h, at 37°C. After several PBS washes, cells were stained with DAPI, for 10 min, at RT, in imaging buffer (PBS 0.5% FCS, 1g/L glucose (#A2494001, ThermoFisher), 2 mM CaCl2, 0.5 mM MgCl2). Following two more PBS washes, cells were then imaged, live, in the imaging buffer.
Microscopy and image analysis
Splenic cDC and MutuDC confocal images were acquired with a laser scanning confocal microscope (LSM780; Zeiss), equipped with 40× Apo objective (numerical aperture: 1.3) and a 1-airy unit pinhole size was used. z stack images were acquired.
Galectin-3 foci quantification
For counting of galectin-3 foci, z-projections of the different stacks were performed, using max intensity criteria. Background signal was subtracted (“rolling = 50”), and threshold applied (Huang threshold) before converting the image to mask. Each cell was defined by a ROI, on which the “analyze particle function” was applied to count the number of foci/cell (criteria used for both MutuDC and splenic cDC: pixel size = 10–700 and circularity = 0.30–1.00). Same criteria were applied to quantify IST1 foci in splenic cDC.
IST1 area analysis
MutuDC were first segmented using the watershed algorithm on maximum intensity z-projections of the different GFP stacks after background signal subtraction (rolling ball, “rolling = 50”). Cell area was measured by applying the “measure” function to individually segmented cells. To measure the area covered by IST1 foci, the macro used for galectin-3 foci quantification was run with modified criteria (pixel size = 10–15000 and circularity = 0.10–1.00). Ratio between IST1 foci area and total cell area was then calculated.
Dextran leakage analysis
Image analysis was performed using Fiji. To quantify dextran release into the cytosol, cells were first segmented using the watershed algorithm on maximum intensity z-projections of the different GFP stacks after background signal subtraction (rolling ball, “rolling = 50”). Maximum intensity z-projections were then performed on the different tetramethylrhodamine stacks and threshold was applied (Huang threshold) to identify intracellular compartments. Cytosol was defined as: total cell area (as defined by segmentation on GFP channel) – area of intracellular compartments. Median of tetramethylrhodamine fluorescence and area were then measured in these compartments.
DAPI+ cells and cells that were not correctly segmented were excluded from the analysis.
Treatment with necroptosis inhibitors
MutuDC were silenced with CHMP2a or CHMP4b-targeting shRNA as previously described. At day 2 post-transduction, MutuDC medium was replaced with medium containing 10 μM GSK′872 (#5303890001, Merck) in absence (for western blotting analysis) or presence (for β-lactamase assay) of puromycin. For immunoblotting kinetics, GSK′872-containing medium was renewed every 24h.
Evaluation of cell death induction by western blotting
To analyze cell death pathways induced in ESCRT-III-silenced cells, total (live and dead) supernatant and adherent cells were collected and counted. 1 × 106 cells were pelleted and lysed in 100 μL 2XSB buffer without DTT (4% SDS (#EU0660, Euromedex), 0.12 M Tris pH 6,8 (#10708976001, Sigma-Aldrich), glycerol 10% (#24388.295, VWR), dash of bromophenol blue (#B-5525, Sigma-Aldrich)) (1 million cells lysed per 100 μL buffer). When needed, DTT (#P2325, ThermoFisher) was added extemporaneously at 100 mM final concentration. Samples were immediately boiled for 5 min at 95°C, quickly spun and frozen at −20°C until use. The following antibodies were used for immunoblotting detection: anti-caspase-3 (#9662, Cell Signaling Technologies), anti-caspase-8 (#9429, Cell Signaling Technologies), anti-caspase-1 (produced in house – Peter Vandenabeele’s lab and Schotte et al. (2004), anti-caspase-11 (#120–10454, Novus Biologicals), anti-cleaved Parp (#9544, Cell Signaling Technologies), anti-Gasdermin-D (Genentech), anti-IL-1β (#GTX74034, Genetex), anti MLKL (#MABC604, Millipore (reducing conditions) or #SAB1302339, Sigma-Aldrich (non-reducing conditions)), anti-phosphoMLKL (#ab196436, Abcam), anti-RIPK3 (#R4277, Sigma-Aldrich), anti-RIPK1 (#610459, BD Biosciences).
Evaluation of ESCRT-III depletion by western blotting
106 MutuDC were lysed in 30 μL RIPA lysis buffer (#89900, ThermoFisher), supplemented with protease inhibitors (#000000011873580001, Sigma-Aldrich). Prior to immunoblotting, proteins were quantified with Pierce BCA Protein Assay Kit (#23250, ThermoFisher), following manufacturer’s instructions. 50 μg total proteins were sampled and supplemented with Laemmli (#1610747, BioRad) and 10% β-mercaptoethanol before heating at 95°C for 5 min. After loading on gel, migration and transfer on membranes (mixed sizes program used), the latter were blocked in TBS (#1706435, BioRad), 5% milk 5%, 0,1% Tween (TBS-T) for one hour at room temperature, and subsequently incubated with rabbit anti-CHMP4b antibody (#42466S, Cell Signaling Technology - 1:500 in TBS-T 5% BSA) or mouse anti-actin antibody (#MAB1501, Millipore – 1:1000 in TBS-T 5% BSA), overnight at 4°C. After numerous washing steps, membranes were incubated for 1h, at room temperature, with the following secondary antibodies: HRP-goat anti-rabbit IgG (111-035-144, Jackson ImmunoResearch Labs) and HRP-goat anti-mouse IgG (#115-035-146, Jackson ImmunoResearch Labs), both diluted 1:10,000 in TBS-T 5% milk.
Rescue experiments
For shRNA-resistant CHMP4b expression studies, a previously described lentiviral expression plasmid carrying RFP657 reporter and puromycin resistance genes (pL-SFFV.Reporter.RFP657.PAC, #61395, Addgene) was used with the following modifications. After AgeI digestion, PacI and AbsI sites were introduced and one of the AgeI sites preserved downstream of the SFFV promoter by polylinker ligation using the following oligos: ACCGGTTTGGGATTAATTAAAATCACCTCGAGGCAGTCCGGT and TGGCCAAACCCTAATTAATTTTAGTGGAGCTCCGTCAGGCCA. In brief, oligos were phosphorylated with T4 PNK (NEB) for 30 min at 37°C, denatured at 95°C for 5 min and then ramped down to 25°C at 5°C per minute. Ligation proceeded overnight at 16°C using a thermocycler, and bacterial transformation of NEB 10-beta competent E. coli (NEB) accomplished by heat shock at 42°C. Minipreps were confirmed by Sanger sequencing. Sequential digestion of modified pL-SFFV-RFP with AgeI and BspEI were carried out at 37°C with column purification between reactions (QIAquick, Qiagen). A codon optimized, shRNA-resistant mouse Chmp4b gBlock was synthesized (IDT) and cloned into pL-SFFV-RFP657 using Gibson assembly (NEB). The Chmp4b gBlock included a stop codon and P2A signal to prevent fusion with RFP while preserving expression of the puromycin resistance gene downstream of the IRES.
Efficiency of the induction of shRNA-resistant Chmp4b expression was monitored using Taqman assay #APRWJAV, designed to specifically detect this shRNA-resistant Chmp4b transcript. In parallel, efficient downregulation of endogenous Chmp4b was assayed using taqman assay #APPRPPX (of note, this custom assay gives identical results than the aforementioned commercial Mm00551493_m1 assay).
MutuDC were simultaneously transduced with lentiviruses encoding CHMP4b-targeting or control shRNAs, and with lentiviruses carrying the empty pL-SFFV.Reporter.RFP657.PAC vector or its resistant CHMP4b-encoding counterpart. MutuDC were transduced with a dose of pL-SFFV.Reporter.RFP657.PAC-encoding viruses that showed efficient shRNA-resistant Chmp4b expression. The chosen virus dose did not alter shRNA-mediated silencing of endogenous Chmp4b, as DC activation observed following ESCRT-III-silencing proved similar between cells transduced only with shRNA, or with shRNA and pL-SFFV.Reporter.RFP657.PAC (empty vector). At day 3 post-transduction, cross-presentation (0.1 × 106 MutuDC/well) or β-lactamase (0.15 × 106 MutuDC/well) assays were then performed as described above.
In parallel, 50,000 MutuDC of each type were plated on poly-L-lysine-coated multiwell chambered coverslips (#C24779, ThermoFisher), left to adhere for 3h, and fixed with 2% PFA for 20min at RT, before blocking, permeabilization and anti-galectin3-PE staining (as previously described).
Detection of antigen export to the cytosol by immunoblotting
Cell fractionation
DC from Flt3-L treated mice were incubated with 0.5mg/mL OVA-Biotin in DC medium (RPMI medium 1640 supplemented with 10% FCS, 50 μM 2-mercaptoethanol, 2 mM L-glutamine, 100 units/mL penicillin and 100 μg/mL streptomycin) for 30 min at 37°C. Cells were washed extensively and further incubated for 2 h in DC medium at 37°C. 20 × 106 DC were used per condition. After extensive washing in homogenization buffer (PBS, 0.25 M sucrose, 10 mM Tris, 1mM EDTA supplemented with proteases inhibitors (Roche), pH 6.8), cells were homogenized with a cell-cracker (HGM Laboratory Equipment) in homogenization buffer. Post-nuclear supernatant was prepared by centrifugation (1000g, 10 min) then ultra-centrifuged (100000g, 1h) to separate cytosolic fractions from intracellular compartments. Fractions were immunoprecipitated with anti-OVA antibody (Abcam) using protein G-sepharose.
Western blot analysis
Total (actin detection) or immunoprecipitated (OVA detection) cytosolic fractions were run on a 4–12% SDS-PAGE gel (Invitrogen) and transferred onto nitrocellulose membrane. Membranes were stained with streptavidin-HRP or polyclonal antibodies against actin (Sigma). Horseradish peroxidase (HRP)–conjugated anti-rabbit IgG antibody (Sigma) was used for actin detection. Signal intensity was quantified using QuantityOne (BioRad).
Endocytic compartment proteomics
Mice. C57BL/6 (B6) mice were bred and maintained under specific pathogen-free conditions at The Walter and Eliza Hall Institute animal breeding facility according to institute guidelines. B6 mice were injected subcutaneously with 5 × 106 B16 melanoma cells secreting murine Fms-like tyrosine kinase ligand (Flt3-L) (Shi et al., 1999) and killed after 9–10 days.
Cell isolation
Total DC were isolated from the spleens of B6 mice as described in (Vremec and Segura, 2013). In brief, splenic cells were digested with Dnase I (Boehringer-Manheim, Mannhein, Germany) and collagenase (Worthington Biochemicals, Freehold, NJ) and enriched for light-density cells by centrifugation in 1.077 g/cm3 Nycodenz (Nycomed Pharma, Oslo, Norway). Cells were isolated after a depletion step using antibodies against CD3 (KT3-1.1), Thy-1 (T24/31.7), Ter 119, Ly6G (RB68C5) and CD45R (RA36B2), followed by incubation with anti-rat IgG-coupled magnetic beads (Dynal, Oslo, Norway) following the manufacturer’s protocol. CD8+ cDC1 were isolated by positive selection using immuno-magnetic beads (MACS, Miltenyi Biotec) after staining with anti-CD8 (YTS 169.4) antibody. Remaining cells were depleted of CD205+CD24high DC after staining with anti-CD205 (NLDC-145) and anti-CD24 (M1/69) antibodies and MACS beads depletion, and finally CD8-cDC2 were isolated by positive selection using MACS beads after staining with anti-CD11b (M1/70) antibody.
Subcellular fractionation
Cells were homogenized with a cell-cracker (HGM Laboratory Equipment) in homogenization buffer (PBS, 0.25 M sucrose, 10 mM Tris, 1mM EDTA supplemented with proteases inhibitors (Roche), pH 6.8). Post-nuclear supernatant was prepared by centrifugation (1000 g, 10 min) and loaded on top of a 10% Percoll (GE Healthcare) solution in homogenisation buffer. After ultra-centrifugation (50,000 g, 45 min), 1 mL fractions were collected. The top 3 fractions (early endosomes) were pooled and concentrated by ultracentrifugation (130,000 g, 1 h). The bottom 2 fractions were pooled and loaded on top of a 45% Percoll solution in homogenisation buffer. After ultra-centrifugation (50,000 g, 45 min), the top 3 fractions (late endosomes) ant the bottom 2 fractions (lysosomes) were pooled and concentrated by ultracentrifugation (130,000 g, 1 h) (Segura et al., 2009).
SDS-PAGE electrophoresis
Samples were run on a 4–12% SDS-PAGE gel (Invitrogen). After electrophoresis, gels were fixed with 40% methanol/7% acetic acid for 30 min, washed 3 times in deionised water and stained in Imperial Protein stain (Pearce).
Gel excision and digestion. For each sample, 50 gel bands were excised from the 1-D gel lane and subjected to automated in-gel reduction, alkylation and tryptic digestion using the MassPREP Station (Micromass). Briefly, gel sections were automatically reduced with 10 mm DTT (Merck) for 30 min, alkylated for 20 min with 25 mm iodoacetic acid (Fluka) and digested with 150 ng trypsin (Worthington) for 4.5 h at 37°C. Extracted peptide solutions (0.1% formic acid) were concentrated to approximately 10 μL by centrifugal lyophilisation using a SpeedVac AES 1010 (Savant).
Mass spectrometry
A 96-well plate containing extracted peptides was loaded into the microwell plate autosampler for injection and fractionation by nanoflow reverse-phase liquid chromatography on a 1200 series LC system (Agilent) using a nanoAcquity C18 150 × 0.15 mm i.d. column (Waters) developed with a linear 60-min gradient with a flow rate of 0.5 μL/min at 45°C from 0 to 100% solvent B in solvent A (as above). The nano HPLC was coupled on-line to an LTQ-Orbitrap mass spectrometer equipped with a nanoelectrospray ion source (ThermoFisher) for automated MS/MS. The Orbitrap was operated in positive ion mode for data-dependent acquisition. Survey MS scans were acquired with the resolution set to a value of 30,000. Real time recalibration by corrections of mass shift was performed by the use of a background ion from ambient air in the C-trap. Up to five most intense ions per cycle were fragmented and analyzed in the linear trap, with target ions already selected for MS/MS being dynamically excluded for 3 min.
Mass spectrometry data analysis
Peak lists were extracted using extract-msn as part of Bioworks 3.3.1 (ThermoFisher). The parameters used to generate the peak lists for the Orbitrap were as follows: minimum mass 700; maximum mass 5000; grouping tolerance 0.01 Da; intermediate scans 1; minimum group count 1; 10 peaks minimum and total ion current of 100. Automatic charge state recognition was used because of the high resolution survey scan (30,000). Peak lists for each LC-MS/MS run were merged into a single MGF file and searched against the IPI (Kersey et al., 2004) mouse and bovine protein sequence database (April 2009) comprising 87,598 sequence entries using the MASCOT v2.2.01 search algorithm (Matrix Science). The search parameters consisted of carboxymethylation of cysteine as a fixed modification (+58 Da), NH2-terminal acetylation (+42 Da) and oxidation of methionine (+16 Da) as variable modifications. A peptide mass tolerance of ±20 ppm, #13C defined as 1, fragment ion mass tolerance of ±0.8 Da, and an allowance for up to two missed tryptic cleavages was used.
The program MSPro was used for parsing and summarizing the Mascot search results for all data files. Peptide spectral matches (PSMs) from the Mascot search results were deemed significant if the PSMs Ions score (IS) was ≥ the Homology score (or Identity score if there was no Homology score). False-discovery rate calculations at the protein level were carried out as previously described (Greening et al., 2009).
Differential protein expression determination
Comparison of the relative abundance of each molecule across the two samples was performed by calculating fold change based on the number of spectral counts of significant PSMs for each protein. As in Segura et al. (2010) significant spectral count fold change ratios (RSc) were determined using a modified formula from a previous serial analysis of gene expression study by Beissbarth et al., (2004).
| (Equation 1) |
Where, n is the number of spectral counts of significant PSMs for a particular protein, t is the total number of spectral counts of significant PSMs for all proteins in a sample, and f a correction factor set to 1.25 (Old et al., 2005).
Quantification and statistical analysis
Statistical tests were performed with the Graphpad Prism software. The nature of the test used is mentioned in figure legends, and exact p-values are shown on figures. In some illustrative panels (Figure S4A), statistical significance was not assessed, and p-values thereby not displayed.
Acknowledgments
We thank Hans Acha-Orbea for the MutuDCs and the animal facility and the PICT-IBiSA@Pasteur Imaging Facility of Institut Curie, a member of the France Bioimaging National Infrastructure (ANR-10-INBS-04). We thank Philippe Benaroch, Nicolas Manel, Franck Perez, and all members of the Immune Responses to Cancer Laboratory for scientific discussions. We thank Pierre-Emmanuel Bonté for generation of the violin plots in Figure S1A, Maria Bottermann for galectin-3-related protocols, Stéphanie Dogniaux for help with molecular biology, and Evan R. Harrell for critical reading of the manuscript. M.G. is supported by a Fondation pour la Recherche Médicale grant (FDT201805005336). S.A. received funding from INSERM, Institut Curie, la Ligue contre le Cancer (Equipe Labellisée Ligue, EL2014.LNCC/SA), Association de Recherche contre le Cancer (ARC), the European Research Council (2013-AdG 340046 DCBIOX), ANR-10-IDEX-0001-02 PSL∗, and ANR-11-LABX-0043. M.B. was supported by an ARC postdoctoral fellowship. E.S. was supported by Marie Curie Actions from the European Commission (project 39408). Research in the group of M.J.M.B. is financially supported by the Vlaams Instituut voor Biotechnologie (VIB), Ghent University, and grants from the Fonds voor Wetenschappelijk Onderzoek Vlaanderen (FWO) (G035320N, G044518N, G0G6618N, and G0I5722N) and from the Flemish Government to Peter Vandenabeele (Methusalem BOF09/01M00709 and BOF16/MET_V/007). J.A.V. received funding from the National Health and Medical Research Council of Australia. P.K. was supported by the Wellcome Trust (101578/Z/13/Z) and Medical Research Council (MC_UP_1201/26).
Author contributions
M.G., M.B., D.C.R., M.J.M.B., E.S., J.A.V., and S.A. designed experiments. M.G., M.B., E.S. (Figures 1B–1D), B.B. (Figure S2D), and M.J.M.B. (Figures 5A–5C, S5A, and S5B) performed experiments. M.B., N.B., D.C.R., S.H.-C., and M.M. helped with in vivo and microscopy experiments. E.A.K. and R.J.S. analyzed proteomics experiments (Figure 1D). M.G., M.B., and M.J.M.B. analyzed the data. M.G., M.B., and S.A. prepared the figures. M.G. and S.A. wrote the manuscript with input from all authors. S.A. and M.G. conceptualized the study. S.A., M.B., P.K., M.J.M.B., and J.A.V. supervised the project.
Declaration of interests
S.A. is shareholder and consultant for Mnemo Therapeutics.
Published: August 16, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111205.
Contributor Information
Marine Gros, Email: marine.mv.gros@gmail.com.
Marianne Burbage, Email: marianne.burbage@curie.fr.
Supplemental information
Data and code availability
Data reported in this paper are available from the lead contact upon request.
This paper does not report original code. However, FIJI macros used to quantify galectin-3 foci, cellular IST1 area or fluorescence intensity in the cytosol versus. intracellular compartments are available from the lead contact upon request.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead contact upon request.
References
- Ackerman A.L., Giodini A., Cresswell P. A role for the endoplasmic reticulum protein retrotranslocation machinery during crosspresentation by dendritic cells. Immunity. 2006;25:607–617. doi: 10.1016/j.immuni.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Adell M.A.Y., Vogel G.F., Pakdel M., Müller M., Lindner H., Hess M.W., Teis D. Coordinated binding of Vps4 to ESCRT-III drives membrane neck constriction during MVB vesicle formation. J. Cell Biol. 2014;205:33–49. doi: 10.1083/jcb.201310114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agmon E., Solon J., Bassereau P., Stockwell B.R. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci. Rep. 2018;8:5155. doi: 10.1038/s41598-018-23408-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aits S., Kricker J., Liu B., Ellegaard A.-M., Hämälistö S., Tvingsholm S., Corcelle-Termeau E., Høgh S., Farkas T., Holm Jonassen A., et al. Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy. 2015;11:1408–1424. doi: 10.1080/15548627.2015.1063871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alloatti A., Kotsias F., Hoffmann E., Amigorena S. Evaluation of Cross-presentation in Bone Marrow-derived Dendritic Cells in vitro and Splenic Dendritic Cells ex vivo Using Antigen-coated Beads. Bio Protoc. 2016;6 doi: 10.21769/BioProtoc.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bache K.G., Stuffers S., Malerød L., Slagsvold T., Raiborg C., Lechardeur D., Wälchli S., Lukacs G.L., Brech A., Stenmark H. The ESCRT-III subunit hVps24 is required for degradation but not silencing of the epidermal growth factor receptor. Mol. Biol. Cell. 2006;17:2513–2523. doi: 10.1091/mbc.e05-10-0915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beissbarth T., Hyde L., Smyth G.K., Job C., Boon W.-M., Tan S.-S., Scott H.S., Speed T.P. Statistical modeling of sequencing errors in SAGE libraries. Bioinformatics. 2004;20 doi: 10.1093/bioinformatics/bth924. i31–39. [DOI] [PubMed] [Google Scholar]
- Bevan M.J. Cross-priming for a secondary cytotoxic response to minor H antigens with H-2 congenic cells which do not cross-react in the cytotoxic assay. J. Exp. Med. 1976;143:1283–1288. doi: 10.1084/jem.143.5.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan M.J. Minor H antigens introduced on H-2 different stimulating cells cross-react at the cytotoxic T cell level during in vivo priming. J. Immunol. 1976;117:2233–2238. [PubMed] [Google Scholar]
- Blander J.M. Regulation of the cell biology of antigen cross-presentation. Annu. Rev. Immunol. 2018;36:717–753. doi: 10.1146/annurev-immunol-041015-055523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bougnères L., Helft J., Tiwari S., Vargas P., Chang B.H.-J., Chan L., Campisi L., Lauvau G., Hugues S., Kumar P., et al. A role for lipid bodies in the cross-presentation of phagocytosed antigens by MHC class I in dendritic cells. Immunity. 2009;31:232–244. doi: 10.1016/j.immuni.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boya P., Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene. 2008;27:6434–6451. doi: 10.1038/onc.2008.310. [DOI] [PubMed] [Google Scholar]
- den Brok M.H., Büll C., Wassink M., de Graaf A.M., Wagenaars J.A., Minderman M., Thakur M., Amigorena S., Rijke E.O., Schrier C.C., Adema G.J. Saponin-based adjuvants induce cross-presentation in dendritic cells by intracellular lipid body formation. Nat. Commun. 2016;7:13324. doi: 10.1038/ncomms13324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgdorf S., Schölz C., Kautz A., Tampé R., Kurts C. Spatial and mechanistic separation of cross-presentation and endogenous antigen presentation. Nat. Immunol. 2008;9:558–566. doi: 10.1038/ni.1601. [DOI] [PubMed] [Google Scholar]
- Canton J., Blees H., Henry C.M., Buck M.D., Schulz O., Rogers N.C., Childs E., Zelenay S., Rhys H., Domart M.-C., et al. The receptor DNGR-1 signals for phagosomal rupture to promote cross-presentation of dead-cell-associated antigens. Nat. Immunol. 2021;22:140–153. doi: 10.1038/s41590-020-00824-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton J.G., Caballe A., Agromayor M., Kloc M., Martin-Serrano J. ESCRT-III governs the Aurora B-mediated abscission checkpoint through CHMP4C. Science. 2012;336:220–225. doi: 10.1126/science.1217180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cebrian I., Visentin G., Blanchard N., Jouve M., Bobard A., Moita C., Enninga J., Moita L.F., Amigorena S., Savina A. Sec22b regulates phagosomal maturation and antigen crosspresentation by dendritic cells. Cell. 2011;147:1355–1368. doi: 10.1016/j.cell.2011.11.021. [DOI] [PubMed] [Google Scholar]
- Chiaruttini N., Redondo-Morata L., Colom A., Humbert F., Lenz M., Scheuring S., Roux A. Relaxation of loaded ESCRT-III spiral springs drives membrane deformation. Cell. 2015;163:866–879. doi: 10.1016/j.cell.2015.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs E., Henry C.M., Canton J., Reis e Sousa C. Maintenance and loss of endocytic organelle integrity: mechanisms and implications for antigen cross-presentation. Open Biol. 2021;11:210194. doi: 10.1098/rsob.210194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz F.M., Colbert J.D., Merino E., Kriegsman B.A., Rock K.L. The biology and underlying mechanisms of cross-presentation of exogenous antigens on MHC-I molecules. Annu. Rev. Immunol. 2017;35:149–176. doi: 10.1146/annurev-immunol-041015-055254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denais C.M., Gilbert R.M., Isermann P., McGregor A.L., te Lindert M., Weigelin B., Davidson P.M., Friedl P., Wolf K., Lammerding J. Nuclear envelope rupture and repair during cancer cell migration. Science. 2016;352:353–358. doi: 10.1126/science.aad7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingjan I., Verboogen D.R., Paardekooper L.M., Revelo N.H., Sittig S.P., Visser L.J., Mollard G.F.v., Henriet S.S., Figdor C.G., Ter Beest M., van den Bogaart G. Lipid peroxidation causes endosomal antigen release for cross-presentation. Sci. Rep. 2016;6:22064. doi: 10.1038/srep22064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingjan I., Paardekooper L.M., Verboogen D.R.J., von Mollard G.F., Ter Beest M., van den Bogaart G. VAMP8-mediated NOX2 recruitment to endosomes is necessary for antigen release. Eur. J. Cell Biol. 2017;96:705–714. doi: 10.1016/j.ejcb.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards A.D., Chaussabel D., Tomlinson S., Schulz O., Sher A., Reis e Sousa C. Relationships among murine CD11c(high) dendritic cell subsets as revealed by baseline gene expression patterns. J. Immunol. 2003;171:47–60. doi: 10.4049/jimmunol.171.1.47. [DOI] [PubMed] [Google Scholar]
- Frank D., Vaux D.L., Murphy J.M., Vince J.E., Lindqvist L.M. Activated MLKL attenuates autophagy following its translocation to intracellular membranes. J. Cell Sci. 2019;132:jcs220996. doi: 10.1242/jcs.220996. [DOI] [PubMed] [Google Scholar]
- Fuertes Marraco S.A., Grosjean F., Duval A., Rosa M., Lavanchy C., Ashok D., Haller S., Otten L.A., Steiner Q.-G., Descombes P., et al. Novel murine dendritic cell lines: a powerful auxiliary tool for dendritic cell research. Front. Immunol. 2012;3:331. doi: 10.3389/fimmu.2012.00331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita H., Umezuki Y., Imamura K., Ishikawa D., Uchimura S., Nara A., Yoshimori T., Hayashizaki Y., Kawai J., Ishidoh K., et al. Mammalian class E Vps proteins, SBP1 and mVps2/CHMP2A, interact with and regulate the function of an AAA-ATPase SKD1/Vps4B. J. Cell Sci. 2004;117:2997–3009. doi: 10.1242/jcs.01170. [DOI] [PubMed] [Google Scholar]
- Garrus J.E., von Schwedler U.K., Pornillos O.W., Morham S.G., Zavitz K.H., Wang H.E., Wettstein D.A., Stray K.M., Côté M., Rich R.L., et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell. 2001;107:55–65. doi: 10.1016/s0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- Gong Y.-N., Guy C., Olauson H., Becker J.U., Yang M., Fitzgerald P., Linkermann A., Green D.R. ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell. 2017;169:286–300.e16. doi: 10.1016/j.cell.2017.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greening D.W., Glenister K.M., Sparrow R.L., Simpson R.J. Enrichment of human platelet membranes for proteomic analysis. Methods Mol Biol. 2009;528:245–258. doi: 10.1007/978-1-60327-310-7_17. [DOI] [PubMed] [Google Scholar]
- Gros M., Amigorena S. Regulation of antigen export to the cytosol during cross-presentation. Front. Immunol. 2019;10:41. doi: 10.3389/fimmu.2019.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grotzke J.E., Kozik P., Morel J.-D., Impens F., Pietrosemoli N., Cresswell P., Amigorena S., Demangel C. Sec61 blockade by mycolactone inhibits antigen cross-presentation independently of endosome-to-cytosol export. Proc. Natl. Acad. Sci. USA. 2017;114:E5910–E5919. doi: 10.1073/pnas.1705242114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson P.I., Roth R., Lin Y., Heuser J.E. Plasma membrane deformation by circular arrays of ESCRT-III protein filaments. J. Cell Biol. 2008;180:389–402. doi: 10.1083/jcb.200707031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand J.M., Tanzer M.C., Lucet I.S., Young S.N., Spall S.K., Sharma P., Pierotti C., Garnier J.-M., Dobson R.C.J., Webb A.I., et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc. Natl. Acad. Sci. USA. 2014;111:15072–15077. doi: 10.1073/pnas.1408987111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann E., Pauwels A.-M., Alloatti A., Kotsias F., Amigorena S. Analysis of phagosomal antigen degradation by flow organellocytometry. Bio. Protoc. 2016;6:e2014. doi: 10.21769/bioprotoc.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howland S.W., Wittrup K.D. Antigen release kinetics in the phagosome are critical to cross-presentation efficiency. J. Immunol. 2008;180:1576–1583. doi: 10.4049/jimmunol.180.3.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai T., Kato Y., Kajiwara C., Mizukami S., Ishige I., Ichiyanagi T., Hikida M., Wang J.-Y., Udono H. Heat shock protein 90 (HSP90) contributes to cytosolic translocation of extracellular antigen for cross-presentation by dendritic cells. Proc. Natl. Acad. Sci. USA. 2011;108:16363–16368. doi: 10.1073/pnas.1108372108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez A.J., Maiuri P., Lafaurie-Janvore J., Divoux S., Piel M., Perez F. ESCRT machinery is required for plasma membrane repair. Science. 2014;343:1247136. doi: 10.1126/science.1247136. [DOI] [PubMed] [Google Scholar]
- Karttunen J., Sanderson S., Shastri N. Detection of rare antigen-presenting cells by the lacZ T-cell activation assay suggests an expression cloning strategy for T-cell antigens. Proc. Natl. Acad. Sci. USA. 1992;89:6020–6024. doi: 10.1073/pnas.89.13.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersey P.J., Duarte J., Williams A., Karavidopoulou Y., Birney E., Apweiler R. The International Protein Index: an integrated database for proteomics experiments. Proteomics. 2004;4:1985–1988. doi: 10.1002/pmic.200300721. [DOI] [PubMed] [Google Scholar]
- Koopmann J.O., Albring J., Hüter E., Bulbuc N., Spee P., Neefjes J., Hämmerling G.J., Momburg F. Export of antigenic peptides from the endoplasmic reticulum intersects with retrograde protein translocation through the Sec61p channel. Immunity. 2000;13:117–127. doi: 10.1016/s1074-7613(00)00013-3. [DOI] [PubMed] [Google Scholar]
- Kovacsovics-Bankowski M., Rock K.L. A phagosome-to-cytosol pathway for exogenous antigens presented on MHC class I molecules. Science. 1995;267:243–246. doi: 10.1126/science.7809629. [DOI] [PubMed] [Google Scholar]
- Kozik P., Gros M., Itzhak D.N., Joannas L., Heurtebise-Chrétien S., Krawczyk P.A., Rodríguez-Silvestre P., Alloatti A., Magalhaes J.G., Del Nery E., et al. Small molecule enhancers of endosome-to-cytosol import augment anti-tumor immunity. Cell Rep. 2020;32:107905. doi: 10.1016/j.celrep.2020.107905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.-A., Beigneux A., Ahmad S.T., Young S.G., Gao F.-B. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr. Biol. 2007;17:1561–1567. doi: 10.1016/j.cub.2007.07.029. [DOI] [PubMed] [Google Scholar]
- Lin M.L., Zhan Y., Proietto A.I., Prato S., Wu L., Heath W.R., Villadangos J.A., Lew A.M. Selective suicide of cross-presenting CD8+ dendritic cells by cytochrome c injection shows functional heterogeneity within this subset. Proc. Natl. Acad. Sci. USA. 2008;105:3029–3034. doi: 10.1073/pnas.0712394105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Luber C.A., Cox J., Lauterbach H., Fancke B., Selbach M., Tschopp J., Akira S., Wiegand M., Hochrein H., O’Keeffe M., Mann M. Quantitative proteomics reveals subset-specific viral recognition in dendritic cells. Immunity. 2010;32:279–289. doi: 10.1016/j.immuni.2010.01.013. [DOI] [PubMed] [Google Scholar]
- Maejima I., Takahashi A., Omori H., Kimura T., Takabatake Y., Saitoh T., Yamamoto A., Hamasaki M., Noda T., Isaka Y., Yoshimori T. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 2013;32:2336–2347. doi: 10.1038/emboj.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamińska A., Bartosik A., Banach-Orłowska M., Pilecka I., Jastrzębski K., Zdżalik-Bielecka D., Castanon I., Poulain M., Neyen C., Wolińska-Nizioł L., et al. ESCRT proteins restrict constitutive NF-κB signaling by trafficking cytokine receptors. Sci. Signal. 2016;9:ra8. doi: 10.1126/scisignal.aad0848. [DOI] [PubMed] [Google Scholar]
- Mandal P., Berger S.B., Pillay S., Moriwaki K., Huang C., Guo H., Lich J.D., Finger J., Kasparcova V., Votta B., et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol. Cell. 2014;56:481–495. doi: 10.1016/j.molcel.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat J., Grueneberg D.A., Yang X., Kim S.Y., Kloepfer A.M., Hinkle G., Piqani B., Eisenhaure T.M., Luo B., Grenier J.K., et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124:1283–1298. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- Moriwaki K., Balaji S., McQuade T., Malhotra N., Kang J., Chan F.K.-M. The necroptosis adaptor RIPK3 promotes injury-induced cytokine expression and tissue repair. Immunity. 2014;41:567–578. doi: 10.1016/j.immuni.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalle S.C., Barreira da Silva R., Zhang H., Decker M., Chalouni C., Xu M., Posthuma G., de Mazière A., Klumperman J., Baz Morelli A., et al. Aquaporin-3 regulates endosome-to-cytosol transfer via lipid peroxidation for cross presentation. PLoS One. 2020;15:e0238484. doi: 10.1371/journal.pone.0238484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H.C., Talledge N., McCullough J., Sharma A., Moss F.R., Iwasa J.H., Vershinin M.D., Sundquist W.I., Frost A. Membrane constriction and thinning by sequential ESCRT-III polymerization. Nat. Struct. Mol. Biol. 2020;27:392–399. doi: 10.1038/s41594-020-0404-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Old W.M., Meyer-Arendt K., Aveline-Wolf L., Pierce K.G., Mendoza A., Sevinsky J.R., Resing K.A., Ahn N.G. Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol. Cell. Proteomics. 2005;4:1487–1502. doi: 10.1074/mcp.M500084-MCP200. [DOI] [PubMed] [Google Scholar]
- Olmos Y., Hodgson L., Mantell J., Verkade P., Carlton J.G. ESCRT-III controls nuclear envelope reformation. Nature. 2015;522:236–239. doi: 10.1038/nature14503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oura J., Tamura Y., Kamiguchi K., Kutomi G., Sahara H., Torigoe T., Himi T., Sato N. Extracellular heat shock protein 90 plays a role in translocating chaperoned antigen from endosome to proteasome for generating antigenic peptide to be cross-presented by dendritic cells. Int. Immunol. 2011;23:223–237. doi: 10.1093/intimm/dxq475. [DOI] [PubMed] [Google Scholar]
- Pace L., Tempez A., Arnold-Schrauf C., Lemaitre F., Bousso P., Fetler L., Sparwasser T., Amigorena S. Regulatory T cells increase the avidity of primary CD8+ T cell responses and promote memory. Science. 2012;338:532–536. doi: 10.1126/science.1227049. [DOI] [PubMed] [Google Scholar]
- Pedrera L., Espiritu R.A., Ros U., Weber J., Schmitt A., Stroh J., Hailfinger S., von Karstedt S., García-Sáez A.J. Ferroptotic pores induce Ca2+ fluxes and ESCRT-III activation to modulate cell death kinetics. Cell Death Differ. 2021;28:1644–1657. doi: 10.1038/s41418-020-00691-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfitzner A.-K., Mercier V., Jiang X., Moser von Filseck J., Baum B., Šarić A., Roux A. An ESCRT-III polymerization sequence drives membrane deformation and fission. Cell. 2020;182:1140–1155.e18. doi: 10.1016/j.cell.2020.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploegh H.L. A lipid-based model for the creation of an escape hatch from the endoplasmic reticulum. Nature. 2007;448:435–438. doi: 10.1038/nature06004. [DOI] [PubMed] [Google Scholar]
- Pooley J.L., Heath W.R., Shortman K. Cutting edge: intravenous soluble antigen is presented to CD4 T cells by CD8- dendritic cells, but cross-presented to CD8 T cells by CD8+ dendritic cells. J. Immunol. 2001;166:5327–5330. doi: 10.4049/jimmunol.166.9.5327. [DOI] [PubMed] [Google Scholar]
- Raab M., Gentili M., de Belly H., Thiam H.R., Vargas P., Jimenez A.J., Lautenschlaeger F., Voituriez R., Lennon-Duménil A.M., Manel N., Piel M. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science. 2016;352:359–362. doi: 10.1126/science.aad7611. [DOI] [PubMed] [Google Scholar]
- Radulovic M., Schink K.O., Wenzel E.M., Nähse V., Bongiovanni A., Lafont F., Stenmark H. ESCRT-mediated lysosome repair precedes lysophagy and promotes cell survival. EMBO J. 2018;37:e99753. doi: 10.15252/embj.201899753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repnik U., Borg Distefano M., Speth M.T., Ng M.Y.W., Progida C., Hoflack B., Gruenberg J., Griffiths G. L-leucyl-L-leucine methyl ester does not release cysteine cathepsins to the cytosol but inactivates them in transiently permeabilized lysosomes. J. Cell Sci. 2017;130:3124–3140. doi: 10.1242/jcs.204529. [DOI] [PubMed] [Google Scholar]
- Rodriguez A., Regnault A., Kleijmeer M., Ricciardi-Castagnoli P., Amigorena S. Selective transport of internalized antigens to the cytosol for MHC class I presentation in dendritic cells. Nat. Cell Biol. 1999;1:362–368. doi: 10.1038/14058. [DOI] [PubMed] [Google Scholar]
- Römisch K. A case for Sec61 channel involvement in ERAD. Trends Biochem. Sci. 2017;42:171–179. doi: 10.1016/j.tibs.2016.10.005. [DOI] [PubMed] [Google Scholar]
- Rühl S., Shkarina K., Demarco B., Heilig R., Santos J.C., Broz P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science. 2018;362:956–960. doi: 10.1126/science.aar7607. [DOI] [PubMed] [Google Scholar]
- Saka H.A., Valdivia R. Emerging roles for lipid droplets in immunity and host-pathogen interactions. Annu. Rev. Cell Dev. Biol. 2012;28:411–437. doi: 10.1146/annurev-cellbio-092910-153958. [DOI] [PubMed] [Google Scholar]
- Saksena S., Wahlman J., Teis D., Johnson A.E., Emr S.D. Functional reconstitution of ESCRT-III assembly and disassembly. Cell. 2009;136:97–109. doi: 10.1016/j.cell.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson S., Shastri N. LacZ inducible, antigen/MHC-specific T cell hybrids. Int. Immunol. 1994;6:369–376. doi: 10.1093/intimm/6.3.369. [DOI] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnorrer P., Behrens G.M.N., Wilson N.S., Pooley J.L., Smith C.M., El-Sukkari D., Davey G., Kupresanin F., Li M., Maraskovsky E., et al. The dominant role of CD8+ dendritic cells in cross-presentation is not dictated by antigen capture. Proc. Natl. Acad. Sci. USA. 2006;103:10729–10734. doi: 10.1073/pnas.0601956103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöneberg J., Lee I.-H., Iwasa J.H., Hurley J.H. Reverse-topology membrane scission by the ESCRT proteins. Nat. Rev. Mol. Cell Biol. 2017;18:5–17. doi: 10.1038/nrm.2016.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotte P., Denecker G., Van Den Broeke A., Vandenabeele P., Cornelis G.R., Beyaert R. Targeting Rac1 by the Yersinia effector protein YopE inhibits caspase-1-mediated maturation and release of interleukin-1beta. J. Biol. Chem. 2004;279:25134–25142. doi: 10.1074/jbc.M401245200. [DOI] [PubMed] [Google Scholar]
- Segura E., Albiston A.L., Wicks I.P., Chai S.Y., Villadangos J.A. Different cross-presentation pathways in steady-state and inflammatory dendritic cells. Proc. Natl. Acad. Sci. USA. 2009;106:20377–20381. doi: 10.1073/pnas.0910295106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segura E., Kapp E., Gupta N., Wong J., Lim J., Ji H., Heath W.R., Simpson R., Villadangos J.A. Differential expression of pathogen-recognition molecules between dendritic cell subsets revealed by plasma membrane proteomic analysis. Mol. Immunol. 2010;47:1765–1773. doi: 10.1016/j.molimm.2010.02.028. [DOI] [PubMed] [Google Scholar]
- Shen Q.-T., Schuh A.L., Zheng Y., Quinney K., Wang L., Hanna M., Mitchell J.C., Otegui M.S., Ahlquist P., Cui Q., Audhya A. Structural analysis and modeling reveals new mechanisms governing ESCRT-III spiral filament assembly. J. Cell Biol. 2014;206:763–777. doi: 10.1083/jcb.201403108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi G.P., Villadangos J.A., Dranoff G., Small C., Gu L., Haley K.J., Riese R., Ploegh H.L., Chapman H.A. Cathepsin S required for normal MHC class II peptide loading and germinal center development. Immunity. 1999;10:197–206. doi: 10.1016/s1074-7613(00)80020-5. [DOI] [PubMed] [Google Scholar]
- Singh R., Cresswell P. Defective cross-presentation of viral antigens in GILT-free mice. Science. 2010;328:1394–1398. doi: 10.1126/science.1189176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowyra M.L., Schlesinger P.H., Naismith T.V., Hanson P.I. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science. 2018;360:eaar5078. doi: 10.1126/science.aar5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Berghe T., Vanlangenakker N., Parthoens E., Deckers W., Devos M., Festjens N., Guerin C.J., Brunk U.T., Declercq W., Vandenabeele P. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010;17:922–930. doi: 10.1038/cdd.2009.184. [DOI] [PubMed] [Google Scholar]
- Vietri M., Schink K.O., Campsteijn C., Wegner C.S., Schultz S.W., Christ L., Thoresen S.B., Brech A., Raiborg C., Stenmark H. Spastin and ESCRT-III coordinate mitotic spindle disassembly and nuclear envelope sealing. Nature. 2015;522:231–235. doi: 10.1038/nature14408. [DOI] [PubMed] [Google Scholar]
- Voorhees R.M., Fernández I.S., Scheres S.H.W., Hegde R.S. Structure of the mammalian ribosome-Sec61 complex to 3.4 Å resolution. Cell. 2014;157:1632–1643. doi: 10.1016/j.cell.2014.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vremec D., Segura E. The purification of large numbers of antigen presenting dendritic cells from mouse spleen. Methods Mol. Biol. 2013;960:327–350. doi: 10.1007/978-1-62703-218-6_24. [DOI] [PubMed] [Google Scholar]
- Vremec D., Shortman K. Dendritic cell subtypes in mouse lymphoid organs: cross-correlation of surface markers, changes with incubation, and differences among thymus, spleen, and lymph nodes. J. Immunol. 1997;159:565–573. [PubMed] [Google Scholar]
- Wong-Ekkabut J., Xu Z., Triampo W., Tang I.-M., Tieleman D.P., Monticelli L. Effect of lipid peroxidation on the properties of lipid bilayers: a molecular dynamics study. Biophys. J. 2007;93:4225–4236. doi: 10.1529/biophysj.107.112565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Wang Y., Zhang Y., He X., Zhong C.-Q., Ni H., Chen X., Liang Y., Wu J., Zhao S., et al. RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nat. Cell Biol. 2018;20:186–197. doi: 10.1038/s41556-017-0022-y. [DOI] [PubMed] [Google Scholar]
- Yatim N., Jusforgues-Saklani H., Orozco S., Schulz O., Barreira da Silva R., Reis e Sousa C., Green D.R., Oberst A., Albert M.L. RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8+ T cells. Science. 2015;350:328–334. doi: 10.1126/science.aad0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S., Kovalenko A., Bogdanov K., Wallach D. MLKL, the protein that mediates necroptosis, also regulates endosomal trafficking and extracellular vesicle generation. Immunity. 2017;47:51–65.e7. doi: 10.1016/j.immuni.2017.06.001. [DOI] [PubMed] [Google Scholar]
- Zehner M., Chasan A.I., Schuette V., Embgenbroich M., Quast T., Kolanus W., Burgdorf S. Mannose receptor polyubiquitination regulates endosomal recruitment of p97 and cytosolic antigen translocation for cross-presentation. Proc. Natl. Acad. Sci. USA. 2011;108:9933–9938. doi: 10.1073/pnas.1102397108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehner M., Marschall A.L., Bos E., Schloetel J.-G., Kreer C., Fehrenschild D., Limmer A., Ossendorp F., Lang T., Koster A.J., et al. The translocon protein Sec61 mediates antigen transport from endosomes in the cytosol for cross-presentation to CD8(+) T cells. Immunity. 2015;42:850–863. doi: 10.1016/j.immuni.2015.04.008. [DOI] [PubMed] [Google Scholar]
- Zhou Y., Bennett T.M., Shiels A. A charged multivesicular body protein (CHMP4B) is required for lens growth and differentiation. Differentiation. 2019;109:16–27. doi: 10.1016/j.diff.2019.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper are available from the lead contact upon request.
This paper does not report original code. However, FIJI macros used to quantify galectin-3 foci, cellular IST1 area or fluorescence intensity in the cytosol versus. intracellular compartments are available from the lead contact upon request.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead contact upon request.