

Abstract
Tau oligomers (oTau) are thought to precede neurofibrillary tangle formation and likely represent one of the toxic species in disease. This study addresses whether mitochondrial reactive oxygen species (ROS) contribute to tau oligomer accumulation. First, we determined whether elevated oxidative stress correlates with aggregation of tau oligomers in the brain and platelets of human Alzheimer’s disease (AD) patient, tauopathy mice, primary cortical neurons from tau mice and human trans-mitochondrial ‘cybrid’ (cytoplasmic hybrid) neuronal cells, whose mitochondria are derived from platelets of patients with sporadic AD- or mild cognitive impairment (MCI)-derived mitochondria. Increased formation of tau oligomers correlates with elevated ROS levels in the hippocampi of AD patients and tauopathy mice, AD- and MCI-derived mitochondria and AD and MCI cybrid cells. Furthermore, scavenging ROS by application of mito-TEMPO/2-(2,2,6,6-Tetramethylpiperidin-1-oxyl-4-ylamino)-2-oxoethyl)triphenylphosphonium chloride, a mitochondria-targeted antioxidant, not only inhibits the generation of mitochondrial ROS and rescues mitochondrial respiratory function but also robustly suppresses tau oligomer accumulation in MCI and AD cybrids as well as cortical neurons from tau mice. These studies provide substantial evidence that mitochondria-mediated oxidative stress contributes to tau oligomer formation and accumulation.
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disease that is associated with the abnormal upregulation of oxidative stress. A key pathological hallmark of AD is the presence of neurofibrillary tangles (NFTs), which are composed of abnormally phosphorylated and aggregated tau (1,2). Elevated phosphorylation and aggregation of tau can reduce tau–microtubule interactions, leading to microtubule abnormalities, dysfunctional axonal transport along microtubules, and neuronal death (3). Tau phosphorylation is modulated by stress conditions such as oxidative stress and alterations in glucose metabolism during hypothermia and starvation (4). Tau oligomers, intermediate species that form prior to NFTs, include various species of tau protein such as dimeric, multimeric, granular and possibly small filamentous aggregates, have deleterious effects on synaptic function and contribute to memory deficiency (5,6).
Mitochondria are a major source of reactive oxygen species (ROS). Oxidative stress is a pathological characteristic of tauopathy and evidence has shown that accumulation of ROS can directly stimulate tau hyperphosphorylation and aggregation (7,8). However, the cause and effect of tau oligomers in the disease process are not fully understood. In this study, we investigated the accumulation of pathological tau oligomers and its relevance to AD pathogenesis, including tau pathology and dysfunctional AD mitochondria to address the following key questions: Is the accumulation of tau oligomers associated with mitochondrial defects and ROS production in AD and a tau-enriched environment? If so, does blockade of mitochondrial ROS eliminate tau oligomer formation and attenuate mitochondrial dysfunction? In this study, we comprehensively analyzed the levels of tau oligomers, ROS and mitochondrial function in AD-affected hippocampus, tauopathy mouse model, primary cortical neurons from tauopathy mice, and mild cognitive impairment (MCI) and AD cybrids as ex vivo models for AD mitochondrial dysfunction. Our studies disclose the link between AD mitochondrial stress and pathological tau oligomer accumulation relevant to AD and tau pathology.
Results
Accumulation of tau oligomers is associated with ROS in the AD brain
Given the accumulation of amyloid beta (Aβ) and abnormal tau in AD-affected brain regions including the hippocampus, we first examined the levels of tau oligomers (oTau) in the AD-affected hippocampus. Immunostaining with the tau oligomeric complex I (TOC1) antibody showed that intensities of oTau-positive signals were significantly elevated in the AD hippocampus (Fig. 1A and B). Increased tau oligomers were mainly present in cortical neurons labeled by neuronal marker MAP-2 (Fig. 1A). Consistent with the immunostaining results, immunodot blotting demonstrated that oTau levels were greatly elevated in the AD hippocampus but not in the cerebellum, which is an AD spared region, as compared with non-AD brains (Fig. 1C–F).
Figure 1.
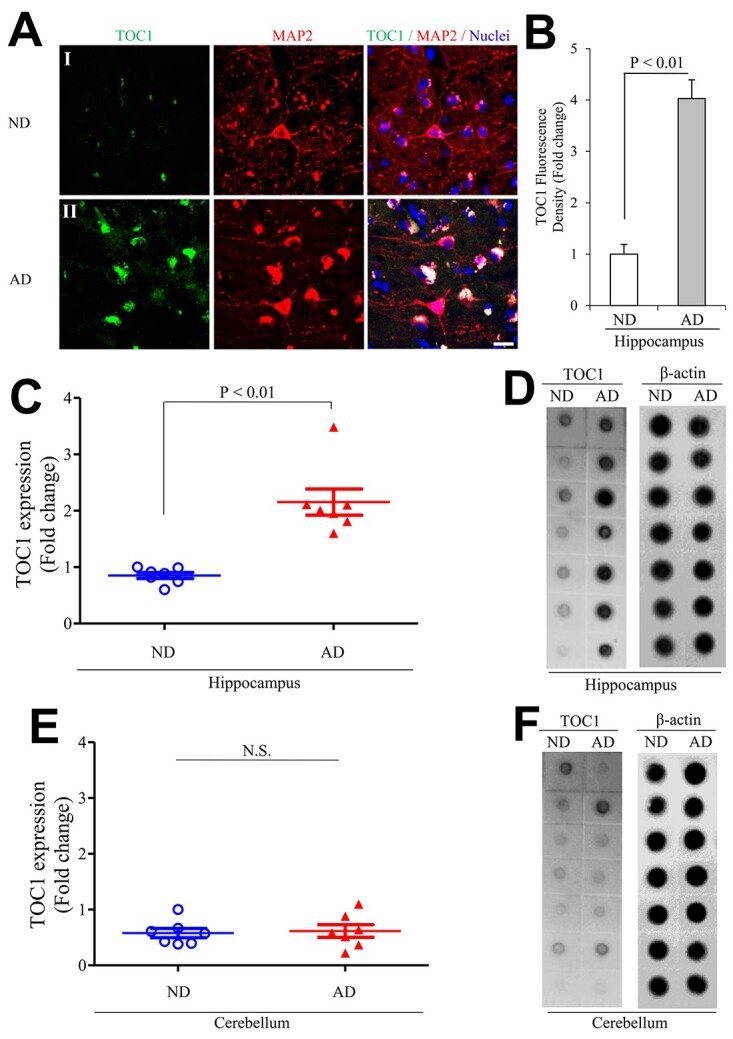
Tau oligomers pathology in human Alzheimer’s disease (AD) hippocampus. (A, B) Tau oligomeric complex 1 (TOC1) immunohistochemistry on AD hippocampus. (A) Representative images showed TOC1 immunohistochemistry on hippocampus (I: ND and II: AD; TOC1: green, MAP2: red). (B) Quantification of TOC1 immunohistochemistry was performed with the hippocampus from the indicated hippocampal sections. n = 4 per group. (C–F) The graph presents quantification of immunodot blotting (D and F) for TOC1 normalized to β-actin on hippocampus (C, D) and cerebellum (E, F) of ND and AD brains. n = 7 per group. The representative immunodot blotting for TOC1 from indicated hippocampal (C and D) and cerebellum (E and F) homogenates, and β-actin served as a loading control.
To determine whether elevation of oTau was correlated to oxidative stress, we quantitatively measured the intracellular ROS levels in the hippocampus with highly specific electron paramagnetic resonance (EPR) spectroscopy. Intracellular ROS levels as indicated by EPR peaks were significantly elevated in the AD hippocampus (Fig. 2A and B). Furthermore, oTau levels by quantification of intensity of immunoreactive oTau dot blots were positively correlated with ROS (Fig. 2C), suggesting a possible link between oTau accumulation and ROS production/accumulation relevant to AD pathology. There are no significant differences in the levels of oTau and ROS between male and females.
Figure 2.

Tau oligomers associate with ROS levels in human AD hippocampus. Quantification of EPR (A) and representative spectra of EPR spectra (B) in the indicated hippocampal homogenous. The peak height in the spectrum indicates levels of ROS. n = 7 per group. (C) Correlation analysis of the relationship between tau oligomer accumulation based on the intensity of immunoreactive oTau dot bots and ROS levels quantified by EPR on hippocampus. n = 14 for the correlation analysis.
Accumulation of tau oligomers associates with ROS in P301S tauopathy mice
Next, we examined tau oligomers, ROS levels and their association with tau pathology in tauopathy mice. Immunostaining revealed increased oTau in the cortex, and hippocampus of 9-month-old P301S mice and their presence in cortical neurons (Fig. 3A and B). Quantification of immunoreactive oTau dot blots revealed robustly elevated oTau in the entorhinal cortex of 6- to 12-month-old and hippocampus of 9- to 12-month-old P301S mice, respectively (Fig. 3C and D), and oTau levels were elevated in an age-dependent manner (Fig. 3C and D). oTau were not elevated at 1-month-old P301S mice (Supplementary Material, Fig. S1A and C), when compared to age-matched nonTg mice. In contrast, phospho-Tau (Ser202/Thr205) and PHF1-Tau (Ser396/Ser404) were detected as early as 1.5-month-old, and both increases with ages, earlier than the increase of tau oligomers (Supplementary Material, Fig. S2).
Figure 3.

Age-dependent Tau oligomers pathology in P301S mutant human tau transgenic mice. (A, B) TOC1 immunohistochemistry on entorhinal cortex and hippocampus from 9-month-old P301S mutant human tau transgenic mice. (A) Representative images showed TOC1 immunohistochemistry on entorhinal cortex (I and II) and hippocampus (III and IV). I, III: nonTg, and II, IV: P301S tau transgenic mice (TOC1: green, MAP2: red). Scale bar = 50 μm. (B) Quantifications of TOC1 immunohistochemistry were performed in the entorhinal cortex (open bar) and hippocampus (black bar) from the indicated mice. n = 5 mice (3 males and 2 females) per group. (C, D) The bar graph presents quantification of immunodot blotting for TOC1 normalized to β-actin on entorhinal cortex (C) and hippocampus (D) of the indicated mice. n = 5 mice (3 males and 2 females) per group. The representative immunodot blotting for TOC1 from indicated entorhinal cortex (C) and hippocampus (D) homogenates, and β-actin served as a loading control.
Accordingly, the intracellular ROS levels indicated by EPR peaks were significantly elevated in the 3- and 6-month-old P301S entorhinal cortex and hippocampus compared to nonTg mice (Fig. 4A and B), but not in 1-month-old P301S mice (Supplementary Material, Fig. S1B and D). Levels of oTau by the quantification of immunoreactive oTau dot blots were positively correlated with ROS levels in both the cortex (Fig. 4C) and hippocampus of P301S mice (Fig. 4D). These data suggest that tau oligomers are associated with aging and oxidative stress relevant to tau pathology. No significant difference in tau oligomers and ROS levels was found between male and female in P301S tau mice (data not shown).
Figure 4.

Tau oligomers associate with ROS levels in P301S mutant human tau transgenic mice. Quantitative data of EPR spectra (A) (up: entorhinal cortex and down: hippocampus) and representative spectra of EPR (B) (left: entorhinal cortex and right: hippocampus) in the indicated mice. The peak height in the spectrum indicates levels of ROS. n = 5 mice (3 males and 2 females) per group. (C, D) Correlation analysis of the relationship between tau oligomer based on the quantification of the intensity of immunoreactive oTau dot bots and ROS levels on entorhinal cortex (C) and hippocampus (D). N = 5 mice (3 males and 2 females) per group.
Scavenging mitochondrial ROS eliminates tau oligomers in cortical tau neurons from tau mice in vitro
To further evaluate the contribution of mitochondrial ROS to tau aggregation, primary cortical neurons cultured from P301S tau mice were treated with mito-TEMPO, a scavenger for mitochondria-derived ROS. Compared to nonTg neurons, cortical neurons cultured from tau mice displayed significantly elevated levels of tau oligomers, with TOC1 staining and expression significantly distributed along the cellular bodies and processes; mito-TEMPO treatment strikingly inhibited these TOC1-positive staining signals (Fig. 5A and B). Treatment with mito-TEMPO almost completely reduced tau oligomer levels to those of the vehicle controls as demonstrated by TOC1 immunodot blotting (Fig. 5C and D).
Figure 5.

Effect of Mito-TEMPO on tau oligomer pathology and ROS levels in hippocampal neurons cultured from tau mice in vitro. (A, B) TOC1 immunocytochemistry 21 days in vitro (DIV) hippocampal neurons. (A) Representative images showed TOC1 immunocytochemistry on hippocampal neurons (TOC1: green, MAP2: red). I: nonTg, II: TAU, and III: TAU + TEMPO. The right panels are the enlarged views of TOC1 in the left panels. (B) Quantification of TOC1 immunocytochemistry in (A). n = 8 neurons per group. Scale bars = 25 μm. (C) The representative immunodot blottings for TOC1 were shown from indicated neurons, and β-actin served as a loading control. (D) The bar graph presents quantifications of immunodot blottings for TOC1 normalized to β-actin of the indicated neurons. n = 3 per group. Complex I (E) activities and ATP levels (F) were determined in indicated groups. Data are expressed as fold change relative to the vehicle group (N = 3 independent experiments). Quantification of EPR (G) and representative spectra of EPR spectra (H) in the indicated hippocampal neurons. The peak height in the spectrum indicates levels of ROS. n = 4 per group. (I) Correlation analysis of the relationship between tau oligomer based on the quantification of immunoreactive oTau immunodot blots for TOC1 and ROS levels on hippocampal neurons. n = 9 for the correlation analysis.
Functionally, mito-TEMPO treatment not only alleviated mitochondrial defects, as shown by increased the activity of a key mitochondrial respiratory enzyme (complex I in Fig. 5E) and ATP levels (Fig. 5F), but also suppressed ROS levels (Fig. 5G and H). Similarly, mito-TEMPO abolishment of tau-mediated oTau formation was positively correlated with reduction in ROS levels in cortical neurons cultured from tau mice (Fig. 5I). These data indicate that scavenging mitochondrial ROS blocks tau- and ad-mediated pathological tau oligomer accumulation and restores mitochondrial function.
Accumulation of tau oligomers is associated with ROS in human MCI and AD cybrids
Mitochondrial dysfunction is one of the early pathological features of AD. Dysfunctional mitochondria produce excessive ROS. Human trans-mitochondrial ‘cybrid’ (cytoplasmic hybrid) neuronal cells whose mitochondria are derived from platelets of patients with sporadic AD or mild cognitive impairment (MCI) exhibit significant changes in mitochondrial structure and function and increases in ROS generation/accumulation (9–11). We therefore utilized MCI and AD cybrids as an ex vivo model to determine the potential impact of MCI- and AD-derived mitochondrial defects on pathological tau oligomers. Levels of tau oligomers significantly increased in differentiated MCI and AD cybrids compared to non-AD controls. AD cybrid cells exhibited higher levels of tau oligomers than MCI cybrids (Fig. 6A), suggesting that accumulation of tau oligomers is associated with progression of mitochondrial perturbation in AD. Similar results were obtained from MCI and AD platelets (Fig. 6B). In addition, application of a mitochondria-targeted antioxidant, mito-Tempo (9,12), significantly reduced both tau oligomer formation (Fig. 6C).
Figure 6.

Tau oligomer pathology in differentiated AD and MCI trans-mitochondrial cybrid cells and human platelets. (A, B) The bar graph presents quantification of immunodot blotting for TOC1 normalized to β-actin on differentiated MCI and AD trans-mitochondrial cybrid cells (A) and human platelets (B). n = 5 for cybrids and 7 for platelets per group. The representative immunodot blottings were shown for TOC1 from indicated cybrids (A) and platelets (B), and β-actin served as a loading control. (C) The bar graph presents quantification of immunodot blotting for TOC1 normalized to β-actin on differentiated MCI and AD trans-mitochondrial cybrid cells with or without mito-TEMPO treatment. The representative immunodot blottings were shown for TOC1 from indicated cybrids and β-actin served as a loading control.
ROS levels were also significantly elevated in differentiated AD and MCI cybrids (Fig. 7A) and their derived platelets (Fig. 7B), which was positively correlated with tau oligomer contents (Fig. 7C and D). In addition, treatment with a mitochondria-targeted antioxidant, mito-Tempo, significantly reduced ROS levels (Fig. 7E) in MCI and AD cybrids. These results support the relevance of increased accumulation of tau oligomers to AD-mediated mitochondrial defects and oxidative stress.
Figure 7.
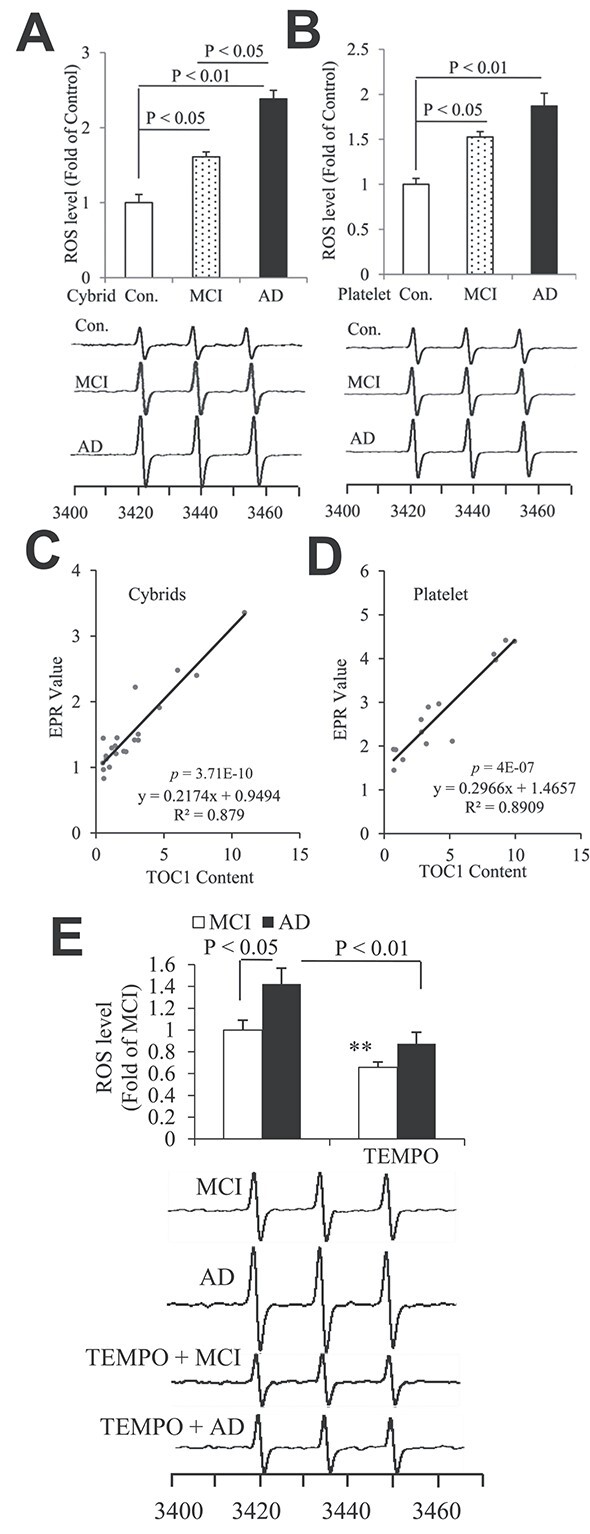
Tau oligomers associate with ROS levels in differentiated AD and MCI trans-mitochondrial cybrid cells and human platelets. Quantifications of EPR spectra and representative spectra of EPR in the indicated cybrids (A) and platelets (B). The peak height in the spectrum indicates levels of ROS. n = 5 for cybrids and 7 for platelets per group. (C, D) Correlation analysis of the relationship between Tau oligomers based on the quantification of immunodot blots for TOC1 and ROS levels on cybrids (C) and platelets (D). n = 15 for cybrids and 21 for platelets for the correlation analysis. (E) The bar graph presents quantification of immunodot blotting for TOC1 normalized to β-actin on differentiated MCI and AD trans-mitochondrial cybrid cells with or without mito-TEMPO treatment. The representative immunodot blottings were shown below for TOC1 from indicated cybrids and β-actin served as a loading control. n = 5 for cybrids per group.
Discussion
Tauopathies are a class of neurodegenerative disorders characterized by abnormal phosphorylation and aggregation of the microtubule-associated protein tau into paired helical filaments (PHFs) or straight filaments, leading to the formation of NFTs. In addition to the filamentous forms that comprise NFTs and SFs tau forms smaller multimeric oligomer species that are thought to represent soluble toxic forms of tau. Recent studies have suggested that tau oligomers can directly cause neuronal dysfunction (13). Upregulated granular tau oligomer levels occur prior to NFT formation and clinical symptoms of AD. Reduction of tau by doxycycline treatment improved memory impairment in P301L tau mice without affecting NFT formation, suggesting an early role for pre-filamentous forms of tau in AD pathogenesis relevant to cognitive decline (14,15).
In this study, we analyzed levels of tau oligomers in the human AD hippocampus, AD-related tauopathy mouse models, and MCI- and AD-derived mitochondria. Levels of tau oligomers were significantly elevated in AD brains, tau overexpressing mice and cortical neurons cultured from tau transgenic mice. Interestingly, tau oligomers accumulated in an age-dependent manner in tau overexpressing mice, with significant elevation starting at 6–7 months of age.
Previous studies show that oxidative stress and mitochondrial abnormalities appear prior to tau pathology (16), and hyperphosphorylation of tau is associated with mitochondrial oxidative stress (7), suggesting that oxidative stress and mitochondrial abnormalities may lead to the formation and accumulation of tangles, tau oligomers and neurodegeneration.
Overproduction of ROS and oxidative stress-mediated cellular perturbation are known to be key players in AD pathogenesis, including tauopathy (17). However, the causes and consequences of pathological tau species, such as toxic tau oligomers, are not fully defined. We observed that accumulation of tau oligomers is significantly elevated and positively correlated to ROS levels in human AD brains, MCI- and AD-derived mitochondria and tauopathy mice. By inhibiting mitochondrial ROS with mito-TEMPO, a mitochondria-targeted antioxidant, a striking reduction of tau oligomer accumulation in cortical neurons cultured from tau mice was achieved. Intriguingly, mito-TEMPO also suppressed AD mitochondria-induced tau oligomer accumulation and rescued mitochondrial respiratory function. Our studies reveal the role of the ROS/mitochondria dysfunction axis in aberrant tau oligomer accumulation. The detailed mechanisms require further investigation. Note that tau oligomers did not significantly increase in AD-spared regions such as the cerebellum when compared to non-AD controls, nor were there significant changes in ROS levels in these spared regions. Thus, our data align with several prior studies showing that the accumulation of tau oligomers is associated with AD pathology (18–22).
In the presence of staurosporine, 12-O-tetradecanoyl phorbol-13 acetate (TPA) or retinoic acid (RA), human SH-SY5Y neuroblastoma cells can differentiate into a mature neuronal phenotype with the appearance of neurite-like processes as the most prominent alterations (18). Differentiated cybrid cells containing patient-derived mitochondria provide an ex vivo model for studying the effects of AD-specific tau oligomer formation. As shown in Figure 6A, levels of tau oligomers were very low in non-AD cybrids. However, levels of tau oligomers significantly increased in differentiated MCI and AD cybrids compared to non-AD controls. AD cybrid cells exhibited higher levels of tau oligomers than MCI cybrids (Fig. 6A), suggesting that accumulation of tau oligomers is associated with progression of mitochondrial perturbation in AD. In addition, treatment with a mitochondria-targeted antioxidant, mito-Tempo (9,12), significantly reduced tau oligomer accumulation and formation (Fig. 6C). These results suggest the relevance of increased accumulation of tau oligomers to AD-mediated mitochondrial defects and oxidative stress.
Taken together, by using human AD samples, human AD patient-derived mitochondria, and tauopathy mouse model, we have provided substantial evidence of the connection between mitochondrial ROS and the accumulation of tau oligomers. Blocking mitochondrial oxidative stress eliminates the accumulation of tau oligomers and improves mitochondrial function relevant to tau pathology. Thus, our findings support the possibility that inhibiting mitochondrial oxidative stress and dysfunctions could be a promising therapeutic target to prevent and treat tauopathies.
Materials and Methods
Human subjects
We obtained hippocampal and cerebellar tissues from individuals with Alzheimer’s disease and age-matched, non-Alzheimer’s disease controls from the New York Brain Bank, Columbia University Medical Center, New York. Detailed information for each of the cases studied is shown in Supplementary Material, Table S1 online. Informed consent was obtained from all subjects.
Creation of cybrid cell lines
MCI, AD and non-MCI/AD cybrid cells were kindly provided by Dr Swerdlow from the University of Kansas Alzheimer’s Disease Center (KUADC) Mitochondrial Genomics and Metabolism Core, Kansas. Cybrid cell lines were created on the human neuroblastoma cell (SH-SY5Y) nuclear background (24). In brief, SH-SY5Y cells were previously depleted of endogenous mtDNA (Rho0 cells), which were fused with platelet cytoplasm from human subjects, and repopulated with mitochondria containing mtDNA from patients or controls as previously described (25). The quantitative real-time polymerase chain reaction showed that the intact mtDNA copies were present in all cybrids without detectable large-scale deletion after many passages of cell proliferation as previously described (9).
MCI, AD patients and non-MCI/AD controls were recruited from the KUADC. Subjects with AD met the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association criteria (26). Non-MCI/AD subjects were cognitively normal and age-matched to MCI and AD subjects. All subjects provided written informed consent to participate in the study. We used five cell lines per group in this study, and the ages of MCI, AD and non-AD platelet donors were 72.6 ± 3.46, 75.8 ± 5.04 and 73.6 ± 2.96 years, respectively. The information about gender, age and disease status of donors is described in Supplementary Material, Table S2 also in our previous studies (9,11,27). We do not have an interest in making cybrids from mutation subjects because presumably the mitochondrial defects would be corrected in cell culture, since the mitochondria are separated from the presence of the nuclear mutation when we do the transfection into the Rho0 cells.
There are several reasons to select SH-SY5Y cells to create MCI and AD cybrid cells: Firstly, they are a commonly used human neuronal line and available in the laboratory when we decided to generate human neuronal cybrid cell line, and secondly, they can be differentiated into neuronal-like cells. Importantly, SH-SY5Y cells have been very successfully transmitted by mitochondria derived from human AD and non-AD subject as a human AD cybrid cell lines that recapitulate specific AD mitochondriopathies (9,28).
Cybrid growth and differentiation
In the presence of staurosporine, TPA or RA, human SH-SY5Y neuroblastoma cells can differentiate into a mature neuronal phenotype with the appearance of neurite-like processes as the most prominent alterations (9,27,29). MCI, AD and non-MCI/AD cybrid cells were grown in T75 tissue culture flasks in Dulbecco’s modified Eagle’s medium supplemented with 10% characterized fetal bovine serum (Gibco BRL, Logan, Utah), 100 μg/ml pyruvate, 50 μg/ml uridine, antibiotic-antimycotic, 100 Units/ml penicillin G and 100 μg/ml streptomycin as previously described (9). Culture media were replaced to the differentiation media [neurobasal media supplemented with 1× B27 (Invitrogen, Carlsbad, CA) and 0.5 mm glutamine, and antibiotic-antimycotic] with 10 nm staurosporine (SAT, Sigma-Aldrich Corp, St Louis, MO). Half of the differentiation media were made fresh with 10 nm SAT and replaced every day as previously described (9,27,29). The differentiated neuronal MCI, AD and non-MCI/AD cybrid cells were used in this study.
Evaluation of intracellular ROS
Evaluation of intracellular ROS levels was conducted by EPR spectroscopy. Brain tissues or cultured neurons were incubated with CMH (cyclic hydroxylamine 1-hydroxy-3-methoxycarbonyl-2, 2, 5, 5-tetramethyl-pyrrolidine, Enzo Life Sciences, NY, 100 μm) for 30 min and then washed three times with cold PBS. Subsequently, brain tissues (same samples used to assess oligomeric tau/TOC1 by immunodot blotting) and neurons were collected and homogenized with 100 μl of PBS for EPR measurement. The EPR spectra were recorded, stored and analyzed with a Bruker EleXsys 540 X-band EPR spectrometer (Billerica, MA) using Bruker Xepr software Xepr (Billerica, MA) (30,31).
Measurement of respiratory chain complex activities and ATP levels
Mitochondrial respiratory complex I activities were measured in neuronal homogenates as described previously (31–33). NADH: ubiquinone oxidoreductase (COX I) enzyme activity was determined in 25 mm potassium buffer containing KCl, Tris–HCl and EDTA (pH 7.4). Homogenates (50 μg protein) were incubated with 2 μg/ml antimycin, 5 mm MgCl2, 2 mm KCN and 65 μm co-enzyme Q1 were and the oxidation of NADH was recorded for 3 min. Subsequently, 2 μg/ml rotenone was added and the absorbance was measured for another 3 min. The change in absorbance was monitored at 340 nm using an Amersham Biosciences Ultrospect 3100 Pro spectrophotometer.
ATP levels were determined using an ATP Bioluminescence Assay Kit (Roche) following the manufacturer’s instruction. Briefly, neurons were collected in the provided lysis buffer, incubated on ice for 30 min and centrifuged at 12 000g for 10 min. ATP levels were then measured in the subsequent supernatants using a luminescence plate reader (Molecular Devices). A 1.6-s delay after substrate injection and 10-s integration time were used.
Immunodot blotting
Brain or cell lysates were prepared and analyzed as described for immunoblotting with the following modifications. Samples were spotted onto the nitrocellulose membrane using a Whatman Minifold I immunodot blotting apparatus. The membranes were blocked and probed with TOC1 (oligomeric tau; 1:1000, mouse IgM; Kanaan Lab, AB 2832939) and β-actin (1:5000, A5441; Sigma-Aldrich). Signal intensity measurements for each dot were expressed as the ratio of oligomeric tau (TOC1 signal) to β-actin. ImageJ software (National Institutes of Health, Bethesda, MD) was used for quantification of the intensity of the developed blots.
Immunohistochemical staining
Brain slices from the indicated Tg mice were subjected to double immunostaining with TOC1 (1:1000) and rabbit anti-microtubule-associated protein 2 (MAP2; 1:5,000, PA5-17646, Thermo Fisher Scientific, Inc.) at 4°C overnight followed by Alexa-Fuor 488 goat anti-mouse IgM- and Alex-Fluor 594 goat anti-rabbit IgG-specific antibodies, respectively. Images were acquired on a Leica SP5 confocal microscope and analyzed using Leica LAS AF software (Leica Wetzlar) and MetaMorph (Molecular Devices) Program.
The information for animal studies, primary neuronal culture and immunoblotting has been described in our previous studies (32,34) and detailed in Supplementary Materials.
Statistical analysis
All data were expressed as the mean ± SEM. Student t-tests were performed for analysis and comparisons between two groups. Data were analyzed by one-way ANOVA for repeated measure analysis using commercially available software (Statview, version 5.0.1, Berkeley, CA), followed by Fisher’s protected least significant difference for post hoc comparisons. P < 0.05 was considered significant.
Data Availability
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Supplementary Material
Acknowledgements
We thank Dr Swerdlow for providing cybrid cells for our study. Generation and characterization of cybrid cells were supported by the University of Kansas Alzheimer’s Disease Center. We thank Justin T. Douglas for assistance in using EPR instrumentation.
Conflict of Interest statement. The authors declare that they do not have any conflict of interest.
Contributor Information
Fang Du, Department of Surgery, Vagelos College of Physicians and Surgeons of Columbia University, New York, NY 10032, USA.
Qing Yu, Department of Surgery, Vagelos College of Physicians and Surgeons of Columbia University, New York, NY 10032, USA.
Nicholas M Kanaan, Department of Translational Neuroscience, Michigan State University College of Human Medicine, MI 49503.
Shirley ShiDu Yan, Department of Surgery, Vagelos College of Physicians and Surgeons of Columbia University, New York, NY 10032, USA; Molecular Pharmacology & Therapeutics, Columbia University, New York, NY 10032, USA.
Funding
National Institutes of Health (NIH)/ National Institute on Aging (NIA) (R37AG037319, RF1AG054320, R01AG053041, R01AG069426); Alzheimer’s Association Research Grant (AARG, 2018-AARG-592230 Alzheimer’s Association).
Authors' contributions
S.S.Y. initiated and supervised the research, designed experiments, developed the concept and wrote the paper. F.D. designed and performed experiments, analyzed data and wrote the paper. Q.Y. performed experiments on mitochondrial function, immunoblotting, analyzed data and assisted in preparation of manuscript. N.M.K provided TOC1 antibody and edited manuscript.
References
- 1. Lee, V.M. and Trojanowski, J.Q. (1999) Neurodegenerative tauopathies: human disease and transgenic mouse models. Neuron, 24, 507–510. [DOI] [PubMed] [Google Scholar]
- 2. Ballatore, C., Lee, V.M. and Trojanowski, J.Q. (2007) Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci., 8, 663–672. [DOI] [PubMed] [Google Scholar]
- 3. Stamer, K., Vogel, R., Thies, E., Mandelkow, E. and Mandelkow, E.M. (2002) Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol., 156, 1051–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Planel, E., Miyasaka, T., Launey, T., Chui, D.H., Tanemura, K., Sato, S., Murayama, O., Ishiguro, K., Tatebayashi, Y. and Takashima, A. (2004) Alterations in glucose metabolism induce hypothermia leading to tau hyperphosphorylation through differential inhibition of kinase and phosphatase activities: implications for Alzheimer's disease. J. Neurosci., 24, 2401–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kaniyappan, S., Chandupatla, R.R., Mandelkow, E.M. and Mandelkow, E. (2017) Extracellular low-n oligomers of tau cause selective synaptotoxicity without affecting cell viability. Alzheimers Dement., 13, 1270–1291. [DOI] [PubMed] [Google Scholar]
- 6. Lasagna-Reeves, C.A., Castillo-Carranza, D.L., Sengupta, U., Clos, A.L., Jackson, G.R. and Kayed, R. (2011) Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener., 6, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Melov, S., Adlard, P.A., Morten, K., Johnson, F., Golden, T.R., Hinerfeld, D., Schilling, B., Mavros, C., Masters, C.L., Volitakis, I.et al. (2007) Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS One, 2, e536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dias-Santagata, D., Fulga, T.A., Duttaroy, A. and Feany, M.B. (2007) Oxidative stress mediates tau-induced neurodegeneration in Drosophila. J. Clin. Invest., 117, 236–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yu, Q., Fang, D., Swerdlow, R.H., Yu, H., Chen, J.X. and Yan, S.S. (2016) Antioxidants rescue mitochondrial transport in differentiated Alzheimer’s disease trans-mitochondrial cybrid cells. J. Alzheimers Dis., 54, 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fang, D., Qing, Y., Yan, S., Chen, D. and Yan, S.S. (2016) Development and dynamic regulation of mitochondrial network in human midbrain dopaminergic neurons differentiated from iPSCs. Stem cell Reports, 7, 678–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Du, F., Yu, Q. and Yan, S.S. (2021) PINK1 activation attenuates impaired neuronal-like differentiation and synaptogenesis and mitochondrial dysfunction in Alzheimer's disease trans-mitochondrial cybrid cells. J. Alzheimers Dis., 81, 1749–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hou, Y., Ghosh, P., Wan, R., Ouyang, X., Cheng, H., Mattson, M.P. and Cheng, A. (2014) Permeability transition pore-mediated mitochondrial superoxide flashes mediate an early inhibitory effect of amyloid beta1–42 on neural progenitor cell proliferation. Neurobiol. Aging, 35, 975–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fa, M., Puzzo, D., Piacentini, R., Staniszewski, A., Zhang, H., Baltrons, M.A., Li Puma, D.D., Chatterjee, I., Li, J., Saeed, F.et al. (2016) Extracellular tau oligomers produce an immediate impairment of LTP and memory. Sci. Rep., 6, 19393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Maeda, S., Sahara, N., Saito, Y., Murayama, S., Ikai, A. and Takashima, A. (2006) Increased levels of granular tau oligomers: an early sign of brain aging and Alzheimer’s disease. Neurosci. Res., 54, 197–201. [DOI] [PubMed] [Google Scholar]
- 15. Santacruz, K., Lewis, J., Spires, T., Paulson, J., Kotilinek, L., Ingelsson, M., Guimaraes, A., DeTure, M., Ramsden, M., McGowan, E.et al. (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science, 309, 476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dumont, M., Stack, C., Elipenahli, C., Jainuddin, S., Gerges, M., Starkova, N.N., Yang, L., Starkov, A.A. and Beal, F. (2011) Behavioral deficit, oxidative stress, and mitochondrial dysfunction precede tau pathology in P301S transgenic mice. FASEB J., 25, 4063–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Elahi, M., Hasan, Z., Motoi, Y., Matsumoto, S.E., Ishiguro, K. and Hattori, N. (2016) Region-specific vulnerability to oxidative stress, neuroinflammation, and tau hyperphosphorylation in experimental diabetes mellitus mice. J. Alzheimers Dis., 51, 1209–1224. [DOI] [PubMed] [Google Scholar]
- 18.Patterson, K.R., Remmers, C., Fu, Y., Brooker, S., Kanaan, N.M., Vana, L., Ward, S., Reyes, J.F., Philibert, K., Glucksman, M.J. et al. (2011) Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. Biol Chem, 286, 23063–23076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ward, S.M., Himmelstein, D.S., Lancia, J.K., Fu, Y., Patterson, K.R. and Binder, L.I. (2013) TOC1: characterization of a selective oligomeric tau antibody. J Alzheimers Dis, 37, 593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maeda, S., Sahara, N., Saito, Y., Murayama, M., Yoshiike, Y., Kim, H., Miyasaka, T., Murayama, S., Ikai, A. and Takashima, A. (2007) Granular tau oligomers as intermediates of tau filaments. Biochemistry, 46, 3856–3861. [DOI] [PubMed] [Google Scholar]
- 21.Koss, D.J., Jones, G., Cranston, A., Gardner, H., Kanaan, N.M. and Platt, B. (2016) Soluble pre-fibrillar tau and beta-amyloid species emerge in early human Alzheimer's disease and track disease progression and cognitive decline. Acta Neuropathol, 132, 875–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tiernan, C.T., Mufson, E.J., Kanaan, N.M. and Counts, S.E. (2018) Tau Oligomer Pathology in Nucleus Basalis Neurons During the Progression of Alzheimer Disease. J Neuropathol Exp Neurol, 77, 246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Prince, J.A. and Oreland, L. (1997) Staurosporine differentiated human SH-SY5Y neuroblastoma cultures exhibit transient apoptosis and trophic factor independence. Brain Res. Bull., 43, 515–523. [DOI] [PubMed] [Google Scholar]
- 24. Miller, S.W., Trimmer, P.A., Parker, W.D., Jr. and Davis, R.E. (1996) Creation and characterization of mitochondrial DNA-depleted cell lines with “neuronal-like” properties. J. Neurochem., 67, 1897–1907. [DOI] [PubMed] [Google Scholar]
- 25. Swerdlow, R.H. (2007) Mitochondria in cybrids containing mtDNA from persons with mitochondriopathies. J. Neurosci. Res., 85, 3416–3428. [DOI] [PubMed] [Google Scholar]
- 26. Albert, M.S., DeKosky, S.T., Dickson, D., Dubois, B., Feldman, H.H., Fox, N.C., Gamst, A., Holtzman, D.M., Jagust, W.J., Petersen, R.C.et al. (2011) The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement., 7, 270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yu, Q., Du, F., Douglas, J.T., Yu, H., Yan, S.S. and Yan, S.F. (2017) Mitochondrial dysfunction triggers synaptic deficits via activation of p38 MAP kinase signaling in differentiated Alzheimer's disease trans-mitochondrial cybrid cells. J. Alzheimers Dis., 59, 223–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Silva, D.F., Selfridge, J.E., Lu, J., E, L., Roy, N., Hutfles, L., Burns, J.M., Michaelis, E.K., Yan, S., Cardoso, S.M.et al. (2013) Bioenergetic flux, mitochondrial mass and mitochondrial morphology dynamics in AD and MCI cybrid cell lines. Hum. Mol. Genet., 22, 3931–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fang, D., Zhang, Z., Li, H., Yu, Q., Douglas, J.T., Bratasz, A., Kuppusamy, P. and Yan, S.S. (2016) Increased electron paramagnetic resonance signal correlates with mitochondrial dysfunction and oxidative stress in an Alzheimer’s disease mouse brain. J. Alzheimers Dis., 51, 571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Du, F., Yu, Q., Yan, S., Zhang, Z., Vangavaragu, J.R., Chen, D., Yan, S.F. and Yan, S.S. (2021) Gain of PITRM1 peptidase in cortical neurons affords protection of mitochondrial and synaptic function in an advanced age mouse model of Alzheimer's disease. Aging Cell, 20, e13368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fang, D., Wang, Y., Zhang, Z., Du, H., Yan, S., Sun, Q., Zhong, C., Wu, L., Vangavaragu, J.R., Yan, S.et al. (2015) Increased neuronal PreP activity reduces Abeta accumulation, attenuates neuroinflammation and improves mitochondrial and synaptic function in Alzheimer disease’s mouse model. Hum. Mol. Genet., 24, 5198–5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Du, F., Yu, Q., Chen, A., Chen, D. and Yan, S.S. (2018) Astrocytes attenuate mitochondrial dysfunctions in human dopaminergic neurons derived from iPSC. Stem Cell Reports, 10, 366–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fang, D., Yan, S., Yu, Q., Chen, D. and Yan, S.S. (2016) Mfn2 is required for mitochondrial development and synapse formation in human induced pluripotent stem cells/hiPSC derived cortical neurons. Sci. Rep., 6, 31462. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.