

ABSTRACT
Obesity is a common comorbidity in patients with asthma, and obese asthma patients present the most refractory phenotype among patients with severe asthma. Similar to the observations in non-obese asthma patients, clinical studies have revealed heterogeneity in obese asthma patients, including the occurrences of T helper (Th)2-high and Th2-low phenotypes. However, the mechanisms underlying obesity-related asthma are not completely understood. Though macroautophagy/autophagy is involved in asthma and obesity, its role in obesity-associated asthma is unknown. We hypothesized that autophagy is involved in the pathogenesis of obese asthma. For our investigations, we used high-fat diet-induced Atg5 (autophagy related 5)-deficient mice and epithelial cell-specific atg5−/− (Scgb1a1/CCSP-atg5−/−) obesity-induced mice. House dust mite (HDM)-sensitized atg5−/− obese mice exhibited marked eosinophilic inflammation and airway hyper-reactivity (AHR), compared to wild-type (WT) obese mice. Analyses of atg5−/− obese mice showed increased levels of Th2 cells but not ILC2s together with elevated expression of Th2 cytokines in the lung. In response to the HDM challenge, activated epithelial autophagy was observed in lean but not obese WT mice. Epithelium-specific deletion of Atg5 induced eosinophilic inflammation in Scgb1a1/CCSP-atg5−/− obese mice, and genetic analyses of epithelial cells from HDM-immunized atg5−/− obesity-induced mice showed an elevated expression of thymic stromal lymphopoietin (TSLP) and IL33. Notably, HDM-sensitized atg5−/− mice developed TSLP- and IL33-dependent eosinophilic inflammation and AHR. Our results suggest that autophagy contributes to the exacerbation of eosinophilic inflammation in obese asthma. Modulations of autophagy may be a therapeutic target in obesity-associated asthma.
Abbreviations: AHR: airway hyper-reactivity; BAL: bronchoalveolar lavage; Cdyn: dynamic compliance; BM: bone marrow; HDM: house dust mite; HFD: high-fat diet; ILC2s: type 2 innate lymphocyte cells; ROS: reactive oxygen species; RL: lung resistance; TSLP: thymic stromal lymphopoietin; TCC: total cell count; WT: wild type.
KEYWORDS: Asthma, autophagy, corticosteroid resistance, eosinophil inflammation, IL33, obesity, thymic stromal lymphopoietin
Introduction
Asthma and obesity are global health problems affecting adults, adolescents, and children [1–3]. Importantly, obesity is a common condition in asthma patients, and nearly 60% of patients with severe asthma are obese [4,5]. Obesity is intertwined with the pathogenesis of asthma via biological, physiological, and environmental factors. Epidemiological studies have revealed that obesity is a risk factor for the development of asthma [6] and vice versa [7]. Obese asthma patients exhibit the most severe phenotype with persistent symptoms, increased risk of hospitalization, and are refractory to standard medications [8–10]. Recently, obese patients with asthma have been shown to exhibit heterogeneous phenotypes similar to those of non-obese asthma patients [8]. In particular, obese asthma patients can be divided into two major phenotypes: late- and early-onset asthma [8]. Patients with late-onset asthma, who exhibit the classical features of obese asthma, are mostly females and have less allergic airway inflammation (non-type 2 phenotype) involving neutrophilic inflammation [11] and oxidative stress [12]. Early-onset asthma, the most severe disease among obese asthma patients, is characterized by the type 2 phenotype, i.e., high expression of markers of allergic inflammation and airway hyper-reactivity (AHR), where eosinophils may play an important role [8]. However, the precise pathophysiology of obesity-associated asthma is not fully elucidated.
Autophagy is a fundamental cellular process that degrades and recycles damaged organelles, microorganisms, and denatured proteins [13–16]. This conserved process is essential for the maintenance of cellular homeostasis and organ function. Autophagy was originally identified as a cellular protective process in response to starvation [17]. Later, a growing body of evidence demonstrated that aberrant changes in autophagy contribute to various diseases, including aging, cancer, cardiovascular disease, diabetes mellitus, and obesity [13]. In addition, autophagy is also involved in asthma and allergic diseases via dendritic cell-, macrophage-, and type 2 innate lymphocyte cell (ILC2)-mediated adaptive and innate immune responses [18–21]. However, studies investigating the involvement of autophagy in obesity-associated asthma are lacking.
Here, we investigated the role of autophagy in the pathogenesis of obese asthma using a high-fat diet (HFD)-induced mouse model of obesity. We showed that aberrant autophagy contributed to deteriorating eosinophilic inflammation and AHR which was thymic stromal lymphopoietin (TSLP) - and IL33-dependent but did not respond to steroids. Defects in autophagy in epithelial cells are responsible for aggravated eosinophilic inflammation in mice with impaired autophagy. This study provides novel information for understanding the biological mechanisms of obese asthma and the importance of autophagy.
Results
Obese mice did not respond to house dust mite exposure
To investigate the role of autophagy in obese asthma, we generated HFD-induced obese mice. WT mice were fed HFD or normal chow for 16–18 weeks (Figure S1A-B), followed by sensitization and intranasal challenge with HDM extract according to the protocol shown in Figure S1C. Two days after the last HDM challenge, lung function was evaluated via direct measurements of lung resistance (RL) and dynamic compliance (Cdyn) as described in the Methods section. Non-sensitized obese mice tended to have higher RL, lower Cdyn, and higher total cell count (TCC) in bronchoalveolar lavage (BAL) than lean mice. Interestingly, HDM-sensitized obese mice exhibited less allergic inflammations, as was evident from the significantly lower RL and higher Cdyn, together with lower eosinophils in BAL than in HDM-sensitized WT lean mice (Figure S1D-E). Interestingly, pharmacological inhibition of autophagy by 3-methyladenine (3-MA) reduced AHR and inflammation in HDM-sensitized WT obese mice (Figure S1F-G).
Autophagy contributed to exacerbating airway inflammation in obese mice
The autophagy pathway consists of a cascade of essential genes, such as autophagy-related gene 5 (ATG5) [14–16,22]. Studies have shown that depletion of ATG5 efficiently disrupts the autophagy pathway; therefore, Atg5-deficient mice are important for studying the autophagy pathway [17,18,20,23]. As constitutively Atg5-deficient mice die soon after birth [17], we used inducible conditional atg5 knockout mice in which tamoxifen injection deletes approximately 80–90% of Atg5 expression (referred to as atg5−/− mice; Figure S2A). Alternatively, the impaired autophagy pathway in atg5−/− obese mice was further validated by assessing increased SQSTM1/p62 levels (Figure S2B).
First, we confirmed that atg5−/− lean mice exhibited higher AHR and eosinophil inflammation than WT lean mice (Figure S3). Then, WT and atg5−/− mice with HFD-induced obesity were sensitized and challenged with HDM (Figure 1A). Notably, HDM-sensitized atg5−/− obese mice exhibited significantly higher AHR, representing higher RL and lower Cdyn than the HDM-challenged WT obese mice (Figure 1B). Further, HDM-challenged atg5−/− obese mice showed significantly higher numbers of TCC, eosinophils, and neutrophils in BAL compared with the WT obese mice (Figure 1C). Histological analyses revealed significantly increased thickening of the bronchial wall and peribronchiolar cell infiltration, together with the presence of PAS+ cells in HDM-challenged atg5−/− obese mice compared with the observations in WT obese mice (Figure 1D-F).
Figure 1.
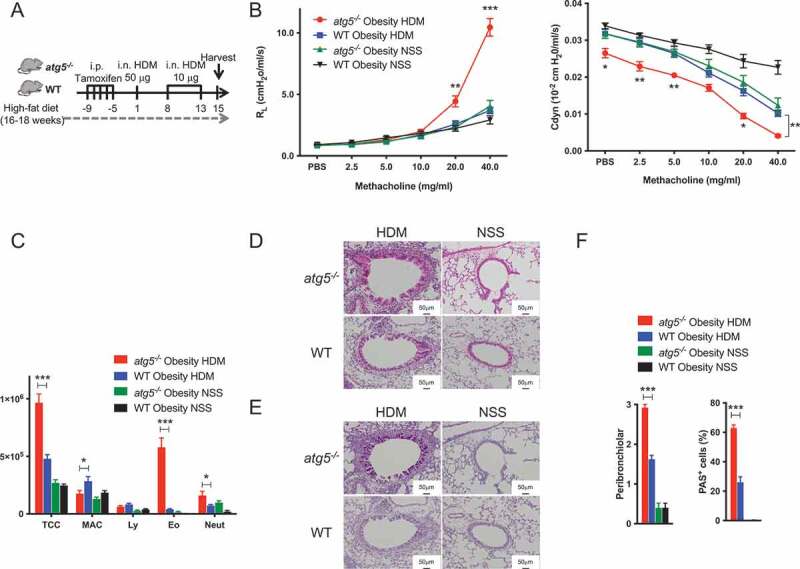
Impaired autophagy exacerbates airway inflammation in house dust mite (HDM)-sensitized obese mice. (A) Experimental timeline. WT and atg5−/− mice were fed with high-fat diet, and then sensitized and challenged with house dust mite (HDM). (B) Lung resistance (RL) and dynamic compliance (Cdyn), (n = 8–10/group) (C) Differential cell counts in BALF (n = 8–10/group). TCC, total cell count; MAC, macrophages; Ly, lymphocytes; Eo, eosinophils; Neut, neutrophils. (D) Hematoxylin and eosin staining. Original magnification: 100 × . (E) PAS staining. Original magnification: 100 × . (F) Quantifications of lung histopathology (n = 3–5/group). Data are representative of three independent experiments. Data are expressed as the mean ± SEM. P-values were calculated using two-way ANOVA test post hoc test with the Tukey’s multiple comparison test. *, P < 0.05 *, **, P < 0.01, ***, P < 0.001.
Next, cytokine levels in the lungs were assessed. Compared to WT obese mice, HDM-challenged atg5−/− obese mice exhibited significantly higher levels of IL5 (interleukin 5), IL13, IL33, and TSLP, along with ADIPOQ/adiponectin downregulation (Figure 2A–C). Meanwhile, the levels of IL17A, IL25, and LEP/leptin did not differ between atg5−/− obese mice and WT obese mice. The proportions of IL5- and IL13-producing CD4+ T cells, but not IL17A-producing CD4+ T cells, were significantly higher in HDM-challenged atg5−/− obese mice compared with those in WT obese mice (Figure 2D). In agreement with these results, GATA-3 and p-STAT6 expression was also elevated in HDM-challenged atg5−/− obese mice (Figure 2E). As both Th2 cells and ILC2s produce IL5 and IL13, we further analyzed ILC2s. As observed in a previous study [20], the number of ILC2s decreased in HDM-challenged atg5−/− obese mice (Figure S4). These results indicated the importance of Th2 cells in the development of severe allergic inflammation in atg5−/− obese mice.
Figure 2.
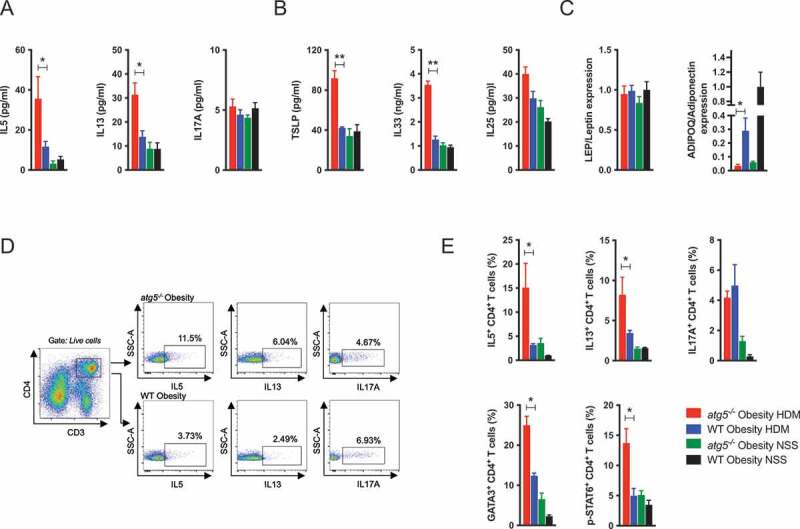
Increase in Th2 cells in HDM-challenged atg5−/− obese mice. (A) Cytokine levels in BALF (n = 5/group). (B) Cytokine levels in lung homogenates (n = 5/group). (C) Relative lung cytokine mRNA expression in lung homogenates (n = 3–7/group). (D) Representative dot plot presentation of cytokine production by CD3+ CD4+ T cells in the lung. (E) Frequencies of IL-5-, IL13-, IL17A-producing CD3+ CD4+ T cells, and GATA3- and p-STAT6-expressing CD3+ CD4+ T cells in the lung (n = 3–5/group). Data are representative of two independent experiments. Data are expressed as the mean ± SEM. P-values were calculated using two-way ANOVA test post hoc test with the Tukey’s multiple comparison test. *, P < 0.05, **, P < 0.01.
House dust mite-challenged atg5−/− obese mice were resistant to steroid treatment
Obese asthma is known to be resistant to steroid treatment [8–10]. In particular, early-onset obese asthma patients exhibited the most severe phenotype with high expression of markers of eosinophilic inflammation and AHR [8]. Further, increased infiltrations of eosinophils in the lungs of obese asthma have been reported [24,25]. Hence, we hypothesized that atg5−/− obese mice might represent obesity-associated asthma with a severe Th2 phenotype and investigated whether mice with HFD-induced obesity were resistant to steroid treatment. HDM-sensitized atg5−/− obese mice were treated with dexamethasone 1 day before and 1 day after the HDM challenge (Figure 3A). Notably, the results showed that dexamethasone did not ameliorate AHR and airway inflammation in HDM-sensitized atg5−/− obese mice (Figure 3B–C). Consistent with the AHR and BAL findings, lung histology showed that dexamethasone did not reduce bronchial wall thickening and the numbers of PAS+ cells in atg5−/− obese mice (Figure S5D-E).
Figure 3.
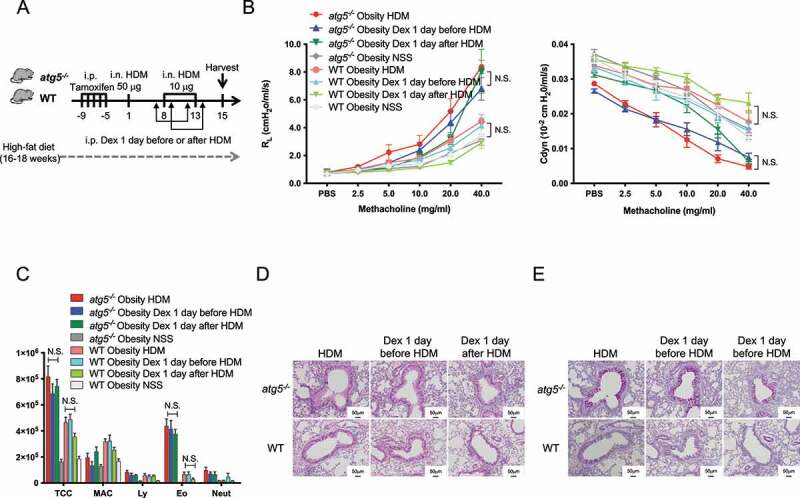
HDM-challenged atg5−/− obese mice were refractory to steroid treatment. (A) Experimental timeline. atg5−/− obese mice and WT obese mice were immunized as described in Figure 1A. The mice were intraperitoneally treated with dexamethasone (1 mg/kg) 1 day before or after HDM challenges. (B) Lung resistance (RL) and dynamic compliance (Cdyn) (C) Differential cell counts in BALF. TCC, total cell count; MAC, macrophages; Ly, lymphocytes; Eo, eosinophils; Neut, neutrophils. (D) Hematoxylin and eosin staining. Original magnification: 100 × . (E) PAS staining. Data are representative of two independent experiments (n = 5–7/group). Data are expressed as the mean ± SEM. P-values were calculated using two-way ANOVA test post hoc test with the Tukey’s multiple comparison test.
Epithelial autophagy contributed to eosinophilic inflammation
As epithelial autophagy is considered to be involved in the pathogenesis of asthma [26,27], we investigated whether lack of autophagy in immune cells or epithelial cells might influence the development of severe asthma. Chimeric mice were generated to assess the role of immune cells and structural cells in autophagy-dependent eosinophilic airway inflammation. Bone marrow (BM) cells from WT or atg5−/− mice were injected into sublethally irradiated atg5−/− mice or WT mice. These mice were fed with HFD and then immunized with HDM (Figure 4A). A part of the BM cells was isolated from CAG-EGFP transgenic mice to determine the reconstitution rate. The results showed a reconstitution rate of approximately 80–90% by donor immune cells in the lungs (Figure 4B). Moreover, atg5−/− BM-injected atg5−/− obese mice (recipient, red) exhibited increased RL and eosinophils in BAL compared to atg5−/− BM-injected WT obese mice (blue, Figure 4C–D). Similarly, another set of chimeric mice in which BM cells from WT mice were injected into irradiated atg5−/− mice (green) exhibited higher AHR and eosinophilic inflammation than WT obese mice generated by injecting WT BM cells (black, Figure 4C–D). HE and PAS staining are shown in Figure 4E–G. Collectively, these results suggested that atg5−/− structural cells are involved in eosinophilic inflammation in obese asthma rather than in immune cells.
Figure 4.
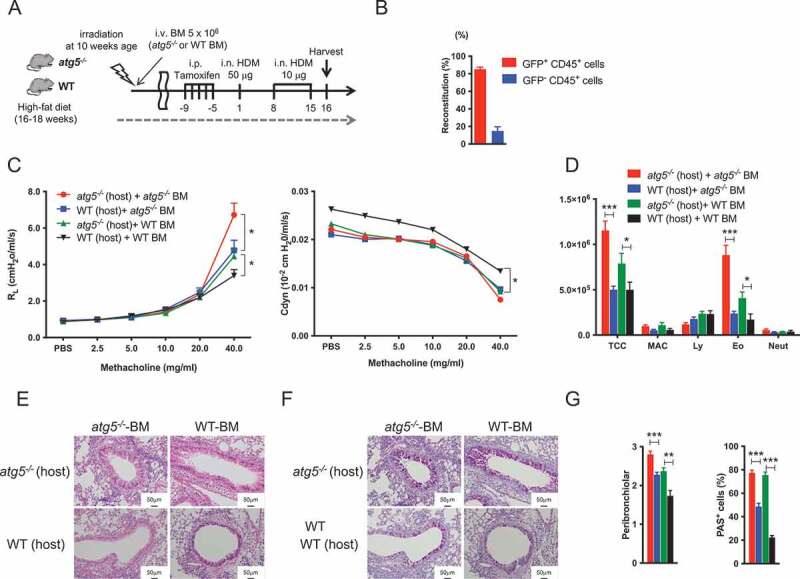
Impaired autophagy in structured cells contribute to autophagy-mediated airway inflammation in obesity asthma. (A) Experimental timeline. WT or atg5−/− bone-marrow (BM) cells were adaptively transferred into irradiated WT or atg5−/− mice. The mice were fed with HFD, and then immunized with HDM. A part of the BM cells was isolated from CAG-EGFP transgenic mice to assess the reconstitution rate. (B) Reconstitution rate. (C) Lung resistance (RL) and dynamic compliance (Cdyn) (D) Differential cell counts in BALF (n = 8–9/group). TCC, total cell count; MAC, macrophages; Ly, lymphocytes; Eo, eosinophils; Neut, neutrophils. (E) Hematoxylin and eosin-staining. Original magnification: 100 × . (F) PAS staining. (G) Quantifications of lung histopathology (n = 5/group). Data are representative of two independent experiments. Data are expressed as the mean ± SEM. P-values were calculated using the Student’s t test (atg5−/− (host) + atg5−/− BM vs. WT (host) + atg5−/−BM, and atg5−/− (host) + WT BM vs. WT (host) + WT BM). *, P < 0.05, **, P < 0.01, ***, P < 0.001.
Among the structural cells of the lung, epithelial cells act as external barriers against foreign antigens and can mediate the innate and adaptive immune responses [28,29]. In this regard, the pivotal role of autophagy in the maintenance of epithelial homeostasis has been reported [30]. Hence, we investigated whether the lack of autophagy in epithelial cells may play a role in the development of persistent eosinophilic inflammation. We generated lung epithelial cell-specific autophagy-deficient mice by cross-matching Atg5flox/flox mice with Scgb1a1/CCSP-Cre mice, in which Atg5 is deleted in the lung epithelial cells under the control of Scgb1a1 (secretoglobin, family 1A, member 1 (uteroglobin)). We observed that the eosinophils in BAL in HDM-sensitized Scgb1a1/CCSP-atg5−/− obese mice were significantly more abundant (Figure 5B), although this was not the case with AHR (Figure 5A).
Figure 5.
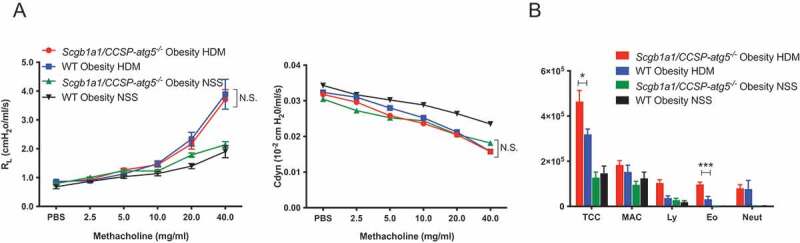
Impaired autophagy in epithelial cells induce eosinophilic inflammation in obese asthma. Scgb1a1/CCSP-atg5−/− obese mice and WT obese mice were immunized as described in Figure 1A. (A) Lung resistance (RL) and dynamic compliance (Cdyn) (B) Differential cell counts in BALF (n = 5–6/group). TCC, total cell count; MAC, macrophages; Ly, lymphocytes; Eo, eosinophils; Neut, neutrophils. Data are representative of two independent experiments. Data are expressed as mean ± SEM. P-values were calculated using two-way ANOVA test post hoc test with the Tukey’s multiple comparison test. *, P < 0.05, **, P < 0.01.
Gene expression analyses in autophagy-deficient epithelial cells
Next, we investigated autophagy levels in lung epithelial cells using GFP-LC3 mice, which allow the analysis of LC3 expression as a marker for autophagosomes [31,32]. Lung epithelial cells were identified as PTPRC/CD45 – PECAM1/CD31 – EPCAM+ cells (Figure 6A), and LC3 expression in lean and obese mice was determined. In agreement with the induction of activated epithelial autophagy in response to IL13 in vivo [27], lung epithelial cells from HDM-sensitized lean WT mice exhibited increased LC3 expression compared with that in the lung epithelial cells of HDM-sensitized obese WT mice (Figure 6A). Confocal microscopic analyses of lung epithelial cells also showed a marked reduction in the GFP-LC3 puncta in HDM-sensitized obese mice compared with the observations in the HDM-sensitized lean mice (Figure 6B), suggesting that epithelial autophagy is induced to maintain epithelial homeostasis in response to allergic inflammation in HDM-sensitized lean mice.
Figure 6.
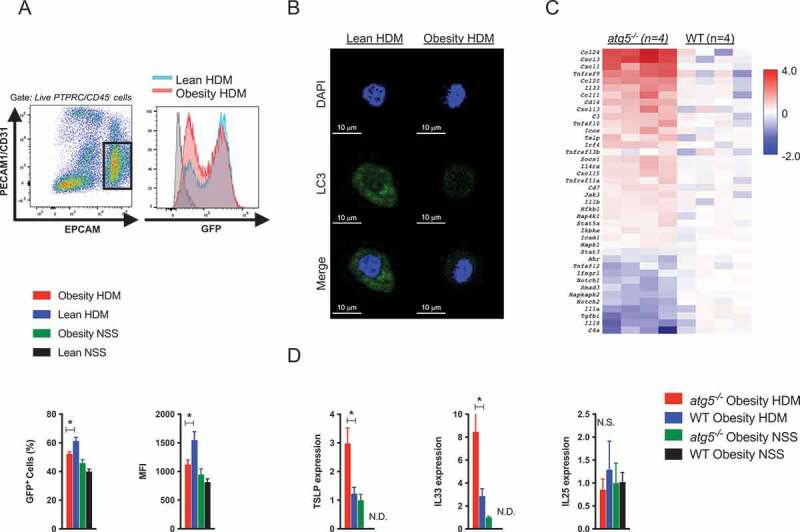
Gene expression analyses in autophagy-deficient epithelial cells. (A) GFP-LC3 mice were immunized as described in Figure E1C. Identification of lung epithelial cells (PTPRC/CD45 – PECAM1/CD31 – EPCAM+ cells). Representative FACS histogram plots of GFP-LC3 expression in lung epithelial cells. Frequencies of GFP-LC3-positive cells and mean fluorescence intensity (MFI) of GFP-LC3 in lung epithelial cells (n = 4–5/group). (B) Confocal microscopy images of isolated lung epithelial cells from GFP-LC3 mice. (C) Heat plot showing alteration of depicted genes in lung epithelial cells in atg5−/− obese mice and WT obese mice (P < 0.05, n = 4). FACS-purified lung epithelial cells from atg5−/− and WT obese mice immunized with HDM were quantified using the Nanostring nCounter technology. (D) Relative mRNA expression of lung epithelial cells (n = 3–5/group). Data are expressed as the mean ± SEM. P-values were calculated using two-way ANOVA test post hoc test with the Tukey’s multiple comparison test. *, P < 0.05.
To investigate the insufficient epithelial autophagy in obesity-associated asthma, we analyzed the gene expression profile of lung epithelial cells in atg5−/− and WT obese mice using Nanostring technology as described in the Methods section. Significant gene expression was depicted using a heat plot (Figure 6C). Eotaxin (CCL24 and CCL11) and inflammatory chemokines (CCL20, CXCL1, and CXCL3), as well as epithelial cell-derived cytokines (IL33 and TSLP), were upregulated in atg5−/− obese mice compared to those in WT obese mice. Furthermore, IL18, IL1A/IL1α, and TGFB/TGF-β levels were reduced in atg5−/− obese mice. Among these, we validated the expression of epithelial cell-derived cytokines. Consistent with the array data, these observations showed increased expression of IL33 and TSLP, but not that of IL25, in epithelial cells from atg5−/− obese mice (Figure 6D).
House dust mite-challenged atg5−/− obese mice developed TSLP- and IL33 -dependent asthma
An increase in Th2 and decrease in ILC2s were observed in atg5−/− obese mice (Figure 2 and Figure S4), while impaired functions of autophagy-deficient ILC2s in response to IL33 have been reported [20]. Here, we investigated TSLP and IL33, as they can activate both type-2 skewed adaptive and innate immune systems. We determined whether AHR and eosinophilic inflammation in atg5−/− obese mice were TSLP- and IL33-dependent. Mice were immunized with HDM and treated with anti-TSLP blocking antibody or anti-IL33 neutralizing antibodies one day before the HDM challenge. As expected, both TSLP and IL33 blockade significantly attenuated AHR and eosinophilic inflammation in atg5−/− obese mice, but not in WT obese mice (Figure 7). Histological analyses also showed that anti-TSLP and anti-IL33 treatment ameliorated airway thickness and reduced the number of infiltrated cells and PAS+ cells in atg5−/− obese mice (Figure 7C, F). Collectively, these results indicated that atg5−/− obese mice developed TSLP- and IL33-dependent AHR with enhanced eosinophil inflammation. Our data suggested that autophagy may contribute significantly to the pathogenesis of severe obese asthma.
Figure 7.
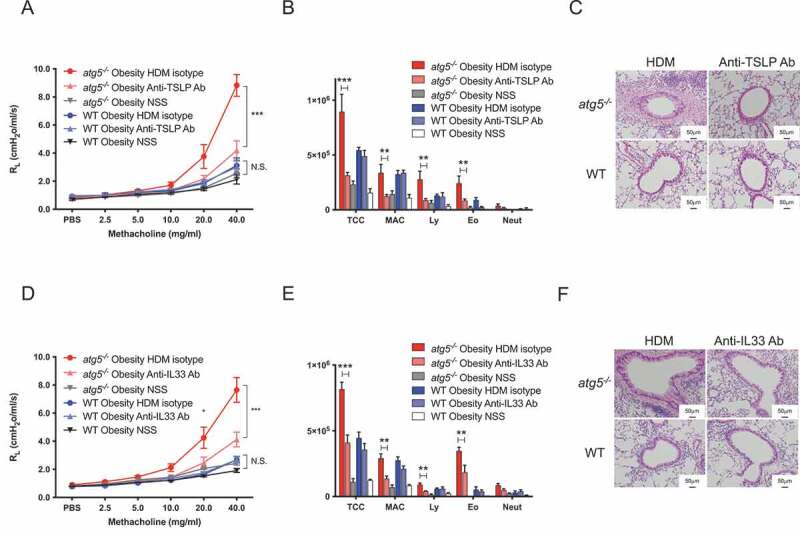
HDM-challenged atg5−/− obese mice develop TSLP-dependent and IL33-dependent asthma. (A) Experimental timeline. atg5−/− obese mice and WT obese mice were immunized as described in Figure 1A. Anti-thymic stromal lymphoprotein (TSLP) antibody (20 μg), anti-IL33 antibody (6 μg), or isotype controls were intranasally administered before HDM challenges. (A and D) Lung resistance (RL), (B and E) Differential cell counts in BALF. TCC, total cell count; MAC, macrophages; Ly, lymphocytes; Eo, eosinophils; Neut, neutrophils. (C and F) Hematoxylin and eosin-staining. Original magnification: 100 × . (E) PAS staining. Data are representative of two independent experiments (n = 5–7/group). Data are expressed as the mean ± SEM. P-values were calculated using two-way ANOVA test post hoc test with the Tukey’s multiple comparison test. *, P < 0.05, **, P < 0.01, ***, P < 0.001.
Discussion
In this study, we investigated the role of autophagy in obese asthma using HFD-induced murine models of obesity. We observed that autophagy disruption in obese mice markedly increased eosinophil-mediated inflammation and elevated AHR in response to HDM sensitization. This autophagy-mediated type 2 inflammation in obese mice was augmented by Th2 cells but not by ILC2s. Furthermore, HDM-sensitized WT obese mice showed lower allergic inflammation. Importantly, epithelial autophagy was activated upon allergic inflammation in HDM-sensitized lean WT mice but not in HDM-sensitized obese WT mice. Conversely, obese mice with specific deletion of Atg5 in lung epithelial cells exhibited enhanced eosinophilic inflammation, and epithelial cells in HDM-sensitized atg5−/− obese mice exhibited an increased expression of TSLP and IL33. Finally, we showed that HDM-sensitized atg5−/− obese mice were refractory to steroid treatment and developed TSLP- and IL33-dependent asthma. Collectively, our findings suggest that the dysregulation of autophagy exacerbates obesity-associated asthma, particularly that with the Th2-high phenotype.
Obesity causes a chronic low-grade inflammatory state that autophagy-associated pathways can potentially modulate [33,34]. Lipid overload can affect autophagy, which can decrease lipophagy and mitochondrial turnover and increase ER stress, low-grade inflammation, and finally lead to insulin resistance, although differences in autophagy regulation can be cell- and organ-specific; autophagy is upregulated in adipose tissue, whereas it is mostly downregulated in the liver, heart and pancreas [34]. However, previous studies have not investigated the effects of HFD on autophagy in the lungs. As described previously, increased autophagosome formation in the epithelium and sputum was observed in patients with asthma [26,35,36]. In agreement with this, we showed that HDM-sensitized lean mice expressed significantly higher levels of LC3 compared with non-sensitized lean mice. Meanwhile, LC3 expressions in HFD treated obese mice (both HDM-sensitized and non-sensitized) were similar to those in non-sensitized lean mice. Collectively, these results suggested that autophagy was activated in response to allergic inflammation, but overnutrition induced by HFD alone did not significantly affect epithelial autophagy levels.
Autophagy is a homeostatic cellular process, perturbations of which lead to various human diseases [13–16]. Recent evidence has suggested that autophagy is involved in asthma and allergy; in addition to activated autophagy in the lungs of asthma patients, autophagy was also involved in the pathogenesis of both childhood and adult asthma with respect to genetic polymorphism of ATG5 [26,37], reduced forced expiratory volume in 1.0 s (FEV1.0) [26], and airway remodeling [38,39]. Importantly, obesity is common in patients with asthma and exacerbates the symptoms of asthma [4,8,9]. Furthermore, although autophagy mediates the shared underlying pathogenesis of obesity and asthma, studies analyzing its role in obese asthma are lacking. Here, we showed for the first time that autophagy exacerbates eosinophilic inflammation using a murine model. Obesity-associated asthma is heterogeneous and includes Th2-high (eosinophilic) and Th2-low asthma (neutrophilic) phenotypes [4,8]. Particularly, Th2-high obese asthma is characterized by early-onset of symptoms and is the most refractory phenotype in patients with severe asthma. Additionally, increased eosinophil accumulation in the lung [40] and elevated submucosal eosinophilia in obese asthma were reported [24,25], suggesting the importance of eosinophilic inflammation in obesity-associated asthma. Collectively, though our murine model did not mimic early-onset asthma, our results suggest the involvement of autophagy in eosinophilic inflammation in obese asthma.
Autophagy plays cell-specific roles in asthma and allergic diseases. Indeed, previously distinct and often opposing effects of autophagy have been reported elsewhere [41–43]. Cell-specific Atg5 deletion in ITGAX/CD11c+ cells augmented lung inflammation with an increase in IL17A, leading to severe neutrophilic asthma in HDM-sensitized mice [18]. atg7−/− myeloid cell lineages exhibited increased eosinophil inflammation in a murine model of chronic rhinosinusitis, possibly mediated by aggravated IL1B and prostaglandin D2 production from macrophages and mast cells, respectively [21]. Conversely, autophagy is essential for the homeostasis and survival of ILC2s [19,20], and Atg5-deficient ILC2s showcase impaired ability to secrete IL5 and IL13, resulting in attenuation of AHR and eosinophil inflammation in IL33-treated mice [20]. Thus, these distinct roles of autophagy point to its ability to modulate different cellular processes, and not only direct control of the turnover of specific proteins [13,14,41]. In this study, we observed that deletion of Atg5 in epithelial cells exacerbated eosinophil inflammation in obese asthma but was not sufficient to induce AHR. Epithelial cells act as the first line of mucosal defense against pathogens and innocuous antigens. Autophagy is induced in epithelial cells together with mucus secretion in response to various stimuli such as IL13 [27,44], IFNG/IFN-γ [45–47], and particulate matter [48]. Recently, autophagy has been determined to be essential for the maintenance of homeostasis of the airway epithelium during steady-state and allergic inflammation [30,41]. Impaired autophagy also leads to epithelial cell dysfunction and lung fibrosis [49]. These pieces of evidence indicate that autophagy plays pivotal roles in maintaining epithelial homeostasis and recovery from epithelial injuries, suggesting that the dysregulation of autophagy in epithelial cells exacerbates allergic airway inflammation.
The role of obesity in development of eosinophilic inflammation in murine models were different by the reports even using same antigens. We found less eosinophilic inflammation in HDM-sensitized WT obese mice, which was similar to previous study [50]. Meanwhile, we detected elevated levels of TSLP and IL33, epithelial cell-derived cytokines, in atg5−/− obese mice, indicative of TSLP and IL33-dependent AHR and eosinophil inflammation. Proteolytic and mechanistic epithelial damage induced by various allergens and pathogens is a key trigger of allergic inflammations [51–53]. Importantly, viral infections, the most common causes of asthma exacerbation, can also modulate the autophagic flux and occasionally hijack the autophagy pathway for their survival [41,54]. Recently, Liu et al. demonstrated that co-exposure of benzopyrene and dermatophagoides group 1 allergen (Der f 1), but not benzopyrene or Der f 1 alone, facilitated the generation of epithelial reactive oxygen species (ROS), a regulator of autophagy, which in turn promoted TSLP and IL33 production, resulting in eosinophil inflammation and AHR [55].
The present study demonstrated alteration of autophagy by genetic deletion and HDM-sensitization. However, obesity could also affect autophagy flux. Consequently, increased SQSTM1/p62 levels, an autophagy substrate, were found in lungs of HDM-sensitized atg5−/− obese mice indicating impaired autophagy pathway. A number of stress signaling pathways, including ER stress, were activated during Atg5 gene deletion, HDM-sensitization and obesity-associated low grade inflammation. Cellular stress induced by external or internal cues activate well-orchestrated processes to mitigate damage caused by such circumstances and maintain cellular homeostasis. The close and complex link between autophagy unfolded protein response (UPR) and mitochondrial functions were noted; both can either be stimulated or inhibited by feedback mechanisms [34,56,57]. Therefore, we speculated that aberrant autophagy in HDM-sensitized atg5−/− obese mice triggered changes in UPR or mitochondrial function, which could modulate autophagy flux, initiating crosstalk between the two processes. Defects in autophagy result in the inability to recycle damaged organelles, potentially causing the release of ROS and other cellular components [58]. Dysregulation of autophagy leads to impaired apoptosis and clearance of dying cells [58]. Considering these essential roles of autophagy in maintaining cellular homeostasis, genetic deletion of Atg5 gene, overnutrition, and allergen exposure synergistically induce massive ROS generation and exacerbate eosinophilic airway inflammation in obese asthma.
The modulation of autophagy appears to be a novel therapeutic strategy for translating our results into a clinical setting. Indeed, viral infection is the most common cause of asthma exacerbation and can disrupt the autophagic flux [41,54]. Metformin, an autophagy inducer, reportedly curtails asthma exacerbation in patients with asthma also having diabetes mellitus; as such patients might be obese [59]. In addition, several autophagy inducers, including rapamycin and simvastatin, also reduced AHR and eosinophilic inflammation in a murine model [60–63]. Conversely, autophagy inhibitors can inhibit allergic inflammation [64], which was further confirmed with the observations in our HDM-sensitized WT obese mice. These bidirectional effects of autophagy on asthma can be explained by their multiple roles. For instance, autophagy is essential for antigen presentation; hence, autophagy inhibitors attenuate allergic inflammation. Meanwhile, rapamycin and metformin preserve energy metabolism by regulating mitophagy and mitochondrial biogenesis [42]. Furthermore, as described above, distinct cell-specific organ-specific roles of autophagy have been reported. Therefore, cell-specific and airway-targeted modulation of autophagy, which ultimately avoids detrimental systemic adverse effects, is required to apply our results in a clinical setting. However, in addition to the known mechanisms of obese asthma [65–67], the present study advances current understanding regarding obese asthma and proposes autophagy and epithelial cell-derived cytokines as therapeutic targets for Th2-high obese asthma.
In conclusion, this study showed that autophagy is involved in exacerbation of obese asthma using HDM-sensitized atg5−/− obese mice. Aberrant autophagy triggered eosinophil inflammation and AHR in a murine model of obese asthma. Importantly, this study identified that epithelial cell-derived cytokines, including TSLP and IL33, are involved in the pathogenesis of this autophagy-mediated disease and may act as therapeutic targets for obesity-associated asthma.
Materials and methods
Mice
Female C57BL/6 mice (4 weeks old) were purchased from Nippon SLC. Atg5flox/flox and GFP-LC3 mice were gifts from Dr. Noboru Mizushima (Tokyo University). The CAG-EGFP transgenic mouse was a gift from Tetsumei Urano (Hamamatsu University School of Medicine). atg5−/− mice and Scgb1a1/CCSP-specific atg5−/− mice were generated by crossing Atg5flox/flox mice with rosa26 Cre ERT mice and Scgb1a1/CCSP-Cre mice, respectively. Mice were screened using polymerase chain reaction (PCR) and atg5flox/flox homozygote Rosa-Cre hemizygote mice and atg5flox/flox homozygote Scgb1a1/CCSP-Cre hemizygote mice were selected for experiments. atg5flox/flox mice were backcrossed with rosa26 Cre ERT mice, and Scgb1a1/CCSP-Cre mice were bred in our facility at Hamamatsu University School of Medicine using protocols approved by the Institutional Animal Care and Use Committee.
Mice (4 weeks old) were fed either a 60% high-fat diet (HFD; D12492) from Research Diets or normal chow (10% fat; PMI Nutrition International, 5L37) for 16 to 18 weeks before the experiments. Mice received tamoxifen (800 μg per mouse per day; Bayer, 341,110,680) intraperitoneally (i.p.) for five consecutive days to induce the deletion of the Atg5 gene. The cellular analyses of BAL and AHR in WT obese mice treated with tamoxifen were similar to those of WT obese mice without tamoxifen (data not shown).
House dust mite-induced AHR and measurement of airway hyper-responsiveness
Mice were sensitized intranasally (i.n.) with 50 μg of HDM (Dermatophagoides. pteronyssinus; Greer Laboratories, XPB82D3A2.5) on day 1, followed by 10 μg of HDM (i.n.) on days 8 and 13. In some experiments, mice were treated with 3-MA (i.p., 300 mg; Sigma-Aldrich, M9281), dexamethasone (i.p., 1 mg/kg; Sigma-Aldrich, D4902) or phosphate-buffered saline (PBS; Gibco, 14,190), anti-mouse IL33 antibody (i.n., 6 μg/mice, clone 396,118; R&D, AF3626) or Goat IgG isotype controls (R&D, AB-108-C), and anti-mouse TSLP antibody (i.n., 20 μg/mice, clone 152,614; R&D, MAB555) or Rat IgG2A isotype control antibody (R&D, MAB006), one day before each HDM challenge. Two days after the last HDM challenge, mice were anesthetized using 300 μl i.p. injection of ketamine (10 mg/ml) and xylazine (1 mg/ml). Measurements of airway resistance and dynamic compliance were conducted using the Fine Pointe RC system (Buxco Research Systems), in which mice were mechanically ventilated using a modified version as described previously [18,68]. Mice were sequentially challenged with aerosolized PBS (baseline), followed by increasing doses of methacholine (2.5 mg/ml, 5.0 mg/ml, 10 mg/ml, 20 mg/ml and 40 mg/ml); Sigma-Aldrich, A2251. RL and Cdyn values were recorded during a 3-min period after each methacholine challenge.
Collection of BAL, lung histology, and lung lysates
After measurements of AHR, BAL cells were obtained as described previously [18,68]. The lung histological section and lung lysate were obtained and quantified. After measurements of AHR, the trachea was cannulated and the lungs were lavaged 3 times with 1 ml ice-cold PBS to collect BAL cells. Transcardial perfusion of the lungs with PBS was performed to remove red blood cells, and the lungs were fixed and harvested for histology using 4% paraformaldehyde in PBS. After fixation, the lungs were embedded in paraffin, cut into 4-µm sections, and stained with hematoxylin-eosin (HE) and periodic acid-Schiff (PAS). In some experiments, the lungs were collected and homogenized in RIPA buffer (Santa Cruz Biotechnology, sc-24,948) or RNA stabilization reagent (Qiagen, 76,104). The homogenates were analyzed for cytokines, as described elsewhere.
An inflammation score was assigned in a blinded fashion according to a previous study [69]. The score of peribronchiolar inflammation from HE staining was determined as follows: 0, normal; 1, few cells; 2, a ring of inflammatory cells one cell layer deep; 3, a ring of inflammatory cells 2–4 cells deep; and 4, a ring of inflammatory cells more than four cells deep. PAS staining was performed by examining at least 20 consecutive fields. Numerical scores for the abundance of PAS-positive goblet cells in each airway were determined as follows: 0, < 5% goblet cells; 1, 5–25%; 2, 25–50%; 3, 50–75%; and 4, > 75%, with 0 being negative and 1–4 being positive for PAS staining.
Identification of lung epithelial cells and ILC2s
Lung epithelial cells were identified as live, PTPRC/CD45 – PECAM1/CD31 – EPCAM+ cells, while lung ILC2 cells were live cells lacking the classical lineage markers (CD3E, PTPRC/CD45R, GSR/Gr-1, ITGAX/CD11c, ITGAM/CD11b, LY76/Ter119, KLRB1/NK1.1, TRB/TCR-β, TRD-TRG/TCR-γδ, and FCER1A/FCεRIα), PTPRC/CD45+, IL7R/CD127+, and IL1RL1/IL33Ra+ populations.
Flow cytometry and intracellular analyses
BAL cells were stained with phycoerythrin (PE)-labeled anti-Singlec-F (clone E50-2440; BD Pharmingen, 562,068), allophycocyanin (APC)-labeled anti-Ly-6 G/Ly-6C (clone RB6-8C5; BioLegend, 108,412), PE-Cy (PE-Cy7)-labeled anti-PTPRC/CD45 (clone 30-F11; BioLegend, 103,114), APC-Cy7-labeled anti-ITAGX/CD11c (clone N418; BioLegend, 117,324), peridinin-chlorophyll-protein complex-Cy5.5 (PerCP-Cy5.5)-labeled anti-CD3E (clone17A2; BioLegend, 100,218), Alexa Fluor-labeled anti-CD19 (clone 6D5; BioLegend, 101,505), and Pacific Blue-labeled anti-ITGAM/CD11b (clone M1/70; Biolegend, 101,224).
The lung epithelial cells were isolated as follows. The lungs were digested with collagenase (Worthington, LS004196) and DNase (Worthington, LS002139) using a GentleMACSTM dissociator (Miltenyi Biotec), and a single cell suspension was prepared. The cells were incubated with anti-mouse PTPRC/CD45-coated magnetic beads (clone 13/2.3; BioLegend, 76,746) and negatively sorted via magnetic cell sorting (BioLegend, 480,019). The cells were stained using the live/dead fixable near-IR dead cell stain kit (Invitrogen, L10119), PE-Cy7-labeled anti-PTPRC/CD45 (clone 30-F11; BioLegend, 103,114), APC-labeled anti- PECAM1/CD31 (clone 390; BioLegend, 102,410), and PE-labeled anti-EPCAM (clone G8.8; BioLegend, 118,205). The lung epithelial cells were sorted as live, PTPRC/CD45–, PECAM1/CD31–, and EPCAM+ cells using FACSARIA SORP (BD Bioscience). For analyses of ILC2 cells, the following antibodies were used: biotinylated anti-mouse lineage (CD3E (145–2C11; BioLegend, 79,751), PTPRC/CD45R (RA3-6B2; BioLegend, 79,752), GSR/Gr-1 (RB6-8C5; BioLegend, 79,750), ITGAX/CD11c (N418; BioLegend, 117,304), ITGAM/CD11b (M1/70; BioLegend, 79,749), LY76/Ter119 (TER-119; BioLegend, 79,748), KLRB1/NK1.1 (PK136; BioLegend, 108,704), TRB/TCR-β (H57-597; BioLegend, 109,204), TRD-TRG/TCR-γδ (BioLegend; 118,103), and FCER1A/FCεRIα (MAR-1; BioLegend, 134,304), Brilliant Violet 510-labeled anti-PTPRC/CD45 (clone30-F11; BioLegend, 103,127), APC-labeled anti-IL7R/CD127 (clone A7R34; BioLegend, 135,011), PE-Cy7-labeled anti-IL7R/CD127 (clone A7R34; BioLegend, 135,014), PerCPCy5.5-labeled anti-IL1RL1/IL33Ra (clone DIH9; BioLegend, 145,312), and streptavidin-FITC (BioLegend, 405,201).
The following antibodies were used for intracellular staining; live/dead fixable near-IR dead cell stain kit (Invitrogen, L34965), PerCP-Cy5.5-labeled anti-CD3E (clone17A2; BioLegend, 100,218), PE-Cy7-labeled anti-CD3E (clone 17A2; BioLegend, 100,219), Pacific Blue-labeled anti-CD4 (clone GK1.5; BioLegend, 100,428), PerCP CD4 (clone RM4-5; BioLegend, 100,537), Brilliant Violet-510 (BV510)-labeled anti-CD44 (clone IM7; BioLegend, 103,043), eFluor 450-labeled anti-PTPRC/CD45 (clone 30 F-11; ebioscience, 40–0451-82), PE-labeled anti-IL5 (clone TRFK5; BoLegend, 504,304), PE-Cy7-labeled anti-IL13 (clone eBio13A; ebioscience, 25–7133-82), APC-labeled anti-IL17A (clone TC11-18H10.1; BioLegend), PE-labeled anti-GATA3 (clone 16E10A23; BioLegend, 653,804), and APC-labeled anti-p-STAT6 (clone CH12S4N; ebioscience, 17–9013-41). Flow cytometry was performed using Gallios (Beckman Coulter) and the data were analyzed using the FlowJo version 8.6 software (TreeStar).
Intracellular staining was performed using a BD Cytofix/Cytoperm kit (BD Bioscience, 554,714) and fixation/permeabilization buffer kit (eBioscience, 88–8824-00) according to the manufacturer’ s instructions.
Gene expression analysis using Nanostring nCounter technology
The difference in the abundance of transcripts between lung epithelial cells purified from HDM-sensitized atg5−/− obese mice or WT obese mice were analyzed using Nanostring nCounter technology (Immunology Panel). Nanostring nCounter technology employs unique fluorescent barcode and enables direct and digital detection of the abundance of transcripts with no amplification. Heat plots were generated using the nSolver software.
Preparation of bone marrow chimeric mice
Mice were irradiated with 600 rad and injected the following day with 5 × 106 BM cells. In some experiments, BM cells were collected from CAG-EGFP mice, and the reconstitution rate was confirmed. Reconstitution of lung leukocytes was approximately 90% in flow cytometry quantification of GFP-labeled CD45+ cells.
Elisa
The levels of cytokines and SQSTM1/p62 were measured by ELISA (Thermo Fisher Scientific,88–7054-22, 88–7137-22, 77–7371-22, 88–7002-22, 88–7333-22, 88–7490-22 and Enzo Life Science, ADI-900-212-0001), according to the manufacturer’s instructions.
Reverse transcription-PCR
Total RNA was extracted using the RNAeasy mini kit (Qiagen, 74,104) and cDNA was synthesized using the HighCapacity cDNA reverse transcription kit (Applied Biosystems, 4,368,814). RT-PCR was performed using StepOnePlus (Applied Biosystems) and the ΔΔCt method [70] was used for data analysis.
Quantification of autophagy levels using flow cytometry and confocal microscopy
GFP-LC3 knock-in transgenic mice were fed HFD or normal diet and then immunized with HDMs as described above (Figure S1). Lung epithelial cells were sorted as described above. MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3) expression was quantified using FACS and confocal microscopy (Leica, TCS SP8).
Statistical analysis
The Student’s t-test and two-way ANOVA test post hoc test with the Tukey’s multiple comparison test were used. P < 0.05 was considered statistically significant. All data are expressed as mean ± SD. Statistical analyses were performed using GraphPad Prism version 6 (GraphPad Software, San Diego, CA, USA)
Supplementary Material
Acknowledgments
We thank Editage for editing a draft of the manuscript.
Funding Statement
This work was supported by a grant-in-aid for scientific research (15H06253, 16K19448, and 19K17632 to YS) from the Japan Society for the Promotion of Science, a GSK Japan Research Grant 2015 from GlaxoSmithKline (A-23 to YS), and a HUSM grant-in-aid from the Hamamatsu University School of Medicine (42351E-101337 to YS).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].(NCD-RisC) NRFC . Trends in adult body-mass index in 200 countries from 1975 to 2014: a pooled analysis of 1698 population-based measurement studies with 19·2 million participants. Lancet. 2016 Apr 2;387(10026):1377–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Afshin A, Forouzanfar MH, Reitsma MB, et al. Health effects of overweight and obesity in 195 countries over 25 years. N Engl J Med. [2017 Jul 6];377(1):13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Masoli M, Fabian D, Holt S, et al. The global burden of asthma: executive summary of the GINA dissemination committee report. Allergy. 2004. May;59(5):469–478. [DOI] [PubMed] [Google Scholar]
- [4].Peters U, Dixon AE, Forno E.. Obesity and asthma. J Allergy Clin Immunol. 2018. Apr;141(4):1169–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Schatz M, Hsu JW, Zeiger RS, et al. Phenotypes determined by cluster analysis in severe or difficult-to-treat asthma. J Allergy Clin Immunol. 2014. Jun;133(6):1549–1556. [DOI] [PubMed] [Google Scholar]
- [6].Beuther DA, Sutherland ER. Overweight, obesity, and incident asthma: a meta-analysis of prospective epidemiologic studies. Am J Respir Crit Care Med. 2007 Apr 1; 175(7):661–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Contreras ZA, Chen Z, Roumeliotaki T, et al. Does early onset asthma increase childhood obesity risk? A pooled analysis of 16 European cohorts. Eur Respir J. 2018. Sep;52(3):1800504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Holguin F, Bleecker ER, Busse WW, et al. Obesity and asthma: an association modified by age of asthma onset. J Allergy Clin Immunol. 2011. Jun;127(6):1486–93 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mosen DM, Schatz M, Magid DJ, et al. The relationship between obesity and asthma severity and control in adults. J Allergy Clin Immunol. 2008. Sep;122(3):507–11 e6. [DOI] [PubMed] [Google Scholar]
- [10].Taylor B, Mannino D, Brown C, et al. Body mass index and asthma severity in the National Asthma Survey. Thorax. 2008. Jan;63(1):14–20. [DOI] [PubMed] [Google Scholar]
- [11].Scott HA, Gibson PG, Garg ML, et al. Airway inflammation is augmented by obesity and fatty acids in asthma. Eur Respir J. 2011;38(3):594–602. [DOI] [PubMed] [Google Scholar]
- [12].Sideleva O, Suratt BT, Black KE, et al. Obesity and asthma: an inflammatory disease of adipose tissue not the airway. Am J Respir Crit Care Med. 2012 Oct 1 186(7):598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013 Feb 14; 368(7):651–662. [DOI] [PubMed] [Google Scholar]
- [14].Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013. Oct;13(10):722–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011 Jan 20; 469(7330):323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ryter SW, Nakahira K, Haspel JA, et al. Autophagy in pulmonary diseases. Annu Rev Physiol. 2012;74:377–401. [DOI] [PubMed] [Google Scholar]
- [17].Kuma A, Hatano M, Matsui M, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004 Dec 23 432(7020):1032–1036. [DOI] [PubMed] [Google Scholar]
- [18].Suzuki Y, Maazi H, Sankaranarayanan I, et al. Lack of autophagy induces steroid-resistant airway inflammation. J Allergy Clin Immunol. 2016. May;137(5):1382–1389.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].O’Sullivan TE, Geary CD, Weizman OE, et al. Atg5 is essential for the development and survival of innate lymphocytes. Cell Rep. 2016 May 31 15(9):1910–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Galle-Treger L, Hurrell BP, Lewis G, et al. Autophagy is critical for group 2 innate lymphoid cell metabolic homeostasis and effector function. J Allergy Clin Immunol. 2020. Feb;145(2):502–517 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Choi GE, Yoon SY, Kim JY, et al. Autophagy deficiency in myeloid cells exacerbates eosinophilic inflammation in chronic rhinosinusitis. J Allergy Clin Immunol. 2017 Dec 7 141(3):938–950.e12. [DOI] [PubMed] [Google Scholar]
- [22].Haspel JA, Choi AM. Autophagy: a core cellular process with emerging links to pulmonary disease. Am J Respir Crit Care Med. 2011 Dec 1; 184(11):1237–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Castillo EF, Dekonenko A, Arko-Mensah J, et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proceedings of the National Academy of Sciences of the United States of America. 2012 Nov 13;109(46):E3168–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Desai D, Newby C, Symon FA, et al. Elevated sputum interleukin-5 and submucosal eosinophilia in obese individuals with severe asthma. Am J Respir Crit Care Med. 2013 Sep 15 188(6):657–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].van der Wiel E, Ten Hacken NH, van Den Berge M, et al. Eosinophilic inflammation in subjects with mild-to-moderate asthma with and without obesity: disparity between sputum and biopsies. Am J Respir Crit Care Med. 2014 May 15 189(10):1281–1284. [DOI] [PubMed] [Google Scholar]
- [26].Poon AH, Chouiali F, Tse SM, et al. Genetic and histologic evidence for autophagy in asthma pathogenesis. J Allergy Clin Immunol. 2012;129(2):569–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dickinson JD, Alevy Y, Malvin NP, et al. IL13 activates autophagy to regulate secretion in airway epithelial cells. Autophagy. 2016;12(2):397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Galli SJ, Tsai M, Piliponsky AM. The development of allergic inflammation. Nature. 2008 Jul 24; 454(7203):445–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lambrecht BN, Hammad H. The airway epithelium in asthma. Nat Med. 2012. May;18(5):684–692. [DOI] [PubMed] [Google Scholar]
- [30].Li K, Li M, Li W, et al. Airway epithelial regeneration requires autophagy and glucose metabolism. Cell Death Dis. 2019 Nov 20 10(12):875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Mizushima N. Chapter 2 methods for monitoring autophagy using GFP‐LC3 transgenic mice Methods Enzymol . Autophagy in Mammalian Systems, Part B. Methods in Enzymology. 2009;452:13–23. [DOI] [PubMed] [Google Scholar]
- [33].Pabon MA, Ma KC, Choi AM. Autophagy and obesity-related lung disease. Am J Respir Cell Mol Biol. 2016. May;54(5):636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhang Y, Sowers JR, Ren J. Targeting autophagy in obesity: from pathophysiology to management. Nat Rev Endocrinol. 2018;14(6):356–376. [DOI] [PubMed] [Google Scholar]
- [35].Liu T, Liu Y, Miller M, et al. Autophagy plays a role in FSTL1-induced epithelial mesenchymal transition and airway remodeling in asthma. Am J Physiol Lung Cell Mol Physiol. 2017 Jul 1 313(1):L27–L40. [DOI] [PubMed] [Google Scholar]
- [36].Ban GY, Pham DL, Trinh TH, et al. Autophagy mechanisms in sputum and peripheral blood cells of patients with severe asthma: a new therapeutic target. Clinical and Experimental Allergy: Journal of the British Society for Allergy and Clinical Immunology. 2016. Jan;46(1):48–59. [DOI] [PubMed] [Google Scholar]
- [37].Martin LJ, Gupta J, Jyothula SS, et al. Functional variant in the autophagy-related 5 gene promotor is associated with childhood asthma. PLoS One. 2012;7(4):e33454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Poon AH, Choy DF, Chouiali F, et al. Increased autophagy-related 5 gene expression is associated with collagen expression in the airways of refractory asthmatics. Front Immunol. 2017;8:355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].McAlinden KD, Deshpande DA, Ghavami S, et al. Autophagy Activation in Asthma Airways Remodeling. Am J Respir Cell Mol Biol. 2019;60(5):541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Farahi N, Loutsios C, Tregay N, et al. In vivo imaging reveals increased eosinophil uptake in the lungs of obese asthmatic patients. J Allergy Clin Immunol. 2018. Nov;142(5):1659–1662 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Painter JD, Galle-Treger L, Akbari O. Role of autophagy in lung inflammation. Front Immunol. 2020;11:1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Theofani E, Xanthou G. Autophagy: a friend or foe in allergic asthma? Int J Mol Sci. 2021 Jun 12; 22(12):6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zeki AA, Yeganeh B, Kenyon NJ, et al. Autophagy in airway diseases: a new frontier in human asthma? Allergy. 2016. Jan;71(1):5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Dickinson JD, Sweeter JM, Warren KJ, et al. Autophagy regulates DUOX1 localization and superoxide production in airway epithelial cells during chronic IL-13 stimulation. Redox Biol. 2018. Apr;14:272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chen YD, Fang YT, Cheng YL, et al. Exophagy of annexin A2 via RAB11, RAB8A and RAB27A in IFN-γ-stimulated lung epithelial cells. Sci Rep. 2017 Jul 18 7(1):5676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Fougeray S, Mami I, Bertho G, et al. Tryptophan depletion and the kinase GCN2 mediate IFN-γ-induced autophagy. J Immunol. 2012. Sep 15;189(6):2954–2964. [DOI] [PubMed] [Google Scholar]
- [47].Wang BF, Cao PP, Wang ZC, et al. Interferon-γ-induced insufficient autophagy contributes to p62-dependent apoptosis of epithelial cells in chronic rhinosinusitis with nasal polyps. Allergy. 2017. Sep;72(9):1384–1397. [DOI] [PubMed] [Google Scholar]
- [48].Chen ZH, Wu YF, Wang PL, et al. Autophagy is essential for ultrafine particle-induced inflammation and mucus hyperproduction in airway epithelium. Autophagy. 2016;12(2):297–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].O’Dwyer DN, Ashley SL, Moore BB. Influences of innate immunity, autophagy, and fibroblast activation in the pathogenesis of lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2016 Sep 1; 311(3):L590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Diaz J, Warren L, Helfner L, et al. Obesity shifts house dust mite-induced airway cellular infiltration from eosinophils to macrophages: effects of glucocorticoid treatment. Immunol Res. 2015. Dec;63(1–3):197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lambrecht BN, Hammad H, Fahy JV. The Cytokines of Asthma. Immunity. 2019 Apr 16; 50(4):975–991. [DOI] [PubMed] [Google Scholar]
- [52].Walker JA, McKenzie ANJ. TH2 cell development and function. Nat Rev Immunol. 2018. Feb;18(2):121–133. [DOI] [PubMed] [Google Scholar]
- [53].Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med. 2012 May 4; 18(5):716–725. [DOI] [PubMed] [Google Scholar]
- [54].Choi Y, Bowman JW, Jung JU. Autophagy during viral infection - a double-edged sword. Nat Rev Microbiol. 2018. Jun;16(6):341–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wang E, Liu X, Tu W, et al. Benzo(a)pyrene facilitates dermatophagoides group 1 (Der f 1)-induced epithelial cytokine release through aryl hydrocarbon receptor in asthma. Allergy. 2019. Sep;74(9):1675–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Rashid HO, Yadav RK, Kim HR, et al. ER stress: autophagy induction, inhibition and selection. Autophagy. 2015 Nov 2 11(11):1956–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Senft D, Ronai ZA. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci. 2015. Mar;40(3):141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science (New York, NY). 2011 Aug 26; 333(6046):1109–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Li CY, Erickson SR, Wu CH. Metformin use and asthma outcomes among patients with concurrent asthma and diabetes. Respirology. 2016. Oct;21(7):1210–1218. [DOI] [PubMed] [Google Scholar]
- [60].Yin Y, Mitson-Salazar A, Wansley DL, et al. Rapamycin preferentially inhibits human IL-5(+) T(H)2-cell proliferation via an mTORC1/S6 kinase-1-dependent pathway. J Allergy Clin Immunol. 2017. May;139(5):1701–1704.e10. [DOI] [PubMed] [Google Scholar]
- [61].Mushaben EM, Kramer EL, Brandt EB, et al. Rapamycin attenuates airway hyperreactivity, goblet cells, and IgE in experimental allergic asthma. J Immunol. 2011. Dec 1;187(11):5756–5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hua W, Liu H, Xia LX, et al. Rapamycin inhibition of eosinophil differentiation attenuates allergic airway inflammation in mice. Respirology. 2015. Oct;20(7):1055–1065. [DOI] [PubMed] [Google Scholar]
- [63].Gu W, Cui R, Ding T, et al. Simvastatin alleviates airway inflammation and remodelling through up-regulation of autophagy in mouse models of asthma. Respirology. 2017. Apr;22(3):533–541. [DOI] [PubMed] [Google Scholar]
- [64].Liu JN, Suh DH, Trinh HK, et al. The role of autophagy in allergic inflammation: a new target for severe asthma. Exp Mol Med. 2016 Jul 1 48(7):e243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Bates JH. Physiological mechanisms of airway hyperresponsiveness in obese asthma. Am J Respir Cell Mol Biol. 2016. May;54(5):618–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Dixon AE, Poynter ME. Mechanisms of asthma in obesity. Pleiotropic aspects of obesity produce distinct asthma phenotypes. Am J Respir Cell Mol Biol. 2016. May;54(5):601–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Shore SA, Cho Y. Obesity and asthma: microbiome-metabolome interactions. Am J Respir Cell Mol Biol. 2016. May;54(5):609–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Galle-Treger L, Suzuki Y, Patel N, et al. Nicotinic acetylcholine receptor agonist attenuates ILC2-dependent airway hyperreactivity. Nat Commun. 2016 Oct 18;7:13202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Tong J, Bandulwala HS, Clay BS, et al. Fas-positive T cells regulate the resolution of airway inflammation in a murine model of asthma. J Exp Med. 2006 May 15 203(5):1173–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Suzuki Y, Suda T, Furuhashi K, et al. Mouse CD11bhigh lung dendritic cells have more potent capability to induce IgA than CD103+ lung dendritic cells in vitro. Am J Respir Cell Mol Biol. 2012. Jun;46(6):773–780. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.