

ABSTRACT
The mitochondrial-anchored deubiquitinating enzyme USP30 (ubiquitin specific peptidase 30) antagonizes PRKN/parkin-mediated mitophagy, making it a potential target for treating Parkinson disease. However, few inhibitors targeting USP30 have been reported. Here, we report a novel peptide (Q14) derived from the transmembrane (TM) domain of USP30 that can target mitochondrial-anchored USP30 directly and increase mitophagy through two intriguing and distinct mechanisms: a novel autoinhibition mechanism in USP30 and accelerated autophagosome formation via the LC3-interacting region (LIR) of the Q14 peptide. We identified the potential binding sites between the Q14 peptide and USP30 and postulated that an allosteric autoinhibition mechanism regulates USP30 activity. Furthermore, the LIR motif in the Q14 peptide offers additional binding with LC3 and accelerated autophagosome formation. The two mechanisms synergistically enhance mitophagy. Our work provides novel insight and direction to the design of inhibitors for USP30 or other deubiquitinating enzymes (DUBs).
Abbreviations: 3-MA: 3-methyladenine; ATTEC: autophagosome-tethering compound; BafA1: bafilomycin A1; BNIP3: BCL2 interacting protein 3; BNIP3L/NIX: BCL2 interacting protein 3 like; CCCP: carbonyl cyanide m-chlorophenyl hydrazone; DMSO: dimethyl sulfoxide; FP: fluorescence polarization; FUNDC1: FUN14 domain containing 1; HCQ: hydroxychloroquine; LIR: LC3-interacting region; MST: microscale thermophoresis; mtDNA: mitochondrial DNA; mtPA-GFP: mitochondria-targeted photoactive fluorescence protein; OMM: outer mitochondrial membrane; PINK1: PTEN induced kinase 1; PRKN/parkin: parkin RBR E3 ubiquitin protein ligase; Rap: rapamycin; SA: streptavidin; TM: transmembrane; Ub: ubiquitin; Ub-AMC: Ub-7-amido-4-methylcoumarin; UPS: ubiquitin-protease system; USP: ubiquitin specific peptidase; USP30: ubiquitin specific peptidase 30.
KEYWORDS: Autoinhibition, mitophagy, peptide inhibitor, transmembrane, USP30
Introduction
Mitochondria are vital organelles that modulate diverse cellular functions such as energy production, metabolism, and apoptosis [1]. Through dynamic processes including fusion and fission, shape, size, number, and physiological function of mitochondria can be regulated precisely, enacting quality control of mitochondria and maintaining cellular homeostasis [2]. When a mitochondrion is subject to stress or damage, it needs to be labeled and removed promptly by a highly selective mechanism to prevent it from polluting the healthy mitochondrial network. The dysregulation of mitochondria can interfere with cellular energy supply, affect biosynthetic pathways, produce excessive reactive oxygen species and cause cell death, and has been closely implicated in Parkinson disease [3].
To maintain mitochondrial health, cells have evolved a process of selective autophagy termed mitophagy, which includes the isolation and subsequent elimination of damaged mitochondria through the lysosome [4]. In mammalian cells, mitophagy can be regulated by several types of mechanisms, including a ubiquitin (ub)-dependent manner via mitochondrial-associated PINK1 (PTEN induced kinase 1) and the E3 ubiquitin ligase PRKN/parkin (parkin RBR E3 ubiquitin protein ligase), a ub-independent manner via mitochondrial proteins such as BNIP3L/NIX (BCL2 interacting protein 3 like), FUNDC1 (FUN14 domain containing 1), BNIP3 (BCL2 interacting protein 3) and a lipid-mediated cargo recognition manner via phospholipids such as cardiolipin and ceramide [5–10]. In the PINK1-PRKN pathway, when the mitochondria are damaged or stressed, PINK1 accumulates at the surface of the outer mitochondrial membrane (OMM), where it initiates a series of modifications (including phosphorylation, multiple conformational changes and binding with Ser65-phosphorylated ubiquitin [p-Ser65-Ub]) on PRKN to promote its stable association with the OMM proteins and activation of its E3 ubiquitin ligase activity [11,12]. Activated PRKN subsequently ubiquitinates several mitochondrial proteins and prevents mitochondrial movement and fusion. Eventually, impaired mitochondria are enveloped by autophagosomal membranes and sent to the lysosome for degradation. Loss of function mutations in both PINK1 and PRKN are involved in Parkinson disease [13,14].
Deubiquitinating enzymes (DUBs) play a critical role in regulating the ubiquitin system by reversing ubiquitin modifications [15,16]. Recent work has identified USP30 (ubiquitin specific peptidase 30), a mitochondrial anchored DUB, as a negative regulator of mitophagy in cells and in vivo [17,18]. USP30 prefers to hydrolyze both unanchored and mitochondrially-conjugated Lys 6- and Lys 11-linked ub chains, which are often synthesized during mitochondrial ubiquitination, and thereby antagonize mitophagy [18–20]. Inhibition of USP30 activity has been considered a potential target for the treatment of Parkinson disease through improving the damaged mitochondria clearance and quality control in neurons [17].
However, few studies of specific inhibitors of USP30 have been reported. These inhibitors include triterpene 15-oxaspiramilactone, N-cyano pyrrolidines, phage-screened dodecapeptides, and racemic phenylalanine derivatives [21–24]. 15-oxaspiramilactone inhibits USP30 by interacting with the cysteine residue in the catalytic domain of USP30 to induce mitochondrial fusion, leading to restoration of normal mitochondrial network and function. However, the specificity and biophysical characterization of this inhibitor have not been reported [21]. A high-throughput screening identified a racemic phenylalanine derivative as a USP30 inhibitor, but only provided preliminary characterization of its specificity and biological activity [24]. Recent work examined the effects of USP30 inhibition with a modified N-cyano pyrrolidine on the proteome and ubiquitylome of neuroblastoma cells [22]. However, the binding mode between the compound and USP30 was not characterized. Therefore, there is an urgent need for developing novel and potent USP30 inhibitors and performing in-depth characterization of their biological activity.
To date, USP30 has been identified as one of only two DUBs (USP19 and USP30) that possess a TM domain. Interestingly, Ye et al. reported that the TM domain of USP19 interacted with its catalytic domain, leading to autoinhibition of its deubiquitinating activity [25]. Autoinhibition is a common allosteric modulation mechanism for maintaining cellular homeostasis [26]. The autoinhibitory segment prevents substrate contacting with the protein before it enters the functional site, making autoinhibition a critical mechanism for ensuring efficiency and precision [26]. A modest shift or rotation of the autoinhibitory segment could expose the active site and relieve autoinhibition [26]. The modulation of autoinhibition has been reported in the ubiquitin-protease system (UPS). For several Homologous to E6AP C terminus (HECT)-type ubiquitin ligases like SMURF2 (SMAD specific E3 ubiquitin protein ligase 2) and NEDD4 (NEDD4 E3 ubiquitin protein ligase), the C2 domain exerts its autoinhibitory function in downregulating the HECT domain activity by interfering with ub thioester formation and disturbing the noncovalent interaction with ub [27,28]. The E3 ligase PRKN adopts an autoinhibited conformation by blocking the E2-ubiquitin binding site on RING1 domain and the catalytic Cys431 of RING2 domain [29]. As for DUBs, besides USP19, several DUBs, including USP7, USP8, USP14, USP25, USP28, and UCHL3 (ubiquitin C-terminal hydrolase L3), exist in an autoinhibited state in which their catalytic activity is precisely controlled through either occluding active sites or mis-aligning catalytic triads [30–36]. Therefore, inspired by the autoinhibitory modulation in the UPS system, we wondered whether USP30 contained an autoinhibitory domain to function as its potential inhibitor.
Here, we first identified a peptide inhibitor derived from the TM domain of USP30 that can perform autoinhibitory functions and induce mitophagy. We performed sequence truncation and alanine mutation analysis to identify the short and potent Q14 peptide inhibitor. The Q14 peptide can easily cross the cell membrane, bind with mitochondrial-anchored USP30 directly, degrade OMM proteins, promote mitochondrial fission, and trigger mitophagy. We characterized the potential binding sites between USP30 and Q14 peptide and proposed an allosteric autoinhibition model for how it regulates USP30 activity. Additionally, we found that Q14 peptide could interact with LC3 and link the mitochondrial-anchored USP30 to the phagophore membrane to accelerate mitophagy. The synergistic effect of these two mechanisms efficiently enhances mitophagy. Our findings provide novel insight and direction to the design of inhibitors for USP30 or other DUBs.
Results
The transmembrane (TM) domain of USP30 inhibits its catalytic activity
Most deubiquitinases are soluble proteins residing in either the cytosol or nucleus, with the exception of USP19 and USP30, which both contain a TM domain. USP19 contains a C-terminal TM domain to localize itself in the membrane of the endoplasmic reticulum (ER), while USP30 is a mitochondrial-anchored deubiquitinase which contains a mitochondrial intermembrane domain, a TM domain and a catalytic domain (Figure 1A) [37]. Previous studies have demonstrated that a synthetic USP19 transmembrane peptide could inhibit USP19 deubiquitinating activity, implying that the TM domain of a deubiquitinase located in a subcellular organelle can regulate its catalytic activity [25]. Inspired by this, we hypothesized that the catalytic activity of USP30 could be restrained by its TM domain. To verify this hypothesis, we carried out sequence alignment between the sequences of USP19 and USP30 using the LALIGN web server and found that their TM domains are highly similar (Figure 1B). We synthesized the TM domain sequence (peptide Q1) and determined whether it could inhibit the catalytic activity of USP30. We bacterially-expressed and then purified human recombinant USP30 that lacks TM domain, but otherwise is full length, for improved solubility [18]. We first tested the catalytic efficiency of USP30 interacting with the substrate Ub-7-amido-4-methylcoumarin (Ub-AMC), where the kcat:KM ratios are consistent with previous reports for USP30 (Fig. S1A). USP30 (15 nM) and increasing amounts of peptide Q1 were incubated with Ub-AMC, and reaction progress was monitored by fluorescence. As shown in Figure 1C, Q1 peptide inhibited USP30 activity with nanomolar IC50 values. Considering the length and solubility of peptide, we truncated the Q1 peptide from the N terminus (Figure 1D). Peptides (100 nM) with different lengths were incubated with USP30 to test the catalytic activity of USP30 (relative to control). The N terminus of the TM domain mildly impacted USP30 activity, while the middle part of the TM domain showed better inhibition of USP30 activity (Figure 1E). We mapped the minimal inhibitory region of Q1 to an 18-amino acid peptide (residues 48–65, hereafter referred to as Q14), which contained a hydrophobic N-terminal region and a basic amino acid-rich C-terminal region (Fig. S1B, S1C). As can be seen from Figure 1F, Q14 peptide enhanced potency by ~4-fold (IC50 = 57.2 nM ± 4.77) relative to Q1. Next, alanine screening was carried out to test which residue was more important for inhibition (Figure 1G). As shown in Figure 1H, we discovered that a specific residue mutation could reduce the inhibitory effect of Q14 peptide on USP30. C-terminal mutations (MT13-MT18) have a greater impact on the inhibition of USP30 enzyme activity, while N-terminal mutations have a relatively minor effect, especially mutations of MT6 and MT9. We segmented the Q14 peptide sequence into C-terminal part (Q23) and N-terminal part (Q24) and compared their inhibitory effects (Figure 1I). The results in Figure 1J showed that Q23 (C terminus of Q14) played a more important role than Q24 (N terminus of Q14) in USP30 inhibition. Peptides truncated from the C terminus (Q24, Q25, Q26) showed a gradually weakened inhibitory effect (Figure 1K). Combined with the inhibitory effect of C-terminal mutations (MT10-MT18), we found that the C terminus of Q14, which was the OMM localization sequence, is necessary and sufficient to confer high-potency inhibition of USP30. The C helix alone and the N-terminal sequence alone did not reach the same inhibitory effect as Q14, indicating that the synergistic effect of the complete N-terminal sequence and the C helix together achieved efficient inhibition of USP30. Likewise, the appropriate amino acids arrangement of Q14 was also vital for suppressing USP30 enzymatic activity as the scramble sequence of Q14 (Qscr) was unable to inhibit USP30 (Fig. S1D). Taken together, our data indicate that the TM domain of USP30 is important for inhibiting USP30 catalytic activity and that the Q14 peptide is the most potent peptide inhibitor among the peptides examined.
Figure 1.
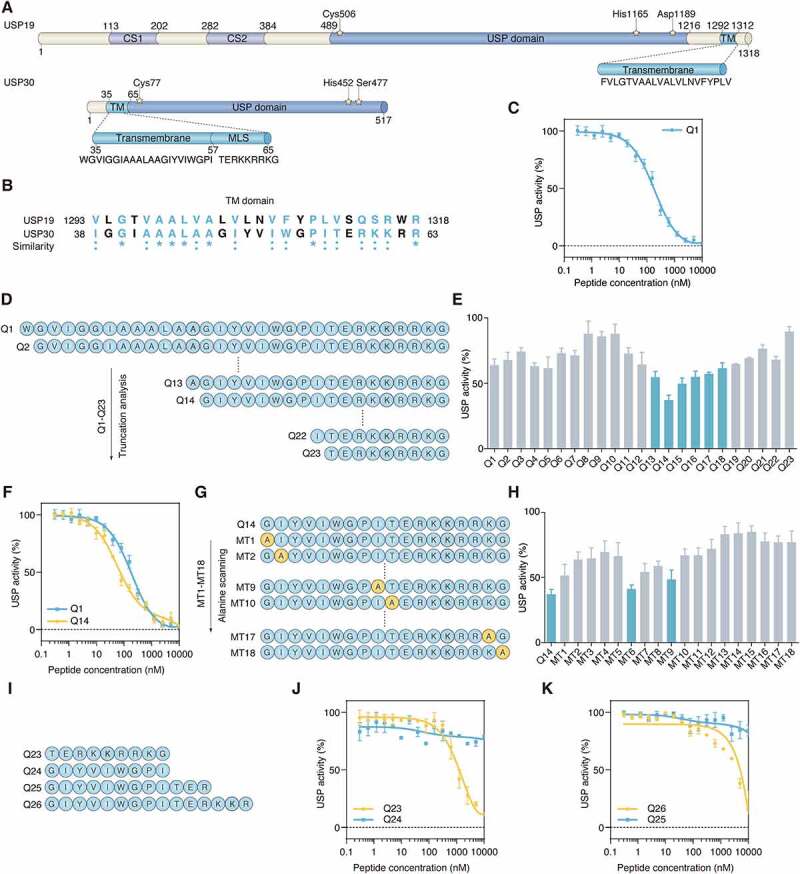
The TM domain of USP30 inhibits its catalytic activity. (A) Schematic representation of protein domain architecture of USP19 and USP30. The positions of the amino acids Cys, His, Asp and Ser in the catalytic triad are indicated and the sequences of TM domain of USP19 and USP30 are listed. CS, CHORD-containing proteins and SGT1; USP, ubiquitin specific peptidase; TM, transmembrane; MLS, mitochondrial localization sequence. (B) The sequence alignment of TM domains of human USP19 and USP30. The similarity between USP19 and USP30 is represented by symbols at the bottom of the proteins. An “*” (asterisk) or “:” (colon) represents an identical or conserved (i.e., not identical but of the same type) amino acid residue. The GenBank accession numbers are as follows: human USP19, NM_006677; human USP30, NM_032663. (C) Inhibition of USP30 activity was shown as dose-response curves for Q1 peptide. The IC50 value was determined as the concentration of peptide that reduced USP activity by 50%. Data are represented as mean ± s.d. of three independent experiments. (D) The schematic diagram showing the peptide truncation analysis. The peptide truncation generates different lengths of peptide. (E) The comparison of USP30 activity inhibition between different length of peptide under 100 nM concentration. Data are represented as mean ± s.e.m. of three independent experiments. (F) Inhibition of USP30 activity was shown as dose-response curves for Q1and Q14 peptides. Data are represented as mean ± s.d. of three independent experiments. (G) Schematic diagram showing the peptide alanine screening analysis. MT1-MT18 peptide was generated by mutating single amino acid to alanine. (H) The USP30 inhibition activity was compared between alanine mutated peptide with 100 nM concentration. 100% USP activity was normalized to the control treatment. Data are represented as mean ± s.e.m. of three independent experiments. (I) Schematic diagram showing the peptide sequences of Q23, Q24, Q25, Q26. (J) Inhibition of USP30 activity was shown as dose-response curves for Q23, Q24 peptide. The C-terminal of TM domain (Q23 peptide) showed more inhibitory potential than N-terminal of TM domain (Q24 peptide). Data are represented as mean ± s.d. of two independent experiments. (K) The comparison of USP30 activity inhibition between Q25 and Q26 peptide. Q26 peptide have a longer length of the C-terminal part of the TM domain than the Q25 peptide. Data are represented as mean ± s.d. of two independent experiments.
Q14 peptide inhibits the deubiquitinating activities of USP30
Previous studies have shown that USP30 is an unusual ubiquitin-specific peptidase (USP)-type deubiquitinating enzyme that prefers compact chain types, with Lys 6 chains processed fastest [18,19]. To assess the inhibitory effect of Q14 peptide against USP30’s ability to digest polyubiquitin chains, different concentrations of Q14 peptide were incubated with recombinant USP30 toward tetramers of Lys 6 and the conversion reaction was monitored in a gel-based assay (Figure 2A). In agreement with the results of the Ub-AMC cleavage assay, hydrolytic ubiquitin chains decreased compared to control as the concentration of Q14 increased. As USP30 is a mitochondrial-anchored deubiquitinase, it is important to also evaluate the deubiquitinating activity of USP30 acting on mitochondrial-anchored ub chains, not just purified ub chains. To verify Q14’s inhibition of deubiquitinating activity of USP30, we used isolated ubiquitinated mitochondria treated with either the uncoupler carbonylcyanide m-chlorophenyl hydrazone (CCCP) or dimethyl sulfoxide (DMSO) as a substrate, then incubated them with purified USP30 and Q14 to assess mitochondrial deubiquitination (Figure 2B). Western blotting probed by anti-ub revealed that USP30 was able to digest ub chains in the mitochondrially-enriched fraction, while Q14 increased the amount of ub chains detected (Figure 2C). Taken together, these data demonstrate that Q14 can inhibit the deubiquitinating activities of USP30 on both purified ubiquitin chains and ubiquitinated mitochondria.
Figure 2.
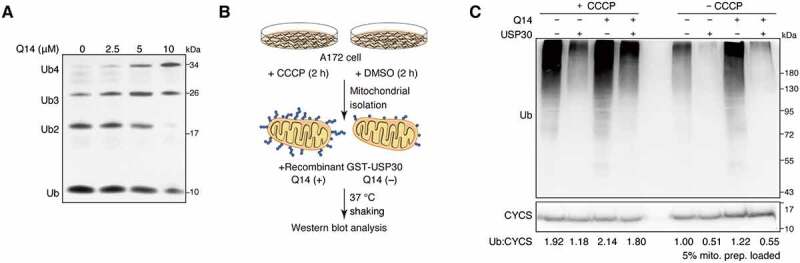
Q14 inhibits the deubiquitinating activities of USP30. (A) Tetrameric ub cleavage inhibition assay. Increasing concentrations of Q14 peptide were incubated with USP30 for 30 min before adding the tetrameric Lys6 ubiquitin chains. After incubating the hydrolysis reaction for 15 min at 37°C, the samples were analyzed by Bis-Tris NuPAGE gels and stained with silver staining. (B) Schematic representation of the experimental setup. Ubiquitinated or normal mitochondria were isolated from CCCP-treated (10 µM, 2 h) or DMSO-treated (2 h) A172 cells. The mitochondrial were incubated with purified GST-USP30 firstly and then peptide Q14 or DMSO were added into them. The reaction was incubated at 37°C with shaking to avoid the mitochondrial settlement. An aliquot for each reaction was used for Western blot analysis to confirm deubiquitination. (C) USP30 and peptide Q14 were incubated with isolated mitochondria that were pre-treated with CCCP or DMSO as shown in Figure 2B. The deubiquitinating activity was assessed by the efficiency of removing the mitochondrial-associated ub chains by Western blot using an anti-ub antibody. A CYCS/cytochrome c antibody was used as a loading control. Representative figure was shown from one of three independent experiments. Full-length gels for A and C are in Fig. S8.
Q14 peptide binds USP30 in vitro
Next, we set out to characterize the interaction between Q14 peptide and USP30. Fluorescence polarization (FP) and microscale thermophoresis (MST) assays were used to assess the binding affinity between Q14 and purified USP30. Fluorescently-labeled Q14 peptide was incubated with a gradient of dilutions of USP30. The FP affinity data showed a Kd value of 37 nM for the Q14 peptide (Figure 3A) and MST affinity data showed 202 nM (Fig. S1E). The Kd values for the Q14 peptide and USP30 as determined by the two biophysical methods above are in the nanomolar range though some differences in binding affinities acquired by different methods. Additionally, we used the streptavidin (SA) affinity-isolation assay to determine the binding affinity of Q14 peptide toward USP30 in cell lysates. We lysed the human A172 glioblastoma cell and incubated the lysates with biotin-labeled Q14 peptide (B-Q14). We observed that B-Q14 peptide could pull down USP30 effectively while DMSO, B-Qscr, and biotin itself could not (Figure 3B). Thermal shift assays are a new developed method for measuring the binding affinity between a protein and a ligand based on the protein thermostability [38]. The melting temperature (Tm) of a protein-ligand complex is higher than the protein alone due to stabilization of the protein by the ligand. As purified USP30 became unstable as temperature increased, we used cell lysate based cellular thermal shift assay to evaluate the binding affinity. As can be seen from Figures 3C and 3D, the incubation of A172 cell lysates with Q14 increased the thermal stability of USP30, whereas control treatments showed no difference. Therefore, these results demonstrate that Q14 peptide can interact with USP30.
Figure 3.
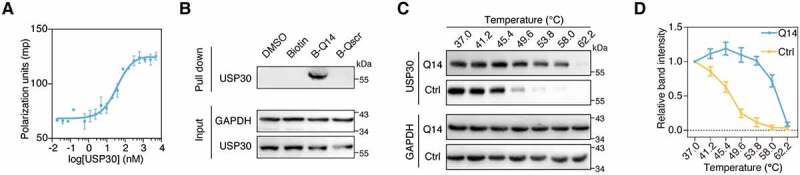
The binding activity of Q14 peptide with USP30. (A) Increasing concentrations of USP30 were incubated with FITC-labeled peptide FITC-Q14 and detected by fluorescence polarization. Data are represented as mean ± s.d. of two independent experiments. (B) A172 cell lysates were incubated with biotin-labeled peptide B-Q14, biotin-labeled peptide-Qscr, biotin and DMSO at 4°C. SA beads were added into the mixture and used for Western blot analysis through an anti-USP30 antibody. GAPDH was used as a loading control. (C) Representative immunoblots for cell lysate based thermal shift assay carried out in A172 cells treated with DMSO or Q14 peptide (50 µM). Western blots were developed using an anti-USP30 antibody. GAPDH was shown as a loading control. (D) Graphical summary of the results of a cell lysate based thermal shift assay of A172 cells treated with DMSO or Q14 peptide (50 µM). Data are presented as mean ± s.e.m. from three independent experiments. Full-length gels for B and C are in Fig. S8.
Q14 peptide targets USP30 in cells
To determine whether Q14 targets USP30 in cells, we carried out a series of assays in A172 cells treated with Q14 peptide. One of the major problems for peptide inhibitors is their inability to cross the cell membrane effectively [39]. To verify that the Q14 peptide could translocate across the plasma membrane of mammalian cells, we first tested the cell permeability of Q14 in A172 cells. We assessed the cellular uptake of the Q14 peptide by flow cytometry (FACS) after stringent tryptic digestion and 0.05% trypan blue treatment. Q14 peptide-treated cells showed more fluorescence compared to DMSO and TAT-FITC treated controls in A172 cells, which suggests that the cellular uptake of Q14 was sufficient for cell-based assays (Figure 4A, Fig. S2A). To corroborate that the Q14 peptide was indeed inhibiting USP30 in cells, we carried out SA-affinity-isolation assays in A172 cells pretreated with B-Q14 peptide. We observed a specific band representing USP30 after treatment with B-Q14 that was not found in cells treated with DMSO, biotin or B-Qscr, which indicated Q14 interacts with USP30 in cells (Figure 4B). To test whether USP30 is the direct target of Q14 peptide in cells, we used a cellular thermal shift assay to measure the interactions between Q14 peptide and USP30 in A172 cells. Incubation of A172 cells with Q14 peptide shifted the Tm of USP30 from 56°C to 60°C (Figure 4C). The increased thermal denaturation temperature confirmed that Q14 directly binds to USP30 in cells. Additionally, we characterized the interaction between Q14 peptide and USP30 using immunofluorescence colocalization assay. Cells incubated with FITC-Q14 peptide displayed significant colocalization with USP30 (Pearson’s coefficient, R > 0.8), which indicated effective cell penetration and USP30 targeting of the Q14 peptide (Figure 4D). Notably, besides the dispersive distribution, FITC-Q14 and USP30 also showed punctate distribution. The puncta were mainly distributed in the perinuclear region, and we wondered whether these puncta were fragmented mitochondria. After incubating the FITC-Q14 peptide with A172 cells, we used MitoTracker red to label mitochondria. We discovered that FITC-Q14 peptide showed partial colocalization with mitochondria (Pearson’s coefficient, R = 0.623) and a high degree of colocalization with punctate mitochondria (Figure 4E), whereas FITC-Qscr and FITC-TAT control peptides showed little colocalization with mitochondria and did not generate punctate mitochondria (Fig. S2B, S2C). Accumulating evidence is demonstrating the importance of mitochondrial fragmentation prior to mitophagy [40,41]. The punctate mitochondria in the FITC-Q14 peptide-treated cells indicate a certain relationship between Q14 peptide and mitochondrial fragmentation. Collectively, the data above indicate that Q14 peptide can interact with USP30 directly in A172 cells.
Figure 4.
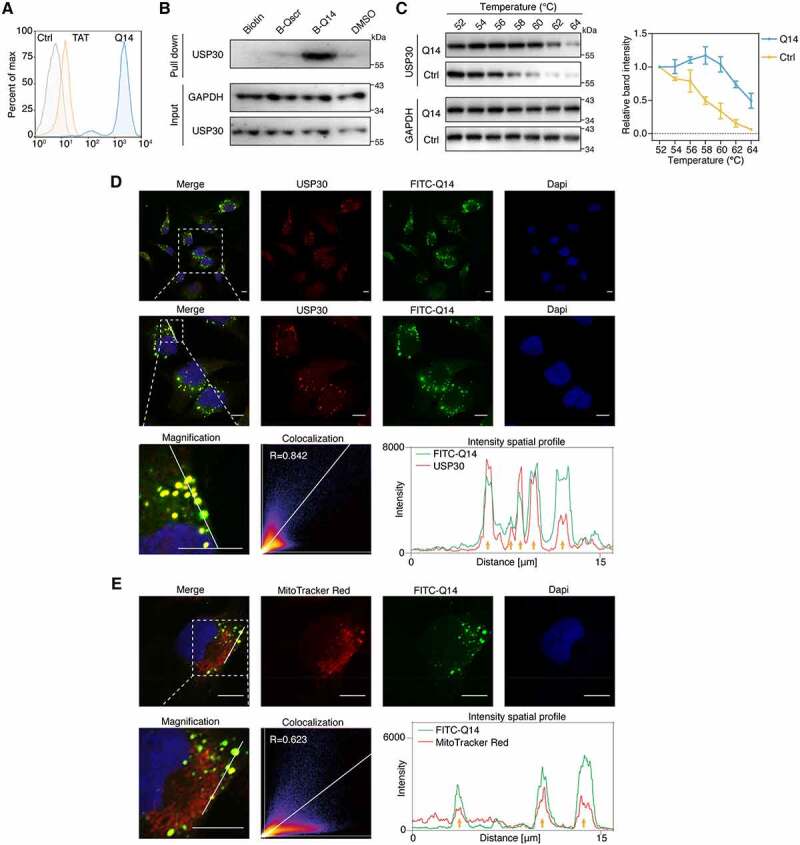
Q14 peptide targets USP30 in cells. (A) FACS analysis illustrates the internalization of FITC-labeled Q14 peptide in A172 cells. Data are representative of two independent experiments. (B) SA-affinity-isolation assay in A172 cells treated with DMSO, biotin, biotin-labeled Q14 peptide (B-Q14, 50 µM) or biotin-labeled scramble peptide (B-Qscr, 50 µM) for 12 h. Western blots from a single experiment were developed with anti-USP30 antibody. GAPDH was used as a loading control. (C) Cellular thermal shift assay was carried out in A172 cells treated with DMSO or Q14 peptide (100 µM) for 12 h. Representative immunoblot was developed with anti-USP30 antibody in a single experiment. GAPDH was used as a loading control. Quantification results are shown on the right. Data are presented as mean ± s.e.m. from three independent experiments. (D) Immunofluorescence showing the colocalization between USP30 and FITC-Q14 peptide. Scale bars: 10 μm. The intensity profiles of USP30 and FITC-Q14 peptide along the white line are plotted on the right panels, with the colocalizing sites marked by yellow arrows. Correlation was determined by Pearson’s coefficient (R value indicated). The white dotted-lined boxes indicate representative fields to be shown in magnification. (E) Immunofluorescence showing the colocalization between FITC-Q14 peptide and MitoTracker Red-marked mitochondrial. Scale bars: 10 μm. The intensity profiles of FITC-Q14 peptide and mitochondrial along the white line are plotted on the right panels, with the colocalizing sites marked by yellow arrows. Correlation was determined by Pearson’s coefficient (R value indicated). The white dotted-lined boxes indicate representative fields to be shown in magnification. Full-length gels for B and C are in Fig. S8.
Q14 peptide increases mitophagy by inhibiting USP30
Since the Q14 peptide binds mitochondrial-anchored USP30 in cells, we next determined whether the Q14 peptide increases mitophagy by inhibiting the deubiquitinating activity of USP30. First, we assessed global, as well as K48- or K63-linked, ubiquitination levels of mitochondria isolated by differential centrifugation from treated A172 cells using Western blotting. Incubation of A172 cells with Q14 peptide for 24 h resulted in a slight increase of mitochondrial ubiquitination, while pretreatment with Q14 peptide for 18 h followed by co-incubation with CCCP for another 6 h increased mitochondrial ubiquitination significantly (Figure 5A, Fig. S3A). These data imply that Q14 peptide can lower the threshold for mitochondrial ubiquitination induced by CCCP. USP30 acts as a brake on mitophagy by deubiquitinating mitochondrial proteins [18,19]. Therefore, we measured the degradation of the mitochondrial proteins TOMM20, TOMM22 and TIMM23 to quantify the extent to which mitophagy was induced by the Q14 peptide. Immunoblotting revealed that Q14 peptide treatment could decrease the TOMM20, TOMM22 and TIMM23 levels and increase the LC3-I to LC3-II transition in a dose- and time-dependent manner (Figure 5B, Fig. S3B). Further, we analyzed the proteins obtained from cell lysates after treatment with DMSO, Q14, CCCP, and Q14+ CCCP. Mitochondrial proteins were modestly degraded when A172 cells were treated with CCCP or Q14 peptide separately, whereas a more dramatic decrease was observed when A172 cells were treated with both Q14 and CCCP, consistent with the mitochondrial ubiquitination results (Figure 5C). Ubiquitination of TOMM20 is regarded as a signal for mitophagy [17]. We therefore isolated the TOMM20 protein from A172 cells by immunoprecipitation after drug treatment and measured the ubiquitination of TOMM20 using an anti-ub antibody. Cotreatment with Q14 peptide and CCCP resulted in enhanced TOMM20 ubiquitination compared with treatment with either alone (Figure 5D). To demonstrate that mitochondria were degraded by an autophagic process, we treated cells with inhibitors or inducers of autophagy and monitored turnover of LC3-II and intensities of endogenous LC3 puncta in A172 cells (Figure 5E, Fig. S3C, S3D). Bafilomycin A1 (BafA1) and hydroxychloroquine (HCQ) are commonly used inhibitors of autophagic flux that function by inhibiting V-ATPase or by raising lysosomal pH to prevent autophagosome-lysosome fusion and endosomal acidification [42]. We observed an additive effect on LC3-II levels and LC3 puncta intensities, indicative of enhanced autophagic flux, in cells treated with Q14 peptide and BafA1 or HCQ compared to those treated with Q14 peptide alone. Similarly, higher LC3-II levels and LC3 puncta intensities were observed when we induced autophagy with rapamycin (Rap) in combination with Q14 peptide compared to treatment with either Q14 or Rap alone. 3-methyladenine (3-MA) is an inhibitor of phosphatidylinositol 3-phosphate kinase (PI3KC3) that affects nucleation of phagophores and thus the formation of autophagosomes [10]. We applied 3-MA (2 mM) to treat cells that were pretreated with Q14 peptide for 12 h and found that 3-MA decreased the LC3-II conversion and formation of autophagosomes induced by the Q14 peptide, suggesting that the Q14 peptide induces the formation of autophagosomes in A172 cells. These results confirm that Q14 peptide indeed induces autophagic activity by elevating autophagic flux and autophagosome formation. Mitochondrial DNA (mtDNA), mitochondrial mass and ATP are important indicators of mitochondrial function that are closely related to mitophagy [43,44]. To confirm that Q14 peptide induces mitophagy beyond changes on protein level, we also detected mtDNA, mitochondrial mass and ATP levels. The mtDNA copy number decreased gradually with increasing concentrations of Q14 peptide, and CCCP-mediated loss of mtDNA was enhanced by Q14 peptide (Figure 5F, Fig. S3E). Besides, we used 10-N-nonyl acridine orange (NAO) to assess the mitochondrial mass [44–46]. NAO flow cytometric analysis revealed that Q14 peptide increased CCCP-induced loss of mitochondrial mass in a dose-dependent manner (Fig. S4A, S4B). Moreover, the levels of ATP changed in a similar manner to mtDNA copy numbers: ATP levels decreased significantly at higher concentrations of Q14 peptide (Figure 5G). Furthermore, we used commercial Mtphagy Dye and Lyso Dye to monitor the occurrence of mitophagy in A172 cells, as previously reported [47,48]. The fusion of mitochondria and lysosome gradually increased with low concentrations (under 50 µM) peptide treatment (Fig. S4C, S4D). When the cells were treated with a high concentration of peptide (100 µM) or cotreated with CCCP, mitochondrial degradation increased, indicating the autophagic process in mitochondrial degradation induced by Q14 peptide (Fig. S4C, S4E). These results suggest that Q14 peptide induces clearance of mtDNA and mitochondrial degradation by mitophagy. Mitochondria are highly dynamic organelles whose morphology is continuously regulated by the balance between fission and fusion [40]. Accumulating evidence emphasizes the importance of mitochondrial fragmentation prior to mitophagy [41,49]. Confocal imaging revealed that A172 cells displayed fragmented and dot- or rod-like mitochondria after incubation with high concentrations of Q14 peptide, which is consistent with the results of the colocalization assay (Figure 5H, Fig. S5A). Importantly, we did not observe any negative impacts on the cell membrane integrity and cell viability (Fig. S5B, S5C). To understand the molecular mechanism of Q14-induced mitochondrial fragmentation, we next investigated the effect of Q14 peptide on key mitochondrial fusion and fission factors [50,51]. To elucidate this question, we first measured the mitochondrial fusion activity in A172 cells using a mitochondria-targeted photoactive fluorescence protein (mtPA-GFP), as previously described [52]. After photoactivation by 405 nm exciting light, mtPA-GFP intensities with regions of interest was tracked by real-time confocal microscopy in 10 min. In control cells, activated mtPA-GFP in mitochondria were found to diffuse to other non-photoactivated mitochondria fastly due to the mitochondria fusion (Figure 5I, Fig. S5D). Strikingly, upon Q14 peptide treatment, the intensity of mtPA-GFP remained stable over 10 min, reflecting slower mitochondrial fusion rate in Q14 peptide treated cells (Figure 5I, Fig. S5D). To further clarify the reason of either inhibition of fusion or activation of fission in fragmentation of mitochondria, we then measured the protein levels of key mitochondrial fusion and fission factors. After 24 h incubation with Q14 peptide, the protein levels of mitochondrial fusion factors MFN1, MFN2, and L-OPA1 (long, 100 kDa) decreased in a dose-dependent manner, whereas the levels of mitochondrial fission factors DNM1L/DRP1 and DNM2 (dynamin 2) were unchanged (Fig. S5E). These findings demonstrate that mitochondrial fragmentation caused by treatment with Q14 peptide is due to decreased mitochondrial fusion with little change in mitochondrial fission. Taken together, our data show that the Q14 peptide induces mitophagy by increasing the degradation of OMM proteins and enhancing mitochondrial fission.
Figure 5.
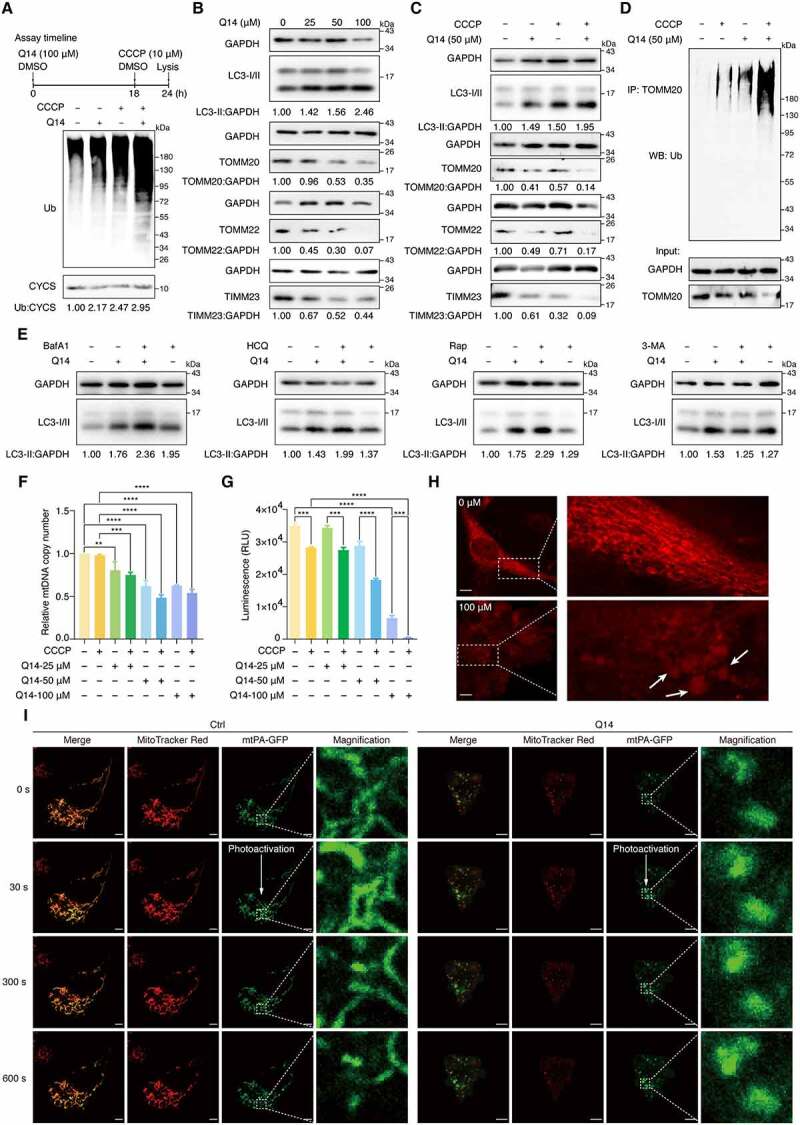
Q14 peptide increased mitophagy by inhibiting USP30 deubiquitinating activity. (A) Deubiquitination of mitochondria was assessed by Western blot analysis using anti-ubiquitin antibody on A172 cells treated with DMSO, CCCP (10 µM, 6 h) and Q14 peptide (50 µM, 24 h). CYCS/cytochrome c was used as a loading control. The relative band intensities are quantified by image-J software and shown below the blots. Protein bands were normalized to CYCS/cytochrome c and then normalized to Ctrl. Assay timeline is shown on the top panel. (B) Immunoblotting for LC3-II, TOMM20, TOMM22 and TIMM23 was assessed in A172 cells incubated with gradient diluted Q14 peptide for 24 h. GAPDH was used as a loading control. The relative band intensities are quantified by image-J software and shown below the blots. Protein bands were normalized to GAPDH and then normalized to Ctrl. (C) A172 cells incubated with 50 µM Q14 peptide and CCCP as the timeline shown and lysed for Western blotting analysis with LC3-II, TOMM20, TOMM22 and TIMM23. GAPDH was used as a loading control. The relative band intensities are quantified by image-J software and shown below the blots. Protein bands were normalized to GAPDH and then normalized to Ctrl. (D) Ubiquitination of TOMM20 was assessed by immunoprecipitation developed with anti-ubiquitin antibody on A172 cells incubated with 50 µM Q14 and CCCP (10 µM, 6 h) for 24 h. (E) Immunoblotting analysis of LC3-II in A172 cells treated with Q14 peptide (50 µM) in the absence and presence of the inhibitors (bafilomycin A1 [100 nM; applied for the final 2 h of the 24 h], hydroxychloroquine [5 μM; applied for the final 2 h of the 24 h], rapamycin [5 μM; applied for the final 12 h of the 24 h] and 3-methyladenine [2 mM; applied for the final 12 h of the 24 h]). The relative band intensity of LC3-II was quantified by image-J software and shown below the blots. Protein bands were normalized to GAPDH and then normalized to Ctrl. (F) Quantification of mitochondrial DNA copy number in A172 cells treated with DMSO, CCCP (10 µM, 6 h) or different concentrations of Q14 peptide. Data was normalized to GAPDH gene and then normalized to Ctrl. Values are means ± s.d. from three independent experiments (n = 3). Statistical differences were determined by one-way ANOVA multiple comparisons using GraphPad Prism 8.0; **P< 0.01, *** P< 0.001, **** P< 0.0001. (G) Quantification of ATP contents in A172 cells treated with DMSO, CCCP (10 µM, 6 h) or different concentrations of Q14 peptide. Data are presented as mean ± s.d. from two independent experiments. Statistical differences were determined by one-way ANOVA multiple comparisons using GraphPad Prism 8.0; *** P< 0.001, **** P< 0.0001. (H) The morphology of mitochondrial was assessed by confocal imaging of A172 cells treated with increasing concentrations of Q14 peptide for 24 h. The fragmented and dot- or rod-like mitochondria are marked by white arrows. The white dotted-lined boxes indicate representative fields to be shown in magnification. Scale bar: 10 μm. (I) Representative images of mitochondrial fusion over time (indicated in second). A172 cells were transfected with mtPA-GFP for 24 h and stained with MitoTracker Red. Where indicated, A172 cells were treated with Q14 peptide (20 μM) for 2 h. mtPA-GFP was photoactivated in a region of interest (dashed white box) and cells were imaged by real-time confocal microscopy in 10 min. Scale bar: 5 µm. Experiments in A, B, C, D and E were repeated three times independently with similar results. Full-length gels for A, B, C, D and E are in Fig. S8.
The LIR motif of Q14 peptide facilitates binding with LC3 and formation of autophagosome
Next, we attempted to understand the molecular mechanism by which Q14 mediates mitophagy. Several mitophagy receptors such as FUNDC1, BNIP3, and BNIP3L/NIX, are recognized by LC3 and its homologues through the canonical LIR motif with a core consensus sequence of [W/F/Y]-X1-X2-[I/L/V] (X1, X2 represent any amino acid). Interestingly, we found that Q14 peptide contains a motif of WxxI in its N-terminal region by using the iLIR database (http://repeat.biol.ucy.ac.cy/iLIR/) (Figure 6A). To elucidate whether Q14 peptide interacts with LC3 via LIR motif, we first generated a series of mutations: single mutant peptides MT2 (I/A), MT4 (V/A), MT6 (W/A) and MT9 (I/A), double mutant peptide Q14-mut (WxxI/AxxA), and two segmented peptides (Q23, Q24). We then labeled the peptides with biotin and carried out SA-affinity-isolation assays in A172 cell lysates. We found that altering the sequence at the LIR motif (MT6, MT9, and Q14-mut), but not at any other motifs (MT2 and MT4), significantly weakened peptide interactions with LC3 and that deletion of the entire LIR motif completely abrogated the interaction, indicating that Q14 peptide binds LC3 via the LIR motif (Figure 6B, 6C). Then, we addressed the critical question of whether the interaction between Q14 peptide and LC3 is functionally important for mitophagy. As expected, we found that mutations in the LIR motif of Q14 peptide caused a remarkable decrease in formation of autophagosomes, implying that Q14 peptide-induced mitophagy is dependent on the interaction with LC3 via the LIR motif (Fig. S6A). To further prove the importance of the LIR motif of the Q14 peptide for mitophagy, we added one or two LIR motifs to the N terminus or C terminus of Q14 peptide. We hypothesized that the extended LIR motifs would facilitate the interaction with LC3 and the autophagosome membrane. Using a lysate-based SA-affinity-isolation, we found that adding two motifs at the N terminus (L2) or C terminus (L4) significantly increased the interaction with LC3 and only slightly affected binding to USP30 (Figure 6D). However, only addition of LIR motifs at the N terminus (L2) enhanced binding with LC3 without any impact on USP30 binding in affinity-isolation experiments with intact cells, which suggests that the performance of the Q14 peptide could be considerably improved by addition of LIR sequences at a suitable site (Figure 6D). Whereas LIR mutation of Q14 peptide significantly impaired the interaction with LC3 and slightly affected binding to USP30 in cells (Fig. S6B). We next investigated the biological impact of these LIR-extended peptides on mitophagy. Consistent with the results of the SA-affinity-isolation assay in intact cells, the L2 peptide induced conversion of LC3-II, reduction of TOMM20, TOMM22 and TIMM23 levels, and enhancement of the total intensities of endogenous LC3 puncta in A172 cells, further confirming the key role of the LIR in Q14 peptide-induced mitophagy (Figure 6E, 6F). Further, the L2 peptide induced mitochondrial fragmentation without affecting peptide co-localization with USP30 (Fig. S6C, S6D), suggesting that the LIR-extended peptides could not only inhibit the USP30 activity to induce mitophagy but also link USP30 on the mitochondria to the autophagosome membrane to promote mitophagy (Figure 6G). Collectively, these data demonstrate the significance of the LIR motif in the Q14 peptide for binding LC3 and promoting autophagosome formation. Addition of LIR motifs at the N terminus of the Q14 peptide might help to optimize USP30 inhibitors to induce mitophagy in A172 cells.
Figure 6.
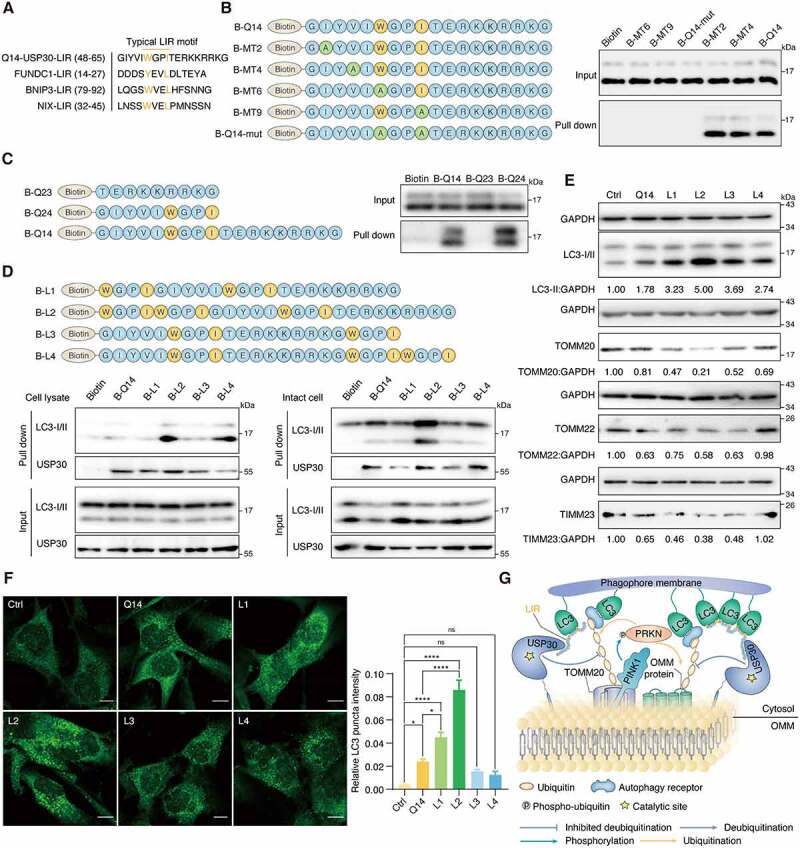
Q14 peptide contains the LIR and interacts with LC3B in A172 cells. (A) Typical LIR motifs of mitophagy related proteins were identified from iLIR database and aligned manually alongside USP30 for comparison. The colored residues indicate highly conserved residues in the putative LIR of USP30. (B) A172 cell lysates were incubated with biotin, single mutant peptide MT2 (I/A), MT4 (V/A), MT6 (W/A) and MT9 (I/A), double mutant peptide Q14-mut (WxxI/AxxA) and biotin-labeled Q14 peptide (B-Q14) at 4°C. SA beads were added into the mixture and used for Western blot analysis through an anti-LC3B antibody. Mutations only at the LIR motif (MT6, MT9, Q14-mut) but not at other motifs (MT2, MT4), significantly weakened their interactions with LC3. The peptide sequences are shown on the left. (C) Peptides contain with LIR motif (B-Q14, B-Q24) could pull down LC3 while the other segment (B-Q23) could not. The peptide sequences are shown on the left. (D) The lysate and intact-cell based pull down assay performed by B-Q14 peptide and peptides with extended LIR motifs (B-L1, B-L2, B-L3, B-L4). The peptide sequences are shown on the top panels. (E) Immunoblotting for LC3-II, TOMM20, TOMM22 and TIMM23 was assessed in A172 cells incubated with peptide (Q14, L1, L2, L3, L4, 50 µM) for 24 h. GAPDH was used as a loading control. The relative band intensities are quantified by image-J software and shown below the blots. (F) Immunofluorescence showing the endogenous LC3 puncta in A172 cells incubated with DMSO, Q14, L1, L2, L3 and L4 peptide. Scale bar: 10 μm. Quantification of the LC3 puncta intensities in the cells. Data are presented as mean ± s.e.m. from at least 300 cells. Data were analyzed by one-way ANOVA (multiple comparisons) using GraphPad Prism 8.0. * P< 0.05, ****p < 0.0001. (G) Q14 peptide binds with USP30 and inhibits its deubiquitinating activity. Besides, through interacting with LC3 by its LIR motif, Q14 peptide links the phagophore membrane to USP30 and ubiquitinated mitochondria, promoting the mitophagy level. Experiments in B, C, D and E were repeated three times independently with similar results. Full-length gels for B, C, D and E are in Fig. S8.
Exploration of the Q14 peptide-USP30 binding site through proximity-promoted protein labeling using a Cys-Met bis-alkylated sulfonium tethered peptide
Next, we identified the binding site between Q14 peptide and USP30. Chemical cross linking is a commonly used method to identify the possible location of ligand binding sites and a practical alternative to crystal structure and homology modeling studies [53–55]. We attempted to obtain the crystal complex of the Q14 peptide bound to USP30 but were unsuccessful due to its poor solubility, and therefore turned to the chemical cross-linking assay. Recently, we developed a method based on bis-alkylation between cysteine and methionine with an on-tether sulfonium center acting as a novel warhead to label cysteine in the vicinity of the binding pocket of the protein of interest (POI) through proximity-triggered cysteine substitution for target identification [56]. Here, we introduced methionine and cysteine to the sites in the Q14 peptide that have less of an effect on inhibition of USP30 enzymatic activity (MT6, MT9) to synthesize the Cys-Met bis-alkylated sulfonium-tethered peptide (MC-lin) (Figure 7A). We incubated this peptide (N-terminal with FAM tag or biotin tag) with USP30 protein (protein/peptide 10/100 µM, 16°C) and visualized conjugation of MC-linker to USP30 over time using in-gel fluorescence or anti-biotin Western blotting (Figure 7B, 7C). Interestingly, the modification sites were mainly located toward the end of fingers subdomain (Cys234 and Cys284) but not in the vicinity of the catalytic center (Cys77) (Figure 7D). Moreover, we synthesized peptide with benzophenones, which could react with majority of natural amino acids under UV (Fig. S7A) [57]. The modification sites identified with matched peaks were Thr306, Val309 and Lys310, which locate toward the end of fingers subdomain nearby the Cys234 and Cys284, further demonstrating that Q14 peptide might function by binding to the sites in the fingers region (Fig. S7B, S7C). These data suggest that the Q14 peptide might function by binding to the sites in the fingers region that are distant from the catalytic center instead of interacting directly with the cysteine in the catalytic center. The USP family exhibits a conserved fingers-palm-thumb architecture that holds ubiquitin. Apart from the thumb-palm catalytic cleft region, the residues at the tip of the fingers also participate in ubiquitin binding [58]. However, the Q14 peptide only weakly inhibits the enzymatic activity of USP2, USP7, and USP8, suggesting that the Q14 peptide might regulate USP30 activity by unique allosteric regulation instead of by simple steric occupancy (Fig. S7D). We then used the CavityPlus web server (http://www.pkumdl.cn:8000/cavityplus/) to detect potential allosteric sites of USP30 and found that the tip of fingers region was a potential allosteric site when the ubiquitin binding pocket was selected as an orthosteric site, confirming the results of chemical cross-linking assay (Figure 7E) [59]. Autoinhibition is an inactivating mechanism in UPS system that guards against spurious activation [25,27–30,60,61]. Similar to how USP19 activity is regulated by subcellular localization, USP30 activity might be regulated by integration of the TM domain into the OMM; integration could shift the protein from the autoinhibited “closed” to the active “open” state. Q14 peptide might exert its inhibitory effects through allosteric regulation at the tip of the fingers subdomain, changing the shape of the finger-palm space of membrane-integrated USP30 (which accommodates ubiquitin) and thus blocking the ubiquitin C terminus from approaching the catalytic center (Figure 8). In conjunction with previous findings from studies examining modulation of autoinhibition in DUBs, our findings suggest that investigating allosteric mechanisms that regulate autoinhibition might be a viable approach to discover potent inhibitors against USP30 or other DUBs.
Figure 7.
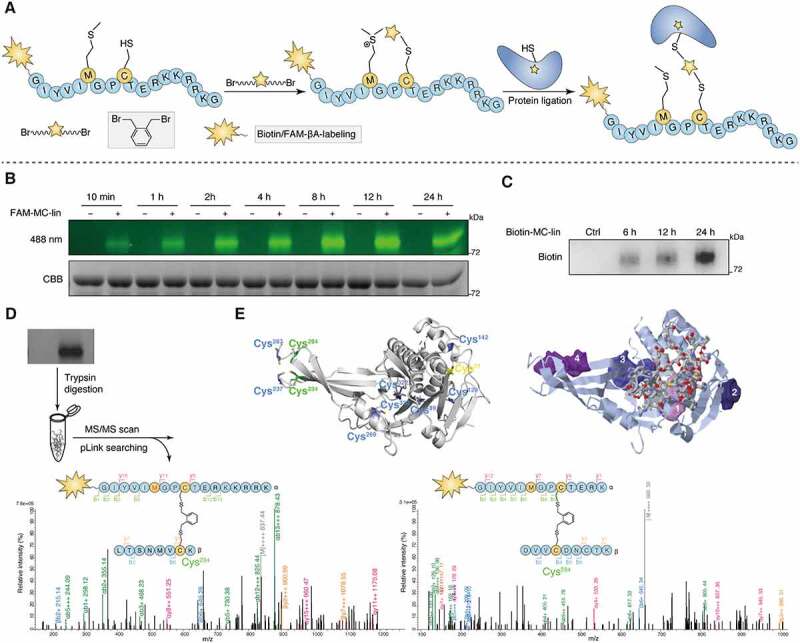
Exploration of the binding site between Q14 and USP30 through proximity promoted protein labeling using Cys-Met bis-alkylated sulfonium tethered peptide. (A) Schematic presentation of the proximity promoted protein labeling using Cys-Met bis-alkylated sulfonium tethered peptide. Mutating the tryptophan and isoleucine to methionine and cysteine separately and Cys-Met bis alkylation was completed by adding the linker o-Xylylene dibromide to peptide. Protein labeling reaction was incubated the alkylated peptide with USP30 protein at 16°C for indicated time. (B) FAM-labeled alkylated peptide (FAM-MC-lin) was incubated with USP30 at 16°C. Sample was collected at indicated time point and analyzed by SDS-PAGE and in-gel fluorescence (488 nm) scanning or Coomassie Brilliant Blue (CBB) staining. (C) Biotin-labeled alkylated peptide (Biotin-MC-lin) was incubated with USP30 at 16°C. Sample was collected at indicated time point and performed by Western blot analysis using anti-biotin antibody. (D) Mass/mass spectrometry analysis using pLink2 for trypsin digested USP30-peptide conjugates indicated that peptide MC-lin binds covalently to C234 and C284 in USP30. Green and red correspond to b- and y- ions of peptide α, respectively. Blue and Orange correspond to b- and y- ions of peptide β. The precursor charge and m/z (gray) are shown in the spectrum. Relative intensity of m/z is plotted. Separate FDR ≤ 5% at PSM level. Variable modification of Oxidation [Met] is in red color. (E) The cysteine distribution of USP30 and potential allosteric binding sites predicted by the online web server CavityPlus. The left figure: all of cysteine in USP30 was colored. The protein labeling cysteines (C234 and C284) were colored green, the enzyme catalytic site cysteine (C77) was colored yellow and other cysteines (C89, C129, C142, C237, C269, C287, C320 and C322) were colored blue. The right figure: prediction of the allosteric binding sites in USP30. The stick region on USP30 indicate the orthosteric site and the other shaded four regions indicate the potential allosteric sites predicted by the online web server CavityPlus. Full-length gels for B and C are in Fig. S8.
Figure 8.
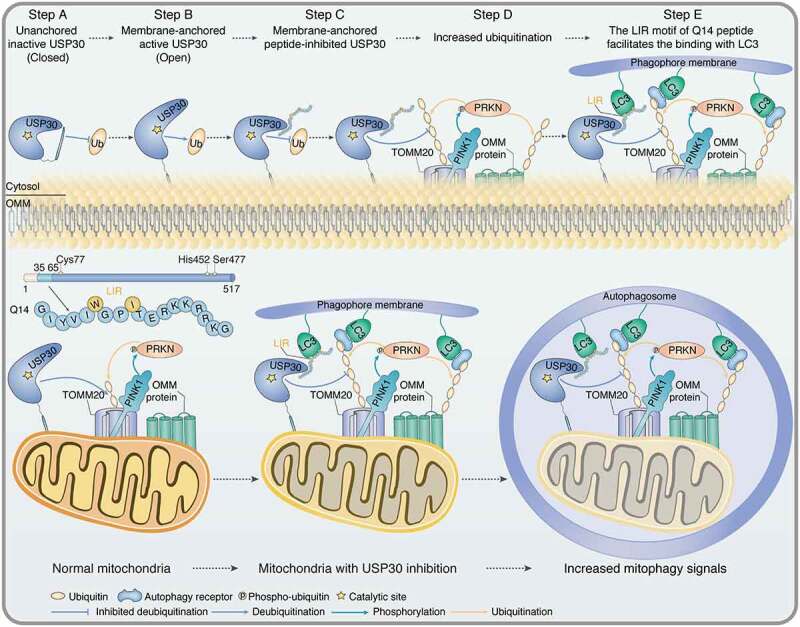
Schematic of the autoinhibitory, mitophagy-inducing model for Q14 peptide. The peptide (Q14) derived from the TM domain of USP30 inhibits its catalytic activity and induces mitophagy. Top: When USP30 is in the cytosol, the TM domain might shield its catalytic center to prevent spurious enzymatic activity (Step A). When the TM domain of USP30 integrates into the OMM, a conformational change triggers exposure of the active catalytic site, thus relieving autoinhibition (Step B). The interaction between Q14 peptide and USP30 deforms the space normally occupied by ubiquitin and prevents the C terminus of ubiquitin from accessing the active site, thus inhibiting USP30 deubiquitinating activity (Step C). Then, Q14 peptide increases ubiquitination of mitochondrial-related proteins by inhibiting the deubiquitinating activity of USP30 (Step D). The LIR motif in Q14 peptide allows it to bind LC3 and accelerate autophagosome formation (Step E). Bottom: Q14 peptide could interact with LC3 and increase mitophagy via the LIR motif. Q14 peptide tethers mitochondrial-anchored USP30 to the autophagosomes by direct binding to USP30 and the key autophagosome protein LC3, further accelerating mitophagy.
Discussion
As a vital organelle in cells, mitochondria play a key role in cellular homeostasis by modulating several cellular functions including energy production, metabolism, and apoptosis. Increasing evidences have shown that mitochondrial dysfunction is closely related to Parkinson disease. To clear damaged mitochondria, cells use mitophagy, a pathway that ensures mitochondrial quality control. Recent studies have indicated that the mitochondrial-anchored deubiquitinating enzymes USP30 could inhibit mitophagy driven by PRKN and PINK1, which are proteins associated with Parkinson disease. Despite its potential as a target for treating Parkinson disease, fewer inhibitors targeting USP30 have been reported.
Here, for the first time, we have shown that a peptide derived from the TM domain of USP30 exerts autoinhibitory function and induces mitophagy. Through a series of sequence truncations and mutational analysis, we identified the Q14 peptide as the shortest and most potent inhibitor of USP30. After efficiently crossing the cell membrane, the Q14 peptide could directly target mitochondrial-anchored USP30, increase degradation of OMM proteins, enhance the mitochondrial fission, and trigger mitophagy. Additionally, we found that the Q14 peptide binds to LC3 via its LIR motif, links mitochondria to the phagophore membrane, and facilitates autophagosome formation. By the proximity-promoted protein labeling, we identified a potential binding site between Q14 peptide and USP30 and proposed an allosteric model for how it modulates USP30 activity (Figure 8). When USP30 is in the cytosol, the TM domain might shield its catalytic center to prevent spurious enzymatic activity. When the TM domain of USP30 integrates into the OMM, a conformational change triggers exposure of the active catalytic site, thus relieving autoinhibition. The interaction between Q14 peptide and USP30 deforms the space normally occupied by ubiquitin and prevents the C terminus of ubiquitin from accessing the active site, thus inhibiting USP30 deubiquitinating activity. These findings reveal an intriguing role for the TM domain of USP30 in regulating deubiquitinating activity through allosteric autoinhibition.
Our results reveal two important insights: the novel autoinhibition mechanism used by USP30 and the accelerated autophagosome formation induced by the LIR motif of the Q14 peptide. First, we discovered that the TM domain-derived Q14 peptide adopts a novel autoinhibitory mode to inhibit the USP30 activity through allosteric regulation at the tip of the fingers subdomain. Autoinhibition is a mechanism adopted by proteins to guard against spurious activation, ensuring that proteins exert their catalytic activity at appropriate places or times [26]. Studies have shown that the full catalytic ability of a USP enzyme not only depends on its active catalytic domain but also depends on its additional domains, the interaction of non-substrate binding ligands, or the interaction between the two [61]. For example, in the case of USP14, two surface loops block the catalytic cleft toward the catalytic center in the absence of ubiquitin binding, and binding with ubiquitin induces the translocation of the loops, thereby allowing the C terminus of ubiquitin to access the catalytic center [32]. In the case of USP7, the catalytic triad residues are misaligned and in an inactive conformation in the resting state, whereas in the active state, the two C-terminal ub-like domains promote their realignment [58]. The subsequent allosteric interaction with guanosine 5ʹ-monophosphate synthase (GMPS) stabilizes the activated state, further enhancing USP7 activity [30]. This conformational flexibility is thought to be necessary to regulate specificity among different USPs. The structural plasticity of the catalytic domain and its susceptibility to regulation make allosteric regulation an important target for the development of specific USP inhibitors. Currently, the reported selective DUB inhibitors are known to function via allosteric regulation, rather than by directly inhibiting the active site, as exemplified by IU1 targeting to USP14 and FT671 targeting to USP7 [62,63]. Analogously, we found that the Q14 peptide exerts autoinhibitory function through allosteric regulation at the end of the fingers subdomain. The fingers appear to be tightened inward so that they can no longer accommodate ubiquitin and the access of the C-terminal tail of ubiquitin to the active site becomes restricted. Unlike the autoinhibitory modes used by the majority of USPs, which involve loops of unknown function, the autoinhibitory segment of USP30 is located within a functional region. Autoinhibition imposed by the TM domain ensures deubiquitinating activity occurs at specific locations in the cell and prevents unintended deubiquitination; this mechanism is quite logical given that USP30 is a membrane-anchored protein. Currently, the major problem for the development of DUB inhibitors is druggability, including not just efficacy but also selectivity. Due to the highly conserved cysteine protease-like activity and low druggability of DUBs, previous studies have mainly focused on covalent inhibitors that react with the catalytic cysteine residue. However, these inhibitors usually have poor selectivity among the DUB family. The design of inhibitors based on the autoinhibition mode proposed here represents a novel approach based on allosteric regulation to discover USP30 inhibitors, which has important implications for the future design of inhibitors against USP30 and other DUBs.
Second, we observed that the Q14 peptide not only directly inhibits USP30 to induce mitophagy but also links USP30 to the phagophore membrane to accelerate mitophagy. The Q14 peptide interacts with LC3 and induces mitophagy via its LIR motif. Further, adding two LIR motifs to the N-terminal domain of the Q14 peptide dramatically increased binding to LC3 and autophagosome formation, suggesting that mitophagy induced by the Q14 peptide might be optimized by addition of two LIR motifs to its N terminus. Similar to the recently reported concept of autophagosome-tethering compound (ATTEC), the Q14 peptide tethers mitochondrial-anchored USP30 to autophagosomes by directly binding USP30 and LC3, further accelerating mitophagy [64]. Different from degradation mechanisms like autophagy-targeting chimera/AUTAC or proteolysis-targeting chimera/PROTAC, which function via ubiquitination, the mechanism of action of the Q14 peptide is a more direct and concise strategy for degrading POIs through autophagy [65]. ATTEC is a novel approach that was initially developed to degrade the mutant HTT protein (mHTT) using a small molecule [64]. To our knowledge, our study is the first to report a peptide-based ATTEC approach, which enriches our understanding of the underlying biology and expands the spectrum of applications for the ATTEC strategy.
In summary, our study identified the Q14 peptide, derived from TM domain of USP30, that effectively inhibits the deubiquitinating activity of USP30 and increases mitophagy through two intriguing and distinct mechanisms, providing new insight into the novel design of USP30 inhibitors. We discovered the potential binding site between the Q14 peptide and USP30 and postulated that an allosteric autoinhibition mechanism regulates USP30 activity. Furthermore, the LIR motif in Q14 peptide allows it to bind LC3 and accelerate autophagosome formation. These two intriguing mechanisms synergistically enhance mitophagy, and we believe that they might provide a powerful approach for the development of novel inhibitors against USP30 or other DUBs, opening up new avenues for DUB drug discovery.
Materials and methods
Peptide synthesis
All amino acids and resins used for peptide synthesis were purchased from GL Biochem (Shanghai, China). Other solvents and reagents were purchased from commercial suppliers including Hanhong Chemical (Shanghai, China), Energy Chemical (Shanghai, China) and Tenglong Logistics (Shenzhen, China) and were used without further purification unless otherwise stated. All peptides were synthesized on Rink-amide-MBHA resin (loading capacity: 0.36 mmol/g; HECHENG S&T Ltd UCHL3, HCRAm04-1-1) through standard Fmoc-based solid-phase peptide synthesis (SPPS). The amino acid symbols represent an L-configuration unless otherwise indicated. Peptides were prepared as follows: The resin was swelled in N, N-dimethylformamide (DMF; Energy Chemical, W610493) and bubbled with N2 for 30 min. Next, Fmoc deprotection was performed by morpholine (50% in DMF) for 30 min. The resin was washed sequentially with dichloromethane (DCM; Energy Chemical, W610154) and DMF for 6 times. Then coupling was conducted by adding the mixture of amino acids (5.0 equivalents (eq) according to initial loading of the resin, unnatural amino acids were used as 3 eq), 2-(1 H-6-chlorobenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HCTU; GL Biochem, GLS171201-00706; 4.9 eq) and N, N-diisopropylethylamine (DIPEA; Energy Chemical, W320014; 10.0 eq) dissolved in DMF to resin for 1–2 h, followed by washing the resin with DCM and DMF for 6 times sequentially. Repeat the coupling step until all the amino acid residues were assembled and then dried under a stream of nitrogen for next step. Acetylation labeling was performed on the resin using acetic anhydride:DIPEA:DMF (1:1.5:10, vol:vol:vol) for 1 h. Biotin labeling was performed on the N-terminal of the peptide on the resin. FITC labeling was performed on the resin with the solution of FITC (Energy Chemical, E080600; isomer I, 4.0 eq) and DIPEA (14.0 eq) in DMF overnight. For cleavage of resin, the final resin was deprotected with a mixture of trifluoroacetic acid (TFA), tri-isopropylsilane (TIS), and water (95:2.5:2.5, vol:vol:vol) for 2.5–3 h and dried by nitrogen. The crude linear peptides were then precipitated with hexane:diethyl ether (1:2 vol:vol) at 4°C, isolated by centrifugation in 13,523 g for 5 min and dissolved in water:acetonitrile (2:1, vol:vol) for the next purification. For sulfonium tethered peptide synthesis, the TRT (trityl) protecting group of cysteine were removed by treating the resin with a solution of TFA:TIS:H2O (3:5:92, vol/vol/vol) for 5 × 10 min, and washed with DCM. On-resin cysteine alkylation was conducted with linkers (2.0 eq) and DIPEA (4.0 eq) in DMF for 3 h with N2 bubbling. The cyclization was finished when the peptide was cleaved from resin treated with TFA:TIS:H2O (95:2.5:2.5) for 2 h. Purification was performed by RP-HPLC (Shimadzu [Kyoto, Japan] and Agilent [Santa Clara, CA, USA] Zorbax SB-Aq: 4.6 × 250 mm, 220 and 254 nm, flow rate 1.0 mL/min) and confirmed by Shimadzu LC-MS 2020 mass spectrometer equipped with Agilent Zorbax SB-Aq column. LC-MS spectra were carried out on SHIMAZU LC-MS 8030 (ESI-MS) in positive ion mode (Data S1). Peptides were quantified by their UV absorbance at 280 nm or 494 nm according to amino acid coefficient contribution.
Antibodies and chemicals
The following primary antibodies were used in assays: anti-ubiquitin (Cell Signaling Technology, 3936S; 1:1000), anti-CYCS/cytochrome c (Proteintech, 10,993-1-AP; 1:1000), anti-USP30 (LSBio, LS-C353592, 1:200; Santa Cruz Biotechnology, sc-515,235, 1:500), anti-GAPDH (Cell Signaling Technology, 2118S; 1:4000), anti-LC3B (Sigma-Aldrich, L7543; 1:1000), anti-TOMM20 (Cell Signaling Technology, 42406S; 1:1000), anti-TIMM23 (Proteintech, 11,123-1-AP; 1:1000), anti-MFN1 (Abcam, ab57602; 1:500), anti-MFN2 (Abcam, ab56889; 1:500), anti-TOMM22 (Abcam, ab57523; 1:1000), anti-OPA1 (Santa Cruz Biotechnology, sc-393,296; 1:500), anti-OPA1 (Cell Signaling Technology, 67589S; 1:1000), anti-DNM1L/Drp1 (Santa Cruz Biotechnology, sc-271,583; 1:500), anti-DNM2/dynamin 2 (Abcam, ab65556; 1:1000). Secondary antibodies for immunoblotting were: anti-mouse IgG, HRP-linked antibody (Cell Signaling Technology, 7076S; 1:2000), anti-rabbit IgG, HRP-linked antibody (Cell Signaling Technology, 7074S; 1:5000), m-IgGκ BP-HRP (Santa Cruz Biotechnology, sc-516,102; 1:1000). Secondary antibodies used in immunofluorescence assay were: Alexa Fluor® 594 AffiniPure Goat Anti-Rabbit IgG (H + L) (Jackson ImmunoResearch, 111–585-003; 1:300), Alexa Fluor® 488 AffiniPure F(ab’)₂ Fragment Goat Anti-Rabbit IgG (H + L) (Jackson ImmunoResearch, 111–546-003; 1:300). Small-molecular compounds such as carbonyl cyanide 3-chlorophenylhydrazone (CCCP; Sigma-Aldrich, C2759), hydrochloroquine (HCQ; MCE, HY-B1370), 3-methyladenine (3-MA; MCE, HY-19312), bafilomycin A1 (BafA1; MCE, HY-100558), rapamycin (Selleck, S1039), MitoTracker Red (Thermo Fisher Scientific, M22425), Fluoroshield with DAPI (Sigma-Aldrich, F6057), and Protease Inhibitor Cocktail (Roche, 04693124001) were purchased from the indicated suppliers.
Cloning, expression and purification of USP30
The cDNA coding for human USP30 was purchased from Sangon Biotech Co. Ltd (G110428). A construct lacking TM domain was inserted into a pGEX-6p-2 vector (GE Healthcare, 28–9546-50) and verified by DNA sequencing from Sangon Biotech Co. Ltd. pGEX-6p-2-USP30 was transformed into Escherichia coli Rosetta (DE3) competent cells (Solarbio, C1420) and incubated at 37°C with aeration until an OD600 0.5–0.6 was reached. Protein expression was induced by addition of 0.5 mM sterile isopropyl-β-D-thiogalactopyranoside/IPTG (Merck, 420,322) to the culture at 18°C for 18 h. Cells were harvested by centrifugation at 3500 g at 4°C for 20 min. Cell pellets were resuspended in PBS (pH 7.3; Solarbio, P1010) with protease inhibitor phenylmethanesulfonyl fluoride (PMSF; Sangon Biotech Co. Ltd, A100754). Cells were lysed by sonication on ice for 100 cycles of 3 s on 4 s off, until the lysate was clear. The lysate was centrifuged at 13,000 g for 30 min at 4°C. The supernatant was decanted and filtered through a 0.22-μm Amicon syringe filter (Millipore, SLGP033RB). After loaded to a GSTrap HP Column (GE Healthcare, 17,528,201) by pumping it onto the column, the column was washed with PBS buffer for 10-volumes and then eluted with elution buffer (50 mM Tris [Sigma-Aldrich, T1503], pH 8.0, 10 mM GSH [Sangon Biotech Co. Ltd, A600229]). The fractions were collected, concentrated and further purified by Superdex 200 (GE Healthcare, 28–9909-44) molecular sieve chromatography in 50 mM HEPES (Sigma-Aldrich, H3375), pH 7.2, 150 mM NaCl (Sigma-Aldrich, S9888), 2 mM DTT (Sigma-Aldrich, 43,815). All the purified proteins were aliquoted and stored at −80°C until used.
AMC cleavage assay
The enzyme activity assays followed a protocol described previously [66,67]. The assays were carried out in a flat-bottom, low-volume 384-well plate (Corning, 3573) in a 30-µL reaction. Inhibition assays were performed in assay buffer (50 mM HEPES, pH 7.5, 0.01% Tween 20 [v:v; Sigma-Aldrich, P7949], 10 mM DTT) by measuring the release of fluorogenic substrate Ub-7-amido-4-methylcoumarin (Ub-AMC; Boston Biochem, U-550). USP30 (15 nM) and serial dilutions of peptides were freshly mixed in assay buffer and incubated at room temperature for 30 min prior to the addition of Ub-AMC. The fluorescence intensities were measured for 30 min by plate reader (Perkin Elmer, Envision, 2104 multilabel reader) with excitation at 340 nm and emission at 450 nm. IC50, defined as the concentration of peptide that inhibits 50% of USP30 activity (non-inhibited USP30 corresponds to 100%) was fitted using the GraphPad Prism 8.0. For kcat:KM determination, a fixed concentration of USP30 was incubated with indicated concentrations of Ub-AMC at room temperature in assay buffer (50 mM HEPES, pH 7.5, 0.01% Tween 20 [v:v], 10 mM DTT). The kinetic parameters determination followed a protocol described previously [66]. The initial reaction velocity (nM:s) was measured at each substrate concentration through determining the linear slope from plotting fluorescence intensity vs. time. The KM and Vmax values were determined by plotting the velocity vs. substrate concentration and using the Michaelis–Menten equation, V0 = Vmax*[S]:(KM + [S]) in GraphPad Prism 8.0. kcat was calculated from the equation kcat = Vmax:[E]o, where [E]o is the total enzyme concentration. For velocity vs. substrate plots that could not be fitted using the Michaelis–Menten equation but could be fitted using a linear line, the kcat:KM value was estimated by dividing the linear slope (nMs−1M−1)by[E]o.
Ubiquitin cleavage assay
The ubiquitin cleavage assays followed a protocol described previously [18]. The Lys 6 ub tetramers (Boston Biochem, UC-15) and USP30 protein (Boston Biochem, E-582) were purchased from Boston Biochem. The ubiquitin cleavage reactions were carried out at 37°C in assay buffer containing with 50 mM HEPES, pH 7.5, 0.01% Tween 20 (v:v), 10 mM DTT, 5 mM MgCl2 (Sigma-Aldrich, M8266). After incubating the 200 nM USP30 protein with indicated concentrations of peptide at room temperature for 30 min, 200 ng Lys 6 ub tetramers was added into the reaction and incubated them at 37°C for 40 min, then the reaction was stopped by adding 4× NuPAGETM LDS Sample Buffer (Thermo Fisher Scientific, NP0007). The samples were loaded onto 4%-12% Bis-Tris NuPAGETM gels (Thermo Fisher Scientific, NP0335BOX) with NuPAGETM MES running buffer (Thermo Fisher Scientific, NP0002) and then silver stained using Protein Stains Kit (Sangon Biotech, C500021). Gels were imaged by the ChemiDoc MP imaging system (Bio-Rad).
Cell culture and treatment
A172 cells were obtained from American Type Culture Collection (CRL-1620) and cultured in Dulbecco’s modified Eagle’s medium (Gibco, 11,995,040) supplemented with 10% (v:v) fetal bovine serum (Hyclone, SH30084.03), in a 5% CO2 incubator at 37°C. Peptide stock solutions were prepared in DMSO (Sigma-Aldrich, D2438) and stored at −20°C and further diluted with the culture media to obtain an optimal range of concentrations for treatments.
Ubiquitinated mitochondria cleavage assay
The ubiquitinated mitochondria cleavage assay followed a protocol described previously [18]. The A172 cells were incubated with 10 µM CCCP or an equivalent of DMSO for 2 h before harvesting. Mitochondria were isolated using the Cell Mitochondria Isolation Kit (Beyotime, C3601) according to the protocol. The isolated mitochondrial pellet was resuspended in PBS buffer with 1 mM DTT for next reaction. Indicated amounts of USP30 and peptide were added into the aliquots of mitochondrial isolated from different condition. Then the reaction was incubated at 37°C with shaking for 5 h at 20 g to prevent the mitochondria from settling down. Then the reaction was stopped by adding 5× SDS Loading Buffer (Sangon Biotech Co. Ltd, C516031) into the tube and used for Western blotting analysis.
Western blotting analysis
A172 cells were seeded in 6-well plates. Peptides were dissolved in 5% Dulbecco’s modified Eagle’s medium containing minimum DMSO. At 60% confluence, the cells were incubated with peptide in the condition as mentioned in article. Cells were rinsed with PBS and harvested using the lysis buffer (50 mM Tris, pH 6.8, 2% SDS [Sigma-Aldrich, L3771], 6% glycerol [Sangon Biotech Co. Ltd, A600232], 1% β-mercaptoethanol [Solarbio, M8210], 0.004% bromophenol blue [Sangon Biotech Co. Ltd, A602230], protease inhibitor cocktail [Roche, 04693124001]). Denatured cellular extracts were resolved by 10% SDS–polyacrylamide gels. Protein bands in the gel were then transferred to nitrocellulose blotting membranes (Pall, 66,485) and blocked in 5% skim milk (BD, 232,100) in TBST (20 mM Tris, pH 8.0, 150 mM NaCl, 0.05% Tween 20 [v:v]) for 1 h at room temperature. Membranes were incubated with the primary antibody overnight at 4°C and then washed with TBST buffer. The HRP-linked secondary antibody diluted with 3% BSA (Sangon Biotech Co. Ltd, A600332) was used for secondary incubation for 1 h at room temperature. After washing with TBST, protein bands were visualized with ECL Blotting Detection Reagents (Thermo Fisher Scientific, 32,106) and ChemiDoc MP imaging system (Bio-Rad).
Fluorescence polarization
Fluorescence polarization experiments were carried out in a flat-bottom, low-volume 384-well plate (Corning, 3573) in a 30-μL reaction. Concentrations of the peptides were determined by 494 nm absorption of FITC. The FITC-labeled peptide was diluted with the assay buffer (50 mM HEPES, pH 7.5, 0.01% Tween 20 [v:v], 10 mM DTT) containing minimum DMSO before the labeled peptide and protein were added sequentially. Purified protein was serially diluted into a solution of FITC-labeled peptides (10 nM) in assay buffer (50 mM HEPES, pH 7.5, 0.01% Tween 20 [v:v], 10 mM DTT). Each experiment was run in triplicate and the fluorescence polarization of the labeled peptides was measured at 25°C using plate reader (Perkin Elmer, Envision) with excitation at 485 nm and emission at 520 nm and then plotted against the concentrations of the protein. The binding affinity (Kd) values were determined by fitting the data using Graphpad Prism 8.0.
Microscale thermophoresis (MST) assay
MST assay was performed using a Monolith NT.115 instrument (NanoTemper Technologies). Concentrations of the peptides were determined by 494 nm absorption of FITC. The FITC-labeled peptide was diluted with the assay buffer (50 mM HEPES, pH 7.5, 0.01% Tween 20 [v:v], 10 mM DTT) containing minimum DMSO before the labeled peptide and protein were added sequentially. Purified protein was serially diluted into a solution of FITC-labeled peptides (10 nM) in assay buffer (50 mM HEPES, pH 7.5, 0.01% Tween 20 [v:v], 10 mM DTT). Samples were loaded into Monolith NT.115 Capillaries (NanoTemper Technologies) and MST measurements were performed using medium MST power and 100% LED power at 25°C. Each experiment was repeated three times. Data analyses were performed using Nanotemper analysis software.
Streptavidin (SA) affinity-isolation assay
For cell lysate Streptavidin (SA) affinity-isolation assay, A172 cells were lysed in lysis buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 0.5% [v:v] NP-40 [Sangon Biotech Co. Ltd, A100109], protease inhibitor cocktail) for 1 h on ice. The cell lysates were cleared by centrifugation at 14,000 g at 4°C for 20 min. Then, the protein concentration of the supernatants was measured by BCA protein assay and BSA as standard. Approximately 500 μg of cell lysates were incubated with biotin-labeled peptide (25 μM) for overnight at 4°C on a vertical roller. For cellular SA-affinity-isolation assay, A172 cells were pretreated with biotin-labeled peptide for the indicated condition and lysed in lysis buffer with concentration determination for the following incubation. Then the mixture was incubated with Streptavidin Magnetic Beads (Thermo Fisher Scientific, 20,359) at 4°C for 4 h. The beads were collected and washed 3 times with lysis buffer. The proteins were eluted by boiling in SDS buffer, and subjected to western analysis.
Cellular thermal shift assay (CETSA)
Cellular thermal shift assay (CETSA) was carried out according to the protocol as previously described [38]. For cell lysate CETSA assay, A172 cells were harvested and washed with PBS. Then the cells were diluted in PBS with complete protease inhibitor cocktail. The cell suspensions were put through a series of freeze-thaw cycles using liquid nitrogen. The cell lysates were isolated from the cell debris by centrifugation at 20,000 g for 20 min at 4°C. The cell lysates were divided into two aliquots and treated with peptide or DMSO separately. After incubation overnight at 4°C, the respective lysates were divided into smaller aliquots (40 µL) and heated separately at indicated temperatures for 3 min followed by cooling for 3 min at 25°C (T100 Thermal Cycler, Bio-rad). After centrifugation at 20,000 g for 20 min at 4°C, the soluble fractions were separated from precipitates and transferred to new microtubes. Then the supernatants were analyzed by Western blotting. For the intact cellular thermal shift assay, A172 cells were incubated with peptide (100 µM) or an equal amount of DMSO for 12 h. Then cells were collected by trypsin digestion and PBS washing. The treated cells were diluted in PBS with complete protease inhibitor cocktail, divided into aliquots and heated as described above. Then the cells were lysed by several freeze-thaw cycles with liquid nitrogen and centrifuged to isolated the soluble fractions. The supernatants were analyzed by Western blot as mentioned above. Blots were quantified using image-J and Graphpad Prism 8.0.
Flow cytometric analysis
A172 cells were seeded overnight in a 24-well culture plate. Then the cells were incubated with FITC-labeled peptides at a concentration of 5 μM in the DMEM with 5% FBS for 4 h at 37°C. After washing with PBS, the cells were exposed to trypsin (0.25%; Gibco, 25,200) digestion (3 min, 37°C). After washing with PBS and 0.05% trypan blue (Sigma-Aldrich, T8154) incubation for 5 min, the cellular fluorescence was analyzed by a BD FACS Calibur flow cytometer (Becton Dickinson) and CellQuest Pro.
Mitochondrial mass was determined by flow cytometry analysis using Nonyl Acridine Orange (NAO; Invitrogen, A-1372). A172 cells were seeded in 24-well plates. Peptides were dissolved in 5% Dulbecco’s modified Eagle’s medium containing minimum DMSO. At 60% confluence, the cells were incubated with peptide in the condition as mentioned in article. After incubation with 2.5 μM NAO for 30 min, the cells were washed with PBS buffer twice and collected for flow cytometry analysis (Attune NxT, Thermofisher Scientific). Ten thousand cells were aquired using channels at the following wavelengths: 488 nm (Ex) and 530 nm (Em).
Mitophagy was evaluated by fluorometry using a Mitophagy Detection Kit according to the manufacturer (Dojindo, MD01). A172 cells were incubated with Mtphagy Dye at a working concentration of 100 nM for 30 min under culture condition. After the washing of the cells with PBS buffer twice, the culture medium containing peptide was added to the well for 24 h incubation. After removing the supernatant, 1 μM Lyso Dye working solution were added to the cells and incubated for 30 min. The cells were washed with PBS buffer twice and collected for flow cytometry analysis (Attune NxT, Thermofisher Scientific). Ten thousand cells were acquired using channels at the following wavelengths: Mtphagy Dye: YL3, 561 nm (Ex) and 695 nm (Em); Lyso Dye: VL2, 405 nm (Ex) and 512 nm (Em).
Immunofluorescence
For colocalization assay, A172 cells were seeded overnight on coverslips in 24-well plates, and then treated with FITC-labeled peptides (5 μM) in DMEM for 4 h at 37°C. Then the cells were digested by trypsin and centrifuged for 3 min at a speed of 233 g. Then incubation with trypan blue (0.05%) for 5 min was used to remove peptides nonspecifically bound on cell surface. After centrifugation for 3 min at 233 g, cells were washed with PBS two times. The cells were re-seeded overnight on coverslips in 24-well plates. After cell attachment, the media containing the peptides was removed followed by washing with PBS three times. The cells were then fixed with 4% paraformaldehyde solution for 15 min followed by washing with PBS three times. After fixation, the cells were treated with 0.25% Triton X-100 (Sigma-Aldrich, T9284) for 15 min followed by being blocked in freshly prepared 3% BSA in TBST for 1 h at room temperature and washed by PBS. Then the cells were incubated with anti-USP30 antibody in 3% BSA in TBST solution at 4°C overnight. The cells were then washed with PBS and were incubated with Alexa Fluor® 594 AffiniPure Goat Anti-Rabbit IgG (H + L) antibody at a dilution rate of 1:400 for 1 h at room temperature. Then coverslips were mounted upside down on slides where previously mounted with Fluoroshield with DAPI. Images were taken on Nikon A1R confocal microscopy under the same parameters and images acquired were further analyzed by imageJ or NIS-Elements Viewer.
For mitochondrial morphology determination or mitochondrial colocalization determination, MitoTracker Red staining was used after peptide treatment as mentioned. MitoTracker Red (50 nM) was added into the cells for 30 min staining. After incubation, the media containing the peptides was removed followed by washing with PBS three times. The cells were then fixed with 4% paraformaldehyde solution for 15 min followed by washing with PBS three times. Then coverslips were mounted upside down on slides where previously mounted with Fluoroshield with DAPI. Images were taken on Nikon A1R confocal microscopy under the same parameters and images acquired were further analyzed by image-J or NIS-Elements Viewer.
For endogenous LC3 puncta measurement, standard immunofluorescence protocol was carried out with anti-LC3 antibody. The LC3 puncta in each sample was counted from at least 300 cells from randomly placed positions within each sample using image-J. The intensities of LC3 puncta were analyzed by Graphpad Prism 8.0.
Photoactivation
For quantification of the mitochondrial fusion rate, A172 cells seeded onto a µ-Slide 8-well chambered coverslip (ibidi, 80,826) were transfected with mtPA-GFP for confocal microscopy. After 24 h, cells were treated as indicated and stained with 50 nM MitoTracker Red for 30 min before imaging. Photoactivation on mitochondria expressing mtPA-GFP (Addgene, 23,348; deposited by Richard Youle) was performed in selected regions of interest using the 405 nm laser with 3% power and 200 iterations by Plan-Apochromat 63x/1.4 Oil DIC objective on the Zeiss LSM980. Images were acquired using 488 nm (mtPA-GFP) and 543 nm (MitoTracker Red) laser for 10 min. The mtPA-GFP fluorescence in ROIs were generated by ZEN software, normalized to the intensity of photoactivated time point and plotted against time. Each experimental group contains at least 10 cells (mean ± s.d.).
Co-immunoprecipitation
For exogenous co-IP, human A172 cells were seeded in 10-cm cell culture dishes. At 60% confluence, the cells were incubated with peptides or DMSO for 24 h at 37°C. Then, the cells were washed twice with ice-cold PBS and lysed in buffer containing 50 mM Tris, pH 7.5, 150 mM sodium chloride, 0.5% (v:v) NP-40, Roche protease inhibitors cocktail for 1 h at 4°C. Cell lysates were cleared by centrifugation at 13,523 g at 4°C for 20 min. Then, the protein concentration of the supernatants was measured by BCA protein assay and BSA as standard. Approximately 1 mg of cell lysates was incubated with 5 μg of anti-TOMM20 overnight at 4°C on a vertical roller. Protein A Sepharose (GE Healthcare, 17–5280-01) was added to the lysates for another 4 h. Beads coated with protein were immunoprecipitated, washed five times with lysis buffer, and applied to 12% SDS–polyacrylamide gels for Western blotting analysis.
mtDNA content determination
A172 cells were seeded in 12-well plates. Peptides were dissolved in 5% Dulbecco’s modified Eagle’s medium containing minimum DMSO. At 60% confluence, the cells were incubated with peptide in the condition as mentioned in article. Genomic DNA used for the analysis of mtDNA content was isolated from A172 cells using TaKaRa MiniBEST Universal Genomic DNA Extraction kit (TaKaRa Bio Inc., 9765). The isolated DNA was evaluated and quantified by a NanoDrop™ 2000/2000 c Spectrophotometers (Thermo Fisher Scientific). The relative mtDNA copy number was determined by qPCR with primers for the mitochondrial MT-RNR2/16S rRNA gene and the housekeeper gene. PCR amplification was performed with real time PCR using SYBR green (Takara, RR420A) in Bio-Rad real-time PCR system. The relative changes for each sample were determined by normalizing to the housekeeper gene levels. Data represent three independent experiments with three technical replicates per experiment.
Primers used were:
MT-RNR2/16S rRNA, forward, 5ʹ-GGTGCAGCCGCTATTAAAGG-3ʹ;
MT-RNR2/16S rRNA, reverse, 5ʹ-ATCATTTACGGGGGAAGGCG-3ʹ;
ACTB, forward, 5ʹ-TGACGTGGACATCCGCAAAG-3ʹ;
ACTB, reverse, 5ʹ-CTGGAAGGTGGACAGCGAGG-3ʹ.
GAPDH, forward, 5ʹ-GGAGCGAGATCCCTCCAAAAT-3ʹ
GAPDH, reverse, 5ʹ-GGCTGTTGTCATACTTCTCATGG-3ʹ
GAPDH-1, forward, 5ʹ-GGAGTTCAACAATGCTATCAGC-3ʹ
GAPDH-1, reverse, 5ʹ-CTTCTGGTAGGTGTGTAGGATC-3ʹ
Measurement of ATP content
The ATP content determination was performed by the Enhanced ATP Assay Kit (Beyotime, S0027) and carried out according to the protocol. A172 cells were seeded in 24-well plate. Peptides were dissolved in 5% Dulbecco’s modified Eagle’s medium containing minimum DMSO. At 60% confluence, the cells were incubated with peptide in the condition as mentioned in article. Cells were rinsed with PBS and harvested using the lysis buffer. The luminescence was measured by plate reader (Perkin Elmer, Envision). The ATP content was calculated from a standard curve for a series of ATP concentrations.
LDH release assay
The LDH release assay was used to detect the cell membrane damage and performed by the LDH release kit (Dojindo). A172 cells were seeded in 96-well plates at a density of 4000 cells per well. Then the cells were treated with 100 μL peptides with 2-fold serial dilution starting at 100 μM for 4 h at 37°C. Lysis buffer was added as positive control and incubated at 37°C for 30 min. Then 100 μL working solutions were added to each well and incubated for 30 min at room temperature. The reaction was stopped by 50 μL stop solutions and detected immediately at 490 nm absorption by a microplate reader (Perkin Elmer, Envision). The LDH release activity was calculated by [(ODsamples-ODblank)/ (ODpositive-ODblank)] × 100.
Cell viability assay
Cell viability was measured by MTT (3-[4, 5-dimethylthiazol-2-yl]-2, 5-diphenylt-etrazolium bromide; Sigma-Aldrich, M2003) assays. Briefly, A172 cells were seeded in 96-well plates at 4000 cells per well in medium supplemented with 10% FBS. Then the cells were treated with DMSO or peptides at the concentrations indicated in the relevant figures for 24 h. MTT (5 mg/mL, 20 μL) in PBS was added and the cells were incubated for another 4 h at 37°C with 5% CO2. DMSO (150 μL) was then added to solubilize the precipitate with 10 min of gentle shaking. Absorbance was measured with a microplate reader (Biotek H1) at 490 nm.
In-gel trypsin digestion assay
In-gel trypsin digestion assay was carried out according to the previously published protocol [56,68]. The protein bands of interest were excised after electrophoresis and cut bands into pieces for destaining. Then the gels were destained by treating the gel pieces with a solution of ammonium bicarbonate:acetonitrile and incubated for 30 min at 37°C. Digested protein fragments were further extracted from gel with 0.5% formic acid:50% acetonitrile and incubated for 15 min and combined with trypsin digestion supernatant and dried for 5–10 min. The treated samples were used for MS/MS analysis. The pLink2 software was used for searching the linkage between peptide and proteins [69].
Statistical analysis
No statistical methods were used to predetermine sample size. For all experiments, the number of replicates (n), mean value, error value, and P value cutoffs are described in the respective figure legends. Error bars are shown for all data points with replicates as a measure of variation with the group. Statistical analysis including one-way ANOVA multiple comparisons was carried out using GraphPad Prism 8.0.
Supplementary Material
Acknowledgments
We thank staff of ShenZhen University for assistance in Mass/mass spectrometry analysis. The USP2, USP7, and USP8 proteins are kind gifts from Prof. Hao Huang, Peking University Shenzhen Graduate School. This work is supported by High-Performance Computing Platform of Peking University.
Funding Statement
This work was supported by the China Postdoctoral Science Foundation [2021M690220]; Natural Science Foundation of China [21778009]; Natural Science Foundation of China [21977010]; Natural Science Foundation of Guangdong Province [2020A1515010522]; Natural Science Foundation of Guangdong Province [2019A1515111184]; National Key Research and Development Program Synthetic Biology Key Special Project of China [2018YFA0902504]; Shenzhen Science and Technology Innovation Committee [JCYJ20170817161908969]; Shenzhen Science and Technology Innovation Committee [JCYJ20180507181527112]; Shenzhen Science and Technology Innovation Committee [JCYJ20180508152213145]; Shenzhen Science and Technology Innovation Committee [JCYJ20170817172023838]; Beijing National Laboratory of Molecular Science open grant [BNLMS20160112]; Shenzhen-Hong Kong Institute of Brain Science-Shenzhen Fundamental Research Institutions grant [2019SHIBS0004]; Natural Science Foundation of Guangdong Province [2019A1515110487]; Shenzhen Science and Technology Innovation Committee [RCJC20200714114433053].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Nunnari J, Suomalainen A.. Mitochondria: in sickness and in health. Cell. 2012;148(6):1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337(6098):1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Saiki S, Sato S, Hattori N. Molecular pathogenesis of Parkinson’s disease: update. J Neurol Neurosurg Psychiatry. 2012;83(4):430–436. [DOI] [PubMed] [Google Scholar]
- [4].Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol. 2018;20(9):1013–1022. [DOI] [PubMed] [Google Scholar]
- [5].Liu L, Feng D, Chen G, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14(2):177–185. [DOI] [PubMed] [Google Scholar]
- [6].Novak I, Kirkin V, McEwan DG, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11(1):45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hanna RA, Quinsay MN, Orogo AM, et al. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. 2012;287(23):19094–19104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chu CT, Ji J, Dagda RK, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15(10):1197–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sentelle RD, Senkal CE, Jiang W, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol. 2012;8(10):831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19(6):349–364. [DOI] [PubMed] [Google Scholar]
- [11].Harper JW, Ordureau A, Heo JM. Building and decoding ubiquitin chains for mitophagy. Nat Rev Mol Cell Biol. 2018;19(2):93–108. [DOI] [PubMed] [Google Scholar]
- [12].Koyano F, Okatsu K, Kosako H, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510(7503):162–166. [DOI] [PubMed] [Google Scholar]
- [13].Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85(2):257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304(5674):1158–1160. [DOI] [PubMed] [Google Scholar]
- [15].Mevissen TET, Komander D. Mechanisms of deubiquitinase specificity and regulation. Annu Rev Biochem. 2017;86(1):159–192. [DOI] [PubMed] [Google Scholar]
- [16].Clague MJ, Urbe S, Komander D. Breaking the chains: deubiquitylating enzyme specificity begets function. Nat Rev Mol Cell Biol. 2019;20(6):338–352. [DOI] [PubMed] [Google Scholar]
- [17].Bingol B, Tea JS, Phu L, et al. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. 2014;510(7505):370–375. [DOI] [PubMed] [Google Scholar]
- [18].Cunningham CN, Baughman JM, Phu L, et al. USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat Cell Biol. 2015;17(2):160–169. [DOI] [PubMed] [Google Scholar]
- [19].Gersch M, Gladkova C, Schubert AF, et al. Mechanism and regulation of the Lys6-selective deubiquitinase USP30. Nat Struct Mol Biol. 2017;24(11):920–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sato Y, Okatsu K, Saeki Y, et al. Structural basis for specific cleavage of Lys6-linked polyubiquitin chains by USP30. Nat Struct Mol Biol. 2017;24(11):911–919. [DOI] [PubMed] [Google Scholar]
- [21].Yue W, Chen Z, Liu H, et al. A small natural molecule promotes mitochondrial fusion through inhibition of the deubiquitinase USP30. Cell Res. 2014;24(4):482–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rusilowicz-Jones EV, Jardine J, Kallinos A, et al. USP30 sets a trigger threshold for PINK1–PARKIN amplification of mitochondrial ubiquitylation. Life Sci Alliance. 2020;3(8):e202000768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bingol B, Corn J, Zhang Y, inventors . USP30 inhibitors and methods of use. International Patent WO2014041111 A1. 2014. Mar 20.
- [24].Kluge AF, Lagu BR, Maiti P, et al. Novel highly selective inhibitors of ubiquitin specific protease 30 (USP30) accelerate mitophagy. Bioorg Med Chem Lett. 2018;28(15):2655–2659. [DOI] [PubMed] [Google Scholar]
- [25].Lee JG, Kim W, Gygi S, et al. Characterization of the deubiquitinating activity of USP19 and its role in endoplasmic reticulum-associated degradation. J Biol Chem. 2014;289(6):3510–3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Nussinov R, Tsai CJ, Jang H. Autoinhibition can identify rare driver mutations and advise pharmacology. FASEB J. 2020;34(1):16–29. [DOI] [PubMed] [Google Scholar]
- [27].Wiesner S, Ogunjimi AA, Wang HR, et al. Autoinhibition of the HECT-type ubiquitin ligase Smurf2 through its C2 domain. Cell. 2007;130(4):651–662. [DOI] [PubMed] [Google Scholar]
- [28].Mari S, Ruetalo N, Maspero E, et al. Structural and functional framework for the autoinhibition of Nedd4-family ubiquitin ligases. Structure. 2014;22(11):1639–1649. [DOI] [PubMed] [Google Scholar]
- [29].Wauer T, Komander D. Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J. 2013;32(15):2099–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Faesen AC, Dirac AM, Shanmugham A, et al. Mechanism of USP7/HAUSP activation by its C-terminal ubiquitin-like domain and allosteric regulation by GMP-synthetase. Mol Cell. 2011;44(1):147–159. [DOI] [PubMed] [Google Scholar]
- [31].Avvakumov GV, Walker JR, Xue S, et al. Amino-terminal dimerization, NRDP1-rhodanese interaction, and inhibited catalytic domain conformation of the ubiquitin-specific protease 8 (USP8). J Biol Chem. 2006;281(49):38061–38070. [DOI] [PubMed] [Google Scholar]
- [32].Hu M, Li P, Song L, et al. Structure and mechanisms of the proteasome-associated deubiquitinating enzyme USP14. EMBO J. 2005;24(21):3747–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Liu B, Sureda-Gómez M, Zhen Y, et al. A quaternary tetramer assembly inhibits the deubiquitinating activity of USP25. Nat Commun. 2018;9(1):4973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sauer F, Klemm T, Kollampally RB, et al. Differential oligomerization of the deubiquitinases USP25 and USP28 regulates their activities. Mol Cell. 2019;74(3):421–435. [DOI] [PubMed] [Google Scholar]
- [35].Gersch M, Wagstaff JL, Toms AV, et al. Distinct USP25 and USP28 oligomerization states regulate deubiquitinating activity. Mol Cell. 2019;74(3):436–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Misaghi S, Galardy PJ, Meester WJ, et al. Structure of the ubiquitin hydrolase UCH-L3 complexed with a suicide substrate. J Biol Chem. 2005;280(2):1512–1520. [DOI] [PubMed] [Google Scholar]
- [37].Nakamura N, Hirose S. Regulation of mitochondrial morphology by USP30, a deubiquitinating enzyme present in the mitochondrial outer membrane. Mol Biol Cell. 2008;19(5):1903–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Martinez Molina D, Jafari R, Ignatushchenko M, et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;341(6141):84–87. [DOI] [PubMed] [Google Scholar]
- [39].Patel LN, Zaro JL, Shen WC. Cell penetrating peptides: intracellular pathways and pharmaceutical perspectives. Pharm Res. 2007;24(11):1977–1992. [DOI] [PubMed] [Google Scholar]
- [40].Gomes LC, Scorrano L. Mitochondrial morphology in mitophagy and macroautophagy. Biochim Biophys Acta. 2013;1833(1):205–212. [DOI] [PubMed] [Google Scholar]
- [41].Twig G, Elorza A, Molina AJ, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27(2):433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Meyer JN, Leuthner TC, Luz AL. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology. 2017;391:42–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jane EP, Premkumar DR, Rajasundaram D, et al. Reversing tozasertib resistance in glioma through inhibition of pyruvate dehydrogenase kinases. Mol Oncol. 2021. DOI: 10.1002/1878-0261.13025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Erbrich U, Septinus M, Naujok A, et al. Hydrophobic acridine dyes for fluorescence staining of mitochondria in living cells. 2. Comparison of staining of living and fixed Hela-cells with NAO and DPPAO. Histochemistry. 1984;80(4):385–388. [DOI] [PubMed] [Google Scholar]
- [46].Petit JM, Maftah A, Ratinaud MH, et al. 10N-nonyl acridine Orange interacts with cardiolipin and allows the quantification of this phospholipid in isolated mitochondria. Eur J Biochem. 1992;209(1):267–273. [DOI] [PubMed] [Google Scholar]
- [47].Nechiporuk T, Kurtz SE, Nikolova O, et al. The TP53 apoptotic network is a primary mediator of resistance to BCL2 inhibition in AML cells. Cancer Discov. 2019;9(7):910–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Iwashita H, Torii S, Nagahora N, et al. Live Cell Imaging of Mitochondrial Autophagy with a Novel Fluorescent Small Molecule. ACS Chem Biol. 2017;12(10):2546–2551. [DOI] [PubMed] [Google Scholar]
- [49].Pryde KR, Smith HL, Chau K-Y, et al. PINK1 disables the anti-fission machinery to segregate damaged mitochondria for mitophagy. J Cell Biol. 2016;213(2):163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. 2012;46(1):265–287. [DOI] [PubMed] [Google Scholar]
- [51].Lee JE, Westrate LM, Wu H, et al. Multiple dynamin family members collaborate to drive mitochondrial division. Nature. 2016;540(7631):139–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lovy A, Molina AJ, Cerqueira FM, et al. A faster, high resolution, mtPA-GFP-based mitochondrial fusion assay acquiring kinetic data of multiple cells in parallel using confocal microscopy. J Vis Exp. 2012;65:e3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Flaxman HA, Chang C-F, Wu H-Y, et al. A binding site hotspot map of the FKBP12–Rapamycin–FRB ternary complex by photoaffinity labeling and mass spectrometry-based proteomics. J Am Chem Soc. 2019;141(30):11759–11764. [DOI] [PubMed] [Google Scholar]
- [54].Ando J, Asanuma M, Dodo K, et al. Alkyne-tag SERS screening and identification of small-molecule-binding sites in protein. J Am Chem Soc. 2016;138(42):13901–13910. [DOI] [PubMed] [Google Scholar]
- [55].Back JW, de Jong L, Muijsers AO, et al. Chemical cross-linking and mass spectrometry for protein structural modeling. J Mol Biol. 2003;331(2):303–313. [DOI] [PubMed] [Google Scholar]
- [56].Wang D, Yu M, Liu N, et al. A sulfonium tethered peptide ligand rapidly and selectively modifies protein cysteine in vicinity. Chem Sci. 2019;10(19):4966–4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tamura T, Hamachi I. Chemistry for covalent modification of endogenous/native proteins: from test tubes to complex biological systems. J Am Chem Soc. 2019;141(7):2782–2799. [DOI] [PubMed] [Google Scholar]
- [58].Hu M, Li P, Li M, et al. Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell. 2002;111(7):1041–1054. [DOI] [PubMed] [Google Scholar]
- [59].Xu Y, Wang S, Hu Q, et al. CavityPlus: a web server for protein cavity detection with pharmacophore modelling, allosteric site identification and covalent ligand binding ability prediction. Nucleic Acids Res. 2018;46(W1):W374–W379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Rouge L, Bainbridge TW, Kwok M, et al. Molecular understanding of USP7 substrate recognition and C-terminal activation. Structure. 2016;24(8):1335–1345. [DOI] [PubMed] [Google Scholar]
- [61].Clerici M, Luna-Vargas MP, Faesen AC, et al. The DUSP-Ubl domain of USP4 enhances its catalytic efficiency by promoting ubiquitin exchange. Nat Commun. 2014;5:5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Wang Y, Jiang Y, Ding S, et al. Small molecule inhibitors reveal allosteric regulation of USP14 via steric blockade. Cell Res. 2018;28(12):1186–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Turnbull AP, Ioannidis S, Krajewski WW, et al. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature. 2017;550(7677):481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Li Z, Wang C, Wang Z, et al. Allele-selective lowering of mutant HTT protein by HTT–LC3 linker compounds. Nature. 2019;575(7781):203–209. [DOI] [PubMed] [Google Scholar]
- [65].Ding Y, Fei Y, Lu B. Emerging new concepts of degrader technologies. Trends Pharmacol Sci. 2020;41(7):464–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Leung I, Dekel A, Shifman JM, et al. Saturation scanning of ubiquitin variants reveals a common hot spot for binding to USP2 and USP21. Proc Natl Acad Sci U S A. 2016;113(31):8705–8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ernst A, Avvakumov G, Tong J, et al. A strategy for modulation of enzymes in the ubiquitin system. Science. 2013;339(6119):590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Huynh ML, Russell P, Walsh B. Tryptic digestion of in-gel proteins for mass spectrometry analysis. Methods Mol Biol. 2009;519:507–513. [DOI] [PubMed] [Google Scholar]
- [69].Chen Z-L, Meng J-M, Cao Y, et al. A high-speed search engine pLink 2 with systematic evaluation for proteome-scale identification of cross-linked peptides. Nat Comm. 2019;10(1):3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.