

Abstract
Introduction:
Immune checkpoint inhibitors (ICIs) are monoclonal antibodies used to activate the immune system against tumor cells. Despite therapeutic benefits, ICIs have the potential to cause immune related adverse events (irAE) such as myocarditis, a rare but serious side effect with up to 50% mortality in affected patients. Histologically, patients with ICI myocarditis have lymphocytic infiltrates in the heart, implicating T-cell-mediated mechanisms. However, the precise pathologic immune subsets and molecular changes in ICI myocarditis are unknown.
Methods:
To identify immune subset(s) associated with ICI myocarditis, we performed time-of-flight mass cytometry (CyTOF) on peripheral blood mononuclear cells from 52 individuals: 29 patients with autoimmune adverse events (irAE) on ICI including 8 patients with ICI myocarditis and 23 healthy controls. We also utilized multi-omics single-cell technology to immunophenotype 30 patients/controls using single-cell RNA sequencing (scRNA-seq), single-cell TCR sequencing and CITE-seq with feature barcoding for surface marker expression confirmation. To correlate between the blood and the heart, we performed scRNA-seq/TCR-seq/CITE-seq on PD-1 deficient mice (MRL/Pdcd1−/−) with spontaneous myocarditis.
Results:
Using these complementary approaches, we found an expansion of cytotoxic CD8+ T effector cells re-expressing CD45RA (Temra CD8+ cells) in ICI myocarditis patients compared to controls. TCR sequencing demonstrated that these CD8+ Temra cells were clonally expanded in myocarditis patients compared to controls. Transcriptomic analysis of these Temra CD8+ clones confirmed a highly activated and cytotoxic phenotype. Longitudinal study demonstrated progression of these Temra CD8+ cells into an exhausted phenotype two months after treatment with glucocorticoids. Differential expression analysis demonstrated elevated expression levels of proinflammatory chemokines (CCL5/CCL4/CCL4L2) in the clonally expanded Temra CD8+ cells, and ligand receptor analysis demonstrated their interactions with innate immune cells, including monocytes/macrophages, dendritic cells, and neutrophils, as well as absence of key anti-inflammatory signals. To complement the human study, we performed scRNA-seq/TCR-seq/CITE-seq in PD-1 deficient mice with spontaneous myocarditis and found analogous expansions of cytotoxic clonal effector CD8+ cells in both blood and hearts of such mice compared to controls.
Conclusions:
Clonal cytotoxic Temra CD8+ cells are significantly increased in the blood of patients with ICI myocarditis, corresponding with an analogous increase in effector cytotoxic CD8+ cells in the blood/hearts of PD-1 deficient mice with myocarditis. These expanded effector CD8+ cells have unique transcriptional changes, including upregulation of chemokines CCL5/CCL4/CCL4L2, which may serve as attractive diagnostic/therapeutic targets for reducing life-threatening cardiac immune-related adverse events in ICI-treated cancer patients.
Keywords: Myocarditis, immune checkpoint inhibitors, immunotherapy, cardio-oncology, cardio-immunology, single-cell multi-omics
INTRODUCTION
Immune checkpoint inhibitors (ICIs) are life-saving monoclonal antibodies that target intrinsic immune regulatory pathways on T-cells (e.g. cytotoxic T-cell antigen-4/CTLA4 or programmed death-1/PD-1) and release the brakes of T-cell cytotoxicity against tumor cells. Despite their beneficial effects, ICIs can cause immune related adverse events (irAE), autoimmune side effects in various organ systems. One particularly concerning irAE is life-threatening myocarditis, a rare but serious side effect of ICI with up to 50% mortality in affected patients leading to significant long term cardiac side effects including arrhythmias and heart failure (1–3).
Myocarditis is a disease with a heterogenous group of etiologies (viral, drug-induced, idiopathic, etc.) and remains poorly understood despite a high mortality rate (4,5). Due to the often fulminant clinical course of myocarditis, there is significant interest in biomarker discovery for both diagnostic and therapeutic purposes (6). In particular, while mouse immune profiling data in autoimmune myocarditis exist (6–8), there remains a significant need for comprehensive human immune phenotyping data to find personalized biomarkers and molecular signatures for myocarditis (6,9). In the special case of ICI-induced myocarditis, it is critical to attain an accurate diagnosis quickly to determine whether to discontinue ICI therapy and start immunosuppressive therapy to minimize cardiac morbidity/mortality (10). Current immunosuppressive therapies, such as glucocorticoids, are largely non-immune cell selective and confer their own nonspecific side effects (11), further highlighting the need to better understand the cellular subsets and mechanisms involved in ICI myocarditis (8).
Histologically, patients with ICI-induced myocarditis have acute lymphocytic infiltrates in the myocardium, implicating T-cell-mediated mechanisms (12–14). Previous mouse studies have reported CD8>CD4 lymphocytic heart infiltration in mice models of PD-1 blockade (14,15), but the specific type of CD8+ cells involved and the mechanisms by which these cells cause pathogenesis in the heart have not been well-understood (7). With the advent of single-cell phenotyping proteomics and transcriptomics, we now have the ability to study the immune repertoire of various tissues in healthy and disease states at high resolution (16–19). This ability to distinguish between individual cell states is particularly important for conditions that affect immune cell subtypes at a broad level, such as in immune checkpoint inhibition (19). Time-of-flight mass cytometry (CyTOF) is a technology that uses heavy-metal isotopes to stain cells and allows for simultaneous analysis of more than 40 different cellular proteins with minimal compensation (16,17,20,21) and has been validated against flow cytometry for many clinical/research applications including the phenotyping of immune cells in cancer clinical trials (22).
In addition to CyTOF, single-cell RNA sequencing (scRNA-seq) can provide high-throughput transcriptomic information of individual cells and allow for cell-type-specific gene expression analysis and molecular signatures (23). This novel technology, when combined with TCR sequencing, can expand our ability to characterize T-cell repertoires of disease states in humans and animal models (24). The combination of CyTOF, scRNA-seq, and single-cell TCR sequencing can therefore be a powerful combination of technologies to allow for high resolution immune phenotyping of myocarditis in the setting of irAE from ICIs.
We present the first comprehensive cellular and transcriptomic profiling of the peripheral blood mononuclear cells of recently diagnosed ICI myocarditis patients and controls using CyTOF, scRNA-seq, and single-cell TCR sequencing. We discovered a unique population of cytotoxic CD8+ T-cells re-expressing CD45RA (Temra) associated specifically with the group of patients with ICI myocarditis compared to control patients with no irAE and those with irAE in other organ systems. We found that this population of Temra CD8+ cells were clonally expanded in ICI myocarditis patients, and longitudinal study of these T-cell clonotypes showed an early activated and cytotoxic phenotype followed by a transition to T-cell exhaustion. Additionally, we found an elevated expression of chemokine transcripts in these Temra CD8+ cells that may facilitate homing to the heart, making them attractive candidates for therapeutic targeting. Last but not least, we corroborated our findings in a mouse model of PD-1 deletion – the MRL/Pdcd1−/− mouse, previously shown by others (14) and confirmed by our laboratory to develop spontaneous myocarditis at 4 weeks of age with 70% penetrance. Using scRNA-seq/TCR-seq/CITE-seq, we demonstrated that MRL/Pdcd1−/− mice have an expansion of clonal effector CD8+ T-cells cells in both their blood and hearts, correlating with the human peripheral blood immunophenotyping data.
RESULTS
Analysis of Immune Cell Populations in ICI Myocarditis using CyTOF Reveals Cytotoxic Temra CD8+ Expansion
Using CyTOF, we analyzed peripheral blood mononuclear cells (PBMCs) from 52 patients categorized into the four patient groups as described in the Methods section (No ICI Group – Healthy patients without cancer and not on ICI from the Stanford FluPrint Database, n=23 (25); Group A – Patients on ICI without irAE, n=8; Group B – Patients on ICI with non-myocarditis irAE, n=13; Group C – Patients on ICI with ICI myocarditis, n=8) (Tables 1, Table S1). All patients were recruited according to a protocol approved by the Stanford Institutional Review Board. A total of 520,000 cells from the peripheral blood of these patients were analyzed using CyTOF (Fig. 1A) with a panel of 33 canonical immune surface markers (Table S2). Unsupervised clustering revealed distinct clusters comprised of all expected immune cell populations including T cells, B cells, monocytes/macrophages, neutrophils, dendritic cells, basophils and natural killer cells (Fig. 1B, Table S3). Feature plots showed robust expression of major immune canonical markers within the expected immune subsets (Fig. 1C). Clusters of cell populations were marked according to differentially expressed markers of each queried cluster as well as established canonical markers of each immune subset (Tables S3–6). Quantification of immune cell distributions across cell clusters showed a relative reduction in circulating T-cells and B cells in the ICI-treated groups (Groups A-C) compared to the non-ICI control group (Fig. 1D). Additionally, there was an increase in circulating monocytes/macrophages in the ICI-treated patients compared to non-ICI-treated patients. Comparison between groups A/B/C demonstrated no significant differences in CD45+ cell subtype proportions (Fig. 1E). We then subsetted CD3+ cells and performed unsupervised clustering, found subclusters of CD3+ cells (Fig. 1F), identified canonical CD3+ gene markers (Fig. 1G, Table S4), and quantified CD3+ subtypes as a proportion of the total CD3+ input number per sample (Fig. 1H–I). We observed no significant differences between the non-ICI treated and ICI-treated groups (Fig. 1H) or between A/B/C (Fig. 1I) in CD8+ or CD4+ T-cell proportions. We then further selected subsets of cells from the CD3+ population – the CD4+ (Fig. 1J) and CD8+ populations (Fig. 1K). Subsequently, we used differential expression analysis and canonical markers of CD4+ (Table S5) and CD8+ (Table S6) subsets to identify cell populations. Quantification of the CD4+ subtypes showed an increase in regulatory T-cells in the ICI-treated compared to non-ICI-treated groups (Fig. 1L), but did not show significant trends among the ICI-treated patient groups (Group A/B/C) (Fig. 1M). Quantification of CD8+ subtypes demonstrated a significant reduction in naïve CD8 cells and increase in the Temra CD8+ (T-effector cells re-expressing CD45RA) CD8+ sub-population in the ICI-treated compared to the non-ICI-treated group. Furthermore, when comparing groups A/B/C, the ICI myocarditis group also demonstrated significantly increased Temra CD8+ cells compared to groups A and B (Fig. 1K). These results demonstrate that ICI-myocarditis (Group C) is associated with a significant increase in Temra CD8+ cells in the peripheral blood compared to ICI-treated patients without side effects (Group A) and with non-myocarditis irAE (Group B).
Table 1.
Demographics of the patient cohorts undergoing CyTOF and single-cell RNA-seq (scRNA-seq) analysis of peripheral blood.
| Healthy Controls No ICI |
Group A ICI + No irAE |
Group B ICI + Non-Myocarditis irAE |
Group C ICI + Myocarditis |
Total | |
|---|---|---|---|---|---|
| Group Size n | 23 (6) | 8 (8) | 13 (8) | 8 (8) | 52 (30) |
| Age in Mean Yrs | 71 (45) | 66 (67) | 64 (60) | 71 (72) | 67 (62) |
| Gender | M: n=17 (n=4) | M: n=6 (n=6) | M: n=10 (n=7) | M: n=7 (n=5) | M: n=40 (n=22) |
| F: n=6 (n=2) | F: n=2 (n=2) | F: n=3 (n=1) | F: n=1 (n=3) | F: n=12 (n=8) | |
| Malignancy | |||||
| Thoracic | 0 (0) | 1 (1) | 4 (2) | 4 (3) | 9 (5) |
| Melanoma | 0 (0) | 2 (2) | 4 (2) | 1 (1) | 7 (5) |
| Head/Neck | 0 (0) | 1 (1) | 1 (1) | 0 (1) | 2 (3) |
| Esophagogastric | 0 (0) | 0 (0) | 1 (1) | 0 (1) | 1 (2) |
| Lymphoma | 0 (0) | 0 (0) | 0 (0) | 1 (1) | 1 (1) |
| Genitourinary | 0 (0) | 4 (4) | 3 (2) | 2 (1) | 10 (7) |
| Recent Immunotherapy | |||||
| PD-1 | 0 (0) | 7 (7) | 11 (7) | 4 (5) | 22 (19) |
| PD-L1 | 0 (0) | 1 (1) | 1 (1) | 2 (1) | 4 (3) |
| PD-1 + CTLA-4 | 0 (0) | 0 (0) | 1 (0) | 2 (2) | 3 (2) |
| Diagnosis | |||||
| Healthy | 23 (6) | 8 (8) | 0 (0) | 0 (0) | 31 (14) |
| Thyroiditis | 0 (0) | 0 (0) | 3 (1) | 0 (0) | 3 (1) |
| Hypophysitis | 0 (0) | 0 (0) | 0 (1) | 0 (0) | 0 (1) |
| Hepatotoxicity | 0 (0) | 0 (0) | 4 (1) | 0 (0) | 4 (1) |
| Vitiligo | 0 (0) | 0 (0) | 1 (2) | 0 (0) | 1 (2) |
| Colitis | 0 (0) | 0 (0) | 3 (1) | 0 (0) | 3 (1) |
| Pneumonitis | 0 (0) | 0 (0) | 2 (1) | 0 (0) | 2 (1) |
| Pericarditis | 0 (0) | 0 (0) | 2 (1) | 0 (0) | 2 (1) |
| Myocarditis | 0 (0) | 0 (0) | 0 (0) | 8 (8) | 8 (8) |
Numbers outside of the parenthesis pertain to patient samples in which CyTOF was performed. Numbers in italics and in parenthesis pertain to patient samples in which scRNA-seq was performed.
Fig. 1. Analysis of Immune Cell Populations in ICI Myocarditis using CyTOF Reveals Cytotoxic Temra CD8+ Expansion.

(A) Workflow showing collection of peripheral blood and processing of single-cell suspensions for mass cytometry (CyTOF). (B) Identification of peripheral blood CD45+ immune cell clusters across all samples (n=8–23 subjects per cohort). (C) Feature plots with canonical markers across CD45+ clusters. (D) Quantification of immune cell distribution across clusters and comparison of non-ICI vs. ICI-treated patients showing a relative increase in monocytes and reduction in T-cells in circulating blood in the ICI-treated patients (consisting of Groups A-C) compared to “No ICI” control group. Average and SEM are shown for each patient group. For each cluster, the average fraction of cells from each patient group is shown, after normalization for total CD45+ input cell numbers per patient. (E) Comparison between groups A/B/C showing no significant difference in CD45+ cell subtype proportions between the ICI-treated groups. (F) Identification of peripheral blood CD3+ immune cell clusters across all samples (n=8–23 subjects per cohort). (G) Feature plots with canonical markers across CD3+ clusters. (H) Quantification of CD3+ cell subtypes across clusters and comparison of non-ICI vs. ICI-treated patient groups showing no significant difference between groups. For each cluster, the average fraction of cells from each patient group is shown, after normalization for total CD3+ input cell numbers per patient. Average and SEM are shown for each patient group. (I) Comparison between groups A/B/C showing no significant difference in CD3+ cell subtype proportions between the ICI-treated groups. (J) Identification of peripheral blood CD4+ immune cell clusters across all samples (n=8–23 subjects per cohort). (K) Identification of peripheral blood CD8+ immune cell clusters across all samples (n=8–23 subjects per cohort). (L) Quantification of CD4+ cell subtypes across clusters comparing non-ICI to ICI-treated groups. For each cluster, the average fraction of cells from each patient group is shown, after normalization for total CD4+ input cell numbers per patient. Statistics are calculated as described above. (M) Comparison between groups A/B/C in CD4+ cell subtype proportions between the ICI-treated groups. (N) Quantification of CD8+ cell subtypes across clusters and comparison of non-ICI vs. ICI-tread patient groups showing a significant increase in the proportion of Temra CD8+ T-cells in the myocarditis group compared to control groups and decrease in naïve CD8+ T-cells. For each cluster, the average fraction of cells from each patient group is shown, after normalization for total CD8+ input cell numbers per patient. (O) Comparison between groups A/B/C in CD8+ cells showing statistically significant increased Temra CD8+ cell proportions in Group C (myocarditis) patients compared to control groups (one-way ANOVA, followed by correction for multiple comparisons by controlling the FDR with a significance threshold of q-value < 0.05*).
Global Analysis of Immune Cell Populations in ICI Myocarditis using scRNA-seq Corroborates CyTOF Results
To confirm our results from CyTOF studies and gain further insights into the molecular characteristics of circulating immune cell subsets that are differentially expanded in ICI-myocarditis patients, we performed scRNA-seq analysis on PBMCs from 30 patients (Table 1). This included healthy control patients without cancer who are not on ICI, n=6 (26) and ICI-treated patients whose blood we collected using a Stanford IRB-approved protocol—Group A, n=8; Group B, n=8; Group C, n=8 (Fig. 2A). We obtained high-quality transcriptomes from a total of 193,000 cells. We observed 14 distinct clusters (Table S8) comprising all expected immune populations, including T-cells, B-cells, monocytes/macrophages, NK cells, dendritic cells and basophils (Fig. 2B). Gene expression feature plots showed presence of major immune canonical markers within the expected immune subsets (Fig. 2C). For genes that exhibit low level of expression at the RNA level (e.g. CD45RA, CD4), feature barcoding using oligonucleotide-tagged antibodies to selected surface receptors (e.g. CD4, CD8, CD45RA) was performed simultaneously on cells captured for scRNA-seq profiling to correlate RNA expression level with protein expression level in the same single cell (Fig. 2D; Table S7). Clusters of cell populations were marked according to differentially expressed markers as well as known canonical markers of each queried cluster (Fig. 2E, Table S8). Similar to the CyTOF data, a relative increase in monocytes and a reduction in circulating T-cells were found in the ICI-treated patients (Groups A-C) compared to the “No ICI” control group (Fig. 2F). No significant changes, other than a slight decrease in basophils, were seen among the ICI-treated groups A/B/C (Fig. 2G).
Fig. 2. Analysis of Immune Cell Populations in ICI Myocarditis Patients using scRNA-seq.
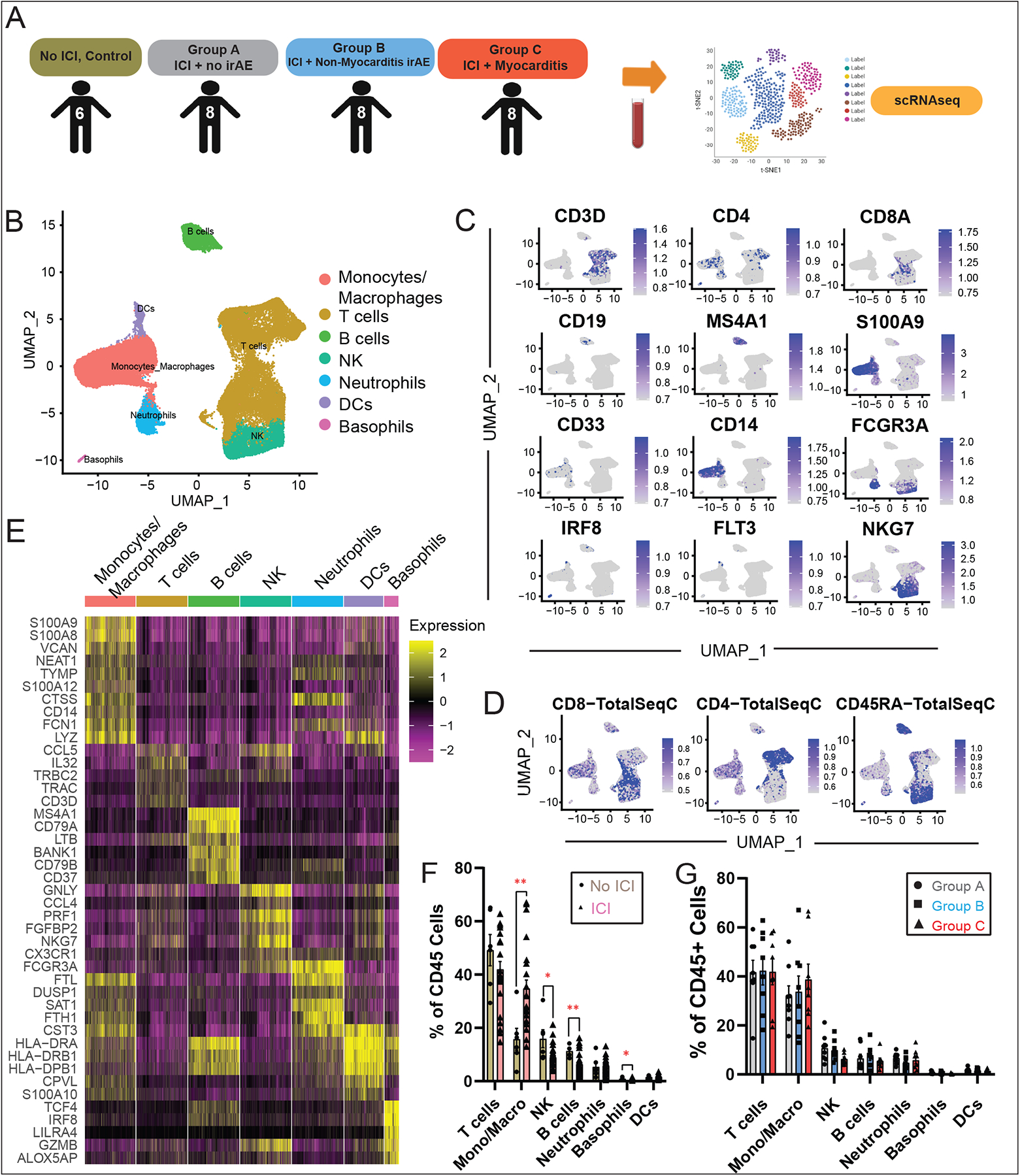
(A) Workflow showing collection of peripheral blood and processing of single-cell suspensions for single-cell RNA-seq. (B) Identification of peripheral blood CD45+ immune cell clusters across all samples (n=6–8 subjects per cohort). (C) Feature plots with canonical markers across CD45+ clusters. (D) Feature barcoding with CITE-seq showing surface markers CD45RA, CD4 and CD8, (E) Heatmap of top differentially expressed genes across clusters. (F) Quantification of CD45+ cell subtypes across clusters and comparison of non-ICI vs. ICI-treated patient groups showing an expansion of monocytes and a relative reduction in circulating T-cells in the ICI-treated groups compared to the “No ICI” control patients, as well as changes in NK, B-cells and basophils. For each cluster, the average fraction of cells from each patient group is shown, after normalization for total CD3+ input cell numbers per patient. Average and SEM are shown for each patient group. (G) Comparisons of CD45+ cell subtypes between groups A/B/C.
We then subsetted CD3+ cells and performed unsupervised clustering to find differentially expressed genes for each cluster (Fig. S1A–D, Table S9) and quantified the frequency of each CD3+ subcluster (Fig. S1E–F). We found no significant differences between the proportion of CD4+ and CD8+ cell populations between non-ICI vs. ICI-treated groups and between groups A/B/C.
Confirmation of Myocarditis-Associated Temra CD8+ Expansion by scRNA-seq
To evaluate changes in CD8+ subpopulations in ICI myocarditis compared to control groups, we subsetted CD8+ cells from our dataset and performed unsupervised clustering on this population (Fig. 3A). Feature plots showed distinct RNA expression patterns of genes such as CCR7, and IL7R that discriminate between different CD8+ subpopulations (Fig. 3B). The presence of the surface marker CD45RA was also detected by feature barcoding using oligo-conjugated antibodies (Fig. 3C). Based on the differential expression of these and the top canonical markers of CD8+ cells, we identified the major CD8+ subpopulations including naïve, central memory, effector memory, and Temra CD8+ cells (Fig. 3D, Table S10). Quantification of the frequency of CD8+ subtypes demonstrated a statistically significant increase in Temra CD8+ population in the ICI-treated compared to the non-ICI-treated groups (Fig. 3E), and in the myocarditis group (Group C) compared to the other ICI-treated control groups A/B (Fig. 3F). These results, like the CyTOF data, demonstrate that ICI myocarditis may be associated with an increase in Temra CD8+ cells in the peripheral blood. Furthermore, these Temra CD8+ cells expressed high levels of cytotoxicity/activation including granzyme B (GZMB) and interleukin-32 (IL-32) as well as the chemokine, CCL5, relative to the other CD8+ cell types (Fig. 3G and 3H). This finding suggests a cytotoxic role for these Temra CD8+ cells.
Fig. 3. Myocarditis-Related Temra CD8+ Expansion Confirmed by scRNA-seq.
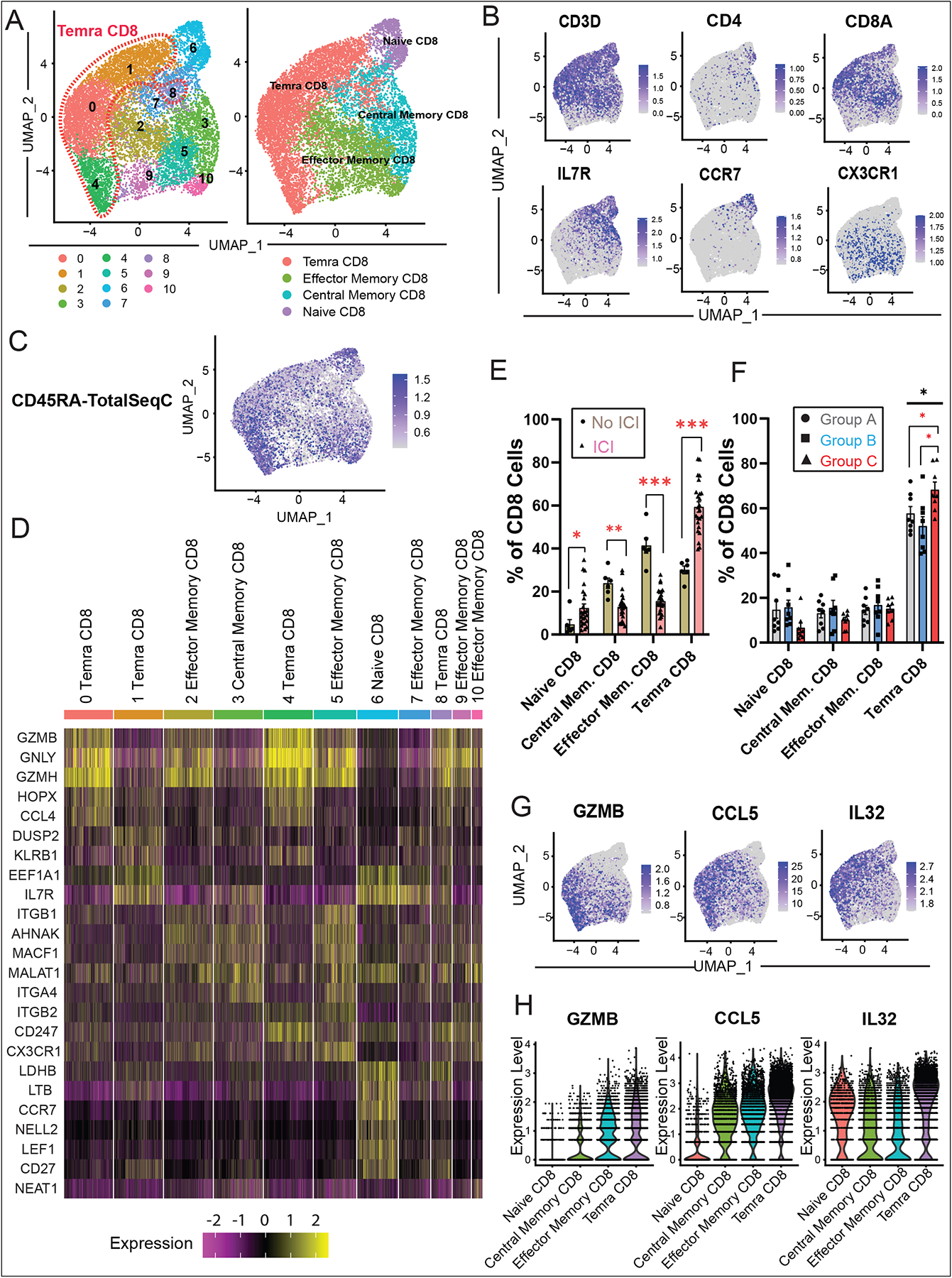
(A) Identification of peripheral blood CD8+ immune cell clusters across all samples (n=6–8 subjects per cohort). (B) RNA feature plots with canonical markers across CD8+ clusters. (C) Feature plots of CD45RA surface protein expression using CITE-seq feature barcoding technology. (D) Heatmap of top differentially expressed genes across clusters. (E) Quantification of CD8+ cell subtypes across clusters and comparison of non-ICI vs. ICI-treated patient groups shows increased proportions of Temra CD8+ T-cells in the myocarditis group compared to controls, as well as other changes across the CD8+ subtypes. For each cluster, the average fraction of cells from each patient group is shown, after normalization for total CD8+ input cell numbers per patient. Average and SEM are shown for each patient group. (F) Quantification of CD8+ cell subtypes across clusters and comparison of A/B/C patient groups shows increased proportions of Temra CD8+ T-cells in the myocarditis group C compared to groups A/B controls (one-way ANOVA, followed by correction for multiple comparisons by controlling the FDR with a significance threshold of q-value < 0.05*). (G) Feature plots displaying expression of GZMB (cytotoxicity marker), CCL5 (pro-inflammatory chemokine), and IL32 (pro-inflammatory interleukin) in the Temra CD8 clusters. (H) Violin plots showing increased log normalized expression of GZMB, CCL5, and IL32 in Temra CD8+ cells compared to the other CD8+ cell types.
To assess whether a unique subpopulation of CD4+ cells may be present in ICI-myocarditis, we evaluated the CD4+ subpopulations in a fashion similar to the CD8+ subpopulation (Fig. S2; Table S11). We did not detect any other significant changes in cell proportions between the patient groups.
Single-Cell TCR Sequencing Reveals Myocarditis-Associated Clonal Expansion of Cytotoxic Temra CD8+ Cell Clusters
Clonal expansion of T-cells occurs after T-cell activation, and has been thought to play a role in autoimmunity (27). In order to interrogate the clonal identities of the expanded T-cells in our ICI myocarditis cohort compared to the other control patient groups, we performed single-cell TCR sequencing on our patient groups A, B, and C using the 10X Genomics V(D)J single-cell TCR sequencing assay to evaluate productive V-J spanning pairs within each patient sample. Most of our CD3+ cells express αβ TCRs (83.4% of sequenced TCRs). For each patient, we identified the top 50 clonotypes and visualized their gene expression signature across patient groups A, B, and C. We found an expansion of the top 50 clonotypes in ICI myocarditis patients (Group C) compared to the control (Group A) and non-myocarditis irAE (Group B) populations (Fig. 4A, B). Furthermore, most of these clonotypes were localized to the Temra CD8+ clusters (Fig. 4A, C). Quantification of the top 50 TCR clonotypes as a fraction of total CD3+ cells showed an enrichment of clonotypes within the Temra CD8+ cell clusters in myocarditis patients (Group C) compared to other groups (Fig. 4C). We further analyzed the abundance of top 50 TCR clonotypes within subclusters of CD8+ cells and found preferential expansion of TCR clonotypes in all Temra CD8+ subclusters (clusters 0, 1, 4, 8; highlighted in red, Fig. 4D), with a significant expansion of clonotypes from myocarditis patients (Group C) in those clusters compared to clonotypes from Groups A/B. These findings indicate the enrichment of several different Temra CD8 clusters in ICI myocarditis patients compared to the other groups, suggesting the possibility that a heart-specific inflammatory signature is present in ICI myocarditis patients.
Fig. 4. Single-Cell TCR Sequencing Reveals Myocarditis-Associated Clonal Expansion of Temra CD8+ Cell Clusters.
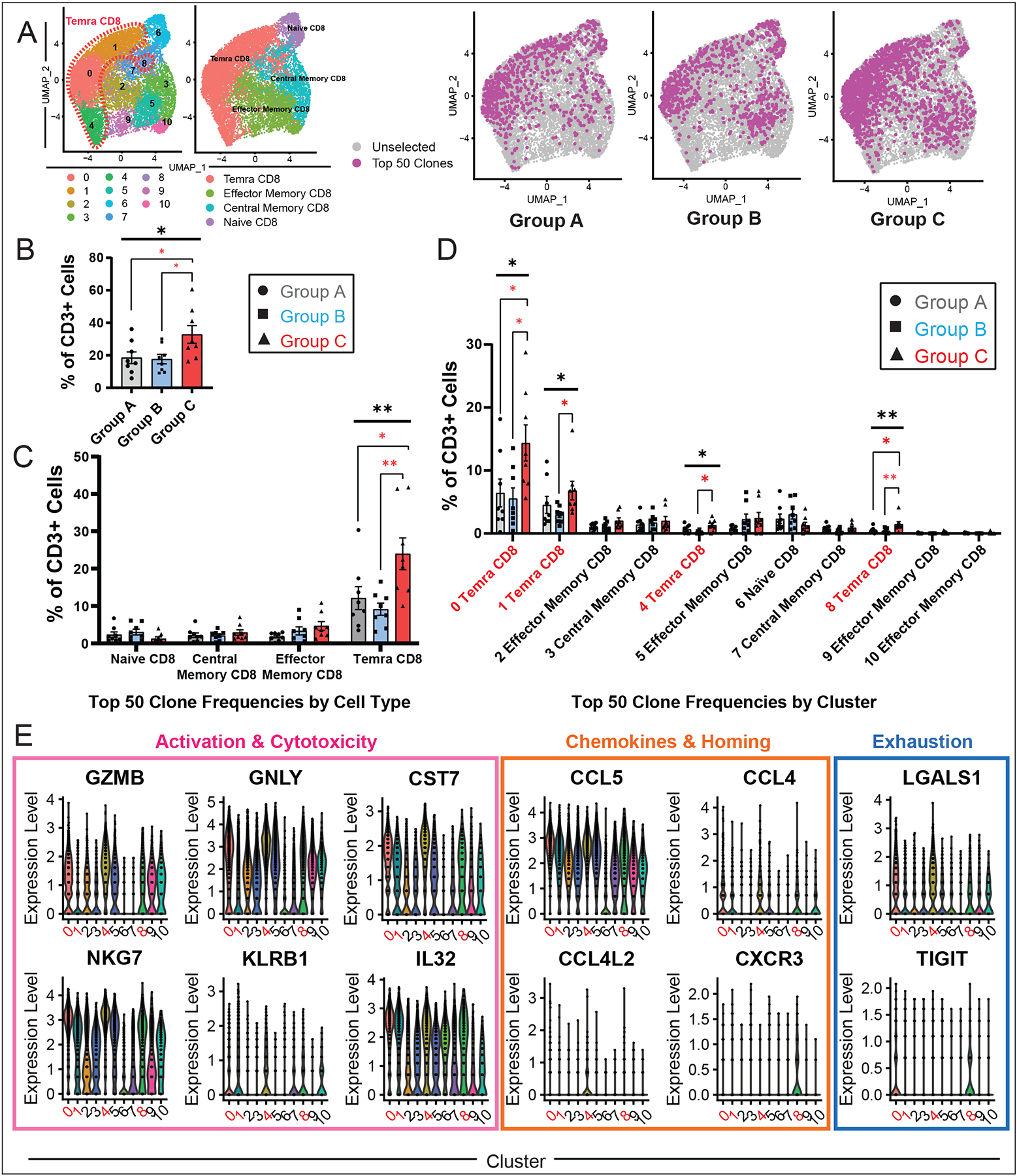
(A) Visualization of top 50 TCR clonotypes across patient groups A, B, and C (n=8 each) showing expansion of clonotypes in group C (myocarditis) patients compared to control groups. (B) Quantification of the top 50 TCR clonotypes as a fraction of total CD3+ cells across patient groups. For each patient in a group, the fraction of top 50 clonotypes was normalized against CD3+ input cell numbers. Average and SEM are shown for each patient group. Statistical analysis compares groups A, B, and C. One-way ANOVA was performed, followed by correction for multiple comparisons by controlling the FDR with a significance threshold of q-value < 0.05*. (C) Quantification of top 50 TCR clonotypes across CD8+ cell subtypes as previously defined in Fig. 3, compared across patient groups A, B, and C showing increase in Temra CD8+ in the myocarditis group C (one-way ANOVA, followed by correction for multiple comparisons by controlling the FDR with a significance threshold of q-value < 0.05*). (D) Quantification of top 50 TCR clonotypes across the 10 CD8+ cell clusters compared across patient groups A, B, and C. All Temra CD8+ clusters (0, 1, 4, 8) show statistically significant expansion in the myocarditis group C and are highlighted in red. Kruskal-Wallis was performed, followed by Dunn’s multiple comparisons test. (E) Violin plots displaying significantly differentially expressed genes across CD8+ T cell clusters comparing log normalized expression of significant genes in myocarditis-associated Temra CD8+ clusters (0, 1, 4, 8) highlighted in red compared to all other clusters. Myocarditis-associated Temra CD8+ clusters show elevated expression levels of classical cytotoxicity/activation genes (GZMB, GNLY, CST7, NKG7, KLRB1, IL32) and increased expression of potentially myocardial-tropic chemokines (CCL5/CCL4/CCL4L2/CXCR3). Myocarditis-associated clusters are also associated with increased expression of select T-cell exhaustion markers (LGALS and TIGIT).
To evaluate transcriptomic differences between different CD8+ cell clusters, particularly the inflammatory signature in the myocarditis-associated Temra CD8+ clusters, we performed differential gene expression analysis (Fig. 4E, Table S12) and visualized the results with violin plots to find that myocarditis-associated clusters 0, 1, 4, and 8 showed a relatively high expression of pro-inflammatory and cytotoxicity markers (GZMB, GNLY, CST7, NKG7, KLRB1, IL32). Myocarditis-associated clusters also showed an increased expression of myocardial-tropic chemokines CCL5/CCL4/CCL4L2 (28,29) as well as the immunotherapy colitis-associated chemokine, CXCR3 (30). Interestingly, the receptors for CCL5, specifically CCR1 and CCR5, have been shown to be robustly expressed in failing and non-failing hearts (29), and CCR5 in particular has had an established role in myocarditis associated with Chagas disease and cardiac autoimmunity (28,31). These transcriptomic findings suggest a phenotypic difference between the myocarditis-associated clusters relative to the non-myocarditis associated clusters in terms of T-cell activation/cytotoxicity. Finally, myocarditis-associated Temra CD8+ clusters also appear to express varying levels of early exhaustion markers and checkpoint genes, such as LGALS1 and TIGIT respectively, suggesting activation of counter-regulatory mechanisms.
Activated and Expanded Temra CD8+ Clones in Myocarditis Transition from an Early Cytotoxic Phenotype to a Late Exhaustion Phenotype Over Time
In order to evaluate the time evolution of clonally-expanded T-cells in ICI myocarditis, we longitudinally analyzed PBMCs from one patient with MRI-confirmed myocarditis at early (t=0 days) and late (t=65 days) timepoints (Fig. 5A). This patient received glucocorticoids (60 mg/day of prednisone) for treatment of his myocarditis on the first day of his diagnosis (after blood collection) and underwent a successful four-week steroid taper with resolution of his cardiac biomarkers (troponin I, measured in ng/mL) by day 25. Cardiac MRI also confirmed the initial presence of late gadolinium enhancement (LGE) with increased T2 signaling suggestive of myocardial edema at day 0 which resolved by day 65 (Fig. 5B). Clonal analysis of CD3+ cells in his peripheral blood in the early and late time points showed an oligoclonal expansion of top 50 TCR clonotypes (47.6% of all CD3+ cells in peripheral blood) at the time of diagnosis of myocarditis which continued to persist (34.7% of CD3+ cells) at the later timepoint. Furthermore, the clonotype with the biggest expansion (defined as “clonotype 1”) accounted for 9.3% of all T-cells in the peripheral blood of this patient at initial diagnosis and decreased only slightly to 8.1% by day 65 despite steroid treatment (Fig. 5C). This data supports the persistence of clonally expanded Temra CD8+ cells in myocarditis patients even after clinical indices of inflammation have resolved.
Fig. 5. Activated and Expanded Cytotoxic Temra CD8+ Clones Persist Two Months after Myocarditis but Exhibit Markers of T-Cell Exhaustion.
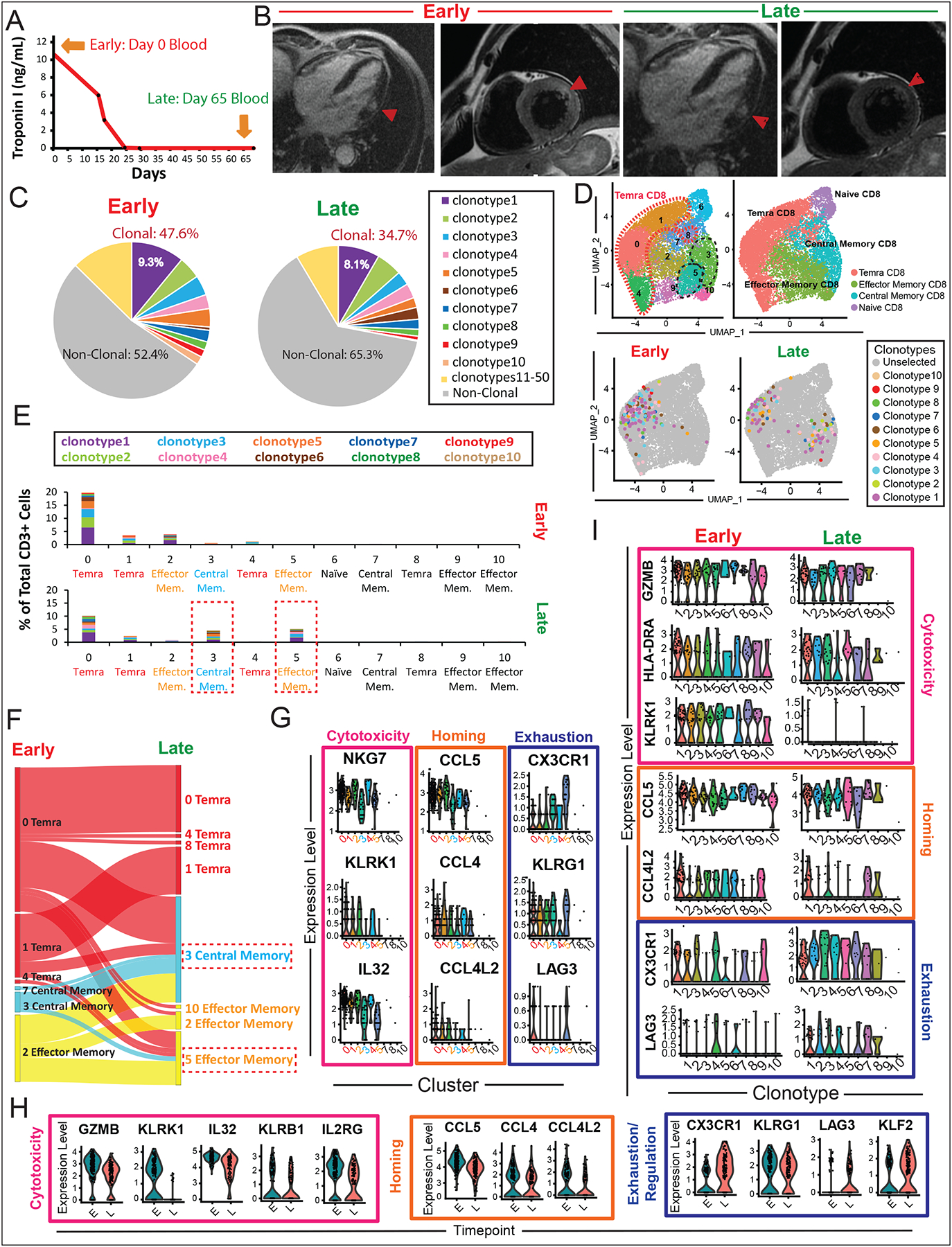
(A) Peripheral blood was collected from a patient with myocarditis (MCE1) at initial myocarditis diagnosis (early timepoint =day 0) and after resolution of clinical myocarditis and troponin I biomarker (late timepoint = day 65). (B) Cardiac magnetic resonance imaging (cMRI) shows myocarditis in the lateral wall at early timepoint (day 0) including myocardial thinning and delayed gadolinium enhancement (DGE) and increased T2 signal consistent with myocardial edema, with resolution myocardial edema by late timepoint (day 65). (C) Clonal analysis of CD3+ cells in peripheral blood by single-cell TCR sequencing shows oligoclonal expansion of top 50 TCR clonotypes (47.6% of all CD3+ cells in peripheral blood) in the early myocarditis timepoint which decreases slightly but persists (34.7% of CD3+ cells) in the late timepoint. Top 10 expanded clonotypes are shown by their individual colors, with top expanded clonotype 1 shown in purple, etc. (D) Visualization of top 10 expanded TCR clonotypes on UMAP showing localization of individual expanded clonotypes in Temra clusters 0, 1, and 4 which translocate to clusters 3 (central memory) and 5 (effector memory) in the late timepoint. (E) Quantification of percentages of top 10 expanded TCR clonotypes in each cluster as a fraction of total CD3+ cells in the patient in the early and late timepoints, showing shift in clonally expanded population from clusters 0, 1, and 4 to clusters 3 and 5 the late timepoint. (F) Sankey diagram showing shift in the top 10 expanded TCR clonotypes from predominantly Temra clusters (0, 1, 4) to central memory cluster 3 and effector memory cluster 5 in the late timepoint. (G) Violin plots showing differential gene expression analysis of the top 10 expanded TCR clonotypes across clusters, showing decreased expression of cytotoxicity genes (IL32, KLRK1, IL32) and homing chemokine genes (CCL5, CCL4, CCl4L2) in clusters 3 and 5 (associated with the late timepoint). In contrast, clusters 3 and 5 exhibited higher expression of T-cell exhaustion genes CX3CR1, KLRG1, and LAG3. (H) Violin plots of differential gene analysis in the early vs. late timepoints, showing relative decrease in T-cell cytotoxicity genes (GZMB, KLRK1, IL32, KLRB1, IL2RG) and homing chemokines (CCL5, CCL4, CCL4L2) in the late compared to early timepoints. In contrast, there was relative increased expression of T-cell exhaustion markers (CX3CR1, KLRG1, LAG3, and KLF2) in the late compared to early timepoint. (I) Violin plots showing differential gene expressions in top 10 expanded individual TCR clonotypes in the early vs. late timepoint, demonstrating similar decrease in cytotoxicity/homing genes and increase in exhaustion genes in the late timepoint.
To assess the transcriptomic changes in the top 10 expanded TCR clonotypes in this patient over time, we tracked the cellular identity of the top 10 expanded TCR clonotypes from initial diagnosis to day 65. We found that these top expanded clonotypes were localized predominantly in clusters 0, 1, 2, and 4 (predominantly Temra CD8+) at the time of diagnosis but translocated to clusters 3 and 5 (predominantly central and effector memory) at day 65 (Fig. 5D). This was confirmed quantitatively (Fig. 5E) and by Sankey diagrams that trace the lineage destination of the clonotypes from the early to late timepoints (Fig. 5F, Fig. S3A).
To better understand the transcriptional differences between early-predominant and late-predominant clusters, we performed differential expression analysis on the top 10 clonotypes across the clusters (Table S13). Among the top differentially expressed markers of each cluster, the early timepoint-associated clusters 0, 1, 2, 4 demonstrated relatively high expression levels of cytotoxicity markers (NKG7, KLRK1, IL32) and homing-associated chemokine genes (CCL5, CCL4 and CCL4L2) compared to the late timepoint-associated clusters 3 and 5 (Fig. 5G; Table S13). Notably, the expression of KLRK1, which encodes the activating cell surface receptor NKG2D known to be expressed on immune cells in rheumatoid arthritis and autoimmune colitis (32), was expressed in clusters 0, 1, 2, and 4. Clusters 3 and 5, in contrast, demonstrated relatively high expression levels of T-cell exhaustion markers (CX3CR1, KLRG1, and LAG3). Intriguingly, the expression of the immune checkpoint, LAG3, generally responsible for suppressive immune responses, was high in cluster 5 but also expressed in myocarditis-associated cluster 0.
Next, we assessed the differential expression of genes between the early and late timepoints in the top 10 clonotypes and similarly found a relative decrease in T-cell cytotoxicity/activation genes (GZMB, KLRK1, IL32, KLRB1, IL2RG), decrease in homing chemokine gene expression (CCL5, CCL4, CCL4L2), and increase in T-cell exhaustion/regulatory genes (CX3CR1, KLRG1, LAG3, KLF2) in the late timepoint compared with early timepoint (Fig. 5H; Table S14). Individual gene expression analysis of the top 10 clonotypes (Fig. 5I, Fig. S3B) demonstrated a similar pattern in decreased expression of cytotoxicity/homing genes and increase in T-cell exhaustion and regulatory markers in the late compared to early timepoints.
Overall, these findings suggest that early cytotoxic transcriptomic programs (elevated GZMB, KLRK1, KLRB1) in these myocarditis-associated clonally-expanded Temra CD8+ T-cells become attenuated with time. Concordant with these changes, an increase in T-cell exhaustion markers (KLRG1, CX3CR1, LAG3, KLF2) was noted in the same Temra CD8+ cells at the late timepoint. Additionally, there is an early robust expression of chemokines (CCL4, CCL5, CCL4L2) that are involved in the recruitment of immune/inflammatory cells to the heart which decreases over time, giving clues to the organ-specific nature of cellular inflammatory programs upregulated with ICI myocarditis.
Ligand-Receptor Analysis of Myocarditis-Associated T Cell Populations Exhibits Interactions with Other Immune Cell Types Through Cardiotropic Chemokines
In order to investigate functional interactions in our Temra CD8+ population of interest with other cell types, we performed differential expression analysis across patient groups A-C for the Temra CD8+ cells (Table S15). The top differentially expressed genes across the patient groups are shown in the feature plots in Fig. 6A, highlighting the upregulation of chemokines, CCL4 and CCL5, as well as cytotoxicity and pro-inflammatory genes, GZMA and IL-32, in the myocarditis cohort (Group C) compared to the other control groups. Quantification of log-fold gene expression levels confirmed this finding (Fig. 6B).
Fig. 6. Gene Expression Programs of Myocarditis-Associated T Cell Populations Exhibit Increased Cardiotropic Chemokines and Markers of Autoimmunity.
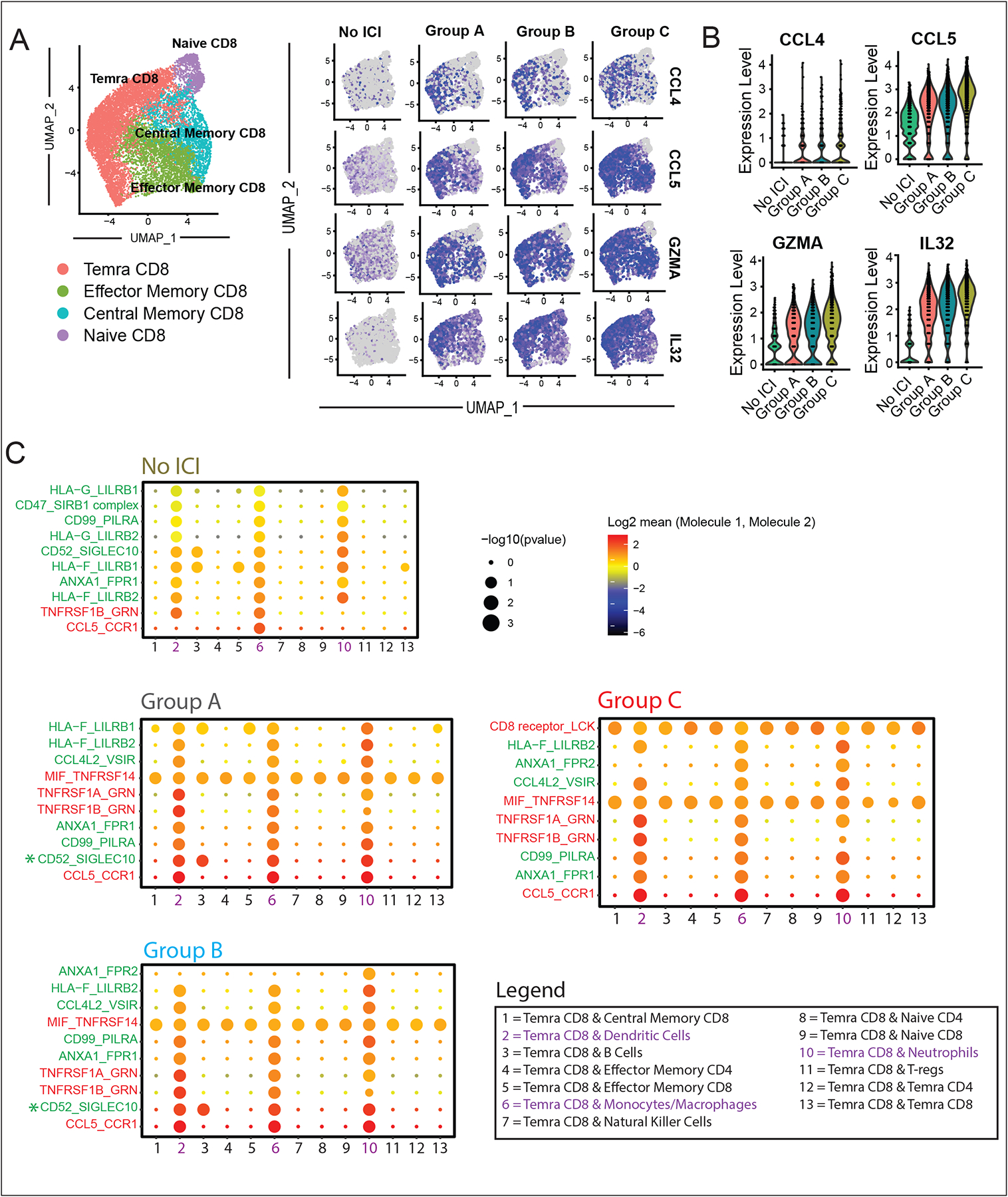
(A) Feature plots showing top differentially expressed genes in CD8+ T-cells across patient groups A, B, and C, showing increased expression of myocardial-tropic chemokines (CCL4 and CCL5) and pro-inflammatory genes (GZMA and IL32). (B) Violin plots showing averaged log-fold expression of top differentially expressed genes across the patient groups A, B and C. (C) Top 10 predicted ligand-receptor interactions of Temra CD8+ cells in each patient cohort. Interaction scores were calculated based on the expression of ligands and corresponding receptors in scRNA-seq data [CellPhoneDB, (https://www.cellphonedb.org/))]. Results highlight the increase in CCL5-CCR1 interactions between Temra CD8 cells and some members of the innate immune system (dendritic cells, monocytes/macrophages, and neutrophils) with ICI treatment. Pro-inflammatory interactions are labeled in red, anti-inflammatory signals in green, and neutral signals in grey. Group C myocarditis interactions lacked T-cell suppressive signal, CD52-SIGLEC10, compared to the other ICI treated groups (A and B).
To examine the potential interactions in vivo of expanded Temra CD8+ cells with other immune cells we used the CellPhoneDB algorithm (33) to identify biologically relevant interactions between known ligand-receptor pairs. CellPhoneDB, a tool which uses a combination of computational approaches and a publicly available database of curated receptors and ligands, allows for the prediction of potential ligand-receptor interactions within a scRNA-seq dataset (Fig. 6C). We applied this algorithm to our CD45+ dataset and identified statistically significant interactions between ligands expressed by our Temra CD8+ cells with members of the innate immune system (dendritic cells, monocytes/macrophages, and neutrophils) via pro-inflammatory crosstalk between CCL5-CCR1 and various members of the TNF superfamily. Interestingly, the non-ICI vs. ICI-treated groups differ in the number of pro-inflammatory interactions (red) compared to anti-inflammatory interactions (green), with higher ratio of pro-inflammatory vs. anti-inflammatory interactions found in the ICI-treated groups. Analysis of myocarditis group C demonstrated a further lack of a key T-cell regulatory interaction between CD52 and SIGLEC10. Patients with type I diabetes, another autoimmune-based disease, have been shown to have reduced numbers of T-cell subsets responsive to SIGLEC10 (34). These data support our hypothesis that ICI treatment increases inflammatory signals between the adaptive and immune systems, and that Temra CD8+ cells are involved in a mostly pro-inflammatory crosstalk with innate immune cells with expression of key inflammatory factors and organ-targeting chemokines.
Analysis of Immune Cell Populations in MRL/Pdcd1−/− Mice with Spontaneous Myocarditis Demonstrates Expansion of Clonal Effector CD8+ T cells in the Heart and Blood
In order to uncover immune changes in the heart during PD-1 deletion induced myocarditis, we utilized a mouse model of PD-1 deletion – the MRL/Pdcd1−/− mouse, previously shown by others (14) and confirmed by our laboratory to develop spontaneous myocarditis at 4 weeks of age with 70% penetrance (Fig. Supp. 4). We performed scRNA-seq/TCR-seq/CITE-seq on MRL/Pdcd1−/− mice and MRL controls (n=6 per genotype) (Fig. 7A). We isolated immune cells from their blood and hearts, and after standard pre-processing steps, retained a final number of ~32,000 high quality immune cells from blood and ~17,000 high quality CD45+ cells from the hearts for analysis. Unsupervised clustering revealed distinct clusters comprised of all expected immune cell populations including T cells, B cells, monocytes/macrophages, neutrophils, dendritic cells, basophils and natural killer cells (Fig. 7B). Clusters of cell populations were marked according to differentially expressed markers of each queried cluster as well as established canonical markers of each immune subset (Table S16). Quantification of immune cell distributions across cell clusters showed a relative increase in monocytes/macrophages as well as T-cells in the hearts of MRL/Pdcd1−/− mice (Fig. 7C). Similar to our ICI-treated patients, we noted a slight decrease (although not statistically significant) in the circulating T-cells in the blood of MRL/Pdcd1−/− compared to MRL mice, although the previously seen increase in circulating monocytes/macrophages in humans was not observed in our mice. We then subsetted CD3+ cells and performed unsupervised clustering, found subclusters of CD3+ cells (Fig. Supp. 5A), identified canonical CD3+ gene markers (Table S17), and quantified CD3+ subtypes as a proportion of the total CD3+ input number per sample. We observed no significant differences between the MRL/Pdcd1−/− and MRL mice in the blood, but observed a statistically significant increase in the proportion of CD4+ and CD8+ cells in the heart (Fig. Supp. 5B). We then further selected subsets of cells from the CD3+ population – the CD4+ (Fig. Supp. 5C, Table S19) and CD8+ populations (Fig. 7D, Table S18). We found no statistically significant differences in the proportions of CD4 in the blood, but saw a statistically significant increase in central memory CD4+ cells and T-regs in the heart. In CD8+ cell subsets, we saw no statistically significant changes but a trend of increasing effector memory CD8+ cells in the blood and a dramatic and statistically significant increase of effector memory CD8+ cells - the closest correlate to Temra CD8+ cells in humans - in the heart. To understand the transcriptomic changes in the setting of PD-1 loss, we performed differential expression analysis between immune cells isolated from the hearts of wildtype MRL mice and MRL/Pdcd1−/− mice (Fig. 7F, Table S20). We found increased expression of cytotoxicity makers (Nkg7), chemokines (Ccl5), and exhaustion markers (Lgals1) in the effector CD8+ T-cell clusters of MRL/Pdcd1−/− mice compared to MRL. We also noted increased expression of the receptor for Ccl5—Ccr5—in the hearts of MRL/Pdcd1−/− mice compared to MRL (Table S20). To evaluate the transcriptomic changes in the effector memory CD8+ cells in the heart compared to other CD8+ cell subtypes, we performed differential expression analysis on the different CD8+ cell subtypes found in the heart and saw increased cytotoxicity genes (Nkg7), chemokines (Ccl5), and exhaustion markers (Lgals1).
Fig. 7. Analysis of Immune Cell Populations in MRL vs. MRL/Pdcd1−/− mice using scRNA-seq.
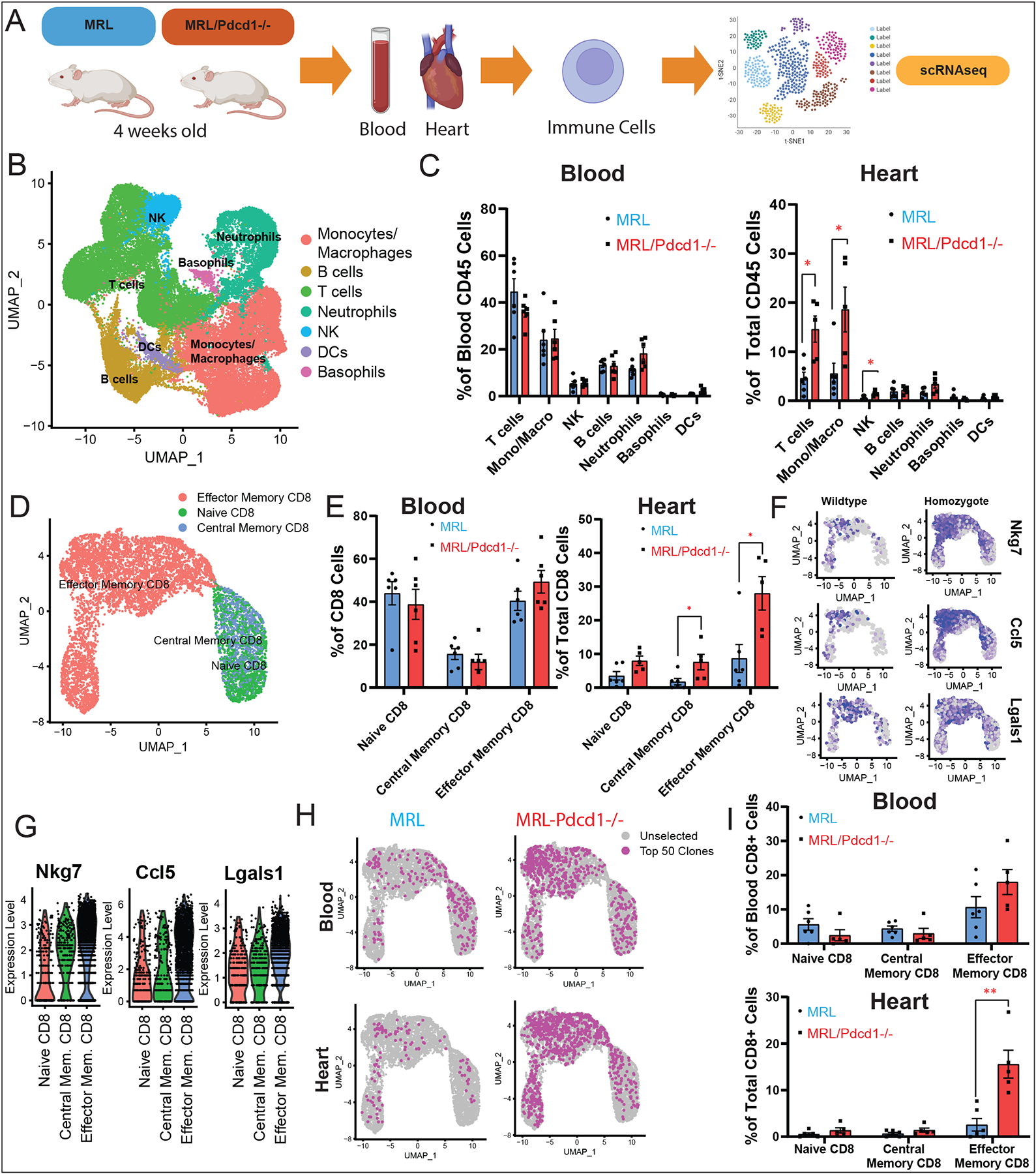
(A) Workflow showing collection of peripheral blood and processing of single-cell suspensions for single-cell RNA-seq from 4-week-old MRL/Pdcd1−/− mice (n=6) with spontaneous myocarditis and control MRL mice (n=6). (B) Identification of CD45+ immune cell clusters from blood and hearts of MRL/Pdcd1−/− and control MRL mice. (C) Quantification of CD45+ cell subtypes across clusters and comparison of MRL vs. MRL/Pdcd1−/− mice show increased macrophages/monocytes as well as T-cells in the hearts of MRL/Pdcd1−/− mice. Average and SEM are shown for each genotype. Statistical analysis compares MRL vs MRL/Pdcd1−/− groups. (D) Identification of CD8+ immune cell clusters from blood and hearts of MRL/Pdcd1−/− and control MRL mice. (E) Quantification of CD8+ cell subtypes across clusters and comparison of MRL vs. MRL/Pdcd1−/− mice. Average and SEM are shown for each genotype. Statistical analysis compares MRL vs MRL/Pdcd1−/− groups. (F) Feature plots showing top differentially expressed genes in CD8+ T-cells among MRL/Pdcd1−/− mice compared to MRL mice, showing increased expression of cytotoxicity markers (Nkg7), myocardial-tropic chemokines (Ccl5), and exhaustion markers (Lgals1) in the effector CD8+ T-cell clusters. (G) Violin plots displaying significantly differentially expressed genes in MRL/Pdcd1−/− compared to MRL mice, including cytotoxicity markers (Nkg7), myocardial-tropic chemokines (Ccl5), and exhaustion markers (Lgals1) in the effector CD8+ T-cell clusters. (H) Visualization of top 50 TCR clonotypes in blood and hearts of MRL and MRL/Pdcd1−/− mice. (I) Quantification of top 50 TCR clonotypes across CD8+ cell subtypes in MRL/Pdcd1−/− mice and MRL control mice.
In order to interrogate the clonal identities of the expanded T-cells in our MRL/Pdcd1−/− mice compared to MRL, we performed single-cell TCR sequencing on immune cells isolated from the blood and hearts of the mice (Fig. 7H). We saw a marked clonal expansion in the effector memory CD8+ cell clusters in both the blood and hearts of MRL/Pdcd1−/− mice compared to MRL (Fig. 7H). We then quantified the cellular identities of the T-cell clonotypes found, and confirmed them to be mostly effector memory CD8+ cells, with a statistically significant expansion of effector memory CD8+ cells found in the hearts of MRL/Pdcd1−/− mice compared to MRL mice. This clonal expansion in effector memory CD8+ cells in the blood/hearts of MRL/Pdcd1−/− mice is correlative with the clonal expansion in blood Temra CD8+ cells found in our ICI myocarditis (Group C) patient samples.
Last but not least, we repeated the ligand-receptor analysis in the blood/hearts of our MRL vs. MRL/Pdcd1−/− mice, and noted similar increased ratio of pro-inflammatory vs. anti-inflammatory interactions between effector CD8+ cells of MRL/Pdcd1−/− with other cell types compared to MRL mice (Fig. Supp. 6). Additionally, we noted a predominance of CCL5 interactions between effector CD8+ T-cells with other immune players in both the blood and heart, suggesting a potentially important role for this chemokine in the setting of PD-1 loss.
DISCUSSION
In this study, we present the first comprehensive profiling of the transcriptome of peripheral blood of patients with ICI myocarditis in comparison to other non-myocarditis irAEs using high resolution single-cell proteomic and transcriptomic profiling techniques. We also correlated our findings using immune cells isolated from the blood and hearts of a correlative mouse model deficient in PD-1 with spontaneous myocarditis. Using both high dimensional mass cytometry (CyTOF) for proteomic data and single-cell RNA-seq for transcriptomic data, we found an expanded population of cytotoxic Temra (effector) CD8+ cells in patients with ICI myocarditis (Group C) compared to all other control groups, including patients without irAEs (Group A) and patients with non-myocarditis irAEs (Group B). Furthermore, we observed that these cytotoxic Temra CD8+ cells are clonally expanded in the blood of myocarditis (Group C) patients compared to the other patient control groups. They also appear to express increased levels of pro-inflammatory activation/cytotoxicity markers (GZMB, GNLY, CST7, NKG7, KLRB1, IL-32) and pro-inflammatory chemokines (CCL5/CCL4/CCL4L2), some of which have an affinity towards chemokine receptors known to be expressed in the heart (CCR1 and CCR5) (29). We believe that these cytotoxic expanded Temra CD8+ cells expressing increased levels of cardio-tropic chemokines play a critical role in the pathogenesis of ICI myocarditis.
Due to its fulminant nature with the risk of heart failure, arrhythmias and death (35), ICI myocarditis remains one of the most feared complications of ICI therapy. Because an understanding of the mechanism of pathogenesis as well as targeted therapeutics for this devastating complication are lacking, our study should provide useful insights into key immune cell subsets and transcriptomic profiles implicated in ICI myocarditis compared to irAE in other organs, thus providing several potential diagnostic and therapeutic targets for this disease.
Our data showing an expansion of cytotoxic CD8+ T-cell populations in ICI-induced myocarditis patients and expansion of effector CD8+ T-cells in the hearts of MRL/Pdcd1−/− mice is consistent with prior human and mouse studies in the field. Post-mortem cardiac muscle histopathology of ICI myocarditis patients have previously demonstrated intense patchy lymphocytic infiltrates within the myocardium of CD8>CD4 T-cells (35). Genetic knockout of the immune checkpoint PD-1 in the MRL-Pdcd−/− mouse model have also demonstrated spontaneous CD8>CD4 lymphocytic infiltrates in the hearts by 4–6 weeks of age (14). However, no prior studies have delineated these cytotoxic T-cells as Temra CD8+ cells and effector CD8+ T-cells respectively, as well as demonstrated their increased clonal expansion in the peripheral blood of ICI myocarditis patients compared to the blood of patients with non-myocarditis irAE. To explain this, we hypothesize that the degree of systemic inflammatory response in ICI myocarditis patients may be greater than in relatively milder irAE cases in group B and in the non-irAE group A, thus leading to a larger effector T cell expansion reflected in the peripheral blood.
Additionally, prior studies have not performed correlative single-cell TCR sequencing in PD-1 deficient mice with myocarditis and shown a similar effector CD8+ clonal expansion in the heart. Interestingly, Temra CD8+ cells have previously been shown to be expanded in the peripheral blood of patients with other types of autoimmune disease—for example, in rheumatoid arthritis (36). However, they have never previously been shown to play a role in myocarditis. Additionally, our in-depth transcriptomic profiling of these Temra CD8+ cells, including the finding of their increased expression of the chemokines CCL5/CCL4/CCL4L2 is novel. Recently published data in mice with TnI-directed autoimmune myocarditis were also found to have elevated levels of expression of CCL3, CCL4 and CCL5 and their chemokine receptors CCR1/CCR2/CCR5 in their hearts (8), suggesting a potential mechanism for T-cell homing to the heart. Our study confirms this finding in PD-1 loss induced myocarditis mice as well as adds the novel finding of a similar discovery in circulating lymphocytes in humans.
In contrast to prior studies reporting increased Th17 effector cell activity in the hearts of two patients with ICI myocarditis (8), we did not observe a consistent increase in Th17 effector cell activity in the peripheral blood of our much larger cohort of patients, although we saw a trend in our CyTOF data. Another study reported increased Th17 cell activity in the blood of patients with autoimmune myocarditis; however, these patients had not been treated with immune checkpoint inhibitors, suggesting subtle differences between these varying types of myocarditis (6). Although our study focused on the peripheral blood of ICI myocarditis patients rather than biopsied heart tissue, we were able to correlate our findings in the heart tissues of a mouse model of PD-1 deficiency-induced myocarditis. Additionally, we have been able to gain insights from this largest known cohort of proteomic and transcriptomic single-cell data from ICI myocarditis patients to date, and make direct nuanced comparisons between several groups – healthy patients not on ICI, patients on ICI who experienced no irAE (Group A), and patients on ICI who experience non-myocarditis irAE (Group B). In our study, we found that clonal Temra CD8+ cell expansion and elevated expression levels of heart-tropic chemokines CCL5/CCL4/CCL4L2 previously shown to play a role in myocardial inflammation (28) can be observed even in the peripheral blood, highlighting them as important peripheral biomarkers in the disease. Furthermore, our ligand-receptor analysis supported the interactions of these Temra CD8+ cells with several innate immune players (monocytes/macrophages, dendritic cells, neutrophils) in the peripheral blood. All of these data provide the basis for generating new hypotheses of ICI myocarditis pathogenesis, suggesting potential mechanisms by which cytotoxic Temra CD8+ may direct immune homing to the heart and recruit other immune cells to cause immune-mediated cardiac damage.
Our findings here also contribute to the broader understanding of ICI immune-mediated toxicities and provide a contrast of ICI myocarditis with irAE in other organ systems. The discovery of clonal expansion of T-cells after checkpoint blockade has been known for some time (37), and may even contribute to the therapeutic benefits of immune checkpoint blockade on tumor regression. However, distinguishing between the therapeutic expansion of clonal T-cell from a pathologic one that leads to autoimmune side effects will be necessary to prevent such toxicity with ICI treatment while retaining the benefits of immune checkpoint blockade. In a recent study using single-cell RNA-seq to examine patients with ICI-mediated colitis, Luoma et al also observed a CD8+ T-cell clonal expansion in the colonic tissues of these patients (30).
Similar to the clonally expanded CD8+ cells in our patient population, the clonally expanded CD8+ cells in their patients were highly cytotoxic and activated (expressing high levels of GZMB and HLA-DRA), but expressed high levels of the mucosal associated chemokine CXCR6, similar to some of our Group B patients. In contrast, the clonally expanded CD8+ cells in our Group C myocarditis patients expressed high levels of chemokines CCL4/CCL4L2/CCL5. The finding of organ-selective chemokine expression in the Temra CD8+ cells that bind to receptors enriched in heart tissues (CCR1/CCR5) of our myocarditis Group C patients may provide clues with regards to who may be at risk for developing cardiac-specific immune related events compared to the general population of patients experiencing irAE. Additionally, T-cells from both our Group C patients and their colitis patients expressed high levels of the immune-cell directing chemokine, CXCR3 (Fig. 4E), suggesting a potential important common pathway of pathogenesis of irAE through this chemokine.
Due to the availability of early and late myocarditis blood samples, our study also provides new insight into the time-course of cellular and molecular changes that occur throughout the onset and resolution of ICI myocarditis. ICI myocarditis has a notoriously unpredictable time course and can occur with a median onset of 17–34 days after initiation of ICI therapy, although cases have been identified anywhere from 1 to 240 days from start of therapy (35,38–40). In our patient, we were able to collect blood at the time of diagnosis of the positive troponin I of 10 ng/mL (t=0) and track to day 65 after a full four-week course of glucocorticoids and resolution of clinical signs of myocarditis. The persistence of elevated clonal CD8+ Temra proportions (although attenuated) even at 65 days after diagnosis and treatment in this patient is intriguing and suggests additional T-cell exhaustion transitional mechanisms may be responsible for decreased pathogenicity over time, rather than singularly steroid-mediated apoptosis of T-cells as previously described (41). The concept of T cell exhaustion is extremely important in immune checkpoint cancer biology, and the ability of ICI to re-invigorate exhausted T-cells to induce cytotoxicity against tumor tissue has been well-established (42). However, in the case of irAE, it may be desirable to reduce the cytotoxicity of T-cells that are damaging the normal tissue. Interestingly, our data suggests a phenotypic shift in the activated CD8+ Temra population, from an early cytotoxic and pro-inflammatory profile to a late exhaustion phenotype expressing known markers of T-cell exhaustion such as KLRG1 (43–45), CX3CR1 (46) and LAG3 (47) after glucocorticoid treatment. Indeed, T cell exhaustion has been described to play a central role in determining the outcome of many autoimmune diseases such as type I diabetes mellitus (48), and thus may also play a role in autoimmunity and response to anti-inflammatory treatment.
One of the challenges of diagnosing ICI myocarditis, and myocarditis at large, is the difficulty and risk of obtaining heart biopsy samples for accurate diagnosis. Our study provides several potential cellular and molecular biomarkers associated with ICI myocarditis in the peripheral blood. Peripheral blood has been shown to be a rich resource of immunologic information which may reflect immune changes at the tissue level, as demonstrated by prior studies from our group (49). Here, we use peripheral blood to glean insights into the state of inflammation in the heart in a rare patient population, with analogous findings in the hearts of PD-1 deficient mice. However, it will be important to adjudicate and verify these biomarker targets in future studies using human heart tissue samples.
Other limitations for our data include the male sex dominance in our data. In larger epidemiologic studies, data have been conflicting regarding the association between ICI myocarditis and sex. Although 67–77% of myocarditis cases across several studies were reported in males (35,39,40), it is unclear whether this may reflect the disproportionate representation of men in ICI trials. Another study utilizing the FDA Adverse Event Reporting System demonstrated a higher incidence of ICI myocarditis in women (51), despite the classic association of autoimmune viral myocarditis with the male sex (52). Thus, these gender differences may be important to study in more detail going forward. Although we adjudicated our data to rule out age and sex differences (Fig. Supp. 8), it would be important to correlate the findings of this novel hypothesis-generating study with much larger confirmatory cohorts in the future.
Our data give rise to important therapeutic targets for the treatment of ICI myocarditis and other ICI-related autoimmune side effects. The expression of T-cell exhaustion markers and upregulation of other immune checkpoints such as LAG3 suggest them as an intriguing therapeutic alternative to steroids. A precedent for this has been seen with abatacept, a CTLA4 agonist which has been used effectively to treat ICI myocarditis (53). Therapeutic targeting and upregulation of other immune checkpoints, however, may have implications for the long-term effectiveness of the ICI from a cancer perspective. Thus, the most intriguing drug targets from our dataset may be the chemokines (CCL4, CCL4L2, and CCL5), which are upregulated in our Temra CD8 dataset and may provide approaches to target myocardial-specific pathways of inflammation while preserving the broader anti-tumor immune pathways activated by ICI. In addition to being expressed on the myocardium as seen in previous studies (29) and in the hearts of our MRL/Pdcd1−/− mice, CCR5 is a receptor expressed broadly on lymphocytes and functions as a port of entry for the HIV virus (54,55). Thus, the well-established CCR5 inhibitor, Maraviroc, already in clinical use for the treatment of patients with HIV with relatively few known side effects, may pose an example of a pharmacologic to explore in patients at risk for or with ICI myocarditis. Maraviroc and other CCR1/CCR5 inhibitors may provide attractive and specific therapeutic options in patients with ICI myocarditis whose current therapeutic options are limited to broad-spectrum glucocorticoids or T-cell suppressive therapies (tacrolimus, antithymocyte globulin, infliximab, etc.) (50). The further investigation of this chemokine pathway in addition to the other therapeutic targets identified in this study may lead to significant improvements in the cardiac safety of patients on ICI.
Lastly, our study contributes to the field of peripheral biomarkers of myocarditis at large. While myocarditis may be due to many different etiologies, there may be a common immune pathway leading to pathogenesis and myocardial damage (8), some of which may be exacerbated in the setting of ICI therapy (7,56). Effector CD8+ T-cells have long been thought to play a role in the pathogenesis of myocarditis (57,58), and these findings have also been reflected in our study in ICI myocarditis patients. Furthermore, we have identified a specialized type of effector CD8+ T-cells – the Temra CD8+ cells—to be particularly associated with ICI myocarditis. To our knowledge, our study is the largest study using single-cell techniques to immunophenotype myocarditis in the blood, utilizing this novel high-resolution technology to better understand peripheral immune changes in the setting of cardiac inflammation.
MATERIALS AND METHODS
Data and materials availability
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure. Anonymized data and materials are publicly available in the following repositories. CyTOF data has been uploaded onto the Mendeley Data repository at doi: 10.17632/cm8pxdt5pm.1 (60). All single-cell RNA-seq, single-cell TCR-seq, and CITE-seq data in this paper have been uploaded onto the NCBI Gene Expression Omnibus (GEO) data repository at accession number GSE180045 (61).
Study Design and Patient Population
All studies were approved by the Stanford Institutional Review Board and performed in accordance with guidelines on human cell research. See full Supplementary Methods for continuation.
Statistical Methods
Statistical comparisons between cell subgroups were performed within each patient or mouse genotype group using standard statistical methods. Specifically, averages were calculated within each patient group and standard error measurements were calculated within each group. The Shapiro-Wilk and Kolmogorov-Smirnov tests of normality were performed on the data sets to determine whether normality could be assumed, prior to proceeding with downstream parametric or non-parametric tests. The non-parametric Mann-Whitney test was used to determine significant cell proportion differences between 1) non-ICI treated vs. ICI treated patient groups and 2) MRL vs. MRL/Pdcd1−/− mice. For comparisons between Group A (no irAE), Group B (non-myocarditis), and Group C (myocarditis) patients, if tests of normality were passed, we performed one-way ANOVA followed by correction for multiple comparisons by controlling the False Discovery Rate (FDR) using the two-stage step-up method of Benjamini, Krieger and Yekutieli with a significance threshold of q-value < 0.05. If tests of normality were not passed, we used the non-parametric Kruskal-Wallis test, followed by comparisons between the individual groups using the post-hoc Dunn’s multiple comparisons test (significance threshold of adjusted p value < 0.05).
Supplementary Material
CLINICAL PERSPECTIVE.
What’s new?
ICI myocarditis is associated with an expansion of specialized effector CD8+ T-cells (Temra CD8+ cells) in the peripheral blood of patients with ICI myocarditis.
These Temra CD8+ cells exhibit markers of increased activation and cytotoxicity, suggesting their potential pathogenic role in ICI myocarditis.
Temra CD8+ cells exhibit increased expression of the heart-tropic chemokines.
This is the first application of deep immunophenotyping with single-cell RNA-seq, single-cell TCR-seq, and CyTOF (mass cytometry) applied large-scale in patients with ICI myocarditis.
What are the clinical implications?
Novel cytotoxic and activated Temra CD8+ cells expanded in the peripheral blood may be a disease biomarker of ICI myocarditis.
Heart-tropic chemokines expressed by Temra CD8+ cells, such as CCL5, may become important therapeutic targets in the prevention and treatment of ICI myocarditis and other types of pathologic cardiac inflammation.
Deep immunophenotyping with single-cell RNA-seq, single-cell TCR-seq, and CyTOF (mass cytometry) can be applied to bio-banked patient populations, such as that used in this study, to gain detailed understanding of immune cell subsets in cardiac and other types of systemic inflammatory disorders.
Acknowledgments:
We thank the following people for allowing access to their patients for recruitment based on the IRB-approved protocol in this manuscript: Dr. Alice Fan, Dr. Sukhimani Padda, Dr. Kavitha Ramchandran, Dr. Dimitrios Colevas, Dr. Sumit Shah, Dr. Maximilian Diehn, Dr. Michael B. Fowler, and Dr. Randall Vagelos. Additionally, we thank the Stanford Cancer Center, Stanford Cardiovascular Research Institute, Stanford Human Immune Monitoring Center (HIMC), and Stanford Department of Immunology and Microbiology for their cooperation and technical support during the course of the work in this manuscript. Acknowledgement also goes to the Howard Hughes Medical Institute for their support and funding of Dr. Davis.
Funding:
NIH/NIGMS grant 1RM1 GM131981-02
American Heart Association - Established Investigator Award
Hoffmann/Schroepfer Foundation
Additional Venture Foundation
Joan and Sanford I. Weill Scholar Fund
Sanofi US, Inc. - iAward grant
NIHLBI grant R01HL13483004
American Heart Association - grant AW849785 - Transformative Award
NIH grant 1F32HL149188-01
Sarnoff Scholar Career Development Award
Howard Hughes Medical Institute
Competing interests:
The authors declare the following potential conflicts of interest. Dr. Sean Wu receives funding from Sanofi. Lih-Ling Lin is an employee of Sanofi. Dr. Ronald Witteles sits on the advisory boards for Pfizer, Alnylam, Ionis/Akcea, Eidos, and Intelia. Dr. Joel Neal serves a consulting or advisory role for: AstraZeneca, Genentech/Roche, Exelixis, Jounce Therapeutics, Takeda Pharmaceuticals, Eli Lilly and Company, Calithera Biosciences, Amgen, Iovance, Biotherapeutics, Blueprint Pharmaceuticals, Regeneron Pharmaceuticals, Natera. Dr. Joel Neal receives research funding from: Genentech/Roche, Merck, Novartis, Boehringer Ingelheim, Exelixis, Nektar Therapeutics, Takeda Pharaceuticals, Adaptimmune, GSK, Janssen, and AbbVie. Dr. Heather Wakelee has grants or contracts from: ACEA Biosciences, Arrys Therapeutics, AstraZeneca/Medimmune, BMS, Celgene, Clovis Oncology, Exelixis, Genentech/Roche, Gilead, Merck, Novartis, Pharmacyclics, Sea Gen, Xcovery. Dr. Wakelee also participates on the data safety monitoring and/or advisory boards of: AstraZeneca, Xcovery, Janssen, Daiichi, Sankyo, Blueprint, Mirati, Helsinn, Merck (not compensated), Genentech/Roche (not compensated). She is President-Elect of IASLC and she sits on the Executive Committee of ECOG-ACRIN.
Non-Standard Abbreviations and Acronyms
- CCL4
Chemokine (C-C motif) ligands 4
- CCL5
Chemokine (C-C motif) ligands 5
- CCL4L2
Chemokine (C-C motif) ligands 4 like 2
- CCR1
Chemokine (C-C Motif) Receptor 1
- CCR5
Chemokine (C-C Motif) Receptor 5
- CITE-seq
Cellular Indexing of Transcriptomes and Epitopes by Sequencing
- CTLA4
Cytotoxic T-Cell Antigen-4
- CXCR3
Chemokine (CXC Motif) Receptor 3
- CXCR6
Chemokine (CXC Motif) Receptor 6
- CX3CR1
Chemokine (C-X3-C Motif) Receptor 1
- CyTOF
Time-of-Flight Mass Cytometry
- FDR
False Discovery Rate
- GEO
Gene Expression Omnibus
- GNLY
Granulysin
- GZMA/B
granzyme A or B
- HLA-DRA
Major Histocompatibility Complex, Class II, DR Alpha
- ICI
Immune Checkpoint Inhibitors
- IL-32
Interleukin-32
- IL2RG
Interleukin 2 Receptor Subunit Gamma
- irAE
immune-related Adverse Events
- IRB
Institutional Review Board
- KLF2
Kruppel Like Factor 2
- KLRB1
Killer Cell Lectin Like Receptor B1
- KLRG1
Killer Cell Lectin Like Receptor G1
- KLRK1
Killer Cell Lectin Like Receptor K1
- LAG3
Lymphocyte Activating 3
- LGALS1
Galectin 1
- LGE
Late Gadolinium Enhancement
- MRI
Magnetic Resonance Imaging
- MRL
Murphy Roths Large
- NKG2D
Nature Killer Cell Granule Protein 2D
- NKG7
Natural Killer Cell Granule Protein 7
- PBMC
Peripheral Blood Mononuclear Cell
- PCA
Principal Component Analysis
- PD-1
Programmed Death-1
- scRNA-seq
Single-Cell RNA Sequencing
- SNN
Shared Nearest Neighbor
- TCR
T Cell Receptor
- TCR-seq
T Cell Receptor Sequencing
- Temra CD8+ Cells
T Effector Memory Cells Re-expressing CD45RA
- Th17
T Helper Cell Subset, Expression IL-17
- TIGIT
T cell immunoreceptor with Ig and ITIM domains
- TNF
Tumor Necrosis Factor
- UMAP
Uniform Manifold Approximation and Projection
Footnotes
Supplemental Materials
See also Supplementary Methods Section for:
Human PBMC Collection and Storage
CyTOF/Mass Cytometry Sample Preparation and Staining
CyTOF Data Pre-Processing and Analysis
Animal Studies
Sample Preparation and Staining for 10x 5' barcoded scRNA-seq with Feature Barcoding and TCR Sequencing
Library Preparation for scRNA-seq
Sequencing of scRNA-seq libraries
Single Cell RNA-seq Data Pre-Processing
Identification of Cell Clusters (includes SCTransform workflow (59))
Annotation of Cell Clusters and Data Visualization
Differential Expression Analysis
T Cell Receptor Data Analysis
Cluster Analysis of Clonally Expanded T-Cells
Ligand-Receptor Expression and Cell Interactions
Data and materials availability:
CyTOF data has been uploaded onto the Mendeley Data repository at doi: 10.17632/cm8pxdt5pm.1. All single-cell RNA-seq, single-cell TCR-seq, and CITE-seq data in this paper have been uploaded onto the NCBI Gene Expression Omnibus (GEO) data repository at accession number GSE180045.
References and Notes
- 1.Moslehi JJ, Salem JE, Sosman JA, Lebrun-Vignes B, Johnson DB. Increased reporting of fatal immune checkpoint inhibitor-associated myocarditis. Lancet. 2018;391:933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mahmood SS, Fradley MG, Cohen JV., Nohria A, Reynolds KL, Heinzerling LM, et al. Myocarditis in Patients Treated With Immune Checkpoint Inhibitors. J Am Coll Cardiol. 2018; 71:1755–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salem J, Manouchehri A, Moey M, Lebrun-vignes B, Bastarache L, Pariente A, et al. Articles Cardiovascular toxicities associated with immune checkpoint inhibitors : an observational, retrospective, pharmacovigilance study. Lancet Oncol. 2018;2045:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caforio ALP. Myocarditis: endomyocardial biopsy and circulating anti-heart autoantibodies are key to diagnosis and personalized etiology-directed treatment. European Heart Journal. 2021;42:1618–1620. [DOI] [PubMed] [Google Scholar]
- 5.Bracamonte-Baran W, Čiháková D. Cardiac autoimmunity: Myocarditis. Advances in Experimental Medicine and Biology. 2017; 1003:187–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blanco-Domínguez R, Sánchez-Díaz R, de la Fuente H, Jiménez-Borreguero LJ, Matesanz-Marín A, Relaño M, et al. A Novel Circulating MicroRNA for the Detection of Acute Myocarditis. N Engl J Med. 2021;384:2014–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grabie N, Lichtman AH, Padera R. T cell checkpoint regulators in the heart. Cardiovasc Res. 2019;115:869–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bockstahler M, Fischer A, Goetzke CC, Neumaier HL, Sauter M, Kespohl M, et al. Heart-Specific Immune Responses in an Animal Model of AutoimmuneRelated Myocarditis Mitigated by an Immunoproteasome Inhibitor and Genetic Ablation. Circulation. 2020;1885–1902. [DOI] [PubMed] [Google Scholar]
- 9.Amioka N, Nakamura K, Kimura T, Ohta-Ogo K, Tanaka T, Toji T, et al. Pathological and clinical effects of interleukin-6 on human myocarditis. J Cardiol. 2021; 2–5. [DOI] [PubMed] [Google Scholar]
- 10.Thavendiranathan P, Zhang L, Zafar A, Drobni ZD, Mahmood SS, Cabral M, et al. Myocardial T1 and T2 Mapping by Magnetic Resonance in Patients With Immune Checkpoint Inhibitor–Associated Myocarditis. J Am Coll Cardiol. 2021;77:1503–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang L, Reynolds KL, Lyon AR, Palaskas N, Neilan TG. The Evolving Immunotherapy Landscape and the Epidemiology, Diagnosis, and Management of Cardiotoxicity: JACC: CardioOncology Primer. JACC CardioOncology. 2021;3:35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson DB, Balko JM, Compton ML, Chalkias S, Gorham J, Xu Y, et al. Fulminant Myocarditis with Combination Immune Checkpoint Blockade. N Engl J Med. 2016;375:1749–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Touat M, Maisonobe T, Knauss S, Ben Hadj Salem O, Hervier B, Auré K, et al. Immune checkpoint inhibitor-related myositis and myocarditis in patients with cancer. Neurology 2018; 985–994. [DOI] [PubMed] [Google Scholar]
- 14.Wang J, Okazaki IM, Yoshida T, Chikuma S, Kato Y, Nakaki F, et al. PD-1 deficiency results in the development of fatal myocarditis in MRL mice. Int Immunol. 2010; 22: 443–452. [DOI] [PubMed] [Google Scholar]
- 15.Tarrio ML, Grabie N, Bu D -x., Sharpe AH, Lichtman AH. PD-1 Protects against Inflammation and Myocyte Damage in T Cell-Mediated Myocarditis. J Immunol. 2012; 188: 4876–4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bendall SC, Simonds EF, Qiu P, Amir ED, Krutzik PO, Finck R, et al. Single-Cell Mass Cytometry of Differential Immune and Drug Responses Across a Human Hematopoietic Continuum. Science. 2011;332:687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newell E, Sigal N, Bendall S, Nolan G, Davis MM. Cytometry by Time-of-Flight Shows Combinatorial Cytokine Expression and Virus-Specific Cell Niches within a Continuum of CD8+ T Cell Phenotypes. Immunity. 2012;36:142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szabo PA, Levitin HM, Miron M, Snyder ME, Senda T, Yuan J, et al. Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease. Nat Commun. 2019;10:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gibellini L, De Biasi S, Porta C, Lo Tartaro D, Depenni R, Pellacani G, et al. Single-Cell Approaches to Profile the Response to Immune Checkpoint Inhibitors. Front Immunol. 2020;11:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leipold M, Maecker H. Phenotyping of Live Human PBMC using CyTOFTM Mass Cytometry. Bio-Protocol. 2015;5:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Russi AE, Brown MA. Mass Cytometry: Single Cells, Many Features. Cell. 2016;165:255–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gadalla R, Noamani B, MacLeod BL, Dickson RJ, Guo M, Xu W, et al. Validation of CyTOF against flow cytometry for immunological studies and monitoring of human cancer clinical trials. Front Oncol. 2019;9:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chattopadhyay PK, Winters AF, Iii WEL, Laino AS, Woods DM. High-Parameter Single-Cell Analysis. Annu Rev Anal Chem. 2019;12:411–30. [DOI] [PubMed] [Google Scholar]
- 24.Han A, Glanville J, Hansmann L, Davis MM. Linking T-cell receptor sequence to functional phenotype at the single-cell level. Nat Biotechnol. 2014; 32:684–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tomic A, Tomic I, Dekker CL, Maecker HT, Davis MM. The FluPRINT dataset, a multidimensional analysis of the influenza vaccine imprint on the immune system. Sci Data. 2019;6:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilk AJ, Rustagi A, Zhao NQ, Roque J, Martínez-Colón GJ, McKechnie JL, et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat Med. 2020; 26: 1070–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Z, Liu Y, Hu S, Zhang M, Shi B, Wang Y. Decreased Treg Cell and TCR Expansion Are Involved in Long-Lasting Graves’ Disease. Front Endocrinol. 2021;12:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Machado FS, Koyama NS, Carregaro V, Ferreira BR, Milanezi CM, Teixeira MM, et al. CCR5 plays a critical role in the development of myocarditis and host protection in mice infected with Trypanosoma cruzi. J Infect Dis. 2005;191:627–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Damås JK, Eiken HG, Øie E, Bjerkeli V, Yndestad A, Ueland T, et al. Myocardial expression of CC- and CXC-chemokines and their receptors in human end-stage heart failure. Cardiovasc Res. 2000;47:778–787. [DOI] [PubMed] [Google Scholar]
- 30.Luoma AM, Suo S, Williams HL, Sharova T, Sullivan K, Manos M, et al. Molecular Pathways of Colon Inflammation Induced by Cancer Immunotherapy. Cell. 2020;182:655–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kelly KM, Tocchetti CG, Lyashkov A, Tarwater PM, Bedja D, Graham DR, et al. CCR5 inhibition prevents cardiac dysfunction in the SIV/macaque model of HIV. J Am Heart Assoc. 2014;3:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wensveen FM, Jelenčić V, Polić B. NKG2D: A master regulator of immune cell responsiveness. Front Immunol. 2018; 9: 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Efremova M, Vento-Tormo M, Teichmann SA, Vento-Tormo R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat Protoc. 2020;15:1484–1506. [DOI] [PubMed] [Google Scholar]
- 34.Bandala-Sanchez E, Zhang Y, Reinwald S, Dromey JA, Lee BH, Qian J, et al. T cell regulation mediated by interaction of soluble CD52 with the inhibitory receptor Siglec-10. Nat Immunol. 2013;14:741–748. [DOI] [PubMed] [Google Scholar]
- 35.Johnson DB, Balko JM, Compton ML, Chalkias S, Gorham J, Xu Y, et al. Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med. 2016;375:1749–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takeshita M, Suzuki K, Kondo Y, Morita R, Okuzono Y, Koga K, et al. Multi-dimensional analysis identified rheumatoid arthritis-driving pathway in human T cell. Ann Rheum Dis. 2019;78:1346–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yost K, Satpathy A, Wells D, Qi Y, Wang C, Kageyama R, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. bioRxiv. 2019;25:648899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Waliany S, Neal JW, Reddy S, Wakelee H, Shah SA, Srinivas S, et al. Myocarditis Surveillance With High-Sensitivity Troponin I During Cancer Treatment With Immune Checkpoint Inhibitors. JACC CardioOncology. 2021;3:137–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mahmood SS, Fradley MG, Cohen JV., Nohria A, Reynolds KL, Heinzerling LM, et al. Myocarditis in Patients Treated With Immune Checkpoint Inhibitors. J Am Coll Cardiol. 2018;71:1755–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salem JE, Manouchehri A, Moey M, Lebrun-Vignes B, Bastarache L, Pariente A, et al. Cardiovascular toxicities associated with immune checkpoint inhibitors: an observational, retrospective, pharmacovigilance study. Lancet Oncol. 2018;19:1579–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arbour KC, Mezquita L, Long N, Rizvi H, Auclin E, Ni A, et al. Impact of baseline steroids on efficacy of programmed cell death-1 and programmed death-ligand 1 blockade in patients with non–small-cell lung cancer. J Clin Oncol. 2018;36:2872–2878. [DOI] [PubMed] [Google Scholar]
- 42.Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell. 2018. Nov 1;175:998–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herndler-Brandstetter D, Ishigame H, Shinnakasu R, Plajer V, Stecher C, Zhao J, et al. KLRG1+ Effector CD8+ T Cells Lose KLRG1, Differentiate into All Memory T Cell Lineages, and Convey Enhanced Protective Immunity. Immunity. 2018;48:716–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Henson SM, Akbar AN. KLRG1-more than a marker for T cell senescence. Age. 2009;31:285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li L, Wan S, Tao K, Wang G, Zhao E. KLRG1 restricts memory T cell antitumor immunity. Oncotarget. 2016;7:61670–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y. Chemokine-chemokine receptor CCL2-CCR2 and CX3CL1-CX3CR1 axis may play a role in the aggravated inflammation in primary biliary cirrhosis. Dig Dis Sci. 2014;59:358–364. [DOI] [PubMed] [Google Scholar]
- 47.Graydon CG, Mohideen S, Fowke KR. LAG3’s Enigmatic Mechanism of Action. Front Immunol. 2021;11:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McKinney EF, Lee JC, Jayne DRW, Lyons PA, Smith KGC. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature. 2015;523:612–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao F, Sikora MJ, Serratelli WS, Fernandes RA, Louis DM, Yao W, et al. Opposing T cell responses in experimental autoimmune encephalomyelitis. Nature. 2019; 572: 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Waliany S, Lee D, Witteles RM, Neal JW, Nguyen P, Davis MM, et al. Immune Checkpoint Inhibitor Cardiotoxicity: Understanding Basic Mechanisms and Clinical Characteristics and Finding a Cure. Annu Rev Pharmacol Toxicol. 2021;61:113–34. [DOI] [PubMed] [Google Scholar]
- 51.Zamami Y, Niimura T, Okada N, Koyama T, Fukushima K, Izawa-Ishizawa Y, et al. Factors Associated with Immune Checkpoint Inhibitor-Related Myocarditis. JAMA Oncology. 2019; 5:1635–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kyto V, Sipila J, Rautava P. Gender differences in myocarditis: a nationwide study in Finland. Eur Heart J. 2013; 34: 3505. [Google Scholar]
- 53.Salem JE, Allenbach Y, Kerneis M. Abatacept for severe immune checkpoint inhibitor–associated myocarditis. N Engl J Med. 2019;380:2377–9. [DOI] [PubMed] [Google Scholar]
- 54.Marques RE, Guabiraba R, Russo RC, Teixeira MM. Targeting CCL5 in inflammation. Expert Opin Ther Targets. 2013;17:1439–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Crawford A, Angelosanto JM, Nadwodny KL, Blackburn SD, Wherry EJ. A role for the chemokine RANTES in regulating CD8 T cell responses during chronic viral infection. PLoS Pathog. 2011;7:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lichtman AH. The heart of the matter: Protection of the myocardium from T cells. Journal of Autoimmunity. 2013; 45: 90–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Henke A, Huber S, Stelzner A, Whitton JL. The role of CD8+ T lymphocytes in coxsackievirus B3-induced myocarditis. J Virol. 1995;69:6720–6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grabie N, Delfs MW, Westrich JR, Love VA, Stavrakis G, Ahmad F, et al. IL-12 is required for differentiation of pathogenic CD8+ T cell effectors that cause myocarditis. J Clin Invest. 2003;111:671–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. bioRxiv. 2019;1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhu Han, Wu Sean (2022). Identification of Pathogenic Immune Cell Subsets in Checkpoint Inhibitor-induced Myocarditis. Mendeley Data. DOI: 10.17632/cm8pxdt5pm.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu Han, Wu Sean (2022). Identification of Pathogenic Immune Cell Subsets in Checkpoint Inhibitor-induced Myocarditis. Geo Expression Omnibus. Accession GSE180045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
CyTOF data has been uploaded onto the Mendeley Data repository at doi: 10.17632/cm8pxdt5pm.1. All single-cell RNA-seq, single-cell TCR-seq, and CITE-seq data in this paper have been uploaded onto the NCBI Gene Expression Omnibus (GEO) data repository at accession number GSE180045.