

Abstract
Background
Cytochrome P450 4F3 (CYP4F3) is an ω‐hydroxylase that oxidizes leukotriene B4 (LTB4), prostaglandins, and fatty acid epoxides. LTB4 is synthesized by leukocytes and acts as a chemoattractant for neutrophils, making it an essential component of the innate immune system. Recently, involvement of the LTB4 pathway was reported in various immunological disorders such as asthma, arthritis, and inflammatory bowel disease. We report a 26‐year‐old female with a complex immune phenotype, mainly marked by exhaustion, muscle weakness, and inflammation‐related conditions. The molecular cause is unknown, and symptoms have been aggravating over the years.
Methods
Whole exome sequencing was performed and validated; flow cytometry and enzyme‐linked immunosorbent assay were used to describe patient's phenotype. Function and impact of the mutation were investigated using molecular analysis: co‐immunoprecipitation, western blot, and enzyme‐linked immunosorbent assay. Capillary electrophoresis with ultraviolet detection was used to detect LTB4 and its metabolite and in silico modelling provided structural information.
Results
We present the first report of a patient with a heterozygous de novo missense mutation c.C1123 > G;p.L375V in CYP4F3 that severely impairs its activity by 50% (P < 0.0001), leading to reduced metabolization of the pro‐inflammatory LTB4. Systemic LTB4 levels (1034.0 ± 75.9 pg/mL) are significantly increased compared with healthy subjects (305.6 ± 57.0 pg/mL, P < 0.001), and immune phenotyping shows increased total CD19+ CD27− naive B cells (25%) and decreased total CD19+ CD27+ IgD− switched memory B cells (19%). The mutant CYP4F3 protein is stable and binding with its electron donors POR and Cytb5 is unaffected (P > 0.9 for both co‐immunoprecipitation with POR and Cytb5). In silico modelling of CYP4F3 in complex with POR and Cytb5 suggests that the loss of catalytic activity of the mutant CYP4F3 is explained by a disruption of an α‐helix that is crucial for the electron shuffling between the electron carriers and CYP4F3. Interestingly, zileuton still inhibits ex vivo LTB4 production in patient's whole blood to 2% of control (P < 0.0001), while montelukast and fluticasone do not (99% and 114% of control, respectively).
Conclusions
A point mutation in the catalytic domain of CYP4F3 is associated with high leukotriene B4 plasma levels and features of a more naive adaptive immune response. Our data provide evidence for the pathogenicity of the CYP4F3 variant as a cause for the observed clinical features in the patient. Inhibitors of the LTB4 pathway such as zileuton show promising effects in blocking LTB4 production and might be used as a future treatment strategy.
Keywords: CYP4F3, LTB4, POR, Cytb5, Exhaustion, Muscle weakness
Introduction
The cytochrome P450 (CYP) superfamily of enzymes are heme‐containing proteins and are involved in a wide variety of pathways such as fatty acid metabolism, cholesterol production, and steroid hormone synthesis. They are also essential for the metabolism of drugs and detoxification of foreign chemicals. 1 The CYP4F subfamily members share 80% amino acid identity and have a highly conserved gene structure. 2 , 3 They are mainly involved in the metabolism of arachidonic acid and xenobiotics. Cytochrome P450 4F3 (CYP4F3) is an ω‐hydroxylase that plays a major role in the metabolism of polyunsaturated fatty acid (PUFA). It specifically oxidizes arachidonic acid (AA), leukotriene B4 (LTB4), and fatty acid epoxides. 4
The CYP4F3 gene undergoes tissue‐specific alternative splicing generating CYP4F3A and CYP4F3B, 4 marked by incorporation of exon 4 or exon 3, respectively. These splice variants retain an identical size but differ in the amino acids 67‐114, leading to a change in substrate‐specificity. CYP4F3A is predominantly expressed in leukocytes, whereas CYP4F3B in liver and kidney. CYP4F3A has the highest affinity of all the CYP4F isoforms for pro‐inflammatory LTB4, and its main role is the ω‐hydroxylation of LTB4 to its inactive metabolite 20‐hydroxy‐leukotriene B4 (20OH‐LTB4). 5 In contrast, CYP4F3B has a 30‐fold lower affinity for LTB4 and is mainly metabolizing AA and ω3 PUFAs. 4 , 6
LTB4 is generated from AA, where AA is converted to leukotriene A4 (LTA4) by 5‐lipoxygenase (5‐LO) and 5‐lipoxygenase activating protein (FLAP), followed by hydrolysis to LTB4. LTB4 is synthesized by leukocytes and acts as a chemoattractant for neutrophils, recruiting them to the site of inflammation. It is therefore considered the initiator of the immune response. 7 The inflammatory process is amplified by LTB4‐secreting neutrophils, attracting other leukocytes, followed by T‐cell attraction and cytokine production. The level of LTB4 is also regulated by its catabolism via ω‐hydroxylation by CYP4F3A to 20OH‐LTB4, which is further converted to the inactive metabolite 20‐carboxy‐LTB4. 8
Cytochrome P450 enzymes require cofactor NADPH as electron source and electron transfer partners for their enzymatic oxidase activity. Binding of LTB4 into the substrate‐binding site of CYP4F3A induces a conformational change that facilitates the transfer of electrons from electron transfer partners such as cytochrome P450 reductase (POR) to the heme group of its active site. POR consists of four domains: the FMN‐binding domain (which interacts with P450 enzymes), the connecting domain, the FAD‐binding domain, and NADPH‐binding domain. POR receives two electrons from NADPH and transfers the electrons via the FAD domain to its FMN domain. 9 , 10 POR and CYP4F3 form complexes via electrostatic interactions, facilitating electron transfer from the FMN domain of POR to the heme group of CYP4F3. 11 Conserved patches of electrostatic and hydrophobic amino acids are specific for each P450 enzyme and contribute to the binding selectivity with POR. 12 Besides POR, cytochrome b5 (Cytb5) can act as an electron donor for cytochrome P450s or mediate P450 reactions through allosteric activation. 13
This study is a follow up of a clinical case, 14 reporting a 26‐year‐old female with a complex immune phenotype marked by muscle weakness, exhaustion, and inflammation‐related conditions (gastritis, joint pain, and exercise‐induced asthma). Whole exome sequencing (WES) identified a de novo CYP4F3 L375V mutation in exon 10, affecting both isoforms CYP4F3A and CYP4F3B. Molecular characterization of this CYP4F3 mutation suggests an effect on LTB4 metabolism by impairing its catalytic activity. Because the LTB4 pathway is involved in many immunological disorders that match our patient's phenotype such as asthma, arthritis, myopathy, and inflammatory bowel disease (IBD), the L375V mutation in CYP4F3 is proposed to play a causative role in the pathology of the patient. We also propose a treatment strategy by blocking the LTB4 pathway.
Methods
Immune phenotyping through flow cytometry
Heparinized blood was collected from the patient and control subjects to isolate peripheral blood mononuclear cells using Lymphoprep (Stemcell Technologies). Control subjects were included using the following criteria: women of Caucasian descent between 20 and 35 years with no known (auto‐)immune diseases, recent infections (<30 days), or pregnancy. This study has been approved by the ethics committee (UZ Leuven, ref. nr. S60344) and has therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. All involved persons gave their informed consent prior to their inclusion in the study. PBMCs were frozen in 10% dimethyl sulfoxide (Sigma) in combination with fetal bovine serum and stored in liquid nitrogen. Thawed PBMCs were stained in phosphate‐buffered saline (PBS) supplemented with 3% fetal bovine serum (FBS) for live‐dead and surface markers for 1 h at 4°C. Anti‐human antibodies included are anti‐CD3 (REA613) (Miltenyi Biotec); anti‐CD14 (M5E5), anti‐CD27 (M‐T271) (both from BioLegend); anti‐IgD (IA6–2) (BD Biosciences); anti‐CCR7 (3D12), anti‐CD45RA (HI 100), anti‐CD8 (SK1), anti‐CD4 (RPA‐T4), anti‐CD3 (UCHT1), anti‐CD19 (HIB19) and anti‐CD14 (61D3) (all from eBioscience). Data were collected on BD CantoII (BD Biosciences) and analysed using FlowJo for Mac version 10.7 (BD Biosciences).
Whole exome sequencing and Sanger sequencing
Whole blood was collected in EDTA tubes. Genomic DNA was isolated using a QIAamp DNA Blood Midi kit (Qiagen), and WES was performed by the Genomics Core (KU Leuven, Belgium). WES and filtering were performed as previously described. 15 Genetic variants were filtered based on the following properties: only coding nonsynonymous variants with CADD score above 10 and absence in dbSNP database (last consulted in 2021). 16 , 17 Validation PCR on DNA extracted from blood cells was done with specific primers spanning the region of the mutation (forward primer: 5′‐CAAGCCAAGGCCAAATCCAAG‐3′, reverse primer: 5′‐AGAGGTGACCTCTCCTTGATG‐3′), followed by Sanger sequencing (LGC Genomics, Berlin, Germany). All blood analyses in this study were performed between age 26 and 27.
Competitive enzyme‐linked immunosorbent assay of LTB4
Whole blood was collected in lithium‐heparine tubes and centrifuged for 15 min at 2500 g. Plasma supernatant was stored at −80°C until analysis. LTB4 Competitive enzyme‐linked immunosorbent assay (ELISA; Elabscience, Texas, USA) was carried out following manufacturer's protocol. Optical density was measured at 450 nm using a Multiskan FC microplate photometer (ThermoFisher Scientific).
Cloning and cell culture
The expression vector of human CY4F3A was developed by inserting Flag‐tagged and Myc‐tagged CYP4F3A cDNA (NM_000896.3) into the pCMV6 vector using KpnI and SacII restriction sites. Vector sequence can be found in supplementary methods. Next, the C1123 > G point mutation was inserted using QuikChange site‐directed mutagenesis (Agilent, Santa Clara, USA). HEK293T cells were kindly provided by the Laboratory of Protein Phosphorylation and Proteomics, KU Leuven and were transfected using X‐tremeGene HP (Sigma‐Aldrich, St Louis, USA).
Leukotriene B4 breakdown and capillary electrophoresis (CE‐UV)
Capillary electrophoresis followed by UV detection (CE‐UV) was done as follows. HEK293T cells were plated in 10 cm plates (Sarstedt, Nümbrecht, Germany) at a density of 2 × 106 cells per plate and transfected with CYP4F3A WT or mutant plasmid using X‐tremeGeneHP. The next day, cells were washed with phosphate‐buffered saline, harvested, pelleted by centrifugation (200× g, 5 min), and washed once with 1 mL of ice‐cold homogenization medium [0.25 M sucrose, 5 mM MOPS (pH 7.2), and 1 mM EDTA (pH 7.2)] and a protease inhibitor mixture (Roche, Bazel, Switzerland). After pelleting, cells were resuspended in 500 μL ice‐cold homogenization medium and homogenized using a cold steel grinder (20 strokes). After low speed centrifugation (1500× g, 10 min), pellet containing nuclear fraction was discarded, and postnuclear supernatant was collected and incubated for 60 min at 37°C in a medium containing 1 mM NADPH, 20 μM LTB4, and 100 mM potassium phosphate buffer (pH 7.4). The lipophilic fraction was extracted using C18 cartridges (Waters, Milford, USA) and eluted with methanol. The background electrolyte consisted of 10 mM Na2B4O7 and 12.5 mM sodium dodecylsulfate, with pH adjusted to 8.3 using 50 mM H3BO3. Samples were injected in a P/ACE™MDQ CE system (Beckman Coulter Instruments, Brea, USA) equipped with a diode array UV–vis detector. The detection wavelength was 270 nm. Uncoated fused silica capillary, 50 μm ID, 40 cm total length, and 30 cm effective length, was used for separation. Instrument control and data analysis were performed using 32 KaratTM software version 5.0 (Beckman Coulter Instruments, Brea, USA). Detailed information can be found in the supporting information.
In silico modelling and co‐evolutionary analysis
The crystal structure of cytochrome P450 (pdb: 5T6Q) was identified as a suitable template for CYP4F3 using Phyre2 and was further modelled using the homology modelling suite in MOE (Molecular Operating Environment, Chemical Computing Group, Canada). 18 , 19 The contact map of the model was created using CMweb. A related human type B cytochrome b5 (cytb5, pdb:3NER) was docked to the CYP4F3 model utilizing HADDOCK with interaction restraints from Ahuja et al. 20 , 21 The EVfold (EVcouplings) server was used to identify co‐evolving residues. 22
Co‐immunoprecipitation and western blot
Co‐immunoprecipitation (co‐IP) of CYP4F3 and POR/cytb5 from protein extracts of HEK293T cells overexpressing these proteins was performed using the Pierce™ HA‐Tag co‐IP Kit (ThermoFisher Scientific) following manufacturer's protocol. Protein elution from the anti‐HA beads was done by heating for 10 min at 100°C. Proteins from input and immunoprecipitated samples were separated on a 4–12% NuPAGE Precast Gel System (ThermoFisher Scientific) and transferred to a 0.45 μM PVDF membrane (GE Healthcare, #10600023, Chicago, USA). Antibodies against GAPDH (Merck Millipore, MAB374, Burlington, USA), HA (Santa Cruz Biotechnology, sc‐7392, Dallas, USA), or Flag (Cell Signalling Technology, #14793, Danvers, USA) and secondary antibodies goat‐anti‐rabbit and rabbit‐anti‐mouse IgG (Dako, P0448 and P0260, Glostrup, Denmark) were used. Detection was performed with an ImageQuant Las4000 using the western lightning Plus‐ECL reagent (Perkin Elmer, Waltham, USA). Western blots were analysed, and signal intensities were quantified using Image Studio Lite (LI‐COR Biosciences, Lincoln, USA). Immunoprecipitated samples were normalized to the corresponding input samples.
Ex vivo assay for LTB4 metabolism
Whole blood (1.75 mL) was incubated with 1 μM montelukast sodium, 100 pM fluticasone propionate and 10 μM zileuton (all from MedChemExpress, Monmouth Junction, USA) at 37°C for 15 min. The concentrations of the compounds mimicked the targeted physiological blood concentrations during treatment. Subsequently, all samples were stimulated with 30 μM Calcium ionophore A23187 (Sigma‐Aldrich) and incubated at 37°C for 30 min. The reaction was terminated by the addition of 12.5 mM EGTA. Samples were centrifuged at 2000× g for 15 min, and supernatants were analysed with the LTB4 ELISA.
Statistical analysis
GraphPad Prism version 8 was used for the statistical analysis. Student's t‐test was used for comparisons between two groups, and multigroup data were analysed by one‐way ANOVA, comparing each sample with the control. The Shapiro–Wilk normality test was used to confirm normal distribution of the samples. P values lower than 0.05 were considered significant and indicated as follows: *P < 0.05, **P < 0.01, ***P < 0.001, ***P < 0.0001. All experiments have been performed in at least three biological replicates unless noted otherwise. The results are expressed as the mean ± SEM.
Results
Description of the patient
At the age of seven, the patient was treated for a worsening asthma with corticosteroid fluticasone propionate. After 6 weeks of high‐dose fluticasone, she experienced increasing fatigue, which was attributed to adrenocortical insufficiency, as described by De Swert et al. 14 She developed corticosteroid myopathy, most severely in arms and legs, forcing her to use a wheelchair for mobility. Discontinuation of fluticasone treatment led to a slow but full recovery, and asthmatic symptoms were relieved with a non‐steroid combination therapy of leukotriene receptor antagonist montelukast and disodium cromoglycate. At age 14, the asthmatic symptoms disappeared, and treatment was discontinued. One year later, she presented with gastritis, which was relieved by using omeprazol, domperidone, and magaldrate. At 21 years, she received a diagnosis of exclusion of chronic fatigue syndrome, marked by lingering fatigue, weakness in the limbs, and joint pain that mainly manifested in the wrists, which were all enhanced by physical activity. Because of persistent exhaustion and muscle weakness, the patient again depended on a wheelchair for mobility. Lung function and electromyography of the four limbs were normal while urinary creatinine was elevated (276 mg/24 h). Symptoms gradually increased over the years, accompanied with signs of depression. At the age of 26, accelerated aging was reported, and the patient remains very prone to inflammation; in the time course of only 1 year (2020), she was diagnosed with lateral epicondylitis, persistent paronychia, irritable bowel syndrome, arthritis, infectious enteritis, herpangina, and vestibular neuritis.
Through the years, the patient developed an indistinct immune phenotype, which is mainly marked by persisting and severe muscle weakness in combination with exhaustion and inflammation‐related conditions. Blood chemistry and haematology parameters do not show any abnormalities that could be linked to the phenotype (Table S1). However, basic immune phenotyping indicate increased total CD19+ B cells (Table S1). Detailed immune phenotyping of PBMCs showed reduced CD14+ total monocyte cell numbers and a 28% increase in CD19+ B cells in the patient compared with healthy subjects (Figure 1A). The B cell compartment has a more naive phenotype with higher levels of CD19+ CD27− B cells and lower levels of switched memory B cells. This more naive B cell phenotype does not compromise immunoglobulins as blood levels appear to be normal (Table S2). When analysing the T cell compartment, total T cell numbers from the index patient do not differ from healthy controls, whereas total naive CD4+ helper T cells and total naive CD8+ cytotoxic T cells are increased as well (Figure 1A).
Figure 1.
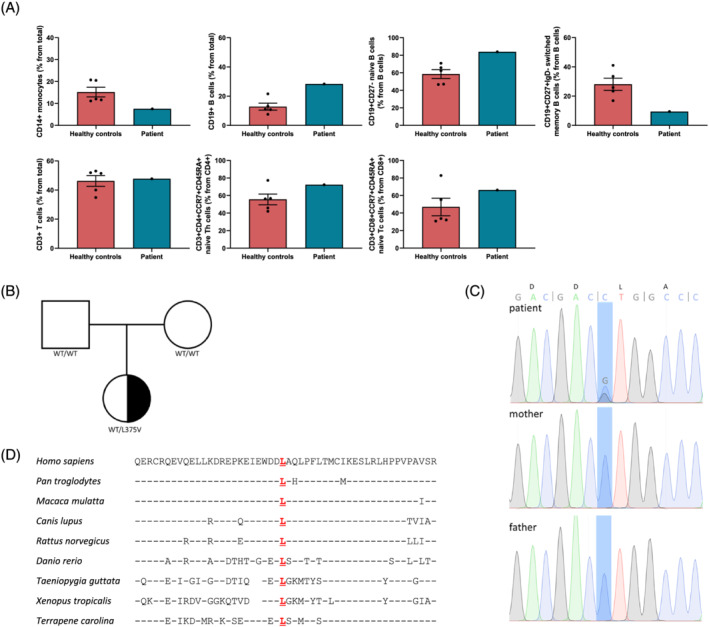
Heterozygous de novo CYP4F3 L375V mutation identified via whole exome sequencing. (A) PBMCs isolated from healthy controls and the index patient were stained and analysed by flow cytometry. Total monocyte cell numbers as a percentage of total cells. Total B cell numbers (CD19+) as a percentage of total cells. Total naive B cell numbers (CD19+ CD27−) as a percentage of total B cells. Total switched memory B cell numbers (CD19+ CD27+ IgD−) as a percentage of total B cells. Total T cell numbers (CD3+) as a percentage of total cells. Total naive CD4+ T cell numbers (CD4+ CCR7+ CD45RA+) as a percentage of total CD4+ T cells. Total naive CD8+ T cell numbers (CD8+ CCR7+ CD45RA+) as a percentage of total CD8+ T cells. Average ±SEM. (B) Pedigree of a family with segregation of the L375V mutant allele. Patient is denoted with a heterozygous de novo L375V mutation in black. White square means male; white circle means female. (C) Sanger sequencing confirming heterozygous L375V mutation in the patient's genomic DNA, while both parents have WT alleles. (D) Multiple sequencing alignment of CYP4F3 displaying conservation of the 351–400 region encompassing the L375V mutation from different organisms. Identical amino acids compared with the Homo sapiens sequence are indicated by dashes; spaces indicate gaps in the alignment. Data were retrieved from Homologene (NCBI) and Ensembl.
Identification of de novo CYP4F3 L375V variant using WES
Whole exome sequencing was performed on genomic DNA extracted from the blood of the patient and her parents. 15 Through filtering for rare de novo or recessive variants with CADD score of more than 10, we identified a novel heterozygous missense mutation in CYP4F3 (NM_000896), c.C1123 > G;p.L375V (Figure 1B and Figure S1a), that was not registered in any public database. The presence of the variant was confirmed via Sanger sequencing (Figure 1C), and functional scores were predicted as damaging by PolyPhen, MutationTaster, SIFT, and CADD (CADD: 10.870) (Figure S1b). 23 The L375V mutation resides in a region of the protein that is strongly conserved among different species, which contributed to the damaging prediction score (Figure 1D).
CYP4F3 L375V has impaired ω‐hydroxylation activity
LTB4 is an essential component of the innate immune system as it attracts leukocytes to the site of inflammation. In order to constrain this inflammatory response, CYP4F3A enzyme metabolizes the pro‐inflammatory molecule LTB4. We postulated that the L375V mutation impairs this activity of CYP4F3A.
First of all, we compared systemic LTB4 levels of the patient to healthy subjects that are sex and age matched using a competitive ELISA. LTB4 levels in plasma of the patient (1034.0 ± 75.9 pg/mL) are significantly increased compared with the control population (305.6 ± 57.0 pg/mL) (Figure 2A).
Figure 2.
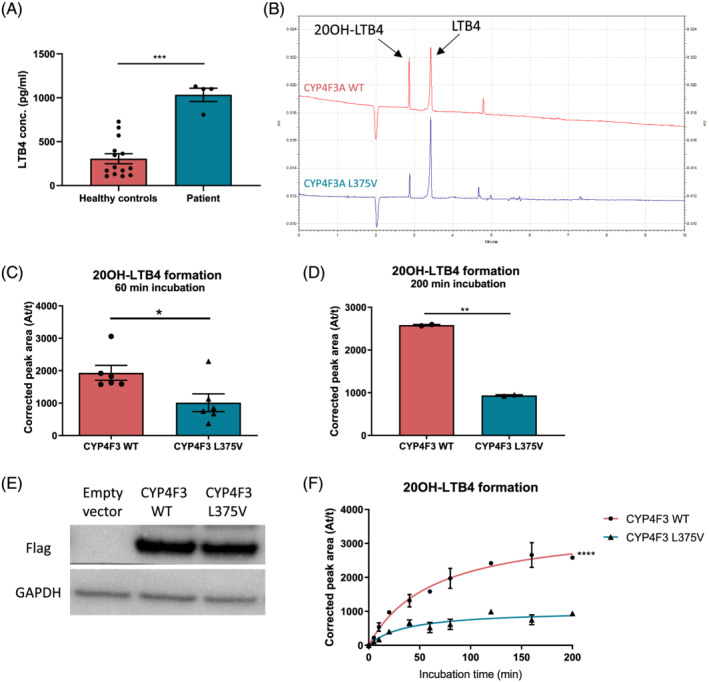
L375V mutation reduces LTB4 ω‐hydroxylation capacity. (A) Quantification of LTB4 levels in plasma from patient and healthy subjects. Each data point represents a different sample. Average ±SEM, one‐way ANOVA, ***P < 0.001. (B) CE‐UV electropherogram of postnuclear supernatant containing CYP4F3 WT or L375V mutant, incubated with 20 μM LTB4 for 60 min at 37°C. BGE for CE‐UV: borate buffer with 12.5 mM SDS at pH 8.3, and detection wavelength is 270 nm. (C) Quantification of CE‐UV detection of 20OH‐LTB4 conversion. WT or L375V CYP4F3 were incubated with LTB4 for 60 min at 37°C (n = 6). Average ±SEM, paired t‐test, *P < 0.05. (D) Quantification of CE‐UV detection of 20OH‐LTB4 conversion. WT or mutant CYP4F3 were incubated with LTB4 for 200 min at 37°C (n = 2). Average ±SEM, paired t‐test, **P < 0.01. (E) Western blot showing protein expression of Flag‐tagged WT and mutant CYP4F3. (F) Quantification of 20OH‐LTB4 formation via CE‐UV detection after incubating postnuclear supernatant containing WT or mutant CYP4F3 with LTB4 for time periods ranging from 0 to 200 min (n = 2). Average ±SEM, nonlinear regression, ****P < 0.0001.
To evaluate the effect of the mutation on enzymatic activity, we developed a protocol that combines in vitro overexpression of the wild type (WT) CYP4F3A or the L375V mutant in HEK293T cells with quantification of LTB4 and 20OH‐LTB4 by capillary electrophoresis (CE‐UV). This allows determination of the impact of the L375V mutation on LTB4 conversion. Postnuclear supernatant containing cell organelles including microsomes from the HEK293T cells overexpressing either WT or L375V mutated CYP4F3A were incubated at 37°C with LTB4 and analysed using CE‐UV. Quality control prior to running the samples ensures accurate measurements of product formation (supporting information and Figure S2a). Incubation of LTB4 with WT or mutant CYP4F3A postnuclear supernatant for 60 min at 37°C resulted in significant differences in product formation (Figure 2B,C), with LTB4 breakdown by mutant CYP4F3 being ±50% less efficient compared with the WT. With 200 min of incubation, mutant CYP4F3 was only able to convert 36% of LTB4 to 20OH‐LTB4 compared with the wild type (Figure 2D). Equal protein levels were checked by western blotting for each of the replicates (Figure 2E). To examine the kinetics of the CYP4F3A activity in more detail, a time course was developed for LTB4 incubation times ranging from 0 to 200 min. With increasing incubation time, the difference between WT and L375V mutant CYP4F3 became more pronounced (Figure 2F).
The observation that the L375V mutation reduces the enzymatic activity of CYP4F3A was also observed using a luciferase‐based P450‐Glo assay for both CYP4F3A and CYP4F3B (Figure S2b and supporting information).
The L375V mutation does not influence binding of CYP4F3 to electron transfer partners
POR and Cytb5 bind P450 proteins and transfer electrons to the catalytic site. In case of CYP4F3, this interaction is known for POR but yet unconfirmed for Cytb5. We constructed a homology model of CYP4F3 based on protein threading, which gave better alignments than homology modelling via sequence similarity. The L375V mutation was highlighted and superimposed with a FMN‐binding domain bound homologues protein (pdb:1BVY) (Figure 3A). 24 First, this model points out that the L375V mutation is not in close proximity of the substrate binding site, indicating that LTB4 binding to CYP4F3A is most likely unaffected. Secondly, the mutation is distant from the heme group; therefore, a direct effect on the enzymatic activity is highly unlikely. However, the L375V mutation is located near the protein–protein interface with the FMN‐domain of POR (Figure 3A). In a docking model of Cytb5 and CYP4F3, Cytb5 fits on the same surface where the FMN‐binding domain of POR binds (Figure 3B). Although an amino acid replacement of leucine to valine is seemingly modest, the leucine at position 375 is very conserved among species (Figure 1D), suggesting that the L375V mutation could affect the interaction interface.
Figure 3.
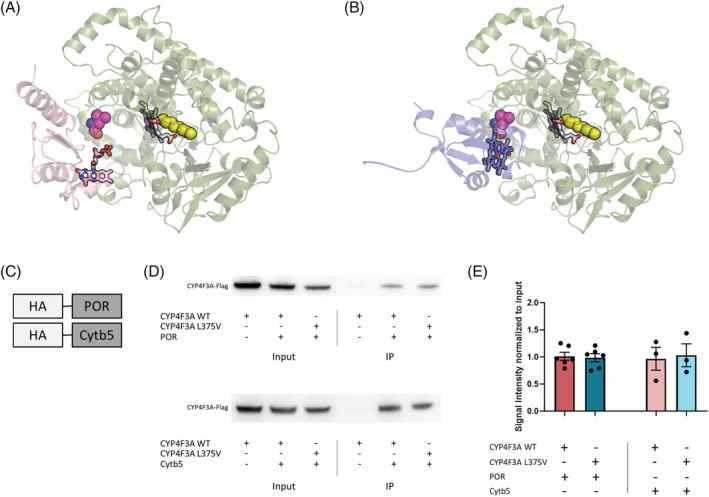
The CYP4F3 interaction with electron transfer partners is unaffected by the L375V mutation. (A) Homology model of CYP4F3 (green) in complex with the FMN‐domain of POR (pink). The substrate binding site of CYP4F3 is depicted in yellow and the heme group is visualized. The L375V mutation (purple) is located in the binding interface of CYP4F3 with POR. Flavin rings of POR are illustrated as well. (B) Homology model of CYP4F3 (green) in complex with Cytb5 (blue). The substrate binding site of CYP4F3 is depicted in yellow and the heme group is visualized. The L375V mutation (purple) is located in the binding interface of CYP4F3 with Cytb5. The heme group of Cytb5 is illustrated in blue. (C) Schematic diagram of HA‐tagged proteins used for the co‐IP assays. Proteins were precipitated using Pierce™ HA‐Tag co‐IP Kit (ThermoFisher scientific). (D) Co‐immunoprecipitated proteins of pulldown with HA‐tagged POR or Cytb5 were detected using western blot. Anti‐Flag antibody was used to detect WT and L375V mutant CYP4F3 (±66 kDa). The total protein fraction before co‐IP is depicted as input, while IP visualizes the immunoprecipitated fraction containing POR or Cytb5 and its bound proteins. (E) Quantification of co‐IP for CYP4F3A with POR (n = 6) and Cytb5 (n = 3). Average ±SEM, one‐way ANOVA.
To determine whether the L375V mutant CYP4F3 can still interact with POR and Cytb5, we performed co‐IP assays. CYP4F3A was co‐precipitated after pulldown of HA‐tagged POR and HA‐tagged Cytb5 (Figure 3C–E). The L375V mutation of CYP4F3A had no influence on the co‐precipitation ratio, which suggests that the mutation does not affect the interaction with POR or Cytb5. In addition, this was also observed for the interaction of CYP4F3B and POR (Figure S3).
In conclusion, our data suggest that the L375V mutation occurs in the interaction interface between CYP4F3 and POR/Cytb5 but does not alter binding to the FMN‐binding domains of the electron carriers POR and Cytb5.
L375V decreases α‐helical stability of the flavin‐heme electron transfer path
We next investigated which amino acids are crucial for electron transfer from flavins of electron carriers like POR and cytb5 to the heme group of CYP4F3. Previous research identified the crucial role of a phenylalanine and a glutamine in the transfer of electrons, which correspond to F461 and Q471 in human CYP4F3 (Figure 4A). 25 Strikingly, when looking more closely at the putative electron transfer path in our model, L375 is in close proximity (8.5 Å) of these residues.
Figure 4.
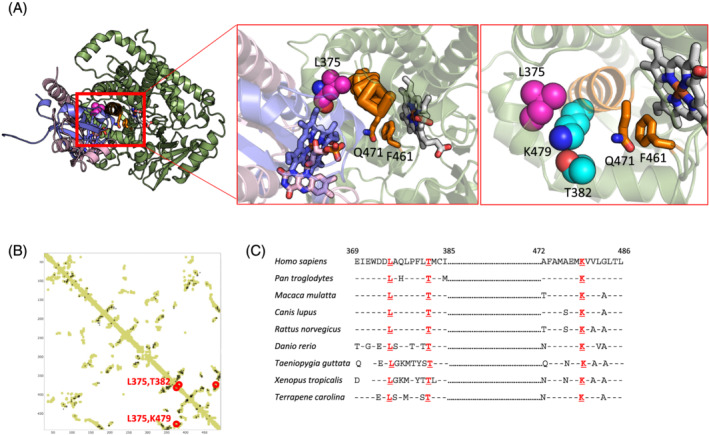
The L375V mutation destabilizes the α‐helix of the electron transfer path. (A) In silico model visualizing CYP4F3 (green) in complex with both POR (pink) and cytochrome b5 (blue) during the electron shuffling process. The heme group is visualized, and the L375V mutation is depicted in purple. The flavin rings of POR and the heme group of Cytb5 (blue) are visualized as well. The residues involved in electron shuffling, F461 and Q471 (orange), and the co‐evolving residues that are forming a triad with L375V, T382 and K479 (light blue) are in close proximity. (B) Co‐evolutionary analysis comparing both the contact map based on our homology model and the co‐evolving residues. Evolutionarily coupled residues T382 and K479 are indicated in red. (C) Multiple sequence alignment of CYP4F3 displaying conservation of the surrounding regions of L375, T382, and K479 residues in CYP4F3 from different organisms. Identical amino acids compared with the Homo sapiens sequence are indicated by dashes; spaces indicate gaps in the alignment. Two regions are separated by dots.
Using co‐evolutionary analysis of CYP4F3 homologues, 22 we identified two residues T382 and K479 that are evolutionarily coupled to L375 as calculated from the sequence with high likelihood (Figure 4B). Both residues are well conserved in the CYP4F3 sequences from various organisms (Figure 4C). In our model, they are in close proximity of L375 (5.0 Å between L375 and T382, and 4.1 Å between L375 and K479). The interactions between T382, K479, and L375 appear to stabilize the α‐helix that carries F461 and Q471 and are involved in electron shuffling (Figure 4A). It is therefore most likely that the L375V mutation found in the patient destabilizes this triad and hence the α‐helix that is involved in the electron transfer between CYP4F3 and the electron carriers POR or cytb5.
Zileuton inhibits LTB4 production in patient's whole blood
Drugs targeting the AA pathway can interact at different stages, such as FLAP inhibitors, 5‐LO inhibitors, LTC4S inhibitors, LTA4H inhibitors, and LTB4 receptor inhibitors (Figure 5A). However, not all these inhibitors affect the production or function of LTB4. We tested the effect of zileuton, an orally active 5‐LO inhibitor obstructing the complete leukotriene pathway, on LTB4 production in whole blood of the patient. Figure 5B clearly shows that zileuton completely inhibited LTB4 production in contrast to the anti‐asthmatic drug montelukast (a blocker of the leukotriene receptor CYSLTR) or fluticasone propionate (a corticosteroid drug) that do not affect LTB4 production in leukocytes when stimulated.
Figure 5.
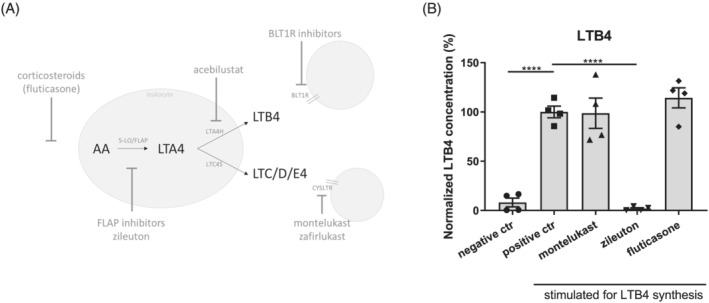
Zileuton inhibits LTB4 production in patient's blood. (A) Overview of LTB4 metabolism in leukocytes, showing sites of inhibition and potential drugs. LTA4H: LTA4 hydrolase, LTC4S: LTC4 synthase, BLT1R: LTB4 receptor 1. (B) Novel ex vivo assay allowing efficient screening of potential drugs targeting LTB4 synthesis. Whole blood was incubated with 1 μM montelukast, 10 μM zileuton, or 100 pM fluticasone to mimic physiological blood concentrations under treatment. LTB4 synthesis is induced by addition of 30 μM calcium ionophore A23187 at 37°C for 30 min, and LTB4 levels were analysed with the LTB4 ELISA (ThermoFisher Scientific) (n = 4). Average ±SEM, one‐way ANOVA, ****P < 0.0001.
Discussion
Here, we report WES of a patient with a complex immune phenotype, mainly marked by myopathy and persistent fatigue, gastritis, arthritis, and exercise‐induced asthma. We identified a de novo heterozygous missense L375V mutation in CYP4F3. Four other germline mutations in the CYP4F3 gene were found on GeneMatcher, 26 but none of the reported variants show any resemblances with our patient's phenotype, which makes our report the first description of a congenital condition associated with a CYP4F3 mutation.
Although most CYP4F family members are well studied, the exact role of CYP4F3 in health and disease is not fully elucidated yet. 27 A study investigating single nucleotide polymorphisms in patients with coeliac disease reported the potential involvement of CYP4F3 SNPs, linking the LTB4 metabolism with a disease marked by inflammation. 28 Also, GWAS studies for Crohn's disease identified SNPs near the CYP4F3 gene. 29 Somatic mutations in CYP4F3 have been reported in the skin, uterus, bronchus and lung cancer, and mutations specifically close to the L375 site seem to be associated with malignancies in the lung, as a L375P mutation reported in a lung tumour (c.T1124 > C;p.L375P) and a nonsense mutation at position 384 (c.1152C > A;p.C384*) in lung adenocarcinoma (Figure S1c).
The high expression of CYP4F3A almost exclusively in neutrophils is in accordance with its specialized function as LTB4 inactivator, which is crucial for the attenuation of excessive inflammation. LTB4 also plays a role in neutrophil polarization, thereby maintaining normal chemotaxis, 29 but as least as important is the dampening of the inflammatory stimulus after proper immune response. LTB4 as a mediator of inflammation has been well established in asthma, arthritis and IBD. 31 , 32 Moreover, neutrophils with reduced LTB4 hydroxylase activity have been identified in IBD patients. 33 Elevated systemic LTB4 has been reported during asthma attacks and the active stage of rheumatoid arthritis in children, 34 , 35 linking active inflammation to high LTB4 blood levels. Strikingly, systemic LTB4 levels are strongly increased in our patient compared with healthy subjects, even when no clear sites of active inflammation were reported at time of blood sampling. This raises the question whether the heterozygous L375V mutation in CYP4F3 affects normal functioning of the innate immune system by impairing LTB4 breakdown, whereby neutrophil chemotaxis and subsequent immune response are dysregulated.
In addition, LTB4 can be produced in skeletal muscle as well where it initiates chemotaxis to damaged tissue and enhances myoblast proliferation and differentiation. 36 This contribution of LTB4 in muscle atrophy and exhaustion is commonly associated with chronical inflammatory diseases. 37 Because our patient presents with inflammation‐related conditions including arthritis, gastritis, exercise‐induced asthma, and muscle weakness, this strongly suggests a causal role for the impaired LTB4 pathway.
Furthermore, neutrophils do not only remove pathogens at the site of inflammation but also mediate diverse immune functions by releasing a broad array of inflammatory stimuli such as chemokines and cytokines. 38 These can recruit and activate cells of the innate and adaptive immune system such as monocytes, T cells, and B cells. Neutrophils can also directly interact with B cells by binding its immunoglobulins to amplify microbial clearance. Therefore, neutrophil dysfunction can increase the susceptibility to infections or can result in the inability to clear existing infections. 39 Immune phenotyping of the patient shows that the adaptive immune system is affected with an increased presence of B cells that have a more naive phenotype and reduced switched memory B cells. Yet immunoglobulin levels do not show abnormal levels. This finding might link increased LTB4 plasma levels to a more naive immune response. A limitation of this study is the fact that only one patient with a genomic CYP4F3 mutation is described. While this allows to make some links between mutation and pathology, it does not allow to conclude on causal relationships.
A critical step of the LTB4 pathway is its metabolism via oxidation by CYP4F3. We found that the L375V mutation severely affects the activity of CYP4F3, because less LTB4 is converted to its metabolite 20OH‐LTB4. Protein expression is not affected because WT and L375V mutant CYP4F3 show equal protein levels using western blot. Our docking models showed that L375 is distant from the LTB4 substrate binding site and the catalytic site, excluding a direct effect of the mutation on substrate binding or enzymatic activity. Also, binding to the electron carriers POR and Cytb5 is unaffected by the L375V mutation. We propose that L375 is important for stabilizing a nearby α‐helix which carries the crucial residues for the electron shuffling process, and that the L375V mutation destabilizes the helix‐mediating electron shuffling, thereby disrupting the catalytic function of CYP4F3. Point mutations affecting electron transfer in CYP enzymes have been reported before, such as polymorphisms in CYP2C8 affecting its hydroxylase function. 40 The same goes for variations in electron transfer partners that disrupt CYP activity. 41
We next sought preclinical evidence for a possible therapeutic strategy that might reduce the pro‐inflammatory LTB4 levels in the patient. Conventional immunosuppressive drugs such as glucocorticoids or NSAIDs do not efficiently suppress the LTB4 pathway and might even aggravate symptoms and worsen muscle strength loss because of the important interplay between AA metabolism and muscle growth. 37 This is in line with the patient's medical history of steroid myopathy. 14 Because the LTB4 pathway is involved in many inflammatory disorders and cancers, there has been an increased interest in the development of LTB4 pathway inhibitors, with LTB4 receptors and FLAP inhibitors as major therapeutic targets 42 (Figure 5A). Unfortunately, although these drugs are currently being tested in clinical trials, they are not available for therapeutic purposes yet. Montelukast, inhibitor of leukotriene receptor CYSLTR, can block downstream effects of LTC4, LTD4, and LTE4 but not of LTB4 as it works via a different receptor (BLT1R). Therefore, montelukast treatment will not be effective in normalizing LTB4 pathway functioning in this patient. Zileuton is a 5‐LO inhibitor and blocks complete leukotriene synthesis including LTB4, 43 as demonstrated ex vivo in whole blood of the patient. Zileuton has been approved for the treatment of asthma, but the broader implications of the LTB4 pathway have extended research on zileuton to other fields like rheumatoid arthritis and cancer. 44 , 45 We suggest that further clinical testing with zileuton (trade name Zyflo) is needed to explore its potential in treating immune phenotypes with involvement of the LTB4 pathway.
Conflict of interests
The authors declare no competing interests in relation to this study.
Supporting information
Figure S1. Whole exome sequencing reveals CYP4F3 p.L375V mutation. A. Visualization of de novo heterozygous missense mutation p.L375V using Integrative Genomics Viewer (IGV). 47% of the patient's reads contain the C1123>G mutation, whereas no mutations were detected in the parents at this site. B. Overview of the software used to predict the possible impact of the L375V substitution on the structure and function of CYP4F3 protein. C. Overview of somatic mutations reported in tumor samples from various malignancies of The Cancer Genome Atlas (TCGA). Variants in very close proximity to our patient's mutation L375V have been indicated by arrows, and are both identified in lung adenocarcinoma from TCGA, dataset TCGA‐LUAD.
Figure S2. Quality control of CE‐UV and P450‐Glo CYP3A4 assay. A. Linear regression showing linearity test of CE‐UV machine. B. P450‐GloTM luciferase assay (V8801, Promega) performed on postnuclear supernatant of HEK293T cells overexpressing CYP4F3A and CYP4F3B, both WT and L375V mutant. Enzyme activity was shown for pro‐luciferin substrate (n=2). Average ±SEM, 1‐way ANOVA.
Figure S3. Co‐IP of POR and CYP4F3B. A. Co‐immunoprecipitated proteins of pulldown with HA‐tagged POR were detected using western blot. Anti‐Flag antibody was used to detect WT and L375V mutant CYP4F3B (±66 kDa). The total protein fraction before co‐IP is depicted as input, while IP visualizes the immunoprecipitated fraction containing POR and its bound proteins.
B. Quantification of co‐IP for CYP4F3B with POR (n=6). Average ±SEM, paired t‐test.
Data S1. Supporting information
Table S1. Hematologic, chemical and immune parameters of patient with heterozygous de novo CYP4F3 L375V mutation. Abnormal values are indicated in grey boxes. RBC: red blood cell, MCV: mean corpuscular volume, MCH: mean corpuscular hemoglobin, MCHC: mean corpuscular hemoglobin concentration, WBC: white blood cell, MPV: mean platelet volume, eGFR: estimated glomerular filtration rate, AST: aspartate aminotransferase, ALT: alanine aminotransferase, Gamma GT: gamma glutamyl transferase, CRP: C‐reactive protein.
Table S2. Determination of blood immunoglobulin levels. Mean values with standard deviation are shown of 3 different blood samplings.
Acknowledgements
We thank all the institutions that provided funding, materials, and/or support for this work. We would also like to thank Dr. Liliane De Swert for her detailed clinical description of the patient, Sofie De Block and Hilde De Bruyn for their excellent technical assistance, and Jeason Haughton for isolating and handling the PBMCs. Claessens F. holds grants from Fonds Wetenschappelijk Onderzoek‐Vlaanderen (GOA9816N, G.0684.12N, and G.0830.13N). This work was also supported by KU Leuven grant C14/19/100. EVN is an FWO fellow (SB grant, 1S22716N). All authors of this manuscript comply with the guidelines of ethical authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. 46
Smeets E., Huang S., Lee X. Y., Van Nieuwenhove E., Helsen C., Handle F., Moris L., El Kharraz S., Eerlings R., Devlies W., Willemsen M., Bücken L., Prezzemolo T., Humblet‐Baron S., Voet A., Rochtus A., Van Schepdael A., de Zegher F., and Claessens F. (2022) A disease‐associated missense mutation in CYP4F3 affects the metabolism of leukotriene B4 via disruption of electron transfer, Journal of Cachexia, Sarcopenia and Muscle, 13, 2242–2253, 10.1002/jcsm.13022
References
- 1. Williams JA, Hyland R, Jones BC, Smith DA, Hurst S, Goosen TC, et al. Drug‐drug interactions for UDP‐glucuronosyltransferase substrates: a pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metab Dispos 2004;32:1201–1208. [DOI] [PubMed] [Google Scholar]
- 2. Bylund J, Finnström N, Oliw EH. Gene expression of a novel cytochrome P450 of the CYP4F subfamily in human seminal vesicles. Biochem Biophys Res Commun 1999;261:169–174. [DOI] [PubMed] [Google Scholar]
- 3. Kikuta Y, Kusunose E, Kusunose M. Prostaglandin and leukotriene omega‐hydroxylases. Prostaglandins Other Lipid Mediat 2002;68‐69:345–362. [DOI] [PubMed] [Google Scholar]
- 4. Christmas P, Jones JP, Patten CJ, Rock DA, Zheng Y, Cheng SM, et al. Alternative splicing determines the function of CYP4F3 by switching substrate specificity. J Biol Chem 2001;276:38166–38172. [DOI] [PubMed] [Google Scholar]
- 5. Kikuta Y, Kusunose E, Endo K, Yamamoto S, Sogawa K, Fujii‐Kuriyama Y, et al. A novel form of cytochrome P‐450 family 4 in human polymorphonuclear leukocytes. cDNA cloning and expression of leukotriene B4 omega‐hydroxylase. J Biol Chem 1993;268:9376–9380. [PubMed] [Google Scholar]
- 6. Fer M, Corcos L, Dréano Y, Plée‐Gautier E, Salaün JP, Berthou F, et al. Cytochromes P450 from family 4 are the main omega hydroxylating enzymes in humans: CYP4F3B is the prominent player in PUFA metabolism. J Lipid Res 2008;49:2379–2389. [DOI] [PubMed] [Google Scholar]
- 7. Kim ND, Chou RC, Seung E, Tager AM, Luster AD. A unique requirement for the leukotriene B4 receptor BLT1 for neutrophil recruitment in inflammatory arthritis. J Exp Med 2006;203:829–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Murphy RC, Gijón MA. Biosynthesis and metabolism of leukotrienes. Biochem J 2007;405:379–395. [DOI] [PubMed] [Google Scholar]
- 9. Iyanagi T. Structure and function of NADPH‐cytochrome P450 reductase and nitric oxide synthase reductase domain. Biochem Biophys Res Commun 2005;338:520–528. [DOI] [PubMed] [Google Scholar]
- 10. Aigrain L, Fatemi F, Frances O, Lescop E, Truan G. Dynamic control of electron transfers in diflavin reductases. Int J Mol Sci 2012;13:15012–15041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Iyanagi T, Xia C, Kim JJ. NADPH‐cytochrome P450 oxidoreductase: prototypic member of the diflavin reductase family. Arch Biochem Biophys 2012;528:72–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bridges A, Gruenke L, Chang YT, Vakser IA, Loew G, Waskell L. Identification of the binding site on cytochrome P450 2B4 for cytochrome b5 and cytochrome P450 reductase. J Biol Chem 1998;273:17036–17049. [DOI] [PubMed] [Google Scholar]
- 13. Porter TD. The roles of cytochrome b5 in cytochrome P450 reactions. J Biochem Mol Toxicol 2002;16:311–316. [DOI] [PubMed] [Google Scholar]
- 14. De Swert LF, Wouters C, de Zegher F. Myopathy in children receiving high‐dose inhaled fluticasone. N Engl J Med 2004;350:1157–1159. [DOI] [PubMed] [Google Scholar]
- 15. Van Nieuwenhove E, Garcia‐Perez JE, Helsen C, Rodriguez PD, van Schouwenburg PA, Dooley J, et al. A kindred with mutant IKAROS and autoimmunity. J Allergy Clin Immunol 2018;142:699, e12–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 2001;29:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 2015;10:845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hsu MH, Baer BR, Rettie AE, Johnson EF. The Crystal Structure of Cytochrome P450 4B1 (CYP4B1) Monooxygenase Complexed with Octane Discloses Several Structural Adaptations for ω‐Hydroxylation. J Biol Chem 2017;292:5610–5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ahuja S, Jahr N, Im SC, Vivekanandan S, Popovych N, Le Clair SV, et al. A model of the membrane‐bound cytochrome b5‐cytochrome P450 complex from NMR and mutagenesis data. J Biol Chem 2013;288:22080–22095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van Zundert GCP, Rodrigues JPGL, Trellet M, Schmitz C, Kastritis PL, Karaca E, et al. The HADDOCK2.2 Web Server: User‐Friendly Integrative Modeling of Biomolecular Complexes. J Mol Biol 2016;428:720–725. [DOI] [PubMed] [Google Scholar]
- 22. Robert S, J. FR , Sikander H, Yichao S, Yevgeniy A, Li Y, et al. EVfold.org: Evolutionary Couplings and Protein 3D Structure Prediction. bioRxiv: 2015.
- 23. Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet 2015;24:2125–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sevrioukova IF, Li H, Zhang H, Peterson JA, Poulos TL. Structure of a cytochrome P450‐redox partner electron‐transfer complex. Proc Natl Acad Sci U S A 1999;96:1863–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hamdane D, Xia C, Im SC, Zhang H, Kim JJ, Waskell L. Structure and function of an NADPH‐cytochrome P450 oxidoreductase in an open conformation capable of reducing cytochrome P450. J Biol Chem 2009;284:11374–11384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat 2015;36:928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Corcos L, Lucas D, Le Jossic‐Corcos C, Dréano Y, Simon B, Plée‐Gautier E, et al. Human cytochrome P450 4F3: structure, functions, and prospects. Drug Metabol Drug Interact 2012;27:63–71. [DOI] [PubMed] [Google Scholar]
- 28. Curley CR, Monsuur AJ, Wapenaar MC, Rioux JD, Wijmenga C. A functional candidate screen for coeliac disease genes. Eur J Hum Genet 2006;14:1215–1222. [DOI] [PubMed] [Google Scholar]
- 29. Costea I, Mack DR, Israel D, Morgan K, Krupoves A, Seidman E, et al. Genes involved in the metabolism of poly‐unsaturated fatty‐acids (PUFA) and risk for Crohn's disease in children & young adults. PLoS One 2010;5:e15672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Afonso PV, Janka‐Junttila M, Lee YJ, McCann CP, Oliver CM, Aamer KA, et al. LTB4 is a signal‐relay molecule during neutrophil chemotaxis. Dev Cell 2012;22:1079–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Flamand N, Mancuso P, Serezani CH, Brock TG. Leukotrienes: mediators that have been typecast as villains. Cell Mol Life Sci 2007;64:2657–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Haeggström JZ, Funk CD. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem Rev 2011;111:5866–5898. [DOI] [PubMed] [Google Scholar]
- 33. Ikehata A, Hiwatashi N, Kinouchi Y, Ito K, Yamazaki H, Toyota T. Leukotriene B4 omega‐hydroxylase activity in polymorphonuclear leukocytes from patients with inflammatory bowel disease. Prostaglandins Leukot Essent Fatty Acids 1993;49:489–494. [DOI] [PubMed] [Google Scholar]
- 34. Shindo K, Matsumoto Y, Hirai Y, Sumitomo M, Amano T, Miyakawa K, et al. Measurement of leukotriene B4 in arterial blood of asthmatic patients during wheezing attacks. J Intern Med 1990;228:91–96. [DOI] [PubMed] [Google Scholar]
- 35. Gürsel T, Firat S, Ercan ZS. Increased serum leukotriene B4 level in the active stage of rheumatoid arthritis in children. Prostaglandins Leukot Essent Fatty Acids 1997;56:205–207. [DOI] [PubMed] [Google Scholar]
- 36. Sun R, Ba X, Cui L, Xue Y, Zeng X. Leukotriene B4 regulates proliferation and differentiation of cultured rat myoblasts via the BLT1 pathway. Mol Cells 2009;27:403–408. [DOI] [PubMed] [Google Scholar]
- 37. Loell I, Alemo Munters L, Pandya J, Zong M, Alexanderson H, Fasth AE, et al. Activated LTB4 pathway in muscle tissue of patients with polymyositis or dermatomyositis. Ann Rheum Dis 2013;72:293–299. [DOI] [PubMed] [Google Scholar]
- 38. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol 2011;11:519–531. [DOI] [PubMed] [Google Scholar]
- 39. Leliefeld PH, Wessels CM, Leenen LP, Koenderman L, Pillay J. The role of neutrophils in immune dysfunction during severe inflammation. Crit Care 2016;20:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Arnold WR, Zelasko S, Meling DD, Sam K, Das A. Polymorphisms of CYP2C8 Alter First‐Electron Transfer Kinetics and Increase Catalytic Uncoupling. Int J Mol Sci 2019;20:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Flück CE, Pandey AV. Impact on CYP19A1 activity by mutations in NADPH cytochrome P450 oxidoreductase. J Steroid Biochem Mol Biol 2017;165:64–70. [DOI] [PubMed] [Google Scholar]
- 42. Gür ZT, Çalışkan B, Banoglu E. Drug discovery approaches targeting 5‐lipoxygenase‐activating protein (FLAP) for inhibition of cellular leukotriene biosynthesis. Eur J Med Chem 2018;153:34–48. [DOI] [PubMed] [Google Scholar]
- 43. Bell RL, Young PR, Albert D, Lanni C, Summers JB, Brooks DW, et al. The discovery and development of zileuton: an orally active 5‐lipoxygenase inhibitor. Int J Immunopharmacol 1992;14:505–510. [DOI] [PubMed] [Google Scholar]
- 44. Weinblatt ME, Kremer JM, Coblyn JS, Helfgott S, Maier AL, Petrillo G, et al. Zileuton, a 5‐lipoxygenase inhibitor in rheumatoid arthritis. J Rheumatol 1992;19:1537–1541. [PubMed] [Google Scholar]
- 45. Meng Z, Cao R, Yang Z, Liu T, Wang Y, Wang X. Inhibitor of 5‐lipoxygenase, zileuton, suppresses prostate cancer metastasis by upregulating E‐cadherin and paxillin. Urology 2013;82:1452:e7–e14. [DOI] [PubMed] [Google Scholar]
- 46. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2021. J Cachexia Sarcopenia Muscle 2021;12:2259–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Whole exome sequencing reveals CYP4F3 p.L375V mutation. A. Visualization of de novo heterozygous missense mutation p.L375V using Integrative Genomics Viewer (IGV). 47% of the patient's reads contain the C1123>G mutation, whereas no mutations were detected in the parents at this site. B. Overview of the software used to predict the possible impact of the L375V substitution on the structure and function of CYP4F3 protein. C. Overview of somatic mutations reported in tumor samples from various malignancies of The Cancer Genome Atlas (TCGA). Variants in very close proximity to our patient's mutation L375V have been indicated by arrows, and are both identified in lung adenocarcinoma from TCGA, dataset TCGA‐LUAD.
Figure S2. Quality control of CE‐UV and P450‐Glo CYP3A4 assay. A. Linear regression showing linearity test of CE‐UV machine. B. P450‐GloTM luciferase assay (V8801, Promega) performed on postnuclear supernatant of HEK293T cells overexpressing CYP4F3A and CYP4F3B, both WT and L375V mutant. Enzyme activity was shown for pro‐luciferin substrate (n=2). Average ±SEM, 1‐way ANOVA.
Figure S3. Co‐IP of POR and CYP4F3B. A. Co‐immunoprecipitated proteins of pulldown with HA‐tagged POR were detected using western blot. Anti‐Flag antibody was used to detect WT and L375V mutant CYP4F3B (±66 kDa). The total protein fraction before co‐IP is depicted as input, while IP visualizes the immunoprecipitated fraction containing POR and its bound proteins.
B. Quantification of co‐IP for CYP4F3B with POR (n=6). Average ±SEM, paired t‐test.
Data S1. Supporting information
Table S1. Hematologic, chemical and immune parameters of patient with heterozygous de novo CYP4F3 L375V mutation. Abnormal values are indicated in grey boxes. RBC: red blood cell, MCV: mean corpuscular volume, MCH: mean corpuscular hemoglobin, MCHC: mean corpuscular hemoglobin concentration, WBC: white blood cell, MPV: mean platelet volume, eGFR: estimated glomerular filtration rate, AST: aspartate aminotransferase, ALT: alanine aminotransferase, Gamma GT: gamma glutamyl transferase, CRP: C‐reactive protein.
Table S2. Determination of blood immunoglobulin levels. Mean values with standard deviation are shown of 3 different blood samplings.