

Abstract
Sarcopenia is a progressive loss of muscle mass and strength with a risk of adverse outcomes such as disability, poor quality of life, and death. Increasing evidence indicates that diminished ability of the muscle to activate satellite cell‐dependent regeneration is one of the factors that might contribute to its development. Skeletal muscle regeneration following myogenic cell death results from the proliferation and differentiation of myogenic stem cells, called satellite cells, located beneath the basal lamina of the muscle fibres. Satellite cell differentiation is not a satellite cell‐autonomous process but depends on signals provided by the surrounding cells. Infiltrating macrophages play a key role in the process partly by clearing the necrotic cell debris, partly by producing cytokines and growth factors that guide myogenesis. At the beginning of the muscle regeneration process, macrophages are pro‐inflammatory, and the cytokines produced by them trigger the proliferation and differentiation of satellite cells. Following the uptake of dead cells, however, a transcriptionally regulated phenotypic change (macrophage polarization) is induced in them resulting in their transformation into healing macrophages that guide resolution of inflammation, completion of myoblast differentiation, myoblast fusion and growth, and return to homeostasis. Impaired efferocytosis results in delayed cell death clearance, delayed macrophage polarization, prolonged inflammation, and impaired muscle regeneration. Thus, proper efferocytosis by macrophages is a determining factor during muscle repair. Here we review that both efferocytosis and myogenesis are dependent on the cell surface phosphatidylserine (PS), and surprisingly, these two processes share a number of common PS receptors and signalling pathways. Based on these findings, we propose that stimulating the function of PS receptors for facilitating muscle repair following injury could be a successful approach, as it would enhance efferocytosis and myogenesis simultaneously. Because increasing evidence indicates a pathophysiological role of impaired efferocytosis in the development of chronic inflammatory conditions, as well as in impaired muscle regeneration both contributing to the development of sarcopenia, improving efferocytosis should be considered also in its management. Again applying or combining those treatments that target PS receptors would be expected to be the most effective, because they would also promote myogenesis. A potential PS receptor‐triggering candidate molecule is milk fat globule‐EGF‐factor 8 (MFG‐E8), which not only stimulates PS‐dependent efferocytosis and myoblast fusion but also promotes extracellular signal‐regulated kinase (ERK) and Akt activation‐mediated cell proliferation and cell cycle progression in myoblasts.
Keywords: Phosphatidylserine, Phosphatidylserine receptor, Efferocytosis, Myogenesis, Myoblast fusion, Muscle regeneration, Sarcopenia, MFG‐E8
Introduction
Contrary to many tissues that repair after an injury, skeletal muscle is capable of complete regeneration, leading to full recovery of its function. Though recently a new myonuclei‐driven regeneration process has been described to handle sarcomere injuries generated during microtraumas, 1 this remarkable regenerative capability is also due to the existence and function of satellite cells, the myogenic stem cells. 2 In the latter case, the regeneration processes begin with degeneration of myofibres and infiltration of inflammatory immune cells (Figure 1). The initial inflammation creates an environment for the activation, proliferation, and differentiation of the satellite cells. Specific depletion of circulating monocytes prior to muscle injury results in a very severe impairment of muscle regeneration demonstrating the crucial involvement of these cells in the muscle repair. 3 The very first studies conducted on muscle regeneration have already indicated that macrophages that are present at the beginning of muscle regeneration (characterized by inflammation, dead cell clearance, and satellite cell differentiation) differ from those that accompany the late phases of the process (characterized by myoblast fusion and myofibre growth). 4 Later it was demonstrated that the late phase healing macrophages are generated from the early phase pro‐inflammatory macrophages 5 and guide the resolution of inflammation, myoblast fusion and growth, fibrosis, vascularization, and return to homeostasis (reviewed in detail in reference 6 ). A recent detailed analysis of these macrophages by determining their mRNA expressions on a single cell level demonstrated the complexity of this transformation process. 7
Figure 1.
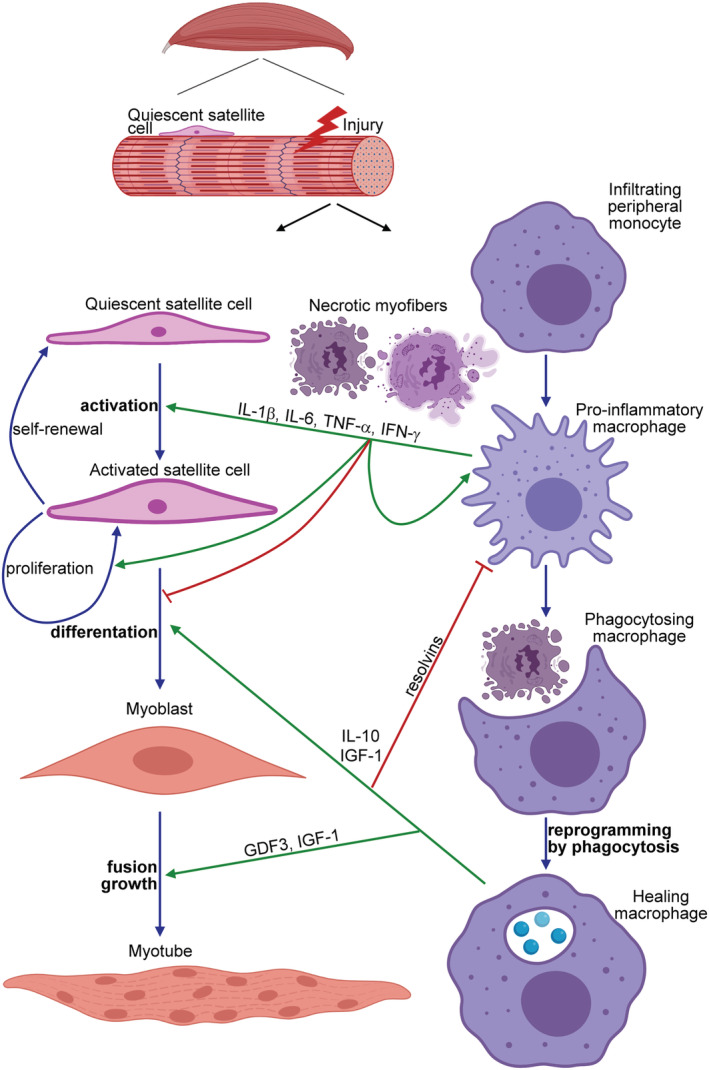
The schematic view of the contribution of macrophages to the myoblast differentiation during skeletal muscle regeneration. Upon induction of necrotic cell death in the skeletal muscle, peripheral monocytes infiltrate the injury site and differentiate into pro‐inflammatory macrophages, and by secreting various pro‐inflammatory cytokines [such as interleukin (IL)‐1β, IL‐6, tumour necrosis factor (TNF)α, or interferon (IFN) γ], they drive the proliferation and differentiation of quiescent satellite cells into myoblasts. In addition, they clear the dead cell debris. Interaction with the apoptotic neutrophils that come first to the injury side but die within hours, and uptake of the dead cells' material during efferocytosis reprogram these pro‐inflammatory macrophages to polarize transcriptionally into healing macrophages. Healing macrophages produce anti‐inflammatory molecules (cytokines and resolvins) to facilitate the resolution of inflammation and growth factors (such as growth differentiation factor 3 and insulin‐like growth factor) to drive the formation of myotubes by promoting both the fusion of myoblasts and the growth of myofibres.
The timed switch between the two main subsets of macrophages, the inflammatory ones that produce pro‐inflammatory cytokines and the healing ones that produce growth and angiogenic factors and anti‐inflammatory mediators, is a key to the proper regeneration process, and it is thought to be induced by the efferocytosis process itself. This idea came from many previous observations that demonstrated that uptake of apoptotic cells induces an anti‐inflammatory phenotype in macrophages. 6 , 8 Accordingly, mice in which macrophage efferocytosis is impaired due to the deletion of a macrophage efferocytosis receptor demonstrate a delayed macrophage phenotypic switch during muscle regeneration leading consequently to an impaired muscle repair. 9 , 10
It has been known for a long time that efferocytosis is a phosphatidylserine (PS)‐dependent process. 11 , 12 Mer proto‐oncogene tyrosine kinase (Mer) is a PS recognizing efferocytosis receptor belonging to the TAM kinase family. 13 While we were investigating the impact of impaired efferocytosis on muscle regeneration by studying Mer null mice, 10 to our surprise, we found that during muscle repair, TAM kinases are required not only for the proper phagocytosis of dead cells by macrophages but also, expressed in myoblasts, for the proper myogenesis. 10 We were aware that two other phagocytic receptors, Brain angiogenesis inhibitor 1 (BAI1) and stabilin‐2, have already been associated with myoblast fusion 14 , 15 and hypothesized that participation of three independent PS‐dependent efferocytosis receptors in the myogenesis cannot be an accidental event.
The appearance of phosphatidylserine on the cell surface
Phosphatidylserine is a glycerophospholipid, which is located exclusively in the cytoplasmic leaflet of the plasma membrane, where it is involved in forming protein recruitment sites needed for the activation of several key signalling pathways, such as protein kinase C, Rapidly Accelerated Fibrosarcoma‐1 (Raf‐1), or Akt signalling. 16 , 17 , 18 The asymmetrical membrane distribution of PS is mediated by ATP‐dependent flippases that translocate PS from the outer to the inner leaflet of the lipid bilayer. 19 When, however, ATP is used up (during necrotic cell death), or when scramblases, enzymes that can randomly distribute PS in the membrane in an ATP‐independent manner, are activated and/or flippases are inactivated, PS can appear in the outer leaflet as well. A number of flippases and scramblases have been identified, and their activity is regulated individually in various cell types. 20 For example, it was shown that the ones that regulate PS exposure during apoptosis are activated by caspase‐mediated cleavage, while the ones that are involved in thrombocyte activation respond to increases in the cytosolic Ca2+ concentration. 20 Increasing evidence indicates that in addition to thrombocyte aggregation and activation, where cell surface PS serves as a docking site for the binding of blood coagulation factors, 21 the appearance of PS on the cell surface is sensed by a group of PS recognition receptors located on the surface of several cell types. 22 All these receptors were reported to control various biological processes in those cells, which express them. 22
Phosphatidylserine located on the apoptotic cell surface is a determinant recognition signal for their uptake by professional or non‐professional phagocytes
After the early discovery that PS is exposed on the surface of apoptotic cells, 23 it was quickly found that PS serves as a determinant recognition signal for the apoptotic cell uptake. This was indicated by the observation that uptake of apoptotic cells can be efficiently inhibited by both PS vesicles and annexin V, a Ca2+‐dependent PS binding protein that can mask exposed PS on the cell surface. 11 , 12 With time, more and more phagocytic receptors were discovered that though are expressed by various phagocyte types, all recognize PS on the surface of apoptotic cells either directly or indirectly by using bridging molecules that link them to the apoptotic cells' PS.
Integrin receptors recognize phosphatidylserine via thrombospondin or MFG‐E8 bridging molecules and use coreceptors
The first receptor that was recognized to serve as a phagocytic receptor for the uptake of apoptotic cells was the vitronectin receptor. 24 The vitronectin receptor is the αvβ3 integrin, a member of the integrin superfamily of adhesion proteins. 25 It was found to work together with its coreceptor CD36, a receptor belonging to the B class of scavenger receptors, and to use thrombospondin as a bridging molecule in the uptake of apoptotic cells. 26 While thrombospondin recognizes PS via its heparin‐binding domain, 27 CD36 was shown to be an oxidized PS receptor. 28 In addition, αvβ3 integrins can use also another bridging molecule, milk fat globule‐EGF‐factor 8 (MFG‐E8), which also binds PS. 29 It uses its RGD motif within its EGF‐like domain to bind to the integrin and contains gamma carboxylated glutamate side chains to link PS in a Ca2+‐dependent manner. The binding of MFG‐E8 to the vitronectin receptor is stabilized by transglutaminase 2 (TG2), another coreceptor of the vitronectin receptor required for the proper phagocytosis of apoptotic cells, but which itself cannot bind PS. 30 MFG‐E8 serves also as a bridging molecule for the αvβ5 integrin, another integrin that has been shown to contribute to the uptake of apoptotic cells. 31 In addition to integrin β3 and β5, β1 integrins were also reported to participate in the efferocytosis process by mediating the signalling initiated by the T‐cell immunoglobulin mucin receptor 4 (Tim4) tethering receptor. 32 Tim4 possesses a metal ion‐dependent pocket that selectively binds PS, but its cytosolic domain is devoid of recognizable intracellular signalling motifs. An integrin β1 ligand has not been identified in this set‐up so far, but TG2 acts as a coreceptor also for integrin β1. 33
TAM kinase receptors recognize phosphatidylserine via the GAS6 or protein S bridging molecules
The TAM kinases (Tyro3/Axl/Mer) form a family of tyrosine kinase receptors. 13 None of them bind PS directly, but they participate in efferocytosis by recognizing PS on the surface of dying cells in a growth arrest‐specific gene 6 (Gas6)‐dependent or protein S‐dependent manner. Their tyrosine kinase domain is required for their function to promote efferocytosis. 34 Gas6 can bind to all three receptors, while protein S does not bind to Axl. Despite a high degree of homology between TAM kinases and their ligands, the ligand‐inducible TAM kinase activation follows a biochemical hierarchy whereby Gas6 preferentially activates Axl with 100–1000× higher binding affinity over Mer, while protein S preferentially activates Tyro3 in both mice and humans. 35 Similar to MFG‐E8, both Gas6 and protein S use γ‐carboxylated glutamic acid side chains to recognize PS. 36
Several scavenger receptors recognize phosphatidylserine in a protein complex with C1q
In addition to the aforementioned bridging molecules, C1q, the first member of the complement cascade pathway, has also been recognized as a PS binding bridging molecule for apoptotic cells. 37 C1q is a 460 kDa hexameric protein comprising six heterotrimeric collagen‐like triple‐helical fibres, each prolonged by a C‐terminal globular region (GR) that supports most, if not all, of the C1 recognition activities. First, it has been demonstrated that C1q mediates efferocytosis through a receptor complex assembled from CD91 and calreticulin, with CD91 being the transmembrane part and calreticulin acting as the C1q‐binding molecule. 37 Later, however, direct interaction between CD91 and C1q has also been demonstrated, 38 and calreticulin was also found to recognize PS. 39
In addition, C1q acts as a bridging molecule also for two additional scavenger receptors belonging to the F family, and which also participate in the efferocytosis process. SCARF1 is a single‐pass type 1 transmembrane protein containing five epidermal growth factor (EGF)‐like cysteine‐rich repeats in its extracellular domain that are used for ligand binding. 40 Another member of this receptor family, multiple EGF‐like domains‐10 (MEGF10), was also found to contribute to the efferocytosis process in a C1q‐dependent manner at least in astrocytes. 41
Phagocytic receptors recognizing phosphatidylserine directly
In addition to phagocytic receptors that require bridging molecules to bind PS, a number of receptors were discovered that themselves recognize PS. With the exception of Tim4, which alone cannot initiate a signalling pathway, and Tim3 that has to be tyrosine phosphorylated first to recruit signalling molecules, 42 , 43 following PS binding, all these PS‐recognizing receptors induce a signalling pathway that contributes to the efficient phagocytosis of apoptotic cells.
Brain angiogenesis inhibitor 1 (BAI1) is a member of the adhesion G protein‐coupled receptor subfamily B. As all members of this receptor family, it consists of a conserved seven‐transmembrane structure and an N‐terminal extracellular domain, 44 but unlike others, it is not a guanine nucleotide exchange factor (GEF) itself. BAI1 recognizes PS via thrombospondin type 1 repeats located in its extracellular domain. 45
Gpr56 is another adhesion G protein‐coupled receptor involved in efferocytosis specifically expressed by microglial cells but not by other types of macrophages. 46 Similar to other members of this receptor family, it also contains an extensive N‐terminal extracellular domain followed by a classical seven‐transmembrane region and a cytoplasmic tail. Within the long extracellular domain, there are two functional domains, named pentraxin/laminin/neurexin/sex‐hormone‐binding‐globulin‐like (PLL) and GPCR autoproteolysis inducing (GAIN) domains. Gpr56 was first shown to bind TG2 in melanoma cells. 47 Later it was demonstrated that it works together also with TG2 in controlling developmental myelination and myelin repair by microglial cells. 48 These cells express a spliced isoform of Gpr56 in which the free GAIN domain recognizes PS. 46
Stabilin‐2 is a multifunctional receptor with a large extracellular domain that consists of seven FAS1 domains, one X‐link domain, and four EGF‐like domain repeats (EGFrps). The latter are responsible for PS binding 49 , 50 and thus initiating the efferocytosis process.
Class B scavenger receptor B type 1 (SR‐B1), a member of the scavenger receptor family, has two transmembrane domains and is localized to the caveolae. In addition to binding several other ligands, it is also a PS binding receptor expressed specifically by testicular Sertoli cells responsible for the phagocytosis of spermatogenic cells undergoing apoptosis 51 and also by infiltrating macrophages engulfing dying muscle cells. 9
In addition to these relatively well‐characterized receptors, three other PS‐recognizing receptors have been recognized to participate in efferocytosis. Receptors of the CD300 family are type I transmembrane proteins that contain a single IgV‐like extracellular domain with two disulfide bonds and intracellular immunoreceptor tyrosine‐based inhibition motifs (ITIMs). 52 Among the seven members of this family, CD300f and CD300b are associated with efferocytosis by promoting the phagocytic capacity of myeloid cells in a PS‐dependent manner. 53
The receptor for advanced glycation end products (RAGE) is a member of the immunoglobulin superfamily of cell‐surface molecules. 54 Although RAGE is primarily a pattern‐recognition receptor, it was shown to recognize also PS and to promote the uptake of apoptotic cells via activating Rac. 55
And finally, lectin‐like oxidized low‐density lipoprotein receptor 1 (LOX‐1) is a type II membrane protein with a C‐type lectin‐like domain that is able to bind to various ligands, including negatively charged phospholipids such as PS. 56 It was shown to mediate phagocytosis of aged red blood cells by endothelial cells. 57
Phagocytic receptors form a phagocytic synapse to activate Rac1 for efferocytosis
Phagocytes in different tissues express different sets of these receptors, but all of them assemble and work together in the phagocytic synapse to mediate tethering and to generate sufficient engulfment signalling once they are exposed to apoptotic cells. 58 Mouse macrophages are able to generate two such engulfment portals simultaneously, and once it is formed, it will be used for the continuous uptake of apoptotic cells. 30 So far, two parallel evolutionarily conserved efferocytosis signalling pathways have been discovered in the initiation of the apoptotic cell uptake, and both converge on the activation of the small GTPase Rac1. 59 One involves the bipartite guanine nucleotide exchange factor (GEF) protein for Rac1 named dedicator of cytokinesis 180 (DOCK180)/engulfment and cell motility protein (ELMO), 60 , 61 while the other is initiated by the adapter protein GULP. 62 Because GULP does not interact with either the GTP‐bound or the GDP‐bound Rac, it is believed that it might regulate a so far unknown Rac‐GTPase activating protein (GAP), especially that it is known to inhibit the GAP protein for Arf. 63 For the functioning of the phagocytic signalling pathway, simultaneous activation of phosphoinositide‐3 kinases (PI3K) is also required to induce the formation of phosphoinositides in the inner leaflet of the plasma membrane. Phosphoinositides guide the recruitment of GEFs for small GTPases including Dock180/ELMO into the phagocytic cup. 64 , 65 Based on the existing literature, the receptors and signalling pathways coupled to the activation of Rac1 are illustrated in Figure 2.
Figure 2.
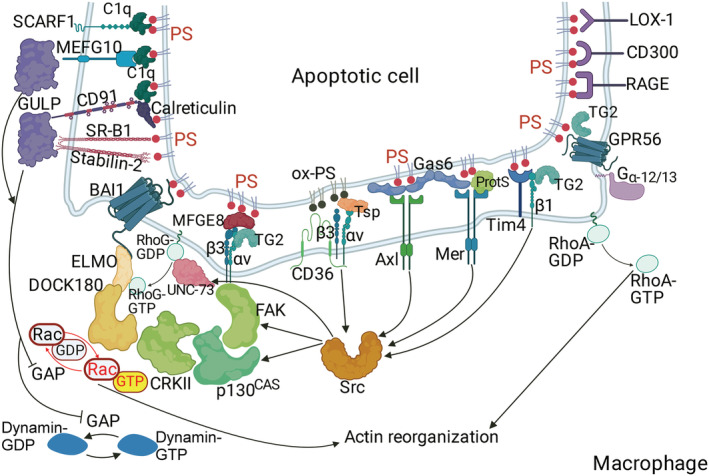
Collaboration between various phagocytosis receptors in the uptake of apoptotic cells. In the initiation of engulfment, increasing the amount of the GTP‐bound Rac1 plays the central role. This is achieved by either enhancing the GDP/GTP exchange on Rac1 catalysed by the Dock180/ELMO complex, the guanine nucleotide exchange factor (GEF) for Rac1, or by attenuating the GTPase activity of Rac1 (which would hydrolyse GTP back to GDP) achieved by the GULP pathway. GULP inhibits very likely the GTPase activating protein (GAP) of Rac1. Some of the phagocytic receptors promote the assembly of the Dock180 and ELMO proteins to generate the functional GEF complex, while others interact with GULP. Sequentially, GTP loading of RhoA and dynamin needs also to be enhanced as they contribute to later phases of efferocytosis. To promote Rac1 activation, integrins trigger simultaneously three distinct signalling pathways involving activation of focal adhesion (FAK), src (SRC), and phosphoinositide‐3 (not shown on the figure) kinases, respectively, leading to the assembly of Dock180/ELMO complex. FAK and SRC do so by activating both the UNC‐73/RhoG and the p130CAS/CrkII pathways via tyrosine phosphorylating the proteins participating in them. Sequential enrichment of phosphoinositides generated by the phosphoinositide‐3 kinases in the inner leaflet of the plasma membrane will then guide the recruitment of the assembled Dock180/ELMO to the phagocytic cup via the phosphoinositide recognizing domains of both proteins. CD36, TG2, and Tim4 act as coreceptors for the integrin receptors, while the TAM kinases Mer and Axl act via enhancing the integrin‐mediated signalling pathways. Simultaneously, following PS binding a trimeric complex is also formed that includes BAI1, ELMO, and DOCK180 that further promotes Rac1 activation (GTP loading). To enhance the effectiveness of all the aforementioned receptors, both CD300f and CD300b act as activators of phosphoinositide‐3‐kinase pathway. Many other phagocytic receptors, on the other hand, contribute to increasing the amount of GTP‐loaded Rac1 via the GULP adaptor protein pathway. These include CD91, SCARF1, stabilin‐2, MEGF10, and SR‐B1. Interestingly, while the bridging molecule C1q links PS only to those phagocytic receptors that are coupled to GULP, the bridging molecules MFG‐E8, protein S (ProtS), Gas6, and thrombospondin (TSP) activate receptors that promote the Dock180/ELMO assembly. GULP, in addition, is also associated with dynamin activation, while spliced Grp56 functions very likely coupled to the Gα12,13/RhoA signalling pathways. The signalling pathways induced by LOX‐1 and RAGE in the context of efferocytosis have not been investigated yet.
In addition to Rac1, the involvement of other GTPases, such as RhoA and Cdc42, has also been demonstrated in the efferocytosis process. These small G proteins function in a temporally controlled fashion in which Rac1 and Cdc42 are activated early to facilitate phagocytic cup formation through actin polymerization followed by RhoA activation, which drives mechanical retraction and phagosome internalization. 59 In addition to the Rho/Rac family of proteins, dynamin‐2, a large GTPase, has also been linked to the phagocytosis process participating in the CD91 and MEFG10‐initiated GULP‐dependent signalling pathways. Dynamin was shown to organize an intracellular vesicle pool and to promote vesicle delivery to phagocytic cups, thus to support pseudopod extension. 66 , 67 , 68
The redundancy of the phagocytic receptors makes it possible that efferocytosis is maintained in the organism, even if at a lower rate when one of these receptors is missing. This lower rate of efferocytosis is sufficient to remove the apoptotic cells generated under steady‐state conditions; however, when a high rate of apoptosis is induced, apoptotic cells are accumulated, converted to secondary necrotic cells, and induce inflammation due to the delayed clearance. Inflammation is prolonged or maintained then due to the fact that proper efferocytosis would also be required for proper macrophage polarization, which normally initiates the resolution of inflammation. As a result, delayed clearance of apoptotic cells in humans leads to the development of various chronic inflammatory diseases. 69 , 70
Phosphatidylserine externalization and Rac activation are also required for proper myoblast fusion
Skeletal muscle fibres are syncytia that are generated via the fusion of myoblasts, to form multinucleated myotubes. On a cellular level, myoblast fusion is initiated by an alignment of myoblast and/or myotube membranes, followed by rearrangements of the actin cytoskeleton at the contact sites and then completed by the fusion of the cellular membranes. In mammals, it occurs during embryogenesis and in the adult, promoting generation, growth, and repair of muscle fibres. 71
The fact that PS might also be required for myoblast differentiation and fusion was suggested first by a report, in which by using the PS binding protein annexin V, the transient appearance of PS was observed at the cell surface of apparently viable myoblasts in the developing heart and skeletal muscle. 72 Later it was demonstrated that during myoblast fusion, PS appears at the fusing cell–cell contact areas independently of caspase activation and almost exclusively only on mononucleated myoblasts in contact with other mononucleated cells or small myotubes containing only a few nuclei. 15 , 73 The need for caspase activation in inducing PS exposure also in myoblasts was recently demonstrated. 74 The formation of multinucleated myotubes from individual differentiating myoblasts could be specifically inhibited by blocking PS via annexin V binding proving a signalling role of PS in the myoblast fusion as well. 75
Studies carried out on the Drosophila muscle development indicated surprisingly that the key intracellular components that control cytoskeletal and particularly actin rearrangements during myoblast fusion similar to efferocytosis are the Rac GTPases, dRac1, and dRac2, and the Rac GEF, dimeric guanine nucleotide exchange factor encoded by dElmo, and myoblast city, a homologue of the vertebrate DOCK1. 76 , 77 Later these proteins were identified as controllers of the vertebrate myoblast fusion process as well. 14 What is more, GULP and dynamin have also been demonstrated to be associated with myoblast fusion. 15 , 78 These data indicated that a common signal triggers and common signalling pathways mediate efferocytosis and myoblast fusion.
Phosphatidylserine recognizing efferocytic receptors guide also myoblast fusion
The aforementioned findings prompted us to search the literature on how general it is that a PS recognizing receptor known to be involved in efferocytosis is also associated with myotube formation or myogenesis. We searched (1) whether the loss of the PS‐dependent phagocytic receptor is known to be associated with defects of myogenesis, especially with muscle fibres characterized by smaller cross‐sectional areas that indicate myotube fusion deficiency during embryogenesis; (2) whether it is known to be associated with impaired muscle repair; and (3) whether down‐regulation of it affects the myoblast fusion of C2C12 or other myoblast cells in the in vitro models of myogenesis. 11 We searched also whether a known PS receptor contributing to myogenesis is also involved in efferocytosis.
The results of our search are summarized in Table 1. To our surprise, we found that nearly all the PS recognizing efferocytosis receptors have already been reported to contribute also to myogenesis. However, these shared receptors do not mediate the membrane fusion itself, 79 although some of the proteins, such as myoferlin, which contribute to membrane fusion, are also PS dependent. 80 Rather, the majority of them participate in the prefusion events, such as myoblast differentiation, cell–cell recognition, adhesion, and cytoskeletal rearrangements that might be needed to generate and then to bring the fusion proteins in proper orientation and proximity between the two fusing cells. 79 Integrin β3 and Rac activity, for example, are required for myoblast differentiation and adhesion, 81 , 82 but integrin β3 has to be down‐regulated prior to fusion. 82 Similar was the finding with thrombospondin‐1, the ligand for integrin β3 receptor. 83 , 84 Although Mer was suggested to contribute to the membrane fusion during fertilization, 85 Mer protein is not expressed by muscle cells. 10 The other TAM kinase efferocytosis receptor (Axl), however, is expressed and involved in myogenesis but is required for myoblast cell survival and also for myoblast growth. 10 In accordance, Mer null mice have normal size myofibres, 10 stabilin‐2 or myoferlin (two fusion‐coupled proteins) null muscles are characterized by small size muscle fibres, 15 , 80 while the Gas6/Axl double knockout mice have decreased hindlimb muscle mass and satellite cell proliferation after injury accompanied by compensatory muscle fibre hypertrophy. 86 , 87 Thus, the muscle phenotype of the lost PS receptor is strongly dependent on its specific contribution to the myogenesis process.
Table 1.
Involvement of phosphatidylserine binding receptors or bridging molecules in efferocytosis, skeletal muscle development and repair, and in vitro myoblast fusion
| Receptor or bridging molecule | Involvement in | |||
|---|---|---|---|---|
| efferocytosis | embryonal myogenesis | skeletal muscle repair | in vitro myoblast fusion | |
| Myoferlin | n.d. | Yes 80 | Yes 80 | Yes 80 |
| Integrin β1 | Yes 32 | Yes 104 | Yes 105 | Yes, 105 no 106 |
| Integrin β3 | Yes 24 | n.d. | Yes 106 | Yes, 81 but has to be down‐regulated 82 |
| Integrin β5 | Yes 31 | n.d. | n.d. | Yes 107 |
| CD36 | Yes 24 , 28 | No effect 108 | Both SCs and macrophages are affected 107 | Yes 109 |
| TG2 | Yes 30 | Yes 91 | Yes 91 | Crosslinking activity is not needed, 90 but the protein yes 91 |
| TIM3 | Yes 43 | n.d. | n.d. | n.d. |
| TIM4 | Yes 43 | n.d. | n.d. | n.d. |
| MFG‐E8 | Yes 29 | n.d. | n.d. | Promotes myoblast differentiation and possibly fusion 88 |
| Tsp‐1 | Yes 24 | No effect 110 | Macrophage‐dependent effect 111 | Promotes adhesion, 83 but inhibits fusion 84 |
| Mer | Yes 34 | Not expressed 10 ¤ | Macrophage‐dependent effect 10 | Not expressed 10 |
| Axl (Gas6) | Yes 35 | Yes 86 , 87 | Yes 87 | Promotes myoblast and myotube survival and growth 10 |
| Tyro‐3 | Yes 35 | Not expressed 10 | n.d. | Not expressed 10 |
| Protein S | Yes 35 , 36 | n.d. | n.d. | Secreted by myoblasts 112 |
| CD91 | Yes 37 , 38 | n.d. | n.d. | n.d. |
| Calreticulin | Yes 37 , 39 | n.d. | n.d. | Secreted by myoblasts 112 |
| C1q | Yes 37 , 38 | n.d. | Negative effect 113 | n.d. |
| SCARF1 | Yes 40 | n.d. | n.d. | n.d. |
| MEGF10 | Yes 41 | Yes 114 | Yes 115 | Yes 116 |
| BAI1 | Yes 44 | Yes 14 | Yes 14 | Yes 14 |
| BAI3 | Not involved | Yes 117 | Yes 117 | Yes 117 , 118 |
| Gpr56 | Yes 46 | No effect 119 | n.d. | Yes 119 |
| Stabilin2 | Yes 49 , 50 | Yes 15 | Yes 15 | Yes 15 , 117 |
| SR‐BI | Yes 9 , 51 | No effect 9 | Macrophage‐dependent effect 9 | n.d. |
| CD300 | Yes 52 , 53 | n.d. | n.d. | n.d. |
| RAGE | Yes 54 , 55 | Increased number of SCs 120 | Yes 120 | Myoblast differentiation, 121 fusion was n.d. |
| LOX‐1 | Yes 56 | n.d. | n.d. | n.d. |
| Annexins | Yes 122 | No effect 123 | Yes 123 | Yes 78 |
| Piezo | n.d. | n.d. | n.d. | Yes 124 |
n.d., not determined.
The data that we collected in Table 1 also indicate that there are several among these PS‐recognizing receptors or their bridging molecules, the role of which was not tested yet in both processes. We propose that they might very likely play a role in the so far non‐investigated side as well. Thus, in addition to macrophages, myoblasts are also known to secrete MFG‐E8, 88 while the myoblast fusion‐coupled myoferlin is known to participate in the lysosomal exocytosis, a process suggested to contribute to the engulfment of apoptotic adipocytes. 89 We propose that the fact that efferocytosis and myoblast fusion share several molecules and evolutionary conserved cellular mechanisms may help us in an in‐depth understanding of these processes, as well as in the identification of new players either in the efferocytosis or in myoblast fusion side. To test this proposal, we decided to investigate whether TG2, an efferocytosis coreceptor, 30 could be involved in the myoblast fusion, although previous studies indicated that its crosslinking activity is not required for it. 90 We found that TG2 null mice display impaired muscle repair. 91 TG2 is required not only for promoting the phenotypic switch of macrophages during muscle regeneration but also for myoblast fusion and growth. Accordingly, similar to other mice that show impaired myoblast fusion, 15 , 80 TG2 null muscles are also characterized by small size myofibres. Our results provide an additional proof for the participation of efferocytosis receptors in both efferocytosis and myogenesis.
Impaired efferocytosis, impaired muscle regeneration, and sarcopenia. Can they be coupled?
Sarcopenia is the progressive loss of muscle mass. The term is often used specifically to denote loss of muscle mass and strength associated with aging. Although the causes of sarcopenia are multifactorial, increasing evidence indicates that impaired satellite cell‐dependent muscle regenerative capacity in the aged might contribute to the development of it. There are multiple suggested mechanisms for the impaired muscle regenerative process in aged muscle including loss of satellite cell number or function, decreased myoblast proliferation, or weakened differentiation states (reviewed in reference 92 ).
Sarcopenia can develop during the course of chronic inflammatory diseases as well, and aging itself is associated with the development of various chronic inflammatory conditions. The mechanisms linking chronic inflammation and sarcopenia were reported to involve increased muscle protein imbalance, cell death, increased muscle adiposity, and also impaired muscle repair and regeneration (reviewed in reference 93 ). A large cohort study that addressed the potential link between impaired muscle regeneration and the development of sarcopenia applied large‐scale transcriptome analysis for studying the skeletal muscle. They found that aging is associated with increased cell death in the skeletal muscle associated with increased satellite cell‐dependent regeneration response in the healthy elderly, while an impaired one in the sarcopenic muscle accompanied by high pro‐inflammatory cytokine levels. Decreased regenerating potential was further investigated on the myoblast side. Using myoblasts obtained from a number of donors across the adult age range, this study demonstrated a decrease of myoblast fusion in older compared with young donors. Moreover, myogenin and troponin I transcription were decreased in the myoblast cultures from the older donors indicating reduced or impaired kinetics of myotube formation. 94
Interestingly, despite the involvement of macrophages in guiding muscle repair and regulating inflammation, in these studies, the age‐related alterations in macrophage functions were not considered in the context of the development of sarcopenia, although age is the most important risk factor for many of the chronic disorders associated with macrophage dysfunction. 95 Thus, impaired macrophage transcription and function have been observed in normal aging, detected as reduced phagocytosis capacity, impaired polarization in vitro, and a loss of wound healing response 96 accompanied with an improper resolution of inflammation. 97 Similarly, chronic inflammation is also known to be associated with impaired resolution of inflammation related to a defective macrophage efferocytosis (reviewed in references 69 , 70 , 97 ). Naturally in humans, it is difficult to demonstrate a decreased efficiency of efferocytosis within the sarcopenic muscle, but when macrophage efferocytosis is generally impaired, it is expected that muscle‐specific macrophages will be similarly affected. And if they are, according to the findings in efferocytosis receptor null mice, 9 , 10 their decreased efferocytosis capacity will influence not only the clearance of dead cells but also resolution of inflammation, myoblast differentiation, and fusion detected to be altered in sarcopenic muscles. Accordingly, the pathophysiologic link between aging, obesity, impaired efferocytosis, abnormal macrophage polarization, chronic inflammation, and the development of sarcopenia is increasingly being recognized. 97 , 98 , 99
Concluding remarks
The data collected in this review highlight that the two central biological processes—efferocytosis by macrophages and myogenesis—that contribute to the successful muscle repair share quite a number of PS‐dependent receptors and signalling pathways. We propose that the involvement of PS receptors in both the myoblast‐related and the macrophage‐related muscle regeneration processes makes them an excellent target in promoting muscle repair following acute muscle injury, because stimulating the function of them would facilitate efferocytosis by macrophages and myogenesis simultaneously. The most logical choice is application of those bridging molecules that target PS receptors coupled to DOCK180/ELMO assembly, such as MFG‐E8. An additional advantage of MFG‐E8 administration would be that it also promotes VEGF‐dependent neovascularization, which is also part of the proper muscle repair. 100 In support of our proposal, administration of the MFG‐E8 bridging molecule successfully facilitated cardiac muscle repair following infarction. 101
Because increasing evidence indicates a pathophysiological role of impaired efferocytosis in the development of chronic inflammatory conditions, as well as in impaired muscle regeneration both contributing to the development of sarcopenia, improving efferocytosis should be considered also in its management. Several methods have already been proposed that could be applied. 69 Again based on our present findings, selecting or combining those that activate PS receptors would be expected to be the most effective, because they would also promote myogenesis. In support of our proposal, after submission of our manuscript, administration of MFG‐E8 was reported to attenuate sarcopenia in a rat model induced by d‐galactose. 102 What is more, a recent report, which investigated the key differentially expressed genes and pathways associated with the progression of sarcopenia, identified MFG‐E8 as a significantly down‐regulated gene in sarcopenic muscles and demonstrated that MFG‐E8 has potential anti‐sarcopenia effects by promoting extracellular signal‐regulated kinase (ERK) and Akt activation‐mediated cell proliferation and cell cycle progression in myoblasts. 103
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This research was funded by the European Union project titled: Institutional Developments for Intelligent Specialization program (grant number: EFOP‐3.6.1‐16‐2016‐00022 ‘Debrecen Venture Catapult Program’), by the National Research, Development and Innovation Office (124244 and 138162), and by the GINOP‐2.3.2‐15‐2016‐00006 project (co‐financed by the European Union and the European Regional Development Fund).
Acknowledgements
The authors acknowledge the technical help of Tamás Varga (Softflow Company, Pécs, Hungary) in the initiation of muscle regeneration studies by our laboratory. Open access funding enabled and organized by Projekt DEAL.
Szondy Z., Al‐Zaeed N., Tarban N., Fige É., Garabuczi É., and Sarang Z. (2022) Involvement of phosphatidylserine receptors in the skeletal muscle regeneration: therapeutic implications, Journal of Cachexia, Sarcopenia and Muscle, 13, 1961–1973, 10.1002/jcsm.13024
References
- 1. Roman W, Pinheiro H, Pimentel MR, Segalés J, Oliveira LM, García‐Domínguez E, et al. Muscle repair after physiological damage relies on nuclear migration for cellular reconstruction. Science 2021;374:355–359. [DOI] [PubMed] [Google Scholar]
- 2. Wang YX, Rudnicki MA. Satellite cells, the engines of muscle repair. Nat Rev Mol Cell Biol 2012;13:127–133. [DOI] [PubMed] [Google Scholar]
- 3. Summan M, Warren GL, Mercer RR, Chapman R, Hulderman T, van Rooijen N, et al. Macrophages and skeletal muscle regeneration: a clodronate‐containing liposome depletion study. Am J Physiol Regul Integr Comp Physiol 2006;290:R1488–R1495. [DOI] [PubMed] [Google Scholar]
- 4. McLennan IS. Degenerating and regenerating skeletal muscles contain several subpopulations of macrophages with distinct spatial and temporal distributions. J Anat 1996;188:17–28. [PMC free article] [PubMed] [Google Scholar]
- 5. Arnold L, Henry A, Poron F, Baba‐Amer Y, van Rooijen N, Plonquet A, et al. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med 2007;204:1071–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chazaud B. Inflammation and skeletal muscle regeneration: leave it to the macrophages. Trends Immunol 2020;6:481–492. [DOI] [PubMed] [Google Scholar]
- 7. Patsalos A, Halasz L, Medina‐Serpas MA, Berger WK, Daniel B, Tzerpos P, et al. A growth factor expressing macrophage subpopulation orchestrates regenerative inflammation via GDF‐15. J Exp Med 2022;219:e20210420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Szondy Z, Sarang Z, Kiss B, Garabuczi É, Köröskényi K. Anti‐inflammatory mechanisms triggered by apoptotic cells during their clearance. Front Immunol 2017;8:909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang J, Qu C, Li T, Cui W, Wang X, Du J. Phagocytosis mediated by scavenger receptor class BI promotes macrophage transition during skeletal muscle regeneration. J Biol Chem 2019;294:15672–15685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Al‐Zaeed N, Budai Z, Szondy Z, Sarang Z. TAM kinase signaling is indispensable for the proper skeletal muscle regeneration process. Cell Death Dis 2021;12:611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol 1992;48:2207–2216. [PubMed] [Google Scholar]
- 12. Krahling S, Callahan MK, Williamson P, Schlegel RA. Exposure of phosphatidylserine is a general feature in the phagocytosis of apoptotic lymphocytes by macrophages. Cell Death Differ 1999;6:183–189. [DOI] [PubMed] [Google Scholar]
- 13. Lemke G. Biology of the TAM receptors. Cold Spring Harb Perspect Biol 2013;5:a009076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hochreiter‐Hufford AE, Lee CS, Kinchen JM, Sokolowski JD, Arandjelovic S, Call JA, et al. Phosphatidylserine receptor BAI1 and apoptotic cells as new promoters of myoblast fusion. Nature 2013;497:263–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Park SY, Yun Y, Lim JS, Kim MJ, Kim SY, Kim JE, et al. Stabilin‐2 modulates the efficiency of myoblast fusion during myogenic differentiation and muscle regeneration. Nat Commun 2016;7:10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Igumenova TI. Dynamics and membrane interactions of protein kinase C. Biochemistry 2015;54:4953–4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Travers T, López CA, Van QN, Neale C, Tonelli M, Stephen AG, et al. Molecular recognition of RAS/RAF complex at the membrane: role of RAF cysteine‐rich domain. Sci Rep 2018;8:8461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang BX, Akbar M, Kevala K, Kim HY. Phosphatidylserine is a critical modulator for Akt activation. J Cell Biol 2011;192:979–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Holthuis JC, Levine TP. Lipid traffic: floppy drives and a superhighway. Nat Rev Mol Cell Biol 2005;6:209–220. [DOI] [PubMed] [Google Scholar]
- 20. Nagata S, Suzuki J, Segawa K, Fujii T. Exposure of phosphatidylserine on the cell surface. Cell Death Differ 2016;23:952–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lentz BR. Exposure of platelet membrane phosphatidylserine regulates blood coagulation. Prog Lipid Res 2003;42:423–438. [DOI] [PubMed] [Google Scholar]
- 22. Naeini MB, Bianconi V, Pirro M, Sahebkar A. The role of phosphatidylserine recognition receptors in multiple biological functions. Cell Mol Biol Lett 2020;25:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martin SJ, Reutelingsperger CPM, McGahon AJ, Rader JA, van Schie RCAA, LaFace DM, et al. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl‐2 and Abl. J Exp Med 1995;182:1545–15567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Savill J, Dransfield I, Hogg N, Haslett C. Vitronectin receptor‐mediated phagocytosis of cells undergoing apoptosis. Nature 1990;343:170–173. [DOI] [PubMed] [Google Scholar]
- 25. Horton MA. The αvβ3 integrin “vitronectin receptor”. Int J Biochem Cell Biol 1997;29:721–725. [DOI] [PubMed] [Google Scholar]
- 26. Stern M, Savill J, Haslett C. Human monocyte‐derived macrophage phagocytosis of senescent eosinophils undergoing apoptosis. Mediation by alpha v beta 3/CD36/thrombospondin recognition mechanism and lack of phlogistic response. Am J Pathol 1996;149:911–921. [PMC free article] [PubMed] [Google Scholar]
- 27. Gayen Betal S, Setty BN. Phosphatidylserine‐positive erythrocytes bind to immobilized and soluble thrombospondin‐1 via its heparin binding domain. Transl Res 2008;152:165–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Greenberg ME, Sun M, Zhang R, Febbraio M, Silverstein R, Hazen SL. Oxidized phosphatidylserine–CD36 interactions play an essential role in macrophage‐dependent phagocytosis of apoptotic cells. J Exp Med 2006;203:2613–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tanaka M. Apoptotic cell clearance by phagocytes. Int Congr Ser 2005;1285:55–59. [Google Scholar]
- 30. Tóth B, Garabuczi E, Sarang Z, Vereb G, Vámosi G, Aeschlimann D, et al. Transglutaminase 2 is needed for the formation of an efficient phagocyte portal in macrophages engulfing apoptotic cells. J Immunol 2009;82:2084–2092. [DOI] [PubMed] [Google Scholar]
- 31. Nandrot EF, Anand M, Almeida D, Atabai K, Sheppard D, Finnemann SC. Essential role for MFG‐E8 as ligand for alphavbeta5 integrin in diurnal retinal phagocytosis. Proc Natl Acad Sci U S A 2007;104:12005–12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Flannagan RS, Canton J, Furuya W, Glogauer M, Grinstein S. The phosphatidylserine receptor TIM4 utilizes integrins as coreceptors to effect phagocytosis. Mol Biol Cell 2014;25:1511–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Akimov SS, Krylov D, Fleischman LF, Belkin AM. Tissue transglutaminase is an integrin‐binding adhesion coreceptor for fibronectin. J Cell Biol 2000;148:825–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dransfield I, Zagórska A, Lew ED, Michail K, Lemke G. Mer receptor tyrosine kinase mediates both tethering and phagocytosis of apoptotic cells. Cell Death Dis 2015;6:e1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tsou WI, Nguyen KQ, Calarese DA, Garforth SJ, Antes AL, Smirnov SV, et al. Receptor tyrosine kinases, TYRO3, AXL, and MER, demonstrate distinct patterns and complex regulation of ligand‐induced activation. J Biol Chem 2014;289:25750–25763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Geng K, Kumar S, Kimani SG, Kholodovych V, Kasikara C, Mizuno K, et al. Requirement of gamma‐carboxyglutamic acid modification and phosphatidylserine binding for the activation of Tyro3, Axl, and Mertk receptors by growth arrest‐specific 6. Front Immunol 2017;8:1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ogden CA, deCathelineau A, Hoffmann PR, Bratton D, Ghebrehiwet B, Fadok VA, et al. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med 2001;194:781–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Duus K, Hansen EW, Tacnet P, Frachet P, Arlaud GJ, Thielens NM, et al. Direct interaction between CD91 and C1q. FEBS J 2010;277:3526–3537. [DOI] [PubMed] [Google Scholar]
- 39. Wijeyesakere SJ, Bedi SK, Huynh D, Raghavan M. The C‐terminal acidic region of calreticulin mediates phosphatidylserine binding and apoptotic cell phagocytosis. J Immunol 2016;196:3896–3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ramirez‐Ortiz ZG, Pendergraft WF 3rd, Prasad A, Byrne MH, Iram T, Blanchette CJ, et al. The scavenger receptor SCARF1 mediates the clearance of apoptotic cells and prevents autoimmunity. Nat Immunol 2013;14:917–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Iram T, Ramirez‐Ortiz Z, Byrne MH, Coleman UA, Kingery ND, Means TK, et al. Megf10 is a receptor for C1Q that mediates clearance of apoptotic cells by astrocytes. J Neurosci 2016;36:5185–5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Toda S, Hanayama R, Nagata S. Two‐step engulfment of apoptotic cells. Mol Cell Biol 2012;32:118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Freeman GJ, Casasnovas JM, Umetsu DT, DeKruyff RH. TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev 2010;235:172–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stephenson JR, Purcell RH, Hall RA. The BAI subfamily of adhesion GPCRs: synaptic regulation and beyond. Trends Pharmacol Sci 2014;35:208–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nishimori H, Shiratsuchi T, Urano T, Kimura Y, Kiyono K, Tatsumi K, et al. A novel brain‐specific p53‐target gene, BAI1, containing thrombospondin type 1 repeats inhibits experimental angiogenesis. Oncogene 1997;15:2145–2150. [DOI] [PubMed] [Google Scholar]
- 46. Li T, Chiou B, Gilman CK, Luo R, Koshi T, Yu D, et al. A splicing isoform of GPR56 mediates microglial synaptic refinement via phosphatidylserine binding. EMBO J 2020;39:e104136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xu L, Begum S, Hearn JD, Hynes RO. GPR56, an atypical G protein‐coupled receptor, binds tissue transglutaminase, TG2, and inhibits melanoma tumor growth and metastasis. Proc Natl Acad Sci U S A 2006;103:9023–9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Giera S, Luo R, Ying Y, Ackerman SD, Jeong SJ, Stoveken HM, et al. Microglial transglutaminase‐2 drives myelination and myelin repair via GPR56/ADGRG1 in oligodendrocyte precursor cells. Elife 2018;7:e33385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Park SY, Kim SY, Jung MY, Bae DJ, Kim IS. Epidermal growth factor‐like domain repeat of stabilin‐2 recognizes phosphatidylserine during cell corpse clearance. Mol Cell Biol 2008;28:5288–5298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Park SY, Jung MY, Kim HJ, Lee SJ, Kim SY, Lee BH, et al. Rapid cell corpse clearance by stabilin‐2, a membrane phosphatidylserine receptor. Cell Death Differ 2008;15:192–201. [DOI] [PubMed] [Google Scholar]
- 51. Kawasaki Y, Nakagawa A, Nagaosa K, Shiratsuchi A, Nakanishi Y. Phosphatidylserine binding of class B scavenger receptor type I, a phagocytosis receptor of testicular sertoli cells. J Biol Chem 2002;277:27559–27566. [DOI] [PubMed] [Google Scholar]
- 52. Clark GJ, Ju X, Tate C , Hart DN. The CD300 family of molecules are evolutionarily significant regulators of leukocyte functions. Trends Immunol 2009;30:209–217. [DOI] [PubMed] [Google Scholar]
- 53. Voss OH, Tian L, Murakami Y, Coligan JE, Krzewski K. Emerging role of CD300 receptors in regulating myeloid cell efferocytosis. Mol Cell Oncol 2015;2:e964625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, et al. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem 1992;267:14998–15004. [PubMed] [Google Scholar]
- 55. He M, Kubo H, Morimoto K, Fujino N, Suzuki T, Takahasi T, et al. Receptor for advanced glycation end products binds to phosphatidylserine and assists in the clearance of apoptotic cells. EMBO Rep 2011;12:358–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Murphy JE, Tacon D, Tedbury PR, Hadden JM, Knowling S, Sawamura T, et al. LOX‐1 scavenger receptor mediates calcium‐dependent recognition of phosphatidylserine and apoptotic cells. Biochem J 2006;393:107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Oka K, Sawamura T, Kikuta K, Itokawa S, Kume N, Kita T, et al. Lectin‐like oxidized low‐density lipoprotein receptor 1 mediates phagocytosis of aged/apoptotic cells in endothelial cells. Proc Natl Acad Sci U S A 1998;95:9535–9540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Barth ND, Marvick JA, Vendrell M, Rossi AG, Dransfield I. “Phagocytic synapse” and clearance of apoptotic cells. Front Immunol 2017;8:1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nakaya M, Kitano M, Matsuda M, Nagata S. Spatiotemporal activation of Rac1 for engulfment of apoptotic cells. Proc Natl Acad Sci U S A 2008;105:9198–9203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wu YC, Horvitz HR. C. elegans phagocytosis and cell‐migration protein CED‐5 is similar to human DOCK180. Nature 1998;392:501–504. [DOI] [PubMed] [Google Scholar]
- 61. Gumienny TL, Brugnera E, Tosello‐Trampont AC, Kinchen JM, Haney LB, Nishiwaki K, et al. CED‐12/ELMO, a novel member of the crkII/Dock180/Rac pathway, is required for phagocytosis and cell migration. Cell 2001;107:27–41. [DOI] [PubMed] [Google Scholar]
- 62. Smits E, Van Criekinge W, Plaetinck G, Bogaert T. The human homologue of Caenorhabditis elegans CED‐6 specifically promotes phagocytosis of apoptotic cells. Curr Biol 1999;9:1351–1354. [DOI] [PubMed] [Google Scholar]
- 63. Ma Z, Nie Z, Luo R, Casanova JE, Ravichandran KS. Regulation of Arf6 and ACAP1 signaling by the PTB‐domain‐containing adaptor protein GULP. Curr Biol 2007;17:722–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kobayashi S, Shirai T, Kiyokawa E, Mochizuki N, Matsuda M, Fukui Y. Membrane recruitment of DOCK180 by binding to PtdIns(3,4,5)P3. Biochem J 2001;354:73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cote JF, Motoyama AB, Bush JA, Vuori K. A novel and evolutionarily conserved PtdIns(3,4,5)P3‐binding domain is necessary for DOCK180 signalling. Nat Cell Biol 2005;7:797–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yu X, Odera S, Chuang CH, Lu N, Zhou Z. C. elegans dynamin mediates the signaling of phagocytic receptor CED‐1 for the engulfment and degradation of apoptotic cells. Dev Cell 2006;10:743–757. [DOI] [PubMed] [Google Scholar]
- 67. Gold ES, Underhill DM, Morrissette NS, Guo J, McNiven MA, Aderem A. Dynamin 2 is required for phagocytosis in macrophages. J Exp Med 1999;190:1849–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Farkas Z, Petric M, Liu X, Herit F, Rajnavölgyi É, Szondy Z, et al. The nucleoside diphosphate kinase NDK‐1/NME1 promotes phagocytosis in concert with DYN‐1/Dynamin. FASEB J 2019;33:11606–11614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Szondy Z, Garabuczi E, Joós G, Tsay GJ, Sarang Z. Impaired clearance of apoptotic cells in chronic inflammatory diseases: therapeutic implications. Front Immunol 2014;5:354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Morioka S, Maueröder C, Ravichandran KS. Driving on the edge: efferocytosis at the interface of homeostasis and pathology. Immunity 2019;50:1149–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Rochlin K, Yu S, Roy S, Baylies MK. Myoblast fusion: when it takes more to make one. Dev Biol 2010;341:66–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Van den Eijnde SM, Boshart L, Reutelingsperger CP, De Zeeuw CI, Vermeij‐Keers C. Phosphatidylserine membrane asymmetry in vivo: a pancellular phenomenon which alters during apoptosis. Cell Death Differ 1997;4:311–316. [DOI] [PubMed] [Google Scholar]
- 73. Jeong J, Conboy IM. Phosphatidylserine directly and positively regulates fusion of myoblasts into myotubes. Biochem Biophys Res Commun 2011;414:9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dehkordi MH, Tashakor A, O'Connell E, Fearnhead HO. Apoptosome‐dependent myotube formation involves activation of caspase‐3 in differentiating myoblasts. Cell Death Dis 2020;11:308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Van den Eijnde SM, van den Hoff MJ, Reutelingsperger CP, van Heerde WL, Henfling ME, Vermeij‐Keers C, et al. Transient expression of phosphatidylserine at cell‐cell contact areas is required for myotube formation. J Cell Sci 2001;114:3631–3642. [DOI] [PubMed] [Google Scholar]
- 76. Luo L, Liao YJ, Jan LY, Jan YN. Distinct morphogenetic functions of similar small GTPases: Drosophila Drac1 is involved in axonal outgrowth and myoblast fusion. Genes Dev 1994;8:1787–1802. [DOI] [PubMed] [Google Scholar]
- 77. Rushton E, Drysdale R, Abmayr SM, Michelson AM, Bate M. Mutations in a novel gene, myoblast city, provide evidence in support of the founder cell hypothesis for Drosophila muscle development. Development 1995;121:1979–1988. [DOI] [PubMed] [Google Scholar]
- 78. Leikina E, Melikov K, Sanyal S, Verma SK, Eun B, Gebert C, et al. Extracellular annexins and dynamin are important for sequential steps in myoblast fusion. J Cell Biol 2013;200:109–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Whitlock JM, Chernomordik LV. Flagging fusion: phosphatidylserine signaling in cell‐cell fusion. J Biol Chem 2021;296:100411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Doherty KR, Cave A, Davis DB, Delmonte AJ, Posey A, Earley JU, et al. Normal myoblast fusion requires myoferlin. Development 2005;132:5565–5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Liu H, Niu A, Chen SE, Li YP. Beta3‐integrin mediates satellite cell differentiation in regenerating mouse muscle. FASEB J 2011;25:1914–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Blaschuk KL, Guérin C, Holland PC. Myoblast alpha v beta3 integrin levels are controlled by transcriptional regulation of expression of the beta3 subunit and down‐regulation of beta3 subunit expression is required for skeletal muscle cell differentiation. Dev Biol 1997;184:266–277. [DOI] [PubMed] [Google Scholar]
- 83. Adams JC, Lawler J. Cell‐type specific adhesive interactions of skeletal myoblasts with thrombospondin‐1. Mol Biol Cell 1994;5:423–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Suárez‐Calvet X, Alonso‐Pérez J, Castellví I, Carrasco‐Rozas A, Fernández‐Simón E, Zamora C, et al. Thrombospondin‐1 mediates muscle damage in brachio‐cervical inflammatory myopathy and systemic sclerosis. Neurol Neuroimmunol Neuroinflamm 2020;7:e694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Rival CM, Xu W, Shankman LS, Morioka S, Arandjelovic S, Lee CS, et al. Phosphatidylserine on viable sperm and phagocytic machinery in oocytes regulate mammalian fertilization. Nat Commun 2019;10:4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Olsen ZE, Mervis MJ, Matsumara MC, Hirschi‐Budge KM, Arroyo JA, Reynolds PR, et al. Gas6‐Axl double knockout in mice decreases skeletal muscle mass despite elevated anabolic intracellular signaling. FASEB J 2020;34:1. [Google Scholar]
- 87. Mervis MJ, Matsumura MC, Olsen ZE, Hirschi‐Budge KM, Reynolds PR, Arroyo JA, et al. The effect of Gas6‐Axl double knockout on satellite cell proliferation and skeletal muscle regeneration after injury. FASEB J 2020;34:1. [Google Scholar]
- 88. Chikazawa M, Shimizu M, Yamauchi Y, Sato R. Bridging molecules are secreted from the skeletal muscle and potentially regulate muscle differentiation. Biochem Biophys Res Commun 2020;522:113–120. [DOI] [PubMed] [Google Scholar]
- 89. Haka AS, Barbosa‐Lorenzi VC, Lee HJ, Falcone DJ, Hudis CA, Dannenberg AJ, et al. Exocytosis of macrophage lysosomes leads to digestion of apoptotic adipocytes and foam cell formation. J Lipid Res 2016;57:980–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Bersten AM, Ahkong QF, Hallinan T, Nelson SJ, Lucy JA. Inhibition of the formation of myotubes in vitro by inhibitors of transglutaminase. Biochim Biophys Acta 1983;762:429–536. [DOI] [PubMed] [Google Scholar]
- 91. Budai Z, Al‐Zaeed N, Szentesi P, Halász H, Csernoch L, Szondy Z, et al. Impaired skeletal muscle development and regeneration in transglutaminase 2 knockout mice. Cell 2021;10:3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Arthur ST, Cooley ID. The effect of physiological stimuli on sarcopenia; impact of Notch and Wnt signaling on impaired aged skeletal muscle repair. Int J Biol Sci 2012;8:731–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Chhetri JK, de Souto Barreto P, Fougère B, Rolland Y, Vellas B, Cesari M. Chronic inflammation and sarcopenia: a regenerative cell therapy perspective. Exp Gerontol 2018;103:115–123. [DOI] [PubMed] [Google Scholar]
- 94. Brzeszczyńska J, Meyer A, McGregor R, Schilb A, Degen S, Tadini V, et al. Alterations in the in vitro and in vivo regulation of muscle regeneration in healthy ageing and the influence of sarcopenia. J Cachexia Sarcopenia Muscle 2018;9:93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Budinger GRS, Kohanski RA, Gan W, Kobor MS, Amaral LA, Armanios M, et al. The intersection of aging biology and the pathobiology of lung diseases: a joint NHLBI/NIA workshop. J Gerontol A Biol Sci Med Sci 2017;72:1492–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Linehan E, Fitzgerald DC. Ageing and the immune system: focus on macrophages. Eur J Microbiol Immunol (Bp) 2015;5:14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Sendama W. The effect of ageing on the resolution of inflammation. Ageing Res Rev 2020;57:101000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Wilson D, Jackson T, Sapey E, Lord JM. Frailty and sarcopenia: the potential role of an aged immune system. Aging Res Rev 2017;36:1–10. [DOI] [PubMed] [Google Scholar]
- 99. Livshits G, Kalinkovich A. Inflammaging as a common ground for the development and maintenance of sarcopenia, obesity, cardiomyopathy and dysbiosis. Aging Res Rev 2019;56:100980. [DOI] [PubMed] [Google Scholar]
- 100. Silvestre JS, Théry C, Hamard G, Boddaert J, Aguilar B, Delcayre A, et al. Lactadherin promotes VEGF‐dependent neovascularization. Nat Med 2005;11:499–506. [DOI] [PubMed] [Google Scholar]
- 101. Nakaya M, Watari K, Tajima M, Nakaya T, Matsuda S, Ohara H, et al. Cardiac myofibroblast engulfment of dead cells facilitates recovery after myocardial infarction. J Clin Invest 2017;127:383–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Le H, Wang R, Wang L, Li L, Ma Y, Zhou S. Bovine milk fat globule epidermal growth factor VIII activates PI3K/Akt signaling pathway and attenuates sarcopenia in rat model induced by D‐galactose. Food Biosci 2021;40:100847. [Google Scholar]
- 103. Li H, Guan K, Liu D, Liu M. Identification of mitochondria‐related hub genes in sarcopenia and functional regulation of MFG‐E8 on ROS‐mediated mitochondrial dysfunction and cell cycle arrest. Food Funct 2022;13:624–638. [DOI] [PubMed] [Google Scholar]
- 104. Schwander M, Leu M, Stumm M, Dorchies OM, Ruegg UT, Schittny J, et al. Beta1 integrins regulate myoblast fusion and sarcomere assembly. Dev Cell 2003;4:673–685. [DOI] [PubMed] [Google Scholar]
- 105. Rozo M, Li L, Fan CM. Targeting β1‐integrin signaling enhances regeneration in aged and dystrophic muscle in mice. Nat Med 2016;22:889–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Hirsch E, Lohikangas L, Gullberg D, Johansson S, Fassler R. Mouse myoblasts can fuse and form a normal sarcomere in the absence of β1 integrin expression. J Cell Sci 1998;111:2397–2409. [DOI] [PubMed] [Google Scholar]
- 107. Sinanan AC, Machell JR, Wynne‐Hughes GT, Hunt NP, Lewis MP. Alpha v beta 3 and alpha v beta 5 integrins and their role in muscle precursor cell adhesion. Biol Cell 2008;100:465–477. [DOI] [PubMed] [Google Scholar]
- 108. Verpoorten S, Sfyri P, Scully D, Mitchell R, Tzimou A, Mougios V, et al. Loss of CD36 protects against diet‐induced obesity but results in impaired muscle stem cell function, delayed muscle regeneration and hepatic steatosis. Acta Physiol (Oxf) 2020;228:e13395. [DOI] [PubMed] [Google Scholar]
- 109. Park SY, Yun Y, Kim IS. CD36 is required for myoblast fusion during myogenic differentiation. Biochem Biophys Res Commun 2012;427:705–710. [DOI] [PubMed] [Google Scholar]
- 110. Malek MH, Olfert MI. Global deletion of thrombospondin‐1 increases cardiac and skeletal muscle capillarity and exercise capacity in mice. Exp Physiol 2009;94:749–760. [DOI] [PubMed] [Google Scholar]
- 111. Bréchot N, Gomez E, Bignon M, Khallou‐Laschet J, Dussiot M, Cazes A, et al. Modulation of macrophage activation state protects tissue from necrosis during critical limb ischemia in thrombospondin‐1‐deficient mice. PLoS ONE 2008;3:e3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Florin A, Lambert C, Sanchez C, Zappia J, Durieux N, Tieppo AN, et al. The secretome of skeletal muscle cells: a systematic review. Osteoarthritis and Cartilage Open 2020;2:100019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Naito AT, Sumida T, Nomura S, Liu ML, Higo T, Nakagawa A, et al. Complement C1q activates canonical Wnt signaling and promotes aging‐related phenotypes. Cell 2012;149:1298–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Logan CV, Lucke B, Pottinger C, Abdelhamed ZA, Parry DA, Szymanska K, et al. Mutations in MEGF10, a regulator of satellite cell myogenesis, cause early onset myopathy, areflexia, respiratory distress and dysphagia (EMARDD). Nat Genet 2011;43:1189–1192. [DOI] [PubMed] [Google Scholar]
- 115. Saha M, Mitsuhashi S, Jones MD, Manko K, Reddy HM, Bruels CC, et al. Consequences of MEGF10 deficiency on myoblast function and Notch1 interactions. Hum Mol Genet 2017;26:2984–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Holterman CE, Le Grand F, Kuang S, Seale P, Rudnicki MA. Megf10 regulates the progression of the satellite cell myogenic program. J Cell Biol 2007;179:911–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Hamoud N, Tran V, Aimi T, Kakegawa W, Lahaie S, Thibault MP, et al. Spatiotemporal regulation of the GPCR activity of BAI3 by C1qL4 and Stabilin‐2 controls myoblast fusion. Nat Commun 2018;9:4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Hamoud N, Tran V, Croteau LP, Kania A, Côté JF. G‐protein coupled receptor BAI3 promotes myoblast fusion in vertebrates. Proc Natl Acad Sci U S A 2014;111:3745–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Wu MP, Doyle JR, Barry B, Beauvais A, Rozkalne A, Piao X, et al. G‐protein coupled receptor 56 promotes myoblast fusion through serum response factor‐ and nuclear factor of activated T‐cell‐mediated signalling but is not essential for muscle development in vivo. FEBS J 2013;280:6097–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Riuzzi F, Sorci G, Sagheddu R, Donato R. HMGB1‐RAGE regulates muscle satellite cell homeostasis through p38‐MAPK‐ and myogenin‐dependent repression of Pax7 transcription. J Cell Sci 2012;125:1440–1454. [DOI] [PubMed] [Google Scholar]
- 121. Riuzzi F, Sorci G, Sagheddu R, Chiappalupi S, Salvadori L, Donato R. RAGE in the pathophysiology of skeletal muscle. J Cachexia Sarcopenia Muscle 2018;9:1213–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Fan X, Krahling S, Smith D, Williamson P, Schlegel RA. Macrophage surface expression of annexins I and II in the phagocytosis of apoptotic lymphocytes. Mol Biol Cell 2004;15:2863–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Leikina E, Defour A, Melikov K, van der Meulen JH, Nagaraju K, Bhuvanendran S. Annexin A1 deficiency does not affect myofiber repair but delays regeneration of injured muscles. Sci Re 2015;5:18246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Tsuchiya M, Hara Y, Okuda M, Itoh K, Nishioka R, Shiomi A, et al. Cell surface flip‐flop of phosphatidylserine is critical for PIEZO1‐mediated myotube formation. Nat Commun 2018;9:2049. [DOI] [PMC free article] [PubMed] [Google Scholar]