

Abstract
Background
Cachexia is frequent, deadly, and untreatable for patients with pancreatic ductal adenocarcinoma (PDAC). The reproductive hormone and cytokine Activin is a mediator of PDAC cachexia, and Activin receptor targeting was clinically tested for cancer cachexia therapy. However, sex‐specific manifestations and mechanisms are poorly understood, constraining development of effective treatments.
Methods
Cachexia phenotypes, muscle gene/protein expression, and effects of the Activin blocker ACVR2B/Fc were assessed in LSL‐Kras G12D/+ , LSL‐Trp53 R172H/+ , and Pdx‐1‐Cre (KPC) mice with autochthonic PDAC. Effects of PDAC and sex hormones were modelled by treating C2C12 myotubes with KPC‐cell conditioned medium (CM) and estradiol. Muscle gene expression by RNAseq and change in muscle from serial CT scans were measured in patients with PDAC.
Results
Despite equivalent tumour latency (median 17 weeks) and mortality (24.5 weeks), male KPC mice showed earlier and more severe cachexia than females. In early PDAC, male gastrocnemius, quadriceps, and tibialis anterior muscles were reduced (−21.7%, −18.9%, and −20.8%, respectively, all P < 0.001), with only gastrocnemius reduced in females (−16%, P < 0.01). Sex differences disappeared in late PDAC. Plasma Activin A was similarly elevated between sexes throughout, while oestrogen and testosterone levels suggested a virilizing effect of PDAC in females. Estradiol partially protected myotubes from KPC‐CM induced atrophy and promoted expression of the potential Activin inhibitor Fstl1. Early‐stage female mice showed greater muscle expression of Activin inhibitors Fst, Fstl1, and Fstl3; this sex difference disappeared by late‐stage PDAC. ACVR2B/Fc initiated in early PDAC preserved muscle and fat only in male KPC mice, with increases of 41.2%, 52.6%, 39.3%, and 348.8%, respectively, in gastrocnemius, quadriceps, tibialis, and fat pad weights vs. vehicle controls, without effect on tumour. No protection was observed in females. At protein and RNA levels, pro‐atrophy pathways were induced more strongly in early‐stage males, with sex differences less evident in late‐stage disease. As with mass, ACVR2B/Fc blunted atrophy‐associated pathways only in males. In patients with resectable PDAC, muscle expression of Activin inhibitors FSTL1, FSLT3, and WFIKKN2/GASP2 were higher in women than men. Overall, among 124 patients on first‐line gemcitabine/nab‐paclitaxel for PDAC, only men displayed muscle loss (P < 0.001); average muscle wasting in men was greater (−6.63 ± 10.70% vs. −1.62 ± 12.00% mean ± SD, P = 0.038) and more rapid (−0.0098 ± 0.0742%/day vs. −0.0466 ± 0.1066%/day, P = 0.017) than in women.
Conclusions
Pancreatic ductal adenocarcinoma cachexia displays sex‐specific phenotypes in mice and humans, with Activin a preferential driver of muscle wasting in males. Sex is a major modulator of cachexia mechanisms. Consideration of sexual dimorphism is essential for discovery and development of effective treatments.
Keywords: Pancreatic cancer, Cachexia, Muscle wasting, Weight loss, Activin, ACVR2B, Sexual dimorphism, Estradiol
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive malignancy with the lowest 5 year survival rate relative to all other solid tumour types that is projected to become the second leading cause of US cancer‐related deaths by 2030. 1 The lethality is partly due to the high penetrance and severity of cachexia. Indeed, mortality rates in pancreatic cancer patients with cachexia are up to 80% per year. 2 Cancer cachexia is well known to be a systemic inflammation‐driven syndrome characterized by super‐induction of pro‐inflammatory cytokines, such as IL‐1β, IL‐6, and TNF‐α leading to metabolic alterations, enhanced catabolism, and reduced anabolism. 3 , 4 As such, patients with cancer cachexia suffer progressive loss of body mass primarily due to muscle and adipose or fat wasting, which severely impairs physical function and general well‐being. 5 Furthermore, cancer patients with cachexia respond to chemotherapy poorly and experience increased treatment toxicity. 6 , 7 Unfortunately, there is no widely approved, effective therapy for this syndrome, excepting anamorelin in Japan. 8
Activins A and B, as well as related family members Myostatin and GDF11, are pluripotent growth and differentiation factors belonging to the TGF‐β superfamily that play critical modulatory roles in inflammation and immunity, including stimulating the production and release of IL‐1β, IL‐6, and TNF‐α. 9 , 10 While data for Myostatin and GDF11 are conflicting or evolving, we and others have demonstrated that Activin is a key cachexia mediator in PDAC. Activin A is overexpressed in PDAC tumours and elevated in blood in humans and orthotopic murine models of PDAC‐cachexia, elevated plasma Activin correlates with PDAC cachexia and mortality, 11 , 12 , 13 , 14 , 15 , 16 , 17 and exogenous Activin induces weight loss and muscle wasting in mice. 14 , 18 Activins signal by forming a ternary complex with type I (ACVR1 and ACVR1B) and type II (ACVR2 and ACVR2B) receptors where type II receptors, through their constitutively active kinase activity, activate type I receptors interacting with the intracellular signalling components of the pathway. 19 , 20 Activin A activity is opposed by Inhibin‐alpha heterodimerization, while Activin A homodimer binding to its receptor is blocked by binding of endogenously expressed inhibitors, including follistatin (FST), follistatin related protein‐1 (FSTL1), follistatin like‐3 (FSTL3), GASP1/WFIKKN1, and GASP2/WFIKKN2. 21 Activin blockade by engineered soluble Activin receptor, ACVR2B/Fc, recombinant Activin A pro‐peptide, or Activin‐receptor blocking antibody induces skeletal muscle hypertrophy and prevents or slows cachexia in mice with cancer or other muscle wasting conditions. 22 , 23 The crucial role of Activin in PDAC cachexia is further reinforced by our prior work, 16 which demonstrates that PDAC induces a systemic Activin response in mice. Further, ACVR2B/Fc treatment reduces tumour growth, prevents weight loss and muscle wasting, and prolongs survival in mice with orthotopic PDAC tumours expressing low amounts of Activin, but does not reduce mortality in tumours expressing high amounts of Activin. Notably, Activin A might also modulate chemo‐resistance or chemotherapy tolerance because increased circulating Activin A associates with poor response to platinum chemotherapy in cancer cachexia patients. 24
Sex has been recognized to impact disease manifestation and therapeutic effects in heart disease, cancer, and other pathological conditions, 25 , 26 , 27 yet few functional data are available for sex effects in cachexia. 28 , 29 , 30 Indeed, sexual dimorphism in basic and translational research is still not being routinely considered, with males being dominantly used in many research fields 29 , 31 including cancer cachexia. Because of this bias, research funding agencies including the US National Institutes of Health are recommending integrating analysis of sex into preclinical research. 32 Carson, Greene, and others have documented sex differences in the ApcMin and LLC models of cachexia, 33 , 34 citing in part differences in interleukin‐6 effects. 35 Here, we aimed to comprehensively characterize cachexia phenotypes, molecular pathways, and sex effects in the genetically engineered mouse model (GEMM) LSL‐Kras G12D/+ , LSL‐Trp53 R172H/+ , and Pdx‐1‐Cre (KPC) mouse of pancreatic cancer, 36 which is characterized by high Activin expression. 16 This model induces tumours via pancreas‐specific expression of mutant Kras and p53 alleles and recapitulates many of the clinical, histopathological, and genomic features of human PDAC, including severe and highly penetrant cachexia as shown here. Furthermore, we evaluated the Activin pathway and sexual dimorphism in patients with pancreatic cancer. Our results add to a growing body of literature demanding a closer look at contributions of biological sex to cancer cachexia.
Materials and methods
Mouse studies
The GEMM KPC mouse model of PDAC is described. 16 , 36 Briefly, the mutant Kras and p53 alleles are activated by Cre‐recombinase expression in the pancreas, leading to autochthonic PDAC. Tissues were collected at two stages: Early—when tumour size was 5–7 mm and body condition score reached 3–6 (where 0 is normal and 9 is consistent with imminent death), and Late—when tumour size was >10 mm and body condition score was ≥8. Body condition was scored according to Indiana University School of Medicine Laboratory Animal Resources criteria as follows: body posture (0—normal, 1—mildly hunched, 2—moderately hunched, and 3—very hunched); activity level (0—normal, 1—slightly reduced, 2—moving slowly, and 3—moving reluctantly or not at all); eye appearance (0—eye closed < 25%, 1—eye closed 25–50%, 2—eye closed 51–75%, and 3—eye closed 76–100%). The soluble Activin receptor ACVR2B/Fc was purified as described previously. 16 Administration of ACVR2B/Fc was initiated when mice met the criteria of early stage and was administered every 5 days (15 mg/kg body weight i.p.) with vehicle control mice receiving phosphate‐buffered saline on the same schedule. Endpoints in KPC mice were compared with no‐tumour genotype controls of age‐ and sex‐matched siblings, co‐housed with KPC mice.
Patients
Studies involving human subjects were approved by the Institutional Review Board of Indiana University. Under IRB protocol 1312105608, rectus abdominus biopsies were taken at the incision site at start of surgery for pancreatic cancer, then flash frozen in liquid nitrogen for storage and processing. Under IRB protocol 1509161309, we analysed all patients with sufficient CT scans who were treated with first‐line gemcitabine/nab‐paclitaxel for advanced/metastatic PDAC at Indiana University from October 2009 to October 2017, using Sliceomatic analysis averaging two L3 slices from CT scans taken nearest the start of chemotherapy and at last follow‐up, similar to our published work. 37
Estradiol effects on Activin inhibitor expression and Myotube size
KPC32908 cell conditioned medium (CM) preparation, C2C12 cell culture, RT‐qPCR, and myotube staining were done as described 16 with some modifications. The CM was collected from the GEMM KPC‐derived cancer cell line KPC32908 in DMEM medium containing 10% foetal bovine serum and 1% penicillin/streptomycin. The KPC32908 CM was diluted in differentiation medium (DM) at a 3:7 ratio, and Estradiol (E2) was added to the diluted KPC32908 CM at final concentrations of 100 or 500 nM. The mixture of CM and E2 was added to 4‐day‐old differentiated C2C12 myotube culture in 12‐well plates, and E2 only was replenished 24 and 48 h later. Total RNA was isolated at 72 h, and endogenous Activin inhibitor mRNA expression was quantified by real‐time PCR. Fold‐change of mRNA was normalized to Inha in growth medium control myotubes. C2C12 myotubes were fixed in 1:1 acetone/methanol at −20°C and blocked in 6% foetal bovine serum in phosphate‐buffered saline, followed by incubation with myosin heavy chain primary antibody (MF 20, DSHB) and staining with Alexa Fluor 488‐conjugated secondary antibody (11029, Invitrogen). Shown are representative images acquired at ×4 using a Lionheart LX imager. Myotube diameters were measured on myotubes in the same localized tiles from the tiled montages across all the four treatment conditions, growth medium, CM, CM + E2 100 nM, and CM + E2 500 nM, to ensure no bias; a total of 425–496 myotubes for each condition were measured.
Enzyme‐linked immunosorbent assay
Estradiol and testosterone concentration in the plasma of KPC and control mice were measured using 17 beta estradiol enzyme‐linked immunosorbent assay (ELISA) Kit (ab108667, Abcam) and Testosterone ELISA Kit (TE187S‐100, Calbiotech), per the manufacturer's protocol. Ratios were calculated for absolute concentration of testosterone divided by estradiol in each individual sample.
Western blotting analysis of muscle lysates
Lysates made from quadriceps skeletal muscles collected from GEMM KPC mice and the sex‐matched and age‐matched genotype control mice at early and late tumour stages were subjected to western blotting. The primary antibodies used were ubiquitin (3933) and phosphor‐Stat3 (9145) from Cell Signalling Technology, Danvers, MA, USA, and GAPDH (ab9484) from Abcam, Cambridge, MA, USA. The secondary antibodies used were IRDye 800CW goat anti‐rabbit IgG (926‐32211) and IRDye 680LT goat anti‐mouse IgG2b‐specific (926‐68052) from LICOR Biosciences, Lincoln, NE, USA. Signal was detected using a LI‐COR Odyssey system.
RNA sequencing and bioinformatics analysis
Mouse quadriceps skeletal muscles were snap frozen in liquid nitrogen and stored at −80°C until use. Muscle tissues were homogenized using the 0.9–2.0 stainless steel beads (SSB14B‐RNA) and the Bullet Blender (Next Advance) and total RNAs were extracted using the RNeasy Mini Kit (74104, Qiagen). RNA sequencing (RNAseq) was performed using the Illumina TruSeq Stranded mRNA standard methods. Total RNA samples were first evaluated for their quantity, and quality using Agilent Bioanalyzer 2100. Five hundred nanograms of total RNA was used for library preparation. Briefly, cDNA library preparation included mRNA enrichment/purification, RNA fragmentation, cDNA synthesis, ligation of index adaptors, and amplification, following the TruSeq Stranded mRNA Sample Preparation Guide, RS‐122‐9004DOC, Part# 15031047 Rev. E (Illumina, Inc.). Each resulting indexed library with average insert size of 150‐200b was quantified and its quality accessed by Qubit and Agilent Bioanalyzer, and multiple libraries pooled in equal molarity. Five microliters of 2 nM pooled libraries per lane were then denatured, neutralized and applied to the cBot for flow cell deposition and cluster amplification, before loading to HiSeq 4000 for 75b paired‐end read sequencing (Illumina, Inc.). Approximately 30 M reads per library were generated. A Phred quality score (Q score) was used to measure the quality of sequencing. More than 90% of the sequencing reads reached Q30 (99.9% base call accuracy).
The reads were mapped to the mouse genome mm10 using STAR (v2.5). RNA‐seq aligner with the following parameter: ‘‐‐outSAMmapqUnique 60’. Uniquely mapped sequencing reads were assigned to GENCODE M15 genes using featureCounts (v1.6.2) with the following parameters: ‘‐s 2 –p –Q 10’. The data were filtered using count per million (CPM) < 0.5 in more than four samples, normalized using TMM (trimmed mean of M values) method and subjected to differential expression analysis using edgeR (v3.20.8). Paired design was used for comparisons by sex, tumour vs. control, early or late stage, and ACVR2B/Fc or vehicle treatment. The data have been deposited to NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE157251 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE157251). Differentially expressed genes (DEGs) defined based on P < 0.05 and linear fold change |FC| > 2 are presented in Supporting Information, Data S1 and S2 . DEGs were used to enrich canonical pathways by the Ingenuity Pathway Analysis programme (http://www.ingenuity.com). The online Venn diagram programme (http://bioinformatics.psb.ugent.be/webtools/Venn/) was used to draw the diagrams revealing the unique and overlapping DEGs between sexes. The comparison analysis tool in Ingenuity Pathway Analysis was used to identify uniquely or commonly altered pathways and regulators between males and females.
For the study of patients, 31 males and 24 females with pancreatic cancer (55 in total), RNA was isolated from rectus abdominis muscle using Qiagen miRNeasy mini kit (Catalogue #: 217004) for total RNA sequencing. cDNA libraries were prepared using Clontech SMARTer RNA Pico Kit v2. Approximately 100 million reads per library was generated. The sequencing data was analysed using Partek® Flow® software Version 9. The raw sequencing files were aligned to hg38 using STAR aligner. BAM files were mapped to GENCODE v29 to generate the raw reads. The raw reads were normalized using CPM. The CPM values generated for each transcript of genes involved in the Activin pathway were compared between males and females.
Statistical analysis
Generally, two group comparisons of parametric data were by unpaired two‐tailed Student's t‐test. Multiple groups were compared by analysis of variance followed by Tukey's post hoc test. Tests of sex effects and interactions were by two‐way analysis of variance. For non‐parametric data (testosterone levels), comparisons were by Kruskal–Wallis test. In the patient study, tests of within‐sex change from baseline in muscle area or radiodensity was by one‐sample t‐test, while comparisons between sexes were tested by unpaired t‐test. For all analyses, significance was set at P < 0.05.
Results
GEMM KPC mice of PDAC develop cachexia in a sex‐specific manner
The GEMM KPC mouse model of PDAC 16 , 36 was used to assess the impact of sex on cachexia symptom onset and progression. As shown in Figure 1 A , male KPC mice ceased gaining lean mass by 20 weeks and fat mass by 11 weeks compared with sex‐matched and age‐matched no‐tumour genotype controls. In contrast, female KPC mice did not lose lean mass; in fact, lean mass was slightly increased over time, which was probably due to tumour mass and splenomegaly (Figure 1 ). In females, fat mass was lost later (at ~20 weeks) than in males. However, there was no sex difference in PDAC tumour latency and survival; male and female KPC mice had the same median latency to tumour palpation of 17 weeks and the same median survival of 24.5 weeks (Figure 1 B ).
Figure 1.
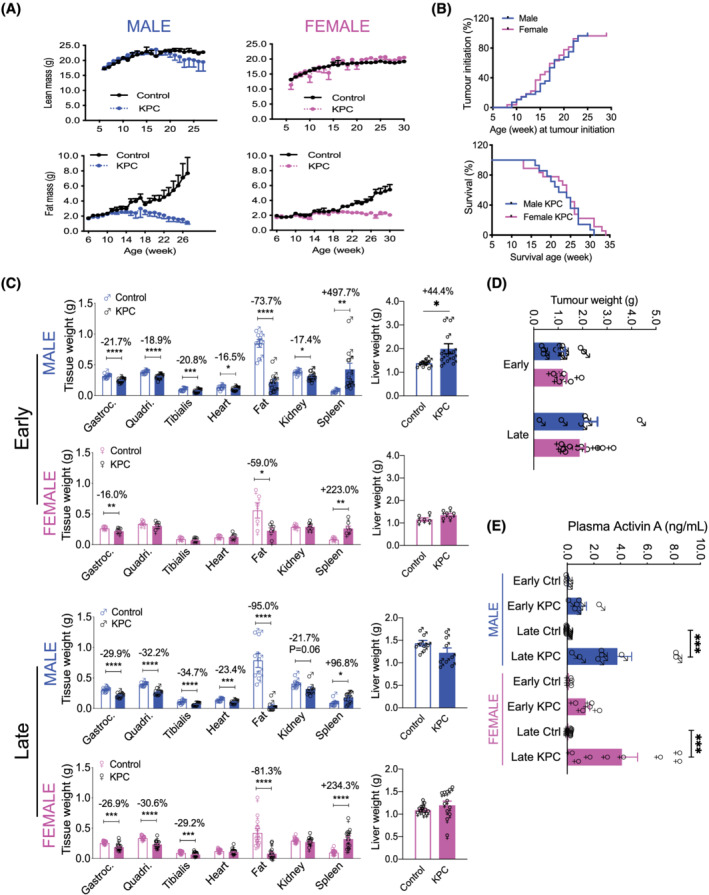
GEMM KPC mice with PDAC develop cachexia in a sex‐specific manner. (A) Male and female KPC and their age‐matched, no‐tumour genotype control mice were monitored for body composition assessed by EchoMRI. Sample size: n = 12–34 per group. (B) Tumour initiation was determined by palpation. For the survival analysis, mice were euthanized when tumour size was >10 mm and body condition score was ≥8. NS (C) Skeletal muscle, heart, adipose and kidney wasting was detectable and greater in males in early‐stage PDAC than in females. Organ wasting was similar in severity between sexes in late‐stage PDAC except spleen; also, neither early nor late PDAC induces heart or kidney wasting in females. ‘Early’ indicates when tumour size was 5–7 mm and/or body condition score reached 3–6 (where 0 is normal and 9 is consistent with imminent death), while ‘late’ indicates when tumour size was >10 mm and body condition score was ≥8. Sample size: n = 5–12 as shown. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. (D) Tumour weights were taken at euthanasia. NS (E) Plasma Activin A concentrations measured by ELISA method using the R&D systems Quantikine human/mouse/rat Activin A kit (Cat# DAC00B). ***P < 0.001 by ANOVA with post‐test.
Organ weights at euthanasia showed sex differences. Most of the organs in the male KPC mice were significantly wasted at the early stage of tumour growth (Figure 1 C , top panel). For example, gastrocnemius, quadriceps, and tibialis anterior muscle were reduced by 21.7% (P < 0.0001), 18.9% (P < 0.0001), and 20.8% (P < 0.001), respectively, while cardiac muscle, epididymal fat, and kidney were reduced by 16.5% (P < 0.05), 73.7% (P < 0.0001), and 17.4% (P < 0.05), respectively. Pronounced hepatosplenomegaly was evident with spleen enlarged by 497.7% (P < 0.01) and liver by 44.4% (P < 0.05). In contrast, early female KPC mice had much less organ wasting with only gastrocnemius reduced by 16.0% (P < 0.01) and fat by 59.0% (P < 0.05), less severe splenomegaly (enlarged by 223.0%; P < 0.01), and no hepatomegaly (second panel). At late stage, both males and females experienced skeletal muscle and fat wasting with similar severity, suggesting a later, rapid wasting progression in females except for heart and kidney which were spared in females throughout tumour progression. However, splenomegaly in males declined from early stage 497.7% (P < 0.01) to late stage 96.8% (P < 0.05) and hepatomegaly from 44.4% (P < 0.05) to normal (top panel vs. third panel), while in females, splenomegaly did not attenuate and hepatomegaly was absent throughout (second vs. bottom). Terminal tumours did not show significant differences in weights between sexes (Figure 1 D ); free water (ascites), determined by EchoMRI, was significantly increased in both male and female KPC mice at late stage (P < 0.01 & P < 0.001) (Figure S1 ).
Because Activin is known to mediate PDAC cancer cachexia, we measured plasma Activin A concentration. Male and female control mice had similar levels of Activin A, while Activin A in male and female KPC mice was elevated by 14.1‐fold and 11.0‐fold, respectively, in early‐stage and 54.4‐fold and 47.7‐fold, respectively, in late‐stage cachexia (Figure 1 E ). However, there were no sex differences, suggesting that the sex differences in cachexia onset and severity was not due to differential presence of circulating Activin protein.
Factors from KPC cells inhibit expression of myocellular Activin inhibitors, which is partly blocked by oestrogen
We then explored mechanisms behind why similarly elevated plasma Activin between sexes failed to exert a cachectic effect in females with early‐stage tumours. We hypothesized that sex differences in PDAC cachexia could be due in part to hormonal regulation of Activin inhibitors and interrogated potential protective effects of estradiol. KPC CM treatment reduced C2C12 myotube diameter (Figure 2CA–2 ), at least partly due to catabolism as demonstrated by induction of the muscle specific E3 ligase gene, Atrogin1 (not shown). KPC CM also down‐regulated the Activin‐binding protein genes Fst and Fstl1, with no effect on Inha, Fstl3, or Wfikkn2 (Figure 2 D ). Treatment with estradiol (E2) blunted loss of myotube diameter (Figure 2 B and 2 C ) and increased only expression of Fstl1 among the Activin inhibitors (Figure 2 D ), although without restoring either to normal. These results suggest a hormonally responsive mechanism of Fstl1 gene expression and a potentially protective effect of oestrogen in PDAC cachexia.
Figure 2.
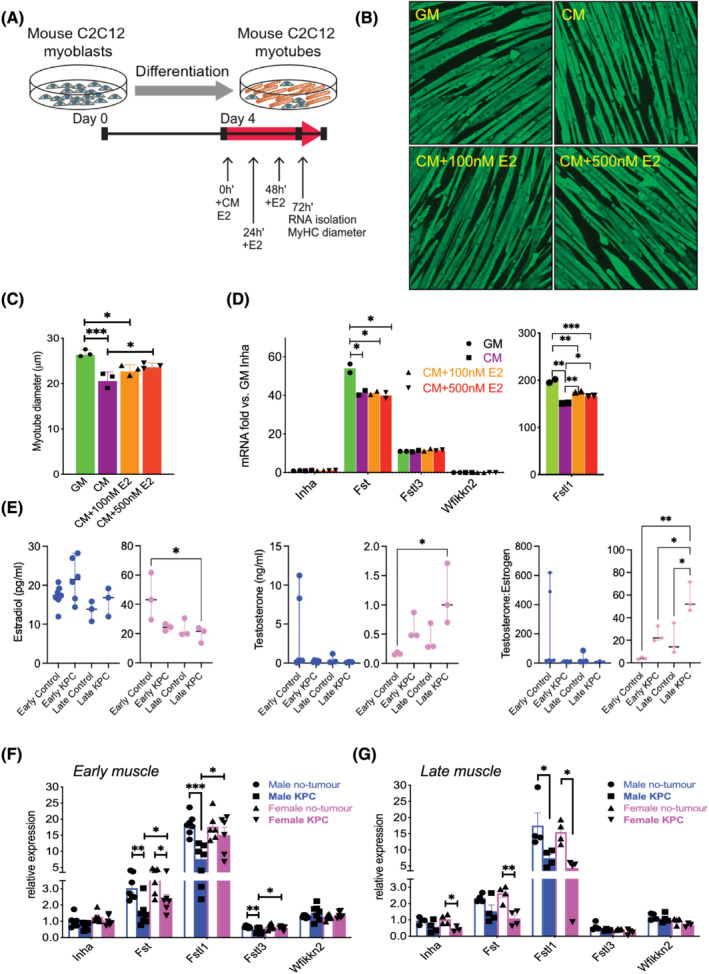
Regulation of Activin pathway members and wasting by PDAC‐produced factors and estradiol (E2). (A) Growth medium (GM) or KPC32908 pancreatic cancer cell‐derived conditioned medium (CM) was added at 30% concentration in differentiation medium (DM), E2 at 100 or 500 nM final concentration to 4‐day‐old differentiated C2C12 myotubes cultured in 12‐well plates, at time 0 h. E2 was replenished at 24 and 48 h. RNA was isolated and myotube diameter was measured at 72 h. (B) C2C12 myotubes were fixed in 1:1 acetone/methanol and blocked in 6% foetal bovine serum phosphate‐buffered saline, followed by incubation with the myosin heavy chain (MyHC) primary antibody and staining with the Alexa Fluor 488‐conjugated secondary antibody. Shown are images acquired at ×4 using the Lionheart LX imager. (C) KPC CM induced atrophy, which was partly blocked by E2. Average myotube diameters were measured from a total of 425–496 myotubes for each group. *P < 0.05, ***P < 0.001. (D) Endogenous Activin inhibitor mRNA expression quantified in C2C12 cultures by real‐time PCR showed Fstl1 was the most abundant among the inhibitors, and was repressed by KPC CM and upregulated by E2. (E) Estradiol (left) and testosterone (middle) concentrations in the plasma of mice were measured by the ELISA method. Testosterone over estradiol ratio was calculated (right). (F) Endogenous Activin inhibitor mRNA in KPC GEMM muscle by real‐time PCR showed sex‐specific and stage‐specific expression in muscle from male and female mice with early‐stage and (G) late‐stage PDAC.
Sex specific changes in sex hormones and Activin binding protein gene expression in muscle
In mice, plasma estradiol levels were similar among across male groups and stages (Figure 2 E ). As expected, estradiol was significantly higher in female control mice than in male control mice. Oestrogen fell significantly from early control levels in late KPC females, although this was not different from late control; testosterone was also significantly elevated only between early controls and late KPC females. Of all female groups, the testosterone : estradiol ratio was elevated in late‐stage female KPC mice vs. all female groups, including same stage controls. These results suggest a virilizing effect of PDAC exposure on female mice.
Consistent with sex‐specific regulation of the Activin pathway, muscle Fst, Fstl, and Fstl3 were all down‐regulated in early male KPC mice (Figure 2 F ). In contrast, only Fst was down‐regulated in early female KPC mouse muscle vs. early controls (Figure 2 F ). Importantly, these three potential Activin inhibitors were also all significantly lower in male mice than in females in early stage PDAC. This relative advantage in females was not sustained. Inha, Fst, and Fstl1 in female KPC mice were all down‐regulated in late cachexia (Figure 2 G ) when sex differences disappeared.
Activin blockade by ACVR2B/Fc preserved muscle and fat in male KPC mice but not in females
Given this evidence for Activin imbalance between the sexes, ACVR2B/Fc, an exogenous Activin blocker, was used to assess the potential sex‐specificity of its therapeutic potential. The treatment schema and the doses administered are shown in Figure 3 A , left and right, respectively. There was no age difference between sexes at the onset of treatment, averaging 19.0 weeks (range 10–26 weeks) in males and 19.2 weeks (range 11–32 weeks) in females (unpaired t‐test, P = 0.8462). Male and female KPC mice received an average of 4.0 and 4.2 doses of ACVR2B/Fc, respectively. Although there was greater variability in females than males, there was no statistically significant difference (unpaired t‐test, P = 0.8100).
Figure 3.
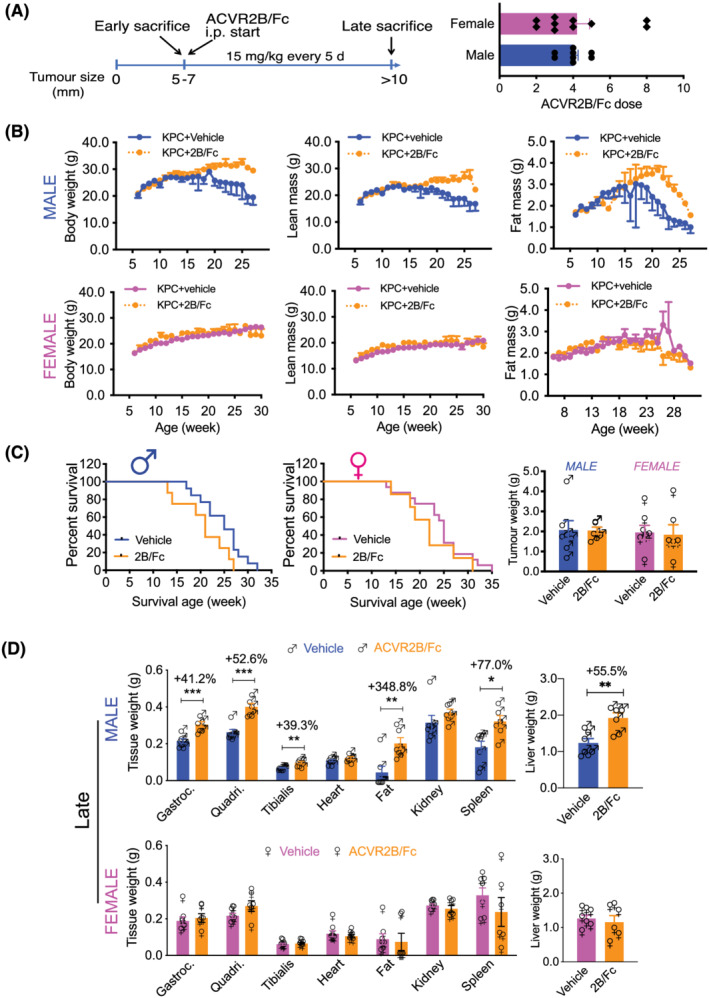
Activin blockade by ACVR2B/Fc preserved muscle and fat in male KPC mice but not in females. (A) Treatment schema and number of doses. Administration of ACVR2B/Fc (2B/Fc) was initiated when mice met early criteria and was administered every 5 days (15 mg/kg body weight i.p.) with vehicle control mice receiving PBS on the same schedule. (B) ACVR2B/Fc prevented wasting with tumour progress in male KPC mice but not the female counterpart. (C) ACVR2B/Fc did not significantly affect survival and tumour weight in either sex. (D) ACVR2B/Fc preserved skeletal muscle and fat mass in males. Sample size: n = 6–11; *P < 0.05, **P < 0.01, ***P < 0.001.
Intraperitoneal administration of ACVR2B/Fc reduced body weight loss and lean and fat mass loss over time in males (Figure 3 B , top), but had no effect in females (bottom). ACVR2B/Fc did not significantly affect survival or tumour weight in either sex (Figure 3 C ). However, ACVR2B/Fc‐treated male KPC mice showed an increase of 41.2% in gastrocnemius (P < 0.001), 52.6% in quadriceps (P < 0.001), 39.3% in tibialis (P < 0.01), and 348.8% in fat (P < 0.01) at late stage compared with the vehicle controls. Heart and kidney were not protected, and spleen and liver were further enlarged by ACVR2B/Fc in males (Figure 3 D , top). In contrast, ACVR2B/Fc did not preserve skeletal muscle or fat mass in females or significantly affect kidney, spleen, or liver weight (bottom). Thus, the protection of skeletal muscle conferred by ACVR2B/Fc was confined to males.
Molecular pathways are unfavourably altered in the muscle of early male KPC mice but not females
Muscle wasting in cancer is mainly mediated by enhanced protein catabolism through the ubiquitin‐proteasome and autophagy‐lysosome pathways, although it is less clear whether these processes occur equally in males and females of PDAC. Here, we show that in early‐stage cachexia, total ubiquitinated proteins were higher in males than in females (P < 0.01) (Figure 4 A ). Furthermore, the pro‐atrophy transcription factor STAT3 38 was phosphorylated (activated) (P < 0.01) in male KPC mice while female KPC mice only trended STAT3 activation (Figure 4 B ).
Figure 4.
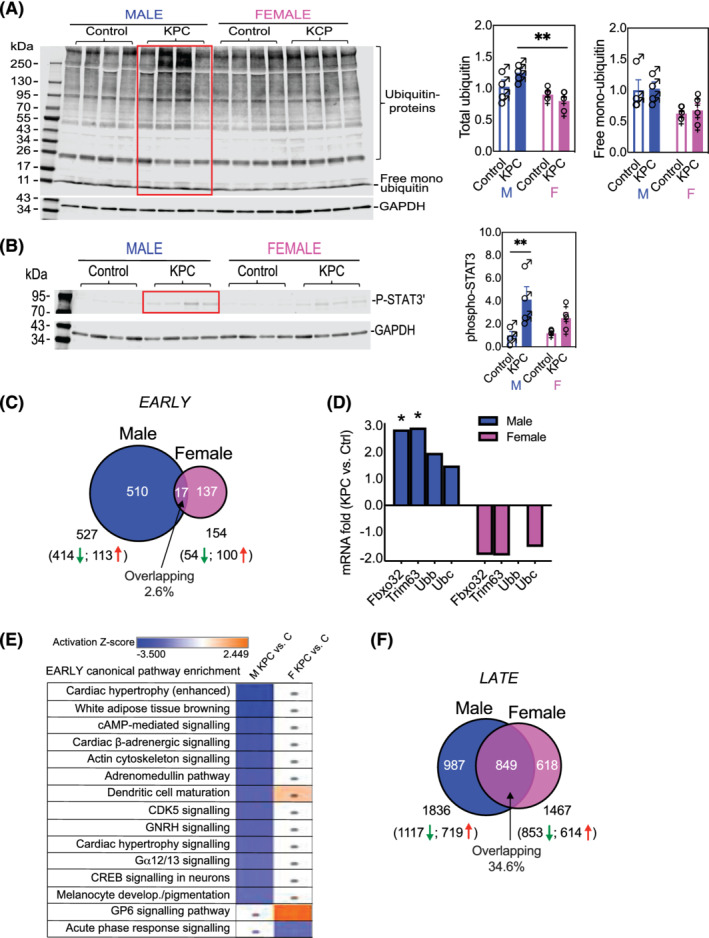
Molecular pathways were unfavourably altered in the muscle of early KPC male mice but fewer changes occurred in females. PDAC increased (A) protein ubiquitination and (B) phosphorylation of the cytokine‐activated transcription factor STAT3 with greater effect on males than females. **P < 0.01. (C) Quadriceps muscles were subjected to total RNA sequencing, and the data are expressed as fold change of KPC vs. control or differentially expressed genes (DEG). Cut‐off for DEG identification: linear fold change |FC| > 2; P < 0.05. There are more dysregulated genes in male KPC mice than female KPC mice at early stage with few overlapping DEGs. (D) PDAC increased the muscle‐specific E3 ligase genes Fbxo32/Atrogin1 and Trim63/MuRF1 in males but not in females. *P < 0.05. (E) Comparison of enriched pathways between male KPC vs. male no‐tumour control or female KPC vs. female no‐tumour control. Each row represents a pathway with blue rectangle indicating inhibition and orange indicating activation determined by |Z| ≥ 2 and P < 0.05. Dots, |Z| < 2. The pathways were sorted based on the male KPC vs. control; the most inhibited pathway is on the top and most activated one on the bottom in this group. All dysregulated pathways were inhibited in male KPC mice while fewer changes were seen in females. (F) RNAseq showed greater overlap between male and female DEGs in late‐stage PDAC cachexia.
To understand the molecular basis more comprehensively for the phenotypic sex differences, we compared the global transcriptome in muscles from KPC mice and their age‐matched and sex‐matched littermate genotype controls by RNAseq. DEGs were defined as those with linear fold change |FC| > 2 and P < 0.05 between KPC mice and the controls. A total of 527 DEGs were identified in early male KPC mice with 414 down‐regulated and 113 up‐regulated; 154 DEGs in early female KPC mice with 54 down‐regulated and 100 up‐regulated. The overlapping DEGs between sexes account for only 2.6% (Figure 4 C ) with the majority either unique to male or female KPC mice. Overall, early PDAC induced dysregulation of 3.4‐fold more genes in males than in females, likely laying the foundation for the earlier and more severe cachexia manifestation. The muscle‐specific E3 ligase genes Fbxo32/Atrogin1 and Trim63/MuRF1 as well as ubiquitin genes Ubb and Ubc in male early KPC mouse muscle were upregulated by 1.5‐fold to 2.9‐fold; in contrast, all but Ubb in female counterparts trended downward by 1.3‐fold to 1.9‐fold (Figure 4 D ). Z‐score of overlap was used to identify the directionality (activation or inhibition) of implicated molecular pathways; |Z| ≥ 2 and P < 0.05 were defined as significant. Many canonical pathways were altered in male KPC mice vs. controls—all inhibited, while fewer were enriched in females (Figure 4 E ).
ACVR2B/Fc prevents the PDAC‐induced dysregulation of many canonical pathways in males but makes fewer changes in females
In late‐stage cachexia, molecular changes were more similar in magnitude, with 1836 DEGs (1117 down‐regulated and 719 up‐regulated) in males and 1467 DEGs (853 down‐regulated and 614 up‐regulated) in females, with 34.6% overlapping between sexes (Figure 4 F ). Males and females shared many dysregulated canonical pathways in late stage (Figure 5 , columns 1 & 2). ACVR2B/Fc treatment reversed many pathways in male KPC mice, such as the top five activated ones and those related to IL‐6, inflammation, cancer progression, gluconeogenesis, and glycolysis (Figure 5 , column 3 vs. 1). Far fewer pathways in females were reversed by ACVR2B/Fc (Figure 5 , column 4 vs. 2). Thus, females did not respond to ACVR2B/Fc therapy either at the organ level or the molecular level.
Figure 5.
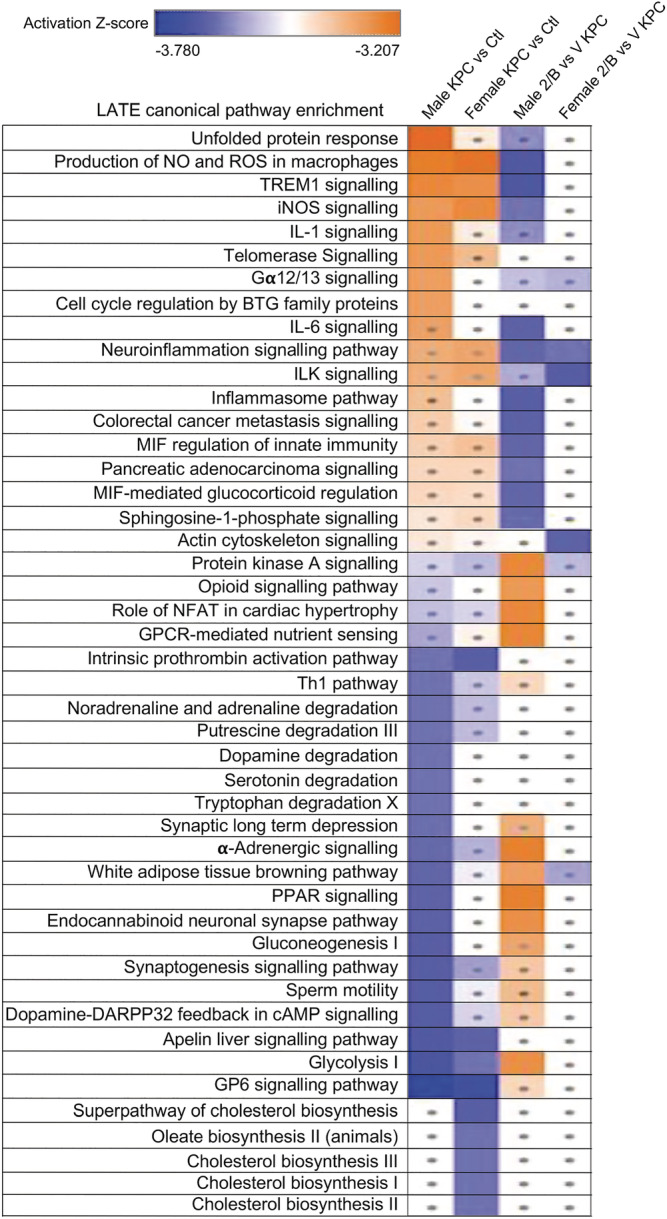
ACVR2B/Fc prevented the PDAC‐induced dysregulation of many atrophy‐associated pathways in males but caused fewer changes in females. Comparison of enriched pathways: Column 1, male KPC vs. male controls; 2, female KPC vs. female controls; 3, male ACVR2B/Fc (2B) treated KPC vs. vehicle treated KPC (V); 4, female ACVR2B/fc (2B) treated KPC vs. vehicle treated KPC (V). Cut‐off for pathway enrichment analysis: |Z| ≥ 2 and P < 0.05. Dots, |Z| < 2. The pathways were sorted based on the male KPC vs. control; the most activated pathway is on the top and the most inhibited pathway is on the bottom in this group.
Sex differences in expression of Activin inhibitors in muscle of patients with PDAC
To determine whether similar sex differences in the muscle molecular response to PDAC might be evident in patients, we quantified Activin inhibitor gene expression in muscle biopsies from patients undergoing surgery with curative intent for resectable PDAC. The endogenous inhibitors FSTL1, FSTL3, and WFIKKN2/GASP2 of the Activin family ligands (Activin, Myostatin, and GDF11) were more abundant in women with PDAC than in men with PDAC (Figure 6 A ). No differences were observed for expression of FST or WFIKKN1 . Notably, as in mouse muscle and C2C12 myotubes, FSTL1 was the most abundant among the mRNAs, suggesting that FSTL1 could plays a key role in resisting cachexia effects of Activin in women with PDAC.
Figure 6.
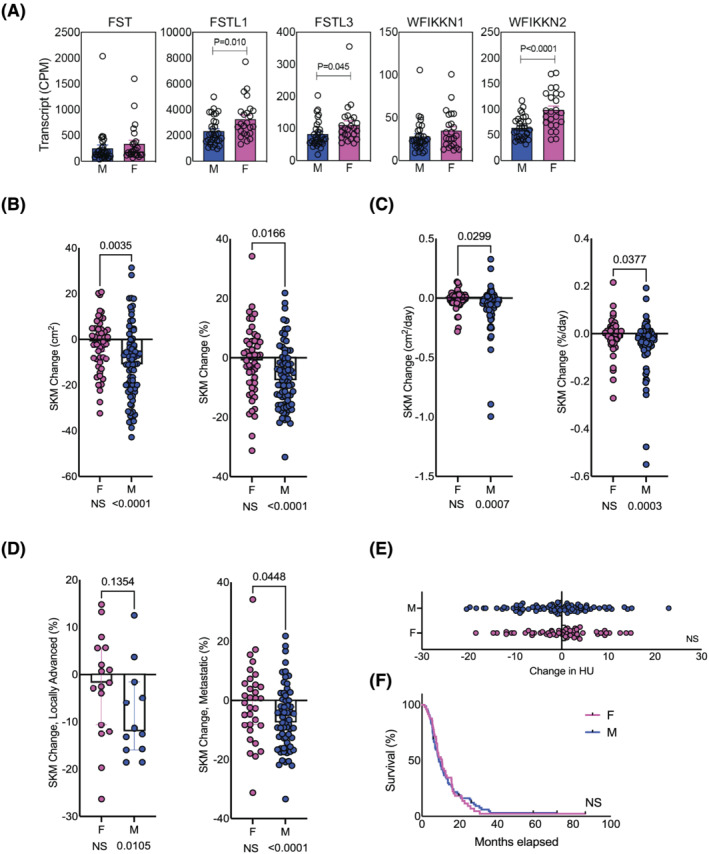
Sexual dimorphism in Activin inhibitor expression (A) and cachexia phenotype (B–F) in two cohorts of patients with PDAC. (A) Cohort 1 shows sex differences in the endogenous inhibitors of the Activin family ligand in the muscles of PDAC patients. Muscle specimens obtained from patients undergoing surgery for pancreatic cancer at Indiana University [n = 31 males (M); n = 24 females (F)] were subjected to RNAseq. FSTL1, FSTL3, and WFIKKN2/GASP2 were more abundant in females. (B–F) Measurement of muscle area in Cohort 2, 124 patients with usable CT scans treated with first‐line gemcitabine/nab‐paclitaxel at Indiana University, shows (B) greater loss of skeletal muscle area and percentage area and (C) greater rate of skeletal muscle loss in men vs. women in the entire cohort. (D) The subset of patients with locally advanced disease at treatment onset showed no difference; however, those with metastatic disease demonstrated sexually dimorphic muscle loss, which was greater in males. (E) Change in skeletal muscle radiodensity was not different between the sexes, nor was survival (F).
Sex differences in muscle wasting in patients on treatment for PDAC
To determine whether cachexia phenotypes might be different between the sexes, we measured change in skeletal muscle from serial CT scans of 124 patients treated with first‐line gemcitabine/nab‐paclitaxel for locally advanced or metastatic PDAC at our institution (Table 1 ). Our analysis demonstrates men lost greater quantity and percentage of skeletal muscle vs. women (Figure 6 B ) and at a higher rate (as defined as change from first to last CT over days of treatment) (Figure 6 C ). This was true when defining change as skeletal muscle area or percentage skeletal muscle area (Figure 6 B and 6 C ), or skeletal muscle index (muscle normalized to height) or percentage skeletal muscle index (not shown). This sex difference was observed in the complete cohort as well as within the subset of patients with metastatic PDAC at diagnosis, although not in the subset of those with locally advanced PDAC at diagnosis (Figure 6 D ), potentially due to small sample size. While some women lost muscle, others gained (mean −1.62%) and average muscle change of −6.63% ± 10.70% was significantly different from 0 only for men (P < 0.001, one‐sample t‐test) (Table 1 ). Change in average muscle radiodensity by Hounsfield units (Figure 6 E ) and survival from diagnosis (Figure 6 F ) were not different between the sexes. Thus, as in mice, patients with PDAC exhibit sexually dimorphic cachexia clinical phenotypes, with men losing more muscle than women.
Table 1.
Characteristics of Cohort 2, patients on first‐line gemcitabine/nab‐paclitaxel
| Female (n = 49) | Male (n = 75) | F vs. M (P) a | |
|---|---|---|---|
| Age, years (SD) | 62.6 (11.98) | 62.4 (10.72) | 0.9085 |
| BMI, kg/m2 (SD) | 24.8 (5.03) | 26.9 (5.14) | 0.0238 |
| Disease response (%) | NS | ||
| Stable | 4 (8.2) | 8 (10.5) | |
| Partial | 27 (55.1) | 40 (52.6) | |
| Progression | 17 (34.5) | 27 (36.8) | |
| Disease extent (% metastatic) | 32 (65.3) | 63 (84.0) | 0.0162 |
| Days between first and last CT scan (range) | 318 (39–1071) | 313 (29–1010) | 0.70 |
| Mean change in muscle area, cm2 (SEM) | −2.45 (11.83) | −10.56*** (16.50) | 0.0035 |
| Mean change in muscle area, % (SEM) | −1.62 (12.00) | −6.63*** (10.70) | 0.0166 |
| Mean rate of change in muscle area, cm2/day (SEM) | −0.014 (0.0758) | −0.075*** (0.1830) | 0.0299 |
| Mean rate of change in muscle area, %/day (SEM) | −0.0098 (0.0742) | −0.0466*** (0.1066) | 0.0377 |
Unpaired t‐test.
P < 0.001, one sample t‐test.
Discussion
Finding effective treatments for cancer cachexia requires understanding mechanisms. Here, we provide evidence for sex‐specific cachexia phenotypes and molecular drivers of muscle wasting, with specific implications for anti‐Activin therapies. Using the KPC genetic mouse model of PDAC, we evaluated peripheral effects of tumour, the molecular alterations driving phenotypes in muscle, and therapeutic effects of blocking the Activin signalling pathway using ACVR2B/Fc. We found that sex significantly impacts all these aspects. Specifically, male PDAC mice experience earlier cachexia; ACVR2B/Fc treatment alleviates cachexia in males but not in females; early alterations in metabolic pathways support the early cachexia manifestation in males; and upregulation of inhibitors of the Activin family ligand partly accounts for the late cachexia‐onset and lower response to ACVR2B/Fc treatment in females. We interrogate these pathways using available data in patients and further show that women express higher levels of Activin inhibitors in muscle than men and that men under treatment for PDAC experience greater and more rapid muscle loss than women. Thus, we conclude that (1) cachexia is sexually dimorphic and should be treated as such in pre‐clinical and clinical studies; (2) anti‐cachexia interventions likely require sex‐specific approaches; (3) early intervention is important; (4) targeting the Activin pathway is a rational therapeutic strategy in male PDAC‐induced cachexia; and (5) enhancing the antagonizing mechanism, potentially through hormone modulation, might be a promising anti‐cachexia approach in both sexes.
Based on our findings and the state of knowledge, we provide a summary and model of the mechanisms mediating sex differences in cancer cachexia manifestations and ACVR2B/Fc therapeutic effects (Figure 7 ). PDAC tumour in both males and females releases Activin A into circulation. In males, PDAC tumours also reduces muscle inhibitor expression, de‐repressing Activin action; the excess Activin A binds to its ACVR2B receptor on the membrane of muscle fibre, activating signalling and early muscle wasting, with wasting later becoming more severe due to tumour growth and increased circulating Activin A. ACVR2B/Fc treatment would block this tumour‐activated Activin in early‐stage disease in males. In contrast, by this model females do not experience early muscle wasting because of the higher level of several endogenous Activin inhibitors. With tumour progression, females experience accelerated muscle wasting ultimately to reach the same severity and mortality as males, potentially due to reduced expression of multiple endogenous inhibitors in the setting of tumour‐induced Activin. This model suggests there is a threshold of tolerance for cachexia factors in females, which once crossed, results in rapid muscle loss and survival endpoints similar to men.
Figure 7.
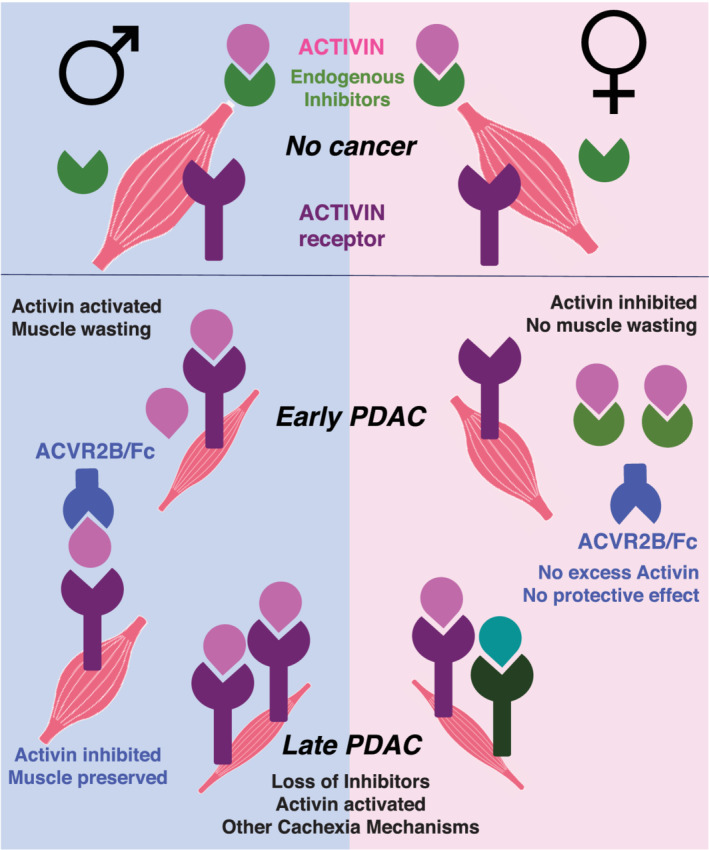
Model for the sex‐specificity of Activin in progression of PDAC cachexia. Based on our findings and the state of knowledge, we provide a summary and model of the mechanisms mediating sex differences in cancer cachexia manifestations and ACVR2B/Fc therapeutic effects (Figure 7 ). In the absence of cancer, Activin signalling on muscle is restrained by FST family, the endogenous Activin family inhibitors. PDAC tumour in both males and females releases Activin A into circulation. In males, PDAC tumours also reduces muscle FST family expression, de‐repressing Activin action; the excess Activin A binds to its ACVR2B receptor on the membrane of muscle fibre, activating signalling and early muscle wasting, with wasting later becoming more severe due to tumour growth and increased circulating Activin A. ACVR2B/Fc treatment would block this tumour‐activated Activin in early stage disease in males. In contrast, females do not experience early muscle wasting because of the higher level of FSTs. With tumour progression, females experience rapid muscle wasting ultimately to reach the same severity as males, potentially due to reduced expression of multiple endogenous inhibitors in the setting of tumour‐induced Activin.
The strengths of this study include the use of the genetic model of PDAC and the use of both an early timepoint when mice were relatively fit and tumour burden was smaller as well as a later timepoint when mice showed morbidity and greater tumour size, thus revealing differences that would not have been evident at an endpoint analysis. Further, phenotypes were assessed against sex‐matched, age‐matched, and in‐cage genotype controls. Molecular analysis confirms the earlier onset of muscle wasting pathways in males, despite similar tumour burden in females. The study also compares the murine data with relevant human data and biospecimens. The patient data, collected on reasonably large sample sizes of people with the same histological diagnosis and treatment regimens, support a similar Activin‐pathway and phenotypic sexual dimorphism in humans as in mice with PDAC.
The limitations of the study include the modest sample sizes in the mouse studies, which constrains our power to detect differences in endpoints such as hormone levels. As well, the focus on Activin signalling and skeletal muscle as endpoints for cachexia is overly simplistic, given the known involvement of other molecular drivers and non‐muscle organs, including liver, adipose, heart, and tumour. Moreover, we functionally queried only one potential sexually dimorphic feature, estradiol, and showed a somewhat modest effect, on C2C12 cells, which are female. A more detailed analysis would require modulating both oestrogen and testosterone in male and female myotubes. Murine quadriceps and human rectus abdominus muscles are different in fibre type composition, and thus the comparison between species is indirect. Moreover, due to our institutional referral pattern and clinical practices, we were unable to query body composition changes, hormone levels, other markers of cachexia, and muscle gene expression in a single cohort of patients. The surgical patients we profiled for gene expression generally receive chemotherapy at distant sites and are thus lost to our follow‐up, while those on chemotherapy were seldom captured in our surgical biorepository due to late presentation and/or timing. Thus, correlation of gene expression to skeletal muscle loss was not possible in the surgical cohort due to lack of CT scans prior to diagnosis of pancreatic cancer; correlation of muscle loss with muscle gene expression was not possible in the retrospective analysis of patients with advanced/metastatic disease on chemotherapy due to lack of muscle biopsies. Nevertheless, these two human studies support sexual dimorphism in the muscle Activin pathway and sex differences in the clinical phenotype of pancreatic cancer cachexia in patients.
Here we document differences in phenotype and offer a plausible molecular pathway leading to sexual dimorphism in PDAC cachexia—Activin released from the tumour and in peripheral tissues by the host response 16 contributes to activation of skeletal muscle wasting, which is initially countered by greater expression of Activin inhibitors in skeletal muscle in females. As in our mice, higher plasma Activin A levels are reported for PDAC patients at later stage and with >5% weight loss, a definition of cachexia, as well as in a mixed cohort of non‐small cell lung cancer and PDAC although results were not sex‐stratified. 11 , 12 , 17 , 39 , 40 Whether Activin acts directly on muscle and/or only on muscle to initiate wasting is unclear, although mice with muscle‐specific expression of a dominant negative ACVR2B show reduced weight loss, suggesting at least part of the effect could be direct. 16 Why Activin inhibitors are more highly expressed in females remains to be determined, but our data in the C2C12 myotube in vitro atrophy model show potential for hormonal regulation of the Activin axis, which orchestrates male and female fertility 41 in addition to pancreatic cancer and cachexia. Of course, the majority of patients in this study are likely to be postmenopausal women and hypogonadal men, given the age of onset of PDAC. Thus, differences between the sexes in the Activin response to PDAC could be due to virilizing effects of PDAC, as opposed to loss of estradiol alone, or to sex chromosome‐specific mechanisms. Moreover, the contributions of other sex‐specific characteristics such as tumour gene expression, body composition, muscle fibre composition, and sickness behaviour (appetite and activity) remain to be determined. Regardless, our results demonstrate that sex is a major consideration for discovery work and development of targeted therapeutics.
Overall, our study supports a growing body of work demonstrating sex as a key consideration for studies of cancer cachexia and for precision therapy for cancer cachexia and suggests re‐evaluation of the design and analysis of cachexia clinical trials using anti‐Activin therapies. 42
Funding
This study was supported by the National Institutes of Health (P01CA236778, R01GM137656, P30CA082709, P30CA023168, and 5P30AR072581), the Veterans Administration (I01BX004177 and I01CX002046), the IU Simon Comprehensive Cancer Center, the Lustgarten Foundation, the Lilly Endowment, Inc., the Adams County Cancer Coalition, and the Walther Cancer Foundation.
Conflict of interests
S.S. is currently an employee of Eli Lilly and Company. T.Z. has previously consulted for Pfizer and is a member of the Scientific Advisory Boards of Emmyon and PeleOs; none of the results reported here relate to that work.
Supporting information
Data S1. Supporting information
Data S2. Supporting information
Acknowledgements
We thank the Indiana University Simon Comprehensive Cancer Center Tissue Procurement and Distribution Core, the Center for Medical Genomics, and the Collaborative Core for Cancer Bioinformatics. The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. 43
Zhong X., Narasimhan A., Silverman L. M., Young A. R., Shahda S., Liu S., Wan J., Liu Y., Koniaris L. G., and Zimmers T. A. (2022) Sex specificity of pancreatic cancer cachexia phenotypes, mechanisms, and treatment in mice and humans: role of Activin, Journal of Cachexia, Sarcopenia and Muscle, 13, 2146–2161, 10.1002/jcsm.12998
References
- 1. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res 2014;74:2913–2921. [DOI] [PubMed] [Google Scholar]
- 2. Hendifar AE, Petzel MQB, Zimmers TA, Denlinger CS, Matrisian LM, Picozzi VJ, et al. Pancreas cancer‐associated weight loss. Oncologist 2019;24:691–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baazim H, Antonio‐Herrera L, Bergthaler A. The interplay of immunology and cachexia in infection and cancer. Nat Rev Immunol 2021; 10.1038/s41577-021-00624-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zimmers TA, Fishel ML, Bonetto A. Stat3 in the systemic inflammation of cancer cachexia. Semin Cell Dev Biol 2016;54:28–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baracos VE, Martin L, Korc M, Guttridge DC, Fearon KCH. Cancer‐associated cachexia. Nat Rev Dis Primers 2018;4:17105. 10.1038/nrdp.2017.105 [DOI] [PubMed] [Google Scholar]
- 6. Huiskamp LFJ, Chargi N, Devriese LA, May AM, Huitema ADR, de Bree R. The predictive value of low skeletal muscle mass assessed on cross‐sectional imaging for anti‐cancer drug toxicity: a systematic review and meta‐analysis. J Clin Med 2020;9. 10.3390/jcm9113780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. da Rocha IMG, Marcadenti A, de Medeiros GOC, Bezerra RA, Rego JFM, Gonzalez MC, et al. Is cachexia associated with chemotherapy toxicities in gastrointestinal cancer patients? A prospective study. J Cachexia Sarcopenia Muscle 2019;10:445–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wakabayashi H, Arai H, Inui A. The regulatory approval of anamorelin for treatment of cachexia in patients with non‐small cell lung cancer, gastric cancer, pancreatic cancer, and colorectal cancer in Japan: facts and numbers. J Cachexia Sarcopenia Muscle 2021;12:14–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Phillips DJ, de Kretser DM, Hedger MP. Activin and related proteins in inflammation: not just interested bystanders. Cytokine Growth Factor Rev 2009;20:153–164. [DOI] [PubMed] [Google Scholar]
- 10. Pettersen K, Andersen S, van der Veen A, Nonstad U, Hatakeyama S, Lambert C, et al. Autocrine activin a signalling in ovarian cancer cells regulates secretion of interleukin 6, autophagy, and cachexia. J Cachexia Sarcopenia Muscle 2019; 10.1002/jcsm.12489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Togashi Y, Kogita A, Sakamoto H, Hayashi H, Terashima M, de Velasco MA, et al. Activin signal promotes cancer progression and is involved in cachexia in a subset of pancreatic cancer. Cancer Lett 2015;356:819–827. [DOI] [PubMed] [Google Scholar]
- 12. Loumaye A, de Barsy M, Nachit M, Lause P, Frateur L, van Maanen A, et al. Role of activin a and myostatin in human cancer cachexia. J Clin Endocrinol Metab 2015;100:2030–2038. [DOI] [PubMed] [Google Scholar]
- 13. Harada K, Shintani Y, Sakamoto Y, Wakatsuki M, Shitsukawa K, Saito S. Serum immunoreactive activin a levels in normal subjects and patients with various diseases. J Clin Endocrinol Metab 1996;81:2125–2130. [DOI] [PubMed] [Google Scholar]
- 14. Chen JL, Walton KL, Qian H, Colgan TD, Hagg A, Watt MJ, et al. Differential effects of interleukin‐6 and activin a in the development of cancer‐associated cachexia. Cancer Res 2016; 10.1158/0008-5472.CAN-15-3152 [DOI] [PubMed] [Google Scholar]
- 15. Chen JL, Walton KL, Winbanks CE, Murphy KT, Thomson RE, Makanji Y, et al. Elevated expression of activins promotes muscle wasting and cachexia. FASEB J 2014;28:1711–1723. [DOI] [PubMed] [Google Scholar]
- 16. Zhong X, Pons M, Poirier C, Jiang Y, Liu J, Sandusky GE, et al. The systemic activin response to pancreatic cancer: implications for effective cancer cachexia therapy. J Cachexia Sarcopenia Muscle 2019;10:1083–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Talar‐Wojnarowska R, Wozniak M, Borkowska A, Olakowski M, Malecka‐Panas E. Clinical significance of activin a and myostatin in patients with pancreatic adenocarcinoma and progressive weight loss. J Physiol Pharmacol 2020;71. 10.26402/jpp.2020.1.10 [DOI] [PubMed] [Google Scholar]
- 18. Chen JL, Walton KL, Al‐Musawi SL, Kelly EK, Qian H, La M, et al. Development of novel activin‐targeted therapeutics. Mol Ther 2015;23:434–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Goebel EJ, Corpina RA, Hinck CS, Czepnik M, Castonguay R, Grenha R, et al. Structural characterization of an activin class ternary receptor complex reveals a third paradigm for receptor specificity. Proc Natl Acad Sci U S A 2019;116:15505–15513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wrana JL, Attisano L, Wieser R, Ventura F, Massague J. Mechanism of activation of the tgf‐beta receptor. Nature 1994;370:341–347. [DOI] [PubMed] [Google Scholar]
- 21. Chang C. Agonists and antagonists of tgf‐beta family ligands. Cold Spring Harb Perspect Biol 2016;8. 10.1101/cshperspect.a021923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Benny Klimek ME, Aydogdu T, Link MJ, Pons M, Koniaris LG, Zimmers TA. Acute inhibition of myostatin‐family proteins preserves skeletal muscle in mouse models of cancer cachexia. Biochem Biophys Res Commun 2010;391:1548–1554. [DOI] [PubMed] [Google Scholar]
- 23. Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, et al. Reversal of cancer cachexia and muscle wasting by actriib antagonism leads to prolonged survival. Cell 2010;142:531–543. [DOI] [PubMed] [Google Scholar]
- 24. Paajanen J, Ilonen I, Lauri H, Järvinen T, Sutinen E, Ollila H, et al. Elevated circulating activin a levels in patients with malignant pleural mesothelioma are related to cancer cachexia and reduced response to platinum‐based chemotherapy. Clin Lung Cancer 2020;21:e142–e150. [DOI] [PubMed] [Google Scholar]
- 25. Group EUCCS , Regitz‐Zagrosek V, Oertelt‐Prigione S, Prescott E, Franconi F, Gerdts E, et al. Gender in cardiovascular diseases: impact on clinical manifestations, management, and outcomes. Eur Heart J 2016;37:24–34. [DOI] [PubMed] [Google Scholar]
- 26. Cosper PF, Leinwand LA. Cancer causes cardiac atrophy and autophagy in a sexually dimorphic manner. Cancer Res 2011;71:1710–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nipp R, Tramontano AC, Kong CY, Pandharipande P, Dowling EC, Schrag D, et al. Disparities in cancer outcomes across age, sex, and race/ethnicity among patients with pancreatic cancer. Cancer Med 2018;7:525–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Montalvo RN, Counts BR, Carson JA. Understanding sex differences in the regulation of cancer‐induced muscle wasting. Curr Opin Support Palliat Care 2018;12:394–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhong X, Zimmers TA. Sex differences in cancer cachexia. Curr Osteoporos Rep 2020;18:646–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Blackburn G, Swan Z, Brantley TJ, Tichy L, Parry T. Biological sex mediates cancer cachexia associated muscle weakness. FASEB J 2020;34:1. [Google Scholar]
- 31. Beery AK, Zucker I. Sex bias in neuroscience and biomedical research. Neurosci Biobehav Rev 2011;35:565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McCullough LD, de Vries GJ, Miller VM, Becker JB, Sandberg K, McCarthy MM. Nih initiative to balance sex of animals in preclinical studies: generative questions to guide policy, implementation, and metrics. Biol Sex Differ 2014;5:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hetzler KL, Hardee JP, Puppa MJ, Narsale AA, Sato S, Davis JM, et al. Sex differences in the relationship of il‐6 signaling to cancer cachexia progression. Biochim Biophys Acta 2015;1852:816–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lim S, Deaver JW, Rosa‐Caldwell ME, Haynie WS, da Silva Morena F, Cabrera AR, et al. Development of metabolic and contractile alterations in development of cancer cachexia in female tumor‐bearing mice. J Appl Physiol (1985) 2022;132:58–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hetzler KL, Hardee JP, LaVoie HA, Murphy EA, Carson JA. Ovarian function's role during cancer cachexia progression in the female mouse. Am J Physiol Endocrinol Metab 2017;312:E447–E459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53r172h and krasg12d cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005;7:469–483. [DOI] [PubMed] [Google Scholar]
- 37. Kays JK, Shahda S, Stanley M, Bell TM, O'Neill BH, Kohli MD, et al. Three cachexia phenotypes and the impact of fat‐only loss on survival in folfirinox therapy for pancreatic cancer. J Cachexia Sarcopenia Muscle 2018;9:673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bonetto A, Aydogdu T, Jin X, Zhang Z, Zhan R, Puzis L, et al. Jak/stat3 pathway inhibition blocks skeletal muscle wasting downstream of il‐6 and in experimental cancer cachexia. Am J Physiol Endocrinol Metab 2012;303:E410–E421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Loumaye A, de Barsy M, Nachit M, Lause P, van Maanen A, Trefois P, et al. Circulating activin a predicts survival in cancer patients. J Cachexia Sarcopenia Muscle 2017;8:768–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lerner L, Gyuris J, Nicoletti R, Gifford J, Krieger B, Jatoi A. Growth differentiating factor‐15 (gdf‐15): a potential biomarker and therapeutic target for cancer‐associated weight loss. Oncol Lett 2016;12:4219–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wijayarathna R, de Kretser DM. Activins in reproductive biology and beyond. Hum Reprod Update 2016;22:342–357. [DOI] [PubMed] [Google Scholar]
- 42. Ma JD, Heavey SF, Revta C, Roeland EJ. Novel investigational biologics for the treatment of cancer cachexia. Expert Opin Biol Ther 2014;14:1113–1120. [DOI] [PubMed] [Google Scholar]
- 43. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2021. J Cachexia Sarcopenia Muscle 2021;12:2259–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting information
Data S2. Supporting information