

CRISPR-mediated DNA damage enriches for cells with escape mutations in a core CRISPR–p53 interactome, which can be suppressed by transient inhibition of p53.
Abstract
Inactivating p53 mutations are the most abundant genetic alterations found in cancer. Here we show that CRISPR/Cas9-induced double-stranded DNA breaks enrich for cells deficient in p53 and in genes of a core CRISPR–p53 tumor suppressor interactome. Such enrichment could predispose to cancer development and thus pose a challenge for clinical CRISPR use. Transient p53 inhibition could suppress the enrichment of cells with these mutations. The level of DNA damage response induced by an sgRNA influenced the enrichment of p53-deficient cells and could be a relevant parameter in sgRNA design to limit cellular enrichment. Furthermore, a dataset of >800 human cancer cell lines identified additional factors influencing the enrichment of p53-mutated cells, including strong baseline CDKN1A expression as a predictor for an active CRISPR–p53 axis. Taken together, these data provide details about p53 biology in the context of CRISPR-induced DNA damage and identify strategies to enable safer CRISPR use.
Significance:
CRISPR-mediated DNA damage enriches for cells with escape mutations in a core CRISPR–p53 interactome, which can be suppressed by transient inhibition of p53.
Introduction
The p53 protein, encoded by the TP53 gene in humans and Trp53 in mice, plays a central role in the cellular response to DNA damage (1, 2). Its activity is often attributed to its role as a transcription factor, controlling the expression of cell cycle and apoptosis-related genes, but also to other, transcription-independent mechanisms (3). The importance of p53 is highlighted by the fact that acquired TP53 mutations are found in more than 50% of human cancers, and that humans and mice with congenital mutations in TP53/Trp53 have a strong susceptibility for cancer development (3–5).
CRISPR is a diverse set of molecular biology tools developed from prokaryotic origins (6). A commonly used application for CRISPR is to inactivate or modify genes (7, 8). This is achieved by delivering a single guide RNA (sgRNA), designed to give specificity for the gene of interest and the Cas9 endonuclease into the nucleus of cells, resulting in double-stranded DNA breaks at the targeted genomic site. Accordingly, CRISPR molecular biology tools are considered to have immense potential for clinical gene therapy use (9). Disease-causing mutations in, for example, congenital monogenic disorders affecting the hematopoietic system are apparent candidates, and CRISPR-based clinical trials for sickle cell anemia and β-thalassemia are ongoing where the patient hematopoietic stem and progenitor cells are modified ex vivo and subsequently returned to the patient (10, 11). CRISPR-mediated modifications of cells used for chimeric antigen receptor (CAR)-based immunotherapy is another clinical setting where CRISPR is likely to have a large impact (12).
Early gene therapy trials have resulted in cancer development (13), and safety concerns related to CRISPR-based gene therapy are being addressed at multiple levels. This predominately relates to CRISPR off-target activity, where, for example, sgRNA design with low off-target activity (14), development of high fidelity Cas9 versions (15), and methods for the evaluation of off-target mutations (16) have been an intense research focus. Another proposed, but less studied, risk is that the CRISPR-mediated DNA damage could give cells with mutations in TP53 a selective advantage and thereby be enriched in a cell population exposed to CRISPR as a part of a therapeutic protocol (17, 18). Notably, TP53 mutations are seen in human embryonic stem cell lines (19), early in the development of certain cancers affecting e.g., skin (20), esophagus (21), and lung (22), and also contribute to clonal hematopoiesis (23, 24) showing that premalignant TP53 mutations can be found in different cell populations of relevance for clinical CRISPR use. In addition, multiple viruses have evolved mechanisms to suppress p53 (25), and thus could potentially interfere with CRISPR use. Here, we present findings showing that cells with mutations in p53 and a CRISPR–p53 interactome are indeed enriched by CRISPR-induced DNA damage. Importantly, we show that the enrichment can be suppressed by transient p53 inhibition, and identify several parameters that influence the enrichment, enabling safe CRISPR use.
Materials and Methods
Additional details are found in Supplementary Materials and Methods, as well as Supplementary Tables S1 (reagents), S2 (sgRNAs and primers), S3 (CRISPR library), and S4 (NGS primers).
Cells
The Hox cell line was generated by transducing bone marrow cells of C57BL/6 Cas9+ GFP+ mice with an estrogen inducible retroviral construct expressing Hoxb8 as described in refs. 26, 27. Hox cells are immortalized by estrogen-regulated Hoxb8 (ER-Hoxb8) and display a granulocyte–monocyte progenitor (GMP) phenotype, as described in Supplementary Figs. S1A and S1B. No cell line authentication was performed to Hox cells.
B16-F10 cell line (mouse melanoma) was purchased from ATCC that performs authentication and used at a low passage number. Cas9 expressing cells were generated by transducing B16-F10 cells with lentiCas9-Blast lentiviral particles. All cells were confirmed Mycoplasma negative before culture.
Trp53 KO Hox and B16-F10 cells were generated by electroporation or transfection, respectively, of a Trp53 targeting sgRNA (Supplementary Table S2, Genomic Location 11:69479559–69479578). To limit artefacts due to selection of specific clones, cells were used as a mixed population strongly dominated by +1 and +2 insertions, and a >95% KO score.
Viral preparation and transduction
Lentiviral particles were generated by transfecting HEK293T cells with transfer plasmids (lentiCas9-Blast; or LentiGuide-Puro-P2A-EGFP_mRFPstuf), as well as pMD2.G, and psPAX2. ER-Hoxb8 retrovirus particles were generated by transfecting HEK293T cells with ER-Hoxb8 and the EcoPac gag-pol-env. Hox or B16 cells were spin-infected and subsequently selected using Puromycin or Blasticidin to remove the noninfected cells.
sgRNA design, electroporation, and transfection
sgRNAs were designed using the Green Listed software (28, 29) utilizing sgRNA design from the Doench mouse library (14). 2′-O-methyl and phosphorothioate stabilized sgRNAs (Supplementary Table S2) were ordered from Sigma-Aldrich or Synthego.
For sgRNA delivery, the Neon Transfection System was used for Hox cells, and Lipofectamine 2000 for B16 cells. The Trp53 siRNA was typically delivered in the same reaction as the sgRNAs.
CRISPR KO genotyping
Purified PCR amplicons were sequenced by Eurofins Genomics. The Sanger sequencing data were analyzed by ICE (https://ice.synthego.com).
Growth curve characterization
Hox cells were cultured with the following interventions: electroporation with a GFP targeting sgRNA, etoposide, or AMG232. Cells were counted each day by flow cytometry (BD Accuri) with CountBright Absolute Counting Beads.
Real-time PCR
RNA was extracted using Direct-zol RNA MiniPrep Kit, and was converted into cDNA using High Capacity RNA-to cDNA Kit. The expression of indicated genes was quantified with a CFX 384 Real-Time PCR machine (Bio-Rad) using TaqMan gene expression FAM assays for Cdkn1a, Bbc3, Pmaip1, Isg15, and Irf7. Expression was normalized by TaqMan gene expression VIC assays for β-actin and gene expression was quantified using the ddCT method.
Apoptosis TUNEL assay
Cells were collected and fixed by PFA at different time points, and the FlowTAC Apoptosis Detection Kit was used to stain apoptotic cells for analysis by flow cytometry (BD Accuri).
Cloning of sgRNAs into lentiviral transfer plasmid and CRISPR screens
sgRNAs with overhangs for the LentiGuide transfer plasmid (Supplementary Table S3) were designed using the Green Listed software (28, 29) using sgRNA design from the Doench mouse library (14) and, for intergenic controls, the Wang mouse library (30). The sgRNA library was ordered from CustomArray as a DNA oligo pool. Cloning, library preparation, and screens were performed as described in Supplementary Materials and Methods. Screens were evaluated by next-generation sequencing (Illumina MiSeq v3 run, 2×75bp reads), and the raw FASTQ data were analyzed by MAGeCK (31). Read counts from CRISPR screens are found in Supplementary Tables S5 and S6.
JAK1/STAT1 signaling assay
Hox cells were cultured with or without mouse Interferon Beta and the Jak1 inhibitor Solcitinib for 7 days. Cells were counted on day 7 by flow cytometry (BD Accuri) using CountBright Absolute Counting Beads.
Competitive coculture assay
Trp53 KO and WT cells were mixed at 1:4 ratio, and subsequently exposed to different interventions: CRISPR - electroporated with sgRNA or transduced with lentivirus and culture for 7 days; or exposed to etoposide or AMG232 and cultured for 7 days; or cultured with Cobalt(II) chloride (CoCl2) for 7 days. For hypoxia experiments, Hox cells were cultured in 1% O2 for 7 days in a Baker InvivO2 Physiological Cell Culture Workstations. For in vivo experiments, B16 cells were transfected with a Ccr1 targeting sgRNA (Supplementary Table S2) or control, and directly injected subcutaneously into C57BL/6 mice, and tumors collected after 21 days. The proportion of Trp53 KO cells was subsequently quantified by sequencing as described in “CRISPR KO genotyping.” The used p53 related inhibitors are described in Supplementary Materials and Methods and Supplementary Table S1.
Flow cytometry analysis
Fresh bone marrow cells from C57BL/6 Cas9+ GFP+ mice and Hox cells were stained with antibodies described in Supplementary Materials and Methods and Supplementary Table S1. After 30 minutes of staining, the cells were washed and analyzed by flow cytometry (BD FACSVerse). FACS FCS files were analyzed by FlowJo version 10 (FlowJo, LLC).
Analysis of data from the depmap portal
sgRNA enrichment [CRISPR (Avana) Public 20Q4 release], mutation profile (Mutation Public 20Q4 release), drug sensitivity (PRISM Repurposing Primary Screen 19Q4 release), and mRNA expression levels (Expression Public 20Q4 release) was extracted December 13, 2020, from the Depmap portal (https://depmap.org/portal/; refs. 32, 33). Connectivity maps, tSNE plots, and transcription factor binding sites were generated as described in Supplementary Materials and Methods.
Statistical analysis
Graphpad Prism versions 8 and 9 were used as indicated in figure legends. Correlation analysis related to the Depmap data were performed with the integrated Depmap data explorer tool.
Results
CRISPR-mediated DNA damage enriches for cells with mutations in Trp53
DNA damage activates p53, which can result in apoptosis and cell-cycle arrest. To study different aspects of how CRISPR-induced DNA damage affects cells, we established an experimental model system comparing CRISPR with a pulse of etoposide (a topoisomerase II inhibitor causing DNA damage and p53 activation; ref. 34), or AMG232 (activating p53 by interfering with the MDM2-p53 interaction; ref. 35; Fig. 1A). Using a cell line derived from mouse hematopoietic stem cells of Cas9+ GFP+ mice (Hox cells; Supplementary Figs. S1A–S1D), we observed that the CRISPR event resulted in partially delayed cell growth (Fig. 1B), apoptosis induction (Fig. 1C), and transcription of Cdkn1a (also known as p21, linked to DNA damage-induced cell-cycle arrest; ref. 36; Fig. 1D), as well as Bbc3, and Pmaip1 (also known as Puma and Noxa, respectively, linked to DNA damage-induced apoptosis; ref. 37; Supplementary Figs. S1E–S1F), although at a lower magnitude compared with treatment with AMG232 or etoposide.
Figure 1.
![Figure 1. CRISPR-mediated DNA damage enriches for cells with mutations in Trp53. A, Model describing how CRISPR and pharmacologic p53 activation used in the study are expected to affect targeted cells. B, Growth characteristics of Hox expanded bone marrow cells (from Cas9+ GFP+ mice), exposed to CRISPR (electroporated with a control or GFP targeting sgRNA; sgGFP), or pulsed for 8h with AMG232, or etoposide (ETOP). C, Kinetic analysis of apoptosis by flow cytometry-based TUNEL assay of Hox cells exposed to CRISPR (electroporated with sgGFP) or etoposide. D, Kinetic qPCR analysis of Cdkn1a expression of Hox cells exposed to CRISPR (electroporated with GFP sgRNA) or etoposide. E, Model describing experimental setup. F, WT and Trp53 KO Hox cells (Cas9+ and GFP+) were mixed and subjected to CRISPR [electroporated with sgGFP, or transduced with nontargeting ctrl (NTC) virus, sgGFP virus, CRISPR library virus], or an 8 hours pulse with etoposide or AMG232. After 7 days in culture, cells were sequenced, and the fraction of Trp53 KO sequences determined. G, WT and Trp53 KO Hox cells were mixed and cultured for seven days in a hypoxia chamber. H, WT and Trp53 KO B16 cells (Cas9+) were mixed, electroporated with control or a Ccr1 targeting sgRNA (sgCcr1), and injected subcutaneously into C57BL/6 mice. Day 21 tumors were collected and analyzed for Trp53 mutations. Data are shown as mean ± SEM, n = 3 (B–D), mean and individual values, n = 3 (F–H). Data are combined from three independently performed experiments (B–D and F–H). **, P < 0.01; ***, P < 0.001; n.s., nonsignificant by two-way ANOVA and Tukey posttest (B–D), one-way ANOVA and Tukey posttest (F–H).](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=9397613_36fig1.jpg)
CRISPR-mediated DNA damage enriches for cells with mutations in Trp53. A, Model describing how CRISPR and pharmacologic p53 activation used in the study are expected to affect targeted cells. B, Growth characteristics of Hox expanded bone marrow cells (from Cas9+ GFP+ mice), exposed to CRISPR (electroporated with a control or GFP targeting sgRNA; sgGFP), or pulsed for 8h with AMG232, or etoposide (ETOP). C, Kinetic analysis of apoptosis by flow cytometry-based TUNEL assay of Hox cells exposed to CRISPR (electroporated with sgGFP) or etoposide. D, Kinetic qPCR analysis of Cdkn1a expression of Hox cells exposed to CRISPR (electroporated with GFP sgRNA) or etoposide. E, Model describing experimental setup. F, WT and Trp53 KO Hox cells (Cas9+ and GFP+) were mixed and subjected to CRISPR [electroporated with sgGFP, or transduced with nontargeting ctrl (NTC) virus, sgGFP virus, CRISPR library virus], or an 8 hours pulse with etoposide or AMG232. After 7 days in culture, cells were sequenced, and the fraction of Trp53 KO sequences determined. G, WT and Trp53 KO Hox cells were mixed and cultured for seven days in a hypoxia chamber. H, WT and Trp53 KO B16 cells (Cas9+) were mixed, electroporated with control or a Ccr1 targeting sgRNA (sgCcr1), and injected subcutaneously into C57BL/6 mice. Day 21 tumors were collected and analyzed for Trp53 mutations. Data are shown as mean ± SEM, n = 3 (B–D), mean and individual values, n = 3 (F–H). Data are combined from three independently performed experiments (B–D and F–H). **, P < 0.01; ***, P < 0.001; n.s., nonsignificant by two-way ANOVA and Tukey posttest (B–D), one-way ANOVA and Tukey posttest (F–H).
To explore if the relatively mild phenotype induced by CRISPR was sufficient to give a selective advantage to cells with mutations in Trp53, we established a competitive assay, where Trp53 KO and WT cells were mixed at a 1:4 ratio and subsequently exposed to CRISPR (electroporation or lentiviral delivery of sgRNA), again comparing it to pharmacologic p53 activation with AMG232 or etoposide, as well as to hypoxia, also known to activate p53 (Fig. 1E; ref. 38). Sequencing the Trp53 locus after seven days in culture revealed that the proportion of Trp53 KO cells in the population did not expand significantly by only being in culture, or by being transduced by nontargeting control (NTC) lentiviral particles. However, the proportion of Trp53 KO cells significantly expanded after being exposed to CRISPR, AMG232, etoposide, or hypoxia (Fig. 1F–G; Supplementary Figs. S1G and S1H, and S2A and S2B). Similar results were obtained by analyzing the Trp53 mutation spectrum of subcutaneous B16 tumors isolated from mice 21 days after injection of B16 cells treated ± CRISPR (Fig. 1H; Supplementary Figs. S2C–S2F). We concluded that CRISPR, as well as other p53 activating interventions, gives cells with mutations in Trp53 a selective advantage in a mixed cell population, resulting in enrichment.
The level of DNA damage response induced by an sgRNA influences the enrichment of Trp53 KO cells
The level of enrichment of Trp53 KO cells mirrored the severity of the cellular phenotype as presented in Fig. 1B–D; Supplementary Figs. S1E and S1F, with both etoposide and AMG232 causing a significantly higher Trp53 KO enrichment concomitant with a more pronounced delay in cell growth, apoptosis, and upregulation of Cdkn1a, Bbc3, and Pmaip1 compared with CRISPR treatment. On the basis of this, we hypothesized that the level of the DNA damage response (DDR) induced by sgRNAs could be a parameter affecting the enrichment of Trp53 KO cells. We designed a set of sgRNAs with different off-target activity and used them alone or in combination at equimolar concentrations (Fig. 2A; Supplementary Figs. S3A and S3B). We found that the level of early CRISPR-induced Cdkn1a transcription was linked to the expected amount of induced DNA damage, based on off-target calculations, and by sgRNAs combined to increase the DDR in a predictable manner (Fig. 2B; Supplementary Fig. S3C). In addition, comparing the Cdkn1a transcription 2 hours after sgRNA electroporation to the enrichment of Trp53 KO sequences 7 days later, we found that the level of early CRISPR-induced Cdkn1a transcription correlated with the enrichment of Trp53 KO cells (Fig. 2C), and that the level of enrichment could be predicted on the basis of the calculated off-target activity for one, or a combination, of sgRNAs (Fig. 2D). Notably, the GFP sgRNA consistently had a higher KO efficiency than the Ccr1 sgRNA (Supplementary Figs. S3D and S3E), despite causing less Trp53 KO enrichment. We concluded that the level of DDR induced by sgRNAs, as measured by Cdkn1a upregulation, affects the enrichment of Trp53 KO cells without direct correlation to the KO efficiency, and thus could be a relevant parameter in sgRNA selection.
Figure 2.
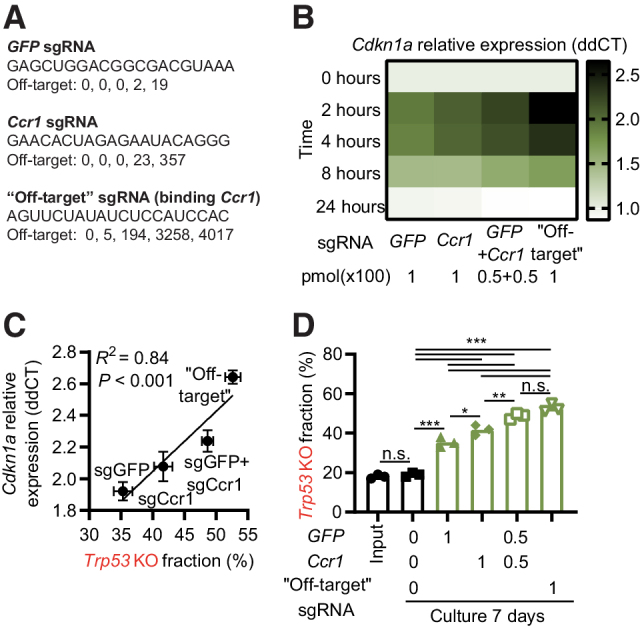
The level of DDR induced by an sgRNA influences the enrichment of Trp53 KO cells. A, Information about sgRNAs used in the figure. Off-target indicates number of targets with 0, 1, 2, 3, and 4 mismatches to the mouse genome as identified by Cas-OFFinder. B, Hox cells (Cas9+ and GFP+) were electroporated with indicated sgRNAs, and Cdkn1a expression analyzed by qPCR at different time points. C, Comparison of the Cdkn1a expression by qPCR 2 hours after electroporation with indicated sgRNAs and enrichment of Trp53 KO sequences day 7. D,Trp53 KO and WT Hox cells (Cas9+ and GFP+) were mixed 1:4 and electroporated by different sgRNAs at the indicated doses (pmol × 100). Seven days later, cells were collected and sequenced to determine the percentage of Trp53 KO sequences. Data presented as heatmap based on the average signal, n = 3 (B), mean ± SEM, n = 3 (C), or mean and individual data, n = 3 (D). Data are combined from three independently performed experiments (B–D). *, P < 0.05; **, P < 0.01; ***, P < 0.001; n.s., nonsignificant by Pearson r correlation and simple linear regression line (C), or one-way ANOVA and Tukey posttest (D).
CRISPR-mediated enrichment of mutations in Trp53 can be inhibited
Since the enrichment of cells with inactive p53 could pose a challenge to the clinical use of CRISPR, we set out to identify strategies to suppress the enrichment. We hypothesized that transient inhibition of p53 or proteins playing an important, nonredundant role in the p53 pathway could be a viable strategy. To this end, we pretreated cells with a selection of potentially relevant inhibitors followed by exposure to CRISPR (Fig. 3A). Using the Hox cells, we found that treating the cells with a Trp53 siRNA completely inhibited the enrichment of cells with Trp53 mutations, whereas use of the other inhibitors did not show any significant activity (Fig. 3A). Similar results were found with B16 cells, although the ATM inhibitor, KU55933, additionally showed partial inhibition of the enrichment (Fig. 3B). Importantly, the inhibition of the enrichment was not explained by the Trp53 siRNA negatively impacting the KO efficiency, as shown targeting GFP (Supplementary Fig. S4A), as well as Ccr1 (Supplementary Figs. S4B–S4E), where the KO efficiency was equivalent (GFP) or even slightly higher (Ccr1) with the Trp53 siRNA addition. We also noted that that the Trp53 siRNA normalized the growth characteristics of cells treated with sgRNAs (Supplementary Fig. S4F). The Trp53 siRNA was additionally able to significantly suppress the enrichment of cells with Trp53 mutations in response to AMG232 and etoposide, although not at the same level as for CRISPR, in line with the stronger DDR response induced by AMG232 and etoposide (Supplementary Figs. S4G and S4H). Notably, the pan-caspase inhibitor Z-VAD, often used as an apoptosis inhibitor, did not significantly suppress the enrichment of cells with Trp53 mutations (Supplementary Fig. S4I). However, the addition of siRNAs targeting both Bax and Bak1, pro-apoptotic members of the Bcl-2 family (39), partly inhibited the enrichment of Trp53 mutated cells, while siRNAs targeting only Bax or Bak1 did not inhibited the enrichment, showing the expected redundancy of these two genes in the apoptosis pathway (Supplementary Figs. S5A and S5B). Similar results were found using Bax and Bak1 KO cells (Supplementary Figs. S5C and S5D), taken together showing that apoptosis is partly involved in the CRISPR-mediated enrichment of Trp53 mutated cells, and that Z-VAD is not sufficiently suppressing the response in the used setting. We concluded that transient inhibition of p53 can limit enrichment of cells with mutations in Trp53 while retaining, or even increasing, the KO efficiency.
Figure 3.

CRISPR-mediated enrichment of mutations in Trp53 can be inhibited. A, Left, inhibitors used in A and B. Right, WT and Trp53 KO Hox cells (Cas9+ and GFP+) were mixed and transduced with a sgGFP virus in the presence of inhibitors. Cells were then cultured for 7 days, followed by quantification of the frequency of Trp53 mutations by sequencing. B, WT and Trp53 KO B16 cells were mixed and transduced with sgGFP virus in the presence of a selection of inhibitors. Cells were then cultured for 7 days, followed by quantification of the frequency of Trp53 mutations by sequencing. Data are shown as mean and individual values, n = 4 (A and B). Data are combined from two independently experiments performed in two duplicates (A and B). ***, P < 0.001 by one-way ANOVA and Tukey posttest (A and B).
CRISPR enriches for low-frequency mutations in tumor suppressor genes
Next, we set out to identify p53-linked genes playing a nonredundant role in the CRISPR-induced DDR. Such genes could represent additional drug targets to modify the CRISPR–p53 response, and, importantly, cells with mutations in such genes could be enriched by CRISPR-mediated DNA damage. We applied a custom CRISPR screen library targeting 395 DNA damage-related genes and controls, with 1640 sgRNAs (Supplementary Table S3) to WT and Trp53 KO cells (Hox and B16 cells). Initially relying on the DDR induced by the delivered sgRNA in the Cas9+ cells, as used previously (17, 40), we corroborated that Trp53 sgRNAs were enriched and that Mdm2 sgRNAs were depleted in a p53-dependent manner, in both Hox and B16 cells (Supplementary Figs. S6A–S6D). In this regard, Mdm2 behaved as an essential gene in Trp53 WT cells, where mutated cells were rapidly lost over time, which was further accelerated by sgRNA delivery (Supplementary Fig. S6E). We also noted that the Hox cells (both Trp53 WT and KO) enriched for sgRNAs relating to type I IFN signaling (Stat1, Jak1, and Ptprc), and, in line with this, were sensitive to induced type I IFN signaling (Supplementary Figs. S6F–S6I; Supplementary Table S7).
We additionally performed a screen using the same sgRNA library, but this time culturing the cells for 14 days after the introduction of the library into the Hox cell population, and then applied a controlled CRISPR DNA damage event by electroporation with sgGFP (Fig. 4A). In this way, we could separate the studied CRISPR event from the introduction of the CRISPR library, and also include control groups treated with AMG232 or etoposide. Comparing the different interventions to control-treated cells, we identified that CRISPR enriched for cells with mutations in Chek2, Trp53, and Cdkn1a (Fig. 4B); AMG232 enriched for cells with mutations in Trp53 and Cdkn1a (Fig. 4C); and etoposide enriched for cells with mutations in Atm, Chek2, Trp53, and Cdkn1a (Fig. 4D). Notably, AMG232 also enriched for mutations in Stat1, and Eif2ak2, two genes related to type I IFN signaling (Fig. 4C), again highlighting this pathway in Hox cells. Upon more detailed analysis of Hox cells, we found that AMG232 caused a significant upregulation of IFN-stimulated genes (Isg15, Irf7) in a p53-dependent manner, linking p53 activation to Type I IFN production in these cells (Supplementary Fig. S7A). Focusing on the ATM–CHEK2–p53–CDKN1A pathway, that links double-stranded DNA damage to cell-cycle arrest (1, 2, 36), we found that CRISPR caused a significant enrichment of sgRNAs targeting all these genes when comparing WT and Trp53 KO cells (Fig. 4E–H). Furthermore, a Trp53 siRNA could significantly suppress the enrichment of cells with mutations in Chek2, Trp53, and Cdkn1a, but not for Atm (for which the enrichment phenotype was the weakest; Fig. 4I; Supplementary Fig. S7B). We did not observe any enrichment of sgRNAs targeting genes related to apoptosis (Fig. 4J; Supplementary Fig. S7C). However, in line with our observations in Supplementary Figs. S5A–S5D, we found that CRISPR could enrich for cells with Bax mutations when performed on a Bak1 KO background (but not on a Bak1 WT background, or a Bak1/Trp53 DKO background), again highlighting the involvement, but redundant nature of individual genes in the apoptosis pathway (Supplementary Figs. S7D–S7H).
Figure 4.

CRISPR enriches for low-frequency mutations in tumor suppressor genes. A, Model describing experimental setup. B–D, Hox cells (Cas9+ and GFP+) were transduced with a custom CRISPR library and cultured for >14 days. Cells were then either exposed to CRISPR (sgGFP electroporation; B), AMG232 8 hours pulse (C), or etoposide 8 hours pulse (D). Cells were subsequently cultured for 7 days, the sgRNA representation analyzed by next-generation sequencing, and enrichment/depletion deconvoluted by MAGeCK. E–H,Z scores of individual sgRNAs (n = 4/gene) for Atm (E), Chek2 (F), Trp53 (G), and Cdkn1a (H) in WT and Trp53 KO Hox cells treated with mock (electroporation without sgRNA), CRISPR (electroporation with sgGFP), AMG232, or etoposide (ETOP). I,Z scores of individual sgRNAs (n = 4/gene) in Hox cells treated with Trp53 siRNA at the same time as being electroporated with sgGFP as described in A. J,Z scores of individual sgRNAs (n = 4/gene) for genes linked to apoptosis. K, Model indicating genes playing a nonredundant role in the DDR. Data presented as volcano plots with Z score (log2-fold change) and adjusted P values (B–D) as calculated by MAGeCK, or mean and individual values for four sgRNAs from the exploratory screen (E–J). *, P < 0.05; n.s., nonsignificant by Mann–Whitney test.
We concluded that (i) the ATM–CHEK2–p53–CDKN1A pathway plays a nonredundant role in the CRISPR-mediated DDR (Fig. 4K), (ii) cells with mutations in these tumor suppressor genes could be enriched in a cell population as CRISPR is applied, and (iii) a Trp53 siRNA can suppress the enrichment of the mutated cells to a large degree.
Enrichment of sgRNAs targeting TP53 and a CRISPR–p53 interactome in full genome CRISPR screens of >800 human cancer cell lines.
To expand our understanding of which mutations could be enriched in a cell population as CRISPR is applied, we used the Depmap portal, containing full genome CRISPR screen data of 808 human cell lines (Public 20Q4 release), as well as baseline gene expression, mutation status, and drug sensitivity data for a large proportion of the same cells. Exploring the CRISPR screen data, we found that 103 of the 808 included cell lines (12.7%) enriched for TP53 sgRNAs as defined by an enrichment score >1. Stratifying these cells based on TP53 mutation status (WT or any type of mutation, making up 32.3% and 67.7% of the cell lines respectively), we found that 94 of the 103 cell lines (91.3%) that enriched for TP53 sgRNAs were confined to the TP53 WT group (Fig. 5A). Anecdotally, we also noted that the cell line with the strongest enrichment for TP53 sgRNAs, of the 808 included cell lines, was a version of the RPE-1 cell line (Supplementary Table S8), which has been used in a significant portion of previous publications related to p53 and CRISPR (17, 40–42). We compared the TP53 sgRNA enrichment to the sensitivity of the cells to p53 modulating drugs. We found a clear correlation between TP53 sgRNA enrichment and the sensitivity to AMG232 (Fig. 5B and C), as well as to Nutlin-3 and CMG097 (both with a similar mode of action as AMG232), but not, for comparison, to the p53 inhibitor Pifithrin-μ (Supplementary Figs. S8A–S8C). Taken together, we concluded that TP53 sgRNA enrichment in the Depmap dataset could be used to identify cells where the CRISPR–p53 pathway is active, and, as a consequence, that correlation to TP53 sgRNA enrichment could identify factors relevant for the pathway. Further supporting this concept, we found that TP53 sgRNA enrichment correlated strongly with MDM2 sgRNA depletion, in line with their opposing functionality (Fig. 5D). Performing the same type of correlation analysis, but on a full genome basis, we identified a list of genes where sgRNA enrichment (+) or depletion (−) correlated with TP53 sgRNA enrichment (Fig. 5E; Supplementary Fig. S8D; Supplementary Table S9). The gene list validated those we had identified in our experimental data, as well as identified additional genes playing a nonredundant TP53-related role in the CRISPR-mediated DDR. Analyzing the top 10 enriched and depleted genes using geneMANIA, we noted a high level of physical interaction between the linked proteins (Fig. 5E). Importantly, using an alternative analysis approach; identifying genes where sgRNA enrichment/depletion correlated with the TP53 mutation status of the cell lines (instead of TP53 sgRNA enrichment, as in Fig. 5E), resulted in a list of genes with a large level of overlap with the list in Fig. 5E (Supplementary Fig. S9; Supplementary Table S10). On the basis of the overlap of these lists, and the experimental data we highlight TP53, TP53BP1, CHEK2, ATM, CDKN1A, USP28, UBE2K, XPO7 as a core CRISPR–p53 tumor suppressor interactome, where cells with inactivating mutations or silencing of these genes (something that is commonly found in cancers; Supplementary Fig. S10; refs. 3, 4, 43–47) could be enriched in cell populations as CRISPR is applied. Furthermore, cells with copy number amplifications, overexpression, or activating mutations of MDM2, PPM1D, MDM4, DDX31, USP7, PPM1G, WDR89, and TERF1 also observed in cancer (48–53), could similarly be enriched by CRISPR. Further analysis additionally identified that mutations in the CRISPR–p53 interactome are not mutually exclusive in a given cell, and that the number of mutations of mutations of these genes are significantly enriched in cell lines that do not show TP53 sgRNA enrichment. (Supplementary Fig. S11; Supplementary Table S11).
Figure 5.
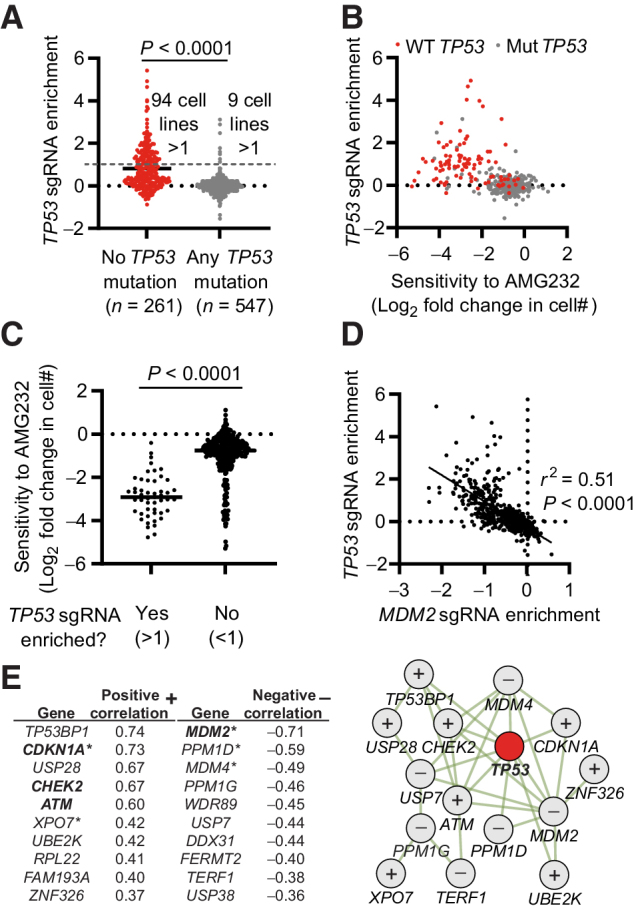
Enrichment of sgRNAs targeting TP53 and a CRISPR–p53 interactome in full genome CRISPR screens of >800 human cancer cell lines. A, Enrichment score of TP53 sgRNAs in 808 cell lines, stratified on the basis of the presence or absence of any mutation in TP53. B, Correlation between the enrichment of TP53 sgRNAs and AMG232 sensitivity. TP53 WT cells are indicated in red. C, Sensitivity to AMG232 in cell lines stratified on the basis of TP53 sgRNA enrichment. D, Correlation between the enrichment of TP53 sgRNAs and MDM2 sgRNAs. E, Left, top 10 genes with the strongest positive (+) and negative (−) correlation with TP53 sgRNA enrichment from full genome CRISPR screens of 808 cell lines. *, genes identified as transcription factor target genes for p53. Bold indicates genes identified experimentally in Fig. 4. Right, physical interactions of genes in E (left) defined by geneMANIA. + indicates genes that positively correlate, and − indicates genes that negatively correlate with TP53 sgRNA enrichment. Data include all available data in the Depmap CRISPR (Avana) 20Q4, Expression Public 20Q4, as well as drug sensitivity (PRISM Repurposing Primary Screen) 19Q4 releases. Each dot represents one cell line (A–D), and the data are based on n = 808 (A, D, and E) n = 408 (B and C). Statistical analysis based on unpaired t test (A and C), Pearson r correlation and simple linear regression line (D), and calculated in Depmap (E).
Gene expression patterns predicting if the CRISPR–p53 axis is active in a cell
Finally, we explored if specific gene expression patterns could predict if the CRISPR–p53 axis is active in a cell, and thus if the cell would enrich for TP53 sgRNAs. On the basis of our data identifying a central, nonredundant role for CDKN1A in the CRISPR-induced response, we used the Depmap dataset to test if a strong baseline expression of CDKN1A could predict if cells would enrich for TP53 sgRNAs. We found that cells that enrich for TP53 sgRNAs, with a few exceptions, had a strong baseline CDKN1A expression (Fig. 6A and B), whereas, for example, TP53 expression itself did not predict cells that would enrich for TP53 sgRNAs (Fig. 6C and D). Correlating strong baseline gene expression with TP53 sgRNA enrichment resulted in a list of genes that, to a large extent, were identified as transcription target genes for p53 (Fig. 6E; Supplementary Table S12), and these genes were shown using geneMANIA to display a strong co-expression pattern (Supplementary Figs. S12A–S12D). Notably, the gene set we identified by analyzing the 800 cell lines largely overlapped with published gene expression patterns induced by CRISPR-mediated DNA damage in specific cell lines (18, 54), as well as genes upregulated in cancers WT for TP53 (55), including ZMAT3 linked to the activity of p53 in c-Myc and Kras-driven tumors (56, 57). Furthermore, a tSNE dimensional reduction analysis based on the expression of the top 10 genes identified that cells broadly clustered on the basis of TP53 mutation status and TP53 sgRNA enrichment (Fig. 6E; Supplementary Fig. S12E). Interestingly, most of the cell lines that enriched for TP53 sgRNAs despite having TP53 mutations, as identified in Fig. 5A, also clustered with the TP53 WT cells based on the expression of the top 10 genes, suggesting that the specific mutations in those cell lines do not abrogate the p53 function sufficiently to cause a phenotype, or that they are misrepresented. Apart from CDKN1A, and partly ZMAT3, the expression gene set list (Fig. 6E) does not overlap with genes identified to play a nonredundant role in the CRISPR–p53 response (Fig. 5E; Supplementary Table S9). Also, knockout cells of the two top hits; Eda2r and Ptchd4, behaved similarly to WT cells in response to CRISPR in a mixed cell population (Supplementary Figs. S12F and S12G). Taken together, these suggest that the upregulated genes are predominately an indication of active p53-mediated transcription in the cell, and not directly involved in the CRISPR response.
Figure 6.
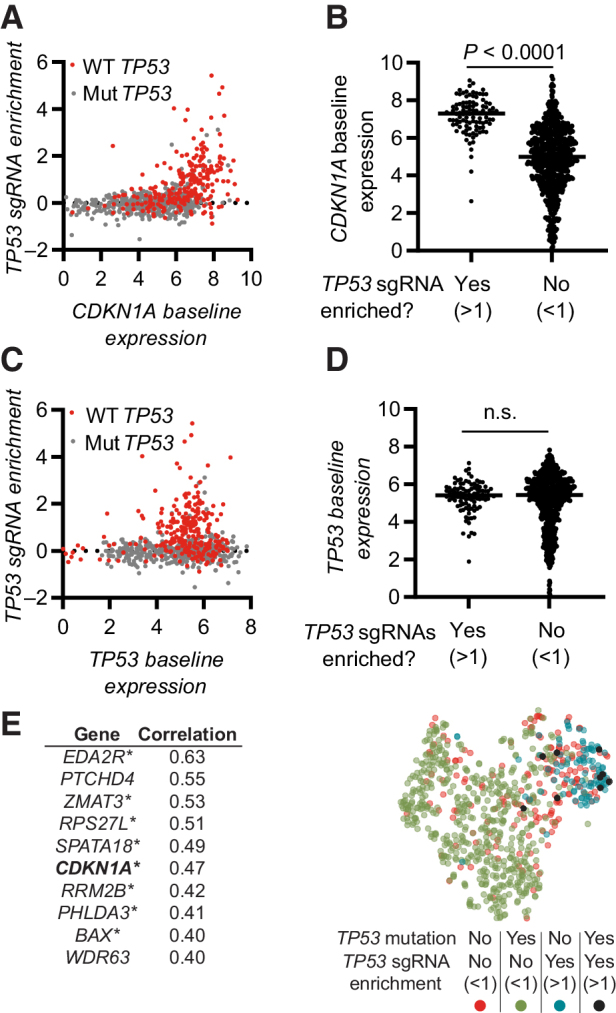
Gene expression patterns predicting if the CRISPR–p53 axis is active in a cell. A, Correlation of TP53 sgRNA enrichment with baseline CDKN1A expression. TP53 WT cells are indicated in red. B, Baseline CDKN1A expression in cells stratified on the basis of the enrichment of TP53 sgRNAs. C, Correlation of TP53 sgRNA enrichment with baseline TP53 expression. TP53 WT cells are indicated in red. D, Baseline TP53 expression in cells stratified on the basis of the enrichment of TP53 sgRNAs. E, Left, top 10 genes with expression correlating with enrichment of TP53 sgRNAs. *, genes identified as transcription factor target genes for p53. Right, tSNE dimensionality reduction analysis of cell based on expression of the 10 genes in E (left). Data include all available overlapping data in the Depmap CRISPR (Avana) 20Q4, Expression Public 20Q4. Each dot represents one cell line (A–D and E), n = 800. Statistical analysis based on unpaired t test (B and D) and calculated in Depmap (E). n.s., nonsignificant.
Discussion
In this study, we set out to improve the potential clinical use of CRISPR by exploring if, and how, cells with inactive p53 mutations are enriched as CRISPR is applied to a cell population. In line with studies by Happaniemi and colleagues (17) and Ihry and colleagues (18), we showed that cells with mutations in p53 are enriched as CRISPR is applied (Fig. 1F), and additionally identified that the enrichment correlates to the level of the induced DDR (Fig. 2), highlighting the induction of CDKN1A expression as a relevant parameter in sgRNA selection.
On the basis of our experimental data and analysis of the Depmap database, we could expand the number of proteins playing a nonredundant role in the CRISPR-induced DDR beyond p53, and identify a core CRISPR–p53 interactome, with genes that display a large level of physical interaction (Fig. 5E). This is of importance, since mutations or duplications of the identified genes, most of which are directly linked to cancer (44–53), have the potential to be enriched as CRISPR is applied. It should also be noted that cells infected with viruses that can suppress the p53 pathway also could gain a competitive advantage in the CRISPR setting. Our findings, thus, identify a set of genes that should be monitored in the clinical CRISPR setting and represent potential drug targets to modify the CRISPR–p53 response.
We observed that genes traditionally linked to p53-induced apoptosis were not identified to play a significant nonredundant role in the enrichment phenotype in either our experimental data, or the Depmap data set. However, we found that the use of siRNAs/sgRNAs to target pro-apoptotic Bak1 and Bax simultaneously, could partly suppress the enrichment of cells with TP53 mutations, but not when targeting only one of the two genes (Supplementary Fig. S5), thus showing the expected redundancy of these genes (39). In the CRISPR screen setting, where one gene is targeted in each cell, only genes that play a nonredundant role in a phenotype are identified, whereas processes where multiple redundant genes performing similar functionality are not identified. This could be seen as a drawback of our study, and for any traditional CRISPR or KO study. However, redundant genes are unlikely to be relevant as drug targets, and would not be enriched by CRISPR, and are thus of less relevance for the scope of this study.
Transient p53 inhibition has been proposed as a strategy to increase CRISPR efficiency (17, 18) and to retain the functionality of targeted cells (54, 58), without affecting genomic stability in the CRISPR context (54). Importantly, we identify that transient p53 inhibition using an siRNA, even when delivered at the same time as sgRNAs, can limit the enrichment of mutations in the CRISPR–p53 tumor suppressor interactome, including Chek2, Trp53, and Cdkn1a (Fig. 4I). As such, our data adds an additional argument supporting the use of transient p53 inhibition in the CRISPR setting. None of the other tested inhibitors showed a robust ability to suppress the enrichment of cells with Trp53 mutations, including Pifithrin-μ and cyclic Pifithrin-α, branded as p53 inhibitors. Several explanations for the absence of activity of the Pifithrin inhibitors exist, include potential stability issues, which could not be controlled for in our studies. Still, our observation is in line with a growing concern challenging the notion of Pifithrin molecules as broad p53 inhibitors (59). Accordingly, there is a need to develop improved p53 inhibitors for experimental and clinical use.
Using the Depmap data sets with extensive characterization of >800 human cancer cells lines, we were additionally able to identify a diverse baseline gene expression profile that predicted cells with an active CRISPR–p53 axis (as defined by cells that enriched for TP53 sgRNAs in a CRISPR screen setting, Fig. 6E). Interestingly, the baseline gene expression pattern also discriminated p53 WT cells and cells with p53 mutations with unexpected resolution. In contrast to the genes identified in the CRISPR–p53 interactome (Fig. 5E), that have a large degree of physical interaction, the genes identified to be upregulated in cells WT for p53, as well as enriching for TP53 sgRNAs, showed a low level of physical interaction and instead an extensive level of co-expression and p53 transcriptional control. Taken together this suggests that the expression of a panel of p53 target genes could be used as a potentially relevant assay to predict how specific cells will respond to p53-activating interventions.
The original observations leading to the identification of p53 were related to transformation induced by the SV40 polyomavirus (60). Although most focus in p53 research relates to cancer, it is noteworthy that SV40 and other viruses have evolved mechanisms to suppress the activity of p53, highlighting the diverse roles of p53. In this regard, we found it interesting that our CRISPR studies in Hox bone marrow cells suggested a role for type I IFN signaling, central to antiviral responses, in the triggered DDR. Although not fully explored here, we additionally identified that AMG232, and to some extent CRISPR, triggers the upregulation of type I IFN response genes in a completely p53 dependent matter (Supplementary Fig. S7A), which taken together experimentally links p53-dependent DDR and stereotypical antiviral responses.
In conclusion, the presented study data provide important details about p53 biology in the context of CRISPR-induced DNA damage and identify strategies that could be implemented to enable safer CRISPR use.
Authors' Disclosures
D.P. Lane reports grants from Swedish Research Council during the conduct of the study and grants from Swedish Research Council outside the submitted work. F. Wermeling reports grants from The Swedish Cancer Society, Swedish Research Council, and Karolinska Institutet during the conduct of the study. No disclosures were reported by the other authors.
Supplementary Material
Acknowledgments
The authors are grateful to Drs. Robert M. Anthony, Kate Jeffrey, Eduardo Villablanca, Laura Plant, and Bernhard Schmierer for their critical review of the manuscript, as well as to Drs. Michael Sundström, Lars Klareskog, Pavitra Kannan, Anna Fogdell-Hahn, Alexander Espinosa, Klas G. Wiman, Lisa Westerberg, and Richard Rosenquist Brandell for important suggestions. psPAX2 (Addgene #12260) and pMD2.G (Addgene #12259) were kind gifts from Dr. Didier Trono. The ER-Hoxb8 and EcoPac plasmids were kind gifts from Dr. Mark P. Kamps. The R-script used to represent individual sgRNAs in Supplementary Figs. S6A and S6B was a kind gift from Dr. Eric Shifrut. This work benefitted from data assembled by the DepMap Consortium, for which the authors are grateful. The authors acknowledge support from the National Genomics Infrastructure in Stockholm funded by Science for Life Laboratory, the Knut and Alice Wallenberg Foundation, and the Swedish Research Council, and SNIC/Uppsala Multidisciplinary Center for Advanced Computational Science for assistance with massively parallel sequencing and access to the UPPMAX computational infrastructure. This research was partly funded by grants from the Swedish Research Council (to F. Wermeling and D.P. Lane), the Swedish Cancer Society, Karolinska Institutet and Magnus Bergvalls stiftelse (to F. Wermeling), the China Scholarship Council (to L. Jiang and Y. Shen), and the Nanyang Technological University–Karolinska Institutet Joint PhD Program (to V.S. Iyer).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Contributions
L. Jiang: Conceptualization, formal analysis, investigation, visualization, methodology, writing–original draft, writing–review and editing. K. Ingelshed: Formal analysis, investigation. Y. Shen: Formal analysis, methodology, writing–review and editing. S.V. Boddul: Methodology, writing–review and editing. V.S. Iyer: Methodology, writing–review and editing. Z. Kasza: Methodology, writing–review and editing. S. Sedimbi: Methodology, writing–review and editing. D.P. Lane: Conceptualization, writing–original draft, writing–review and editing. F. Wermeling: Conceptualization, resources, supervision, writing–original draft, project administration.
References
- 1. Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Res 1991;51:6304–11. [PubMed] [Google Scholar]
- 2. Levine AJ. p53, the cellular gatekeeper for growth and division. Cell 1997;88:323–31. [DOI] [PubMed] [Google Scholar]
- 3. Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol 2007;8:275–83. [DOI] [PubMed] [Google Scholar]
- 4. Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science 1991;253:49–53. [DOI] [PubMed] [Google Scholar]
- 5. Kotler E, Segal E, Oren M. Functional characterization of the p53 "mutome. Mol Cell Oncol 2018;5:e1511207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Makarova KS, Wolf YI, Iranzo J, Shmakov SA, Alkhnbashi OS, Brouns SJJ, et al. Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol 2020;18:67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012;337:816–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-guided human genome engineering via Cas9. Science 2013;339:823–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Porteus MH. A new class of medicines through DNA Editing. N Engl J Med 2019;380:947–59. [DOI] [PubMed] [Google Scholar]
- 10. Frangoul H, Altshuler D, Cappellini MD, Chen YS, Domm J, Eustace BK, et al. CRISPR-Cas9 gene editing for sickle cell disease and beta-Thalassemia. N Engl J Med 2020. [DOI] [PubMed] [Google Scholar]
- 11. Ledford H. CRISPR gene therapy shows promise against blood diseases. Nature 2020;588:383. [DOI] [PubMed] [Google Scholar]
- 12. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017;543:113–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003;302:415–9. [DOI] [PubMed] [Google Scholar]
- 14. Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 2016;34:184–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016;529:490–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 2015;33:187–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med 2018;24:927–30. [DOI] [PubMed] [Google Scholar]
- 18. Ihry RJ, Worringer KA, Salick MR, Frias E, Ho D, Theriault K, et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat Med 2018;24:939–46. [DOI] [PubMed] [Google Scholar]
- 19. Merkle FT, Ghosh S, Kamitaki N, Mitchell J, Avior Y, Mello C, et al. Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature 2017;545:229–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ananthaswamy HN, Loughlin SM, Cox P, Evans RL, Ullrich SE, Kripke ML. Sunlight and skin cancer: inhibition of p53 mutations in UV-irradiated mouse skin by sunscreens. Nat Med 1997;3:510–4. [DOI] [PubMed] [Google Scholar]
- 21. Gao H, Wang LD, Zhou Q, Hong JY, Huang TY, Yang CS. p53 tumor suppressor gene mutation in early esophageal precancerous lesions and carcinoma among high-risk populations in Henan, China. Cancer Res 1994;54:4342–6. [PubMed] [Google Scholar]
- 22. Sozzi G, Miozzo M, Donghi R, Pilotti S, Cariani CT, Pastorino U, et al. Deletions of 17p and p53 mutations in preneoplastic lesions of the lung. Cancer Res 1992;52:6079–82. [PubMed] [Google Scholar]
- 23. Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014;20:1472–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen S, Wang Q, Yu H, Capitano ML, Vemula S, Nabinger SC, et al. Mutant p53 drives clonal hematopoiesis through modulating epigenetic pathway. Nat Commun 2019;10:5649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cardozo CM, Hainaut P. Viral strategies for circumventing p53: the case of severe acute respiratory syndrome coronavirus. Curr Opin Oncol 2021;33:149–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang GG, Calvo KR, Pasillas MP, Sykes DB, Hacker H, Kamps MP. Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8. Nat Methods 2006;3:287–93. [DOI] [PubMed] [Google Scholar]
- 27. Panda SK, Wigerblad G, Jiang L, Jimenez-Andrade Y, Iyer VS, Shen Y, et al. IL-4 controls activated neutrophil FcgammaR2b expression and migration into inflamed joints. Proc Natl Acad Sci U S A 2020;117:3103–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Panda SK, Boddul SV, Jimenez-Andrade GY, Jiang L, Kasza Z, Fernandez-Ricaud L, et al. Green listed-a CRISPR screen tool. Bioinformatics 2017;33:1099–100. [DOI] [PubMed] [Google Scholar]
- 29. Iyer VS, Jiang L, Shen Y, Boddul SV, Panda SK, Kasza Z, et al. Designing custom CRISPR libraries for hypothesis-driven drug target discovery. Comput Struct Biotechnol J 2020;18:2237–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang T, Yu H, Hughes NW, Liu B, Kendirli A, Klein K, et al. Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic Ras. Cell 2017;168:890–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 2014;15:554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dempster JM, Rossen J, Kazachkova M, Pan J, Kugener G, Root DE, et al. Extracting biological insights from the project achilles genome-scale CRISPR screens in cancer cell lines. bioRxiv 2019:720243.
- 33. Ghandi M, Huang FW, Jane-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019;569:503–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Karpinich NO, Tafani M, Rothman RJ, Russo MA, Farber JL. The course of etoposide-induced apoptosis from damage to DNA and p53 activation to mitochondrial release of cytochrome c. J Biol Chem 2002;277:16547–52. [DOI] [PubMed] [Google Scholar]
- 35. Canon J, Osgood T, Olson SH, Saiki AY, Robertson R, Yu D, et al. The MDM2 inhibitor AMG 232 demonstrates robust antitumor efficacy and potentiates the activity of p53-inducing cytotoxic agents. Mol Cancer Ther 2015;14:649–58. [DOI] [PubMed] [Google Scholar]
- 36. Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998;282:1497–501. [DOI] [PubMed] [Google Scholar]
- 37. Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 2003;302:1036–8. [DOI] [PubMed] [Google Scholar]
- 38. Alarcón R, Koumenis C, Geyer RK, Maki CG, Giaccia AJ. Hypoxia induces p53 accumulation through MDM2 down-regulation and inhibition of E6-mediated degradation. Cancer Res 1999;59:6046–51. [PubMed] [Google Scholar]
- 39. Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell 2000;6:1389–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bowden AR, Morales-Juarez DA, Sczaniecka-Clift M, Agudo MM, Lukashchuk N, Thomas JC, et al. Parallel CRISPR-Cas9 screens clarify impacts of p53 on screen performance. Elife 2020;9:e55325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Geisinger JM, Stearns T. CRISPR/Cas9 treatment causes extended TP53-dependent cell cycle arrest in human cells. Nucleic Acids Res 2020;48:9067–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brown KR, Mair B, Soste M, Moffat J. CRISPR screens are feasible in TP53 wild-type cells. Mol Syst Biol 2019;15:e8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lane DP. Cancer. p53, guardian of the genome. Nature 1992;358:15–6. [DOI] [PubMed] [Google Scholar]
- 44. Cuella-Martin R, Oliveira C, Lockstone HE, Snellenberg S, Grolmusova N, Chapman JR. 53BP1 integrates DNA repair and p53-dependent cell fate decisions via distinct mechanisms. Mol Cell 2016;64:51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bell DW, Varley JM, Szydlo TE, Kang DH, Wahrer DC, Shannon KE, et al. Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome. Science 1999;286:2528–31. [DOI] [PubMed] [Google Scholar]
- 46. Choi M, Kipps T, Kurzrock R. ATM mutations in cancer: Therapeutic implications. Mol Cancer Ther 2016;15:1781–91. [DOI] [PubMed] [Google Scholar]
- 47. Cazier JB, Rao SR, McLean CM, Walker AK, Wright BJ, Jaeger EE, et al. Whole-genome sequencing of bladder cancers reveals somatic CDKN1A mutations and clinicopathological associations with mutation burden. Nat Commun 2014;5:3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 1992;358:80–3. [DOI] [PubMed] [Google Scholar]
- 49. Momand J, Jung D, Wilczynski S, Niland J. The MDM2 gene amplification database. Nucleic Acids Res 1998;26:3453–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bulavin DV, Demidov ON, Saito S, Kauraniemi P, Phillips C, Amundson SA, et al. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat Genet 2002;31:210–5. [DOI] [PubMed] [Google Scholar]
- 51. Riemenschneider MJ, Buschges R, Wolter M, Reifenberger J, Bostrom J, Kraus JA, et al. Amplification and overexpression of the MDM4 (MDMX) gene from 1q32 in a subset of malignant gliomas without TP53 mutation or MDM2 amplification. Cancer Res 1999;59:6091–6. [PubMed] [Google Scholar]
- 52. Daizumoto K, Yoshimaru T, Matsushita Y, Fukawa T, Uehara H, Ono M, et al. A DDX31/Mutant-p53/EGFR axis promotes multistep progression of muscle-invasive Bladder cancer. Cancer Res 2018;78:2233–47. [DOI] [PubMed] [Google Scholar]
- 53. Song MS, Salmena L, Carracedo A, Egia A, Lo-Coco F, Teruya-Feldstein J, et al. The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature 2008;455:813–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schiroli G, Conti A, Ferrari S, Volpe LD, Jacob A, Albano L, et al. Precise gene editing preserves hematopoietic stem cell function following transient p53-mediated DNA damage response. Cell Stem Cell 2019;24:551–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Donehower LA, Soussi T, Korkut A, Liu Y, Schultz A, Cardenas M, et al. Integrated analysis of TP53 gene and pathway alterations in The Cancer Genome Atlas. Cell Rep 2019;28:1370–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Janic A, Valente LJ, Wakefield MJ, Di Stefano L, Milla L, Wilcox S, et al. DNA repair processes are critical mediators of p53-dependent tumor suppression. Nat Med 2018;24:947–53. [DOI] [PubMed] [Google Scholar]
- 57. Bieging-Rolett KT, Kaiser AM, Morgens DW, Boutelle AM, Seoane JA, Van Nostrand EL, et al. Zmat3 is a key splicing regulator in the p53 tumor suppression program. Mol Cell 2020;80:452–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ferrari S, Jacob A, Beretta S, Unali G, Albano L, Vavassori V, et al. Efficient gene editing of human long-term hematopoietic stem cells validated by clonal tracking. Nat Biotechnol 2020;38:1298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhu J, Singh M, Selivanova G, Peuget S. Pifithrin-α alters p53 post-translational modifications pattern and differentially inhibits p53 target genes. Sci Rep 2020;10:1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer 2009;9:749–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.