

Hypoxia induces HIF2α-mediated overexpression of dihydropyrimidine dehydrogenase in TAMs, leading to chemoresistance to 5-FU in colon cancers.
Abstract
Colorectal adenocarcinoma is a leading cause of death worldwide, and immune infiltration in colorectal tumors has been recognized recently as an important pathophysiologic event. In this context, tumor-associated macrophages (TAM) have been related to chemoresistance to 5-fluorouracil (5-FU), the first-line chemotherapeutic agent used in treating colorectal cancers. Nevertheless, the details of this chemoresistance mechanism are still poorly elucidated. In the current study, we report that macrophages specifically overexpress dihydropyrimidine dehydrogenase (DPD) in hypoxia, leading to macrophage-induced chemoresistance to 5-FU via inactivation of the drug. Hypoxia-induced macrophage DPD expression was controlled by HIF2α. TAMs constituted the main contributors to DPD activity in human colorectal primary or secondary tumors, while cancer cells did not express significant levels of DPD. In addition, contrary to humans, macrophages in mice do not express DPD. Together, these findings shed light on the role of TAMs in promoting chemoresistance in colorectal cancers and identify potential new therapeutic targets.
Significance:
Hypoxia induces HIF2α-mediated overexpression of dihydropyrimidine dehydrogenase in TAMs, leading to chemoresistance to 5-FU in colon cancers.
Introduction
Colorectal cancers are a leading cause of death worldwide and constitute the third cancer-related cause of death in the United States (1). Chemotherapy is one of the tools used to treat these tumors; however, some patients do not respond well to treatment, resulting in poor prognosis. This chemoresistance is caused by various mechanisms such as drug inactivation, drug efflux from targeted cells, and modifications of target cells (2, 3). Interestingly, the importance of tumor microenvironment in chemoresistance has recently garnered attention. The tumor immune microenvironment, notably through its innate immune part that is mainly composed of tumor-associated macrophages (TAM), deserves particular attention (4, 5). TAMs have been associated with bad prognosis in the case of various solid tumors (6) and have been shown to orchestrate a defective immune response to tumors (7). It has been suggested that TAMs are reprogrammed by cancerous cells to secondarily become supporting elements of tumor growth (8). The involvement of macrophages in colorectal cancer has been controversial, and only recently has the association between CD163+ TAMs and a poorer prognosis been recognized (9, 10). Their implication in chemoresistance, particularly against 5-fluorouracil (5-FU), a first-line chemotherapy in colorectal cancer, has been reported previously (11, 12). This suggests that targeting macrophages could be an effective way to increase treatment efficiency. However, the precise mechanisms by which TAMs participate in creating chemoresistance in human colorectal tumors are still poorly understood. The underappreciated impact of hypoxia on macrophage biology (13) and the increasingly recognized role of hypoxia in resistance to anticancer treatments and cancer relapse (14), lead us to reassess the role of hypoxic macrophages in chemoresistance. On the basis of the abundance of macrophages in colorectal cancer, we hypothesized that hypoxia could directly modulate macrophage involvement in 5-FU resistance.
Materials and Methods
Cell culture
RAW264.7, CT-26, RKO, and HT-29 were purchased from ATCC. RAW were maintained in high-glucose DMEM (Gibco) supplemented with 10% FBS (Gibco) at 37°C, CT-26 and RKO were maintained in RPMI (Gibco) supplemented with 10% FBS (Gibco) at 37°C, and HT-29 were maintained in McCoy's medium (Gibco) supplemented with 10% FBS (Gibco) at 37°C. All cells were routinely tested for Mycoplasma contamination using MycoAlert detection kit (Lonza). All cells have been used in the following year of their reception.
Human samples
Human blood samples from healthy deidentified donors were obtained from EFS (French national blood service) as part of an authorized protocol (CODECOH DC-2018–3114). Donors gave signed consent for use of their blood in this study. Tumor samples were obtained from the department of pathology of the university hospital of Grenoble as part of a declared sample collection AC-2014-2949. Patient selection criteria were a diagnostic of colorectal adenocarcinoma and tissue samples availability for the primary tumor and liver metastasis. Clinical characteristics of the patients are reported in the table (Supplementary Table). All patients gave their signed consent for this study as part of an authorized protocol (INDS MR4916160120) and the study was conducted in accordance with the Declaration of Helsinki.
Animals
Eight-week-old Balb/c female mice were obtained from Charles River. Animals were housed and bred at Plateforme de Haute Technologie Animale (PHTA) UGA core facility (Grenoble, France), EU0197, agreement C38-51610006, under specific pathogen–free conditions, temperature-controlled environment with a 12-hour light/dark cycle and ad libitum access to water and diet. Animal housing and procedures were conducted in accordance with the recommendations from the Direction des Services Vétérinaires, Ministry of Agriculture of France, according to European Communities Council Directive 2010/63/EU and according to recommendations for health monitoring from the Federation of European Laboratory Animal Science Associations. Protocols involving animals were reviewed by the local ethic committee “Comité d'Ethique pour l'Expérimentation Animale no. #12, Cometh-Grenoble” and approved by the Ministry of Research under the authorization number (January 2020) APAFIS#22660-2019103110209599.
Human macrophage differentiation from monocytes
Monocytes were isolated from leukoreduction system chambers of healthy EFS donors using differential centrifugation (Histopaque 1077, Sigma) to obtain peripheral blood mononuclear cells. CD14+ microbeads (Miltenyi Biotec) were used to select monocytes according to the manufacturer's instructions. Monocytes were plated in RPMI (Life Technologies) supplemented with 10% SAB (Sigma), 10 mmol/L HEPES (Life Technologies), MEM Non-essential amino acids (Life Technologies) and 25 ng/mL MCSF (Miltenyi Biotec). Differentiation was obtained after 6 days of culture. Hypoxic cultures were performed in a hypoxic chamber authorizing an oxygen partial pressure control (HypoxyLab).
Bone marrow–derived macrophage differentiation
Bone marrow was extracted from the femurs of Balb/c mice in RPMI and then filtered by a 70 μm cell strainer. Cells were washed in RPMI and cultured in RPMI (Gibco) supplemented with 10% of FBS (Gibco) and mouse MCSF at 25 ng/mL (Miltenyi Biotec) for 6 days. Medium was refreshed at day 3. Differentiation was assessed by flow cytometry through membrane expression analysis of F4/80.
Macrophage conditioned medium
Macrophages were cultured at 1 × 106 cells per well in 12-well plates with RPMI supplemented with 10% SAB with DMSO (vehicle), Gimeracil (Sigma-Aldrich) 1 μg/mL, 5-FU (ACCORD HEALTHCARE) 0.1 μg/mL, 5-FU 1 μg/mL, 5-FU 0.1 μg/mL + Gimeracil 1 μg/mL or 1 μg/mL + Gimeracil 1 μg/mL during 24 hours in normoxia and hypoxia. The produced macrophage conditioned medium (MCM) was added for 48 hours to HT-29 and RKO, which were plated previously at 3 × 105 cells per well during 24 hours. Then cancer cells were collected, counted and the mortality rate assessed by flow cytometry (Annexin V and 7-AAD).
RNAi
siRNAs (GE Dharmacon) were transfected at a final concentration of 50 nmol/L using Lipofectamine (RNAiMAx, Life Technologies).
Expression of human dihydropyrimidine dehydrogenase in mice macrophages
RAW264.7 were transduced using lentivirus particles with the open reading frame (ORF) of the human dihydropyrimidine dehydrogenase (DPD) gene (mGFP-tagged) inserted in a pLenti-C-mGFP-P2A-Puro plasmid (Origen Technologies). Control RAW were obtained using the lentivirus particles containing the plasmid without the DPD ORF sequence (pLenti-C-mGFP-P2A-Puro).
RNA isolation and qPCR analysis for gene expression
Cells were directly lyzed and RNA was extracted using the NucleoSpin RNA kit components (Macherey Nagel) according to the manufacturer's instructions. Reverse transcription was performed using the iScript Ready-to-use cDNA supermix components (Bio-Rad). qPCR was then performed with the iTaq universal SYBR green supermix components (Bio-Rad) on a CFX96 (Bio-Rad). Quantification was performed using the ΔΔCt method.
Immunoblotting
Cells were lyzed in RIPA buffer supplemented with antiprotease inhibitors (AEBSF 4 mmol/L, Pepstatine A 1 mmol/L, and Leupeptine 0.4 mmol/L; Sigma-Aldrich) and HIF-hydroxylase inhibitor (DMOG 1 mmol/L, Sigma-Aldrich). Proteins were quantified by BCA assay (Thermo Fisher Scientific) and 15 μg of total protein were run on SDS-PAGE gels. Proteins were transferred from SDS-PAGE gels to polyvinylidene difluoride membrane (Bio-Rad), blocked with TBS-Tween supplemented with 5% milk, primary antibodies were incubated at 1 μg/mL overnight 4°C. After washing with TBS, the membrane was incubated with a horseradish peroxidase-conjugated secondary antibody (Jackson Immunoresearch). Signal was detected by chemoluminescence (Chemi-Doc Imaging System, Bio-Rad) after exposition to West Pico ECL (Thermo Fisher Scientific).
Proteomics
Cells are directly lyzed in Laemmli buffer and prepared and analyzed as described previously (13). Briefly, the protein equivalent of 300,000 cells for each sample was loaded on NuPAGE Bis-Tris 4%–12% acrylamide gels (Life Technologies). Electrophoretic migration was controlled to allow each protein sample to be split into six gel bands. Gels were stained with R-250 Coomassie blue (Bio-Rad) before excising protein bands. Gel slices were washed then dehydrated with 100% acetonitrile (Merck Millipore), incubated with 10 mmol/L DTT (dithiothreitol, Merck Millipore), followed by 55 mmol/L iodoacetamide (Merck Millipore) in the dark. Alkylation was stopped by adding 10 mmol/L DTT in 25 mmol/L ammonium bicarbonate. Proteins were digested overnight at 37°C with Trypsin/Lys-C Mix (Promega) according to manufacturer's instructions. After extraction, fractions were pooled, dried, and stored at −80°C until further analysis. The dried extracted peptides were resuspended and analyzed by online nano-LC (Ultimate 3000, Thermo Fisher Scientific) directly linked to an impact IITM Hybrid Quadrupole Time of-Flight (QTOF) instrument fitted with a CaptiveSpray ion source (Bruker Daltonics). All data were analyzed using MaxQuant software (version 1.5.2.8) and the Andromeda search engine. The FDR was set to 1% for both proteins and peptides, and a minimum length of seven amino acids was set. MaxQuant scores peptide identifications based on a search with an initial permissible mass deviation for the precursor ion of up to 0.07 Da after time-dependent recalibration of the precursor masses. Fragment mass deviation was allowed up to 40 ppm. The Andromeda search engine was used to match MS-MS spectra against the Uniprot human database (https://www.uniprot.org/). Enzyme specificity was set as C terminal to Arg and Lys, cleavage at proline bonds and a maximum of two missed cleavages were allowed. Carbamidomethylation of cysteine was selected as a fixed modification, whereas N-terminal protein acetylation and methionine oxidation were selected as variable modifications. The “match between runs” feature of MaxQuant was used to transfer identification information to other LC/MS-MS runs based on ion masses and retention times (maximum deviation 0.7 minutes); this feature was also used in quantification experiments. Quantifications were performed using the label-free algorithms. A minimum peptide ratio counts of two and at least one “razor peptide” were required for quantification. The label-free quantification metric was used to perform relative quantification between proteins identified in different biological conditions, protein intensities were normalized on the basis of the MaxQuant “protein group.txt” output (reflecting a normalized protein quantity deduced from all peptide intensity values). Potential contaminants and reverse proteins were strictly excluded from further analysis. Three analytic replicates from three independent biological samples (donors) were analyzed for each normoxic and hypoxic conditions. Missing values were deduced from a normal distribution (width: 0.3; down shift: 1.8) using the Perseus (version 1.5.5.3) after data acquisition package contained in MaxQuant (www.maxquant.org). Data were further analyzed using JMP software (v.13.0.0, SAS Institute Inc.). Proteins were classed according to the paired Welch test difference (difference between the mean value for triplicate MS-MS analyses for the two compared conditions), and the median fold change between the two compared conditions.
Immunochemistry
A total of 3-μm-thick consecutive tissue sections were prepared from formalin-fixed and paraffin-embedded tissues. Deparaffinization, rehydration, antigen retrieval, and peroxidase blocking were performed on a fully automated system BENCHMARK ULTRA (Roche) according to manufacturer recommendations. The sections were incubated with the following primary antibodies: anti-CD68 clone Kp1 (Dako) and anti-DPD (Thermo Fisher Scientific). Revelation was performed using the Ultraview DAB revelation kit (Roche). Nuclei were counterstained with hematoxylin solution (Dako). Images were captured using an APERIO ATS scanner (Leica).
Immunofluorescence
Formalin-fixed and paraffin-embedded human tissue samples were sectioned at 3 μm thickness. Samples were deparaffinized by xylene and hydrated by baths of decreasing concentrations of ethanol. Antigen retrieval was achieved using IHC-TekTM Epitope Retrieval Steamer Set (IW-1102, IHCworld) in IHC-TekTM Epitope Retrieval (IW-1100, IHCworld) for 40 minutes. Nonspecific binding sites were blocked by 1% BSA in PBS. Samples were incubated with the primary antibodies: Monoclonal Mouse anti-Human CD68 clone PG-M1 at 0.4 μg/mL, Mouse anti-Human CD163 clone EDHu-1 at 10 μg/mL, anti-Human HIF2α at 4 μg/mL, and DPD polyclonal antibody at 3 μg/mL for 1 hour at room temperature followed by an incubation of secondary antibodies: Alexa Fluor 488 goat anti-mouse IgG (H+L) and Alexa Fluor 546 goat anti-rabbit IgG (H+L) both at 4 μg/mL for 30 minutes at room temperature. Nuclei were stained by Hoechst 33342 at 5 μg/mL for 5 minutes at room temperature. Images were captured under ×20 magnification using ApoTome microscope (Carl Zeiss) equipped with a camera AxioCam MRm, collected by AxioVision software and analyzed using ImageJ software.
Flow cytometry
Flow cytometry data were acquired on an Accuri C6 (BD) flow cytometer. The reagents used were: AnnexinV-FITC, mouse anti-F4/80-PE clone REA126 and mouse anti-human CD11b-FITC from Miltenyi Biotech and 7-AAD staining solution from BD Pharmingen. Doublet cells were gated out by comparing forward scatter signal height (FSC-H) and area (FSC-A). At least 10,000 events were collected in the analysis gate. Median fluorescence intensity was determined using Accuri C6 software (BD).
Cancer cell line mRNA DPYD expression
Cancer cell line gene expression data were collected from the Cancer Cell Line Encyclopedia (CCLE; https://portals.broadinstitute.org/ccle). Briefly, sequencing was performed on the Illumina HiSeq 2000 or HiSeq 2500 instruments, with sequence coverage of no less than 100 million paired 101 nucleotides-long reads per sample. RNA-sequencing (RNA-seq) reads were aligned to the B37 version of human genome using TopHat version 1.4. Gene and exon-level RPKM values were calculated using pipeline developed for the GTEx project (15, 16). The cell lines were classified on the basis of their tissue origin resulting in 24 different groups. The number of cell lines in each group is indicated. The histogram plot was generated using the JMP software (SAS).
Mice and human mRNA DPYD expression analysis
Genevestigator 7.5.0 (https://genevestigator.com/gv/) is a search engine that summarizes datasets in metaprofiles. GENEVESTIGATOR integrates manually curated and quality-controlled gene expression data from public repositories (17). In this study, the Condition Tools Search was used to obtain DPD mRNA levels obtained from human (Homo sapiens) and mice (Mus musculus) in various anatomic parts. Mean of logarithmic level of expression obtained from AFFIMETRIX expression microarrays was used to generate a cell plot from selected results related to macrophages and monocytes. The lowest and the highest expression results in both series (human and mice) were used to evaluate the expression level in the dataset. Cell plot was generated using the JMP software (SAS).
DPD activity measurements
Because DPD is involved in the hydrogenation of uracil (U) into dihydrouracil (UH2), DPD activity was indirectly evaluated in the cell culture supernatants by determining the concentration of U and its metabolite UH2 followed by the calculation of the UH2/U ratio. These analyses were performed in the Pharmacology Laboratory of Institut Claudius-Regaud (France) using an high performance liquid chromatography (HPLC) system composed of Alliance 2695 and diode array detector 2996 (Waters), according to a previously described method (18). Uracil (U), dihydrouracil (UH2), 5-FU, ammonium sulfate 99%, acetonitrile (ACN) gradient chromasolv for HPLC and 2-propanol were purchased from Sigma. Ethyl acetate Scharlau was of HPLC grade and purchased from ICS. Water from Milli-Q Advantage A10 and MultiScreen-HV 96-well plates were used (Merck Millipore). Calibration ranges were 3.125–200 ng/mL for U and 25–500 ng/mL for UH2 and 5-FU (5 μg/mL) was used as an internal standard. We have validated that gimeracil does not interfere with 5-FU measurements, using calibrated 5-FU samples with and without gimeracil. For the 5-FU kinetics experiments, no internal standard was added to the samples and the amount of 5-FU in the supernatant was quantified by the peak area corresponding to 5-FU.
Quantification and statistical analysis
Statistics were performed using Graph Pad Prism 7 (Graph Pad Software Inc). When two groups were compared, we used a two-tailed Student t test for a normal distribution and a Mann–Whitney nonparametric test otherwise. When more than two groups were compared, we used a one-way ANOVA analysis with a Tukey post hoc test. Tumor growth curves were analyzed using two-ways ANOVA using the open access TumorGrowth software (19). All error bars represent means with SEM. All group numbers and explanation of significant values are presented within the figure legends.
Data availabilty
All mass spectrometry proteomics data were deposited on the Proteome- Xchange Consortium website (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository, dataset identifier: PXD006354.
See Supplementary Materials and Methods for further details.
Results
Macrophages confer a chemoresistance to 5-FU in a low-oxygen environment
To evaluate the effect that tumor-infiltrating macrophages have on chemotherapy, we analyzed the impact of the coculture with macrophages on cancer cells growth. We chose two human colorectal cancer cell lines, sensitive to 5-FU, differing from their genetic background to avoid any specific genetic cell line involvement: RKO (p53 and APC WT) and HT-29 (p53 and APC mutated) cells. To examine the role of oxygen, we performed these experiments in normoxia (N = 18.6% O2) and in hypoxia (H = 25 mm Hg 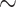 3% O2). We observed that in hypoxia, monocyte-derived macrophages (MDM) confer a chemoresistance toward 5-FU that is not observed in normoxia (Fig. 1A). To determine whether MDM act directly on 5-FU, we used MCM containing 5-FU or none. It has been previously reported that macrophages are resistant to 5-FU (20). Nonconditioned 5-FU strongly inhibited HT-29 and RKO proliferation independently of oxygen concentration (Fig. 1B and C). MCM without 5-FU had little effect on the proliferation of HT-29 (Fig. 1B) and RKO (Fig. 1C) cells. Meanwhile, MCM with 5-FU at 1 μg/mL inhibited cell growth in normoxia but not in hypoxia, advocating for a protection against 5-FU inhibition mediated by hypoxic MDM (Fig. 1B and C). As RKO cells were found sensitive to a lower concentration of 5-FU (0.1 μg/mL), MDM were able to protect RKO against 5-FU at this concentration, not only in hypoxia but also in normoxia (Fig. 1C). This observation led us to consider an inactivation mechanism of 5-FU driven by macrophages with an increased efficiency in hypoxia. A previously proposed mechanism for macrophage-induced chemoresistance in colorectal cancer was related to their ability to secrete IL6, leading to an inhibition of cancer cell apoptosis (11). We first tried to verify whether we could confirm the presence of IL6 in our conditioned medium (CM) by human MDMs in normoxia and in hypoxia and found no spontaneous secretion (<10 pg/mL) of IL6 (Supplementary Fig. S1A). As the conditioning by MDM provided a complete protection, we then hypothesized that a direct action of macrophages on 5-FU was the likely mechanism. To obtain a molecular explanation of the differing effect under various oxygen concentrations, we performed a proteomic analysis of human macrophages in hypoxia compared with normoxia. Our quantitative proteomic approach revealed that DPD is strongly overexpressed in hypoxia (Fig. 1D). DPD (coded by the DPYD gene) is the rate-limiting enzyme of the pyrimidine degradation pathway. DPD adds two hydrogen atoms to uracil, with NADPH as an obligatory cofactor, leading to dihydrouracil, which is secondarily degraded to β-alanine under the control of DPYS and UPB1 (Fig. 1E). 5-FU is a fluorinated analog of uracil reduced by DPD to 5-fluorodihydrouracil, an inactive compound (Fig. 1F). We confirmed through immunoblotting the increased expression of DPD in hypoxia when compared with the basal expression in normoxia (Supplementary Fig. S1B). Because the putative action of DPD on 5-FU is related to its enzymatic activity, we also confirmed that DPD expression in hypoxic MDM was functional, leading to an increase of the dihydrouracil to uracil ratio in the extracellular medium (Fig. 1G). To precise the role of macrophages in the 5-FU chemoresistance, we analyzed DPD expression in RKO and HT-29 cancer cells. We found no significant level of expression of DPD at the protein level, neither in HT-29 nor in RKO cells, irrespective of the oxygen concentration (Supplementary Fig. S2A and S2B). Furthermore, we found a downregulation of the DPYD mRNA in HT-29 and in RKO, emphasizing a defective transcription of the DPYD gene (Supplementary Fig. S2C). It has been reported that the transcription factor PU.1 drives the expression of DPYD and that EZH2 is responsible for the histone H3K27 trimethylation at the DPYD promotor site, leading to its downregulation in colon cancer cells (21). We thus confirmed that the pharmacologic inhibition of the methyltransferase EZH2 by the specific inhibitor GSK126 led to a detectable level of DPYD mRNA in RKO cells (Supplementary Fig. S2D).
3% O2). We observed that in hypoxia, monocyte-derived macrophages (MDM) confer a chemoresistance toward 5-FU that is not observed in normoxia (Fig. 1A). To determine whether MDM act directly on 5-FU, we used MCM containing 5-FU or none. It has been previously reported that macrophages are resistant to 5-FU (20). Nonconditioned 5-FU strongly inhibited HT-29 and RKO proliferation independently of oxygen concentration (Fig. 1B and C). MCM without 5-FU had little effect on the proliferation of HT-29 (Fig. 1B) and RKO (Fig. 1C) cells. Meanwhile, MCM with 5-FU at 1 μg/mL inhibited cell growth in normoxia but not in hypoxia, advocating for a protection against 5-FU inhibition mediated by hypoxic MDM (Fig. 1B and C). As RKO cells were found sensitive to a lower concentration of 5-FU (0.1 μg/mL), MDM were able to protect RKO against 5-FU at this concentration, not only in hypoxia but also in normoxia (Fig. 1C). This observation led us to consider an inactivation mechanism of 5-FU driven by macrophages with an increased efficiency in hypoxia. A previously proposed mechanism for macrophage-induced chemoresistance in colorectal cancer was related to their ability to secrete IL6, leading to an inhibition of cancer cell apoptosis (11). We first tried to verify whether we could confirm the presence of IL6 in our conditioned medium (CM) by human MDMs in normoxia and in hypoxia and found no spontaneous secretion (<10 pg/mL) of IL6 (Supplementary Fig. S1A). As the conditioning by MDM provided a complete protection, we then hypothesized that a direct action of macrophages on 5-FU was the likely mechanism. To obtain a molecular explanation of the differing effect under various oxygen concentrations, we performed a proteomic analysis of human macrophages in hypoxia compared with normoxia. Our quantitative proteomic approach revealed that DPD is strongly overexpressed in hypoxia (Fig. 1D). DPD (coded by the DPYD gene) is the rate-limiting enzyme of the pyrimidine degradation pathway. DPD adds two hydrogen atoms to uracil, with NADPH as an obligatory cofactor, leading to dihydrouracil, which is secondarily degraded to β-alanine under the control of DPYS and UPB1 (Fig. 1E). 5-FU is a fluorinated analog of uracil reduced by DPD to 5-fluorodihydrouracil, an inactive compound (Fig. 1F). We confirmed through immunoblotting the increased expression of DPD in hypoxia when compared with the basal expression in normoxia (Supplementary Fig. S1B). Because the putative action of DPD on 5-FU is related to its enzymatic activity, we also confirmed that DPD expression in hypoxic MDM was functional, leading to an increase of the dihydrouracil to uracil ratio in the extracellular medium (Fig. 1G). To precise the role of macrophages in the 5-FU chemoresistance, we analyzed DPD expression in RKO and HT-29 cancer cells. We found no significant level of expression of DPD at the protein level, neither in HT-29 nor in RKO cells, irrespective of the oxygen concentration (Supplementary Fig. S2A and S2B). Furthermore, we found a downregulation of the DPYD mRNA in HT-29 and in RKO, emphasizing a defective transcription of the DPYD gene (Supplementary Fig. S2C). It has been reported that the transcription factor PU.1 drives the expression of DPYD and that EZH2 is responsible for the histone H3K27 trimethylation at the DPYD promotor site, leading to its downregulation in colon cancer cells (21). We thus confirmed that the pharmacologic inhibition of the methyltransferase EZH2 by the specific inhibitor GSK126 led to a detectable level of DPYD mRNA in RKO cells (Supplementary Fig. S2D).
Figure 1.

Macrophages confer a chemoresistance to 5-FU in hypoxia. A, Top left, induction of death in RKO cells by 5-FU in a cocultured assay with macrophages in normoxia (blue) and hypoxia (red). 5-FU was used at 1 μg/mL. Dead cancer cells were defined as CD11b-AnnexinV+7-AAD± cells by flow cytometry. The gating strategy is represented on the bottom left. Dead cell quantification is represented on the right (n = 4). B, Growth inhibition of HT-29 cells by MCM(Ø), 5-FU, and CM(5-FU) in normoxia (blue), and in hypoxia (red), 5-FU was used at 1 μg/mL (n = 3). C, Growth inhibition of RKO cells by MCM(Ø), 5-FU, and MCM (5-FU) in normoxia (blue) and in hypoxia (red) 5-FU was used at 0.1 and 1 μg/mL (n = 3). D, Protein heatmap of macrophages in hypoxia and normoxia. Proteins were selected by a fold change > 2 and P < 0.01. Proteins were organized according to descending mean z-score of hypoxic proteins. E, Schematic presentation of the rate-limiting steps of the pyrimidine degradation pathway involving DPD. F, Chemical structures of uracil and 5-FU. G, Dihydrouracil/uracil ratio measured by HPLC in the macrophage supernatant from macrophages cultured in normoxia and hypoxia (n = 3). Statistical significance was determined using a one-way ANOVA analysis with Tukey post hoc test (A–C). Statistical significance was performed using a two-tailed paired t test (G). Error bars, mean 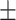 SEM.*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, nonsignificant.
SEM.*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, nonsignificant.
Chemoresistance to 5-FU is driven by increased DPD activity in hypoxic macrophages
To confirm the inactivation of 5-FU by macrophage's DPD and its potential clinical relevance, we analyzed the kinetics of 5-FU degradation in normoxia and hypoxia. As 5-FU is a small molecule, its diffusion in tissues is quite high. Indeed, following 4 days of oral ingestion of 5-FU, the plasmatic concentration was found to be approximately 1 μg/mL (22). The mean 5-FU concentration was 0.411 ± 0.381 (μg/g of tissue) in the tumor portions of the specimens and 0.180 ± 0.206 (μg/g of tissue) in the normal portions in colorectal cancers (23). As TAMs represent 2%–10% of cells in colorectal cancer tumors, especially localized in the invasion front (9) and because 1 g of tissue typically contains approximately 108 cells (24), a reasonable estimated ratio in colorectal cancer is approximately 106 macrophages/g of tissue. According to 5-FU reported concentration, an estimated relevant ratio in colorectal cancer is 1 μg of 5-FU/106 macrophages. We interestingly found that 2 μg of 5-FU could be eliminated by 106 MDM in hypoxia in less than 24 hours and that normoxic MDM were unable to completely eliminate this quantity in 48 hours (Fig. 2A). We validated that 5-FU degradation was due to DPD catalytic activity using gimeracil, a specific inhibitor of DPD (Fig. 2B and C). 5-FU induced death in HT-29 and RKO cells irrespective of the concentration of oxygen and the presence of gimeracil, demonstrating that no significant DPD activity is present in these cells (Fig. 2D and F; Supplementary Fig. S2A and S2B). Whereas MCM with 5-FU at 1 μg/mL induced cell death in HT-29 cells in normoxia, its cytotoxic effect dramatically decreased in hypoxia and this was reverted by gimeracil inhibition of DPD activity in MDM (Fig. 2D). Following these results, 5-FU chemoresistance appears to be based solely on DPD activity in macrophages promoted by hypoxia. We observed a similar result using a three-dimensional (3D) tumor model growth of HT-29 exposed to MCM (Fig. 2E; Supplementary Fig. S3A), and we confirmed the generality of this mechanism in RKO cells (Fig. 2F). To confirm that oxygen was the main factor controlling DPD expression in hypoxia, we verified that DPD was not induced or repressed by 5-FU itself (Fig. 2G). We also explored whether cancerous cells can modulate DPD expression in macrophages. Using a transwell coculture assay between cancerous cells and macrophages, we did not find any modulation in DPD expression in MDM (Fig. 2H). Similarly, we observed no induction of DPD expression in cancer cells when exposed to MCM (Supplementary Fig. S3B). In addition, we further confirmed that when applying medium conditioned from MDM with DPD loss of function through siRNA depletion, the cancer cell sensitivity to 5-FU was restored (Fig. 2I). To assess the efficiency of DPD degradation of 5-FU, we also performed a direct coculture assay between RKO cancerous cells and MDM. We found that MDM protected RKO cells from 5-FU in a DPD enzymatic activity dependent manner. Indeed, DPD inhibition by gimeracil restored RKO sensitivity to 5-FU (Supplementary Fig. S3C).
Figure 2.

Chemoresistance to 5-FU is driven by DPD activity in macrophages. A, Kinetics of 5-FU degradation by macrophages obtained by HPLC. 5-FU initial concentration was 1 μg/mL. Normoxia, blue; hypoxia, red. 5-FU without macrophages was stable during the 48 hours period of study in normoxia (full black circle) and in hypoxia (empty black circle; n = 3). B, Chemical structure of gimeracil, a specific inhibitor of DPD. C, 5-FU degradation due to DPD activity in macrophages inhibited by gimeracil at 48 hours. 5-FU initial concentration was 1 μg/mL. Normoxia, blue; hypoxia, red (n = 3). D, Induction of death in HT-29 in normoxia (blue) and in hypoxia (red) by nonconditioned medium (square) and conditioned medium (triangle). Dead cells were defined as AnnexinV+ cells in flow cytometry. 5-FU was used at 1 μg/mL and gimeracil at 1 μg/mL (n = 4). E, Inhibition of growth and death induction in 3D tumoroïd of HT-29 cells in normoxia and hypoxia. Tumoroïds were exposed to MCM (vehicle), MCM (5-FU 1 μg/mL), and MCM (5-FU 1 μg/mL + gimeracil 1 μg/mL). Pictures were obtained with a phase contrast microscope. Scale bar, 200 μm (n = 8). F, Induction of death in RKO in normoxia (blue) and in hypoxia (red) by non-conditioned medium (square) and conditioned medium (triangle). Dead cells were defined as AnnexinV+ cells in flow cytometry. 5-FU was used at 1 μg/mL and gimeracil at 1 μg/mL (n = 3). G, Immunoblot of DPD expression in macrophages exposed to 5-FU at 50 μg/mL during 48 hours (n = 3). H, Immunoblot of DPD expression in macrophages transwell cocultured with HT-29 or RKO in normoxia and hypoxia (n = 3). I, Immunoblot analysis of DPD expression in hypoxic macrophages transiently transfected with siRNA against DPD and nonsilencing control siRNA (left). MCM from macrophages treated with siRNA against DPD restore sensitivity to 5-FU in RKO cells (middle; representative of three independent experiments). Quantification of induced apoptosis was done with flow cytometry (right; n = 3). Statistical significance was determined using a one-way ANOVA analysis with Tukey post hoc test (A, C, D, and F) and by paired Student t test for (I). Error bars, mean 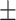 SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, nonsignificant.
SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, nonsignificant.
DPD expression in hypoxic macrophages is under the control of HIF2α
We discovered that a decreased oxygen concentration was able to increase the expression of DPD in human macrophages. To gain further insight into this, we carried out the transition of macrophages to various oxygen environments, to study the way DPD is controlled. We observed that DPD expression was inversely correlated to oxygen levels during the transitions (Fig. 3A and B). The evolution of DPD expression was then analyzed with the help of a first-order differential equation (Fig. 3C). This analysis revealed that the DPD half-life does not decrease during transition from hypoxia to normoxia, excluding DPD degradation as the only mechanism controlling DPD expression (Fig. 3C). We have confirmed this analysis using proteasome inhibitors (MG132 and bortezomib), which do not modify DPD decreased expression during a hypoxia to normoxia transition (Supplementary Fig S4A). These results suggested a synthesis control of DPD expression rather than a degradation control. We then noted that hypoxic transitions were associated with the production of a functional DPD resulting in an increased dihydrouracil/uracil ratio in the extracellular medium (Supplementary Fig. S4B). Besides, we observed that profound hypoxic conditions (7 mm Hg 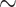 1% O2), similarly to moderate hypoxic conditions, induced DPD overexpression (Supplementary Fig. S4C). As hypoxic-induced factors HIF1α and HIF2α are known to be stabilized during hypoxic transitions, we checked whether their stabilization could be implicated in DPD overexpression. We found that HIF1α stabilization in hypoxic human macrophages was not involved in DPD increased expression, as siRNA-mediated depletion of HIF1α did not modify DPD protein synthesis in hypoxia (Fig. 3D). Using siRNA depletion, we demonstrated that DPD overexpression in hypoxia is under the control of a HIF2α-dependent mechanism (Fig. 3E). We next sought to determine whether the expression of DPD is transcriptionally controlled when macrophages are exposed to low-oxygen environments. To do so, we analyzed mRNA levels of oxygen-sensitive genes in macrophages (VEGFA, NDRG1, P4HA1, SLC2A1) in the transition from normoxia to hypoxia or from hypoxia to normoxia. We discovered that the DPYD mRNA level did not present any significant variation of its level of expression compared with oxygen responsive genes (Fig. 3F). We confirmed the absence of transcriptional control by inhibiting the synthesis of new mRNAs with actinomycin D and found no effect on DPD protein synthesis during a hypoxic transition (Fig. 3G). We further confirmed the translation-mediated control expression of DPD using translation inhibition by cycloheximide (Supplementary Fig. S4D). This absence of correlation between the mRNA level and the protein expression level suggested a HIF2a-related mechanism independent of its direct transcription factor activity. Recently, it has been demonstrated that the initial steps of protein synthesis such as the binding of the eukaryotic translational initiation factor E (eIF4E), part of the eIF4F initiation complex, to mRNA are repressed in hypoxia. Another complex, involving eIF4E2 (an homolog of eIF4E normally involved in translation inhibition in normoxia), RBM4 (RNA binding protein 4), and HIF2α has been proposed to interact with mRNA mediating a selective cap-dependent protein synthesis in low oxygen environments (25, 26). Interestingly, the participation of HIF2α in this complex is independent of its transcription factor activity (26). Using these results, we depleted the expression of eIF4E and eIF4E2 in macrophages using specific siRNAs and found that hypoxia-induced DPD synthesis is an eIF4E2-dependent process (Fig. 3H). These results suggested that DPD expression in hypoxia is controlled by HIF2α independently of its transcription factor activity in an eIF4E2-dependent mechanism.
1% O2), similarly to moderate hypoxic conditions, induced DPD overexpression (Supplementary Fig. S4C). As hypoxic-induced factors HIF1α and HIF2α are known to be stabilized during hypoxic transitions, we checked whether their stabilization could be implicated in DPD overexpression. We found that HIF1α stabilization in hypoxic human macrophages was not involved in DPD increased expression, as siRNA-mediated depletion of HIF1α did not modify DPD protein synthesis in hypoxia (Fig. 3D). Using siRNA depletion, we demonstrated that DPD overexpression in hypoxia is under the control of a HIF2α-dependent mechanism (Fig. 3E). We next sought to determine whether the expression of DPD is transcriptionally controlled when macrophages are exposed to low-oxygen environments. To do so, we analyzed mRNA levels of oxygen-sensitive genes in macrophages (VEGFA, NDRG1, P4HA1, SLC2A1) in the transition from normoxia to hypoxia or from hypoxia to normoxia. We discovered that the DPYD mRNA level did not present any significant variation of its level of expression compared with oxygen responsive genes (Fig. 3F). We confirmed the absence of transcriptional control by inhibiting the synthesis of new mRNAs with actinomycin D and found no effect on DPD protein synthesis during a hypoxic transition (Fig. 3G). We further confirmed the translation-mediated control expression of DPD using translation inhibition by cycloheximide (Supplementary Fig. S4D). This absence of correlation between the mRNA level and the protein expression level suggested a HIF2a-related mechanism independent of its direct transcription factor activity. Recently, it has been demonstrated that the initial steps of protein synthesis such as the binding of the eukaryotic translational initiation factor E (eIF4E), part of the eIF4F initiation complex, to mRNA are repressed in hypoxia. Another complex, involving eIF4E2 (an homolog of eIF4E normally involved in translation inhibition in normoxia), RBM4 (RNA binding protein 4), and HIF2α has been proposed to interact with mRNA mediating a selective cap-dependent protein synthesis in low oxygen environments (25, 26). Interestingly, the participation of HIF2α in this complex is independent of its transcription factor activity (26). Using these results, we depleted the expression of eIF4E and eIF4E2 in macrophages using specific siRNAs and found that hypoxia-induced DPD synthesis is an eIF4E2-dependent process (Fig. 3H). These results suggested that DPD expression in hypoxia is controlled by HIF2α independently of its transcription factor activity in an eIF4E2-dependent mechanism.
Figure 3.

DPD expression in hypoxic macrophages is under the control of HIF2α and eIF4E2. A, Immunoblot of DPD expression in normoxia (N) and hypoxia (H) and during transition from normoxia to hypoxia (NH) and hypoxia to normoxia (HN; n = 4). B, Quantification of the expression of DPD from immunoblots of A for normoxia-to-hypoxia and hypoxia-to-normoxia transitions (n = 4). C, Left, mathematical model fitting DPD expression curves from B. Right, DPD half-life extracted from the model. D, Immunoblot analysis of DPD and HIF1α in macrophages exposed to hypoxia (7 mm Hg) for 6 hours under siRNA silencing of HIF1α. β-Actin was used as loading control (n = 3). E, Immunoblot of DPD expression in normoxia and during normoxia-to-hypoxia transition (hypoxia PO2 = 7 mm Hg) under siRNA silencing of HIF2α (n = 3). F, mRNA expression ratio NH/N and HN/H transitions (hypoxia PO2 = 25 mm Hg) determined by qPCR for the following genes: DPYD, VEGFA, NDRG1, P4HA1, and SLC2A1. Macrophages were previously cultured in normoxia or hypoxia (n = 3). G, Immunoblot of DPD expression during normoxia-to-hypoxia (hypoxia PO2 = 7 mm Hg) transition with macrophages previously exposed to actinomycin D at 1 μg/mL for 20 minutes (n = 3). H, Immunoblot of DPD expression during normoxia-to-hypoxia transition (hypoxia PO2 = 7 mm Hg) under siRNA silencing of eIF4E2 and eIF4E (n = 3). Statistical significance was determined using a one-way ANOVA analysis with Tukey post hoc test (E and H). Error bars, mean  SEM. *, P < 0.05; **, P < 0.01; ns, nonsignificant.
SEM. *, P < 0.05; **, P < 0.01; ns, nonsignificant.
TAMs in human colon cancer tissues harbor the principal component of DPD expression in tumors
We have demonstrated that DPD expression in macrophages confers a chemoresistance to 5-FU. To assess the clinical relevance of this result, we further determined the relative expression of DPD in various cellular populations found in colorectal tumors and colorectal liver metastasis. We first used RNA-seq analysis in various cancer cell lines from the CCLE and confirmed that the 59 cancer cell lines originating from colon cancer presented the lowest level of expression for DPYD when compared with other types of cancers (Fig. 4A). This result confirmed what we had observed for two colon cancer cell lines RKO and HT-29 (Supplementary Fig. S2C), and emphasized a preeminent role of macrophages in DPD-induced chemoresistance in tumors. We further analyzed tissue samples from patients suffering from colorectal cancer with liver metastasis and found that the strongest expression of DPD was found in areas with a predominance of CD68+ macrophages (Fig. 4B). Tumor cells did not present a significant level of DPD expression in metastasis when compared with neighboring TAMs (Fig. 4C). Furthermore, in primary tumors, macrophages were also found to express the highest level of DPD, with no detectable expression found in cancer cells (Fig. 4D). We further confirmed that macrophages represent the main source of DPD expression, by showing that strongly DPD+ cells were also CD68+ using an immunofluorescence coexpression analysis, both in liver metastasis (Fig. 4E) and primary tumors (Fig. 4F). Because CD68 has been found to be less specific than previously thought as a macrophage marker (27), we confirmed our results using the CD163 macrophage marker. We confirmed that DPD+ cells are CD163+ TAMs in liver metastasis and primary tumors (Supplementary Fig. S5A and S5B). These results are consistent with previous IHC analysis of colorectal cancer tissues, demonstrating that cancer cells do not express DPD whereas normal cells that are morphologically similar to macrophages present a strong expression (28). We further confirmed that DPD expressing cells in primary tumors and liver metastasis also express HIF2α (Supplementary Fig. S5C and S5D). All these results indicate that DPD expression in colorectal cancers at the primary site and liver metastasis belongs to macrophages, under the control of oxygen.
Figure 4.

Macrophages harbor the main DPD expression in colorectal cancer. A, RNA-seq analysis of DPYD expression in various cancer cell lines from the CCLE. Colon cancer cell lines are in red. B, Immunochemistry analysis of CD68 (top) and DPD (bottom) expression in liver metastasis of colorectal cancer (n = 15). Scale bar, 200 μm. C, Immunochemistry analysis of DPD expression in various cell populations in liver metastasis. Red arrowheads, macrophages; black arrows, metastatic cancerous cells (n = 15; zoomed image scale bar, 60 μm). D, Immunochemistry analysis of DPD expression in primary tumors. Red arrowheads, macrophages; black arrows, cancer cells; black arrowhead, tripolar mitosis of a cancer cell (n = 15; zoomed image scale bar, 60 μm). E, Left, immunofluorescence staining in liver metastatic tissues, with CD68 in green, DPD in red, and nuclei stained by Hoescht in blue (n = 4). Scale bar, 50 μm. Right, quantification of CD68+ cells in the group of DPD-expressing cells (n = 4 patients). F, Immunofluorescence staining in primary tumors, with CD68 in green, DPD in red, nuclei stained by Hoescht in blue (n = 4). Scale bar, 50 μm. Right, quantification of CD68+ cells in the group of DPD-expressing cells (n = 4 patients).
Rodents' macrophages do not express significant levels of DPD
To assess the generality of the oxygen control of DPD expression in macrophages we checked whether this mechanism still holds in rodents. Surprisingly, we found that mice bone marrow–derived macrophages (BMDM) do not express a significant level of mouse DPD in normoxia or hypoxia despite the presence of the protein in the mouse liver (Fig. 5A). No detectable level of dpyd mRNA was found in BMDM from BALB/c mice (Supplementary Fig. S6A). We used open datasets from microarrays to compare the expression of DPYD mRNA levels in humans to those in mice. We found that DPYD presented the highest levels of expression in the monocyte/macrophages lineage in humans, contrary to what was found in mice where macrophages expressed few mRNA dpyd molecules compared with other cellular lineages (Fig. 5B). This observation suggested a repression of the mRNA synthesis in mice macrophages. We discovered that the RAW264.7 mice macrophage cell line presented the same pattern with no protein expression in normoxia or hypoxia, confirmed by the absence of DPD enzymatic acitivity (Fig. 5C). And this was correlated with the absence of mRNA of dpyd in these cells (Supplementary Fig. S6B). We then confirmed the epigenetic control of gene expression using 5-aza-2′deoxycytidine (decitabine), a DNA hypomethylation agent. Indeed, decitabine led to a strong increase in dpyd mRNA level of expression in treated RAW macrophages compared with nontreated cells (Fig. 5D). This result suggested that DNA methylation in mice is, at least in part, responsible for dpyd repressed expression. To further confirm the generality of this finding we analyzed DPYD mRNA expression in TAMs from colorectal cancers and breast cancers in mice and confirm the low level of expression of DPYD in mice macrophages (Supplementary Fig. S7).
Figure 5.

Mice macrophages do not express DPD. A, BMDMs were differentiated in normoxia and hypoxia. F4/80 was studied by flow cytometry (left) and mouse DPD (mDPD) expression by immunoblot (right; n = 4). Mouse liver was used as a positive control for mDPD. B, Microarray analysis of DPYD mRNA expression in monocytes/macrophage populations in mice and humans. In each group, the highest and lowest level of expression was used to scale the heatmap. C, RAW264.7 macrophages were cultivated in normoxia and in hypoxia. mDPD expression was studied by immunoblot. No production of dihydrouracil was found in RAW supernatant using HPLC. nd, nondetected. D,dpyd mRNA level of expression in RAW macrophages exposed to decitabine at 5 μmol/L during 24 hours (n = 3).
Transduced human DPD in mice macrophages leads to 5-FU chemoresistance in vivo
To obtain a mice model mimicking the human DPD expression in macrophages, we transduced the ORF of the human DPYD gene under the control of a cytomegalovirus promoter incorporated into a lentivirus to obtain “DPD-humanized” mice macrophages (Fig. 6A). CM of mice macrophages expressing DPD were able to confer chemoresistance to CT-26 (a mice colon cancer cell line that does not express DPD) demonstrating the functionality of the transduced DPD (Fig. 6B). We also observed that wild-type macrophages were associated with a weak decrease of 5-FU–induced growth inhibition compared with macrophages expressing DPD, demonstrating that the DPD-induced chemoresistance mechanism is probably the most efficient one (Fig. 6B). To confirm the relationship between DPD expression in macrophages and chemoresistance in colorectal cancers, a tumor assay in mice was performed. CT-26 and RAW macrophages expressing or not human DPD were implanted into flanks of BALB/c mice. Ten days after the implantation, 5-FU was injected intraperitoneally at 25 mg/kg during 5 days for 2 consecutive weeks (Fig. 6C). We confirmed that tumors harboring macrophages expressing DPD were more resistant to 5-FU than the control tumors with wild-type macrophages (Fig. 6D–F), indicating that DPD expression in TAMs promotes chemoresistance in vivo.
Figure 6.

Transduced human DPD in mice macrophages leads to 5-FU chemoresistance in vivo. A, Left, immunoblot of RAW macrophages transduced to express GFP and human DPD (hDPD)-GFP. Right, transduced GFP and hDPD-GFP proteins levels of expression analyzed by flow cytometry. B, Growth inhibition of CT-26, after 48 hours, under the presence of CM containing 5-FU (0.1 μg/mL) exposed to macrophages WT, expressing GFP or hDPD-GFP for 24 hours. Gimeracil was used to block hDPD activity at 1 μg/mL. C, Tumor assay was performed on female Balb/c mice of 7 weeks. A total of 106 CT-26 and 106 RAW were implanted subcutaneously. After 10 days, daily bolus of 5-FU 25 mg/kg was injected intraperitoneally according to the timeline represented. D, Tumor growth was followed during the protocol (n = 7 in each group). E, Tumor size was determined at day 21 (last day of 5-FU injection protocol). F, Tumor weight was determined at day 24 (n = 7 in each group). Statistical significance was determined using a one-way ANOVA analysis with Tukey post hoc test (B), a two-way ANOVA analysis for tumor growth curves (D), and a Mann–Withney test (E and F). Error bars, mean  SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Discussion
In recent years, the immune system has become a key element in the understanding of the mechanisms involved in the tumor interaction with its surrounding healthy tissue as well as a provider of new therapeutic strategies. The tumor immune microenvironment is composed of various types of immune cells. Nevertheless, TAMs usually represent quantitatively the largest population found in solid cancers. TAMs are involved in tumor growth, immune evasion, neoangiogenesis, and treatment resistance. Using depletion methods, a large number of studies have reported an increased chemosensitivity when macrophages are removed from the tumor (5). Furthermore, coculture studies have revealed macrophage-mediated resistance mechanisms to various anti-cancer drugs such as paclitaxel, doxorubicin, etoposide, or gemcitabine (29, 30). Specifically, depletion of MHCIIlo TAMs leads to an increased sensitivity to taxol-induced DNA damage and apoptosis (31). Another key point is that TAMs were mainly found in hypoxic areas where they can further favor hypoxia by secreting VEGFA, leading to the formation of an abnormal vasculature (32). Accordingly, mechanisms involving macrophages-induced chemoresistance rely usually on the secretions of factors by macrophages, such as pyrimidine nucleosides (deoxycytidine) inhibiting gemcitabine induction of apoptosis in pancreatic ductal adenocarcinoma (33). In colorectal cancer, the implication of macrophages in chemoresistance has also been suggested on the basis of in vitro and in vivo studies. The proposed mechanisms are diverse but also involve secreted factors. For example, it has been proposed that IL6 secreted by macrophages can stimulate STAT3 in cancer cells inducing the inhibition of the RAB22A/BCL2 pathway through miR-204-5p expression, thereby leading to chemoresistance to 5-FU (11). Similarly the secretion of putrescin, a member of the polyamine family, by macrophages was shown to suppress the JNK/Caspase 3 pathway in cancer cells, providing a protection against 5-FU (12).
To understand the involvement of TAMs in chemoresistance in colorectal cancer, we designed the current study to incorporate oxygen concentration as a key environmental parameter. We previously reported that the oxygen availability in macrophages' environment greatly influences their immune functions such as their ability to clear apoptotic cells (13). As colon tissues are naturally exposed to levels of oxygen that are usually lower than 5% O2 (34) with values that could reach even lower values (<1% O2) in tumors, oxygen appears as a fundamental parameter to understand macrophage involvement in chemoresistance. We found that hypoxic macrophages provide a chemoresistance to 5-FU when cocultured with cancer cells contrary to normoxic macrophages (Fig. 1A). We then verified whether a secreted factor by human macrophages could provide a chemoresistance and finally found a direct effect of hypoxic macrophages on 5-FU (Fig. 1B and C). Our molecular analysis revealed that the DPD, an enzyme of the pyrimidine catabolism pathway, is overexpressed in hypoxic human macrophages providing a direct chemoresistance mechanism relying on its enzymatic activity (Fig. 2A, C, D, E, F, and I; Supplementary Fig. S3). It is known for more than thirty years that DPD expression in the liver limits 5-FU biodisponibility (35) and its expression in peripheral mononuclear cells is the gold standard to detect defects in DPD activity due to mutations to prevent 5-FU intolerance in patients (36). Despite this knowledge, no systematic analysis of the control of DPD expression and functions in human macrophages in the tumor microenvironment was performed before this study. We have discovered that in hypoxia, DPD is under the control of HIF2α independently of its transcription factor activity (Fig. 3E–G). This control was also found to be under the control of eIF4E2 (Fig. 3H). This protein has been proposed to be part of a translation initiation complex, comprising HIF2α, which is only active in hypoxia (25, 26). This complex is one of the mechanisms involved in the adaptive protein synthesis in hypoxia, mitigating the global shutdown of translation taking place in this context (37).
DPD expression in macrophages seemed to be particularly relevant in colorectal cancer where cancer cells present a low expression level of the protein in primary tumors as well as in liver metastasis (Fig. 4; Supplementary Fig. S5A and S5B). This general feature seems to rely on the epigenetic control of DPD expression in cancer cells. Indeed, many colon cancer cells lines have been noted to harbor a histone H3K27me3 mark that blocks the fixation of the transcription factor PU.1, leading to the inhibition of DPD mRNA transcription (21). We further found that macrophages in mice do not express DPD due to a repression of its transcriptional expression (Fig. 5). This finding forced us to reevaluate previous in vivo models that were used to assess the involvement of macrophages in colorectal cancer, as DPD expression in macrophages was lacking in these models. Indeed, we showed the importance of DPD activity in macrophages and found that it represents the main quantitative source of degradation of 5-FU in human colorectal tumors. To demonstrate the relevance of this mechanism to chemoresistance, we designed an in vivo model using mice macrophages expressing the transduced human DPD. This model offered us the possibility to validate the importance of DPD expression in TAMs leading to 5-FU chemoresistance. Supporting these results, previous clinical studies have suggested that DPD mRNA expression is a marker of chemoresistance in colorectal cancer. These studies usually assumed that DPD expression is mainly found in cancer cells, contrary to what we have observed. Nevertheless, the mRNA level in the bulk of the tumor is correlated with a low response to 5-FU confirming its relevance (38–41). The results obtained in our study suggest that the main predictive factor for 5-FU response is DPD expression in macrophages located in tumors and liver metastasis. That expression is probably important in the invasive front, where TAMs seem to concentrate (9). Furthermore, the invasion front is known to be a hypoxic area in colorectal cancers (42). Because the mechanism identified in this study relied on quantitative expression of DPD by macrophages, the assessment of the spatial heterogeneity of DPD expression will be necessary to stratify patients in various response groups for chemotherapy (43). Thus this study constitutes an important progress in the understanding of the role of the tumor immune environment in chemoresistance to 5-FU in colorectal cancer. Finally, further clinical studies are needed to confirm the clinical relevance of these findings and validate DPD expression in macrophages as a new predictive marker of response to 5-FU–based treatments.
Authors' Disclosures
A. Millet reports grants from Inserm ATIP Avenir grant, Fondation AGIR pour les maladies chroniques, Ligue nationale contre le cancer, European Union's Horizon 2020, and Fondation MSD-Avenir during the conduct of the study. No disclosures were reported by the other authors.
Supplementary Material
supplementary materials and supplementary figure legends
supplementary Table
Supplementary figures S1 to S7
Acknowledgments
A. Millet is supported by the ATIP/Avenir program (Inserm and La ligue nationale contre le cancer). M. Malier is supported by the APMC Fondation (Agir pour les Maladies Chroniques). K. Gharzeddine is supported by the ITN (International Training Network) Phys2Biomed project, which is funded from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 812772. This work is supported by the ERiCAN program of Fondation MSD-Avenir (Reference DS-2018-0015). The authors gratefully acknowledge Florent Chuffart from the EpiMed core facility (http://epimed.univ-grenoble-alpes.fr) for his support and assistance in this work. The authors would like to thank Xavier Fonrose, Edwige Col, and Floriane Laurent for their technical assistance. The authors thank the zoo technicians of PHTA facility for animal housing and care. They thank Lemoigne, Federspiel, and Plasse from the University Hospital of Grenoble-Alpes for kindly providing clinical grade 5-FU.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Contributions
M. Malier: Investigation, writing–review and editing. K. Gharzeddine: Investigation, writing–review and editing. M.-H. Laverriere: Investigation. S. Marsili: Investigation. F. Thomas: Investigation. T. Decaens: Supervision. G.S. Roth: Validation, investigation. A. Millet: Conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, validation, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing.
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70:7–30. [DOI] [PubMed] [Google Scholar]
- 2. Marin JJG, Sanchez de Medina F, Castaño B, Bujanda L, Romero MR, Martinez-Augustin O, et al. Chemoprevention, chemotherapy, and chemoresistance in colorectal cancer. Drug Metab Rev 2012;44:148–72. [DOI] [PubMed] [Google Scholar]
- 3. Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature 2019;575:299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 2018;24:541–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015;27:462–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang M, McKay D, Pollard JW, Lewis CE. Diverse functions of macrophages in different tumor microenvironments. Cancer Res 2018;78:5492–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol 2019;19:369–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Aras S, Zaidi MR. TAMeless traitors: macrophages in cancer progression and metastasis. Br J Cancer 2017;117:1583–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pinto ML, Rios E, Durães C, Ribeiro R, Machado JC, Mantovani A, et al. The two faces of tumor-associated macrophages and their clinical significance in colorectal cancer. Front Immunol 2019;10:1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ye L, Zhang T, Kang Z, Guo G, Sun Y, Lin K, et al. Tumor-infiltrating immune cells act as a marker for prognosis in colorectal cancer. Front Immunol 2019;10:2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yin Y, Yao S, Hu Y, Feng Y, Li M, Bian Z, et al. The Immune-microenvironment confers chemoresistance of colorectal cancer through macrophage-derived IL6. Clin Cancer Res 2017;23:7375–87. [DOI] [PubMed] [Google Scholar]
- 12. Zhang X, Chen Y, Hao L, Hou A, Chen X, Li Y, et al. Macrophages induce resistance to 5-fluorouracil chemotherapy in colorectal cancer through the release of putrescine. Cancer Lett 2016;381:305–13. [DOI] [PubMed] [Google Scholar]
- 13. Court M, Petre G, Atifi ME, Millet A. Proteomic signature reveals modulation of human macrophage polarization and functions under differing environmental oxygen conditions. Mol Cell Proteomics 2017;16:2153–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Henze A-T, Mazzone M. The impact of hypoxia on tumor-associated macrophages. J Clin Invest 2016;126:3672–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modeling of anticancer drug sensitivity. Nature 2012;483:603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, et al. Defining a cancer dependency map. Cell 2017;170:564–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hruz T, Laule O, Szabo G, Wessendorp F, Bleuler S, Oertle L, et al. Genevestigator V3: a reference expression database for the meta-analysis of transcriptomes. Adv Bioinforma 2008;2008:420747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thomas F, Hennebelle I, Delmas C, Lochon I, Dhelens C, Tixidre CG, et al. Genotyping of a family with a novel deleterious DPYD mutation supports the pretherapeutic screening of DPD deficiency with dihydrouracil/uracil ratio. Clin Pharmacol Ther 2016;99:235–42. [DOI] [PubMed] [Google Scholar]
- 19. Enot DP, Vacchelli E, Jacquelot N, Zitvogel L, Kroemer G. TumGrowth: an open-access web tool for the statistical analysis of tumor growth curves. OncoImmunology 2018;7:e1462431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Větvička V, Bilej M, Kincade PW. Resistance of macrophages to 5-fluorouracil treatment. Immunopharmacology 1990;19:131–8. [DOI] [PubMed] [Google Scholar]
- 21. Wu R, Nie Q, Tapper EE, Jerde CR, Dunlap GS, Shrestha S, et al. Histone H3K27 trimethylation modulates 5-fluorouracil resistance by inhibiting PU.1 binding to the DPYD promoter. Cancer Res 2016;76:6362–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zheng J-F, Wang H-D. 5-Fluorouracil concentration in blood, liver and tumor tissues and apoptosis of tumor cells after preoperative oral 5′-deoxy-5-fluorouridine in patients with hepatocellular carcinoma. World J Gastroenterol 2005;11:3944–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tanaka-Nozaki M, Onda M, Tanaka N, Kato S. Variations in 5-fluorouracil concentrations of colorectal tissues as compared with Dihydropyrimidine Dehydrogenase (DPD) enzyme activities and DPD messenger RNA levels. Clin Cancer Res 2001;7:2783–7. [PubMed] [Google Scholar]
- 24. Wilson ZE, Rostami-Hodjegan A, Burn JL, Tooley A, Boyle J, Ellis SW, et al. Inter-individual variability in levels of human microsomal protein and hepatocellularity per gram of liver. Br J Clin Pharmacol 2003;56:433–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ho JJD, Wang M, Audas TE, Kwon D, Carlsson SK, Timpano S, et al. Systemic reprogramming of translation efficiencies on oxygen stimulus. Cell Rep 2016;14:1293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Uniacke J, Holterman CE, Lachance G, Franovic A, Jacob MD, Fabian MR, et al. An oxygen-regulated switch in the protein synthesis machinery. Nature 2012;486:126–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ruffell B, Au A, Rugo HS, Esserman LJ, Hwang ES, Coussens LM. Leukocyte composition of human breast cancer. Proc Natl Acad Sci U S A 2012;109:2796–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kamoshida S, Shiogama K, Matsuoka H, Matsuyama A, Shimomura R, Inada K, et al. Immunohistochemical demonstration of dihydropyrimidine dehydrogenase in normal and cancerous tissues. Acta Histochem Cytochem 2003;36:471–9. [Google Scholar]
- 29. Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res 2013;73:1128–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K, et al. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev 2011;25:2465–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Olson OC, Kim H, Quail DF, Foley EA, Joyce JA. Tumor-associated macrophages suppress the cytotoxic activity of antimitotic agents. Cell Rep 2017;19:101–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer 2008;8:618–31. [DOI] [PubMed] [Google Scholar]
- 33. Halbrook CJ, Pontious C, Kovalenko I, Lapienyte L, Dreyer S, Lee H-J, et al. Macrophage-released pyrimidines inhibit gemcitabine therapy in pancreatic cancer. Cell Metab 2019;29:1390–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Keeley TP, Mann GE. Defining physiological normoxia for improved translation of cell physiology to animal models and humans. Physiol Rev 2018;99:161–234. [DOI] [PubMed] [Google Scholar]
- 35. Diasio RB, Harris BE. Clinical pharmacology of 5-fluorouracil. Clin Pharmacokinet 1989;16:215–37. [DOI] [PubMed] [Google Scholar]
- 36. Chazal M, Etienne MC, Renée N, Bourgeon A, Richelme H, Milano G. Link between dihydropyrimidine dehydrogenase activity in peripheral blood mononuclear cells and liver. Clin Cancer Res 1996;2:507–10. [PubMed] [Google Scholar]
- 37. Lee P, Chandel NS, Simon MC. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol 2020;21:268–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ichikawa W, Uetake H, Shirota Y, Yamada H, Nishi N, Nihei Z, et al. Combination of dihydropyrimidine dehydrogenase and thymidylate synthase gene expressions in primary tumors as predictive parameters for the efficacy of fluoropyrimidine-based chemotherapy for metastatic colorectal cancer. Clin Cancer Res 2003;9:786–91. [PubMed] [Google Scholar]
- 39. Salonga D, Danenberg KD, Johnson M, Metzger R, Groshen S, Tsao-Wei DD, et al. Colorectal tumors responding to 5-fluorouracil have low gene expression levels of dihydropyrimidine dehydrogenase, thymidylate synthase, and thymidine phosphorylase. Clin Cancer Res 2000;6:1322–7. [PubMed] [Google Scholar]
- 40. Shirota Y, Ichikawa W, Uetake H, Yamada H, Nihei Z, Sugihara K. Intratumoral dihydropyrimidine dehydrogenase messenger RNA level reflects tumor progression in human colorectal cancer. Ann Surg Oncol 2002;9:599–603. [DOI] [PubMed] [Google Scholar]
- 41. Soong R, Shah N, Salto-Tellez M, Tai BC, Soo RA, Han HC, et al. Prognostic significance of thymidylate synthase, dihydropyrimidine dehydrogenase and thymidine phosphorylase protein expression in colorectal cancer patients treated with or without 5-fluorouracil-based chemotherapy. Ann Oncol 2008;19:915–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Righi A, Sarotto I, Casorzo L, Cavalchini S, Frangipane E, Risio M. Tumour budding is associated with hypoxia at the advancing front of colorectal cancer. Histopathology 2015;66:982–90. [DOI] [PubMed] [Google Scholar]
- 43. Marusyk A, Janiszewska M, Polyak K. Intratumor heterogeneity: the rosetta stone of therapy resistance. Cancer Cell 2020;37:471–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary materials and supplementary figure legends
supplementary Table
Supplementary figures S1 to S7