

PLCB1 functions as an oncogenic driver in cholangiocarcinoma development that confers an actionable therapeutic vulnerability to AKT inhibition.
Abstract
As a member of the phospholipase family, phospholipase C beta 1 (PLCB1) is involved in phospholipid hydrolysis and is frequently upregulated in human cancer. However, little is known about the role of PLCB1 in cholangiocarcinoma (CCA). In this study, we uncover a role for PLCB1 in CCA progression and identify the underlying mechanisms. Both human CCA tissues and CCA cell lines expressed high levels of PLCB1. PLCB1 promoted tumor development and growth in various CCA mouse models, including transposon-based tumorigenesis models. PLCB1 activated PI3K/AKT signaling to induce CCA cells to undergo epithelial-to-mesenchymal transition (EMT). Mechanistically, PABPC1 interacted with PLCB1 and PI3K to amplify PLCB1-mediated EMT via PI3K/AKT/GSK3β/Snail signaling. Ectopic PLCB1 induced resistance to treatment with gemcitabine combined with cisplatin, which could be reversed by the AKT inhibitor MK2206. PLCB1 expression was regulated by miR-26b-5p through direct interaction with PLCB1 3′UTR. Collectively, these data identify a PLCB1–PI3K–AKT signaling axis vital for CCA development and EMT, suggesting that AKT can be used as a therapeutic target to overcome chemotherapy resistance in CCA patients with high PLCB1 expression.
Significance:
PLCB1 functions as an oncogenic driver in cholangiocarcinoma development that confers an actionable therapeutic vulnerability to AKT inhibition.
Graphical Abstract
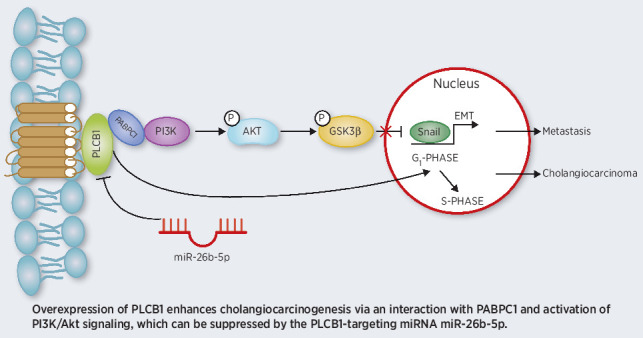
Introduction
Cholangiocarcinoma (CCA), an epithelial cell malignancy originating from the bile duct, is associated with an extremely short survival time and is the second most common primary liver cancer worldwide (1, 2). Although new modalities of chemotherapy (3) and immunotherapy (4) are being applied broadly, the accepted effective treatment is still surgical resection (5). Nevertheless, due to insidious early symptoms and fast tumor progression, patients with CCA often do not present to the clinic until after the optimal resection time has passed (6). Hence, it is extremely urgent to illuminate the underlying mechanisms of CCA progression to find novel therapeutic strategies.
Phospholipase C, β1 (PLCB1) is a member of the phospholipase family of proteins whose main function is to catalyze the formation of inositol 1,4,5-trisphosphate (IP3) and diacylglycerol from phosphatidylinositol 4,5-bisphosphate (PIP2; ref. 7). Calcium is required for this reaction, and PLCB1 plays a pivotal role in the intracellular transduction of many extracellular signals (8, 9). PLCB1 has been reported to be associated with several diseases, such as schizophrenia (10), epileptic encephalopathy (11), and myotonic dystrophy (12). PLCB1 is also involved in the cell cycle and cell proliferation (13), and it was shown to be a candidate tumor promoter in small cell lung carcinoma (14), breast cancer (15), and colorectal carcinoma (16). However, the biological function of PLCB1 in CCA is not clear. Therefore, we first demonstrated that PLCB1 was a critical promoter of human CCA and explored its mechanism in the progression of CCA.
MicroRNAs (miRNA) are a class of noncoding single-stranded RNAs with a length of 22 nucleotides encoded by endogenous genes, that can regulate posttranscriptional gene expression and silence an extensive range of target genes. As regulatory factors of oncogenic or suppressive genes, miRNAs can act as therapeutic targets or biomarkers for the diagnosis of cancer (17). To date, miR-26b-5p has been shown to be associated with multiple cancers, such as Burkitt lymphoma (18), non–small cell lung cancer (19), and breast cancer (20). However, we discovered for the first time that miR-26b-5p could suppress the progression of CCA by targeting PLCB1.
PABPC1 is one of the functional protein members of the polyadenylate-binding protein conserved gene family that participates in poly(A) shortening, ribosome recruitment, and translation initiation (21). There is accumulating evidence that PABPC1 is related to several cancer types, such as prostate cancer (22), hepatocellular carcinoma (23), and leukemia (24). Via further research, we found that PABPC1 could directly interact with PLCB1. Given that epithelial-to-mesenchymal transition (EMT) is an important mechanism of malignant tumor metastasis and contributes pathologically to fibrosis and cancer progression, we also verified that EMT-related markers can be changed by PLCB1 in CCA. Furthermore, there is evidence from other studies that EMT plays a vital role in the spread of CCA cells (25, 26). Ultimately, we demonstrated that PABPC1 could facilitate PI3K/AKT/GSK3β/snail signaling downstream of PLCB1 and regulate the PLCB1-mediated EMT in CCA.
Materials and Methods
Patients and tissue samples
From 2010 to 2018, 60 CCA and matching noncancerous border tissue samples were acquired during routine surgeries at the First Affiliated Hospital of Harbin Medical University (Harbin, China). The histopathologic diagnosis used World Health Organization criteria. The patients who participated in the study did not receive radiotherapy, chemotherapy, or immunotherapy before or after surgery. Tumors were classified according to the sixth edition of the tumor node metastasis (TNM) classification system published by the International Union Against Cancer. Tumor differentiation was classified according to the classification proposed by Edmondson and Steiner. Ethical approval was obtained from the First Affiliated Hospital of Harbin Medical University Research Ethics Committee and written informed consent was obtained from each patient. The detailed clinicopathologic characteristics of the 60 CCA specimens used in this study are listed in Supplementary Table S1.
Ethics approval and consent to participate
Tissue samples were obtained from patients with hepatobiliary surgery who agreed to participate in the research at The First Affiliated Hospital of Harbin Medical University (Harbin, China).
Cell lines and cell culture
Normal primary human biliary epithelial cells (HIBEpiC) were gained from ScienCell Research Laboratories. The immortalized hepatocyte cell line (Chang) was purchased from the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, China. The HuCCT1 cell line was kindly provided by the Cancer Cell Repository of the Tohoku University in Japan. CCLP1 and KMBC cell lines were purchased from the BeNa Culture Collection. RBE and HCCC9810 cell lines were purchased from Shanghai Bioleaf Biotech Corporation. RBE, HCCC9810, CCLP1, and HuCCT1 were authenticated using short tandem repeat analysis. The cancer cell lines used in our study are derived from CCA. All cell lines were cultured in Dulbecco's modified Eagle's medium or Roswell Park Memorial Institute (RPMI) 1640 supplemented with 10% v/v fetal bovine serum and 1% antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin) at 37°C in a 5% CO2 incubator.
Duolink PLA
First, the samples were deposited onto glass slides. Cells were fixed with 4% (w/v) paraformaldehyde for 5 minutes, and washed three times with PBS. Nonadherent cells were detached with 0.25% Triton-X100 for 5 minutes, and then cells were washed with 0.05% PBST solution. The Duolink PLA fluorescence protocol (Sigma-Aldrich) was followed according to the instructions provided by the manufacturer. Fluorescence or confocal microscopy was used to image the samples.
Hydrodynamic tail-vein injection
For hydrodynamic liver injection, plasmid DNA suspended in 2 mL saline was injected via the tail vein for 5 to 7 seconds into 7-week-old female C57BL/6 mice and female FVB/N (H2d) mice. The amount of DNA injected into each mouse was 20 μg for sgPTEN, 20 μg for sgP53, 5.5 μg for pCMV-Sleeping Beauty transposase, and 20 μg for PLCB1 or empty vector control. Vectors for hydrodynamic tail-vein injection were prepared using the EndoFreeMaxi Kit (Qiagen). All surviving mice were sacrificed 8 weeks after hydrodynamic injection. The incidence and tumor volume of CCA were assessed.
In vivo efficacy studies
Firstly, 5 × 106 cells suspended in 150 μL PBS were subcutaneously injected into the flank of male BALB/c athymic nude mice (4–6-week-old). When the tumors reached a volume of 0.2–0.3 cm3, the mice were divided into two groups (five mice per group): Gem + Cis + DMSO (200 mg/kg gemcitabine, 5 mg/kg cisplatin, intraperitoneal injection) and Gem + Cis + MK2206 (as Gem + Cis, with the addition of 60 mg/kg MK2206, intraperitoneal injection). All drugs were administered on experimental days (ED) 1, 5, 9, and 13. Tumor volumes were calculated using the following formula: volume = (length × width2)/2.
Statistical analysis
Statistical analysis was performed using Statistical Package for the Social Sciences (SPSS) 23.0 software (SPSS) and the GraphPad Prism software package (v.8.0). Results are presented as means ± standard deviation. Differences between the groups were analyzed using Student t tests, and P < 0.05 was considered statistically significant.
Availability of data and materials
All data generated and analyzed during this study are included in this published article and its additional file.
Results
PLCB1 is aberrantly expressed in human CCA
To determine the genes involved in the development of CCA, we screened more than 900 metabolism-related genes from the Kyoto Encyclopedia of Genes and Genomes database. However, only 12 genes had statistically significant differential expression and survival prognoses (Fig. 1A; Supplementary Fig. S1A and S1B). Next, we regarded PLCB1 as our candidate gene and investigated its function in CCA. To investigate the relationship between PLCB1 and CCA, the expression of PLCB1 was examined in human CCA tissue and compared with that in corresponding tumor border tissue. In Fig. 1B and C, we found that PLCB1 was expressed at markedly higher levels in CCA tissue than in border tissues. Analysis of IHC data indicated that expression of PLCB1 was high in paraffin sections from CCA patients. In CCA tissues, there was a total positive proportion of 75% (45/60), which was much higher than the 26% (13/50) observed in border liver tissue and 20% (2/10) in normal bile duct samples (Fig. 1D). Moreover, data derived from public gene-expression databases confirmed that PLCB1 expression was strikingly higher in human CCA tissue than in tissue adjacent to the cancer (Supplementary Fig. S2A–S2C). Furthermore, PLCB1 protein and mRNA levels were observed to increase gradually from normal liver (Chang) and normal human intrahepatic biliary (HIBEpiC) cell lines to CCA cell lines with low metastatic potential (CCLP1, RBE) to CCA cell lines with high metastatic potential (KMBC, QBC-939, HCCC-9810, and HuCCT1; Fig. 1E and F), suggesting that PLCB1 might be related to the metastatic potential of CCA.
Figure 1.

Overexpression of PLCB1 in human CCA. A, Heat map presented candidate metabolic genes from The Cancer Genome Atlas and Kyoto Encyclopedia of Genes and Genomes databases and these genes had statistical significance in differential expression and survival prognosis. CHOL, CCA tissues. B,PLCB1 mRNA levels were increased in 60 human CCA tissues compared with 50 corresponding normal liver samples and 10 normal bile duct samples. ****, P < 0.0001, based on Student t test. C, Typical images of PLCB1 protein levels in 60 paired human CCA and adjacent tissues. T, tumors; N, adjacent normal tissues. D, Typical images of PLCB1 IHC staining in CCA, normal liver tissues, and normal bile duct tissues. Scale bars, 100× = 400 μm; 400× = 100 μm. E,PLCB1 mRNA levels in CCA cells compared with normal liver (Chang) and normal human intrahepatic biliary (HIBEpiC) cell lines were evaluated by qRT-PCR. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001, based on the one-way ANOVA. F, Western blot indicated PLCB1 protein levels in CCA cells were higher than normal liver (Chang) and normal human intrahepatic biliary (HIBEpiC) cell lines. G, CCA patients with lymph node metastasis presented higher PLCB1 mRNA levels. ****, P < 0.0001, based on Student t test. H, Typical images of PLCB1 protein levels in CCA patients with or without lymph node metastasis. LM, lymph node metastasis; N0 LM, no lymph node metastasis. I, The influence of PLCB1 strong or weak staining on OS was analyzed by Kaplan–Meier plot. OS, overall survival. J, Typical IHC images of the relationship between expression of PLCB1 and Ki-67 in clinical CCA samples. Scale bars, 100× = 400 μm; 400× = 100 μm. All experiments were implemented three times and data are presented as mean ± SD.
To investigate the relationship between PLCB1 and clinical characteristics, we performed statistical analyses and revealed that the expression of PLCB1 was correlated with cancer stage (P = 0.001), lymph node status (P = 0.031), and metastasis (P = 0.011; Supplementary Fig. S2D; Supplementary Table S1). Eight paired independent CCA samples were then investigated to demonstrate that PLCB1 expression levels in patients presenting with lymph node metastasis were higher than those in patients without metastasis (Fig. 1G and H). We hypothesized that PLCB1 was related to the survival and recurrence of patients with CCA. According to IHC staining, CCA patients were divided into PLCB1-high and PLCB1-low expression groups. Kaplan–Meier curve analysis verified that patients with strong PLCB1 staining exhibited a decreased trend in overall survival (OS) and disease-free time (DFS) compared with those with weak PLCB1 staining (Fig. 1I; Supplementary Fig. S2E). Analysis of publicly available gene-expression data identified consistent trends in OS and DFS (Supplementary Fig. S2F). To investigate the prognostic significance of PLCB1, we applied Cox regression analysis of OS and DFS and found that PLCB1 was an independent prognostic factor for OS in CCA patients (Supplementary Table S2). Next, we hypothesized that there was a correlation between PLCB1 levels and tumor proliferation markers in CCA patient samples. Overall, 32 of 45 CCA samples with high PLCB1 expression exhibited a high level of Ki-67, whereas 12 of 15 CCA samples with low PLCB1 expression had a low level of Ki-67, suggesting that PLCB1 expression is correlated with CCA cell proliferation (Fig. 1J; Supplementary Fig. S2G). These results indicated that PLCB1 expression is frequently upregulated in human CCA and is correlated with poor clinical outcomes.
PLCB1 facilitates the proliferation and oncogenicity of CCA cells by enhancing the G1–S transition in vitro and in vivo
To explore whether PLCB1 facilitates the proliferation and oncogenicity of CCA cells, RBE and CCLP1 CCA cell lines with low PLCB1 expression were infected with a lentivirus overexpressing PLCB1, whereas HuCCT1 and KMBC cell lines with high PLCB1 levels were infected with three shRNAs (PLCB1-KD1, PLCB1-KD2, and PLCB1-KD3) to knock down PLCB1. After transfection, we verified the efficiency of overexpression and knockdown at the transcription and protein levels (Supplementary Fig. S3A). HuCCT1 PLCB1-KD3 and KMBC PLCB1-KD3 cells were chosen for further experiments due to their high knockdown efficiencies. We measured the proliferation ability of stable CCA cell lines, determining that PLCB1 overexpression increased the proliferation ability of RBE and CCLP1cells, whereas PLCB1 knockdown had the opposite effect on HuCCT1 and KMBC cells (Fig. 2A; Supplementary Fig. S3B). Similarly, the colony number of RBE and CCLP1 cells was significantly increased by overexpressed PLCB1, whereas PLCB1 KD3 resulted in reduced colony numbers in HuCCT1 and KMBC cells (Fig. 2B). To explore how the growth of CCA cells was affected by PLCB1, cell-cycle analysis was conducted, which showed that the G1–S transition was accelerated in RBE-PLCB1 and CCLP1-PLCB1 cells, but was significantly inhibited in HuCCT1 PLCB1 KD3 and KMBC PLCB1 KD3 cells (Supplementary Fig. S4A). We next investigated the expression of cell-cycle regulators, cyclin D1, D2, D3, E1, and E2, and CDK2. cyclins D1, D2, and D3 were increased following PLCB1 overexpression, whereas PLCB1 KD3 had the opposite effect. Cyclin E1, and E2 and CDK2 were not significantly affected by PLCB1 (Supplementary Fig. S4B). Results obtained by qRT-PCR at the transcription level supported the above changes (Supplementary Fig. S4B). Additionally, we explored whether PLCB1 could contribute to CCA cell growth in vivo with two mouse models (10 mice/group) based on subcutaneous and orthotopic injection of RBE cells. The data indicated that PLCB1 increased the tumor volume, whereas knocking down PLCB1 almost completely abolished tumor formation (Fig. 2C; Supplementary Fig. S5A–S5C). To further confirm the increased tumorigenicity CCA cells overexpressing PLCB1, pX330 vectors coexpressing sgRNAs targeting PTEN/P53 and Cas9 and plasmids coexpressing PLCB1 were constructed. We established a CCA mouse model (27) by hydrodynamic injection with vector/sgPTEN/sgP53 and PLCB1/sgPTEN/sgP53 (20 mice/group). The data indicated that PLCB1 overexpression significantly increased the incidence of CCA, tumor volume and tumor burden caused by sgPTEN and sgP53 (Fig. 2D; Supplementary Fig. S5D). Subsequently, in subcutaneous tumor tissues, PLCB1 overexpression upregulated the key markers of the G1–S transition, whereas PLCB1 knockdown downregulated these markers (Supplementary Fig. S6A). We also evaluated Ki-67 expression, which was upregulated in the RBE-PLCB1 group and downregulated in the HuCCT1-PLCB1-KD3 group relative to their respective controls (Supplementary Fig. S6B). These results revealed that PLCB1 facilitates CCA cell proliferation in vitro and tumorigenesis in vivo.
Figure 2.

PLCB1 is important for the proliferation and oncogenicity of CCA cells. A, PLCB1 overexpression and knockdown could affect proliferation abilities of indicated CCA cell lines via Edu assay analysis. **, P < 0.01; ***, P < 0.001, based on Student t test. Scale bars, 100× = 400 μm. B, Typical images of colony formation assays and colony counts. *, P < 0.1; **, P < 0.01; ***, P < 0.001, based on Student t test. C, Typical images of subcutaneous xenograft mice models and hematoxylin and eosin staining. Scale bars, 400× = 100 μm. D, Typical images of bioluminescence pictures, hematoxylin and eosin (H&E), and CK19 staining of vector/sgPTEN/sgP53 and PLCB1/sgPTEN/sgP53 C57BL mice livers at 8 weeks after hydrodynamic injection respectively. ****, P < 0.0001, based on Student t test. Scale bars, 100× = 400 μm. All experiments were implemented three times, and data are presented as mean ± SD.
PLCB1 promotes the motility, migration, and invasion of CCA cells in vitro and in vivo
We then explored whether PLCB1 could facilitate CCA cell migration and invasion in vitro and metastasis in vivo. As expected, PLCB1 overexpression increased RBE and CCLP1 cell migration, whereas PLCB1 knockdown reduced HuCCT1 and KMBC cell migration (Fig. 3A). Matrigel-coated (for invasion) and Matrigel-free (for migration) Transwell assays also showed that overexpressing PLCB1 increased the invasion and migration of RBE and CCLP1 cells, whereas silencing PLCB1 had the opposite effect on HuCCT1 and KMBC cells (Fig. 3B; Supplementary Fig. S6C). Three animal models were established for in vivo investigation by injecting stably transfected cells (RBE-PLCB1, HuCCT1-PLCB1-KD3, and controls). Liver metastasis, peritoneal cavity metastasis, and lung metastasis were measured (10 mice/group). The number of tumor nodules in the liver was increased in mice implanted with RBE-PLCB1 cells compared with the control group and was reduced in mice implanted with HuCCT1-PLCB1-KD3 cells compared with vector control cells 4 weeks after the operation (Supplementary Fig. S6D). Analysis of the peritoneal cavity metastasis model also showed that PLCB1 overexpression increased the number of metastatic nodules, whereas PLCB1 knockdown reduced the number of nodules (Supplementary Fig. S6E). We next injected stably transfected cells into nude mice via the tail vein and monitored the development of metastatic nodules in the lungs. After 6 weeks, the number of nodules and incidence rate were increased with RBE-PLCB1 cells and reduced with HuCCT1-PLCB1-KD3 cells compared with the respective control cells (Fig. 3C). The presence of pulmonary metastatic nodules was also tested after dissecting lung tissue (Fig. 3C). These data provide evidence that PLCB1 is involved in promoting CCA metastasis.
Figure 3.

PLCB1 is important for the migration and invasion of CCA cells. A, Wound-healing assays for indicated cell lines showed motility changes with PLCB1 expression. ***, P < 0.001; ****, P < 0.0001, based on Student t test. Scale bars, 100× = 400 μm. B, Typical images of Matrigel-coated (for invasion) and Matrigel-uncoated (for migration) transwell assays for indicated cell lines. Scale bars, 100× = 400 μm. C, Typical images of bioluminescence pictures, specimens, and hematoxylin and eosin staining of lung metastasis for indicated CCA cell lines. ***, P < 0.001, based on Student t test. Scale bars, 400× = 100 μm. All experiments were implemented three times, and data are presented as mean ± SD.
PLCB1 induces EMT in CCA, and snail is essential for PLCB1-mediated CCA metastasis
EMT is a crucial process for the invasion and metastasis of malignant tumors, so we sought to investigate the relationship between PLCB1 and EMT in CCA. We found that CCA samples with low PLCB1 expression had higher E-cadherin levels than those with high PLCB1 expression. Concurrently, N-cadherin and vimentin had an inverse expression pattern, suggesting that PLCB1 is correlated with EMT markers (Fig. 4A; Supplementary Fig. S7A). Proteins and mRNAs of CCA human samples exhibited identical effects (Supplementary Fig. S7B). Analyzing the effects of PLCB1 changes on EMT-related markers of CCA cell lines, we observed that PLCB1 overexpression in RBE and CCLP1 cells reduced the expression of E-cadherin and increased the expression of N-cadherin and vimentin. The opposite results were obtained in HuCCT1-PLCB1-KD3 and KMBC-PLCB1-KD3 cells (Fig. 4B and C; Supplementary Figs. S7C and S8A). Previous studies have demonstrated that EMT markers can be regulated by transcription factors such as Snail, Slug, Twist, and ZEB1/2 (28, 29). We next investigated whether the protein and mRNA levels of these transcription factors were affected by PLCB1. Our results revealed that only snail protein and mRNA levels were significantly upregulated after PLCB1 overexpression, whereas the opposite was observed after PLCB1 knockdown (Fig. 4B; Supplementary Fig. S7C). Consistent changes were obtained by immunofluorescence staining (Supplementary Fig. S8A). The analysis of proteins and mRNAs extracted from tumor-forming tissue in nude mice was consistent with the above in vitro experiments (Supplementary Fig. S8B and S8C). The role of snail in PLCB1-mediated EMT progression was then examined by overexpressing or knocking down snail. The use of a lentivirus encoding a snail shRNA in RBE-PLCB1 and CCLP1-PLCB1 cells restored E-cadherin expression and reduced N-cadherin and vimentin expression. The use of a lentivirus encoding a snail overexpression construct in HuCCT1-PLCB1-KD3 and KMBC-PLCB1-KD3 cells reversed the upregulation of E-cadherin and decreased N-cadherin and vimentin expression (Fig. 4D). Knockdown of snail reduced the migration and invasion of RBE-PLCB1 and CCLP1-PLCB1 cells, and, the overexpression of snail enhanced the migration and invasion of HuCCT1-PLCB1-KD3 and KMBC-PLCB1-KD3 cells (Fig. 4E; Supplementary Fig. S8D). Furthermore, the liver metastatic nodules in mice injected with RBE-PLCB1-sh Snail cells were fewer and smaller than those in mice injected with RBE-PLCB1-sh control cells. Conversely, the PLCB1 knockdown–mediated decrease in liver metastatic nodules with HuCCT1 cells was significantly reversed by snail overexpression. These data were further supported by the liver sections in mice (Supplementary Fig. S9A). Our results indicate that snail plays a critical role in PLCB1-induced EMT and metastasis in CAA.
Figure 4.

Snail is essential for PLCB1-mediated EMT in CCA. A, Typical images of IHC staining in CCA tissues presented expression of PLCB1 was associated with the EMT-related biomarkers. Scale bars, 100× = 400 μm; 400× = 100 μm. B, Western blot showed the expression of EMT-associated proteins and transcription factors. C, Typical IF images of the expression of E-cadherin and vimentin for indicated CCA cell lines. Scale bars, 400× = 100 μm. D, Western blot presented that snail overexpression and knockdown restored the levels of the EMT-related markers and was affected by PLCB1 in indicated CCA cell lines. Sh Con, snail knockdown negative control. Sh Snail, snail knockdown. Con, snail overexpression negative control. Snail, snail overexpression. E, Typical images of migration and invasion assays demonstrated that upregulated and downregulated snail restored the number of the RBE-PLCB1 group and the HuCCT1-PLCB1-KD3 group. Scale bars, 100× = 400 μm. All experiments were implemented three times, and data are presented as mean ± SD.
PLCB1 stimulates the PI3K/AKT/GSK3β/snail signaling pathway to regulate EMT
Because PLCB1 induced CCA metastasis, we aimed to determine the mechanism by which PLCB1 regulates EMT. We measured the abundance of multiple proteins and their phosphorylated forms via a protein microarray (Full Moon Microsystems) and found that phosphorylation of AKT and ERK was regulated by PLCB1, which prioritized the pathway proteins related to tumor metastasis and invasion (Fig. 5A). Subsequently, these results were confirmed in other CCA cell lines (Fig. 5B). In addition, IHC staining in C57BL and FVB/N (H2d) mice spontaneous tumor tissues further verified that PLCB1 overexpression could significantly enhance the phosphorylation of AKT (Fig. 5C). Therefore, we conducted experiments to determine whether PLCB1 regulated CCA cell metastasis via the AKT or ERK signaling pathways. MK2206, a specific small-molecule inhibitor of AKT, reversed the downregulation of E-cadherin and upregulation of N-cadherin and vimentin in both RBE-PLCB1 and CCLP1-PLCB1 cells (Fig. 5D). In addition, the number of lung metastatic nodules in mice (10 mice/group) implanted with PLCB1-overexpressing RBE cells was reduced by MK2206 (Supplementary Fig. S9B). However, U1026 and Senkyunolide I, an ERK inhibitor and agonist, respectively, had no impact on the levels of the proteins mentioned above (Supplementary Fig. S9C and S9D). To rule out the possibility of off-target effects of MK2206, lentiviruses encoding AKT shRNA or overexpression constructs were used. The AKT shRNA lentivirus had similar effects to MK2206, and the lentivirus that stably overexpressed AKT had opposite effects (Fig. 5E; Supplementary Fig. S10A). To confirm that AKT is vital for PLCB1 to induce the EMT process, specific small-molecule inhibitors of PKCθ and GRK2 (sotrastaurin and GSK180736A) were used to verify that they had no impact on the levels of EMT-related proteins, which were affected by PLCB1 (Supplementary Fig. S10B and S10C). Furthermore, we observed that the effects of PLCB1 in normal bile duct and liver cell lines were similar to the effects in CCA cell lines (Supplementary Fig. S10D). Overexpression of PLCB1 enhanced the levels of total and phosphorylated β-catenin and β-catenin nuclear accumulation (Supplementary Fig. S11A–S11C). In short, our data demonstrate that the AKT pathway is vital for the PLCB1-induced EMT process in CCA.
Figure 5.

The PI3K/AKT/GSK3β/snail axis is vital for PLCB1-mediated EMT. A, Phosphoproteome array analysis of the expression changes of 157 phospho-specific proteins and 147 non-phospho pairs of 16 cell signaling pathways upon PLCB1 overexpression in HuCCT1 cells. Phosphorylation levels increased by FC (fold changes) ≥ 1.2 compared between HuCCT1 PLCB1 cell lines and HuCCT1 cell lines are shown in the histogram. The phosphorylated proteins of the PI3K/AKT and ERK pathways, which we paid more attention, are labeled red. B, Western blot analysis of key proteins in the PI3K/AKT and ERK pathways for indicated cell lines. C, Typical images of IHC staining in C57BL and FVB/N (H2d) mice spontaneous tumor tissues presented PLCB1 overexpression enhanced the expression level of pAKT. Scale bars, 100× = 400 μm; 400× = 100 μm. D, The expression levels of key proteins of the PI3K/AKT pathway and EMT process for indicated CCA cell lines were determined by Western blot after the treatment of 0.2 μmol/L MK2206. E, The expression levels of key proteins of the PI3K/AKT pathway and EMT process in the indicated cell lines were determined by Western blot after stably transfected AKT knockdown lentivirus. All experiments were implemented three times, and data are presented as mean ± SD.
PABPC1 interacts with PLCB1 and regulates the PLCB1-mediated PI3K/AKT/GSK3β/snail signaling pathway and metastasis in CCA
To better understand the mechanism of the PLCB1-affected PI3K/AKT/GSK3β/snail axis, we characterized the protein interaction profile of PLCB1 in RBE and HuCCT1 cell lines by mass spectrometry. To eliminate the heterogeneity between CCA cells, we identified 15 candidate proteins that were identified in both two mass spectrometry analyses (Supplementary Fig. S12A; Supplementary Table S3). We found that PABPC1 was highly expressed in human CCA tissue and was significantly increased compared with border tissue (Supplementary Fig. S12B and S12C). These results agreed with those from the gene-expression database (Supplementary Fig. S12D and S12E). We also verified that the protein and mRNA levels of PABPC1 in CCA cell lines were significantly higher than in normal cell lines (Supplementary Fig. S12F). Both endogenous and exogenous PLCB1 interacted with PABPC1, and endogenous and exogenous PABPC1 were able to interact with PLCB1, indicating that there was a correlation between PLCB1 and PABPC1 (Fig. 6A). Using IF staining, we demonstrated that PLCB1 and PABPC1 were both localized in the cytoplasm (Fig. 6B). To test whether the proteins could directly interact, we observed that endogenous and exogenous PLCB1 could directly interact with PABPC1 (Fig. 6C). We hypothesized that PABPC1 regulates the PLCB1-mediated PI3K/AKT/GSK3β/snail signaling pathway and EMT process. Lentiviruses with encoding PABPC1 overexpression or knockdown constructs were transfected into HuCCT1-PLCB1-KD3 or RBE-PLCB1 cells, respectively. Figure 6D shows that PABPC1 activated the PI3K/AKT/GSK3β/snail pathway and the EMT process in HuCCT1-PLCB1-KD3 cells. In contrast, the knockdown of PABPC1 reduced the activation of the AKT pathway and reversed the EMT process in RBE-PLCB1 cells (Fig. 6E). To understand the mechanism by which PABPC1 regulates the PLCB1-mediated PI3K/AKT pathway and EMT process, we investigated how endogenous and exogenous PABPC1 interact with PI3K (Supplementary Fig. S13A and S13B). In addition, to better understand whether PABPC1 is related to lymph node metastasis in patients with CCA, we verified that CCA tissues with lymph node metastasis had higher protein levels of PABPC1 than those without (Supplementary Fig. S13C). These findings indicated that PABPC1 can interact with PLCB1 and regulate the PLCB1-mediated AKT pathway and EMT process.
Figure 6.

PABPC1 is closely related to PLCB1-mediated PI3K/AKT/GSK3β/snail signaling in CCA. A, Endogenous and exogenous coimmunoprecipitation indicated that PLCB1 could interact with PABPC1. B, Colocalization between PLCB1 and PABPC1 was explored by immunofluorescence in the indicated CCA cell lines. Scale bars, 400× = 100 μm. C, Typical images of the Duolink PLA experiment predicted PABPC1 directly interacted with PLCB1 via the fluorescence microscope. Scale bars, 400× = 100 μm. D and E, The expression levels of key proteins of the PI3K/AKT pathway and EMT process for indicated CCA cell lines were determined by Western blot after stable PABPC1 knockdown and overexpression lentiviral transfection. All experiments were implemented three times, and data are presented as mean ± SD.
MiR-26b-5p targets PLCB1 and inhibits PLCB1-mediated metastasis in CCA
Two public CCA data sets with sample sizes of more than 100 cases, namely, the MSK-IMPACT study (n = 195; ref. 30) and the Shanghai study (n = 103; ref. 31), were retrieved from the cBioPortal database. PLCB1 was not mutated in any of these samples, nor did it have DNA copy-number variation (Supplementary Fig. S14A and S14B). Therefore, we aimed to determine whether downregulation of miRNAs that inhibit and degrade PLCB1 leads to increased PLCB1 expression. To explore the upstream regulatory mechanism of PLCB1 overexpression in CCA, we used TargetScan, starBase, and OncomiR to identify candidate miRNAs. We focused on miRNA-26b-5p, miRNA-20a-5p, and miRNA-17–5p, which were expressed at low levels in CCA samples, for further study (Fig. 7A). Only miRNA-26b-5p was statistically significant in human CCA samples (Fig. 7B; Supplementary Fig. S15A). Similarly, the level of miR-26b-5p in HIBEpiC and Chang cell lines was higher than in CCA cell lines (Fig. 7C). To confirm the direct interaction between miR-26b-5p and the 3′UTR of PLCB1 mRNA, we constructed wild-type and mutant sequences of the 3′UTR for luciferase-reporter assays (Fig. 7D). Luciferase activity was suppressed by miR-26b-5p in HEK293T cells transfected with wild-type PLCB1–3′ UTR, whereas there was no difference in the mutant group (Fig. 7E). We therefore concluded that miR-26b-5p was a direct target of PLCB1. We next downregulated or upregulated miRNA-26b-5p with inhibitors in RBE cells or with mimics in HuCCT1 cells. To validate that miR-26b-5p participates in the CCA-promoting function of PLCB1, rescue experiments were performed. These revealed that the knockdown or overexpression of PLCB1 could restore the activation of the AKT pathway and alter the expression of EMT-related proteins in RBE cell lines treated with miR-26b-5p inhibitors or in HuCCT1 cell lines caused by miR-26b-5p mimics (Fig. 7F and G). Moreover, the knockdown or overexpression of PLCB1 reversed the changes in invasion and migration induced by miR-26b-5p inhibitors in RBE cells or by miR-26b-5p mimics in HuCCT1 cells (Fig. 7H; Supplementary Fig. S15B). The results of lung metastasis experiments in nude mice were consistent with those of in vitro experiments (Supplementary Fig. S15C). Taken together, these results indicate that miR-26b-5p can act on PLCB1 and prevent PLCB1-mediated metastasis in CCA.
Figure 7.

miR-26b-5p targets PLCB1 in a PI3K/AKT/GSK3β/snail manner. A, Analysis using TargetScan, starBase, and OncomiR databases revealed several candidate miRNAs that might regulate PLCB1. B, miR-26b-5p expression was analyzed by qRT-PCR. ****, P < 0.0001, based on Student t test. C, The expression levels of miR-26b-5p in CCA cell lines and normal bile duct and liver cell lines. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001, based on the one-way ANOVA. D, Binding sites of miR-26b-5p in wild-type (WT) 3′-untranslated region (UTR) of PLCB1 and corresponding mutant type were constructed. E, Luciferase-reporter assays showed that luciferase activity of HEK293T transfected with WT 3′-UTR was obviously inhibited by miR-26b-5p overexpression. **, P < 0.01, based on Student t test. F, The expression of the PI3K/AKT pathway and EMT-related markers was analyzed by Western blot after transfecting with miR-26b-5p inhibitor NC (negative control), miR-26b-5p inhibitor, miR-26b-5p inhibitor plus PLCB1 vector, and miR-26b-5p inhibitor plus PLCB1-KD3. G, The expression of the PI3K/AKT pathway and EMT-related markers was analyzed by Western blot after transfecting with miR-26b-5p mimic NC (negative control), miR-26b-5p mimic, miR-26b-5p mimic plus PLCB1 Con, and miR-26b-5p mimic plus PLCB1. H, Migration and invasion assays were performed in HuCCT1 and RBE cell lines transfected with miR-26b-5p inhibitor NC (negative control), miR-26b-5p inhibitor, miR-26b-5p inhibitor plus PLCB1 vector, miR-26b-5p inhibitor plus PLCB1-KD3, miR-26b-5p mimic NC (negative control), miR-26b-5p mimic, miR-26b-5p mimic plus PLCB1 Con, and miR-26b-5p mimic plus PLCB1. Scale bars, 100× = 400 μm. All experiments were implemented three times, and data are presented as mean ± SD.
MK2206 can reverse the chemotherapy resistance to gemcitabine/cisplatin induced by ectopic PLCB1
A growing body of evidence suggests that the efficacy of gemcitabine/cisplatin in the treatment of CCA can be enhanced by inhibiting the AKT pathway (32, 33). Our findings suggest that the AKT pathway is vital for PLCB1-mediated EMT in CCA. We hypothesized that MK2206 could reverse the chemotherapy resistance to gemcitabine/cisplatin induced by ectopic PLCB1. Establishing an in vitro treatment model, we determined that MK2206 significantly enhanced the inhibitory effects of gemcitabine plus cisplatin on the proliferation of HuCCT1 and KMBC cell lines, which have high PLCB1 expression. However, cotreatment with MK2206 and gemcitabine plus cisplatin had only minor effects on RBE and CCLP1 cell lines, which have low PLCB1 expression (Supplementary Fig. S16A). Similar results were obtained from RBE and CCLP1 cell lines overexpressing PLCB1 (Supplementary Fig. S16B). We also established an in vivo therapeutic mouse model, and the results obtained from the mouse model were consistent with those obtained from in vitro experiments (Supplementary Fig. S17A and S17B). To summarize, MK2206 can reverse the chemotherapeutic resistance to gemcitabine/cisplatin induced by ectopic PLCB1.
The combination of PLCB1 with PABPC1, pAKT, or miR-26b-5p indicates a poor prognosis for CCA
Given the regulatory relationship between PLCB1 and PABPC1, pAKT, and miR-26b-5p obtained above, we next analyzed their abundance in CCA samples. We found positive correlations between PLCB1 and PABPC1 and between PLCB1 and pAKT and a negative correlation between PLCB1 and miR-26b-5p (Fig. 8A–C). In addition, patients presenting with PLCB1 and PABPC1 or pAKT coexpression had comparatively worse prognoses (Fig. 8D and E; Supplementary Fig. S18A and S18B). Patients with high PLCB1 and low miR-26b-5p exhibited similarly poor outcomes (Fig. 8F; Supplementary Fig. S18C). In short, the combination of two parameters increases the prognostic value compared with PLCB1 alone.
Figure 8.

Analysis of clinical prognosis in combination of PLCB1 with PABPC1, pAKT, or miR-26b-5p. A, Typical images of IHC staining analysis of PLCB1 and pAKT expression in 60 CCA human tissues. Scale bars, 100× = 400 μm; 400× = 100 μm. B, Typical images of IHC staining analysis of PLCB1 and PABPC1 expression in 60 CCA human tissues. Scale bars, 100× = 400 μm; 400× = 100 μm. C, Correlation analysis indicated that PLCB1 was positively correlated with pAKT or PABPC1 and was an inverse correlation with miR-26b-5p. Pearson correlation analysis was used. D, Kaplan–Meier analysis of fraction survival in patients with variable expressions of PLCB1 and pAKT. E, Kaplan–Meier analysis of fraction survival in patients with variable expressions of PLCB1 and PABPC1. F, Kaplan–Meier analysis of fraction survival in patients with variable expressions of PLCB1 and miR-26b-5p. All experiments were implemented three times, and data are presented as mean ± SD.
Discussion
PLCB1, which exists in the cytoplasm of cells and is located on chromosome 20p12, is one of the significant members of the phospholipase family. PLCB1 plays an important role in intracellular signal transduction, and aberrant signal transduction is one of the significant causes of oncogenesis and cancer progression (34, 35). The upregulation of PLCB1 has been reported in several types of malignant tumors. Li and colleagues (36) verified that PLCB1 was upregulated and related to cell proliferation in hepatocellular carcinoma (HCC), but they did not explore in any depth the mechanism by which PLCB1 activates the ERK pathway in HCC. Similarly, although Wang and colleagues (37) reported the diagnostic and prognostic value of mRNA expression of the phospholipase C β family genes in hepatitis B virus–associated HCC, they did not explore the mechanisms of PLCB1-mediated proliferation and metastasis in HCC. It is well known that HCC and CCA have different biological characteristics. Although the incidence of CCA is lower than that of HCC, CCA has a worse prognosis and shorter median survival time. Moreover, CCA has no effective targeted drugs such as sorafenib for HCC.
In our research, via The Cancer Genome Atlas and GEO databases, we found that PLCB1 was highly expressed in CCA. We also demonstrated that PLCB1 was upregulated in CCA tissues and cell lines. Hence, we propose that PLCB1 has an effect on the occurrence and development of CCA. By analyzing the relationship between PLCB1 and clinical characteristics, survival, and recurrence, we found significant differences in TNM stage, lymph node status, distant metastasis, and prognosis between patients with CCA who exhibited high PLCB1 expression and those patients who exhibited low PLCB1 expression. Therefore, PLCB1 was identified as a novel oncogene and independent prognostic biomarker for CCA. To discover the pathophysiologic and clinical significance and latent mechanism of PLCB1 in CCA, both loss-of-function and gain-of-function experiments were performed, which showed that knockdown of PLCB1 inhibited and overexpression of PLCB1-stimulated CCA proliferation, motility, invasion, and metastasis in vitro and in vivo. To probe the potential mechanism by which PLCB1 enhances CCA proliferation, we performed a series of in vitro and in vivo experiments to verify that the differential expression of PLCB1 could affect the proliferation and tumorigenicity of CCA cells and impact the G1–S transition. The mechanism by which PLCB1 contributes to CCA proliferation has not been well explored, and we hope to determine this mechanism in future research. EMT has been shown to significantly accelerate metastasis in epithelium-derived carcinomas, including CCA (38, 39). We hypothesized that PLCB1 facilitated CCA metastasis by regulating EMT. Using in vitro and in vivo metastasis-related experiments, we demonstrated that PLCB1 could affect the protein and mRNA levels of EMT-related markers in CCA cell lines. Previous studies have demonstrated that EMT markers can be regulated by transcription factors such as Snail, Slug, Twist, and ZEB1/2 (28, 29). Via screening these EMT-related transcription factors, our findings revealed that only snail protein and mRNA levels were significantly affected by PLCB1 overexpression or knockdown. Subsequently, in order to discover the signaling pathway by which PLCB1 affected snail, multiple pathways were screened using a protein microarray. Although PLCB1 influenced multiple proteins, PKCθ and GRK2 were the cytoplasmic kinase whose expression were most affected by PLCB1. However, our findings revealed that PKCθ and GRK2 did not affect PLCB1-mediated EMT in CCA cells. Previous studies have demonstrated that the AKT and ERK pathways are closely related to malignant tumor metastasis (40, 41). Therefore, we focused on pAKT and p-ERK. Although PLCB1 affected the level of p-ERK, the activation of ERK was not critical for the PLCB1-mediated EMT process. PLCB1 might affect CCA proliferation via the ERK pathway, but further experiments are required to verify this hypothesis. In addition, our findings showed that PI3K/AKT signaling pathway activation and its downstream effects were essential for PLCB1-mediated EMT in CCA. Although AKT1 is a known oncogene and regulates many processes, including cell proliferation, cell survival, and the cell cycle, we paid close attention to the role of AKT in PLCB1-mediated EMT (42–44). Then, to better understand the manner by which PLCB1 affected PI3K/AKT signaling, we identified and verified poly(A) binding protein cytoplasmic 1 (PABPC1) as a PLCB1-binding protein using mass spectrometry, co-IP, and Duolink PLA experiments. PABPC1, which is shuttled between the nucleus and cytoplasm, can bind specifically to the poly(A) tail of mRNAs to regulate mRNA metabolism (45). PABPC1 is also associated with a variety of malignant tumors. Likewise, we discovered that PABPC1 was upregulated in human CCA tissue and cell lines and could regulate the PLCB1-mediated PI3K/AKT pathway, EMT, and CCA metastasis.
We then explored the reasons for PLCB1 overexpression in CCA. Two public CCA data sets with sample sizes of more than 100 cases, namely, the MSK-IMPACT study (n = 195; ref. 27) and Shanghai study (n = 103; ref. 28), were retrieved from the cBioPortal database. PLCB1 was not mutated in any of these samples, nor did it have DNA copy-number variation. Therefore, we aimed to determine whether downregulated miRNAs that inhibit and degrade PLCB1 lead to PLCB1 overexpression. Accumulating evidence demonstrates that miRNAs have instrumental functions in tumor development by suppressing translation and degrading mRNAs (46). Here, via the TargetScan, starBase, and OncomiR databases, we identified 11 miRNAs that had potential binding sites for PLCB1. However, only miR-26b-5p was significantly downregulated in CCA samples. Using in vitro and in vivo experiments, we found that miR-26b-5p directly targeted PLCB1 to negatively regulate PLCB1 expression and that miR-26b-5p blocked PLCB1-mediated EMT and CCA metastasis. A growing body of evidence suggests that the efficacy of gemcitabine/cisplatin chemotherapy for CCA can be enhanced by inhibiting the AKT pathway (30, 31). Using in vitro and in vivo therapeutic models, we found that the small-molecule AKT inhibitor MK2206 could reverse the chemotherapy resistance to gemcitabine/cisplatin induced by overexpression of PLCB1 in CCA. Finally, we used correlation analyses to determine that the combination of PLCB1 and PABPC1, pAKT, or miR-26b-5p could be used to predict prognosis for patients with CCA, which may have guiding significance for future clinical practice. To our knowledge, our study provides the first evidence that PLCB1 is a crucial oncogene in CCA and is involved in CCA proliferation and metastasis. We revealed that PLCB1 could induce CCA metastasis by activating the PI3K/AKT signaling pathway. PABPC1 became a bridge between PLCB1 and PI3K and regulated PLCB1-mediated EMT. In addition, miR-26b-5p targeted PLCB1 and negatively regulated PLCB1-mediated EMT and CCA metastasis. MK2206 could inhibit the chemotherapy resistance to gemcitabine/cisplatin induced by PLCB1 overexpression. The combination of PLCB1 and PABPC1, pAKT, or miR-26b-5p was predictive of poor prognosis for patients with CCA. We propose that CCA patients with high PLCB1 can be treated with AKT inhibitors combined with gemcitabine and cisplatin. We also hope to develop specific inhibitors for PLCB1 and apply them in clinical trials. Although cancer stem cells are closely related to metastasis, there are no reports in the literature on PLCB1 in cancer stem cells. Therefore, in future studies, we will investigate the role of PLCB1 in cancer stem cells.
In conclusion, our study demonstrates that the PLCB1–PI3K–AKT axis plays a crucial role in CCA development. Our study provides a basis for understanding the mechanism of CCA progression and suggests new prognostic biomarkers for CCA patients.
Authors' Disclosures
No disclosures were reported.
Supplementary Material
Supplementary Figure S1-S18
Clinical characteristics of 60 CCA patients depending on PLCB1 expression levels
Univariate and multivariate analyses of factors associated with survival in CCA patients.
Information of PLCB1 associated RNA binding proteins
Supplementary methods
Acknowledgments
The authors thank Qingsong Hu for daily discussions and helpful suggestions. This study was supported by the National Key R&D Program of China (grant no. 2019YFA0709300), the National Natural Science Foundation of China (grant nos. 81772588, U19A2008, 81972307, and 81773194), China Postdoctoral Science Foundation (2020M671911, 2020TQ0313), and the Natural Science Foundation of Anhui Province (2008085QH376).
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Contributions
S. Liang: Conceptualization, data curation, formal analysis, investigation, visualization, methodology, writing–original draft, writing–review and editing. H. Guo: Data curation, and methodology. K. Ma: Data curation, methodology. X. Li: Formal analysis and methodology. D. Wu: Formal analysis and methodology. Y. Wang: Formal analysis and methodology. W. Wang: Software. S. Zhang: Software. Y. Cui: Investigation. Y. Liu: Visualization. L. Sun: Visualization. B. Zhang: Investigation. M. Xin: Methodology. N. Zhang: Methodology. H. Zhou: Visualization. Y. Liu: Conceptualization, funding acquisition, writing–original draft, writing–review and editing. J. Wang: Conceptualization, funding acquisition, writing–review and editing. L. Liu: Conceptualization, supervision, funding acquisition, validation, project administration, writing–review and editing.
References
- 1. Razumilava N, Gores GJ. Cholangiocarcinoma. Lancet 2014;383:2168–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bridgewater J, Galle PR, Khan SA, Llovet JM, Park JW, Patel T, et al. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J Hepatol 2014;60:1268–89. [DOI] [PubMed] [Google Scholar]
- 3. Valle J, Wasan H, Palmer DH, Cunningham D, Anthoney A, Maraveyas A, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med 2010;362:1273–81. [DOI] [PubMed] [Google Scholar]
- 4. Rizvi S, Khan SA, Hallemeier CL, Kelley RK, Gores GJ. Cholangiocarcinoma - evolving concepts and therapeutic strategies. Nat Rev Clin Oncol 2018;15:95–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jarnagin WR, Fong Y, DeMatteo RP, Gonen M, Burke EC, Bodniewicz BJ, et al. Staging, resectability, and outcome in 225 patients with hilar cholangiocarcinoma. Ann Surg 2001;234:507–17; discussion 17–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Skipworth JR, Olde Damink SW, Imber C, Bridgewater J, Pereira SP, Malago M. Surgical, neo-adjuvant and adjuvant management strategies in biliary tract cancer. Aliment Pharmacol Ther 2011;34:1063–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Martelli AM, Fiume R, Faenza I, Tabellini G, Evangelista C, Bortul R, et al. Nuclear phosphoinositide specific phospholipase C (PI-PLC)-beta 1: a central intermediary in nuclear lipid-dependent signal transduction. Histol Histopathol 2005;20:1251–60. [DOI] [PubMed] [Google Scholar]
- 8. Spyridakis S, Leondaritis G, Nakos G, Lekka ME, Galanopoulou D. A specific phospholipase C activity regulates phosphatidylinositol levels in lung surfactant of patients with acute respiratory distress syndrome. Am J Respir Cell Mol Biol 2010;42:357–62. [DOI] [PubMed] [Google Scholar]
- 9. Ngoh A, McTague A, Wentzensen IM, Meyer E, Applegate C, Kossoff EH, et al. Severe infantile epileptic encephalopathy due to mutations in PLCB1: expansion of the genotypic and phenotypic disease spectrum. Dev Med Child Neurol 2014;56:1124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Poduri A, Chopra SS, Neilan EG, Elhosary PC, Kurian MA, Meyer E, et al. Homozygous PLCB1 deletion associated with malignant migrating partial seizures in infancy. Epilepsia 2012;53:e146–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McTague A, Appleton R, Avula S, Cross JH, King MD, Jacques TS, et al. Migrating partial seizures of infancy: expansion of the electroclinical, radiological and pathological disease spectrum. Brain 2013;136:1578–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Faenza I, Blalock W, Bavelloni A, Schoser B, Fiume R, Pacella S, et al. A role for PLCbeta1 in myotonic dystrophies type 1 and 2. FASEB J 2012;26:3042–8. [DOI] [PubMed] [Google Scholar]
- 13. Bavelloni A, Dmitrienko GI, Goodfellow VJ, Ghavami A, Piazzi M, Blalock W, et al. PLCbeta1a and PLCbeta1b selective regulation and cyclin D3 modulation reduced by kinamycin F during k562 cell differentiation. J Cell Physiol 2015;230:587–94. [DOI] [PubMed] [Google Scholar]
- 14. Strassheim D, Shafer SH, Phelps SH, Williams CL. Small cell lung carcinoma exhibits greater phospholipase C-beta1 expression and edelfosine resistance compared with non-small cell lung carcinoma. Cancer Res 2000;60:2730–6. [PubMed] [Google Scholar]
- 15. Sengelaub CA, Navrazhina K, Ross JB, Halberg N, Tavazoie SF. PTPRN2 and PLCβ1 promote metastatic breast cancer cell migration through PI(4,5)P2-dependent actin remodeling. EMBO J 2016;35:62–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nomoto K, Tomita N, Miyake M, Xhu DB, LoGerfo PR, Weinstein IB. Expression of phospholipases gamma 1, beta 1, and delta 1 in primary human colon carcinomas and colon carcinoma cell lines. Mol Carcinog 1995;12:146–52. [DOI] [PubMed] [Google Scholar]
- 17. Chen Y, Gao DY, Huang L. In vivo delivery of miRNAs for cancer therapy: challenges and strategies. Adv Drug Deliv Rev 2015;81:128–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Niu F, Kazimierska M, Nolte IM, Terpstra MM, de Jong D, Koerts J, et al. The miR-26b-5p/KPNA2 axis is an important regulator of Burkitt lymphoma cell growth. Cancers 2020;12:1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang R, Niu Z, Pei H, Peng Z. Long noncoding RNA LINC00657 induced by SP1 contributes to the non-small cell lung cancer progression through targeting miR-26b-5p/COMMD8 axis. J Cell Physiol 2020;235:3340–9. [DOI] [PubMed] [Google Scholar]
- 20. Jia J, Shi Y, Chen L, Lai W, Yan B, Jiang Y, et al. Decrease in lymphoid specific helicase and 5-hydroxymethylcytosine is associated with metastasis and genome instability. Theranostics 2017;7:3920–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kühn U, Wahle E. Structure and function of poly(A) binding proteins. Biochim Biophys Acta 2004;1678:67–84. [DOI] [PubMed] [Google Scholar]
- 22. Schaid DJ, McDonnell SK, FitzGerald LM, DeRycke L, Fogarty Z, Giles GG, et al. Two-stage study of familial prostate cancer by whole-exome sequencing and custom capture identifies 10 novel genes associated with the risk of prostate cancer. Eur Urol 2021;79:353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang H, Sheng C, Yin Y, Wen S, Yang G, Cheng Z, et al. PABPC1 interacts with AGO2 and is responsible for the microRNA mediated gene silencing in high grade hepatocellular carcinoma. Cancer Lett 2015;367:49–57. [DOI] [PubMed] [Google Scholar]
- 24. Graetz D, Crews KR, Azzato EM, Singh RK, Raimondi S, Mason J, et al. Leukemic presentation of ALK-positive anaplastic large cell lymphoma with a novel partner, poly(A) binding protein cytoplasmic 1 (PABPC1), responding to single-agent crizotinib. Haematologica 2019;104:e218–e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vaquero J, Guedj N, Clapéron A, Nguyen Ho-Bouldoires TH, Paradis V, Fouassier L. Epithelial-mesenchymal transition in cholangiocarcinoma: from clinical evidence to regulatory networks. J Hepatol 2017;66:424–41. [DOI] [PubMed] [Google Scholar]
- 26. Qian Y, Yao W, Yang T, Yang Y, Liu Y, Shen Q, et al. aPKC-ι/P-Sp1/Snail signaling induces epithelial-mesenchymal transition and immunosuppression in cholangiocarcinoma. Hepatology 2017;66:1165–82. [DOI] [PubMed] [Google Scholar]
- 27. Xue W, Chen S, Yin H, Tammela T, Papagiannakopoulos T, Joshi NS, et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 2014;514:380–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stemmler MP, Eccles RL, Brabletz S, Brabletz T. Non-redundant functions of EMT transcription factors. Nat Cell Biol 2019;21:102–12. [DOI] [PubMed] [Google Scholar]
- 29. Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol 2014;16:488–94. [DOI] [PubMed] [Google Scholar]
- 30. Lowery MA, Ptashkin R, Jordan E, Berger MF, Zehir A, Capanu M, et al. Comprehensive molecular profiling of intrahepatic and extrahepatic cholangiocarcinomas: potential targets for intervention. Clin Cancer Res 2018;24:4154–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zou S, Li J, Zhou H, Frech C, Jiang X, Chu JS, et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat Commun 2014;5:5696. [DOI] [PubMed] [Google Scholar]
- 32. Jang DK, Lee YG, Chan Chae Y, Lee JK, Paik WH, Lee SH, et al. GDC-0980 (apitolisib) treatment with gemcitabine and/or cisplatin synergistically reduces cholangiocarcinoma cell growth by suppressing the PI3K/Akt/mTOR pathway. Biochem Biophys Res Commun 2020;529:1242–8. [DOI] [PubMed] [Google Scholar]
- 33. Zakharia K, Miyabe K, Wang Y, Wu D, Moser CD, Borad MJ, et al. Preclinical in vitro and in vivo evidence of an antitumor effect of CX-4945, a casein kinase II inhibitor, in cholangiocarcinoma. Transl Oncol 2019;12:143–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Perugorria MJ, Olaizola P, Labiano I, Esparza-Baquer A, Marzioni M, Marin JJG, et al. Wnt-beta-catenin signalling in liver development, health and disease. Nat Rev Gastroenterol Hepatol 2019;16:121–36. [DOI] [PubMed] [Google Scholar]
- 35. Morell CM, Strazzabosco M. Notch signaling and new therapeutic options in liver disease. J Hepatol 2014;60:885–90. [DOI] [PubMed] [Google Scholar]
- 36. Li J, Zhao X, Wang D, He W, Zhang S, Cao W, et al. Up-regulated expression of phospholipase C, β1 is associated with tumor cell proliferation and poor prognosis in hepatocellular carcinoma. Onco Targets Ther 2016;9:1697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang X, Huang K, Zeng X, Liu Z, Liao X, Yang C, et al. Diagnostic and prognostic value of mRNA expression of phospholipase C β family genes in hepatitis B virus-associated hepatocellular carcinoma. Oncol Rep 2019;41:2855–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev 2013;27:2192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nitta T, Mitsuhashi T, Hatanaka Y, Miyamoto M, Oba K, Tsuchikawa T, et al. Prognostic significance of epithelial-mesenchymal transition-related markers in extrahepatic cholangiocarcinoma: comprehensive immunohistochemical study using a tissue microarray. Br J Cancer 2014;111:1363–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tiemin P, Fanzheng M, Peng X, Jihua H, Ruipeng S, Yaliang L, et al. MUC13 promotes intrahepatic cholangiocarcinoma progression via EGFR/PI3K/AKT pathways. J Hepatol 2020;72:761–73. [DOI] [PubMed] [Google Scholar]
- 41. Wang C, Maass T, Krupp M, Thieringer F, Strand S, Wörns MA, et al. A systems biology perspective on cholangiocellular carcinoma development: focus on MAPK-signaling and the extracellular environment. J Hepatol 2009;50:1122–31. [DOI] [PubMed] [Google Scholar]
- 42. Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature 2001;411:355–65. [DOI] [PubMed] [Google Scholar]
- 43. Schmitz KJ, Lang H, Wohlschlaeger J, Sotiropoulos GC, Reis H, Schmid KW, et al. AKT and ERK1/2 signaling in intrahepatic cholangiocarcinoma. World J Gastroenterol 2007;13:6470–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim AH, Khursigara G, Sun X, Franke TF, Chao MV. Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol Cell Biol 2001;21:893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Grosset C, Chen CY, Xu N, Sonenberg N, Jacquemin-Sablon H, Shyu AB. A mechanism for translationally coupled mRNA turnover: interaction between the poly(A) tail and a c-fos RNA coding determinant via a protein complex. Cell 2000;103:29–40. [DOI] [PubMed] [Google Scholar]
- 46. Hammond SM. An overview of microRNAs. Adv Drug Deliv Rev 2015;87:3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1-S18
Clinical characteristics of 60 CCA patients depending on PLCB1 expression levels
Univariate and multivariate analyses of factors associated with survival in CCA patients.
Information of PLCB1 associated RNA binding proteins
Supplementary methods
Data Availability Statement
All data generated and analyzed during this study are included in this published article and its additional file.