

FGFR pathway activation inhibits IFNγ signaling in tumor cells, and FGFR inhibition with lenvatinib enhances antitumor immunity and the activity of anti-PD-1 antibodies.
Abstract
Combination therapies consisting of immune checkpoint inhibitors plus anti-VEGF therapy show enhanced antitumor activity and are approved treatments for patients with renal cell carcinoma (RCC). The immunosuppressive roles of VEGF in the tumor microenvironment are well studied, but those of FGF/FGFR signaling remain largely unknown. Lenvatinib is a receptor tyrosine kinase inhibitor that targets both VEGFR and FGFR. Here, we examine the antitumor activity of anti-PD-1 mAb combined with either lenvatinib or axitinib, a VEGFR-selective inhibitor, in RCC. Both combination treatments showed greater antitumor activity and longer survival in mouse models versus either single agent treatment, whereas anti-PD-1 mAb plus lenvatinib had enhanced antitumor activity compared with anti-PD-1 mAb plus axitinib. Flow cytometry analysis showed that lenvatinib decreased the population of tumor-associated macrophages and increased that of IFNγ-positive CD8+ T cells. Activation of FGFR signaling inhibited the IFNγ-stimulated JAK/STAT signaling pathway and decreased expression of its target genes, including B2M, CXCL10, and PD-L1. Furthermore, inhibition of FGFR signaling by lenvatinib restored the tumor response to IFNγ stimulation in mouse and human RCC cell lines. These preclinical results reveal novel roles of tumor FGFR signaling in the regulation of cancer immunity through inhibition of the IFNγ pathway, and the inhibitory activity of lenvatinib against FGFRs likely contributes to the enhanced antitumor activity of combination treatment comprising lenvatinib plus anti-PD-1 mAb.
Significance:
FGFR pathway activation inhibits IFNγ signaling in tumor cells, and FGFR inhibition with lenvatinib enhances antitumor immunity and the activity of anti-PD-1 antibodies.
Graphical Abstract
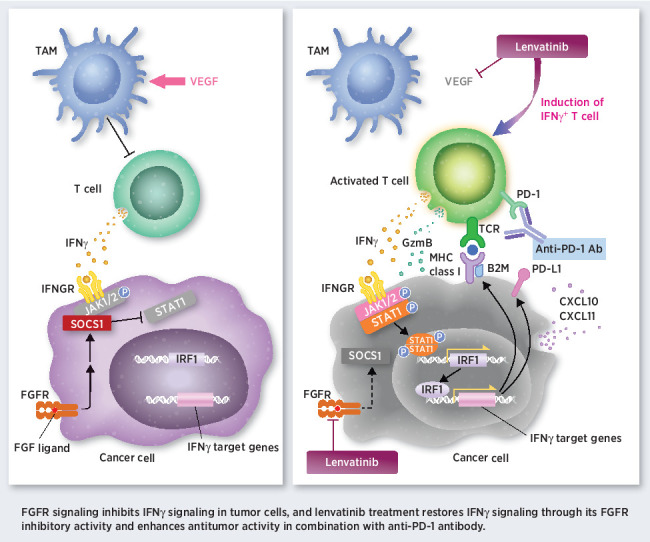
Introduction
Immunotherapy has revolutionized cancer treatment. Both anti-programmed cell death 1 (PD-1) and antiprogramed death-ligand 1 (PD-L1) mAbs show durable responses and their use has improved survival for patients with many types of solid tumors. However, because of intrinsic or acquired resistances to immunotherapies, the overall response rates of single-agent treatment with PD-1 blockade are modest (1). To overcome the mechanism by which tumors become resistant to PD-1 blockade and, hence, to improve the treatment efficacy of PD-1 blockade, the combination of PD-1 blockade with other anticancer therapies is under investigation in many of clinical trials (1–4). Among them, PD-1 blockade combined with either an anti-CTLA4 (CTL-associated protein 4) mAb or anti-VEGF therapy have been approved for clinical use (1, 5–9).
Lenvatinib mesilate (lenvatinib) is an orally administered, multitarget, tyrosine kinase inhibitor that selectively inhibits VEGFRs 1–3, FGFRs 1–4, platelet-derived growth factor receptor α, RET, and KIT (10–12). Lenvatinib is currently used as monotherapy for patients with radioactive-iodine–refractory differentiated thyroid cancer in the United States and European Union (13), for unresectable thyroid cancer in Japan (14), and for unresectable hepatocellular carcinoma in the United States, European Union, and Japan (15). In the United States and European Union, the combination of lenvatinib plus everolimus is approved for the treatment of patients with advanced renal cell carcinoma (RCC) after one prior antiangiogenic therapy (16).
Our previous preclinical studies in syngeneic mouse tumor models demonstrated that lenvatinib has both antiangiogenic and immunomodulatory activity, and combination treatment with lenvatinib plus anti-PD-1 mAb activated IFN signaling to exert enhanced antitumor effects (17, 18). IFNγ signaling in cancer cells orchestrates anticancer immunity, thus increasing tumor immunogenicity, facilitating recognition by cytotoxic CD8+ T cells, and causing tumor rejection by the host immune system (19). Combination therapy comprising lenvatinib plus pembrolizumab, a humanized monoclonal anti-PD-1 mAb, has now been approved in the United States, Canada, and Australia for patients with advanced endometrial carcinoma that is not microsatellite instability-high or mismatch repair–deficient who have disease progression following prior systemic therapy and are not candidates for curative surgery or radiation (20, 21), and multiple phase III clinical trials of this combination are ongoing (e.g., NCT02811861, NCT03884101).
VEGF is well known as the master regulator of angiogenesis. In addition, VEGF plays immunosuppressive roles in the tumor microenvironment by promoting the proliferation of regulatory T cells (Treg), expansion of myeloid-derived suppressor cells (MDSC), recruitment of monocytes from bone marrow, and infiltration of macrophages into tumor tissues (22–24). Moreover, VEGF inhibits T-cell development and dendritic cell maturation (25–27). The activation of FGFR signaling contributes to tumor progression by: enhancing tumor-cell proliferation, survival, and angiogenesis; promoting resistance to anticancer therapy (28). Contrary to VEGFR signaling, the immunomodulatory effects of FGFR signaling in tumor cells and the consequences of FGFR inhibition regarding the antitumor activity of anti-PD-1 mAb remain largely unknown.
In this study, we evaluated antitumor activity of combination treatment with lenvatinib plus anti-PD-1 mAb or a selective VEGFR inhibitor, axitinib plus anti-PD-1 mAb in a mouse RAG syngeneic RCC model, in which the tumor microenvironment features abundant tumor-associated macrophages (TAM) and tumor-infiltrating T cells. In addition, we investigated cross-talk between FGFR- and IFNγ-signaling pathways in cancer cells to examine whether lenvatinib—by inhibiting FGFR—modulates the IFNγ pathway in tumor cells to result in anticancer activity.
Materials and Methods
Compounds
Lenvatinib mesilate and E7090 succinate were synthesized at Eisai Co., Ltd.. Axitinib was purchased from Selleck Chemicals.
Cell lines
Mouse RCC RAG cell line is a culture-adapted cell line derived from a spontaneous renal adenocarcinoma in mouse (29). RAG cells and human RCC 786-O cells were obtained from the ATCC. Human hepatocellular carcinoma JHH-7 (30) and HuH7 cells were obtained from the Japanese Collection of Research Bioresources Cell Bank. Human endometrial cancer MFE280 cells were obtained from the German Collection of Microorganisms and Cell Cultures.
All cell lines in this study were authenticated by means of short tandem repeat profiling. Cell lines were cultured at 37°C in 5% CO2 in the supplier's recommended media supplemented with 10% FBS (RAG, 786-O, JHH-7, and HuH7 cells) or 20% FBS (MFE280), and 100 U/mL penicillin and 100 μg/mL streptomycin (p/s). All cell lines were confirmed to be negative for Mycoplasma by using a VenorGem Advance Mycoplasma Detection Kit (Minerva Biolabs). All experiments were performed within 1 month after thawing early-passage cells.
In vivo RAG tumor models
All animal experiments in this study were approved and conducted in accordance with the Institutional Animal Care and Use Committee guidelines of Eisai Co., Ltd. Female BALB/c mice were obtained from Charles River Laboratories Japan.
To evaluate antitumor activity by using RAG cells in syngeneic mice, RAG cells were adapted to grow subcutaneously in BALB/c mice as described in Supplementary Materials and Methods. In vivo adapted RAG cells (2.5 × 106 cells in 0.1 mL Hank's Balanced Salt Solution per animal) were inoculated subcutaneously into the right flank of 5 to 7 weeks old mice. Tumor dimensions were measured twice weekly by using a caliper, and tumor volume (TV) was calculated as 0.5 × length × width2.
In vivo antitumor activity in the RAG tumor model
Mice were randomly allocated to treatment groups when the tumor became a palpable size (day 1); the average TV reached 70–110 mm3. Lenvatinib was dissolved in 3 mmol/L HCl, and axitinib was suspended in 0.5% carboxymethyl cellulose solution. Lenvatinib and axitinib, at the doses indicated in the figure legends, were administered orally once daily and twice daily, respectively, for the indicated periods. Anti-mouse PD-1 mAb (clone RMP1–14), anti-mouse IFNγ mAb (clone R4–6A2), isotype control for IFNγ mAb (clone HRPN), anti-mouse CSF1R mAb (clone AFS98), and its isotype control (clone 2A3) were purchased from Bio X Cell. All antibodies were diluted in PBS, and mice were dosed as follows: anti-PD-1 mAb, 200 μg/mouse or 10 mg/kg twice weekly for the indicated periods; anti-IFNγ mAb and its isotype control mAb, 300 μg/mouse, beginning 2 days before random allocation into groups (day 1) and twice weekly thereafter for 4 weeks; anti-CSF1R mAb and its isotype control mAb, 300 μg/mouse, beginning 1 day before random allocation into groups (day 1) and twice weekly thereafter for 2 weeks. Mice were euthanized when TV exceeded 2,000 mm3 or when body weight relative to that on day 1 was less than 0.7. For experiments involving IFNγ blockade, the TVs of mice that were found dead or euthanized during the treatment period were imputed by last observation carried forward.
To analyze mouse survival, survival (in days) was defined for each mouse as the duration from day 1 until the day when a mouse was euthanized or found dead. A survival curve was generated for each group by using GraphPad Prism (GraphPad Software).
Waterfall plot of tumor responses in the RAG tumor model
The change in TV at time t (ΔTV) was expressed as a percentage of the volume at baseline (TV1) as follows: ΔTV = 100% × [(TVt − TV1)/TV1]. The best response was defined as the smallest value of ΔTV when t ≥ day 8. Response was evaluated according to modified RECIST for mouse studies (31) and categorized as described in Table legend. The difference in response rate (responders vs. nonresponders) between groups was analyzed by using Fisher exact test.
Flow cytometry analysis
Tumor tissue was dissociated into single cells using a Tumor Dissociation Kit and gentleMACS Dissociator (Miltenyi Biotec). CD45+ leukocytes were isolated from cell suspensions by using microbeads for mouse tumor-infiltrating lymphocytes (TIL; CD45; Miltenyi Biotec) and an OctoMACS Separator (Miltenyi Biotec). After washing and filtration, nonspecific binding was blocked by using Mouse BD Fc Block (BD Biosciences), and CD45+ cells were stained with antibodies against each immune cell population as described in figure legends and Supplementary Materials and Methods. Cells were analyzed through flow cytometry (BD LSRFortessa X-20 or FACSymphony A5; BD Biosciences), and the resulting data were analyzed by using Cytobank software version 7.3.0 (Cytobank Inc.).
T-cell coculture assay
Tumor tissues and spleens were resected from Balb/c mice bearing RAG tumors 37 days after subcutaneous inoculations of tumor cells. Naïve T cells were isolated from the spleens by using a Pan T Cell Isolation Kit II, mouse (Miltenyi Biotec). Single cell suspensions of RAG tumors were prepared using a Tumor Dissociation Kit and gentleMACS Dissociator (Miltenyi Biotec). F4/80+ cells (TAMs), Ly6G+ cells and Ly6G− Gr-1int (Gr-1intermediate) cells were isolated from the single-cell suspensions of RAG tumors by using Anti-F4/80 MicroBeads UltraPure, mouse and a mouse Myeloid-Derived Suppressor Cells Isolation Kit (Miltenyi Biotec), respectively. The isolated T cells were labeled with CMFDA dye (CellTracker, Thermo Fisher Scientific), stimulated with Dynabeads Mouse T-Activator CD3/CD28 (Thermo Fisher Scientific), and then plated at 1.0 × 105 cells/well in round-bottom 96-well plates containing complete medium (RPMI1640, 10% FBS, 50 μmol/L 2-mercaptoethanol). The isolated TAMs, Ly6G+ cells, and Ly6G− Gr-1int cells were added at indicated ratios and were cocultured with T cells at 37°C in 5% CO2. After 4 days, cells were stained with antibodies against CD3 and CD4 (BD Biosciences), CD8, and CD45 (BioLegend) and DAPI (Dojindo) for 20 minutes. The proliferation of CMFDA-labeled T cells was analyzed through flow cytometry.
Western blot analysis
RAG and MFE280 cells were seeded at 3 × 105 cells/well in 6-well plates and cultured for 23 hours. Cells were first treated with DMSO (control), lenvatinib at 1 or 3 μmol/L, or E7090 at 1 μmol/L (RAG) or 0.3 μmol/L (MFE280) for 1 hour. Then RAG cells were treated with 10 ng/mL mouse basic FGF (bFGF; R&D Systems) for 5 minutes or 24 hours and MFE280 cells were treated with human bFGF (R&D Systems) for 24 hours. Treated cells were finally stimulated with 5 ng/mL mouse or human IFNγ (Fujifilm Wako) for 24 hours. For detection of phosphorylation of FRS2 in MFE280 cells, cells were seeded at 3 × 105 cells/well in 6-well plates and cultured for 23 hours. Then cells were starved in EMEM supplemented with 0.1% FBS for 18 hours and treated with DMSO control, lenvatinib at 1 or 3 μmol/L, or E7090 0.3 μmol/L for 1 hour then with 10 ng/mL human bFGF for 5 minutes. Cells were lysed in 0.25% Triton X100, 50 mmol/L Tris-HCl (pH 7.5), 150 mmol/L NaCl lysis buffer and prepared for Western blotting.
For in vivo tumor samples, mice bearing RAG tumors were received lenvatinib or axitinib at the indicated doses for 2 weeks, and were euthanized at 2 hours after the final administration (day 15). Tumors were harvested and 2 whole-tumor samples from different mice were pooled and lysed as a single sample by using a Qproteome Cell Compartment Kit (QIAGEN) and the cytosolic fractions were prepared for Western blotting.
The lysed samples were electrophoresed in 5% to 20% or 15% polyacrylamide gels. Separated proteins were transferred onto nitrocellulose membranes, which were then incubated individually with the following primary antibodies: phospho-FRS2 (Tyr436), phospho-Stat1 (Tyr701), Stat1, GAPDH (Cell Signaling Technology), PD-L1, B2M, SOCS1 (Abcam), and FRS2 (R&D Systems). All the primary antibodies were diluted 1:1,000. Secondary antibodies were horseradish peroxidase (HRP) conjugates (1:2,000; Cell Signaling Technology), and chemiluminescence was detected by using Immobilon Western Chemiluminescent HRP Substrate (Millipore) and Fusion FX (Vilber Lourmat).
ELISA
RAG and 786-O cells were seeded at 2.5 × 104 cells/well and 1.5 × 104 cells/well, respectively, in 24-well plates; HuH7 cells were seeded at 2.5 × 105 cells/well in 6-well plates; plated cells were cultured overnight. Cells were then treated with lenvatinib, E7090, bFGF, and IFNγ as described for Western blot analysis, and culture supernatants were collected. The concentration of CXCL10 was assessed by using an IP-10 (CXCL10) SimpleStep ELISA Kit (Abcam) in accordance with the manufacturer's instructions; absorbance at 450 nm was measured by using a microplate reader (SpectraMax 190, Molecular Devices).
Immunofluorescence staining
Mice bearing RAG tumors were treated with lenvatinib and PD-1 Ab at indicated doses for 2 weeks. On day 15, the mice were euthanized, tumors were harvested, and tissue samples were embedded in OCT compound (Sakura Finetek) and frozen. Cryosections were fixed with cold methanol; stained with anti-PD-L1 antibody (Abcam) and anti-IFN regulatory factor 1 (IRF1) antibody (Cell Signaling Technology) as primary antibodies; and incubated with Alexa Fluor488–conjugated anti-rabbit IgG or Alexa Fluor594–conjugated anti-rat IgG, respectively, as secondary antibodies. Hoechst dye was used for nuclear counterstaining. Entire immunofluorescence images were scanned by using a NanoZoomer (Hamamatsu Photonics), and the PD-L1–positive and IRF1-positive areas were analyzed by using the Object Colocalization FL v1.0 algorithm of HALO image analysis platform (Indica Labs).
Statistical analysis
Statistical analyses were performed by using GraphPad Prism version 8.01 or 8.3.1. P values less than 0.05 were considered significant.
Results
Antitumor activity and mouse survival in the RAG model with combination treatment of lenvatinib and anti-PD-1 mAb
We evaluated the antitumor activities of lenvatinib and a selective VEGFR inhibitor, axitinib (32), when combined with anti-mouse PD-1 mAb in a RAG, mouse RCC syngeneic tumor model (Fig. 1). We started the treatments when tumors became palpable (∼90 mm3, day 1). Single-agent treatment (days 1–29) using lenvatinib (10 mg/kg, once daily), axitinib (10 mg/kg, twice daily), or anti-PD-1 mAb (200 μg/animal, twice weekly) all suppressed tumor growth, but the effect of anti-PD-1 mAb was weaker than that of either lenvatinib or axitinib (Fig. 1A; Supplementary Fig. S1A). Combination treatments in which anti-PD-1 mAb was added to lenvatinib or axitinib showed enhanced antitumor activity compared with each monotherapy. However, only the group treated with lenvatinib plus anti-PD-1 mAb showed regression of the average TV (purple line in Supplementary Fig. S1A). Regardless of treatment group, none of the mice showed severe body weight loss compared with the control group (Supplementary Fig. S1B). Treatment with anti-PD-1 mAb alone or with axitinib and anti-PD-1 mAb in combination caused complete tumor regression (nonpalpable tumors) in 2 of the 20 mice treated, whereas the combination of lenvatinib plus anti-PD-1 mAb achieved complete tumor regression in 7 of the 20 mice until the end of the observation period (day 92; Fig. 1A). Plotting the greatest percentage of tumor shrinkage achieved in each mouse (waterfall plot) revealed a strong response to the lenvatinib plus anti-PD-1 mAb treatment (Fig. 1B). When the mice in each group were categorized as responders (those where TV decreased more than 50%) and nonresponders (those where TV decreased 50% or less), tumor response was greater for lenvatinib plus anti-PD-1 mAb (responders: 70%) than for all other treatment groups (responders: 0%–15%; Table 1).
Figure 1.

Antitumor activity of combination treatment with lenvatinib plus anti-PD-1 mAb or axitinib plus anti-PD-1 mAb in the RAG syngeneic tumor model. Mice bearing RAG tumors were allocated into treatment groups on day 1—when TVs were approximately 90 mm3—and were orally treated with lenvatinib at 10 mg/kg once daily or axitinib at 10 mg/kg twice daily, and were intraperitoneally injected with anti-PD-1 mAb at 200 μg/mouse twice weekly for 4 weeks. Data from two independent experiments (n = 20) were combined. A, TVs of individual mice. Black arrows, treatment period. B, Waterfall plots showing the greatest percent change of individual TVs from baseline after day 8. C, Kaplan–Meier plots of mouse survival. Top left, control, lenvatinib, anti-PD-1 mAb, and lenvatinib plus anti-PD-1 mAb treatment groups. Top right, control, axitinib, anti-PD-1 mAb, and axitinib plus anti-PD-1 mAb treatment groups. Survival (in days) was defined as the time from day 1 until the day when a mouse was euthanized or found dead. *, P < 0.05; ***, P < 0.001 vs. control group (log-rank test with Bonferroni correction); #, P < 0.05; ##, P < 0.01; ####, P < 0.0001 (log-rank test between groups). Bottom, median survival time of mice in each treatment group and HR (95% CI) compared with control group. N.R., not reached.
Table 1.
Summary of tumor responses in the RAG model.
| Tumor response | Control | Lenvatinib | Axitinib | PD-1 Ab | LEN+ PD-1 Ab | Axi+ PD-1 Ab |
|---|---|---|---|---|---|---|
| PD (≥30%) | 95% | 35% | 80% | 85% | 15% | 60% |
| SD (≥−50%, <30%) | 5% | 60% | 20% | 0% | 15% | 25% |
| PR (≥−95%, <−50%) | 0% | 5% | 0% | 5% | 20% | 5% |
| CR (<−95%) | 0% | 0% | 0% | 10% | 50% | 10% |
| Nonresponder (PD+SD) | 100% | 95% | 100% | 85% | 30% | 85% |
| Responder (PR+CR) | 0% | 5% | 0% | 15% | 70% | 15% |
| P (vs. LEN + PD-1 Ab) | <0.0001 | <0.0001 | <0.0001 | 0.0011 | — | 0.0011 |
Note: Tumor responses were defined as follows: complete response (CR), tumor volume (TV) decreased by more than 95%; partial response (PR), TV decreased by 50% to 95%; stable disease (SD), TV decreased by 30% to 50%; progressive disease (PD), TV not otherwise categorized. Nonresponders are mice categorized as having as PD and SD, and responders are those defined as having PR and CR. The difference in response rate (responders vs. nonresponders) between groups was analyzed by Fisher exact test.
Abbreviation: LEN, lenvatinib.
Survival curves for mice with each treatment group for combination with lenvatinib or axitinib in the RAG model are shown in Fig. 1C. Single-agent treatment with either lenvatinib, axitinib, or anti-PD-1 mAb prolonged survival compared with the control group. In addition, both combination treatments (i.e., lenvatinib plus anti-PD-1 mAb, axitinib plus anti-PD-1 mAb) increased survival compared with those of the control and monotherapy groups. Median survival of control group was 28 days, and those of lenvatinib, axitinib, anti-PD-1 mAb, lenvatinib plus anti-PD-1 mAb, and axitinib plus anti-PD-1 mAb-groups were 47.5 days, 46 days, 37 days, not reached [> 92 days], and 53 days, respectively (Fig. 1C, bottom). Notably, mice that received the lenvatinib plus anti-PD-1 mAb combination (purple line in Fig. 1C, top left) survived longer than those given axitinib plus anti-PD-1 mAb [tan line in Fig. 1C, top right; HR, 0.3146; 95% confidence interval (CI), 0.1421–0.6966; P = 0.0015].
Next, we evaluated microvessel density in RAG tumors and found that the antiangiogenic activities of lenvatinib (10 mg/kg daily) and axitinib (10 mg/kg twice daily) were comparable (Supplementary Fig. S1C). Because increases in plasma FGF23 are a well-known pharmacodynamic biomarker of FGFR inhibition (33–35), we measured the plasma FGF23 levels of treated mice to assess the FGFR inhibitory activities of lenvatinib and axitinib (Supplementary Fig. S2). Whereas treatment with lenvatinib (10 or 30 mg/kg) or E7090 (25 mg/kg), a selective FGFR inhibitor (36), increased plasma FGF23 levels in mice, axitinib (5, 10, or 30 mg/kg) did not. These results indicated both lenvatinib and axitinib inhibited angiogenesis at 10 mg/kg, but only lenvatinib inhibited FGFR signaling in vivo. These results demonstrate that the combination of lenvatinib plus anti-PD-1 mAb caused strong tumor shrinkage and prolonged survival time of mice compared with the axitinib plus anti-PD-1 mAb combination in the RAG model. Moreover, these results suggest that FGFR inhibition of lenvatinib may play an important role in enhanced antitumor activity of combination with anti-PD-1 mAb (nonpalpable tumors, response rate, and survival of mice) in vivo beyond angiogenesis inhibition.
Immunomodulatory activity of lenvatinib in the RAG model
To investigate the mechanism underlying the antitumor activity of lenvatinib plus anti-PD-1 mAb in the RAG model, we analyzed various immune cell populations by using flow cytometry (Fig. 2). To this end, we isolated CD45+ TILs from resected tumors and evaluated them according to the gating strategy shown in Supplementary Fig. S3. As we previously reported for the mouse colon CT26 model (18), the population of TAMs (gated as CD45+ CD11b+ Ly6G− Ly6C− F4/80+) was significantly smaller in the lenvatinib-treated mice compared with the control group. Anti-PD-1 mAb also decreased the TAM population, but less so than lenvatinib alone, and the combination of lenvatinib plus anti-PD-1 mAb further decreased the number of TAMs compared with anti-PD-1 mAb treatment alone (Fig. 2A). The proportion of polymorphonuclear MDSCs (PMN-MDSC, gated as CD45+ CD11b+ Ly6G+ Ly6Clow) did not differ between treatment groups, and the proportion of monocytic MDSCs (M-MDSC, gated as CD45+ CD11b+ Ly6G− Ly6C+) increased after treatment with lenvatinib alone or in combination with anti-PD-1 Ab (Fig. 2A).
Figure 2.

Immunomodulatory activity of lenvatinib, anti-PD-1 mAb, and lenvatinib plus anti-PD-1 mAb in RAG tumors. Mice bearing RAG tumors were allocated into treatment groups on day 1—when TVs were approximately 80 and 110 mm3 (two independent experiments)—and treated with lenvatinib at 10 mg/kg once daily or anti-PD-1 mAb at 10 mg/kg twice weekly (or both) for 2 weeks; tumors were resected on day 15. CD45+ TILs were isolated from tumor tissue, and populations of immune cells were analyzed by flow cytometry. The gating strategy is shown in Supplementary Fig. S3. A, Percentages of TAM, PMN-MDSC, and M-MDSC in CD45+ TILs. B, CD8+ T-cell populations. Percentages of CD8+ T cells in CD45+ TILs, IFNγ+ CD8+ T cells, GzmB+ CD8+ T cells, and PD1+ CD8+ T cells in CD8+ T cells. C, CD4+ T-cell populations. Percentages of CD4+ T cells in CD45+ TILs, IFNγ+ CD4+ T cells, and Treg (CD25+ CD127− CD4+ T cells) in CD4+ T cells. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 versus control group; #, P < 0.05; ###, P < 0.001; ####, P < 0.0001 versus combination treatment group (Dunnett multiple comparison test). Data were combined from two independent experiments (n = 10). LEN, lenvatinib; Treg, regulatory T cells.
Regarding CD8+ T cells (gated as CD45+ CD11b− CD11c− B220− CD3+ CD8+), mice treated with either anti-PD-1 mAb only or lenvatinib plus anti-PD-1 mAb had larger CD8+ T cell populations than the control group (Fig. 2B). Similarly, lenvatinib—either as monotherapy or in combination with anti-PD-1 mAb—increased the numbers of IFNγ+ CD8+, or GzmB+ CD8+ T cells relative to controls, and the population of IFNγ+ CD8+ T cells following lenvatinib plus anti-PD-1 mAb treatment was larger than that for each single-agent treatment. None of the treatments had any effect on the proportion of PD-1+ CD8+ T cells. Treatment with lenvatinib or in combination with anti-PD-1 Ab increased the population of CD4+ T cells (gated as CD45+ CD11b− CD11c− B220− CD3+ CD4+; Fig. 2C). In addition, treatment with anti-PD-1 mAb or combination with lenvatinib increased the number of IFNγ-expressing effector CD4+ T cells, whereas treatment with lenvatinib decreased the proportion of Tregs (gated as CD45+ CD3+ CD4+ CD25+ CD127−; Fig. 2C; refs. 37, 38).
To evaluate the immunosuppressive role of TAMs in the RAG model, we used an anti-CSF1R antibody to deplete the TAM population (Fig. 3A and B) and depletion of TAMs was confirmed by flow cytometry analysis (Fig. 3A). TAM depletion did not change the population of CD8+ T cells, but increased the proportion of activated CD8+ T cells (PD-1+ CD25+ CD8+ T cells and PD-1+ Tim3+ CD8+ T cells). And TAM depletion also led to decrease in populations of CD4+ T cells, including Tregs (Fig. 3A). These results were confirmed through qRT-PCR analyses, in which gene expression levels of TAM markers (Csf1r and Cx3cr1), a myeloid cell marker (Itgam), a Treg marker (Foxp3), and the immunosuppressive molecule Tgfb1 were all decreased after TAM depletion (Fig. 3B), indicating that TAMs play immunosuppressive roles in RAG tumors and that decreasing the TAM population may induce the activation of CD8+ T cells.
Figure 3.

Immunomodulatory activity of TAM in the RAG model. A and B, Mice bearing RAG tumors were injected intraperitoneally with anti-CSF1R mAb or control IgG2a at 300 μg/animal 1 day before allocation (i.e., day 0). On day 1, when TVs were approximately 101 mm3, anti-CSF1R mAb or control IgG2a at 300 μg/animal were administered twice weekly thereafter for 2 weeks. A, CD45+ TILs were isolated from tumor tissue, and populations of immune cells were analyzed by flow cytometry. The gating strategy is shown in Supplementary Fig. S3. Percentages of TAMs, CD8+ T cells in CD45+ TILs, and PD-1+ Tim3+ CD8+ T cells and PD-1+ CD25+ CD8+ T cells in CD8+ T cells. Percentages of CD4+ T cells in CD45+ TILs and Treg (CD25+ CD127− CD4+ T cells) in CD4+ T cells. N = 8. *, P < 0.05; **, P < 0.01; ****, P < 0.0001, unpaired t test between groups. B, Expression levels of the indicated genes determined through qRT-PCR analysis. Data were normalized by the Gapdh gene. Data are presented as means + SEM (N = 8). **, P < 0.01; ****, P < 0.0001, unpaired t test between groups. C and D, T-cell coculture assay with F4/80+ cells (TAMs) isolated from RAG tumors was performed at the indicated ratios (T cells: F4/80+ cells) as described in Materials and Methods. C, Histograms of CMFDA-labeled CD8+ T cells cocultured with F4/80+ cells. D, Proliferation of CD8+ T cells and CD4+ T cells cocultured with F4/80+ cells compared with T-cell single culture (T cell only). Data are presented as means ± SEM of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (Dunnett multiple comparison test) versus T cell only group.
We further investigated the immunosuppressive activity of the TAMs and MDSCs in RAG tumors through an ex vivo coculture experiment with T cells (Fig. 3C and D; Supplementary Fig. S4). T cells were isolated from mouse spleen, TAMs were isolated as F4/80+ cells, and MDSCs were isolated as either Ly6G+ or Ly6G− Gr-1int (Gr-1intermediate) cells from RAG tumors. The purities of the isolated cell populations were confirmed by flow cytometry (Supplementary Fig. S4A–S4D). CD3+ cells were highly purified (97%) among the isolated T cells (Supplementary Fig. S4A). F4/80+ cells and Ly6G+ cells were highly concentrated as TAMs (87%) and PMN-MDSCs (93%), respectively (Supplementary Fig. S4B and S4C). The Ly6G− Gr-1Int population comprised 68% of the M-MDSCs and 31% of TAMs among CD45+ TILs (Supplementary Fig. S4D). Isolated T cells were labeled with CMFDA dye, stimulated with anti-CD3/CD28 magnetic beads, and cocultured for 4 days with TAMs (F4/80+ cells), Ly6G+ cells, or Ly6G− Gr-1int cells. The proliferation of T cells after coculture was analyzed via flow cytometry. Coculture with TAMs (F4/80+ cells) suppressed the proliferation of CD8+ T cells at a ratio of 1:1 or 3:1 (Fig. 3C and D, left) and CD4+ T cells (Fig. 3D, right). However, coculture of PMN-MDSCs (Ly6G+ cells) with stimulated T cells at a ratio of 1:1 or 3:1 (T cells:Ly6G+ cells) had no effect on the expansion of CD8+ T cells (Supplementary Fig. S4E and S4G). Coculture of Ly6G− Gr-1int cells (enriched M-MDSCs containing a small population of TAMs) with stimulated T cells at a 1:1 ratio, but not at 3:1 (T cells:M-MDSC–rich cells) inhibited the expansion of CD8+ T cells (Supplementary Fig S4F and S4G), as well as CD4+ T cells (Supplementary Fig S4H).
These results suggest that lenvatinib and anti-PD-1 mAb mainly affect different immune cell populations (i.e., TAM, Treg, CD4+ T cells, and CD8+ T cells) in the RAG model. In addition, lenvatinib single treatment increased the population of GzmB+ CD8+ T cells, and combination treatment with anti-PD-1 mAb further increased the population of IFNγ-expressing CD8+ T cells more than either single treatment.
Inhibition of IFNγ signaling due to activation of FGFR-signaling pathways
Next, we explored the interactions between the IFNγ-signaling and FGFR-signaling pathways in RAG cells using Western blot analysis. Treatment of RAG cells with bFGF induced the phosphorylation of FRS2 (Fig. 4A), which is a direct substrate of FGFR (39). Conversely, E7090 (1 μmol/L), a selective FGFR inhibitor, clearly inhibited the phosphorylation of FRS2—as did lenvatinib, in a dose-dependent manner (Fig. 4A). In contrast, IFNγ treatment of RAG cells induced the phosphorylation of STAT1 (Tyr701) and the expression of its downstream molecules B2M (β2 microglobulin) and PD-L1 (Fig. 4B). IFNγ stimulation likewise increased total STAT1 expression through the positive feedback loop in the IFNγ/JAK/STAT pathway (40). Activation of the FGFR signaling pathway with bFGF treatment inhibited IFNγ-induced STAT1 phosphorylation and the expression of B2M and PD-L1 (Fig. 4B). Because lenvatinib inhibits FGFR, treatment with lenvatinib canceled the inhibitory effects of FGFR signaling on IFNγ-signaling pathways and restored STAT1 phosphorylation and the expression of B2M and PD-L1 (Fig. 4B). SOCS1 (suppressor of cytokine signaling 1) is a potent inhibitor of the JAK/STAT signaling pathway (41), and the expression of SOCS1 is upregulated by the activation of FGFR signaling and disrupted IFNγ-signaling pathways in rat chondrocytes (42). We confirmed that bFGF treatment increased SOCS1 expression in RAG cells and that lenvatinib treatment suppressed this induction (Fig. 4B). Using flow cytometry analysis, we examined the cell-surface expression of PD-L1 on RAG cells (Fig. 4C). As same as the results of Western blot analysis, bFGF treatment suppressed IFNγ-induced expression of PD-L1, whereas treatment with lenvatinib or E7090 restored the PD-L1 expression. In addition, consistent with previous reports (19), IFNγ treatment increased the CXCL10 levels in the culture medium of RAG cells (Fig. 4D); bFGF suppressed this induction of CXCL10, and treatment with either lenvatinib or E7090 to inhibit FGFR restored CXCL10 levels in RAG culture supernatants.
Figure 4.

Inhibitory effect of FGFR signaling on IFNγ-signaling pathways in RAG cells. A and B. Experimental schemes (top) and Western blot analysis using the indicated antibodies (bottom). The housekeeping molecule GAPDH was used as a loading control. A, Effects of lenvatinib and E7090 on the FGFR-signaling pathway. RAG cells were treated first with lenvatinib at 1 or 3 μmol/L or with E7090 at 1 μmol/L for 1 hour and then treated with bFGF 10 ng/mL for 5 minutes. B, Effects of FGFR signaling on the IFNγ-signaling pathway. RAG cells were treated first with lenvatinib at 1 or 3 μmol/L for 1 hour and then with bFGF 10 ng/mL for 23 hours. After those treatments, cells were stimulated with IFNγ 5 ng/mL for 24 hours. C, Effects of FGFR signaling on expression levels of IFNγ-induced cell surface PD-L1. RAG cells were treated first with lenvatinib at 1 or 3 μmol/L or with E7090 at 1 μmol/L for 1 hour, after which, all cells were treated with bFGF 10 ng/mL for 23 hours and then stimulated with IFNγ 5 ng/mL for 24 hours. Cell surface expression level of PD-L1 was analyzed by using flow cytometry. Data are presented as means + SEM (n = 3). ***, P < 0.001; ****, P < 0.0001, unpaired t test between groups. ####, P < 0.0001, Dunnett multiple comparison test versus bFGF+IFNγ-treated group (blue bar). †††, P < 0.001, unpaired t test versus bFGF+IFNγ-treated group (blue bar). D, Effects of FGFR signaling on expression levels of IFNγ-induced CXCL10 in culture supernatant. RAG cells were treated as same as C. CXCL10 levels in culture supernatants were analyzed through ELISA. Data are presented as means + SEM (n = 4). ****, P < 0.0001, unpaired t test versus bFGF+IFNγ-treated group (blue bar). ####, P < 0.0001, Dunnett multiple comparison test versus bFGF+IFNγ-treated group (blue bar). LEN, lenvatinib; N.D., not determined (i.e., below the lower limit of detection).
Next, we further evaluated the cross-talk between the IFNγ- and FGF-signaling pathways through qRT–PCR analyses of gene targets of IFNγ, including B2m, H2-k1, Cxcl10, Cxcl11, and Ifit2 in RAG cells (Supplementary Fig. S5). As expected, IFNγ stimulation upregulated the expression levels of IFNγ target genes, which then were inhibited by bFGF treatment. Although the change in levels of gene expression varied somewhat, depending on the gene (e.g., Cxcl11, Ifit2) and drugs (lenvatinib or E7090), FGFR inhibitors broadly increased the expression levels of induced IFNγ-target genes, which had been suppressed by bFGF treatment (Supplementary Fig. S5).
We then evaluated the cross-talk between IFNγ- and FGF-signaling pathways in cultured human cancer cell lines (Fig. 5). MFE280 cells harbor the FGFR2 S252W mutation, which increases ligand binding affinity to FGFR2 and activates FGFR signaling pathway (43). Western blot analyses demonstrated that constitutive phosphorylation of FRS2, which is further activated by exogenous bFGF, and treatment with either E7090 or lenvatinib inhibited the phosphorylation of FRS2 in the human endometrial cancer cell line MFE280 (Fig. 5A). In MFE280 cells, bFGF treatment weakly suppressed the phosphorylation of STAT1 and the expression levels of B2M and PD-L1, whereas lenvatinib treatment increased the phosphorylation of STAT1 and expression of B2M and PD-L1 (Fig. 5B). Because MFE280 cells showed constitutively activated phosphorylated FRS2 in the absence of exogenous bFGF treatment (Fig. 5A), the suppression of IFNγ-signaling by bFGF-treatment might be weak. These results were further confirmed when we treated MFE280 cells with IFNγ in the absence of exogenous bFGF (Fig. 5C). In this experiment, IFNγ treatment increased the phosphorylation of STAT1 and the production of B2M and PD-L1, and the addition of lenvatinib further upregulated IFNγ-induced B2M and PD-L1 levels. Consistent with qRT-PCR results in RAG cells (Supplementary Fig. S5), expression levels of B2M, CXCL10, CXCL11, and CD274 (PD-L1) in MFE280 cells were upregulated by IFNγ stimulation, inhibited by bFGF treatment, and restored after lenvatinib treatment (Supplementary Fig. S6A, top). In addition, regardless of exogenous bFGF treatment, the addition of FGFR inhibitors further upregulated the expression of these genes increased with IFNγ in MFE280 (Supplementary Fig. S6A).
Figure 5.

Inhibitory effect of FGFR signaling on the IFNγ signaling pathway in human cancer cells. A–C, Western blot analysis in MFE280 human endometrial cancer cells using the indicated antibodies; GAPDH was included as a loading control. A, MFE280 cells were starved in medium containing 0.1% FBS for 18 hours, and then treated first with lenvatinib at 1 or 3 μmol/L or with E7090 at 0.3 μmol/L for 1 hour and finally with bFGF (10 ng/mL) for 5 minutes. B, MFE280 cells were pretreated with lenvatinib at 1 or 3 μmol/L for 1 hour and then treated with bFGF (10 ng/mL) for 23 hours. Finally, cells were stimulated with IFNγ (5 ng/mL) for 24 hours. C, MFE280 cells were treated with lenvatinib at 1 or 3 μmol/L for 24 hours and then stimulated with IFNγ (5 ng/mL) for 24 hours. D and E, CXCL10 levels in culture supernatants were analyzed by ELISA. Data are presented as means + SEM of three independent experiments. D, Human renal cell carcinoma 786-O cells were treated first with lenvatinib at 1 or 3 μmol/L or with E7090 at 0.3 μmol/L for 1 hour and then with bFGF (10 ng/mL) for 23 hours. Finally, cells were stimulated with IFNγ (5 ng/mL) for 24 hours. *, P < 0.05; **, P < 0.01, unpaired t test versus bFGF+IFNγ-treated group (blue bar); ###, P < 0.001, Dunnett multiple-comparison test versus bFGF+IFNγ-treated group (blue bar). E, Human hepatocellular carcinoma HuH7 cells were treated first with lenvatinib at 1 or 3 μmol/L or with E7090 at 0.3 μmol/L for 24 hours and then stimulated with IFNγ (5 ng/mL) for 24 hours. ***, P < 0.001, unpaired t test versus IFNγ-treated group (red bar). ##, P < 0.01; ###, P < 0.001, Dunnett multiple comparison test versus IFNγ-treated group (red bar). LEN, lenvatinib; N.D., not determined (i.e., below the lower limit of detection).
In the human hepatocellular carcinoma JHH-7 cells, which highly express FGF-19 (44), the FGFR-inhibitory activity of lenvatinib further induced the expression of IFNγ-stimulated target genes (Supplementary Fig. S6B). E7090 also increased IFNγ-induced gene expression in JHH-7 cells, but to lower levels than lenvatinib, because E7090 has weak inhibitory activity against FGFR4 compared with FGFR1–3 that is addressed further in the Discussion.
Furthermore, we investigated the IFNγ-induced CXCL10 expression in human RCC 786-O cells; this line harbors a mutated VHL gene, which is frequently mutated in RCCs. Lenvatinib and E7090 restored the IFNγ-induced CXCL10 expression in 786-O cells that had been suppressed by bFGF treatment (Fig. 5D). In addition, in HuH7 human hepatocellular carcinoma cells (Fig. 5E), in which FGF19 is also overexpressed and FGFR signaling is intrinsically activated (45), treatment with lenvatinib or E7090 further increased IFNγ-stimulated CXCL10 expression compared with IFNγ-stimulation alone (Fig. 5E).
Cross-talk between IFNγ- and FGF-signaling pathways in the RAG tumor microenvironment
To investigate cross-talk between IFNγ- and FGF-signaling pathways in vivo, we used immunofluorescence staining of PD-L1 and IRF1 expression in RAG tumors (Fig. 6A and B). Anti-PD-1 mAb monotherapy increased the PD-L1–positive area, which further increased numerically after combination treatment with lenvatinib plus anti-PD-1 mAb (Fig. 6A). In contrast, the area associated with IRF1, a pivotal transcription factor in IFNγ signaling (19), increased only in response to combination treatment with lenvatinib plus anti-PD-1 mAb (Fig. 6B).
Figure 6.

Immunofluorescence and Western blot analysis of IFNγ-signaling pathways in RAG tumors. Mice bearing RAG tumors were treated with lenvatinib at 10 mg/kg once daily, with anti-PD-1 mAb at 200 μg/animal twice weekly, or with both agents for 2 weeks. A and B, Representative immunofluorescence images (left) and quantified positive areas (right) for each treatment group are shown for PD-L1 (A) and IRF1 (B). Bars, 100 μm. Data are presented as means + SEM (n = 8). *, P < 0.05; ***, P < 0.001; ****, P < 0.0001 versus control group; #, P < 0.05; ###, P < 0.001 versus combination treatment group (Dunnett multiple comparison test). C, Western blot analysis of IFNγ-signaling pathways. Mice bearing RAG tumors were treated with lenvatinib at 10 mg/kg once daily or with axitinib at 5 or 10 mg/kg twice daily for 2 weeks. Tumor lysate from each treatment group underwent Western blot analysis using the indicated antibodies; GAPDH was included as a loading control. LEN, lenvatinib.
We examined the effects of lenvatinib and axitinib, a selective inhibitor of VEGFR, on JAK/STAT pathway signaling and the expression of its downstream molecules in RAG tumors by Western blot analysis (Fig. 6C). Lenvatinib treatment induced the total expression and phosphorylation (Tyr701) of STAT1 and upregulated the expression of downstream molecules of IFNγ signaling, including B2M and PD-L1 (Fig. 6C), whereas STAT1 and PD-L1 expression was, at most, weakly induced in RAG tumors from axitinib-treated mice (Fig. 6C). Expression levels of B2M were equivalently increased by both lenvatinib and axitinib. In comparison, treatment with lenvatinib—but not axitinib—decreased the expression of SOCS1, a negative regulator of the JAK/STAT pathway, in RAG tumors. These results suggest that, in addition to its inhibitory effects on angiogenesis (Supplementary Fig. S1C), lenvatinib effectively reactivated IFNγ-signaling pathways, which were inhibited via activation of FGFR signaling pathway in RAG tumors in vivo (Fig. 6C).
Role of IFNγ in the antitumor activity of combination treatment comprising lenvatinib and anti-PD-1 mAb in the RAG model
To investigate the role of IFNγ in the antitumor activity of lenvatinib plus anti-PD-1 mAb, we used an inhibitory anti-IFNγ mAb in the RAG model (Fig. 7A and B; Supplementary Fig. S7A and S7B). Severe body weight loss was not observed in any treatment groups compared with the control group (Fig. 7B). In the control (IgG1-treated) group (Fig. 7A, left and Supplementary Fig. S7B, left), combination treatment with lenvatinib plus anti-PD-1 mAb showed greater antitumor activity than each single-agent treatment. In contrast, after application of an anti-IFNγ blocking mAb, lenvatinib plus anti-PD-1 was equivalent to lenvatinib-only treatment in antitumor activity (Fig. 7A, right). These results demonstrate that the enhanced antitumor activity of lenvatinib plus anti-PD-1 mAb is dependent on an active IFNγ-signaling pathway.
Figure 7.

Effects of anti-IFNγ mAb on the antitumor activity of treatment with lenvatinib only, anti-PD-1 mAb only, or their combination in the RAG model. Mice bearing RAG tumors were injected intraperitoneally with anti-IFNγ mAb or control IgG1 at 300 μg/animal 2 days before allocation to treatment groups (i.e., day −1) and twice weekly thereafter. On day 1, when TVs were approximately 90 mm3, mice were allocated randomly into treatment groups. Lenvatinib was orally administered at 10 mg/kg once daily, and anti-PD-1 mAb was injected intraperitoneally at 200 μg/mouse twice weekly for 4 weeks. A, TVs of control IgG-treated (left) and anti-IFNγ mAb–treated (right) groups. Data are presented as means ± SEM (n = 8). *, P < 0.05; **, P < 0.01; ****, P < 0.0001 versus control group; ##, P < 0.01; ####, P < 0.0001 versus combination treatment animals (Dunnett multiple comparison test after logarithmic transformation). TVs of mice that were found dead or were euthanized preemptively during the treatment period were imputed by last observation carried forward (control IgG1: 3 mice in control group, 4 mice in anti-PD-1 mAb group; anti-IFNγ mAb: 1 mouse in control group, 1 mouse in anti-PD-1 mAb group). B, Relative body weight of control IgG-treated (left) and anti-IFNγ mAb-treated group (right). Data are presented as means ± SEM. Combo, combination; LEN, lenvatinib.
Discussion
In this study, we discovered that the activation of FGFR signaling inhibits the IFNγ-stimulated JAK/STAT signaling pathway and that treatment with lenvatinib releases the FGFR-induced inhibition of JAK/STAT signaling and reinstates the tumor response to IFNγ activation in mouse RCC RAG cells and four human cultured cancer cells (Figs. 4 and 5; Supplementary Figs. S5 and S6). Mutations in JAK, genetic loss of B2M, and activation of various oncogenic signals (e.g., mutations in genes related to WNT/β-catenin, BRAF/MAPK, and CDK4/6) in tumor cells, have been reported as mechanisms underlying the resistance to immune checkpoint inhibitors in preclinical models and patients with cancer (46). Our results suggest that FGFR signaling in tumor cells is another resistance mechanism to immune checkpoint inhibition, that is, PD-1 blockade. In addition, our data demonstrate that—in addition to its inhibitory activity against VEGFR2 signaling—lenvatinib's ability to inhibit FGFR provides a rationale for its combination with anti-PD-1 mAb to overcome resistance to immune checkpoint inhibitors.
High-level expression of PD-L1 is widely known to be associated with poor prognosis in a variety of solid tumors (47). Conversely, decreases in PD-L1 expression due to epigenetic and posttranscriptional regulation or mutations in JAK are acknowledged as a key mechanism of resistance to PD-1 blockade (48). IFNγ stimulation upregulates the expression of chemokines and of genes encoding antigen-presentation components, which play important roles in the recognition of cancer cells by cytotoxic CD8+ T cells and in the immunologic rejection of malignancy (19). In particular, IFNγ activated the JAK/STAT pathway and upregulated the expression of IRF1 and B2M, thus likely positively regulating antigen presentation and cancer immunity (Fig. 6). In addition to its effects on IRF1 and B2M, IFNγ signaling effectively upregulated the expression of PD-L1, that is considered to be an escape mechanism from overactivation of immune systems. However, this induction of PD-L1 might contribute to the enhanced antitumor activity of combination treatment with lenvatinib and anti-PD-1 mAb, compared with that due to selective anti-VEGF therapy, through the inhibitory activity of lenvatinib against FGFR signaling (Figs. 4–6).
In the RAG model, combining lenvatinib, which blocks signaling through both VEGFR and FGFR, with anti-PD-1 mAb provided superior antitumor activity and prolonged survival compared with combination treatment with axitinib, a selective inhibitor of VEGFR, plus anti-PD-1 mAb (Fig. 1; Supplementary Fig. S1). In clinical trials, lenvatinib treatment increased blood levels of VEGF and FGF23 (35, 49), which are pharmacodynamics biomarkers of VEGFR and FGFR inhibition, respectively (33, 34, 50, 51). Lenvatinib at 10 mg/kg was the minimal dose that increased blood levels of VEGF and FGF23 in a preclinical mouse model (52), and lenvatinib used in the current study in mice might be relevant to that used in clinical trials. We chose to use an axitinib at a dose of 10 mg/kg according to a previous report (32), in which preclinical pharmacokinetic analysis indicated that axitinib primarily targets VEGFR. Axitinib at 10 mg/kg yielded comparable antiangiogenic activity versus lenvatinib at 10 mg/kg (Supplementary Fig. S1C). Furthermore, we verified that selective inhibition of VEGFR with axitinib at 5–30 mg/kg axitinib did not inhibit the in vivo assessment of FGFR signaling via plasma levels of FGF23 (Supplementary Fig. S2). According to these data, we treated mice with axitinib at 10 mg/kg as a selective VEGFR inhibitor in the RAG model.
We evaluated the immunosuppressive activity of three myeloid-derived populations (F4/80+ cells, Ly6G+ cells, and Ly6G− Gr-1int cells) that we isolated from RAG tumors (Fig. 3C and D; Supplementary Fig. S4E–S4H). Among the three populations, Ly6G+ cells (PMN-MDSCs) had no immunosuppressive activity against the proliferation of T cells in our coculture assay. F4/80+ cells (TAMs) and Ly6G− Gr-1int cells (60% M-MDSCs; 30% TAMs) differed in their immunosuppressive activities. These results suggested that myeloid cells contain subpopulations that differ in their immunosuppressive activities against T cells and that the F4/80+ population (TAMs) showed clear immunosuppressive activity in the mouse RCC RAG model.
The combination treatment of lenvatinib and anti-PD-1 Ab demonstrated strong antitumor activity and prominent tumor shrinkage in the RAG model (Fig. 1). However, the population of M-MDSCs increased after both lenvatinib alone and combination with lenvatinib plus anti-PD-1 Ab, although in both cases, the M-MDSC population was smaller than the TAM population (Fig. 2A). In TBP-3743 murine anaplastic thyroid cancer orthotopic model, lenvatinib treatment increased the population of PMN-MDSCs, and the addition of anti-Gr-1 antibody (MDSC-depletion antibody) improved the antitumor effect of lenvatinib (53). Increases in M-MDSCs might be a resistance mechanism to the lenvatinib–anti-PD-1 Ab combination. It may be interesting to test whether the depletion of M-MDSC in the RAG model enhances the antitumor activity of combination treatment with lenvatinib and anti-PD-1 Ab to overcome the resistance mechanism.
In human hepatocellular carcinoma cell lines that highly express FGF19, such as HuH7 and JHH7 (44, 45), FGFR4 is constitutively activated, and this activation might inhibit the expression of IFNγ target genes and molecules. We found that both lenvatinib and E7090 further increased the expression of IFNγ-regulated genes and molecules in both HuH7 and JHH-7 cells, while in JHH-7 cells, the induction of IFNγ-target genes was more prominent with lenvatinib than with E7090 (Supplementary Fig. S6B). This outcome may reflect differences in the tyrosine kinase inhibitory profiles between lenvatinib (pan-FGFR1–4) and E7090 (selective against FGFR1–3), because in JHH-7 cells, the FGF signaling pathway might be activated primarily via FGFR4.
Lenvatinib and E7090 had different effects on the expression level of Cxcl11 compared with Ifit2 (Supplementary Fig. S5) in RAG cells, which mainly express FGFR1 and FGFR2. Cross-talk among IFNγ and other RTKs rather than FGFR may contribute to specifically regulating the expression levels of IFNγ-regulated genes, because the different FGFR kinase inhibition profiles of lenvatinib and E7090 do not explain, and a further investigation is needed to explain the varied expression patterns of Cxcl11 and Ifit2 in RAG cells.
Western blot analysis revealed that treatment for 2 weeks with lenvatinib—but not axitinib—induced the expression and phosphorylation of STAT1 and the expression of PD-L1 and B2M in RAG tumors (Fig. 6C). In addition, whereas the control group showed constitutive expression of SOCS1, lenvatinib—but not axitinib—decreased SOCS1 expression in RAG tumors (Fig. 6C). Because adding bFGF increased the expression of SOCS1 in cultured RAG cells (Fig. 4B), these results might indicate that FGFR signaling is constitutively active in RAG tumors in vivo. In previous studies, chronic VEGF signal blockade led to upregulation of FGF ligands in the RIP–TAG2 (pancreatic neuroendocrine tumor) model and a human head-and-neck squamous cell carcinoma xenograft model (54, 55). Recently, we demonstrated that under conditions of chronic VEGF inhibition, pericytes expressed high levels of FGF2 and FGFR2 signaling was activated in anti–VEGF-therapy-resistant B16F10 and Renca tumor models (56). These results suggest that VEGFR inhibitors may upregulate FGF ligand expression and FGFR signaling in the RAG model in vivo.
Average TVs with anti-PD-1 Ab treatment under IFNγ-blocking condition was numerically small to that with the control group (Fig. 7A). In spider plot analysis (Supplementary Fig. S7A, bottom), 1 mouse occasionally had a small tumor with anti-PD-1 Ab treatment with IFNγ-blocking Ab. Another mouse with a relatively small tumor (<800 mm3) was found dead on day 19, as mice with RAG tumors sometimes die unexpectedly due to ulceration on their tumor, and TVs from that mouse were imputed to analysis until day 29. These factors all might contribute to the smaller average TV with anti-PD-1 Ab under IFNγ-blocking conditions (Fig. 7A). Contrary, antitumor activity of combination treatment with lenvatinib plus anti-PD-1 Ab was suppressed by IFNγ blocking (Fig. 7A), even though 1–2 mice occasionally also had smaller tumors with anti-PD-1 Ab (Supplementary Fig S7A bottom and S7B, right).
In conclusion, our findings indicate that cross-talk occurs between the IFNγ- and FGF-signaling pathways in tumor cells, in which FGFR signaling inhibits the expression of genes and molecules induced through IFNγ signaling. Treatment with lenvatinib inhibited the FGFR signaling in tumor cells and restored the expression of IFNγ-target molecules that had been suppressed through FGFR signaling. Accordingly, activation of the IFNγ/JAK/STAT pathway upregulates not only the expression of antigen presentation components and chemokines but also PD-L1, which allows suppression of cancer immunity, and then tumor cells to evade from a host immune system. In such circumstances, combination treatment comprising lenvatinib plus PD-1 blockade enhances antitumor activity, as we showed in the RAG model. Therefore, when combined with anti-PD-1 mAb, lenvatinib's dual inhibition of both VEGFR and FGFR yielded more potent antitumor activity than using a selective VEGFR inhibitor with anti-PD-1 mAb. Further investigation in a clinical setting is needed to confirm this unique mechanism of lenvatinib in patients with cancer.
Authors' Disclosures
Y. Kato reports a patent for WO2016140717A1 pending. Y. Funahashi reports a patent for WO2002-032872 issued. No disclosures were reported by the other authors.
Supplementary Material
Supplementary materials and methods
Supplementary Figure S1-S7
Acknowledgments
This research was funded by Eisai Co., Ltd., Ibaraki, Japan, and Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.
The authors thank Kyoko Nishibata, Kimiyo Tabata, Naoko Hata Sugi, Masahiro Bando, Taisuke Hoshi, Yuki Niwa, Yusaku Hori, Kazuhiko Yamada, Yukinori Minoshima, Taro Semba, and Saori Ikeda, and Sunplanet for fruitful discussions and/or their support in conducting the experiments.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Contributions
Y. Adachi: Conceptualization, validation, investigation, methodology, writing–original draft, project administration, writing–review and editing. H. Kamiyama: Validation, investigation, methodology, writing–review and editing. K. Ichikawa: Validation, investigation, writing–review and editing. S. Fukushima: Validation, investigation, methodology, writing–review and editing. Y. Ozawa: Validation, investigation, writing–review and editing. S. Yamaguchi: Validation, investigation, methodology, writing–review and editing. S. Goda: Validation, investigation, writing–review and editing. T. Kimura: Methodology, writing–review and editing. K. Kodama: Methodology, writing–review and editing. M. Matsuki: Supervision, methodology, writing–review and editing. S. Watanabe Miyano: Supervision, writing–review and editing. A. Yokoi: Supervision, writing–review and editing. Y. Kato: Conceptualization, supervision, methodology, project administration, writing–review and editing. Y. Funahashi: Conceptualization, supervision, writing–review and editing.
References
- 1. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017;168:707–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tang J, Shalabi A, Hubbard-Lucey V. Comprehensive analysis of the clinical immuno-oncology landscape. Ann Oncol 2018;29:84–91. [DOI] [PubMed] [Google Scholar]
- 3. Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol 2018;15:325–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Khan KA, Kerbel RS. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat Rev Clin Oncol 2018;15:310–24. [DOI] [PubMed] [Google Scholar]
- 5. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rini BI, Plimack ER, Stus V, Gafanov R, Hawkins R, Nosov D, et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med 2019;380:1116–27. [DOI] [PubMed] [Google Scholar]
- 7. Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L, Campbell MT, et al. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med 2019;380:1103–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Motzer RJ, Tannir NM, McDermott DF, Arén Frontera O, Melichar B, Choueiri TK, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med 2018;378:1277–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cheng A-L, Qin S, Ikeda M, Galle P, Ducreux M, Zhu A, et al. IMbrave150: efficacy and safety results from a ph III study evaluating atezolizumab (atezo)+ bevacizumab (bev) vs sorafenib (Sor) as first treatment (tx) for patients (pts) with unresectable hepatocellular carcinoma (HCC). Ann Oncol 2019;30:ix186–ix7. [Google Scholar]
- 10. Matsui J, Yamamoto Y, Funahashi Y, Tsuruoka A, Watanabe T, Wakabayashi T, et al. E7080, a novel inhibitor that targets multiple kinases, has potent antitumor activities against stem cell factor producing human small cell lung cancer H146, based on angiogenesis inhibition. Int J Cancer 2008;122:664–71. [DOI] [PubMed] [Google Scholar]
- 11. Tohyama O, Matsui J, Kodama K, Hata-Sugi N, Kimura T, Okamoto K, et al. Antitumor activity of lenvatinib (e7080): an angiogenesis inhibitor that targets multiple receptor tyrosine kinases in preclinical human thyroid cancer models. J Thyroid Res 2014;2014:638747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yamamoto Y, Matsui J, Matsushima T, Obaishi H, Miyazaki K, Nakamura K, et al. Lenvatinib, an angiogenesis inhibitor targeting VEGFR/FGFR, shows broad antitumor activity in human tumor xenograft models associated with microvessel density and pericyte coverage. Vasc Cell 2014;6:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schlumberger M, Tahara M, Wirth LJ, Robinson B, Brose MS, Elisei R, et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N Engl J Med 2015;372:621–30. [DOI] [PubMed] [Google Scholar]
- 14. Takahashi S, Kiyota N, Yamazaki T, Chayahara N, Nakano K, Inagaki L, et al. A Phase II study of the safety and efficacy of lenvatinib in patients with advanced thyroid cancer. Future Oncol 2019;15:717–26. [DOI] [PubMed] [Google Scholar]
- 15. Kudo M, Finn RS, Qin S, Han K-H, Ikeda K, Piscaglia F, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet 2018;391:1163–73. [DOI] [PubMed] [Google Scholar]
- 16. Motzer RJ, Hutson TE, Glen H, Michaelson MD, Molina A, Eisen T, et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. Lancet Oncol 2015;16:1473–82. [DOI] [PubMed] [Google Scholar]
- 17. Kimura T, Kato Y, Ozawa Y, Kodama K, Ito J, Ichikawa K, et al. Immunomodulatory activity of lenvatinib contributes to antitumor activity in the Hepa1–6 hepatocellular carcinoma model. Cancer Sci 2018;109:3993–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kato Y, Tabata K, Kimura T, Yachie-Kinoshita A, Ozawa Y, Yamada K, et al. Lenvatinib plus anti-PD-1 antibody combination treatment activates CD8+ T cells through reduction of tumor-associated macrophage and activation of the interferon pathway. PLoS One 2019;14:e0212513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Castro F, Cardoso AP, Gonçalves RM, Serre K, Oliveira MJ. Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front Immunol 2018;9:847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Makker V, Rasco D, Vogelzang NJ, Brose MS, Cohn AL, Mier J, et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer: an interim analysis of a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol 2019;20:711–8. [DOI] [PubMed] [Google Scholar]
- 21. Taylor MH, Lee C-H, Makker V, Rasco D, Dutcus CE, Wu J, et al. Phase IB/II trial of lenvatinib plus pembrolizumab in patients with advanced renal cell carcinoma, endometrial cancer, and other selected advanced solid tumors. J Clin Oncol 2020;38:1154–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Terme M, Pernot S, Marcheteau E, Sandoval F, Benhamouda N, Colussi O, et al. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res 2013;73:539–49. [DOI] [PubMed] [Google Scholar]
- 23. Varney ML, Johansson SL, Singh RK. Tumour-associated macrophage infiltration, neovascularization and aggressiveness in malignant melanoma: role of monocyte chemotactic protein-1 and vascular endothelial growth factor-A. Melanoma Res 2005;15:417–25. [DOI] [PubMed] [Google Scholar]
- 24. Roland CL, Dineen SP, Lynn KD, Sullivan LA, Dellinger MT, Sadegh L, et al. Inhibition of vascular endothelial growth factor reduces angiogenesis and modulates immune cell infiltration of orthotopic breast cancer xenografts. Mol Cancer Ther 2009;8:1761–71. [DOI] [PubMed] [Google Scholar]
- 25. Ohm JE, Gabrilovich DI, Sempowski GD, Kisseleva E, Parman KS, Nadaf S, et al. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood 2003;101:4878–86. [DOI] [PubMed] [Google Scholar]
- 26. Oyama T, Ran S, Ishida T, Nadaf S, Kerr L, Carbone DP, et al. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-κB activation in hemopoietic progenitor cells. J Immunol 1998;160:1224–32. [PubMed] [Google Scholar]
- 27. Ott PA, Hodi FS, Buchbinder EI. Inhibition of immune checkpoints and vascular endothelial growth factor as combination therapy for metastatic melanoma: an overview of rationale, preclinical evidence, and initial clinical data. Front Oncol 2015;5:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Babina IS, Turner NC. Advances and challenges in targeting FGFR signalling in cancer. Nat Rev Cancer 2017;17:318–32. [DOI] [PubMed] [Google Scholar]
- 29. Klebe RJ, Chen T-R, Ruddle FH. Controlled production of proliferating somatic cell hybrids. J Cell Biol 1970;45:74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Homma S, Nagamori S, Fujise K, Hasumura S, Sujino H, Matsuura T, et al. Establishment and characterization of a human hepatocellular carcinoma cell line JHH-7 producing alpha-fetoprotein and carcinoembryonic antigen–changes in secretion of AFP and CEA from JHH-7 cells after heat treatment. Hum Cell 1990;3:152–7. [PubMed] [Google Scholar]
- 31. Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med 2015;21:1318–25. [DOI] [PubMed] [Google Scholar]
- 32. Hu-Lowe DD, Zou HY, Grazzini ML, Hallin ME, Wickman GR, Amundson K, et al. Nonclinical antiangiogenesis and antitumor activities of axitinib (AG-013736), an oral, potent, and selective inhibitor of vascular endothelial growth factor receptor tyrosine kinases 1, 2, 3. Clin Cancer Res 2008;14:7272–83. [DOI] [PubMed] [Google Scholar]
- 33. Wöhrle S, Bonny O, Beluch N, Gaulis S, Stamm C, Scheibler M, et al. FGF receptors control vitamin D and phosphate homeostasis by mediating renal FGF-23 signaling and regulating FGF-23 expression in bone. J Bone Miner Res 2011;26:2486–97. [DOI] [PubMed] [Google Scholar]
- 34. Kim KB, Chesney J, Robinson D, Gardner H, Shi MM, Kirkwood JM. Phase I/II and pharmacodynamic study of dovitinib (TKI258), an inhibitor of fibroblast growth factor receptors and VEGF receptors, in patients with advanced melanoma. Clin Cancer Res 2011;17:7451–61. [DOI] [PubMed] [Google Scholar]
- 35. Tahara M, Schlumberger M, Elisei R, Habra MA, Kiyota N, Paschke R, et al. Exploratory analysis of biomarkers associated with clinical outcomes from the study of lenvatinib in differentiated cancer of the thyroid. Eur J Cancer 2017;75:213–21. [DOI] [PubMed] [Google Scholar]
- 36. Miyano SW, Yamamoto Y, Kodama K, Miyajima Y, Mikamoto M, Nakagawa T, et al. E7090, a novel selective inhibitor of fibroblast growth factor receptors, displays potent antitumor activity and prolongs survival in preclinical models. Mol Cancer Ther 2016;15:2630–9. [DOI] [PubMed] [Google Scholar]
- 37. Miller AM, Lundberg K, Özenci V, Banham AH, Hellström M, Egevad L, et al. CD4+ CD25high T cells are enriched in the tumor and peripheral blood of prostate cancer patients. J Immunol 2006;177:7398–405. [DOI] [PubMed] [Google Scholar]
- 38. Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med 2006;203:1701–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kouhara H, Hadari Y, Spivak-Kroizman T, Schilling J, Bar-Sagi D, Lax I, et al. A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell 1997;89:693–702. [DOI] [PubMed] [Google Scholar]
- 40. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep 2017;19:1189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liau NP, Laktyushin A, Lucet IS, Murphy JM, Yao S, Whitlock E, et al. The molecular basis of JAK/STAT inhibition by SOCS1. Nat Commun 2018;9:1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Krejci P, Prochazkova J, Bryja V, Jelinkova P, Pejchalova K, Kozubik A, et al. Fibroblast growth factor inhibits interferon γ-STAT1 and interleukin 6-STAT3 signaling in chondrocytes. Cell Signal 2009;21:151–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yu K, Herr AB, Waksman G, Ornitz DM. Loss of fibroblast growth factor receptor 2 ligand-binding specificity in Apert syndrome. Proc Natl Acad Sci U S A 2000;97:14536–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Futami T, Okada H, Kihara R, Kawase T, Nakayama A, Suzuki T, et al. ASP5878, a novel inhibitor of FGFR1, 2, 3, and 4, inhibits the growth of FGF19-expressing hepatocellular carcinoma. Mol Cancer Ther 2017;16:68–75. [DOI] [PubMed] [Google Scholar]
- 45. Matsuki M, Hoshi T, Yamamoto Y, Ikemori-Kawada M, Minoshima Y, Funahashi Y, et al. Lenvatinib inhibits angiogenesis and tumor fibroblast growth factor signaling pathways in human hepatocellular carcinoma models. Cancer Med 2018;7:2641–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kalbasi A, Ribas A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol 2020;20:25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wu P, Wu D, Li L, Chai Y, Huang J. PD-L1 and survival in solid tumors: a meta-analysis. PLoS One 2015;10:e0131403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov 2017;7:188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Finn RS, Kudo M, Cheng A-L, Wyrwicz L, Ngan R, Blanc J-F, et al. LBA30Analysis of serum biomarkers (BM) in patients (pts) from a phase 3 study of lenvatinib (LEN) vs sorafenib (SOR) as first-line treatment for unresectable hepatocellular carcinoma (uHCC). Ann Oncol 2017;28. [Google Scholar]
- 50. Burstein HJ, Elias AD, Rugo HS, Cobleigh MA, Wolff AC, Eisenberg PD, et al. Phase II study of sunitinib malate, an oral multitargeted tyrosine kinase inhibitor, in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J Clin Oncol 2008;26:1810–6. [DOI] [PubMed] [Google Scholar]
- 51. Ebos JM, Lee CR, Christensen JG, Mutsaers AJ, Kerbel RS. Multiple circulating proangiogenic factors induced by sunitinib malate are tumor-independent and correlate with antitumor efficacy. Proc Natl Acad Sci U S A 2007;104:17069–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ichikawa K, Miyano S, Adachi Y, Matsuki M, Okamoto K, Matsui J. Lenvatinib suppresses angiogenesis through the inhibition of both the VEGFR and FGFR signaling pathways. Glob J Cancer Ther 2016;2:019–24. [Google Scholar]
- 53. Gunda V, Gigliotti B, Ashry T, Ndishabandi D, McCarthy M, Zhou Z, et al. Anti-PD-1/PD-L1 therapy augments lenvatinib's efficacy by favorably altering the immune microenvironment of murine anaplastic thyroid cancer. Int J Cancer 2019;144:2266–78. [DOI] [PubMed] [Google Scholar]
- 54. Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005;8:299–309. [DOI] [PubMed] [Google Scholar]
- 55. Gyanchandani R, Alves MVO, Myers JN, Kim S. A proangiogenic signature is revealed in FGF-mediated bevacizumab-resistant head and neck squamous cell carcinoma. Mol Cancer Res 2013;11:1585–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ichikawa K, Watanabe Miyano S, Minoshima Y, Matsui J, Funahashi Y. Activated FGF2 signaling pathway in tumor vasculature is essential for acquired resistance to anti-VEGF therapy. Sci Rep 2020;10:2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials and methods
Supplementary Figure S1-S7