

Abstract
Trophoblast cell surface antigen 2 (TROP2) is highly expressed on various epithelial tumors and correlates with poor prognosis. We developed the novel TROP2-directed antibody–drug conjugate (ADC), datopotamab deruxtecan (Dato-DXd, DS-1062a), with a potent DNA topoisomerase I inhibitor (DXd), and evaluated its antitumor activity and safety profiles in preclinical models.
The pharmacologic activity and mechanism of action of Dato-DXd were investigated in several human cancer cell lines and xenograft mouse models including patient-derived xenograft (PDX) models. Safety profiles were also assessed in rats and cynomolgus monkeys.
Dato-DXd bound specifically to TROP2 and was internalized into tumor cells followed by intracellular trafficking to lysosome and DXd release, which induced DNA damage and apoptosis in TROP2-expressing tumor cells in vitro. Dato-DXd exhibited in vivo antitumor activity with DNA damage induced by the accumulated DXd in TROP2-expressing xenograft tumors, but neither isotype control IgG-ADC nor anti-TROP2 antibody had this effect. Dato-DXd also showed potent antitumor activity with tumor regression in several TROP2-expressing xenograft tumors including NSCLC PDX models. Safety profiles of Dato-DXd in rats and cynomolgus monkeys were acceptable.
Dato-DXd demonstrated potent antitumor activity against TROP2-expressing tumors by efficient payload delivery into tumors and acceptable safety profiles in preclinical models. These results suggest Dato-DXd could be a valuable treatment option for patients with TROP2-expressing tumors in the clinical setting.
Graphical Abstract

Introduction
Antibody–drug conjugate (ADC) is an antibody conjugated with cytotoxic drugs via a chemical linker. It is a rapidly growing cancer therapeutic class which has a broader therapeutic window compared with conventional cancer chemotherapeutic drugs. ADC in cancer therapy is designed to bind to cancer-associated cell-surface antigens and internalize into cancer cells. Then the cytotoxic drugs are released into the cytoplasm leading to the target cell death. Although the concept of ADC is clear and straightforward, it is still challenging to develop an ideal ADC with the appropriate combination of antibody, linker and cytotoxic drug, and commercially available ADCs are still limited (1).
Recently, we developed a novel ADC technology platform with a novel DNA topoisomerase I (Topo I) inhibitor, DXd, and a cleavable tetrapeptide-based linker (2, 3). Trastuzumab deruxtecan (ENHERTU, T-DXd, DS-8201), a novel HER2-directed ADC with this DXd-ADC technology, demonstrated potent antitumor activity with acceptable safety profiles in preclinical models and patients with HER2-positive cancers (2, 4–8). Trastuzumab deruxtecan has received an accelerated approval from FDA and PMDA for patients with unresectable or metastatic HER2-positive breast cancer who have received two or more prior anti-HER2-based regimens in the metastatic setting. The DXd-ADC technology has desirable characteristics for ADC including a highly potent payload (DXd) with a short systemic half-life, a cleavable linker designed to be tumor selective which is stable in circulation, and an average drug-to-antibody ratio (DAR) up to 8 being optimized for each target. These features enable the wide therapeutic window of the ADC with high antitumor potency and less systemic toxicity (9). In the preclinical setting, the DXd-ADC technology has been effective when applied to target other cancer-associated antigens as well (10–12). These results suggest that the DXd-ADC technology is widely applicable to various target antigens for cancer therapy.
Trophoblast cell surface antigen 2 (TROP2), also known as tumor-associated calcium signal transducer 2, is a 36-kDa single pass transmembrane protein highly expressed in a variety of epithelial cancers. Although the physiologic function of TROP2 is still under investigation, it has been demonstrated that TROP2 is involved in multiple intracellular signaling including MAPK and PI3K/AKT pathways which are associated with proliferation, migration, and invasion of cancer cells (13–15). Overexpression of TROP2 correlates with poor prognosis in several types of cancers including breast cancer and non–small cell lung cancer (NSCLC; refs. 16–19). These characteristics make TROP2 an attractive target for cancer therapy.
Sacituzumab govitecan (TRODELVY, IMMU-132), a TROP2-directed ADC with a Topo I inhibitor SN-38 (an active metabolite of irinotecan), has recently received an accelerated approval from the FDA for adult patients with metastatic triple-negative breast cancer who received at least two prior therapies for metastatic disease, based on results from its phase I/II study (20). Sacituzumab govitecan has shown promising efficacy in patients with several types of TROP2-expressing cancers (21–24) and in preclinical models (25). However, its half-life in patients is approximately 11–15 hours in plasma, and accordingly, frequent dosing is required and the most commonly observed side effects are neutropenia and diarrhea which are similar to irinotecan (26).
TROP2 is also expressed in some normal epithelial tissues including skin and esophagus (27). PF-06664178, a terminated TROP2-directed ADC with a novel tubulin inhibitor Aur0101, induced skin rash and mucosal inflammation as dose-limiting toxicities in phase I study in adult patients with advanced solid tumors (28). Similar toxicity signals were reported in preclinical studies of PF-06664178 using cynomolgus monkeys (29). Although there is a report that TROP2 expression is invariably upregulated in various tumor types regardless of the baseline expression level of TROP2 in their normal tissues (30) and the toxicologic sensitivity of ADCs depends on what payload is used, careful drug design is essential for TROP2-directed therapy to minimize the risk of possible on-target toxicity in TROP2-expressing normal tissues.
To provide a valuable therapeutic option for patients with TROP2-expressing tumors, here we generated datopotamab deruxtecan (Dato-DXd, DS-1062a), the novel TROP2-directed ADC with a potent Topo I inhibitor, DXd, by applying the DXd-ADC technology platform with the optimized DAR of 4, which is expected to maximize the therapeutic window. In this article, we describe the preclinical profiles of Dato-DXd, including its mechanisms of action, antitumor activity, pharmacokinetics, and safety profiles in rats and monkeys.
Materials and Methods
Antibodies and ADCs
Datopotamab is a human IgG1 mAb (Supplementary Fig. S1) generated by humanization of the mouse mAb against human TROP2, which was obtained by immunization of mice with NCI-H322 human lung adenocarcinoma cells followed by screening of the mAbs that internalized into tumor cells using Adv-FZ33 and DT3C technology (31–33). Dato-DXd was synthesized according to the procedure published previously (2). The drug distribution was analyzed by hydrophobic interaction chromatography. Control ADC was synthesized in the same manner as Dato-DXd using a matched isotype control mAb with a comparable DAR.
Cell lines
The human pancreatic cancer cell lines BxPC-3 and CFPAC-1, the human gastric cancer cell line NCI-N87, the human pharyngeal cancer cell line FaDu, the human ovarian cancer cell line CaOV3, the human breast cancer cell line HCC1806, and the human lung cancer cell lines Calu-3, Calu-6, HCC827, and NCI-H2170, the Chinese hamster ovary cell line CHO-K1 were purchased from ATCC. The human lung cancer cell line COR-L23 was purchased from the European Collection of Authenticated Cell Cultures. The human lung cancer cell lines EBC-1 and LK-2 were purchased from the Health Science Research Resources Bank. All cell lines were cultured with the appropriate media (RPMI1640 for BxPC-3, NCI-N87, HCC1806, HCC827, NCI-H2170, COR-L23, and LK-2, EMEM or MEM supplemented with MEM non-essential amino acids solution and 1 mmol/L sodium pyruvate for FaDu, Calu-3, Calu-6, and EBC-1, IMDM for CFPAC-1, DMEM for CaOV3, and F-12 for CHO-K1) supplemented with 10% heat-inactivated FBS at 37°C and 5% CO2 atmosphere.
Binding specificity
Binding specificity of Dato-DXd was analyzed by ELISA using recombinant His-tagged human TROP2 and EpCAM proteins (Sino Biological Inc.). See “Supplementary Materials and Methods” for further information.
Species cross-reactivity
Species cross-reactivity of Dato-DXd was analyzed by cell-based ELISA using CHO-K1 cells transiently expressing FLAG-tagged human, cynomolgus monkey, rat and mouse TROP2. See “Supplementary Materials and Methods” for further information.
In vitro stability in plasma
The percent drug release of DXd from 100 μg/mL of Dato-DXd in mouse, rat, monkey, and human plasma was evaluated at 37°C for up to 21 days. The percent drug release (%) was calculated as the ratio of the released DXd determined by LC/MS-MS to the hypothetical total DXd conjugated to Dato-DXd.
Pharmacokinetics in cynomolgus monkey and xenograft model mouse
Dato-DXd was intravenously administered at 6 mg/kg to male cynomolgus monkeys, or at 10 mg/kg to NCI-N87 xenograft model mice. Plasma concentrations of Dato-DXd, total antibody, and DXd, and tumor concentrations of DXd were measured up to 21 days postdose. The pharmacokinetic parameters were estimated by noncompartmental analysis with Phoenix WinNonlin (Ver. 6.3, Certara). All animal experiments performed in this study were approved by the Institutional Animal Care and Use Committee at Daiichi Sankyo Co., Ltd.
Bioanalysis
All the DXd concentrations were determined with a validated LC/MS-MS method as described previously (34, 35). The lower limit of quantitation (LLOQ) was 0.100 ng/mL. Concentrations of Dato-DXd and the total antibody in animal plasma were determined with a validated ligand-binding assay method using human TROP2 protein and anti-DXd antibody or anti-human IgG antibody as described previously (LLOQ, 0.01 μg/mL; refs. 34, 35).
Flow cytometry
Cells were stained with mouse anti-human TROP2 mAb or isotype control mAb and FITC-conjugated anti-mouse antibody, and then analyzed by a flow cytometer. The number of binding sites of mouse anti-human TROP2 mAb were calculated using QIFI kit (DAKO Denmark A/S) according to the manufacturer's instructions.
Cytotoxic assay
Cells were seeded in 96-well microplates at 2,000 cells per well. After overnight incubation, Dato-DXd, datopotamab, control ADC or DXd in the dilution range of 0.32–1,000 nmol/L was added to the designated well. Cell viability was evaluated 6 days after the treatment using a CellTiter-Glo Luminescent Cell Viability Assay (Promega Corp.) and a microplate reader according to the manufacturer's instructions. All the readings were normalized to the percentage of the value derived from control untreated wells and the GI50 values were calculated by a Sigmoid Emax model.
Internalization assay
The internalization assay was conducted according to the procedure of Austin and colleagues (36). Briefly, the detached cells were stained with Alexa Fluor 488-labeled Dato-DXd (10 μg/mL) for 1 hour at 4°C. After washing, the cells were incubated for 0 (preincubation), 15, 30, 60, 120, and 180 minutes at 37°C, and the washed cells were fixed with 1% paraformaldehyde overnight at 4°C in the dark. The fixed cells were washed and treated with anti-Alexa488 antibody (10 μg/mL) for quenching the fluorescence on the cell surface or nontreated to detect the whole signals, and analyzed by a flow cytometer. Internalization rate (%) was calculated using the following equation: [1 – (Na – Qa)/(Na – Na × Qi / Ni)] × 100, Na: Mean fluorescence intensity (MFI) of the sample at each incubation time without quenching, Qa: MFI of sample at each incubation time with quenching, Ni: MFI of the preincubation sample without quenching, Qi: MFI of the preincubation sample with quenching.
DXd release assay
Cells were seeded in 6-well plates at 3 × 105 cells/well. After overnight culture, the cells were treated with 100 nmol/L Dato-DXd and the culture media was collected after 24 hours incubation and DXd concentration was determined.
Fluorescence imaging by confocal microscopy
The detached cells were stained, washed, and incubated in the same manner as described in “Internalization assay.” The cells were spread onto glass slides using a cytocentrifuge machine Cytospin 4 (Thermo Fisher Scientific). The cells were fixed with 10% neutral buffered formalin for 10 minutes at room temperature, permeabilized with 0.1% Triton X-100/PBS for 10 minutes at room temperature, and stained with anti-LAMP2 mAb (Abcam) at 1 μg/mL and anti-mouse IgG Alex Fluor Plus 555 (Thermo Fisher Scientific) at 2 μg/mL using an autostainer Bond-RX (Leica Biosystems). After mounting, the cells with the mounting media containing DAPI (Thermo Fisher Scientific), fluorescent images were acquired by sequential scanning of excitations at 405/488/555 nm and emissions at 410-485/493-555/560-635 nm using confocal microscope TCS-SP8 STED 3X (Leica Microsystems). Stimulated emission depletion (STED) images were also acquired by sequential scanning of excitations at 488/555 nm and emissions at 493-555/560-635 nm using the STED function with a 660 nm laser.
Simple Western analysis
Cells were lysed in RIPA buffer supplemented with protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). The equal amounts of homogenized lysates were subjected to Simple Western analysis using the WES system (ProteinSimple) and antibodies to detect H2AX, γH2AX, Chk1, pChk1, KAP1, pKAP1, cleaved PARP and cleaved caspase-3, according to the manufacturer's instructions. See “Supplementary Materials and Methods” for further information.
Cell line–derived xenograft studies
NCI-N87 model was established by injecting 1 × 107 cells suspended in saline, Calu-3 and Calu-6 models were established by injecting 6 × 106 cells suspended in Matrigel, HCC827 model was established by injecting 5 × 106 cells suspended in Matrigel, LK-2 model was established by injecting 1 × 106 cells suspended in Matrigel/saline (1:1), NCI-H2170 model was established by injecting 5 × 106 cells suspended in Matrigel/saline (1:1), and EBC-1 model was established by injecting 5 × 106 cells suspended in saline, subcutaneously into female CAnN.Cg-Foxn1nu/CrlCrlj mice (Charles River Laboratories, Inc.), respectively. ABS buffer containing 10 mmol/L acetate buffer (pH 5.5) and 5% sorbitol was used for the vehicle control group and the diluent of the test substances.
When the tumor volume reached approximately 150–300 mm3, the tumor-bearing mice were assigned to the vehicle control group, the treatment groups and the satellite sampling groups, and Dato-DXd or other test substances were administered intravenously once on day 0. The tumor volume defined as 1/2 × length × width2 was measured twice a week. The antitumor activity was evaluated approximately 3 weeks after the administration, where tumor growth inhibition (TGI, %) was calculated according to the formula of 100 × [1 – (average tumor volume of the treatment group)/(average tumor volume of the vehicle control group)], and tumor volumes were compared between the vehicle control group and the treatment groups.
Patient-derived xenograft studies
CTG-0163, CTG-0838, and CTG-1014 studies were performed by Champions Oncology Inc. Models were established by inoculating tumor fragments derived from patients with NSCLC, which were maintained in host mice, subcutaneously into female Hsd:Athymic Nude-Foxn1nu mice (Envigo). A total of 10 mmol/L histidine buffer (pH 6.0) containing 9% sucrose and 0.02% polysorbate 80 was used for the vehicle control group and the diluent of the test substances.
IHC
Tumor tissues excised from cell line–derived xenograft (CDX) and patient-derived xenograft (PDX) models were fixed in 10% neutral buffered formalin and embedded in paraffin, and 4 μm thickness of tissue sections were used for IHC against TROP2, γH2AX, and/or pKAP1. See “Supplementary Materials and Methods” for further information.
Toxicity studies in rats and monkeys
Dato-DXd was intravenously administered once every 3 weeks for a total of 3 months to Crl:CD(SD) rats, the non-cross reactive species to Dato-DXd, or cynomolgus monkeys, the cross reactive species. Clinical signs, body weight, food consumption, and clinical pathology were monitored throughout the study. Necropsy was performed on the day after the last administration. The reversibility of the toxic changes was assessed in a subsequent 2-month recovery period in both rats and monkeys.
Statistical analysis
All statistical analysis except for toxicity studies was performed using SAS System Release 9.1.3 and 9.2 (SAS Institute Inc.). Statistical analysis in toxicity studies was performed using TOXS (Fujitsu Limited) for the rat study and MiTOX System (Mitsui Zosen Systems Research Inc.) for the monkey study. All GI50 and EC50 values were determined by a Sigmoid Emax model. Dunnett multiple comparison tests was used to compare the vehicle control group and the treatment groups in CDX model studies, and unpaired t test was used to compare the vehicle group and the treatment group in PDX model studies. Spearman rank correlation coefficients were calculated by comparing the amount of the released DXd and the cell-surface expression level of TROP2 in each cancer cell line.
Results
Structure and characterization of Dato-DXd
Dato-DXd is an ADC, composed of a recombinant humanized anti-TROP2 IgG1 mAb conjugated with a Topo I inhibitor (DXd) via a tetrapeptide-based linker to reduced cysteine residues at the interchain disulfide bonds of datopotamab (Fig. 1A). The tetrapeptide-based linker is enzymatically cleavable and designed to release DXd after proteolytic processing by lysosomal enzymes such as cathepsins (2). On average, the target number of drug-linker to one antibody molecule is 4 (Fig. 1B).
Figure 1.

Structures and characteristics of Dato-DXd. A, Schematic structure of Dato-DXd. B, DAR distribution of Dato-DXd. C, Binding specificity of Dato-DXd against human TROP family proteins. Recombinant proteins were incubated with Dato-DXd or isotype control ADC (control ADC) and binding activities were measured by ELISA. Each value represents the mean and SD of triplicates. D, Species cross-reactivity of Dato-DXd. CHO-K1 cells expressing human, cynomolgus monkey, rat and mouse TROP2 were incubated with Dato-DXd and binding activities were measured by cell-based ELISA. Each value represents the mean and SD of triplicates. E,In vitro stability of Dato-DXd in plasma. The release rate at each timepoint was calculated using the mean concentration of the released DXd (N = 3). F, Pharmacokinetics of Dato-DXd in cynomolgus monkeys. Dato-DXd was intravenously administered to cynomolgus monkeys at the dose of 6 mg/kg and the concentrations of Dato-DXd, total antibody and DXd in plasma were determined. Each value represents the mean and SD (N = 3).
To confirm the binding specificity of Dato-DXd, we assessed its binding activity to human TROP family proteins, EpCAM (TROP1) and TROP2, by ELISA. Dato-DXd bound to human TROP2, but no binding was detected to human EpCAM (Fig. 1C). Appropriate coating of both proteins on the immunoplates was confirmed by detecting them with anti-His mAb (Supplementary Fig. S2A). We also assessed the species cross-reactivity of Dato-DXd by cell-based ELISA assay using CHO-K1 cells expressing human, cynomolgus monkey, rat and mouse TROP2, respectively. Dato-DXd showed similar binding affinity to both human and cynomolgus monkey TROP2 with the dissociation constant (Kd) values of 0.74 and 0.65 nmol/L, respectively. On the other hand, no binding was detected to rat and mouse TROP2 (Fig. 1D). Similar expression levels of transfected genes were confirmed by anti-FLAG mAb in each cell (Supplementary Fig. S2B). These results indicate that Dato-DXd binds specifically to TROP2 among TROP family proteins and cynomolgus monkey is an appropriate species for preclinical pharmacokinetic, and toxicologic analysis of Dato-DXd with similar binding affinity to human.
We also confirmed the stability of Dato-DXd in plasma in vitro and in vivo. The release rate of DXd from Dato-DXd in human, cynomolgus monkey, rat and mouse plasma ranged from 1.4% to 5.5% on day 21 (Fig. 1E). In cynomolgus monkeys after single intravenous administration of Dato-DXd at 6 mg/kg, there was no clear difference in the concentration of Dato-DXd and total antibody in plasma, and a low level of the released DXd was detected only at the limited early timepoints (Fig. 1F). These results indicate that Dato-DXd is stable in plasma.
Inhibition of cancer cell growth by Dato-DXd
The in vitro cell growth inhibitory activity of Dato-DXd against several cancer cell lines from multiple tumor types were evaluated. TROP2 expression on the cell surface of the cancer cell lines, FaDu (Pharynx), BxPC-3 (Pancreas), Caov-3 (Ovary), NCI-N87 (Stomach), CFPAC-1 (Pancreas), HCC1806 (Breast), COR-L23 (Lung), LK-2 (Lung), and Calu-6 (Lung) were quantitatively analyzed by flow cytometry using QIFI kit (Table 1). Dato-DXd remarkably reduced the cell growth of TROP2-high cell lines including FaDu, BxPC-3, Caov-3, NCI-N87, CFPAC-1, HCC1806, and COR-L23 which are expressing TROP2 at more than 9.7 × 104 /cell surface and had GI50 values of 0.48–7.8 nmol/L, but did not against TROP2-low cell lines including LK-2 and Calu-6 with less TROP2 expression than 8.0 × 103 /cell surface and had GI50 values of >100 nmol/L (Table 1). All of the cell lines were sensitive to DXd with GI50 values of 0.38–2.1 nmol/L, and both of datopotamab and control ADC did not inhibit the growth of any of the cell lines with GI50 values of >100 nmol/L (Table 1). These results indicate that Dato-DXd is effective in the inhibition of cancer cell growth from multiple tumor types and that it requires TROP2 expression on the cell surface.
Table 1.
In vitro cell growth inhibitory activity of Dato-DXd.
| TROP2 expression | Dato-DXd | Datopotamab | Control ADC | DXd | ||
|---|---|---|---|---|---|---|
| Cell name | Tumor type | (No. on the cell surface) | GI50 (nmol/L) | GI50 (nmol/L) | GI50 (nmol/L) | GI50 (nmol/L) |
| FaDu | Pharynx | 4.2 × 105 | 0.48 | >100 | >100 | 0.42 |
| BxPC-3 | Pancreas | 3.0 × 105 | 0.58 | >100 | >100 | 0.99 |
| Caov-3 | Ovary | 2.4 × 105 | 0.57 | >100 | >100 | 0.77 |
| NCI-N87 | Gastric | 2.0 × 105 | 7.8 | >100 | >100 | 2.1 |
| CFPAC-1 | Pancreas | 1.8 × 105 | 1.2 | >100 | >100 | 1.9 |
| HCC1806 | Breast | 1.4 × 105 | 2.5 | >100 | >100 | 0.38 |
| COR-L23 | Lung (large cell carcinoma) | 9.7 × 104 | 0.92 | >100 | >100 | 0.67 |
| LK-2 | Lung (squamous cell carcinoma) | 8.0 × 103 | >100 | >100 | >100 | 1.3 |
| Calu-6 | Lung (anaplastic carcinoma) | <1.7 × 103 | >100 | >100 | >100 | 0.98 |
Note: GI50 values are represented as the average of two independent experiments of triplicates. The values more than 100 nmol/L are represented as >100 in the table.
Internalization and intracellular trafficking of Dato-DXd in cancer cells
An ADC is expected to internalize into cancer cells after binding to the cell-surface antigens and then be trafficked to a lysosome for release of its payload. The internalization of Dato-DXd and the DXd release were evaluated in TROP2-expressing cancer cell lines in parallel with the cell growth inhibitory activity. Dato-DXd efficiently internalized with an average rate of 68.5% at 3 hours after binding to the cell-surface TROP2 in all the cell lines tested (Fig. 2A). The amount of the released DXd in the culture media at 24 hours after the Dato-DXd treatment tended to correlate with the cell-surface TROP2 level of each cell line (Fig. 2B). Intracellular trafficking of Dato-DXd was further analyzed in BxPC-3 cells by confocal microscopy. Dato-DXd internalized time-dependently, aggregated in perinuclear regions, and partly colocalized with the lysosomal marker LAMP-2 in the cells (Fig. 2C). Similar results on intracellular trafficking of Dato-DXd were also obtained in NCI-N87 cells (Supplementary Fig. S3). These observations support the expected molecular dynamics that Dato-DXd binds to cell-surface TROP2 and internalizes into tumor cells, and is trafficked to lysosome leading to DXd release.
Figure 2.

Dato-DXd dynamics and effects on cancer cells. A, Internalization rate of Dato-DXd. Each cancer cell line was treated with Alexa 488-labeled Dato-DXd and the internalization rates were measured as described in Materials and Methods. Each data represent the mean and SD of three or four independent experiments. B, Correlation between TROP2 expression and DXd release. TROP2 expression level on each cancer cell line was determined by flow cytometry. The DXd concentration in the culture media at 24 hours after the treatment with 100 nmol/L Dato-DXd was determined by LC/MS-MS and the average of two independent experiments of triplicates were used for correlation analysis (Spearman rank correlation coefficient: ρ = 0.60). C, Intracellular trafficking of Dato-DXd to lysosome. BxPC-3 cells treated with Alexa 488-labeled Dato-DXd (green) were costained with anti-LAMP2 antibody (red) and DAPI (blue), and analyzed by confocal microscopy. Bars represent 10 μm for confocal images (top) and STED images (bottom). D, DNA damage and apoptosis induced by Dato-DXd. NCI-N87 cells treated with Dato-DXd or controls for up to 72 hours were analyzed by Western blot analysis for DNA damage markers (γH2AX, pKAP1, and pChk1) and apoptosis markers (cleaved PARP and caspase 3).
Induction of DNA damage and apoptosis in cancer cells by Dato-DXd
It is presumed the cytotoxic effect of Dato-DXd is mainly caused by DXd because datopotamab did not inhibit the growth of cancer cells (Table 1). DXd is an exatecan-derivative Topo I inhibitor which is known to induce DNA damage and apoptosis in cancer cells (2, 10). To verify the contribution of DXd to the cytotoxic effect of Dato-DXd, we evaluated the phosphorylation of H2AX, KAP1, and Chk1 as DNA damage markers and the cleavage of caspase 3 and PARP as apoptosis markers in NCI-N87 cells after the treatment with Dato-DXd. Dato-DXd induced the phosphorylation of H2AX, KAP1 and Chk1 and the cleavage of caspase 3 and PARP, respectively and time-dependently as well as DXd, but neither datopotamab nor control ADC induced these changes (Fig. 2D). These results indicate that the induction of the DNA damage and apoptosis by the DXd released from Dato-DXd is one of the cytotoxic mechanisms of Dato-DXd against cancer cells.
Antitumor activity and pharmacokinetic profiles of Dato-DXd in xenograft model
Next, we evaluated the antitumor activity and the pharmacokinetic profiles of Dato-DXd using NCI-N87 xenograft mouse model in which TROP2 expression was detected in tumor by IHC analysis (Supplementary Fig. S4A). Single-dose intravenous administration of Dato-DXd at 10 mg/kg significantly inhibited tumor growth with the TGI of 96% (P < 0.001), but neither datopotamab nor control ADC inhibited tumor growth at the same dose (Fig. 3A). There was no clear difference in plasma concentration of Dato-DXd and total Ab, and a low level of DXd in plasma was detected only at the limited early timepoints (Fig. 3B). On the other hand, DXd was accumulated in tumor after the administration of Dato-DXd (Fig. 3C). Although the plasma concentrations of Dato-DXd and control ADC were similar (Fig. 3D), the tumor concentration of DXd on day 1 after the administration of Dato-DXd was higher than that of control ADC (Fig. 3E). In accordance with the accumulation of DXd in tumor, the phosphorylation of H2AX, which is a DNA damage marker, was induced time-dependently until day 7 in tumors treated with Dato-DXd, but not in tumors treated with control ADC or datopotamab (Fig. 3F). Similar results were also obtained for the phosphorylation of KAP1, which is another DNA damage marker, in tumors treated with Dato-DXd, control ADC and datopotamab (Supplementary Fig. S4B). These results suggest that the treatment of TROP2-expressing tumors with Dato-DXd leads to DXd accumulation and DNA damage in tumor cells, and then results in tumor growth inhibition.
Figure 3.

Pharmacokinetic and pharmacodynamic analysis of Dato-DXd in NCI-N87 xenograft mouse model. Mice inoculated with NCI-N87 cells were intravenously administered with Dato-DXd, control ADC, datopotamab (10 mg/kg) or vehicle on day 0. A, Antitumor activity of Dato-DXd and controls. Each value represents the mean and SE (N = 6), and statistically significant difference compared with the vehicle control analyzed by Dunnett multiple comparison test (*, P < 0.01). Plasma concentration of Dato-DXd, total Ab, and DXd (B), and tumor concentration of DXd (C) in NCI-N87 xenograft mice treated with Dato-DXd. Each value represents the mean and SD (N = 3). Plasma concentration of ADC, total Ab or DXd on day 1 in NCI-N87 xenograft mice treated with Dato-DXd, control ADC or Datopotamab (D), and tumor concentration of DXd on day 1 in NCI-N87 xenograft mice treated with Dato-DXd or control ADC (E). Each value represents the mean and SD (N = 3). F, IHC analysis for γH2AX on NCI-N87 xenograft tumors treated with Dato-DXd, control ADC, or datopotamab. Each value represents the mean (red bar) and individual data (light blue; N = 3). Representative images for pretreatment tumor, tumors treated with Dato-DXd on days 1, 3, and 7, and tumors treated with control ADC or datopotamab on day 1 were also shown.
Antitumor activity of Dato-DXd in several NSCLC CDX and PDX models
NSCLC is a promising target indication of Dato-DXd because high TROP2 expression has been observed in lung cancer (64% of adenocarcinomas and 75% of squamous cell carcinomas; ref. 37). We evaluated the antitumor activity of Dato-DXd in several NSCLC CDX and PDX models. Dato-DXd significantly inhibited the growth of TROP2-high tumors in Calu-3 model (TROP2 H-score 145) with TGI of 85% (P < 0.001), NCI-H2170 model (TROP2 H-score 115) with TGI of 95% (P < 0.001), HCC827 model (TROP2 H-score 220) with TGI of 90% (P < 0.001), and EBC-1 model (TROP2 H-score 133) with TGI of 100% (P < 0.001), but did not inhibit the growth of TROP2-low tumors in LK-2 model (TROP2 H-score 20) and Calu-6 model (TROP2 H-score 0; Fig. 4A) in accordance with the in vitro cell growth inhibitory activity shown in Table 1. In addition, Dato-DXd induced significant tumor growth inhibition in TROP2-high NSCLC PDX models, including CTG-0163 model (TROP2 H-score 262) with TGI of 77% (P < 0.001), CTG-0838 model (TROP2 H-score 163) with TGI of 98% in (P < 0.001), and CTG-1014 model (TROP2 H-score 252) with TGI of 95% (P < 0.001; Fig. 4B). There was no obvious body weight loss or unforeseen event observed in mice treated with Dato-DXd in all xenograft mouse models in this study. These results support the therapeutic potential of Dato-DXd against TROP2-expressing tumors including NSCLC.
Figure 4.
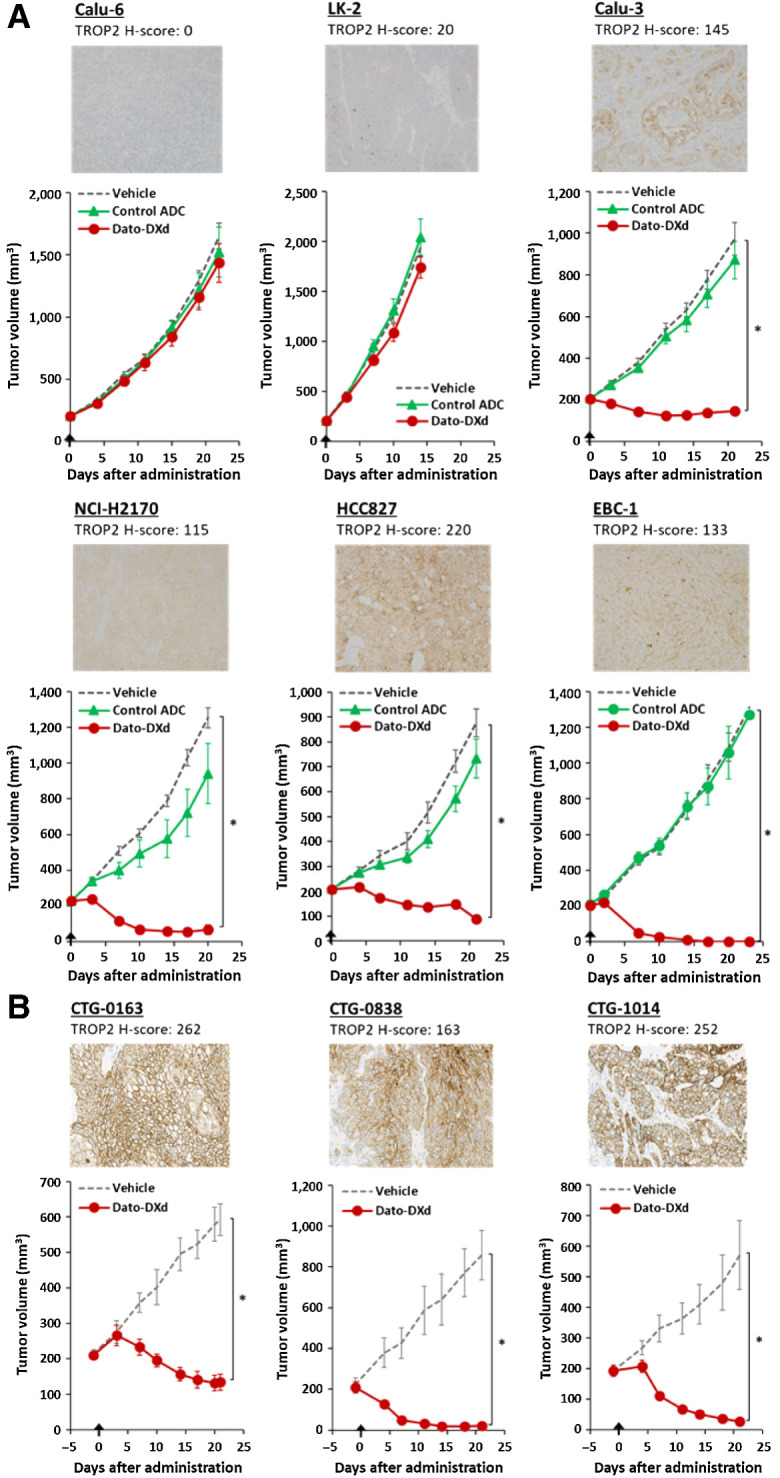
Antitumor activity of Dato-DXd in NSCLC xenograft models. A, NSCLC CDX mice were intravenously administered with Dato-DXd, control ADC (10 mg/kg) or vehicle on day 0. Each value represents the mean and SE (N = 6), and statistically significant difference compared with the vehicle control analyzed by Dunnett multiple comparison test (*, P < 0.01). Representative TROP2 IHC images and H-scores for each model were also shown. B, NSCLC PDX mice were intravenously administered with Dato-DXd (10 mg/kg) or vehicle on day 0. Each value of tumor volume represents the mean and SE (N = 5), and statistically significant difference compared with the vehicle control analyzed by unpaired t test (*, P < 0.01). Representative TROP2 IHC images and H-scores for each model were also shown.
Safety profiles of Dato-DXd
Nonclinical safety studies consisted of 3-month intermittent dose toxicity studies in rats and cynomolgus monkeys (Table 2). In the rat study, no deaths or life-threatening toxicities were observed at up to 200 mg/kg, the highest dose tested. In the monkey study, severe toxicity was limited to pulmonary toxicity at ≥30 mg/kg which was pathologically characterized by cell infiltration, edema, and fibrosis, and the incidence was low (1 and 2 animals of each 10 animals at 30 and 80 mg/kg, respectively). Although major safety concerns in the clinical use of Topo I inhibitors are gastrointestinal toxicity and bone marrow toxicity, Dato-DXd induced slight intestinal or hematopoietic toxicity in rats and monkeys and did not pronounce severe changes even at the maximum feasible doses, presumably due to reduced off-target toxicity by the stable linker adopted for Dato-DXd. The histopathologic findings in the skin and cornea, which are known to express TROP2, were also noted at ≥30 mg/kg in monkeys. The main skin lesion was pigmentation in the epidermis, and the corneal lesion included single-cell necrosis and pigmentation in the epithelium. The lesions in the skin and cornea except for the pigmentation showed reversibility after treatment withdrawal. Therefore, the highest non-severely toxic dose (HNSTD) for monkeys was concluded as 10 mg/kg on the basis of the pulmonary findings. Accordingly, Dato-DXd was well tolerated in rats and monkeys following the repeated administration corresponding to the clinical regimen, and the nonclinical safety profile warranted clinical investigation.
Table 2.
Summary of nonclinical safety profiles of Dato-DXd.
| Species | Crl:CD(SD) rats | Cynomolgus monkeys |
|---|---|---|
| Regimens | 0, 20, 60, and 200 mg/kg | 0, 10, 30 and 80 mg/kg |
| Intravenous, every 3 weeks for 3 months (five times in total) | Intravenous, every 3 weeks for 3 months (five times in total) | |
| No. of animals | 10/sex/group (main): all dose groups | 3/sex/group (main): all dose groups |
| 5/sex/group (2-month recovery): 0 and 200 mg/kg | 2/sex/group (2-month recovery): 30 and 80 mg/kg | |
| Lethal dose | >200 mg/kg | >80 mg/kg |
| Body weight | 20 mg/kg: normal | 10 mg/kg: normal |
| ≥60 mg/kg: decrease | ≥30 mg/kg: decrease | |
| Hematology | ≤60 mg/kg: normal | ≤80 mg/kg: normal |
| 200 mg/kg: decreased WBC, RET, NEU, and LYM | ||
| Blood chemistry | ≤60 mg/kg: normal | ≤80 mg/kg: normal |
| 200 mg/kg: decreased ALB and increased UN | ||
| Target organs and tissues | ≥20 mg/kg: thymus | ≥10 mg/kg: intestines |
| ≥60 mg/kg: kidney, intestines, incisor | ≥30 mg/kg: lung, cornea, skin, thymus, liver | |
| 200 mg/kg: lung, skin, bone marrow, spleen, testis, epididymis, mammary gland, ovary, vagina | 80 mg/kg: kidney, joint cartilage | |
| STD10/HNSTD | STD10: >200 mg/kg | HNSTD: 10 mg/kg |
Abbreviations: ALB, albumin; ADA, anti-drug antibody; HNSTD, highest non-severely toxic dose; LYM, lymphocyte; NEU, neutrophils; RET, reticulocyte; STD10, severely toxic dose in 10% of animals; UN, urea nitrogen; WBC, white blood cell.
Discussion
In this study, we demonstrated the TROP2-specific antitumor activity of Dato-DXd in preclinical models of various tumor types. Both in vitro and in vivo studies, Dato-DXd showed potent antitumor activity by induction of DNA damage and apoptosis in TROP2-high tumor models but not in TROP2-low tumor models, where neither datopotamab nor control ADC showed antitumor activity in any TROP2 tumor models (Table 1; Figs. 2D, 3A, 3F, and 4A). In accordance with the concept of ADC, Dato-DXd efficiently internalized into TROP2-expressing cancer cells and was trafficked to lysosome leading to the DXd release (Fig. 2A–C). However, because there could be unidentified factors which potentially affect the molecular dynamics and antitumor activity of Dato-DXd in cancer cells, the threshold of TROP2 expression level needed for Dato-DXd activity is still under investigation. In addition, the difference in efficacy and safety profiles from other TROP2-directed ADCs, such as sacituzumab govitecan and PF-06664178, should be clarified in more details.
We selected a DAR of 4 for Dato-DXd as an optimal design of TROP2-directed DXd-ADC to maximize the therapeutic window (Fig. 1A and B). Our exploratory evaluation revealed that TROP2-directed DXd-ADC with higher DAR (DAR 7) showed a narrower therapeutic window due to less tolerable safety profiles in cynomolgus monkeys compared with that with DAR of 4. In the monkey study with DAR of 7, severe toxicity was observed in the skin and esophagus (Supplementary Table S1) in which TROP2 expresses on the epithelial cells in spite of mild skin toxicity and lack of esophagus lesion in monkeys treated with DAR of 4. Although the difference in the effect of Dato-DXd on cancer cells and normal cells is still under investigation, it is speculated that TROP2 overexpression in cancer cells and the tetrapeptide-based linker which is designed for tumor-selective cleavage (9, 38) may contribute to preferential delivery and release of DXd in cancer cells. In addition, rapid proliferation of cancer cells may lead to the higher sensitivity to topoisomerase inhibitors including DXd versus relatively static or less-proliferative normal cells (39).
Regarding the details of molecular dynamics of Dato-DXd, although the internalization rate at 3 hours after binding to the cell-surface TROP2 was more than 50% in all of the cell lines analyzed here, there was variation in the initial and maximum rate among the cell lines (Fig. 2A). In addition, the amount of the released DXd showed a trend of positive correlation with the cell-surface TROP2 level in the cell lines analyzed here (Spearman rank correlation coefficient: ρ = 0.60); however, again there was variation among the cell lines as well (Fig. 2B). These variation in the internalization and release of DXd might be due to the difference in the factors related to intracellular trafficking pathways such as clathlin- or caveolin-mediated endocytosis, or lysosomal enzyme activities (40). Recycling and turnover of the TROP2 protein could also affect the DXd release rate. Further analysis is ongoing to clarify the detailed dynamics of Dato-DXd in cancer cells.
In addition to the efficiency of DXd delivery into cancer cells, the sensitivity of cancer cells to DXd can be another key factor which defines the potency of Dato-DXd. DXd showed potent cell growth inhibitory activity against all of the cancer cell lines across tumor types used in this study with GI50 values of 0.38–2.1 nmol/L (Table 1). In addition, it has been demonstrated that DXd has more potent inhibitory activity of Topo I than SN-38, an active metabolite of irinotecan (2). Irinotecan is effective against a broad range of solid tumors including colorectal, pancreatic, lung, and ovarian cancers (41), most of which overexpress TROP2. Therefore, Dato-DXd can potentially have greater antitumor efficacy via TROP2-directed DXd delivery into tumor cells than conventional chemotherapy with irinotecan in a broad range of TROP2-expressing tumors. It has also been suggested that the sensitivity to Topo I inhibitors, such as irinotecan and topotecan, is affected by several factors including Topo I expression, active DNA replication and transcription, homologous recombination deficiency, and the expression levels of DNA damage repair (DDR)-associated genes such as PARP, ATR, and SLFN11 based on the mechanism of action (42). Ongoing exploratory analyses are assessing if the response and resistance to DXd could be affected by these factors.
DXd-ADCs have a short systemic half-life payload, DXd, with a systemically stable but tumor-selective cleavable linker, which enables preferable pharmacokinetic profiles for tumor-selective payload delivery. The pharmacokinetic profiles of Dato-DXd in cynomolgus monkeys were comparable with trastuzumab deruxtecan (2) and stability in plasma was similar to other DXd-ADCs as well, as shown in the in vitro and in vivo analysis (Fig. 1E and F). The terminal elimination half-life (T1/2) of Dato-DXd at 6 mg/kg in cynomolgus monkeys was 45.12 ± 13.92 hours to be compared with the T1/2 of sacituzumab govitecan in human at 8 and 10 mg/kg that were 14.4 ± 3.3 and 11.7 ± 3.3 hours, respectively. The most common dose-limiting events related to sacituzumab govitecan in patients are neutropenia (26), which was also shown as a dose-limiting toxicity in monkeys (25). It is expected that Dato-DXd could potentially improve the efficiency of payload delivery into tumor cells and reduce the systemic toxicity possibly caused by released payload in circulation based on the difference in the stability of both drugs. In fact, Dato-DXd induced no severe hematopoietic changes even at the maximum feasible doses of 80 mg/kg in cynomolgus monkeys (Table 2) which is presumably due to the stable linker and less payload release in circulation. This advantage in potentially less hematotoxicity might allow the opportunity of the possible combination therapy of Dato-DXd. For example, the combination of Topo I inhibitors and PARP inhibitors are attractive because both drugs are targeting the DDR pathway and promising preclinical results have been reported (43); however, it is still challenging due to the overlap of the hematologic side effects (42, 44, 45). For that reason, Dato-DXd could offer improved clinical outcomes in combination therapy with PARP inhibitors because of its less myelosuppressive side effects. Moreover, uridine 5′-diphosphate glucuronosyltransferase (UGT)-dependent metabolism of DXd was not observed in human liver microsomes, while the glucuronidation of SN-38, which involved in the major dose-limiting toxicity of irinotecan (46), was affected by UGT activity. It could also result in the different safety profiles of both drugs in patients.
Another attractive opportunity of combination therapy for Dato-DXd is immunotherapy because it is well known that chemotherapy including Topo I inhibitors can enhance the effects of immune checkpoint inhibitors (ICI; ref. 47). In preclinical syngeneic mouse tumor models, it has been demonstrated that DXd-ADCs including trastuzumab deruxtecan sensitize tumors to ICIs possibly through enhanced antitumor immunity caused by the delivered DXd payload (48, 49). A similar combination effect is expected with Dato-DXd and ICIs and preclinical evaluation is ongoing.
In summary, we generated Dato-DXd, a novel TROP2-directed ADC with DXd-ADC technology, and demonstrated its potent antitumor activity and acceptable safety profiles in preclinical models. These results suggest that Dato-DXd could be a valuable therapy with a potential to benefit patients with a broad range of TROP2-expressing tumors including NSCLC in the clinical setting. Clinical evaluation of Dato-DXd in a first-in-human phase I study in patients with advanced solid tumors is in progress (ClinicalTrials.gov identifier: NCT03401385).
Authors' Disclosures
D. Okajima reports a patent for Anti-TROP2 antibody-drug conjugate issued. S. Takahashi reports a patent for Anti-TROP2 antibody–drug conjugate issued. M. Yamaguchi reports grants and personal fees from Daiich-Sankyo; grants from Japan Society for the Promotion of Science during and outside the submitted work; in addition, M. Yamaguchi has a patent for Anti-TROP2 antibody-drug conjugate issued. T. Agatsuma reports a patent for Anti-TROP2 antibody-drug conjugate issued. No disclosures were reported by the other authors.
Authors' Contributions
D. Okajima: Conceptualization, data curation, formal analysis, validation, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing. S. Yasuda: Conceptualization, data curation, formal analysis, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. T. Maejima: Conceptualization, data curation, formal analysis, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. T. Karibe: Conceptualization, data curation, formal analysis, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. K. Sakurai: Conceptualization, data curation, formal analysis, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. T. Aida: Conceptualization, writing–review and editing. T. Toki: Conceptualization, writing–review and editing. J. Yamaguchi: Data curation, formal analysis, validation, investigation, methodology. M. Kitamura: Data curation, formal analysis, validation, investigation, methodology. R. Kamei: Data curation, formal analysis, validation, investigation, methodology. T. Fujitani: Data curation, formal analysis, validation, investigation, methodology. T. Honda: Data curation, formal analysis, validation, investigation, methodology. T. Shibutani: Data curation, validation, investigation, methodology. S. Muramatsu: Data curation, validation, investigation, methodology. T. Nakada: Resources, data curation, validation, investigation, methodology, writing–review and editing. R. Goto: Resources, data curation, validation, investigation, methodology. S. Takahashi: Resources, discussion, acquisition and analysis of the mouse anti-TROP2 monoclonal antibody. M. Yamaguchi: Resources, discussion, acquisition and analysis of the mouse anti-TROP2 monoclonal antibody. H. Hamada: Resources, discussion, acquisition and analysis of the mouse anti-TROP2 monoclonal antibody. Y. Noguchi: Conceptualization, project administration, writing–review and editing. M. Murakami: Supervision, funding acquisition. Y. Abe: Supervision, funding acquisition, writing–review and editing. T. Agatsuma: Supervision, funding acquisition, writing–review and editing.
Supplementary Material
Acknowledgments
The authors would like to thank all the researchers in Daiichi Sankyo and the external collaborators who provided their expertise and assistance in the studies.
This article is featured in Highlights of This Issue, p. 2315
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Therapeutics Online (http://mct.aacrjournals.org/).
References
- 1. Chau CH, Steeg TS, Figg WD. Antibody-drug conjugates for cancer. Lancet 2019;394:793–804. [DOI] [PubMed] [Google Scholar]
- 2. Ogitani Y, Aida T, Hagihara K, Yamaguchi J, Ishii C, Harada N, et al. DS-8201a, a novel HER2-targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T-DM1. Clin Cancer Res 2016;22:5097–108. [DOI] [PubMed] [Google Scholar]
- 3. Nakada T, Masuda T, Naito H, Yoshida M, Ashida S, Morita K, et al. Novel antibody drug conjugates containing exatecan derivative-based cytotoxic payloads. Bioorg Med Chem Lett 2016;26:1542–5. [DOI] [PubMed] [Google Scholar]
- 4. Doi T, Shitara K, Naito Y, Shimomura A, Fujiwara Y, Yonemori K, et al. Safety, pharmacokinetics, and antitumour activity of trastuzumab deruxtecan (DS-8201), a HER2-targeting antibody-drug conjugate, in patients with advanced breast and gastric or gastro-oesophageal tumours: a phase 1 dose-escalation study. Lancet Oncol 2017;18:1512–22. [DOI] [PubMed] [Google Scholar]
- 5. Tamura K, Tsurutani J, Takahashi S, Iwata H, Krop IE, Redfern C, et al. Trastuzumab deruxtecan (DS-8201a) in patients with advanced HER2-positive breast cancer previously treated with trastuzumab emtansine: a dose-expansion, phase 1 study. Lancet Oncol 2019;20:816–26. [DOI] [PubMed] [Google Scholar]
- 6. Shitara K, Iwata H, Takahashi S, Tamura K, Park H, Modi S, et al. Trastuzumab deruxtecan (DS-8201a) in patients with advanced HER2-positive gastric cancer: a dose-expansion, phase 1 study. Lancet Oncol 2019;20:827–36. [DOI] [PubMed] [Google Scholar]
- 7. Modi S, Saura C, Yamashita T, Park YH, Kim SB, Tamura K, et al. Trastuzumab deruxtecan in previously treated HER2-positive breast cancer. N Engl J Med 2020;382:610–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tsurutani J, Iwata H, Krop I, Jänne PA, Doi T, Takahashi S, et al. Targeting HER2 with trastuzumab deruxtecan: a dose-expansion, phase I study in multiple advanced solid tumors. Cancer Discov 2020;10:688–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nakada T, Sugihara K, Jikoh T, Abe Y, Agatsuma T. The latest research and development into the antibody-drug conjugate, [fam-] trastuzumab deruxtecan (DS-8201a), for HER2 cancer therapy. Chem Pharm Bull 2019;67:173–85. [DOI] [PubMed] [Google Scholar]
- 10. Hashimoto Y, Koyama K, Kamai Y, Hirotani K, Ogitani Y, Zembutsu A, et al. A novel HER3-targeting antibody-drug conjugate, U3–1402, exhibits potent therapeutic efficacy through the delivery of cytotoxic payload by efficient internalization. Clin Cancer Res 2019;25:7151–61. [DOI] [PubMed] [Google Scholar]
- 11. Ogitani Y, Abe Y, Iguchi T, Yamaguchi J, Terauchi T, Kitamura M, et al. Wide application of a novel topoisomerase I inhibitor-based drug conjugation technology. Bioorg Med Chem Lett 2016;26:5069–72. [DOI] [PubMed] [Google Scholar]
- 12. Iida K, Abdelhamid Ahmed AH, Nagatsuma AK, Shibutani T, Yasuda S, Kitamura M, et al. Identification and therapeutic targeting of GPR20, selectively expressed in gastrointestinal stromal tumors, with DS-6157a, a first-in-class antibody-drug conjugate. Cancer Discov 2021;11:1508–23. [DOI] [PubMed] [Google Scholar]
- 13. Cubas R, Zhang S, Li M, Chen C, Yao Q. Trop2 expression contributes to tumor pathogenesis by activating the ERK MAPK pathway. Mol Cancer 2010;9:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guan H, Guo Z, Liang W, Li H, Wei G, Xu L, et al. Trop2 enhances invasion of thyroid cancer by inducing MMP2 through ERK and JNK pathways. BMC Cancer 2017;17:486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guerra E, Trerotola M, Tripaldi R, Aloisi AL, Simeone P, Sacchetti A, et al. Trop-2 induces tumor growth through AKT and determines sensitivity to AKT inhibitors. Clin Cancer Res 2016;22:4197–205. [DOI] [PubMed] [Google Scholar]
- 16. Lin H, Huang JF, Qiu JR, Zhang HL, Tang XJ, Li H, et al. Significantly upregulated TACSTD2 and cyclin D1 correlate with poor prognosis of invasive ductal breast cancer. Exp Mol Pathol 2013;94:73–8. [DOI] [PubMed] [Google Scholar]
- 17. Kobayashi H, Minami Y, Anami Y, Kondou Y, Iijima T, Kano J, et al. Expression of the GA733 gene family and its relationship to prognosis in pulmonary adenocarcinoma. Virchows Arch 2010;457:69–76. [DOI] [PubMed] [Google Scholar]
- 18. Mühlmann G, Spizzo G, Gostner J, Zitt M, Maier H, Moser P, et al. TROP2 expression as prognostic marker for gastric carcinoma. J Clin Pathol 2009;62:152–8. [DOI] [PubMed] [Google Scholar]
- 19. Fong D, Spizzo G, Gostner JM, Gastl G, Moser P, Krammel C, et al. TROP2: a novel prognostic marker in squamous cell carcinoma of the oral cavity. Mod Pathol 2008;21:186–91. [DOI] [PubMed] [Google Scholar]
- 20. Bardia A, Mayer IA, Vahdat LT, Tolaney SM, Isakoff SJ, Diamond JR, et al. Sacituzumab govitecan-hziy in refractory metastatic triple-negative breast cancer. N Engl J Med 2019;380:741–51. [DOI] [PubMed] [Google Scholar]
- 21. Starodub AN, Ocean AJ, Shah MA, Guarino MJ, Picozzi VJ Jr, Vahdat LT, et al. First-in-human trial of a novel anti-Trop-2 antibody-SN-38 conjugate, sacituzumab govitecan, for the treatment of diverse metastatic solid tumors. Clin Cancer Res 2015;21:3870–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Faltas B, Goldenberg DM, Ocean AJ, Govindan SV, Wilhelm F, Sharkey RM, et al. Sacituzumab govitecan, a novel antibody–drug conjugate, in patients with metastatic platinum-resistant urothelial carcinoma. Clin Genitourin Cancer 2016;14:e75–9. [DOI] [PubMed] [Google Scholar]
- 23. Heist RS, Guarino MJ, Masters G, Purcell WT, Starodub AN, Horn L, et al. Therapy of advanced non-small-cell lung cancer with an SN-38-anti-Trop-2 drug conjugate, sacituzumab govitecan. J Clin Oncol 2017;35:2790–7. [DOI] [PubMed] [Google Scholar]
- 24. Gray JE, Heist RS, Starodub AN, Camidge DR, Kio EA, Masters GA, et al. Therapy of small cell lung cancer (SCLC) with a topoisomerase-I-inhibiting antibody-drug conjugate (ADC) targeting Trop-2, sacituzumab govitecan. Clin Cancer Res 2017;23:5711–9. [DOI] [PubMed] [Google Scholar]
- 25. Cardillo TM, Govindan SV, Sharkey RM, Trisal P, Goldenberg DM. Humanized anti-Trop-2 IgG-SN-38 conjugate for effective treatment of diverse epithelial cancers: preclinical studies in human cancer xenograft models and monkeys. Clin Cancer Res 2011;17:3157–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ocean AJ, Starodub AN, Bardia A, Vahdat LT, Isakoff SJ, Guarino M, et al. Sacituzumab govitecan (IMMU-132), an anti-Trop-2-SN-38 antibody-drug conjugate for the treatment of diverse epithelial cancers: safety and pharmacokinetics. Cancer 2017;123:3843–54. [DOI] [PubMed] [Google Scholar]
- 27. Stepan LP, Trueblood ES, Hale K, Babcook J, Borges L, Sutherland CL. Expression of Trop2 cell surface glycoprotein in normal and tumor tissues: potential implications as a cancer therapeutic target. J Histochem Cytochem 2011;59:701–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. King GT, Eaton KD, Beagle BR, Zopf CJ, Wong GY, Krupka HI, et al. A phase 1, dose-escalation study of PF-06664178, an anti-Trop-2/Aur0101 antibody-drug conjugate in patients with advanced or metastatic solid tumors. Invest New Drugs 2018;36:836–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Strop P, Tran TT, Dorywalska M, Delaria K, Dushin R, et al. RN927C, a site-specific Trop-2 antibody-drug conjugate (ADC) with enhanced stability, is highly efficacious in preclinical solid tumor models. Mol Cancer Ther 2016;15:2698–708. [DOI] [PubMed] [Google Scholar]
- 30. Trerotola M, Cantanelli P, Guerra E, Tripaldi R, Aloisi AL, Bonasera V, et al. Upregulation of Trop-2 quantitatively stimulates human cancer growth. Oncogene 2013;32:222–33. [DOI] [PubMed] [Google Scholar]
- 31. Yamaguchi M, Nishii Y, Nakamura K, Aoki H, Hirai S, Uchida H, et al. Development of a sensitive screening method for selecting monoclonal antibodies to be internalized by cells. Biochem Biophys Res Commun 2014;454:600–3. [DOI] [PubMed] [Google Scholar]
- 32. Nishii Y, Yamaguchi M, Kimura Y, Hasegawa T, Aburatani H, Uchida H, et al. A newly developed anti-Mucin 13 monoclonal antibody targets pancreatic ductal adenocarcinoma cells. Int J Oncol 2015;46:1781–7. [DOI] [PubMed] [Google Scholar]
- 33. Yamaguchi M, Hirai S, Sumi T, Tanaka Y, Tada M, Nishii Y, et al. Angiotensin-converting enzyme 2 is a potential therapeutic target for EGFR-mutant lung adenocarcinoma. Biochem Biophys Res Commun 2017;487:613–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nagai Y, Oitate M, Shiozawa H, Ando O. Comprehensive preclinical pharmacokinetic evaluations of trastuzumab deruxtecan (DS-8201a), a HER2-targeting antibody-drug conjugate, in cynomolgus monkeys. Xenobiotica 2019;49:1086–96. [DOI] [PubMed] [Google Scholar]
- 35. Okamoto H, Oitate M, Hagihara K, Shiozawa H, Furuta Y, Ogitani Y, et al. Pharmacokinetics of trastuzumab deruxtecan (T-DXd), a novel anti-HER2 antibody-drug conjugate, in HER2-positive tumour-bearing mice. Xenobiotica 2020;50:1242–50. [DOI] [PubMed] [Google Scholar]
- 36. Austin CD, De Mazière AM, Pisacane PI, van Dijk SM, Eigenbrot C, Sliwkowski MX, et al. Endocytosis and sorting of ErbB2 and the site of action of cancer therapeutics trastuzumab and geldanamycin. Mol Biol Cell 2004;15:5268–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Inamura K, Yokouchi Y, Kobayashi M, Ninomiya H, Sakakibara R, Subat S, et al. Association of tumor TROP2 expression with prognosis varies among lung cancer subtypes. Oncotarget 2017;8:28725–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shiose Y, Ochi Y, Kuga H, Yamashita F, Hashida M. Relationship between drug release of DE-310, macromolecular prodrug of DX-8951f, and cathepsins activity in several tumors. Biol Pharm Bull 2007;30:2365–70. [DOI] [PubMed] [Google Scholar]
- 39. Ramaker RC, Lasseigne BN, Hardigan AA, Palacio L, Gunther DS, Myers RM, et al. RNA sequencing-based cell proliferation analysis across 19 cancers identifies a subset of proliferation-informative cancers with a common survival signature. Oncotarget 2017;8:38668–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chalouni C, Doll S. Fate of antibody-drug conjugates in cancer cells. J Exp Clin Cancer Res 2018;37:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. De Man FM, Goey AKL, Van Schaik RHN, Mathijssen RHJ, Bins S. Individualization of irinotecan treatment: a review of pharmacokinetics, pharmacodynamics, and pharmacogenetics. Clin Pharmacokinet 2018;57:1229–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Thomas A, Pommier Y. Targeting topoisomerase I in the era of precision medicine. Clin Cancer Res 2019;25:6581–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Murai J, Zhang Y, Morris J, Ji J, Takeda S, Doroshow JH, et al. Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J Pharmacol Exp Ther 2014;349:408–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen EX, Jonker DJ, Siu LL, McKeever K, Keller D, Wells J, et al. A phase I study of olaparib and irinotecan in patients with colorectal cancer: Canadian Cancer Trials Group IND 187. Invest New Drugs 2016;34:450–7. [DOI] [PubMed] [Google Scholar]
- 45. Wahner Hendrickson AE, Menefee ME, Hartmann LC, Long HJ, Northfelt DW, Reid JM, et al. A phase I clinical trial of the poly(ADP-ribose) polymerase inhibitor veliparib and weekly topotecan in patients with solid tumors. Clin Cancer Res 2018;24:744–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gagné JF, Montminy V, Belanger P, Journault K, Gaucher G, Guillemette C. Common human UGT1A polymorphisms and the altered metabolism of irinotecan active metabolite 7-ethyl-10-hydroxycamptothecin (SN-38). Mol Pharmacol 2002;62:608–17. [DOI] [PubMed] [Google Scholar]
- 47. McKenzie JA, Mbofung RM, Malu S, Zhang M, Ashkin E, Devi S, et al. The effect of topoisomerase I inhibitors on the efficacy of T-cell-based cancer immunotherapy. J Natl Cancer Inst 2018;110:777–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Iwata TN, Ishii C, Ishida S, Ogitani Y, Wada T, Agatsuma T. A HER2-targeting antibody-drug conjugate, trastuzumab deruxtecan (DS-8201a), enhances antitumor immunity in a mouse model. Mol Cancer Ther 2018;17:1494–503. [DOI] [PubMed] [Google Scholar]
- 49. Haratani K, Yonesaka K, Takamura S, Maenishi O, Kato R, Takegawa N, et al. U3–1402 sensitizes HER3-expressing tumors to PD-1 blockade by immune activation. J Clin Invest 2020;130:374–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.