

This study describes compartmentalization of base excision repair during mitosis that is dependent on NEIL3, APE1, and POLB to repair oxidative damage accumulating at telomeres in hepatocellular carcinoma.
Abstract
Patients with hepatocellular carcinoma (HCC) suffer from few treatment options and poor survival rates. Here we report that endonuclease VIII-like protein 3 (NEIL3) is overexpressed in HCC and correlates with poor survival. All six HCC cell lines investigated were dependent on NEIL3 catalytic activity for survival and prevention of senescence, while NEIL3 was dispensable for nontransformed cells. NEIL3-depleted HCC cell lines accumulated oxidative DNA lesions specifically at telomeres, resulting in telomere dysfunctional foci and 53BP1 foci formation. Following oxidative DNA damage during mitosis, NEIL3 relocated to telomeres and recruited apurinic endonuclease 1 (APE1), indicating activation of base excision repair. META-FISH revealed that NEIL3, but not NEIL1 or NEIL2, is required to initiate APE1 and polymerase beta (POLB)-dependent base excision repair at oxidized telomeres. Repeated exposure of NEIL3-depleted cells to oxidizing damage induced chromatin bridges and damaged telomeres. These results demonstrate a novel function for NEIL3 in repair of oxidative DNA damage at telomeres in mitosis, which is important to prevent senescence of HCC cells. Furthermore, these data suggest that NEIL3 could be a target for therapeutic intervention for HCC.
Significance:
This study describes compartmentalization of base excision repair during mitosis that is dependent on NEIL3, APE1, and POLB to repair oxidative damage accumulating at telomeres in hepatocellular carcinoma.
Graphical Abstract
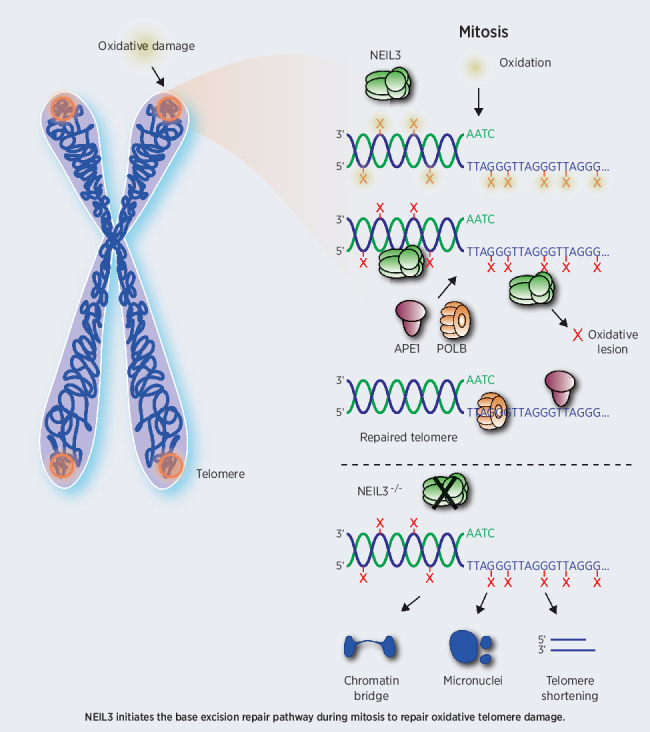
Introduction
Hepatocellular carcinoma (HCC) is the most common type of primary liver cancer and the third leading cause of cancer-related death worldwide (1, 2). Most new HCC cases and HCC-related mortalities occur in Africa, China, and Southeast Asia, where it is a reasonably common disease. HCC is commonly caused by chronic liver diseases, such as HBV- and HCV-related infection, alcoholic, metabolically and dietary-induced fatty liver disease, autoimmune or chronic cholesteric diseases (3, 4). Chronic liver diseases contribute to accumulation of reactive oxygen species (ROS) and inflammation, leading to cirrhosis and eventually HCC (5). Because of mild and nonspecific symptoms at early stages, many patients with HCC are diagnosed at advanced stages, where there are limited treatment options, are commonly drug resistant and have high recurrence rates. Although many approaches have been suggested to treat HCC [e.g., surgery, transarterial chemoembolization (TACE), immunotherapy and targeted therapies], effective drugs, and nonsurgical treatment for patients with HCC remain very limited (6–8).
Cancers have deregulated metabolism, redox homeostasis and DNA damage response and repair (DDR), which all contribute to deregulated or excessive ROS production (9), which irreversibly induce cell-cycle arrest, apoptosis and various oncogenic pathways, altogether fueling genome instability (9, 10). 8-oxodG is the most abundant oxidized lesion in cells and is prone to further oxidation, producing highly mutagenic hydantoin lesions such as spiroimindiohydantoin (Sp) and guanidinohydantoin (Gh), typically repaired through the base excision repair pathway (BER; refs. 11–13).
ROS production increases during G2–M-phase (14) and induction of oxidative damage in this phase arrests cells in pro-metaphase (15), suggesting that the level of endogenous DNA damage varies during the cell cycle and have more severe effects in certain phases. It is well established that there is differential DNA repair at telomeres (16). Telomeres are repeated TTAGGG sequence, which form G-quadruplex at the end of chromosome and are protected by the Shelterin complex to ensure the natural ends of chromosome are not mistaken as DNA damage sites (5, 17, 18). Telomeres are sensitive to oxidative damage, resulting in cell senescence, chromosome fusion, and apoptosis (19, 20). While BER pathways are described to be active at telomeres, information about these processes or potential differential roles of glycosylases are generally lacking.
The Nei endonuclease VIII-like 3 (NEIL3) is a monofunctional glycosylase that belongs to the Fpg/Nei family and functions in the BER pathway. NEIL3 preferentially recognizes G-quadruplexes and hydantoin lesions (such as Sp and Gh; refs. 12, 21). A role of NEIL3 has been illustrated in fibroblast proliferation, telomere maintenance, DNA interstrand cross-link unhooking and autoimmunity (22–25). It has been shown that NEIL3 functions in S phase to maintain telomere integrity. Silencing NEIL3 resulted in telomere loss, telomere fusion, extra telomere signals, and telomere associations between chromosomes (25). However, the increase of NEIL3 expression in G2–M phase and the observation that oxidative stress has profound effects in mitosis, suggests that NEIL3 has a crucial role in DNA repair in this phase.
Here, we find that HCC has high NEIL3 levels and that NEIL3-dependent BER is required for preventing accumulation of telomere-specific lesions in mitosis of HCC, which is required to prevent genomic instability and promote survival of HCC.
Materials and Methods
Clinical specimens
All samples were collected in Department of Liver Surgery, Renji Hospital, Shanghai Jiao Tong University School of Medicine. Tumor and nontumor tissues were all collected from 202 patients with primary HCC undergoing hepatectomy between 2010 and 2015. We randomly selected 80 pairs of tumors and paired nontumor specimens to measure NEIL3 expression level with IHC staining. 102 pairs of tumor and nontumor tissues were collected for RNA isolation. 120 patients with HCC were collected for tissue microarray (TMA) and patients were followed up regularly.
For the RNA isolation, three groups (normal liver, tumor and cirrhotic liver tissues; n = 20 for each group) were obtained from patients undergoing liver transplantation between 2016 and 2018 at Department of Liver Surgery, Renji Hospital. All specimens were collected using the same standardization process and were confirmed by pathologic examination. Written informed consent form was acquired from all patients before surgery and protocols were approved by the Institutional Review Board of Shanghai Jiao Tong University and Ethics Committee of Renji Hospital for the use of samples. The study was conducted in accordance with the Declaration of Helsinki and approved by Ethics Committee of Renji Hospital. The Ethical permit number is KY2019–114.
Data collection and analysis from the database
mRNA sequencing data and clinical data for normal tissue, cirrhotic tissue, tumor tissue and paired nontumor tissue was acquired from The Cancer Genome Atlas Liver Hepatocellular Carcinoma (TCGA LIHC) database (https://tcga-data.nci.nih.gov/), GEO database (GSE25097) and IST Online (http://ist.medisapiens.com/).
Heatmaps, genes clustering, and sample clustering were performed with MeV software. Kaplan–Meier analysis for overall survival and recurrence and Student t test were carried out with GraphPad Prism version 8.
HCC TMA IHC and analysis
TMA with 120 HCC tissues was used in this study. All the samples were applied to evaluate the prognostic value of NEIL3 based on their detailed survival data. The sections were dewaxed with xylene, gradually hydrated, and then boiled in 10 mmol/L citrate buffer (pH 6.0) for 5 minutes for antigen retrieval. The sections were blocked with goat serum, incubated with primary antibody overnight at 4°C. Then TMA was incubated with 1:200 secondary antibody in blocking buffer at room temperature following for 1 hour followed by DAPI staining for 10 minutes.
Images for each patient were taken with a confocal microscope. The NEIL3 integrated intensity in the nucleus was calculated with cell profiler. Patients were divided into two groups referring to the median. Kaplan–Meier analysis for overall survival and recurrence were carried out with GraphPad Prism software.
Cell lines and cell culture
Human HCC cell line HEP3B and osteosarcoma cell line U2-OS were purchased from ECACC in 2016 and 2013 respectively and immortalized epithelial cell line hTERT RPE-1 were purchased from ATCC in 2016. Huh7, SMMC-7721, MHCC97H, MHCC97L, L-02 and HEPG2 cells were purchased from Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences in 2013 and were preserved in the department of Liver Surgery, Renji Hospital, Shanghai, China. All cells were identified with STR PCR, and tested Mycoplasma negative with PCR or MycoAlert Mycoplasma Detection Kit before the experiment and all cells used in the experiments were within 15 passages from thawing.
HEP3B cells were cultured in minimum essential media (MEM, Gibco, 11095080) supplemented with 1% NEAA (Sigma Aldrich, M7145) and 10% FBS (Gibco, 12662029). Huh7, hTERT RPE-1, SMMC-7721, MHCC-97L, MHCC-97H, and L-02 cells were cultured in DMEM (Gibco, 11995065) with 10% FBS (Gibco, 12662029). All cells were cultured in a humidified incubator containing 5% CO2 at 37°C.
Proliferation assays
Growth curve
1,000 cells were seeded in triplicates in a 96-well plate. Cell viability was measured for 5 consecutive days with resazurin and results were plotted with GraphPad Prism. Clonogenic assay: Cells were cultured to 30–40 confluency and transfected with siRNA overnight. Cells were seeded at 300 cells/well in 6-well plates the following day and cultured in complete media (refreshed every 3 days). Colonies were counted manually after 14 days.
For the clonogenic survival assay with paclitaxel and doxorubicin, cells were first transfected with siRNA, the day after reseeded in 6-well plates and on the day 3 treated with the compounds. Fresh media was added every fourth day and colonies were counted after 12 days. For the IR experiment, cells were irradiated on day 3 with the TOMO Therapy Hi·Art radiation source.
siRNA transfections
HEP3B and Huh7 cell were seeded in 6-well polystyrene microplates and incubated until they reached 30% to 40% confluence. Cells were transiently transfected with small interfering RNA (siRNA) at a final concentration of 10 nmol/L with INTERFERin transfection reagent according to manufacturer's instructions. The cells were cultured for 48–72 hours, followed by quantification of mRNA using real-time qPCR or Western blot. The sequences that were used for the siRNA transfections are summarized in Supplementary Table S1.
All siRNAs were purchased from Qiagen, except POT1 that was purchased from Eurofins Genomics.
Flow cytometric analysis of apoptosis
To quantify apoptosis, Huh7 and HEP3B cells were transfected with negative control siRNA or siNEIL3 s2 or s4 for 72 hours. After transfection, the cells were harvested by trypsinization, washed twice with PBS and stained with Annexin V: FITC Apoptosis Detection Kit (#556547, BD Biosciences), following the manufacturer's protocol. Apoptosis (Annexin V−, PI+) or necrosis (Annexin V+, PI+) cells were gated and analyzed with FlowJo.
Antibodies
The information on the primary antibodies used is summarised in Supplementary Table S2.
Quantitative real-time PCR
A standard TRIzol method (Invitrogen) was used to extract total RNA from tissues and cultured cells and to synthesize complementary DNA. RT-PCR was performed using SYBR Premix Ex Taq (Takara) in an ABI PRISM 7900HT sequence detector. Two housekeeping genes, β-actin, and GAPDH, were used as endogenous control and the primer sequences used in this paper are summarised in Supplementary Table S3.
Western blot analysis
HCC cell line extracts were prepared for immunoblotting analyses after washing with PBS and scraped in lysis buffer (10 mmol/L HEPES pH 7.1, 50 mmol/L NaCl, 0.3 mol/L sucrose, 0.1 mmol/L EDTA, 0.5% Triton X-100, 1 mmol/L DTT and protease inhibitor cocktail). Samples were kept on ice for 30 minutes and centrifuged at 15,000 × g for 15 minutes. Supernatant was collected and 4x sample buffer (Invitrogen) was added with 100 mmol/L DTT (Sigma). and samples were denatured at 95°C for 10 minutes. Proteins were separated on 4%–12% Bis-Tris acrylamide gels in MES running buffer (Invitrogen). After separation, proteins were transferred to Hybond ECL nitrocellulose membranes (GE Healthcare) followed by blocking with 5% milk in TBS-Tween 20. Blots were probed with primary and secondary antibodies in blocking solution before image acquisition with Odyssey Fc.
EdU staining
Cells were seeded on a coverslip and incubated with 10 μmol/L EdU (Invitrogen, A10044) in culture media for 30 minutes before the experiment. Cells were fixed with 4% PFA in PBS and permeabilized with 0.5% Triton in PBS. Click-it reaction buffer were prepared according to protocol: 1 mmol/L CuSO4, 10 μmol/L Alexa Fluor 488 azide (Invitrogen, A10266), 100 mmol/L Tris (pH 7.5), and 100 mmol/L ascorbic acid (Sigma, A92902; ref. 26). After 30-minute incubation with reaction buffer, cells were washed three times with PBS. Blocking and antibody staining was performed after EdU staining.
Enzymes and inhibitors
Endonuclease VIII enzyme was purchased from New England Biolabs. Human NEIL1 was purified in bacteria by expression of the construct pET28hNEIL1 (kind gift from Susan Wallace) in BL21 (DE3) T1R pRARE2 at 18°C overnight. Bacteria were lyzed using sonication and the resulting lysate was centrifuged and filtered. C-terminally His-tagged human NEIL1 was purified using affinity chromatography using HisTrap HP (GE Healthcare). Fractions containing human NEIL1 eluted from the HisTrap column were pooled and protein was further purified using gel filtration on HiLoad 16/60 Superdex 200 (GE Healthcare). The purity of the protein was analyzed using SDS-PAGE.
Proteinase K and RNase A were purchased from Thermo Fisher.
Inhibitors used are as follows: vincristine (Sigma), Colcemid (Gibco), RO-3306 (CDK1 inhibitor, MedChemExpress), Reversine (GSK923295, Axon MedChem), APE1 inhibitor (Sigma, CAS 6960–45–8), PARP inhibitor (olaparib, SelleckChem, 763113–22–0).
Modified comet assay
Cells were suspended in 0.5% low melting point agarose in PBS and transferred onto a frosted glass microscope slide precoated with a layer of 0.5% normal melting point agarose. Slides were immersed in lysis solution (2.5 mol/L NaCl, 100 mmol/L EDTA, 10 mmol/L Tris, 1% sodium lauryl sarcosinate, 10% DMSO, and 1% Triton X-100 (pH 10) at 4°C overnight. Cells were washed with enzyme assay buffer (40 mmol/L HEPES pH 8.0, 0.1 M KCl, 0.5 mmol/L EDTA and 0.2 mg/mL BSA) three times and incubated with NEIL1 or Endonuclease VIII in enzyme assay buffer or buffer alone for 30 minutes at 37°C. Electrophoresis buffer (0.3 mol/L NaOH and 1 mmol/L EDTA) was precooled to 4 degrees and slides were incubated in electrophoresis buffer for 20 utesmin. Electrophoresis was run at 300 mA, 25 V for 30 minutes in a Comet Assay tank (Thistle Scientific). Slides were washed in neutralization buffer (0.4 mol/L Tris-HCl pH 7.5) and counterstained with 5 mmol/L YOYO-1 dye (Invitrogen). Images were acquired with a 20× objective in a Zeiss LSM 510 confocal microscope and tail moment was quantified using Comet IV software. At least 100 comets per sample were analyzed.
Telomere qPCR
Cells were seeded in a 6-well plate and DNA extraction was performed as reported before (27). The DNA concentration was measured with nanodrop and aliquots of DNA (200 ng each) from each sample were air-dried at 65°C for 1 hour. Enzyme assay buffer (25 mmol/L Tris-HCl pH = 8, 15 mmol/L NaCl, 2 mmol/L MgCl2 and 0.0025% Tween-20) was prepared before experiment. Each sample was incubated with NEIL1 or Endonuclease VIII in enzyme assay buffer or buffer alone at 37°C for 2 hours. qPCR was performed with SYBR Premix Ex Taq (Takara) in an ABI PRIS 7900HT sequence detector. One-way ANOVA analysis was performed with GraphPad Prism software.
Telomere primers: F: CGGTTTGTTTGGGTTTGGGTTTGGGTTTGGGTTTGGGTT; R: GGCTTGCCTTACCCTTACCCTTACCCTTACCCTTACCCT; The single copy gene, 36B4, primers: F: CAGCAAGTGGGAAGGTGTAATCC; R: CCCATTCTATCATCAACGGGTACAA.
The NEIL3 overexpression constructs and site-directed mutagenesis
Full-length NEIL3 was inserted at the Sal1 and Not1 restriction sites of pENTR1A-3xFlag vector by PCR amplification of with Phusion HF DNA polymerase (Thermo Fisher Scientific), NEIL3 untagged clone (NM_078248, Origene) and forward primer 5′-TATAGTCGACACCATGGTGGAAGGACCAGGCTGTACTCTG-3′ and reverse primer 5′-TATAGCGGCCGCGCATCCAGGAATAATTTTTATTCCTGGCCC-3′. To make the NEIL3–3xFlag pENTR1A vector siRNA resistant, PCR based site-directed mutagenesis was used to introduce silent mutations in NEIL3 corresponding to the siNEIL3 #4 sequence by substitution of three nucleotides using primers 5′-AAAGCTGCAACCCTGGATATAGCAACAGTGAACTTCAAAT-3′ and 5′-TTAATTTGAAGTTCACTGTTGCTATATCCAGGGTTGCAGC-3′. The K81A mutation was also introduced by site-directed mutagenesis using primers 5′-GTGGAAACTTTGGGGGCCGAGCTCTTTATGTACTTTGGACC-3′ and 5′-GTACATAAAGAGCTCGGCCCCCAAAGTTTCCACGCCACTGT-3′.
The resulting plasmids were transferred into pLenti CMV Blast DEST plasmid (Addgene #17451) by LR clonase (Invitrogen). All plasmids were verified by Sanger DNA sequencing of the inserted DNA.
The pENTR1A-3xFlag plasmid was made by inserting a 3xFlag-tag (amino acids DYKDHDGDYKDHDIDYKDDDDK) at the Xho1 and Xba1 restriction sites of pENTR1A no ccDB (Addgene #17398). pLenti CMV Blast DEST (706–1) and pENTR1A no ccDB (w48–1) were gifts from Eric Campeau and Paul Kaufman (Addgene plasmids #17398 and #17451).
Immunofluorescence and FISH
Cells were seeded on coverslips in 24-well plates (10,000–15,000 cells per well) for observation with confocal microscopy. The cells were transfected with siRNA for 72 hours or treated with different drugs. Pre-extraction was performed with 0.1% Triton in PBS for 1 minutes. Then cells were fixed with 3.7% PFA for 10 minutes followed by cold methanol (−20°C) permeabilization for 10 minutes. Cells were washed with PBS twice and blocked for 1 hour at room temperature with blocking solution (3% BSA complemented with 0.1% Tween-20 in PBS). Cells were incubated with the primary antibody in blocking solution overnight. Cells were washed three times with PBS-Tween 20 (0.1%) and secondary antibody was diluted in blocking solution for 1 hour at room temperature in dark. Cells were washed three times with PBS-Tween 20 (0.1%). Cells were refixed with 3.7% PFA, dehydrated with graded ethanol (70%, 90%, 100%) and air dried to complete. Alexa488-labeled C-rich telomere probe (Eurogentec, PN-TC060–005) or Cy3-labeled centromere probe (Eurogentec, PN-CN050–005) was diluted in hybridization buffer and incubated with cells at 80°C for 5 minutes followed by incubation at room temperature for 60 minutes. After hybridization, cells were washed with PNA wash A (70% formamide, 10 mmol/L Tris-Cl pH 7.5) twice followed by three washes with PNA wash B (50 mmol/L Tris-Cl pH7.5, 150 mmol/L NaCl, 0.8% Tween-20). DAPI (Invitrogen) was added to PNA wash B in the second wash to counterstain DNA. Coverslips were dehydrated with graded ethanol, air dried, and mounted with prolonged gold (Life Technologies, P36934). Images were acquired in a Zeiss LSM-780 confocal microscope with the 40× oil objective. Foci and colocalization were calculated with Cell Profiler.
Metaphase spread FISH
Metaphase FISH (META-FISH) were performed according to protocol (28). Cells were synchronized with RO-3306 before the experiment. Cells were washed five times with PBS and cultured in complete media with 50 ng/mL Colcemid for 1.5–2.5 hours to accumulate cells in mitosis. Cells were trypsinized and suspended in hypotonic buffer (2% Trisodium citrate/2% KCl in ddH2O) at 100,000–200,000/mL for 5 minutes. 500 μL of each sample was centrifuged with Thermo Shandon Cytospin 4 at 2,000 rpm for 10 minutes.
Cells were fixed immediately after centrifuge for 10-minutes with 3.7% PFA followed by KCM buffer for 10 minutes to permeabilize the cells. Cells were blocked in ABDIL buffer with RNase A for at least 15 minutes at 37°C. Primary antibody was diluted in ABDIL buffer and incubated at room temperature for 1 hour. Cells were washed with PBS-Tween 20 (0.1%) three times and secondary antibody was diluted in ABDIL buffer and incubated at room temperature for 0.5 hour. Telomere PNA probe hybridization is performed as described in FISH. At least 20 cells in META-FISH or 200 cells in FISH were analyzed in each sample.
Results
NEIL3 dysregulation correlates with unfavorable prognosis in HCC
To identify potential DDR genes that correlated with poor prognosis in HCC, we analyzed five major DNA repair pathways, including BER, nucleoid excision repair, mismatch repair, homologues recombination and non-homologous end-joining pathways, in publicly available databases. All of the DDR pathways were consistently overexpressed in cancer as compared with paired noncancer tissues but heterogeneously activated within HCC (Supplementary Fig. S1A and S1B). We established a prognosis risk model with genes in the 5 major DDR pathways in which NEIL3, MUTYH, XRCC5, GTF2H1, CUL4A1, and RFC3 were found to be independent markers for overall survival (OS) and NEIL3, APEX1, ERCC8, DDB1, and MLH1 markers for progression-free survival (PFS) in patients with HCC (Supplementary Fig. S1C–S1F). The OS and PFS risk score were calculated according to their HR and was significantly correlated with OS and PFS, respectively (Supplementary Fig. S1G and S1H). Furthermore, we analyzed all of the genes in the prognosis risk model and NEIL3 was found to be the most promising prognostic marker in HCC, being significant for both OS and PFS (Supplementary Fig. S1I).
Interestingly, we found that NEIL3 expression is high in the fetal liver, thymus and the bone marrow and significantly reduced in adult liver tissues (GSE2361 database and IST online, Supplementary Fig. S2A). The NEIL3 expression increases from normal, nontumor tissue to cirrhotic tissue and peaks in tumor tissue (GSE25097 database, Supplementary Fig. S2B). To determine whether the high level of NEIL3 is functionally relevant for tumor survival, we compared the tissues from the HCC cohort with paired nontumor tissues and correlated with advanced TNM staging, accelerated proliferation and found unfavorable outcome such as reduced OS and PFS (TCGA database, Fig. 1A and B; Supplementary Fig. S2C and S2D; ref. 29).
Figure 1.

NEIL3 overexpression correlates with unfavorable outcome in HCC. A, NEIL3 mRNA overexpression correlates with reduced overall survival and progression-free survival (PFS). TCGA LIHC cohort, Cox regression from 370 patients. B, NEIL3 mRNA is overexpressed in HCC compared with nontumor tissue. TCGA LIHC cohort, medians, and quartiles from 50 patients each. C, NEIL3 mRNA is overexpressed in HCC compared with nontumor tissue in Renji HCC cohort. Results from 102 patients; Student t test. D, NEIL3 mRNA level increases from normal tissue, cirrhotic liver tissue to HCC tissue. Medians and quartiles from 20 patients each. E, IHC staining quantification (average optical density, AOD) showed that NEIL3 increases in HCC compared with paired nontumor tissue. Means ± SEM from Renji #1 Cohort, 80 patients. F, Images of NEIL3 staining in different TNM stages. Scale bar, 200 μm. G, Percentage of patients in different TNM stages with high or low NEIL3 level. (n = 82). H, NEIL3 level in different TNM stages. Medians and SD from 84 patients. I, NEIL3 overexpression correlates with reduced overall survival and PFS in Renji #2 Cohort. Cox regression analysis from 82 patients. *, P < 0.05; **, P < 0.01; ****, P < 0.0001; Student t test.
To validate these results, we used a separate database with a cohort of 202 patients with HCC collected in Renji Hospital in between 2010–2015. By RT-qPCR analysis, we could observe an increase in NEIL3 mRNA expression in HCC tissues compared with paired nontumor tissue and increased from normal liver, to cirrhosis and tumor tissue (Fig. 1C and D; Supplementary Fig. S2E). Furthermore, by IHC analysis, we found that the NEIL3 expression was negatively correlated with survival and progression-free survival of patients with HCC and positively correlates with the advances in TNM staging (Fig. 1E–I; Supplementary Fig. S2F and S2G; Table 1).
Table 1.
Comparison of clinicopathologic profiles between low and high NEIL3 expression in patients with HCC from Renji Hospital Cohort.
| NEIL3 level | |||||||
|---|---|---|---|---|---|---|---|
| Low (n = 43) | High (n = 42) | ||||||
| Variables | No. of patients | % | No. of patients | % | OR | 95% CI | P Value |
| Age, yrb | |||||||
| <50 | 15 | 34.9% | 16 | 38.1% | 0.871 | 0.360–2.107 | 0.758 |
| ≥50 | 28 | 65.1% | 26 | 61.9% | |||
| Genderb | |||||||
| Female | 9 | 20.9% | 9 | 21.4% | 0.971 | 0.343–2.748 | 0.955 |
| Male | 34 | 79.1% | 33 | 78.6% | |||
| HBV-DNAb | |||||||
| <500 copies/mL | 21 | 60.0% | 13 | 50.0% | 1.500 | 0.539–4.176 | 0.437 |
| ≥500 copies/mL | 14 | 40.0% | 13 | 50.0% | |||
| Cirrhosisb | |||||||
| Absent | 5 | 11.6% | 7 | 17.1% | 0.639 | 0.185–2.203 | 0.476 |
| Present | 38 | 88.4% | 34 | 82.9% | |||
| Alpha-fetoproteina,b | |||||||
| ≤500 ng/mL | 24 | 60.0% | 15 | 36.6% | 2.600 | 1.061–6.374 | 0.035 |
| >500 ng/mL | 16 | 40.0% | 26 | 63.4% | |||
| Tumor sizeb | |||||||
| ≤5 cm | 23 | 53.5% | 15 | 36.6% | 1.993 | 0.832–4.774 | 0.120 |
| >5 cm | 20 | 46.5% | 26 | 63.4% | |||
| Multinodular tumorb | |||||||
| No | 34 | 79.1% | 30 | 73.2% | 1.385 | 0.505–3.797 | 0.526 |
| Yes | 9 | 20.9% | 11 | 26.8% | |||
| Histological gradeb | |||||||
| I–II | 27 | 62.8% | 25 | 61.0% | 1.080 | 0.448–2.606 | 0.864 |
| III–IV | 16 | 37.2% | 16 | 39.0% | |||
| BCLC stagea,b | |||||||
| A | 26 | 60.5% | 13 | 31.7% | 3.294 | 1.342–8.086 | 0.008 |
| B–D | 17 | 39.5% | 28 | 68.3% | |||
| pTNM stagea,b | |||||||
| I–II | 28 | 65.1% | 17 | 41.5% | 2.635 | 1.090–6.371 | 0.030 |
| III–IV | 15 | 34.9% | 24 | 58.5% | |||
Abbreviations: BCLC, Barcelona Clinic Liver Cancer staging; HBV-DNA, hepatitis B virus DNA; lymph node, metastasis classification; pTNM, pathologic tumor.
a P < 0.05, χ2 test significant.
b χ 2 test.
HCC cell lines require NEIL3 for cell proliferation in vitro
We hypothesized that the high NEIL3 levels in HCC may relate to a functional role in cancer cell survival and we wanted to further investigate the role of NEIL3 in cell survival of HCC cell lines. Silencing NEIL3 by siRNA (Supplementary Fig. S3A) reduced the clonogenic ability for all of the 6 HCC cells tested but had no significant effect on normal L-02 liver cells or nontransformed RPE-1 hTERT retinal cells (Fig. 2A). NEIL3 depletion in HEP3B and Huh7 cells significantly reduced cell viability (Fig. 2B; Supplementary Fig. S3B and S3C) and the proliferation of HEP3B cells, measured by EdU staining, was slightly reduced after NEIL3 silencing (Supplementary Fig. S3D). In HEP3B cells, overexpressing WT NEIL3-flag, but not catalytically dead mutant K81A, increased clonogenic outgrowth and cell viability (Fig. 2C and D; Supplementary Fig. S3E). This indicates that the catalytic activity is important for the observed effect on cell proliferation.
Figure 2.

NEIL3 modulates proliferative potency of liver cancer cells and protects against senescence. A, Clonogenic survival of 6 liver cancer cell lines and two normal cell lines (L-02 and hTERT RPE-1). Cells were transfected with NEIL3 siRNA s2, NEIL3 siRNA s4, or nontargeting control (siCtrl). Means ± SEM from three independent experiments. B, The proliferation curves of HEP3B cells. Cells were transfected with NEIL3 siRNA s2, s4, or nontargeting control (siCtrl). Means ± SEM from three independent experiments. C, The proliferation curves of HEP3B cells with empty vector (EV), wild-type (WT), and catalytic dead (K81A) FLAG-tagged NEIL3. Means ± SEM from three independent experiments. D, Clonogenic survival of HEP3B cells overexpressing wild-type (WT) and catalytic dead (K81A) FLAG-tagged NEIL3. Means ± SEM from three independent experiments. E, Images of β-gal staining in wild-type HEP3B (left) and in HEP3B cells–overexpressing wild-type (WT) and catalytic dead (K81A) FLAG-tagged NEIL3 (right). Cells were transfected with NEIL3 siRNA s2, s4, or nontargeting control (siCtrl). Scale bar, 100 μm. F, Quantification of β-gal–positive cells per field in wild-type HEP3B (top) and HEP3B cells overexpressing wild-type (WT) and catalytic dead (K81A) FLAG-tagged NEIL3 (bottom). Means ± SEM from three independent experiments; at least 200 cells were analyzed. *, P < 0.05; **, P < 0.01; ***, P < 0.001; Student t test.
To investigate whether NEIL3 knockdown would cause cell death in HCC cells, we measured apoptosis with Annexin V and PI staining and necrosis with Cytox blue staining. However, we did not find any significant differences in apoptosis or necrosis in NEIL3-depleted cells compared with wild-type cells (Supplementary Fig. S3F). In contrast, when we analyzed NEIL3-silenced HEP3B and Huh7 cells in the β-galactosidase (β-gal) senescence assay, we could observe increased percentage of β-gal–positive cells as compared with control cells (Fig. 2E and F; Supplementary Fig. S3G and S3H). By site-directed mutagenesis, we designed a rescue system in which both wild-type (WT) and catalytically dead mutant (K81A) NEIL3-flag vectors are sensitive to NEIL3 siRNA sequence 2 (s2) but resistant to siRNA sequence 4 (s4) (Supplementary Fig. S3I). Overexpression of WT NEIL3, but not the catalytic-dead mutant, could rescue the induced senescence (Fig. 2E and F). We could also show that NEIL3 knockdown induced senescence by using p21 and p16INK4 antibodies (Supplementary Fig. S3J–S3L). Furthermore, NEIL3 knockdown did not increase the sensitivity to commonly used cancer treatments such as ionizing radiation (IR), paclitaxel and doxorubicin in a clonogenic survival assay (Supplementary Fig. S3M and S3N).
NEIL3 maintains telomere integrity by removing oxidized lesions
Here, we wanted to identify the molecular mechanism of NEIL3 in maintaining genome stability in HCC. NEIL3 has been shown to have a broad substrate specificity (30), to be active at telomeres and to be highly expressed in the G2–M phase (25), suggesting that it could have a role for BER in mitosis. We started by validating that silencing of NEIL3 induces DNA double-strand breaks (DSB) in cancer cells (31). After siRNA-mediated NEIL3 knockdown in HEP3B and Huh7 cells, we observed an increase in 53BP1 foci (Supplementary Fig. S4A and S4B). Overexpressing WT but not the catalytically dead mutant could rescue the 53BP1 foci formation after NEIL3 knockdown (Supplementary Fig. S4C). To validate that the catalytic activity of NEIL3 is needed to repair oxidized lesion in liver cancer cells, we performed a modified comet assay with endonuclease VIII and NEIL1. Both enzymes recognize and excise Sp and Gh lesions in DNA, similarly to NEIL3 (30, 32), and treatment with recombinant enzymes would introduce nicks in the DNA that would be detected in the alkaline comet assay. We validated the assay by treatment of HEP3B cells with 1 and 5 mmol/L KBrO3 for 2 hours, and this treatment induced increased tail moment after NEIL1 or Endo VIII digestion as compared to undigested control (Supplementary Fig. S5A). NEIL3 knockdown in HEP3B or Huh7 cells dramatically increased the amount of NEIL1 and Endo VIII–dependent lesions, indicating that NEIL3 is involved in removing endogenous oxidized lesions in nuclear DNA (Fig. 3A and B; Supplementary Fig. S5B–S5E). As for 53BP1 foci, oxidized lesions could be rescued by overexpressing of WT NEIL3 but not by the K81A mutant (Fig. 3C and D; Supplementary Fig. S5F and S5G). Furthermore, overexpression of WT NEIL3 could reduce the basal level of oxidized lesions in HEP3B cells, while the catalytic mutant could not, indicating that the catalytic activity of NEIL3 is needed for removing oxidized lesions in cells (Fig. 3E and F).
Figure 3.

NEIL3 silencing induces oxidative damage on telomeres. A and B, Images (A) and quantification (B) of modified comet assay with Endo VIII in HEP3B cells after NEIL3 silencing. Medians and quartiles; at least 100 cells were analyzed in each group. C and D, Images (C) and quantification (D) of modified comet assay with Endo VIII in HEP3B cells overexpressing wild-type (WT) and catalytic dead (K81A) FLAG-tagged NEIL3. Cells were transfected with NEIL3 siRNA s2, s4, or nontargeting control (siCtrl). Medians and quartiles; at least 100 cells were analyzed in each group. E and F, Images (E) and quantification (F) of modified comet assay with Endo VIII in HEP3B overexpressing EV, NEIL3 WT, or NEIL3 K81A. Medians and quartiles; at least 100 cells were analyaed in each group. G, Images of 53BP1 (red) and telomeres (TTAGGG; green) in HEP3B cells by anti-53BP1 IF and telo-FISH. Cells were transfected with NEIL3 siRNA s2, s4, or nontargeting control (siCtrl). H, Quantification of TIFs (53BP1/telomere colocalization) in HEP3B and Huh7 cells after NEIL3 silencing. Means ± SEM of three repeats; at least 200 cells were analyzed in each group. I, Quantification of TIFs in HEP3B cells overexpressing wild-type (WT) and catalytic dead (K81A) FLAG-tagged NEIL3 after NEIL3 silencing. Means ± SEM of three repeats; at least 200 cells were analyzed in each group. J, Telomere qPCR with Endo VIII or NEIL1 incubation in HEP3B cells. Cells were transfected with NEIL3 siRNA s2, s4, or nontargeting control (siCtrl). K, Telomere qPCR with Endo VIII incubation in NEIL3 WT- or K81A-expressing HEP3B cells. Cells were transfected with NEIL3 siRNA s2, s4, or nontargeting control (siCtrl). Means ± SEM of three repeats. Scale bars: A, C, and E, 50 μm; G, 5 μm. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; Student t test.
It's been shown that NEIL3 knockdown can induce both replication stress and telomere instability (25, 33). To distinguish between these possibilities, we measured colocalization of DNA lesions by 53BP1 foci and determined whether these preferentially located to collapsed replication forks by EdU co-staining or to telomere dysfunction foci (TIF), respectively. No significant increase in 53BP1/EdU colocalization could be observed while the TIFs increased after silencing of NEIL3 in HEP3B and Huh7 cells (Fig. 3G and H; Supplementary Fig. S5H and S5I). The increase in TIFs after NEIL3 siRNA could be rescued by WT NEIL3 overexpression, but not K81A mutant (Fig. 3I), demonstrating NEIL3 catalytic activity is required to supress TIFs. To analyze whether NEIL3 is involved in repair of damaged telomeres, we performed a telomere qPCR assay on genomic DNA incubated with recombinant Endo VIII or NEIL1 enzymes to measure the oxidative lesions on telomeres (34). Briefly, after siRNA transfection, genomic DNA was isolated and incubated with Endo VIII or NEIL1 enzymes that would excise the damaged base and introduce a nick in the DNA. In a subsequent qPCR reaction, telomeres with many nicks would have reduced amplification of the DNA and a lower signal. We observed decreased telomere amplification in the NEIL3 silenced group, indicating that NEIL3 knockdown increased oxidized lesions on telomeres (Fig. 3J). The increase in damaged telomeres after NEIL3 siRNA silencing could be rescued by overexpression of WT NEIL3, but not the K81A mutant (Fig. 3K). It has been shown that prolonged mitotic arrest, for example by microtubule inhibition by vincristine, induce ROS (35). However, treatment with the microtubule inhibitor vincristine did not further decrease the telomere level after NEIL3 silencing (Supplementary Fig. S5J). We also noted that silencing of NEIL3 caused a slight decrease in relative telomere intensity (telomere/centromere intensity; Supplementary Fig. S5K and S5L), suggesting that NEIL3 could be involved in telomere length maintenance.
NEIL3 relocates to damaged telomeres and recruits APE1 during mitosis
In a previous study, it was suggested that the expression of NEIL3 is regulated via the Ras dependent ERK-MAP kinase pathway and that the NEIL3 level peaks in G2–M phase (36). To understand if NEIL3 is recruited to damaged telomeres in G2-phase or in mitosis, we arrested cells in G2 phase with the CDK1 inhibitor RO-3306 or in mitosis with vincristine, followed by immunofluorescence analysis. Results show that vincristine treatment induced an increase in NEIL3 foci colocated to telomeres and increased NEIL3-positive TIFs (Fig. 4A and B). The accumulation of ROS in mitosis can be prevented by cotreatment with reversine, an Mps1 inhibitor that prevents spindle assembly checkpoint (SAC)-mediated mitotic arrest (37). Here we found that cotreatment with reversine prevented recruitment of NEIL3 to damaged telomeres, demonstrating that a mitotic arrest is required for NEIL3 recruitment to TIFs. In contrast, NEIL3 foci was not induced by RO-3306-mediated G2 phase arrest (Fig. 4B), showing that NEIL3 is involved in repair during mitosis but not in the G2 phase of the cell cycle. We could also observe a similar recruitment of NEIL3 to telomeres and to TIFs after inducing telomere damage by silencing the Shelterin complex protein POT1 or TPP1 (Supplementary Fig. S6A–S6C), which induced unprotected telomeres. Altogether, these data show that NEIL3 is recruited to telomeres when the DDR is activated.
Figure 4.

NEIL3 recruits APE1 to TIFs during mitosis. A, Images of NEIL3 foci (red) and telomere (TTAGGG; green) colocalization by anti-NEIL3 IF and telo-FISH in HEP3B cells. Cells were treated with DMSO, 24-hour vincristine (VCR; 20 nmol/L), 24-hour vincristine (20 nmol/L) + 24-hour Reversine (REV; 0.5 μmol/L) or 24-hour RO-3306 (10 μmol/L). Scale bar, 5 μm. B, Quantification of NEIL3 foci–positive cells (top left), TIFs (top right), NEIL3 on telomere (bottom left), and NEIL3-positive TIFs (bottom right) in HEP3B cells. Means ± SEM; at least 200 cells were analyzed. C, Images of NEIL3 foci (red), telomere (TTAGGG, green), and γH2AX foci (cyan) colocalization by anti-NEIL3, PNA telC probe, and anti-γH2AX IF in HEP3B metaphase spread. Cells were treated with 24-hour vincristine (20 nmol/L) or synchronized in 1-hour colcemid (20 ng/mL) with 30 min KBrO3 (5 mmol/L) or DMSO. White arrowheads, NEIL3/telomere/γH2AX colocalization. Scale bar, 5 μm. D, Quantification of relative TIFs (left), NEIL3 on telomere (middle), or NEIL3-positive TIFs (right) in HEP3B metaphase spread. Means ± SEM; at least 20 metaphase cells were analyzed in each group. E, Quantification of APE1 foci on telomere (left) and APE1/NEIL3 colocalization (right) in HEP3B metaphase spread. Cells were treated with 24-hour vincristine (20 nmol/L) or synchronized in 1-hour colcemid (20 ng/mL) with 30-minute KBrO3 (5 mmol/L) or DMSO. Means ± SEM; at least 20 metaphase cells were analyzed in each group. F, Quantification of APE1 on telomere (left) and APE1-positive TIFs (right) after NEIL1, NEIL2, or NEIL3 silencing in HEP3B metaphase spread. Means ± SEM; at least 20 metaphase cells were analyzed in each group. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Next, we wanted to understand if NEIL3 is recruited to damaged telomeres in metaphase cells after induction of DNA damage. With the META-FISH assay, we could show increased recruitment of NEIL3 to TIFs after KBrO3 treatment during mitosis or prolonged vincristine treatment (which induce ROS and TIFs) in both HEP3B and U2OS cells (Fig. 4C and D; Supplementary Fig. S6D). Interestingly, the BER pathway protein AP endonuclease (APE1) was also recruited to TIFs in metaphase cells and APE1 colocalized with NEIL3 after inducing TIFs by KBrO3, both in HEP3B and U2OS cells (Fig. 4E; Supplementary Fig. S6E). To explore whether the APE1 recruitment to TIFs during mitosis is NEIL dependent, we silenced NEIL1, NEIL2, and NEIL3 respectively (Supplementary Fig. S6C) and measured recruitment of APE1 to TIFs by the META-FISH assay. The result showed that recruitment of APE1 during mitosis is impaired after NEIL3, but not after NEIL1 or NEIL2, silencing (Fig. 4F; Supplementary Fig. S6F). Furthermore, the APE1 recruitment to damaged telomeres during mitosis was rescued by overexpression of WT NEIL3 but not by the catalytically dead mutant (Supplementary Fig. S6G).
Telomere DNA damage is repaired during mitosis by base excision repair
Next, we wanted to further understand the role of NEIL3 in telomere maintenance and repair during mitosis. It has been shown that the later steps of DSB repair are inhibited during mitosis to prevent telomere fusions (38). However, whether DNA single-strand breaks or oxidized lesions are repaired during mitosis has not been explored. Here we focused on the role of BER during mitosis by treating HEP3B cells with KBrO3 at different time points while synchronizing cells to mitosis. In brief, cells were synchronized with RO-3306 in the G2 phase of the cell cycle and followed by colcemid treatment to accumulate cells in metaphase. Cells were either kept in colcemid as negative control (group 0), cotreated with KBrO3 from at the beginning (group B, C) or toward the end (group A) of the colcemid treatment. Cells would either recover in control media (group B) or media supplemented with APE1 inhibitor or PARP inhibitor (group C) (Fig. 5A). BER at telomeres was measured by Meta-FISH assay and the number of APE1-positive TIFs was analyzed. Treatment with KBrO3 (group A) increased both the number of TIFs and the number of APE1-positive TIFs in mitotic cells as expected and both were reduced after repair in control media for 1 hour (group B). Treatment with APE1 inhibitor or the PARP inhibitor olaparib prevented repair of the DNA damage (Fig. 5B and C). Similar results could be observed in telomerase negative U2OS cells subjected to a 2-hour repair period (Supplementary Fig. S7A and S7B). In addition, similar results were observed when analyzing the repair of XRCC1-positive TIFs in mitotic HEP3B cells (Supplementary Fig. S7C and S7D).
Figure 5.

NEIL3 repairs telomere damage through base excision repair in mitotic cells. A, Schematic of mitotic telomere damage repair assay. Cells were treated with 10 μmol/L RO-3306 for 16 hours to synchronize cells in G2 phase. No damage was induced in group 0 and oxidative damage was induced in groups A–C. Group A represents no repair, group B 1 hour of repair, and group C 1 hour of repair in the presence of inhibitors. DNA repair at telomeres were analyzed by META-FISH. B, Images of APE1 foci (red), telomere PNA probe (TTAGGG, green), and γH2AX (cyan) by anti-APE1, anti-γH2AX IF, and telo-FISH in HEP3B metaphase spread. Cells were treated as indicated in A with 10 μmol/L APE1 inhibitor or 10 μmol/L PARP inhibitor (olaparib). White arrowheads, APE1/telomere/γH2AX colocalization. Scale bar, 5 μm. C, Quantification of TIFs (left) and APE1-positive TIFs (right) in HEP3B metaphase spread described in B. Means ± SEM; at least 20 metaphase cells were analyzed in each group. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. D, Quantification of TIFs (top) and APE1-positive TIFs (bottom) in HEP3B metaphase spread described in A. Cells were transfected with NT siRNA and siRNA targeting PolB or NEIL3 for 72 hours. Means ± SEM; at least 20 metaphase cells were analyzed in each group. *, P < 0.05; **, P < 0.01; ***, P < 0.001. E, Telomere qPCR of NEIL1 digestion in HEP3B cells transfected with NEIL3 siRNA s2, s4, or nontargeting control (siCtrl) for 72 hours. After transfection, cells were synchronized in G1 with 2 mmol/L thymidine, followed by synchronization in G2 with RO-3306. Cells were released into mitosis and treated with 2.5 mmol/L KBrO3 for 30 minutes, followed by recovery for 60 minutes. Controls are shown with no treatment (untreated) and with no recovery period (no repair). Values were normalized to untreated siCtrl and represent the mean ± SEM from three experiments. ns, nonsignificant; **, P < 0.01, one-way ANOVA analysis.
It has been shown that XRCC1 interacts with polymerase beta (PolB) to coordinate efficient BER (39) and that PolB is essential for the excision step of both short- and long-patch BER (40, 41). To demonstrate that the repair of telomere DNA damage mitotic cells is dependent on PolB and NEIL3, we silenced these genes in HEP3B cells and performed the Meta-FISH telomere DNA damage repair assay with KBrO3 treatment (Supplementary Fig. S7E). The result show that cells failed to repair the mitotic TIFs after silencing of PolB or NEIL3 (Fig. 5D, top). The APE1 positive TIFs were repaired in cells transfected with nontargeting siRNA while the siPolB-transfected cells did not repair the damaged telomeres (Fig. 5D, bottom). Surprisingly, in NEIL3-silenced cells, the recruitment of APE1 to the TIFs was impaired, but APE1-positive TIFs that were induced were not repaired (Fig. 5D, bottom). This indicates that the repair of KBrO3 induced TIFs in mitotic cells are both BER and NEIL3-dependent and that the recruitment of APE1 to damaged telomeres is NEIL3-dependent.
To validate that NEIL3 is involved repair of oxidized lesion at the telomere during the M-phase, we transfected cells with NEIL3 siRNA and synchronized cells with thymidine block for 24 hours followed by RO-3306 G2-phase arrest for additional 18 hours. Cells were released into mitosis in the presence of colcemid and treated with 2.5 mmol/L potassium bromate for 30 minutes. Samples were recovered for 1 hour before the oxidized lesions at the telomeres were measured using the telomere qPCR assay with NEIL1 digestion. The relative telomere signal in samples was reduced after potassium bromate treatment and in control siRNA transfected cells, the oxidized lesions was partially repaired after the 1 hour of recovery (Fig. 5E). In contrast, the NEIL3 siRNA–transfected samples did not repair to the same degree compared with the control. This would further support the model that NEIL3 is involved in repair of oxidized lesions in mitosis.
NEIL3 maintains genome stability and prevents telomere shortening
Previously, it has been shown that unrepaired telomere lesions can lead to genomic instability and senescence (42). To explore the outcome of failure in repairing oxidative telomere damage induced in mitosis, we silenced NEIL3 in HEP3B and U2OS cells and synchronized them in mitosis by RO-3306 and colcemid. Oxidative DNA damage was induced by KBrO3 treatment for 30 minutes followed by release for 3 hours as shown in Fig. 6A. Enhanced genome instability, characterized by the formation of micronuclei and chromatin bridges, was observed in NEIL3-silenced cells challenged with KBrO3, but not in control siRNA transfected cells nor in uninduced, NEIL3-silenced cells (Fig. 6B and C; Supplementary Fig. S7F).
Figure 6.

NEIL3 maintains telomere integrity during long-term oxidative damage. A, Schematic of mitotic telomere damage release assay. B, Micronucleus in HEP3B cells. Left, images of DAPI (blue), telomere (green), and centromere (red) in HEP3B. White arrowhead, micronucleus. Right, quantification of micronuclei number per 100 cells in HEP3B cells treated as in A. Means ± SEM; at least 200 cells were analyzed in each group. C, Chromatin bridges in HEP3B cells. Left, images of DAPI (blue), telomere (green), and centromere (red) in interphase cells (left top) and mitotic cells (left bottom). White arrowheads, chromatin bridge. Right, quantification of number of micronuclei per 100 cells in HEP3B cells described in A. Cells were transfected with NEIL3 siRNA s2 and s4 or nontargeting siRNA (siCtrl). Means ± SEM; at least 200 cells were analyzed in each group. D, Schematic of chronic induction of telomere damage. E, Measurement of telomere length with qPCR. Telomere length of HEP3B cells overexpressing empty vector (EV), NEIL3 WT, and NEIL3 K81A was measured after 0, 6, or 12 days of treatment with KBrO3, as indicated in D. Means ± SEM of three repeats. F, Telomere and nontelomere qPCR after Endo VIII incubation. HEP3B cells overexpressing empty vector, NEIL3 WT, and NEIL3 K81A were treated as indicated in G. Telomere damage (left) and nontelomere damage (right) were measured with qPCR. Means ± SEM of three repeats. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Scale bars, 5 μm.
To demonstrate the protective role of NEIL3 in cells undergoing long-term oxidative stress, we treated HEP3B cells, overexpressing WT or K81A NEIL3 or the empty vector control, with 1 mmol/L KBrO3 for 30-minute periods, once a day for 6 or 12 consecutive days (Fig. 6D). By relative telomere length (T/S ratio) measurements, we could show that this treatment induced telomere shortening and that overexpression of WT NEIL3, but not the K81A mutant, could prevent the telomere shortening (Fig. 6E). Furthermore, to assess whether long-term exposure to KBrO3 accumulates oxidative damage at telomeres and if overexpression of NEIL3 could protect cells from damage, we measured the uncleaved telomeric or nontelomeric DNA by qPCR after 2-hour incubation with endonuclease VIII. Although no difference was observed in the nontelomeric DNA level (Fig. 6F), telomere levels significantly increased in NEIL3 WT–overexpressing HEP3B cells, and decreased in NEIL3 K81A mutant–overexpressing cells and in control HEP3B cells (Fig. 6G). These findings indicate NEIL3 preferentially prevents oxidative lesions at telomeres (compared with nontelomere DNA) under chronic oxidative stress.
Discussion
As there are limited treatment options and poor survival outcomes for patients with HCC there is a large unmet clinical need of new anticancer therapies. Inducing DNA damage by radio- or chemotherapy is a current treatment for HCC. The development of PARP inhibitors in BRCA1- or BRCA2-mutated cancers demonstrate that targeting the DDR may be an effective way of generating cancer-specific DNA damage in a highly tolerable way (43, 44). Here we wanted to identify if targeting a specific nonessential DDR protein could offer a potential therapy in HCC.
Here, we describe that NEIL3 was upregulated in many HCC and that the level of NEIL3 correlated to poor prognosis. It has been reported that NEIL3 is essential in many biological contexts, such as fibroblast proliferation, telomere maintenance in the S phase of the cell cycle, DNA interstrand cross-link unhooking, and autoimmune diseases (22–25). However, the role of NEIL3 in cancer, especially in liver cancer is unclear. Here we found the elevated levels of NEIL3 in tumor correlated with unfavorable outcome in both the TCGA HCC database and our own validation HCC cohort from the Renji hospital. This is in agreement with other studies showing NEIL3 is overexpressed in many cancer tissues, including malignant melanoma (45), compared with normal tissue (46). In this study, we also show that NEIL3, and its catalytic activity, is important for liver cancer cell proliferation and to prevent premature senescence. These data support that a small-molecule inhibitor of NEIL3 could be a potential therapeutic target for HCC.
In previous studies, it was shown that later steps of DDR are inhibited during mitosis, although the primary signal events, such as Histone modifications, still occur (38). It is believed that the DDR signaling is haltered to prevent telomere fusions in mitotic cells. This would, on the other hand leave damaged DNA unrepaired until the G1 phase of the cell cycle, forming 53BP1 bodies that are eventually repaired. Here, we show NEIL3-dependent BER occur in mitosis. Generally, the BER process is less reliant on the DDR signaling than the DSB repair pathways and repair of oxidative damage on telomere during mitosis appears important to promote survival. Here, we showed that the relocation of NEIL3 to telomere damage sites is damage-oriented rather than Shelterin complex-dependent. We also demonstrated that the recruitment of NEIL3 to TIFs also occurs during mitosis and that NEIL3 and its catalytic ability, but not NEIL1 or NEIL2, can recruit APE1 to TIFs during mitosis, indicating NEIL3 is involved in the initiation of BER pathway on telomeres.
To further address the mechanism of BER on telomeres during mitosis, we designed a mitotic telomere repair assay by inducing oxidative damage then leaving cells to recover while arrested in mitosis. Results showed a significant decrease in TIFs after repair, which could be prevented by either APE1 inhibitor or PARP inhibitor treatment. Silencing NEIL3 or PolB abolished the mitotic BER on telomeres. Taken together, our result proves that oxidative damage on telomeres can be repaired by NEIL3 initiated PARP-dependent BER pathway. The level of ROS as well as the oxidation of biomolecules increases during mitosis in freely cycling cancer cells (14) and many antimitotic drugs can induce elevated ROS (47), at least partly by targeting mitochondria during prolonged mitotic arrest (48). Indeed, oxidative stress can induce mitotic arrest, perhaps through oxidation of cysteine residues in mitotic proteins (48). Because the telomeres are more susceptible to DNA damage compared with the rest of the genome, especially during mitosis, the cell needs to repair toxic lesions during mitosis to prevent aneuploidy and genomic instability. In a previous study, it was shown that NEIL3 interacts with APE1, FEN1, and TRF1 by its C-terminal domain (25) and in this study we could show that NEIL3 is needed for APE1 recruitment to damaged telomeres. This suggest that NEIL3 has unique features combining DNA glycosylase activity of oxidized nucleotides and protein binding domains to recruit other DNA repair proteins to the telomere and could thus act as a scaffold in BER of telomere damage during mitosis.
It has been shown in vitro that NEIL3 has a broad substrate specificity that includes hydantoins, such as Sp and Gh, as well as thymine glycol (Tg), FapyA and FapyG (12, 49). NEIL3 can recognize oxidized lesion in single-stranded regions such as G-quadruplexes, bubbles and single-stranded DNA, typical structures for telomere and promoter regions. We have demonstrated by both modified comet assay and telomere qPCR, using recombinant hNEIL1 and ecoEndoVIII enzymes, that NEIL3 eliminates oxidized lesions on telomeres. Introducing the K81A point mutation in the catalytic domain of NEIL3, reducing its activity, prevented the repair of the lesions. These results indicate that NEIL3 functions in removing oxidized DNA damage in telomeres as well as in genomic DNA. Because NEIL3, hNEIL1 and ecoEndoVIII all have broad substrate specificity, it is still unclear exactly which lesions are recognized by NEIL3 at the telomere.
In conclusion, we described NEIL3 as an independent prognosis marker and a novel molecular target in HCC proliferation and progression. Mechanistically, we showed that NEIL3 relocates and recruits APE1 to TIFs during mitosis and that TIFs can be repaired in a NEIL3-dependent BER pathway. Removing NEIL3 had effects on cell growth but did not induce cell death; however, combining the knockdown with an oxidizing reagent showed a more pronounced effect on cell viability and genomic stability, suggesting that the endogenous damage in cancer cells is not enough to cause cell death. This could be important in a therapeutic perspective when developing NEIL3 inhibitors, and perhaps a combination treatment with a NEIL3 inhibitor and oxidizing compounds (e.g., elesclomol) could prove to be a good strategy to induce ROS and prevent its repair in the tumor. In summary, these findings suggest that NEIL3 and telomere damage repair pathway are promising targets for cancer treatment and drug discovery.
Authors' Disclosures
H. Gad reports grants from European Research Council during the conduct of the study. U. Berglund reports Ulrika Warpman Berglund is member of the board of Thomas Helleday Foundation (THF) and chairman of the board of Oxcia AB at the time of the work of this article. Oxcia AB and THF are responsible for clinical development of a MTH1 inhibitor. No disclosures were reported by the other authors.
Supplementary Material
Acknowledgments
The authors thank the PSF facility for purification of the NEIL1 protein and Dr. Susan Wallace for the NEIL1 expression construct. They thank Dr. Eric Campeau and Dr. Paul Kaufman for the pLenti CMV Blast DEST (706-1) and pENTR1A no ccDB (w48-1) plasmids. Financial support was given by the European Research Council (695376-TAROX to T. Helleday), Swedish Research Council 2015-00162 (to T. Helleday), Swedish Cancer Society (CAN 2018/600 to T. Helleday), the Swedish Children's Cancer Foundation (to T. Helleday), the Swedish Pain Relief Foundation (to T. Helleday). W410170015, Cohort Study of HCC and Liver Diseases, Double First-Class Foundation, Shanghai Jiao Tong University (to Q. Xia); 2017ZZ01018, Overall Leverage Clinical Medicine Center, NHFPC Foundation (to Q. Xia); 2017YFC0908100, Cohort Study of HCC and Liver Diseases, National Key R&D Program of China (to Q. Xia).
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Contributions
Z. Zhao: Conceptualization, formal analysis, investigation, visualization, writing–original draft, writing–review and editing. H. Gad: Conceptualization, resources, supervision, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing. C. Benitez-Buelga: Resources, supervision, methodology, writing–review and editing. K. Sanjiv: Supervision, methodology, writing–review and editing. H. Xiangwei: Supervision, investigation. H. Kang: Supervision, investigation. M. Feng: Supervision, investigation. Z. Zhao: Supervision, investigation. U.W. Berglund: Resources, supervision, funding acquisition, writing–review and editing. Q. Xia: Resources, supervision, funding acquisition, writing–review and editing. T. Helleday: Conceptualization, resources, supervision, funding acquisition, project administration, writing–review and editing.
References
- 1. El-Serag HB. Hepatocellular carcinoma. N Engl J Med 2011;365:1118–27. [DOI] [PubMed] [Google Scholar]
- 2. Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet 2018;391:1301–14. [DOI] [PubMed] [Google Scholar]
- 3. Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology 2004;127:S35–50. [DOI] [PubMed] [Google Scholar]
- 4. Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet 2012;379:1245–55. [DOI] [PubMed] [Google Scholar]
- 5. Ko E, Seo HW, Jung G. Telomere length and reactive oxygen species levels are positively associated with a high risk of mortality and recurrence in hepatocellular carcinoma. Hepatology 2018;67:1378–91. [DOI] [PubMed] [Google Scholar]
- 6. Inarrairaegui M, Melero I, Sangro B. Immunotherapy of hepatocellular carcinoma: facts and hopes. Clin Cancer Res 2018;24:1518–24. [DOI] [PubMed] [Google Scholar]
- 7. Bouattour M, Raymond E, Qin S, Cheng AL, Stammberger U, Locatelli G, et al. Recent developments of c-Met as a therapeutic target in hepatocellular carcinoma. Hepatology 2018;67:1132–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Galle PR, Tovoli F, Foerster F, Worns MA, Cucchetti A, Bolondi L. The treatment of intermediate stage tumours beyond TACE: from surgery to systemic therapy. J Hepatol 2017;67:173–83. [DOI] [PubMed] [Google Scholar]
- 9. Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell 2012;48:158–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rao CV, Asch AS, Yamada HY. Frequently mutated genes/pathways and genomic instability as prevention targets in liver cancer. Carcinogenesis 2017;38:2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang SF, Chang CW, Wei RJ, Shiue YL, Wang SN, Yeh YT. Involvement of DNA damage response pathways in hepatocellular carcinoma. Biomed Res Int 2014;2014:153867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhou J, Fleming AM, Averill AM, Burrows CJ, Wallace SS. The NEIL glycosylases remove oxidized guanine lesions from telomeric and promoter quadruplex DNA structures. Nucleic Acids Res 2015;43:7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hazra TK, Muller JG, Manuel RC, Burrows CJ, Lloyd RS, Mitra S. Repair of hydantoins, one electron oxidation product of 8-oxoguanine, by DNA glycosylases of Escherichia coli. Nucleic Acids Res 2001;29:1967–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Patterson JC, Joughin BA, van de Kooij B, Lim DC, Lauffenburger DA, Yaffe MB. ROS and oxidative stress are elevated in mitosis during asynchronous cell cycle progression and are exacerbated by mitotic arrest. Cell Syst 2019;8:163–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang GF, Dong Q, Bai Y, Yuan J, Xu Q, Cao C, et al. Oxidative stress induces mitotic arrest by inhibiting Aurora A-involved mitotic spindle formation. Free Radic Biol Med 2017;103:177–87. [DOI] [PubMed] [Google Scholar]
- 16. de Lange T. Shelterin-mediated telomere protection. Annu Rev Genet 2018;52:223–47. [DOI] [PubMed] [Google Scholar]
- 17. Bandaria JN, Qin P, Berk V, Chu S, Yildiz A. Shelterin protects chromosome ends by compacting telomeric chromatin. Cell 2016;164:735–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maciejowski J, de Lange T. Telomeres in cancer: tumour suppression and genome instability. Nat Rev Mol Cell Biol 2017;18:175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barnes RP, Fouquerel E, Opresko PL. The impact of oxidative DNA damage and stress on telomere homeostasis. Mech Ageing Dev 2019;177:37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ahmed W, Lingner J. Impact of oxidative stress on telomere biology. Differentiation 2018;99:21–7. [DOI] [PubMed] [Google Scholar]
- 21. Krokeide SZ, Laerdahl JK, Salah M, Luna L, Cederkvist FH, Fleming AM, et al. Human NEIL3 is mainly a monofunctional DNA glycosylase removing spiroimindiohydantoin and guanidinohydantoin. DNA Repair 2013;12:1159–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Massaad MJ, Zhou J, Tsuchimoto D, Chou J, Jabara H, Janssen E, et al. Deficiency of base excision repair enzyme NEIL3 drives increased predisposition to autoimmunity. J Clin Invest 2016;126:4219–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Olsen MB, Hildrestrand GA, Scheffler K, Vinge LE, Alfsnes K, Palibrk V, et al. NEIL3-dependent regulation of cardiac fibroblast proliferation prevents myocardial rupture. Cell Rep 2017;18:82–92. [DOI] [PubMed] [Google Scholar]
- 24. Semlow DR, Zhang J, Budzowska M, Drohat AC, Walter JC. Replication-dependent unhooking of DNA interstrand cross-links by the NEIL3 glycosylase. Cell 2016;167:498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhou J, Chan J, Lambele M, Yusufzai T, Stumpff J, Opresko PL, et al. NEIL3 repairs telomere damage during S phase to secure chromosome segregation at mitosis. Cell Rep 2017;20:2044–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Salic A, Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci U S A 2008;105:2415–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. O'Callaghan NJ, Fenech M. A quantitative PCR method for measuring absolute telomere length. Biol Proced Online 2011;13:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cesare AJ, Heaphy CM, O'Sullivan RJ. Visualization of telomere integrity and function in vitro and in vivo using immunofluorescence techniques. Curr Protoc Cytom 2015;73:12 40 1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thienpont B, Steinbacher J, Zhao H, D'Anna F, Kuchnio A, Ploumakis A, et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 2016;537:63–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhou J, Liu M, Fleming AM, Burrows CJ, Wallace SS. Neil3 and NEIL1 DNA glycosylases remove oxidative damages from quadruplex DNA and exhibit preferences for lesions in the telomeric sequence context. J Biol Chem 2013;288:27263–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Klattenhoff AW, Thakur M, Chu CS, Ray D, Habib SL, Kidane D. Loss of NEIL3 DNA glycosylase markedly increases replication associated double strand breaks and enhances sensitivity to ATR inhibitor in glioblastoma cells. Oncotarget 2017;8:112942–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gasparutto D, Muller E, Boiteux S, Cadet J. Excision of the oxidatively formed 5-hydroxyhydantoin and 5-hydroxy-5-methylhydantoin pyrimidine lesions by Escherichia coli and Saccharomyces cerevisiae DNA N-glycosylases. Biochim Biophys Acta 2009;1790:16–24. [DOI] [PubMed] [Google Scholar]
- 33. Yang Z, Nejad MI, Varela JG, Price NE, Wang Y, Gates KS. A role for the base excision repair enzyme NEIL3 in replication-dependent repair of interstrand DNA cross-links derived from psoralen and abasic sites. DNA Repair 2017;52:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cawthon RM. Telomere length measurement by a novel monochrome multiplex quantitative PCR method. Nucleic Acids Res 2009;37:e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hua X, Sanjiv K, Gad H, Pham T, Gokturk C, Rasti A, et al. Karonudib is a promising anticancer therapy in hepatocellular carcinoma. Ther Adv Med Oncol 2019;11:1758835919866960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Neurauter CG, Luna L, Bjoras M. Release from quiescence stimulates the expression of human NEIL3 under the control of the Ras dependent ERK-MAP kinase pathway. DNA Repair 2012;11:401–9. [DOI] [PubMed] [Google Scholar]
- 37. Rudd SG, Gad H, Sanjiv K, Amaral N, Hagenkort A, Groth P, et al. MTH1 inhibitor TH588 disturbs mitotic progression and induces mitosis-dependent accumulation of genomic 8-oxodG. Cancer Res 2020;80:3530–41. [DOI] [PubMed] [Google Scholar]
- 38. Orthwein A, Fradet-Turcotte A, Noordermeer SM, Canny MD, Brun CM, Strecker J, et al. Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science 2014;344:189–93. [DOI] [PubMed] [Google Scholar]
- 39. Whitehouse CJ, Taylor RM, Thistlethwaite A, Zhang H, Karimi-Busheri F, Lasko DD, et al. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell 2001;104:107–17. [DOI] [PubMed] [Google Scholar]
- 40. Dianov GL, Prasad R, Wilson SH, Bohr VA. Role of DNA polymerase beta in the excision step of long patch mammalian base excision repair. J Biol Chem 1999;274:13741–3. [DOI] [PubMed] [Google Scholar]
- 41. Sobol RW, Horton JK, Kuhn R, Gu H, Singhal RK, Prasad R, et al. Requirement of mammalian DNA polymerase-beta in base-excision repair. Nature 1996;379:183–6. [DOI] [PubMed] [Google Scholar]
- 42. O'Sullivan RJ, Karlseder J. Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol 2010;11:171–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005;434:913–7. [DOI] [PubMed] [Google Scholar]
- 44. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005;434:917–21. [DOI] [PubMed] [Google Scholar]
- 45. Hildrestrand GA, Neurauter CG, Diep DB, Castellanos CG, Krauss S, Bjoras M, et al. Expression patterns of Neil3 during embryonic brain development and neoplasia. BMC Neurosci 2009;10:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kauffmann A, Rosselli F, Lazar V, Winnepenninckx V, Mansuet-Lupo A, Dessen P, et al. High expression of DNA repair pathways is associated with metastasis in melanoma patients. Oncogene 2008;27:565–73. [DOI] [PubMed] [Google Scholar]
- 47. Alexandre J, Hu Y, Lu W, Pelicano H, Huang P. Novel action of paclitaxel against cancer cells: bystander effect mediated by reactive oxygen species. Cancer Res 2007;67:3512–7. [DOI] [PubMed] [Google Scholar]
- 48. Domenech E, Maestre C, Esteban-Martinez L, Partida D, Pascual R, Fernandez-Miranda G, et al. AMPK and PFKFB3 mediate glycolysis and survival in response to mitophagy during mitotic arrest. Nat Cell Biol 2015;17:1304–16. [DOI] [PubMed] [Google Scholar]
- 49. Regnell CE, Hildrestrand GA, Sejersted Y, Medin T, Moldestad O, Rolseth V, et al. Hippocampal adult neurogenesis is maintained by Neil3-dependent repair of oxidative DNA lesions in neural progenitor cells. Cell Rep 2012;2:503–10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.