

Abstract
Since 2011, with the approval of crizotinib and subsequent approval of four additional targeted therapies, anaplastic lymphoma kinase (ALK) inhibitors have become important treatments for a subset of patients with lung cancer. Each generation of ALK inhibitor showed improvements in terms of central nervous system (CNS) penetration and potency against wild-type (WT) ALK, yet a key continued limitation is their susceptibility to resistance from ALK active-site mutations. The solvent front mutation (G1202R) and gatekeeper mutation (L1196M) are major resistance mechanisms to the first two generations of inhibitors while patients treated with the third-generation ALK inhibitor lorlatinib often experience progressive disease with multiple mutations on the same allele (mutations in cis, compound mutations). TPX-0131 is a compact macrocyclic molecule designed to fit within the ATP-binding boundary to inhibit ALK fusion proteins. In cellular assays, TPX-0131 was more potent than all five approved ALK inhibitors against WT ALK and many types of ALK resistance mutations, e.g., G1202R, L1196M, and compound mutations. In biochemical assays, TPX-0131 potently inhibited (IC50 <10 nmol/L) WT ALK and 26 ALK mutants (single and compound mutations). TPX-0131, but not lorlatinib, caused complete tumor regression in ALK (G1202R) and ALK compound mutation-dependent xenograft models. Following repeat oral administration of TPX-0131 to rats, brain levels of TPX-0131 were approximately 66% of those observed in plasma. Taken together, preclinical studies show that TPX-0131 is a CNS-penetrant, next-generation ALK inhibitor that has potency against WT ALK and a spectrum of acquired resistance mutations, especially the G1202R solvent front mutation and compound mutations, for which there are currently no effective therapies.
Graphical Abstract
Introduction
Chromosomal rearrangements of the anaplastic lymphoma kinase (ALK) gene produce oncogenic fusion proteins, which occur in 3% to 5% of patients with non–small cell lung cancer (NSCLC; ALK+ NSCLC; refs. 1, 2). These fusion proteins exhibit aberrant dimerization or oligomerization that results in constitutive ALK activation (3, 4). The fusion of ALK with echinoderm microtubule-associated protein-like 4 (EML4) gene to form EML4-ALK was found to be highly oncogenic in preclinical NSCLC models (4). Targeting the kinase domain of EML4-ALK with small-molecule ALK inhibitors results in clinical benefit for patients with ALK+ NSCLC (5, 6). To date, three generations of ALK inhibitors have been approved, comprising first- (crizotinib), second- (alectinib, brigatinib, ceritinib), and third-generation (lorlatinib) therapies. However, the durability of responses to these therapies can be abrogated, in part, due to the emergence of ALK mutations that interfere with drug binding (5, 6). Crizotinib was approved in 2011 for patients with ALK+ NSCLC and has a 74% objective response rate in the first-line setting (7). However, crizotinib treatment can lead to resistant mutations in the ALK active site, for example, G1269A and C1156Y, and to the gatekeeper residue L1196M (8, 9), which limit its durability of response. Second-generation ALK inhibitors (alectinib, brigatinib, ceritinib) are approved for use in patients with ALK+ NSCLC but are also susceptible to resistance from ALK mutations such as solvent front mutations (e.g., G1202R), which are found in 33% to 37% of relapsed patients, I1171 mutations in the hydrophobic regulatory spine (24%–26% of patients), and the L1196M gatekeeper mutation (17%–22% of patients; refs. 8, 10, 11). The third-generation ALK inhibitor lorlatinib is approved for patients with ALK+ NSCLC who have been previously treated with crizotinib and at least one other ALK inhibitor, or after first-line treatment with either alectinib or ceritinib (11, 12). However, ALK mutations are detected in 76% of plasma specimens from patients whose disease progressed on lorlatinib treatment: L1196M (38%), G1202R (28%), D1203N (24%), F1174C/L (14%), and I1171X (14%; ref. 11). These mutations are a mixture of single mutations and compound mutations with compound mutations found in 35% to 48% of treated patients (11, 13). The reemergence of single mutations such as L1196M and G1202R may be due to lorlatinib's moderate potency against these mutations (L1196M IC50 18–30 nmol/L, G1202R IC50 37–63 nmol/L; refs. 13, 14). Therefore, a central nervous system (CNS)-penetrant, highly potent wild-type (WT) ALK inhibitor that is not susceptible to resistance from mutations that arise from treatment with first-, second-, and third-generation ALK inhibitors is needed.
TPX-0131 is compact macrocyclic inhibitor designed to fit completely in the ATP-binding pocket with a minimal binding interface to achieve potent inhibition of WT ALK and reduce the susceptibility to a broad range of ALK drug-resistant mutations (solvent front, gatekeeper, hinge region, and compound mutations). Using a combination of biochemical, cellular, and in vivo preclinical assessments, TPX-0131 has been shown to be a CNS-penetrant molecule that potently inhibits WT ALK and a broad array of clinically relevant ALK mutants that limit the utility of previous generations of ALK inhibitors.
Materials and Methods
Reagents and chemicals
Crizotinib, ceritinib, alectinib, and lorlatinib were purchased from Selleckchem. Brigatinib was purchased from MedChem Express.
Preparation of (4S)-4-(difluoromethyl)-8-fluoro-13,13-dimethyl-3,4,13,14-tetrahydro-6H-18,1-(metheno)[1,4]oxazino[3,4-i]pyrazolo[4,3-f][1,4,8,10]benzoxatriazacyclotridecin-15(12H)-one (TPX-0131)
The synthetic scheme is described in Supplementary Fig. S1. Tert-butyl [1-(2-(chloromethyl)-4-fluorophenoxy)-2-methylpropan-2-yl)carbamate; 25 mg, 76 μmol)] was dissolved in dimethylformamide (0.5 mL) at room temperature. Cs2CO3 (66 mg, 201 μmol) was added followed by ethyl (3S)-3-(difluoromethyl)-3,4-dihydro-2H-pyrazolo[1,2]pyrimido[2,4-d](1,4)oxazine-6-carboxylate (20 mg, 67 μmol). The mixture was stirred at 22°C for 18 hours. Reaction was diluted with dichloromethane (5 mL) and cooled. The solution was filtered, and the filtrate was concentrated under reduced pressure. Flash column chromatography (Teledyne ISCO system, 12 g, 20%–80% ethyl acetate in hexanes) to afford 1 (23.4 mg, 39 μmol, 59% yield) to a solution of 1 (23.4 mg, 39 μmol) in tetrahydrofuran (1 mL), ethanol (2 mL), and methanol (1 mL) at ambient temperature was added aqueous LiOH (2 mol/L, 2 mL). The mixture was stirred at 22°C for 36 hours, cooled to −20°C, then quenched with aqueous HCl solution (2.0 mol/L, 2.1 mL) to acidic, diluted with water (10 mL), and the mixture was extracted with dichloromethane (3 × 10 mL). The combined organic layer was washed with brine and then dried over Na2SO4. Salts were filtered and the filtrate was concentrated under reduced pressure and dried under high vacuum, used directly without further purification and assuming quantitative yield. Crude was dissolved in anhydrous dichloromethane (2 mL), and 4 mol/L HCl in dioxane (2 mL) was added and stirred for 45 minutes at 22°C. It was then concentrated to dryness under reduced pressure followed by high vacuum treatment. Anhydrous dichloromethane (1.90 mL) was added followed by Hunig's base (51 mg, 394 μmol, 69 μL) and pentafluorophenyl diphenylphosphinate (FDPP; 23 mg, 59 μmol) in one portion, let stir for 18 hours, and then quenched reaction with 2 mol/L Na2CO3 solution (5 mL). The mixture was stirred for 5 minutes and then extracted with dichloromethane (3 × 10 mL). Combined organic extracts were dried with Na2SO4 and concentrated under reduced pressure. Flash chromatography (Teledyne ISCO system, silica 12 g, 0%–10% methanol in dichloromethane) provided TPX-0131 (10.8 mg, 24 μmol, 61% yield). TPX-0131 was characterized by 1H nuclear magnetic resonance (NMR; DMSO-d6, 500 MHz) δ 9.14 (s, 1H) 8.66 (s, 1H) 8.00 (s, 1H) 7.49 (dd, 1H, J = 9.16, 2.86 Hz) 7.00 - 7.16 (m, 2H) 6.36 - 6.64 (m, 1H) 5.60 (d, 1H, J = 14.89 Hz) 5.04 (br t, 1H, J = 10.02 Hz) 4.67 (d, 1H, J = 12.03 Hz) 4.34 - 4.41 (m, 1H) 4.30 (d, 1H, J = 14.89 Hz) 4.09 (d, 1H, J = 9.74 Hz) 3.88 (d, 1H, J = 9.74 Hz) 1.63 (s, 3H) 1.48 (s, 3H). In addition, TPX-0131 was evaluated by low-resolution mass spectrometry and shown to have the expected mass [M+H]+m/z = 448.17, calc. for C21H21F3N5O3+ = 448.16.
Molecular modeling
Molecular modeling was performed in Maestro (Schrodinger Inc.) using Prime MM-GBSA with the OPLS4 force field. The ALK crystal structure was from the PDB database (PDB 2XP2).
Biochemical kinase analysis
The biochemical kinase assays were performed at Reaction Biology Corporation following previously described procedures (15). Specific kinase/substrate pairs along with required cofactors were prepared in reaction buffer (20 mmol/L HEPES pH 7.5, 10 mmol/L MgCl2, 1 mmol/L EGTA, 0.02% Brij35, 0.02 mg/mL BSA, 0.1 mmol/L Na3VO4, 2 mmol/L dithiothreitol, 1% DMSO). Compounds were delivered into the reaction, followed about 20 minutes later by addition of a mixture of ATP (Sigma) and γ-[33P]-ATP (Perkin Elmer) to a final concentration of 10 μmol/L. Reactions were carried out at room temperature for 120 minutes, followed by spotting of the reactions onto P81 ion exchange filter paper (Whatman Inc.). Unbound phosphate was removed by extensive washing of filters in 0.75% phosphoric acid. After subtraction of background derived from control reactions containing inactive enzyme, kinase activity data was expressed as the percent remaining kinase activity in test samples compared with vehicle (DMSO) reactions. IC50 values and curve fits were obtained using GraphPad Prism software (GraphPad, Inc.).
Cell lines and cell culture
Ba/F3 cells (a murine IL3-dependent pro–B cell line) were maintained in RPMI1640 supplemented with 10% FBS, 1 ng/mL of mouse IL3, and 100 U/mL of penicillin/streptomycin. Ba/F3 stable cell lines were maintained in RPMI1640 supplemented with 10% FBS, 100 U/mL of penicillin, and 0.5 μg/mL puromycin solution.
Cloning and creation of stable Ba/F3 cell lines
The WT EML4-ALK gene (variant 1) and its mutations (G1202R, L1196M, L1198F, G1269A, G1269S, I1171N, I1171S, I1171T, G1202R/L1196M, G1202R/L1198F, G1202R/C1156Y, L1196M/L1198F, L1198F/I1171N, G1202R/G1269A, G1202R/G1269A/L1204V, G1202R/G1269A/L1198F) were synthesized at GenScript and cloned into pCDH-CMV-MCS-EF1-Puro plasmid (System Biosciences, Inc.). Ba/F3 cells lines containing WT EML4-ALK or its mutations (G1202R, L1196M, L1198F, G1269A, G1269S, I1171N, I1171S, I1171T, G1202R/L1196M, G1202R/L1198F, G1202R/C1156Y, L1196M/L1198F, L1198F/I1171N, G1202R/G1269A, G1202R/G1269A/L1204V, G1202R/G1269A/L1198F) were generated by transducing Ba/F3 cells with lentivirus containing WT or mutant EML4-ALK. Stable cell lines were selected by puromycin treatment, followed by IL3 withdrawal. Briefly, 5 × 106 Ba/F3 cells were transduced with lentivirus supernatant in the presence of 8 μg/mL protamine sulfate. The transduced cells were subsequently selected with 1 μg/mL puromycin in the presence of IL3-containing medium RPMI-1640 plus 10% FBS. After 10 to 12 days of selection, the surviving cells were further selected for IL3-independent growth.
Cell proliferation assays
Two-thousand Ba/F3 cells harboring EML4-ALK (WT or mutant variants) were seeded per well in 384-well white plates for 24 hours, and then treated with compounds for 72 hours (at 37°C, 5% CO2). Cell proliferation was measured using CellTiter-Glo luciferase-based ATP-detection assay (Promega) following the manufacture's protocol. IC50 determinations were performed using GraphPad Prism software (GraphPad, Inc.).
Immunoblotting for cellular kinase phosphorylation assays
Half a million cells (Ba/F3 EML4-ALK WT, G1202R, G1202R/L1196M, and G1202R/L1198F) per well were seeded in 24-well plate for 24 hours, and then treated with compound for 4 hours. Cells were collected after treatment and lysed in radioimmunoprecipitation assay buffer (50 mmol/L Tris, pH 7.4, 150 mmol/L NaCl, 1% NP 40, 0.5% deoxycholate, 0.1% SDS) supplemented with 10 mmol/L EDTA, 1× Halt Protease and Phosphatase Inhibitor (Thermo Scientific). Protein lysates (approximately 20 μg) were resolved on 4%–12% Bolt Bis-Tris precast gels with MES running buffer (Life Technologies), transferred to nitrocellulose membranes using Trans-Blot Turbo Transfer System (Bio-Rad), and detected with antibodies targeting phosphorylated ALK (Y1282/1283), ALK (Y1604), total ALK, and actin (Cell Signaling Technology). Antibodies were typically incubated overnight at 4°C with gentle shaking, followed by washes and incubation with the appropriate horseradish peroxidase (HRP)–conjugated secondary antibodies. Membranes were incubated with chemiluminescent substrate for 5 minutes at room temperature (SuperSignal West Femto; Thermo Scientific). The chemiluminescent images were acquired with a C-DiGit Imaging System (LI-COR Biosciences). The relative density of the chemiluminescent bands was quantified via Image Studio Digits from LI-COR.
In vivo xenograft studies
All animal studies were conducted in accordance with the guidelines published in the Guide for the Care and Use of Laboratory Animals. Mice were maintained and used in accordance with animal protocol EB17–010 [approved by Explora BioLabs' Institutional Animal Care and Use Committee (IACUC)]. Female SCID/beige mice (5–8 weeks old) were obtained from Charles River Laboratory and were housed in Innovive IVC disposable cages on HEPA-filtered ventilated racks with ad libitum access to rodent chow and water. Five million cells in 100 μL serum-free medium supplemented with 50% Matrigel (Corning, Inc.) were implanted subcutaneously in the right flank region of each mouse. Tumor size and body weight were measured about four times per week. Tumor volume was calculated as length × width2 × 0.5. Mice were randomized by tumor size into treatment groups when tumor volume reached about 150 to 200 mm3. TPX-0131 formulated in vehicle [0.5% carboxymethyl cellulose (CMC) and 1% Tween-80 in water] was administered orally at predetermined dose levels twice a day. Vehicle was administered using the same regimen as TPX-0131 and was deemed the experimental negative control. Lorlatinib was used as a reference for efficacy evaluation.
Tumor growth inhibition (TGI) was calculated as follows:
If TVt ≥TV0, TGI = 100%×(1-(TVt -TV0)/(CVt-CV0))
If TVt<TV0, TGI = 100%×(2-TVt/TV0)
Where TV0 was the mean tumor volume in the treatment group at the beginning of the treatment, TVt was the mean tumor volume in the treatment group at the end of the treatment, CV0 was the mean tumor volume in the negative control group at the beginning of the treatment, and CVt was the mean tumor volume in the negative control group at the end of the treatment. A TGI >100% indicates tumor regression; TGI = 200% is equivalent to complete tumor regression. Statistical analyses were performed using GraphPad Prism 8 and P < 0.05 was considered a statistically significant difference. The pharmacodynamic and the corresponding free plasma concentrations of TPX-0131 were evaluated in mice bearing the Ba/F3 cell-derived xenograft tumors harboring an EML4-ALK fusion with G1202R/L1196M mutations. Samples were taken at 2 hours and 12 hours for pharmacokinetic and pharmacodynamic analyses (immunoblotting). For immunoblotting analysis of the phosphorylated ALK fusion proteins, tumor samples were collected by snap freezing in liquid nitrogen and processed in RIPA buffer. Immunoblotting analyses were performed as described above.
Pharmacokinetics and brain distribution properties of TPX-0131
Daily oral doses of 10 mg/kg TPX-0131 were administered for 7 consecutive days to male Sprague Dawley rats. Pharmacokinetic profiles using individual and mean plasma concentrations of TPX-0131 were evaluated on study Days 1 and 7 after dosing. Terminal TPX-0131 concentrations (1, 4, and 24 hours postdose on study day 7) in plasma, cerebrospinal fluid, and brain tissue samples were analyzed by LC/MS-MS to assess brain tissue distribution. A detailed description of the methods for pharmacokinetic analyses and determining rat brain penetration can be found in Supplementary Analysis.
Results
Design of TPX-0131
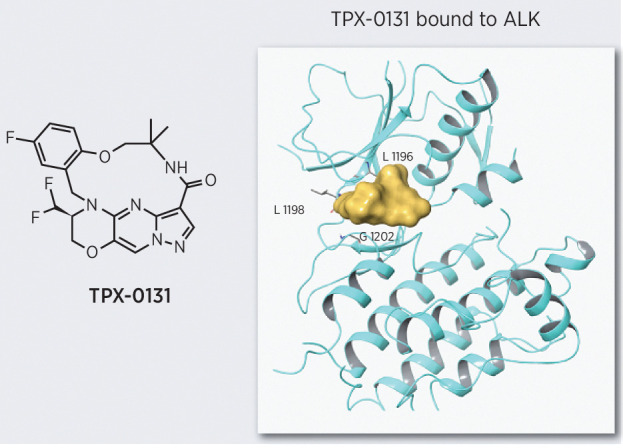
The most prevalent resistance mutations to second-generation ALK inhibitors (e.g., alectinib, brigatinib) are in the solvent front region (e.g., G1202; ref. 16). Molecular modeling illustrates that second-generation ALK inhibitors have structures extending out of the active site into the solvent front, which is consistent with observed resistance mutations in this region (Fig. 1A). The third-generation ALK inhibitor lorlatinib has a pyrazole ring that extends directly over the G1202 residue, which may contribute to its reduced potency against solvent front mutations. ALK compound mutations consist of multiple alterations in the binding site that together obstruct binding and reduce potency. TPX-0131 (Fig. 1B) was designed to avoid a broad array of resistance mutations, especially solvent front and compound mutations. TPX-0131 has a conformationally constrained and compact macrocyclic structure that binds completely inside the adenine binding site of ATP and avoids the solvent front area. Conformational analysis indicates that TPX-0131 adopts a low-energy conformation very similar to the bound conformation, minimizing entropic penalties upon binding to ALK. Modeling studies predict that TPX-0131 binding in the ALK active site can accommodate the bulkier arginine side chain in the solvent front without steric interference, allowing it to remain active against solvent front, hinge, and gatekeeper mutations (Fig. 1A).
Figure 1.

Design of TPX-0131. A, Molecular modeling of alectinib, brigatinib, and lorlatinib in the ALK active site shows that they extend into the solvent front area. TPX-0131 has a conformationally constrained, compact, macrocyclic structure, which is modeled to bind completely inside the ATP adenosine-binding site and avoid gatekeeper and solvent front regions. B, The molecular structure of TPX-0131. MW, molecular weight.
Enzymatic kinase activities against WT and mutant ALK variants
Biochemical characterization of TPX-0131 potency against WT and mutant ALK was assessed in a panel of enzymatic assays with recombinant ALK kinase domains performed at 10 μM ATP (Table 1). TPX-0131 potently inhibited WT ALK (IC50 = 1.4 nmol/L) and 26 ALK resistance mutations. TPX-0131 inhibited C1156Y, E1210K/S1206C, L1198F/C1156Y, L1196M/L1198F, E1210K, L1196M, T1151M, deleted G1202, S1206R, G1202R/L1198F, F1174L, F1245C, R1275Q, and G1202R ALK mutations with IC50 values of <1 nmol/L. TPX-0131 had IC50 values of 1 to 2 nmol/L for the following ALK mutations: L1198F, L1152R, F1174S, T1151-L1152 insT, V1180L, G1269A, F1174C. TPX-0131 was less active against ALK mutations including I1171N, L1152P, D1203N, D1203N/E1210K, and G1269S, with IC50 values of 2 to 7 nmol/L. Additionally, TPX-0131 was determined to be a selective ALK inhibitor by evaluating its potency toward a panel of 373 kinases (Supplementary Table S1). From this analysis, TPX-0131 has molecular properties (three dimensional shape, key binding interactions) that enable selective inhibition of ALK. Taken together, TPX-0131 was highly potent against a broad spectrum of ALK drug-resistant mutations.
Table 1.
Evaluation of TPX-0131 in biochemical assays of WT and mutant ALK variants.
| Kinase | IC50 (nmol/L) | Kinase | IC50 (nmol/L) | Kinase | IC50 (nmol/L) |
|---|---|---|---|---|---|
| WT | 1.4 | S1206R | 0.5 | T1151-L1152insT | 1.2 |
| C1156Y | 0.2 | G1202R/L1198F | 0.6 | V1180L | 1.6 |
| E1210K/S1206C | 0.2 | F1174L | 0.7 | G1269A | 1.6 |
| L1198F/C1156Y | 0.2 | F1245C | 0.7 | F1174C | 1.8 |
| L1196M/L1198F | 0.2 | R1275Q | 0.8 | I1171N | 2.3 |
| E1210K | 0.3 | G1202R | 0.9 | L1152P | 2.9 |
| L1196M | 0.3 | L1198F | 1.0 | D1203N | 4.4 |
| T1151M | 0.4 | L1152R | 1.1 | D1203N/E1210K | 6.3 |
| Deleted G1202 | 0.5 | F1174S | 1.2 | G1269S | 6.6 |
Note: The biochemical kinase assays were performed in the presence of 10 μmol/L ATP following previously described procedures (15).
Cellular potency of TPX-0131
To enable comparisons of TPX-0131 with previous generations of ALK inhibitors, a panel of matched cell lines was created that are dependent on ALK resistance mutations found in patients as well as other mutations that may arise in the clinic. Ba/F3 cells were engineered to express the oncogenic EML4-ALK variant 1 fusion protein, as well as EML4-ALK with either single mutations (G1202R, L1196M, L1198F, G1269A, G1269S, I1171N/S/T) or compound mutations (L1196M/L1198F, L1198F/C1156Y, L1198F/I1171N, G1202R/C1156Y, G1202R/L1196M, G1202R/L1198F, G1202R/G1269A, G1202R/G1269A/L1204V, G1202R/G1269A/L1198F). TPX-0131 potency was benchmarked against three generations of ALK inhibitors (crizotinib, ceritinib, alectinib, brigatinib, and lorlatinib). The initial assessment of cellular potency was in proliferation assays (Table 2). All tested ALK compounds had minimum activity (IC50 > 800 nmol/L) against parental Ba/F3 cells which means that proliferation potency values for Ba/F3 cells harboring EML4-ALK measure inhibition of ALK, not other aspects of the assay (Supplementary Table S2). TPX-0131 was the most potent inhibitor of cells harboring the WT EML4-ALK fusion. TPX-0131 WT potency (IC50 = 0.4 nmol/L) was two-fold more potent than lorlatinib (IC50 = 0.8 nmol/L) and significantly more potent relative to other ALK inhibitors: crizotinib (IC50 = 50 nmol/L), alectinib (IC50 = 7.4 nmol/L), brigatinib (IC50 = 12 nmol/L), and ceritinib (IC50 = 3.9 nmol/L). Toward cells harboring EML4-ALK with the G1202R solvent front mutation, TPX-0131 demonstrated the most potent inhibition of cell proliferation (IC50 = 0.2 nmol/L), which was at least 260-fold more potent than any other tested ALK inhibitor. TPX-0131 was 11- to 550-fold more potent toward the gatekeeper mutation (L1196M) than previous generations of ALK inhibitors. TPX-0131 was the most potent inhibitor against the L1198F mutation in the hinge region with an IC50 <0.2 nmol/L and was 90- to 3,000-fold more potent than other ALK inhibitors. TPX-0131 was moderately potent against cells harboring the EML4-ALK G1269A mutation (IC50 = 13 nmol/L) and was less active against a serine mutation at this position (IC50 = 701 nmol/L). TPX-0131 was not highly potent against single I1171N/S/T mutations (IC50 189 – 516 nmol/L). Taken together, TPX-0131 potently inhibits WT EML4-ALK and EML4-ALK harboring a range of point mutations with significantly greater potency against many key resistance mutations, such as solvent front, gatekeeper, and hinge region mutations, relative to previous generations of ALK inhibitors.
Table 2.
Inhibitory activity (IC50) of TPX-0131 and other ALK inhibitors in Ba/F3 cell proliferation assays against single and compound EML4-ALK mutations.
| Cell proliferation IC50 value (nmol/L) | ||||||
|---|---|---|---|---|---|---|
| EML4-ALK | TPX-0131 | Crizotinib | Alectinib | Brigatinib | Ceritinib | Lorlatinib |
| Single EML4-ALK mutations | ||||||
| WT | 0.4 | 50 | 7.4 | 12 | 3.9 | 0.8 |
| I1171N | 516 | 254 | 4,310 | 49 | 72 | 48 |
| I1171S | 189 | 188 | 306 | 31 | 27 | 31 |
| I1171T | 316 | 232 | 210 | 33 | 29 | 25 |
| L1196M | 0.5 | 274 | 50 | 21 | 5.4 | 38 |
| L1198F | <0.2 | 18 | 397 | 74 | 618 | 30 |
| G1202R | 0.2 | 434 | 2,690 | 188 | 329 | 52 |
| G1269A | 13 | 451 | 197 | 20 | 15 | 49 |
| G1269S | 701 | 1,390 | 671 | 46 | 97 | 191 |
| Compound EML4-ALK mutations | ||||||
| L1196M/L1198F | <0.2 | 252 | 2,250 | 253 | 1,410 | 1,310 |
| L1198F/C1156Y | <0.2 | 19.3 | 776 | 102 | 1,310 | 140 |
| L1198F/I1171N | 1.6 | 626 | 236 | 55.1 | 64.1 | 78.7 |
| G1202R/C1156Y | 0.2 | 745 | 2,420 | 810 | 1,300 | 521 |
| G1202R/L1196M | 0.7 | 808 | >10,000 | 1,100 | 1,260 | 4,780 |
| G1202R/L1198F | <0.2 | 188 | 3,000 | 2,040 | 2,010 | 1,710 |
| G1202R/G1269A | 9.9 | 705 | 7,200 | 164 | 303 | 636 |
| G1202R/G1269A/L1204V | 14.9 | 634 | 6,740 | 176 | 345 | 673 |
| G1202R/G1269A/L1198F | 0.2 | 596 | >10,000 | 907 | 1,670 | 6,330 |
Note: IC50 values were determined from three independent replicates.
In cell proliferation assays, TPX-0131 was the most potent inhibitor against a range of EML4-ALK compound mutations (Table 2). TPX-0131 inhibited six of nine compound mutations with IC50 < 1 nmol/L, had IC50 < 10 nmol/L for two mutations, and IC50 = 14.9 nmol/L for the ALK G1202R/G1269A/L1204V triple mutation. Previous generations of ALK inhibitors did not potently inhibit any of the nine EML4-ALK compound mutations tested (IC50 19–10,000 nmol/L). Of the 45 inhibitor/compound mutation combinations tested using previous generations of ALK inhibitors (nine assays, five ALK inhibitors), only four had an IC50<100 nmol/L. In comparison with previous generations ALK inhibitors, TPX-0131 was the only ALK inhibitor with significant potency against the entire panel of compound EML4-ALK mutations evaluated.
To confirm the results obtained from the cell proliferation assays, TPX-0131 and select other ALK inhibitors were evaluated in assays measuring pharmacodynamic modulation of ALK (autophosphorylation). TPX-0131 suppressed autophosphorylation of Tyr1604 and Tyr1282/1283 residues of ALK oncogenic fusion proteins in engineered stable cell lines expressing WT or mutant ALK fusion proteins (Fig. 2). TPX-0131 exhibited comparable activity to lorlatinib in suppressing WT EML4-ALK phosphorylation with an IC50 value of approximately 3 to 10 nmol/L. TPX-0131 was a potent inhibitor of ALK autophosphorylation in Ba/F3 cells expressing EML4-ALK G1202R solvent front, EML4-ALK G1202R/L1196M, or EML4-ALK G1202R/L1198F mutations, with IC50 values of approximately 3 to 10 nmol/L. Lorlatinib was much less potent at inhibiting ALK autophosphorylation in cells harboring G1202R, G1202R/L1196M, and G1202R/L1198 mutations, with IC50 values >100 nmol/L. The potency values in these in vitro pharmacodynamic assays correlated well with results from the cell proliferation assays and demonstrate potent inhibition of both single and compound EML4-ALK resistance mutations by TPX-0131.
Figure 2.

The effect of TPX-0131 on phosphorylation of ALK in engineered Ba/F3 cell models containing WT or mutated EML4-ALK as measured by immunoblotting for ALK and phospho-ALK relative to a control protein (actin). TPX-0131 had similar potency to lorlatinib in suppressing WT EML4-ALK phosphorylation but significantly more potency against an EML4-ALK fusion carrying G1202R and compound mutations including G1202R/L1198F and G1202R/L1196M.
Evaluation of TPX-0131 in xenograft tumor models
TGI and the pharmacodynamic modulation of ALK were performed in cell-derived xenograft (CDX) models for clinically relevant ALK mutants that limit the utility of previous generations of ALK inhibitors (e.g. SFM, compound mutations). In a Ba/F3 CDX of EML4-ALK fusion harboring the G1202R solvent front mutation, TPX-0131 treatment at 2, 5, and 10 mg/kg BID (twice daily) resulted in dose-dependent TGI of 64%, 120%, and 200% (complete regression), respectively (Fig. 3A). The mean free plasma trough concentration of TPX-0131 was 13.7 nmol/L at the dose level of 10 mg/kg twice daily. Treatment of these tumor-bearing mice with lorlatinib at 5 mg/kg twice daily resulted in 158% TGI. In a Ba/F3 CDX model harboring an EML4-ALK fusion with the G1202R/L1198F solvent front and hinge region compound mutation, treatment with 2, 5, and 10 mg/kg of TPX-0131 twice daily resulted in complete tumor regression at all dose levels (200% TGI); in contrast, lorlatinib treatment at 5 mg/kg twice daily resulted in 31% TGI (Fig. 3B). In a Ba/F3 CDX harboring an EML4-ALK fusion with the G1202R/L1196M solvent front and gatekeeper compound mutations, TPX-0131 treatment at 0.6, 2, 5, and 10 mg/kg twice daily resulted in dose-dependent efficacy with TGIs of 19%, 44%, 83%, and 200% (complete regression), respectively. Lorlatinib treatment at 5 mg/kg BID resulted in 18% TGI (Fig. 3C). No body weight loss was observed during treatment with TPX-0131 (Supplementary Fig. S2).
Figure 3.

Evaluation of efficacy of TPX-0131 in CDX models in SCID/beige mice administered TPX-0131 via oral gavage twice daily for seven consecutive days. A, Antitumor effect of TPX-0131 on Ba/F3 cell–derived xenograft model with an EML4-ALK G1202R fusion. B, Antitumor effect of TPX-0131 on Ba/F3 cell-derived xenograft model with an EML4-ALK G1202R/L1198F fusion. C, Antitumor effect of TPX-0131 on Ba/F3 cell-derived xenograft model with an EML4-ALK G1202R/L1196M fusion. Waterfall plots for each model represents the degree of xenograft response for each mouse. BID, twice daily. It should be noted that 5 mg/kg dosing of TPX-0131 and lorlatinib in mouse models result in different unbound exposures (e.g., 12 hours postdose TPX-0131, 8 nmol/L; lorlatinib, 358 nmol/L).
The correlation of inhibition of ALK phosphorylation (Tyr1282/1283) was evaluated as a function of TPX-0131 plasma exposure analysis using the Ba/F3 CDX model harboring the EML4-ALK fusion with the G1202R/L1196M compound mutation (Fig. 4; Supplementary Fig. S3). For 2 and 5 mg/kg doses of TPX-0131, near complete suppression of phospho-ALK (92%–95%) was observed (Supplementary Fig. S3). However, 12 hours after single-dose TPX-0131 administration, the level phospho-ALK suppression was reduced (0%–63%) consistent with the use of a BID dosing regimen in mouse models (Supplementary Fig. S3). TPX-0131 exhibited more than 90% phosphorylation inhibition of EML4-ALK G1202R/L1196M fusion at a mean free plasma concentration of 19.5 nmol/L (Fig. 4). Tumor growth inhibition correlates with TPX-0131 exposure and suppression of ALK phosphorylation (Figs. 3 and 4). Taken together, TPX-0131 demonstrated marked antitumor effects in Ba/F3 CDX models of ALK resistance mutations such as the G1202R solvent front mutation, the G1202R/L1198F compound mutation, and the gatekeeper/solvent front compound mutation G1202R/L1196M. Furthermore, in vivo efficacy correlated with suppression of ALK phosphorylation.
Figure 4.
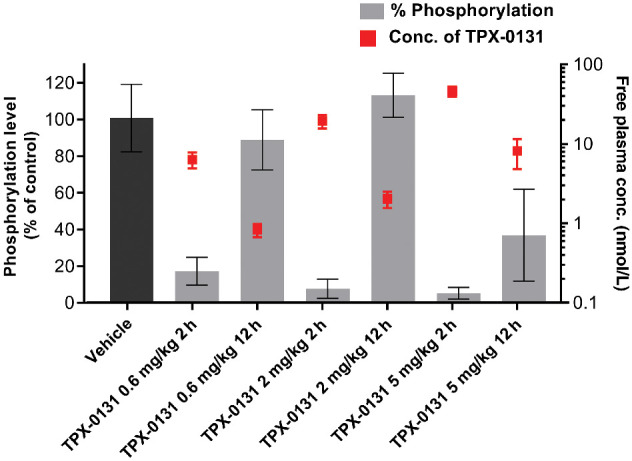
Pharmacokinetic/pharmacodynamic analysis of TPX-0131 in the EML4-ALK G1202R/L1196M xenograft model. Bar graph quantitates the degree of phosphorylation (Tyr1282/12833) modulation of ALK as a function of dose. The TPX-0131 unbound exposure is shown as red squares. Higher TPX-0131 exposure correlates with lower levels of ALK phosphorylation. Primary data can be found in the supplemental information (Supplementary Fig. S3).
Pharmacokinetics and brain distribution properties of TPX-0131
The pharmacokinetics and brain distribution of TPX-0131 were investigated in Sprague Dawley rats following repeat oral administration (Supplemental Analysis). The pharmacokinetic profile of TPX-0131 in male Sprague Dawley rats was characterized by rapid absorption and a long elimination half-life following a single oral administration. Following repeat oral administration of TPX-0131 (10 mg/kg/day) to rats for 7 consecutive days, brain levels of TPX-0131 were approximately 66% of those observed in plasma. These preclinical study results suggest that TPX-0131 has the potential to cross the blood–brain barrier in humans.
Discussion
Despite the approval of three generations of ALK inhibitors, disease progression for patients with ALK+ NSCLC often occurs due to treatment-resistant ALK mutations. The patterns of drug-resistant mutations are dependent on the molecular properties of the individual ALK inhibitors. Resistance to crizotinib arises, in part, through mutations in the ALK kinase domain such as L1196M, G1269A, and C1156Y (8). The second-generation ALK inhibitors alectinib, brigatinib, and ceritinib are effective against crizotinib resistance mutations but are susceptible to mutations in the solvent front region, which occur in approximately a third of patients that progress with ALK mutations (11). As such, clinicians use a sequence of ALK inhibitors (17). However, the sequence of ALK inhibitor administration can be complex and depends on the ALK mutant sensitivity profile. The complexity of inhibitor-specific responses is integrally connected to an inhibitor's molecular structure. TPX-0131, a compact macrocyclic molecule, was rationally designed based on the crystal structure of ALK to efficiently target the active kinase conformation and circumvent the steric interference conferred by resistance mutations, especially the solvent front and compound mutations (Fig. 1). By targeting the minimal binding interface of ATP in the ALK active site with a small, conformationally constrained macrocyclic structure, TPX-0131 is highly potent against wild-type ALK and many of the ALK mutations that limit the effectiveness of prior-generation inhibitors and, therefore, it has the potential to become an important new therapy for both first-line treatment as well as for patients that progress on prior therapies.
Solvent front mutations are a frequent mechanism of resistance to both the first-generation inhibitor crizotinib and second-generation inhibitors (alectinib, brigatinib, ceritinib). The solvent front region occurs at the C-terminal end of the hinge and forms a hydrophobic interaction with the kinase β1 sheet. Prior generations of ALK inhibitors have substituents that extend into this region to achieve potent ALK inhibition; however, bulky mutations (e.g., G1202R) clash with these inhibitors, preventing potent inhibition (Fig. 1). Lorlatinib has a smaller group (pyrazole ring) extending into the solvent front region compared with previous generations of ALK inhibitors, but it resides directly over the G1202 residue. Although clinical responses in patients harboring solvent front mutations such as G1202R can be achieved with lorlatinib, these mutations are often detected in patients with relapsed disease, indicating that lorlatinib may not be optimally effective against this class of resistance mutations (11). In the current study, lorlatinib had moderate potency against the G1202R solvent front mutation (IC50 = 52 nmol/L), consistent with previous studies (13, 14). Lorlatinib is reported to achieve Cave approximately 155 nmol/L unbound exposure in patients (AUC0–24h 5650 ng*h/mL, 66% plasma protein binding; FDA label) which is less than both EC50 and EC90 unbound exposure values reported for inhibition of ALK(G1202R) phosphorylation in a preclinical xenograft model (190 nmol/L, 682 nmol/L) as well as the unbound exposure necessary for maximum antitumor efficacy in that preclinical model (624 nmol/L, 53%–86% regression; ref. 14). In comparison, TPX-0131 caused complete regression (200% TGI) of an EML4-ALK(G1202R) xenograft model at 13.7 nmol/L mean free plasma trough concentration (Fig. 3). The more complete tumor growth regression observed with TPX-0131 treatment may be due to the 260-fold greater cellular potency of TPX-0131 relative to lorlatinib (Table 2). Absolute potency – not just the potency relative to inhibitor exposure – may contribute toward overall in vivo efficacy due to time-dependent changes in target engagement in the dynamic environment of the human body where both drug and target concentrations vary as a function of time (18–20). In addition to superior potency against the G1202R solvent front mutation, TPX-0131 is more potent than previous generations of ALK inhibitors against WT ALK and ALK point mutations (Table 2). Against the L1196M gatekeeper mutation, TPX-0131 is 76-fold more potent than lorlatinib and 11- to 550-fold more potent than the first two generations of inhibitors. TPX-0131 is 90- to 3000-fold more potent than other ALK inhibitors against L1198F mutations. Even though TPX-0131 has a small binding “footprint” in the active site, we report that it has attenuated potency to I1171 mutations in the αC helix (Table 2). Taken together, the greater potency of TPX-0131 toward WT ALK and common ALK mutations has the potential to translate into enhanced efficacy in patients.
An emerging mechanism of resistance in patients with ALK+ NSCLC is from compound ALK mutations. Both preclinical and clinical data have shown that compound mutations can emerge after sequential treatment with multiple ALK inhibitors or treatment with lorlatinib (e.g., 35%–48% of patients relapsing after lorlatinib treatment; refs. 11, 13). The ALK G1202R/L1196M compound mutation has been observed in patients with treatment-resistant disease (13). TPX-0131 is potent against the G1202R/L1196M compound mutation in cell proliferation assays (IC50 = 0.7 nmol/L) while other ALK inhibitors had modest to no measurable potency. In the G1202R/L1196M xenograft tumor model, TPX-0131 (10 mg/kg twice daily) caused complete tumor regression while lorlatinib treatment did not lead to significant TGI at a dose that produced regression in other ALK-dependent models (14). TPX-0131 was also potent in cellular assessments of eight other compound mutations. Interestingly, TPX-0131 is over 300- fold more potent toward the L1198F/I1171N compound mutation relative to the I1171N point mutation. TPX-0131 was the only one of the five approved ALK inhibitors tested that had potent activity against a spectrum of compound mutations.
TPX-0131 is a compact macrocycle designed to address important unmet medical needs for patients with ALK+ NSCLC. TPX-0131 has potent preclinical activity against WT and mutant ALK in vitro and in vivo. The compact macrocyclic structure allows TPX-0131 to target the active kinase conformation and overcome a broad spectrum of clinical resistant ALK mutations including solvent front and compound mutations. TPX-0131 demonstrated a high level of CNS penetration in an in vivo preclinical model and therefore has the potential to cross the blood–brain barrier in humans. Based on in vitro and in vivo preclinical studies, TPX-0131 is differentiated from previous generations of ALK inhibitors by both its breadth and degree of potency against a range of clinically relevant ALK resistance mutations and represents a new generation of ALK inhibitor that addresses a critical unmet medical need. A phase I/II clinical study of TPX-0131 focused on pretreated patients with ALK+ NSCLC is currently being initiated (NCT04849273).
Authors' Disclosures
No disclosures were reported.
Acknowledgments
All work was funded by Turning Point Therapeutics. We thank Drs. Stephanie Correia, Krishna Allamneni, and Szu-Wei Lee for providing valuable scientific insight in the development of the manuscript.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
This article is featured in Highlights of This Issue, p. 1497
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Therapeutics Online (http://mct.aacrjournals.org/).
Authors' Contributions
B.W. Murray: Conceptualization, data curation, formal analysis, writing–original draft, writing–review and editing. D. Zhai: Conceptualization, resources, formal analysis, supervision, methodology, writing–original draft. W. Deng: Conceptualization, formal analysis, supervision, writing–original draft. X. Zhang: Resources, data curation, formal analysis, methodology. J. Ung: Resources, data curation, investigation, methodology. V. Nguyen: Resources, data curation, formal analysis, methodology. H. Zhang: Resources, data curation, formal analysis, validation, methodology. M. Barrera: Resources, data curation, investigation, methodology. A. Parra: Resources, data curation, formal analysis, methodology. J. Cowell: Resources, data curation, supervision, investigation, methodology. D.J. Lee: Resources, data curation, investigation. H. Aloysius: Conceptualization, data curation, formal analysis, investigation, methodology, writing–original draft. E. Rogers: Conceptualization, formal analysis, supervision, investigation, methodology, writing–original draft.
References
- 1. Pikor LA, Ramnarine VR, Lam S, Lam WL. Genetic alterations defining NSCLC subtypes and their therapeutic implications. Lung Cancer 2013;82:179–89. [DOI] [PubMed] [Google Scholar]
- 2. Devarakonda S, Morgensztern D, Govindan R. Genomic alterations in lung adenocarcinoma. Lancet Oncol 2015;16:e342–51. [DOI] [PubMed] [Google Scholar]
- 3. Bischof D, Pulford K, Mason DY, Morris SW. Role of the nucleophosmin (NPM) portion of the non-Hodgkin's lymphoma-associated NPM-anaplastic lymphoma kinase fusion protein in oncogenesis. Mol Cell Biol 1997;17:2312–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561–6. [DOI] [PubMed] [Google Scholar]
- 5. Golding B, Luu A, Jones R, Viloria-Petit AM. The function and therapeutic targeting of anaplastic lymphoma kinase (ALK) in non-small cell lung cancer (NSCLC). Mol Cancer 2018;17:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lin JJ, Riely GJ, Shaw AT. Targeting ALK: precision medicine takes on drug resistance. Cancer Discov 2017;7:137–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med 2014;371:2167–77. [DOI] [PubMed] [Google Scholar]
- 8. Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discov 2016;6:1118–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Katayama R, Lovly CM, Shaw AT. Therapeutic targeting of anaplastic lymphoma kinase in lung cancer: a paradigm for precision cancer medicine. Clin Cancer Res 2015;21:2227–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dagogo-Jack I, Brannon AR, Ferris LA, Campbell CD, Lin JJ, Schultz KR, et al. Tracking the evolution of resistance to ALK tyrosine kinase inhibitors through longitudinal analysis of circulating tumor DNA. JCO Precis Oncol 2018 Jan 23 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dagogo-Jack I, Rooney M, Lin JJ, Nagy RJ, Yeap BY, Hubbeling H, et al. Treatment with next-generation ALK inhibitors fuels plasma ALK mutation diversity. Clin Cancer Res 2019;25:6662–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shaw AT, Felip E, Bauer TM, Besse B, Navarro A, Postel-Vinay S, et al. Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: an international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol 2017;18:1590–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yoda S, Lin JJ, Lawrence MS, Burke BJ, Friboulet L, Langenbucher A, et al. Sequential ALK inhibitors can select for lorlatinib-resistant compound ALK mutations in ALK-positive lung cancer. Cancer Discov 2018;8:714–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zou HY, Friboulet L, Kodack DP, Engstrom LD, Li Q, West M, et al. PF-06463922, an ALK/ROS1 inhibitor, overcomes resistance to first and second generation ALK inhibitors in preclinical models. Cancer Cell 2015;28:70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol 2011;29:1039–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shaw AT, Solomon BJ, Besse B, Bauer TM, Lin CC, Soo RA, et al. ALK resistance mutations and efficacy of lorlatinib in advanced anaplastic lymphoma kinase-positive non-small-cell lung cancer. J Clin Oncol 2019;37:1370–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McCusker MG, Russo A, Scilla KA, Mehra R, Rolfo C. How I treat ALK-positive non-small cell lung cancer. ESMO Open 2019;4:e000524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Peters SA, Petersson C, Blaukat A, Halle JP, Dolgos H. Prediction of active human dose: learnings from 20 years of Merck KGaA experience, illustrated by case studies. Drug Discov Today 2020;25:909–19. [DOI] [PubMed] [Google Scholar]
- 19. Tonge PJ. Drug-target kinetics in drug discovery. ACS Chem Neurosci 2018;9:29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chuang YC, Huang BY, Chang HW, Yang CN. Molecular modeling of ALK L1198F and/or G1202R mutations to determine differential crizotinib sensitivity. Sci Rep 2019;9:11390. [DOI] [PMC free article] [PubMed] [Google Scholar]