

Abstract
The protein arginine methyltransferase 5 (PRMT5) methylates a variety of proteins involved in splicing, multiple signal transduction pathways, epigenetic control of gene expression, and mechanisms leading to protein expression required for cellular proliferation. Dysregulation of PRMT5 is associated with clinical features of several cancers, including lymphomas, lung cancer, and breast cancer. Here, we describe the characterization of JNJ-64619178, a novel, selective, and potent PRMT5 inhibitor, currently in clinical trials for patients with advanced solid tumors, non-Hodgkin's lymphoma, and lower-risk myelodysplastic syndrome. JNJ-64619178 demonstrated a prolonged inhibition of PRMT5 and potent antiproliferative activity in subsets of cancer cell lines derived from various histologies, including lung, breast, pancreatic, and hematological malignancies. In primary acute myelogenous leukemia samples, the presence of splicing factor mutations correlated with a higher ex vivo sensitivity to JNJ-64619178. Furthermore, the potent and unique mechanism of inhibition of JNJ-64619178, combined with highly optimized pharmacological properties, led to efficient tumor growth inhibition and regression in several xenograft models in vivo, with once-daily or intermittent oral-dosing schedules. An increase in splicing burden was observed upon JNJ-64619178 treatment. Overall, these observations support the continued clinical evaluation of JNJ-64619178 in patients with aberrant PRMT5 activity–driven tumors.
Graphical Abstract

Introduction
Arginine methylation is a post-translational modification regulating numerous biological processes such as cell signaling, transcription, DNA damage, and mRNA splicing (1). Nine protein arginine methyltransferases (PRMT) catalyzing the transfer of a methyl group from S-adenosylmethionine (SAM) to arginine residues on protein substrates, have been identified in humans. PRMTs are classified in three groups, with overlapping and diverse functions: Type I (asymmetric dimethylarginine, aDMA), type II (symmetric dimethylarginine, sDMA), and type III (monomethylarginine). In mammals, PRMT5 represents the main type II arginine methyltransferase for sDMA of both histones and non-histone proteins (2). PRMT5 binds to methylosome protein 50 (MEP50) forming a stable and functionally active complex (3, 4). Recently, pICIn and RioK were identified to bind the PRMT5/MEP50 complex in a mutually exclusive manner, promoting distinct protein substrate recruitment and pleiotropic modulation of cellular functions orchestrated by PRMT5 (3, 4).
PRMT5 inhibition may be of therapeutic value to treat hematological cancers like acute myelogenous leukemia (AML; refs. 5, 6) and lung carcinoma (7). Furthermore, elevated expression of PRMT5 and its cofactor, MEP50, in NSCLC is highly correlated with poor survival (8). Therapeutic rationales for PRMT5 inhibition based on genetic evidence are emerging. Besides its epigenetic role, PRMT5 regulates homologous recombination and the DNA damage response (DDR; refs. 9, 10) and methylates several spliceosome constituents (11) as well as RNA-binding proteins, including splicing factors, regulating constitutive and alternative splicing events (12, 13). PRMT5 inhibition may, therefore, be a synthetic vulnerability in tumors addicted to splicing alterations and might increase their neoantigen presentation, thereby providing a new therapeutic rationale to combine PRMT5 inhibitors with immunotherapy approaches (14). Moreover, Marjon and colleagues (15) reported PRMT5 as a vulnerability in methylthioadenosine phosphorylase (MTAP)–deficient cancers. The MTAP gene is proximal and co-deleted with CDKN2A, the most commonly deleted gene in human cancer. MTAP deletion increases cellular concentrations of its substrate MTA, which binds to and is a weak inhibitor of PRMT5, leading to suggested increased sensitivity of these cells to PRMT5 inhibition (15) and creating a novel MTAP-deleted cancer cell-specific target: The PRMT5–MTA complex (16).
PRMT5-dependent sDMA post-translational modification may not be actively reversed because an undisputable counterpart to PRMT5-demethylating sDMA has not been identified (17). Therefore, the consequences of sDMA modification persist until the proteins are degraded and non-sDMA proteins are newly synthesized, suggesting that small-molecule PRMT5 inhibitors may require different pharmacological properties than kinases inhibitors, whose biological activities can be reversed by phosphatases.
Here, we describe the discovery and characterization of a new selective and potent PRMT5 inhibitor, JNJ-64619178 [International Patent Number: WO/2017/032840 A1 (https://patents.google.com/patent/WO2017032840A1/fi) Example 80, CAS 2086772–26–9], which occupies the SAM and the substrate pockets of PRMT5 and demonstrates a long-residence–binding time resulting in extended trapping of PRMT5/MEP50 in a catalytically inactive state. This mode-of-inhibition (MOI) enables potent and long-lasting target engagement, confirmed by in vitro compound washout experiments and in vivo drug holidays, leading to dose-dependent cancer cell death in multiple preclinical cancer models. Pharmacological PRMT5 inhibition by JNJ-64619178 led to an increase in alternatively spliced events in a panel of histologically different cancer cell lines. Importantly, the presence of splicing factor mutations has been associated with an antiproliferative effect following ex vivo JNJ-64619178 treatment in primary AML samples. We developed a novel PRMT5 inhibitor, JNJ-64619178, with unique pharmacological properties supporting its continued clinical development in tumors addicted to altered splicing events, fostering patient-tailored and precision medicine approaches (clinicaltrials.gov Identifier: NCT03573310).
Materials and Methods
Chemicals, proteins, and reagents
Full-length PRMT5 (NP_006100) and MEP50 (NP_077007) were purified as a 1:1 PRMT5:MEP50 protein complex. Full-length human recombinant histone H2A (NM_021052, Cat#HMT-11–146) was purchased from Reaction Biology Corporation, SAM (Cat#13956) and S-Adenosyl–L-homocysteine (SAH; Cat#13603) from Cayman Chemical Company, and Cycloheximide (CHX) from Merck–Millipore (Cat#239764). References to the preparation of JNJ-64619178 and Cpd-2, as well as analytic data of additional compounds (Cpd-3, 4, 5 and 6); PRMT5:MEP50 expression and purification; siRNAs; antibodies for immunoblotting (Odyssey, Li-COR); and WES technology (ProteinSimple) are described previously in Supplementary Materials and Methods.
Enzymatic assays, MOI, binding kinetics, and crystallography
The enzyme activity of PRMT5:MEP50 and MOI studies with JNJ-64619178 were measured by following the production/prevention of SAH formation by RapidFire Mass Spectrometry (MS; ref. 18). Binding kinetics between PRMT5 inhibitors and Avi-tagged PRMT5:MEP50 protein, immobilized on streptavidin coated sensors, were determined by surface plasmon resonance spectroscopy (Biacore System, GE Healthcare). Cellular binding of JNJ-64619178 and Cpd-2 to PRMT5 was measured by a modified cellular thermal shift assay (19). PRMT5:MEP50 was co-crystallized with JNJ-64619178 and the refined structure was deposited in the Protein Data Bank (PDB) with the accession code: PDB=6RLQ. Detailed protocols are listed in Supplementary Materials and Methods.
Cellular target profiling
Cellular target profiling (20) was applied to identify protein binders of JNJ-64619178 with corresponding binding affinities (Kd values) from SILAC-encoded NCI-H1048 cells by chemical proteomic analysis. Experimental details, analysis, and visualization approaches are described previously in Supplementary Materials and Methods.
Cell lines
All cell lines were authenticated by suppliers using short tandem repeat analysis and kept in culture for ≤15 passages, but not >3 months. Cells were cultured in standard conditions (37°C, 5% CO2, 95% humidity) in medium containing 10% (v/v) fetal bovine serum and were regularly tested for absence of Mycoplasma contaminations.
Assay protocols
RNA isolation, RNA sequencing (RNA-seq), splicing analysis, isogenic cell line generation, cell proliferation, and determination of 50% cell growth inhibition (GI50), assays and protocols are described previously in Supplementary Materials and Methods. The Sequence Read Archive (SRA) ID for the deposited RNA sequence is PRJNA760893.
Mouse xenograft studies
All experiments were performed in accordance with the European Communities Council Directives (2010/63/EU) and approved by the local IACUC and ethical committee of Janssen Pharmaceutica, and the Janssen Research and Development, LLC, and Charles River Discovery Services Institutional Animal Care and Use Committees. Protocols and data analyses are described in Supplementary Materials and Methods.
Results
Discovery of JNJ-64619178, a potent PRMT5 inhibitor with slow off-rate kinetics
SAM mimetics have been described previously as efficient inhibitors of the PRMT5/MEP50 complex (3, 21–25). From a focused adenosine derivative compound library, designed on the basis of the co-crystallized structure of human PRMT5/MEP50 with a SAM analog (3), we identified a 5′ spiro bisamine lead (26) that underwent Structure Activity Relationship (SAR) iterations and pharmacochemical optimizations leading to JNJ-64619178 (Fig. 1A) identification.
Figure 1.

JNJ-64619178 potently inhibits the PRMT5/MEP50 complex with a long-residence–binding time mechanism through interaction with PRMT5's SAM and substrate binding pockets. A, JNJ-64619178 chemical structure. B, Inhibition of PRMT5 by JNJ-64619178 was studied in a reaction mixture of 10 μmol/L SAM, 1 μmol/L histone H2A, and 0.156 nmol/L enzyme in the absence or presence of JNJ-64619178 at different concentrations as depicted in Fig. 1B. Production of SAdenosyl–L-homocysteine (SAH) was followed as a function of reaction time. Reaction progress curves were fit to Eq. 1 to derive the inhibition rate constant kobs at each compound concentration. C, Plot of derived kobs versus JNJ-64619178 concentration was fit to a hyperbolic function of Eq. 2. Binding and dissociation curves to and from immobilized PRMT5/MEP50 complex were measured by surface plasmon resonance (SPR for (D) JNJ-64619178 and (E) Cpd-2. PRMT5/MEP50 complex was immobilized on a CM5 sensor chip and increasing concentrations of JNJ-64619178 or Cpd-2 (1.56–100 nmol/L) were added to determine the binding kinetics. Compound association to and dissociation from the immobilized PRMT5/MEP50 complex, described previously as SPR response units (RU), were plotted against time (s). F, X-ray co-crystal structure of JNJ-64619178 bound to the active site of the PRMT5:MEP50 complex (PDB ID: 6RLQ). JNJ-64619178 is shown in yellow and key PRMT5 interaction residues are shown as gray sticks. Oxygen atoms are colored red, nitrogen atoms blue, and the bromine atom in maroon. Hydrogen bonds are depicted as black-dashed lines and the halogen bond as an orange-dashed line. The binding site surface of PRMT5 is depicted and the putative SAM-binding site is colored in ivory whereas the substrate-binding site is colored in cyan.
Investigation of JNJ-64619178 kinetic–binding properties revealed a time-dependent inhibition of PRMT5/MEP50 complex (Fig. 1B). In detail, the first-order rate constants (kobs) of inhibition were derived by fitting the reaction progress curves in presence of multiple concentrations of the inhibitor (0.24–20.8 nmol/L). Data fitting to the plot kobs versus inhibitor concentration (Fig. 1C), indicated a two-step inhibition mechanism (Scheme A, Supplementary Materials and Methods), in which rapid binding of the inhibitor to the enzyme (EI) was followed by a slow isomerization step that resulted in a stable complex, EI*. The compound dissociation constant (k4) was determined to be nearly 0, indicating very slow release of JNJ-64619178 from the enzyme. The rate constant of the isomerization step, k3, has been assessed as 4.7×10−4 s−1. The compound concentration required to reach the half-maximal rate of the enzyme inhibition, 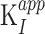 , was 0.772 ± 0.417 nmol/L under the defined assay conditions. Finally, the compound inhibition potency was calculated to be 6.1×105 M−1s−1.
, was 0.772 ± 0.417 nmol/L under the defined assay conditions. Finally, the compound inhibition potency was calculated to be 6.1×105 M−1s−1.
To further assess the mode-of-binding of JNJ-64619178 and of a close analogue, compound-2 (Cpd2, structure in Supplementary Fig. S1), Surface Plasmon Resonance (SPR)–binding studies were performed with the immobilized PRMT5/MEP50 complex. JNJ-64619178 demonstrated a slower off-rate binding kinetics versus Cpd-2 (Fig. 1D–E). JNJ-64619178 and Cpd-2 association constants [ka (M−1s−1)] of 6.31 × 105 and 3.47 ×106 and dissociation constants [kd (s−1)] of <3.96 ×10−4 and 1.14 ×10−2 were measured, as well as the equilibrium dissociation constants (Kd) of <0.628 and 3.28 nmol/L, respectively. The derived half-life (t1/2) for Cpd-2 binding to PRMT5/MEP50 was 1.01 minutes, whereas it could not be derived for JNJ-64619178 under the tested conditions (max 10 minutes sensorgram) confirming the dissociation constant data (k4 ∼0) observed in the kinetic study. In contrast with the reversible inhibitor Cpd-2, JNJ-64619178 showed binding properties consistent with a pseudo-irreversible PRMT5 inhibitory mechanism, based on a very slow Kd, in the absence of potential reactive groups required for covalent binding.
A co-crystal structure of JNJ-64619178 bound to PRMT5/MEP50 with a 2.3Å resolution (PDBID: 6RLQ) was obtained (Fig. 1F). The structure revealed that JNJ-64619178 binds non-covalently in the PRMT5 SAM-binding site, with its bromo-aminoquinoline moiety extended toward the substrate-binding site, allowing for bidentate hydrogen bonding interactions with the catalytic Glu444 residue. On the basis of molecular similarity, Cpd-2 should also engage the Glu444 residue, but in a monodentate configuration (due to lack of an amine substituent on the quinoline moiety), which may partially contribute toward its relatively lower affinity or faster dissociation rate compared with JNJ-64619178. Furthermore, the bromine atom of JNJ-64619178 formed a halogen interaction with the backbone oxygen atom of Ser578 residue.
JNJ-64619178 is a highly selective PRMT5 inhibitor
Selectivity of JNJ-64619178 was assessed against 37 purified recombinant human arginine (Arg) and lysine (Lys) methyltransferases, including 4 DNA methyltransferases (Fig. 2A), to exclude potential off-target effects. Remarkably, high JNJ-64619178 concentration (10 μmol/L) inhibited the PRMT5/MEP50 enzymatic complex by >80%, whereas other closely related Arg methyltransferases, such as PRMT1 and PRMT7, were not or minimally inhibited (<15% inhibition). No Lys methyltransferases were inhibited at >15%.
Figure 2.

JNJ-64619178 is a selective, time-dependent, and potent inhibitor of PRMT5 in cells and JNJ-64619178-mediated inhibition of symmetric arginine dimethylation of SmD1/3 proteins (SmD1/3-Me2) is a marker of target engagement, but not cancer cell sensitivity. A, Phylogenic trees depicting percentage (%) inhibition of recombinant human SAM arginine (Arg) and lysine (Lys) methyltransferases by JNJ-64619178 in vitro. Small gray circles indicate enzymes with <15% inhibition at 10 μmol/L JNJ-64619178, whereas the enlarged circle marks enzyme (in this case only PRMT5/MEP50 complex) with >80% inhibition at 10 μmol/L JNJ-64619178. B, Affinity (Kd value) of JNJ-64619178 binding to cellular proteins determined by competitive pull-down quantitative chemical proteomics. Extrapolated Kd value was assigned to FECH, marked with an asterix (*) due to partial competitive binding with JNJ-64619178. C, Cellular inhibition kinetics of Sym-Arg dimethylation of SmD1/3 proteins (SmD1/3-Me2) in NCI-H1048 cells at various time points following continued exposure of 0 (DMSO), 1 or 10 nmol/L of JNJ-64619178. D, Inhibition of human lung cancer cell line proliferation as measured by MTT after 6-day continuous treatment with JNJ-64619178 at multiple concentrations. GI50 values are listed on graph. GI50 values for COLO-699 and DMS-53 were set as the highest compound concentration tested (1.0 μmol/L), as 50% inhibition was not reached under the conditions tested. Error bars represent standard deviation (SD) from the mean. Exponentially growing (E) NCI-H1048 and (F) COLO-699 cancer cells were treated with DMSO or JNJ-64619178. Line graphs depict inhibition of cell proliferation (MTT assay, OD540) at several JNJ-64619178 concentrations. Hatched bars depict symmetric arginine dimethylation of SmD1/3 (SmD1/3-Me2, 10 nmol/L JNJ-64619178) monitored by immunoblotting. Quantification was normalized to β-actin. Error bars represent mean ± standard deviation (SD).
Cellular target profiling by chemical proteomic analysis was performed (20). An affinity matrix carrying two immobilized aminopropyl–linker derivatives of JNJ-64619178 (Cpd-3 and Cpd-4, Supplementary Fig. S1) was challenged with human small-cell lung carcinoma (SCLC) NCI-H1048 tumor cell line protein extract for cellular targets enrichment (protein binders). Additional equilibrium competition experiments with JNJ-64619178 and less active compounds (Cpd-5 and Cpd-6, Supplementary Fig. S1), followed by quantitative MS analysis of eluted material were performed. The binding constants (Kd) of the enriched proteins with their respective binding affinities are represented in Fig. 2B (Supplementary Fig. S2 for Cpd-5 and Cpd-6). Notably, only three cellular high-affinity binders to JNJ-64619178 were identified: PRMT5 (Kd ≤1 nmol/L), MEP50 (Kd ∼ 0.4 nmol/L), and the serine/threonine protein kinase RIO1 (RIOK1, Kd ≤0.01 nmol/L). These three proteins are known to be constituents of the methylosome, and PRMT5 is most likely the unique direct binder of JNJ-64619178, as confirmed by SPR and visualized by crystallography (Fig. 1D and F). In fact, RIOK1 and MEP50 have been previously identified as PRMT5-interacting proteins (4, 27) and therefore likely co-purified with PRMT5. Interestingly, other interactors like pICln or COPR5 were not identified as high-affinity binders in the NCI-H1048 cell model used. The pICln and RIOK1 proteins were identified to bind the PRMT5/MEP50 complex in a mutually exclusive manner (27), suggesting that in NCI-H1048 cells, RIOK1 may preferentially and with potentially higher affinity bind to PRMT5, competing against pICln or COPR5 to form PRMT5 complexes. NAD(P)H dehydrogenase [quinone] 1 (NQO1) and ferrochelatase (FECH) were also detected in the enrichment procedure. These are unlikely to be PRMT5 binders as they are frequently identified as false-positive hits in chemical proteomic analyses (28, 29). Overall, we demonstrated that in a cellular context, JNJ-64619178 selectively binds to PRMT5.
JNJ-64619178 functionally inhibits the PRMT5/MEP50 complex impairing cancer cell proliferation
To confirm that PRMT5 is a targetable vulnerability in lung cancer cell lines (7), we performed PRMT5 genetic knockdown (KD) and pharmacological inhibition studies and analyzed phenotypical and functional readouts in A549 lung cancer cells. We first confirmed that siRNA-mediated PRMT5 KD decreased the arginine methylation of the two previously reported PRMT5 targets SmD1 and SmD3 (SmD1/3; ref. 30; Supplementary Fig. S3A). SmD1/3 methylation was shown to be dependent on PRMT5 catalytic activity as the genetic ablation effect was reversed by the expression of siRNA-resistant wild-type (WT), but not a catalytically deficient mutant form of PRMT5 (Supplementary Fig. S3A–S3B). Although we demonstrated that JNJ-64619178 inhibited general nuclear arginine dimethylation by IHC assay on A549 cells (IC50 = 0.2 nmol/L; ref. 28), we measured the kinetics of the pharmacological PRMT5 inhibition in NCI-H1048 cells by monitoring the levels of dimethylated SmD1/3 (SmD1/3-Me2), a well-defined pharmacodynamic marker of PRMT5 activity, following compound treatment (Fig. 2C). After 48 hours of continuous compound exposure, JNJ-64619178 inhibited dimethylation of SmD1/3 (reduction of SmD1/3-Me2 levels) by >50% and 80% at 1 and 10 nmol/L concentrations, respectively. Very low to undetectable levels of SmD1/3-Me2 were observed after 96 hours of continuous exposure to JNJ-64619178 at 1 and 10 nmol/L concentrations. No changes in PRMT5, MEP50, and SmD3 protein levels were observed. These data suggest that a prolonged PRMT5 inhibition is required to achieve a complete SmD1/3 dimethylation inhibition.
To assess the functional consequences of PRMT5 blockage on JNJ-64619178 treatment, a panel of 6 lung cancer cell lines was exposed to increasing concentrations of the inhibitor for 6 consecutive days. JNJ-64619178 treatment resulted in a robust inhibition of cell proliferation in 4/6 cell lines screened, as assessed by a MTT assay (Fig. 2D). In detail, NCI-H520, HCC-78, NCI-H1048, and A427 cells were particularly sensitive to JNJ-64619178, with GI50s ranging from 0.4 to 1.9 nmol/L, whereas COLO-699 and DMS-53 cell lines were less sensitive, bearing GI50 values >1.0 μmol/L (50% inhibition was not reached at 1.0 μmol/L, the highest concentration tested).
These findings demonstrate that JNJ-64619178 inhibits PRMT5-mediated Sym-Arg dimethylation of SmD1/3 proteins in a time-dependent manner and has a strong antiproliferative effect in a subset of lung cancer cell lines.
SmD1/3-me2 is a marker of JNJ-64619178-mediated PRMT5 engagement, but not a surrogate of cell growth inhibition
The correlation between SmD1/3-Me2 levels and inhibition of cell proliferation was assessed over time in JNJ-64619178 sensitive (NCI-H1048, Fig. 2E and Supplementary Fig. S4A) and insensitive (COLO699, Fig. 2F and Supplementary Fig. S4B) human lung cancer cells. Under conditions of exponential proliferation, SmD1/3Me2 levels were reduced by >80% at 72 hours and undetectable thereafter in both cell lines upon treatment with 10 nmol/L JNJ-64619178. Though the reduction in SmD1/3Me2 levels was associated with inhibition of NCI-H1048 cancer cell proliferation compared with vehicle, JNJ-64619178 had no effect on proliferation in the COLO-699 cell line (Fig. 2F), even after 200 hours exposure at the highest concentration tested (50 nmol/L). In addition, in the sensitive cell line (NCI-H1048), although the level of SmD1/3-Me2 was decreased upon treatment under reduced proliferation conditions (Supplementary Fig. S4C), the viability was unaffected (Supplementary Fig. S5), demonstrating that SmD1/3-Me2 is a marker of PRMT5 target engagement but not necessarily a marker of cellular viability.
Extended JNJ-64619178 binding to cellular PRMT5 is associated with prolonged target engagement and downstream pathway suppression
To the best of our knowledge, no PRMT5 counteracting demethylases have been identified. Jumonji domain-containing 6 (JMJD6) was reported to catalyze N-methyl-arginine residue demethylation on the N-terminus of the human histones H3 and H4. However, this finding has been subject to conflicting reports (17) and our results of a long-lasting PRMT5 methylation–dependent mark of protein upon inhibition of PRMT5 support the absence or low activity of a PRMT5-specific demethylase. Moreover, although a short protein half-life has been demonstrated for transfected tag-PRMT5 in HEK293 cells (31), our data using CHX, an inhibitor of protein synthesis, have shown that endogenous PRMT5 demonstrated a long protein half-life in assessed lung cell lines, with protein stability for at least 48 hours upon CHX treatment (Supplementary Fig. S6).
Hence, a prolonged PRMT5 inhibition to elicit significant antiproliferative activity and cancer cell death may be necessary. Indeed, the long-residence binding time of JNJ-64619178 to PRMT5 (Fig. 1B–D) suggested that brief compound exposure might be sufficient for prolonged PRMT5 inhibition in cells. On the basis of these findings, we compared the effects of short-term and continuous compound exposures on PRMT5 stability in a cellular thermal melt shift assay. We defined the heat-shock conditions (60°C for 3 minutes) where compound binding stabilizes soluble PRMT5 and prevents its misfolding and aggregation. In compound test samples, following heat-shock and centrifugation, aggregated, compound-free PRMT5 in the pellet fraction and compound-stabilized PRMT5 in the soluble cell lysate fraction are detected by immunoblotting. A 1-hour treatment of NCI-H1048 cells with JNJ-64619178 or Cpd-2, demonstrating a shorter t1/2 binding to PRMT5 (Fig. 1E), followed by heat-shock, resulted in stabilization of PRMT5 as evidenced by its presence in the soluble lysate fraction (Fig. 3A and Supplementary Fig. S7A). As expected, PRMT5 was not detected in the soluble fraction from DMSO-treated cells. Similarly, PRMT5 was stabilized following heat-shock in NCI-H1048 cells treated continuously for 72 hours with JNJ-64619178 or Cpd-2. However, when cells were treated for 1 hour, followed by a washout step and then subjected to heat-shock 72 hours later, soluble PRMT5 was detected only in JNJ-64619178, and not in Cpd-2–treated cells. Comparable results were obtained in HCC-78 cells (Supplementary Fig. S7B and S7C). Although Cpd-2 exhibited a considerably shorter binding t1/2 than JNJ-64619178 in vitro (Fig. 1E), rapid dissociation and re-binding when compound is in excess may stabilize cellular PRMT5 and prevent aggregation during the heat-shock treatment. PRMT5 stabilization by JNJ-64619178 after washout revealed a prolonged binding time in cells and demonstrated that a short (1 hour) cellular exposure period was sufficient to efficiently trap and inhibit PRMT5 for up to 72 hours.
Figure 3.

JNJ-64619178 promotes cellular PRMT5 thermal shift stabilization and leads to prolonged pathway inhibition after compound washout. A, NCI-H1048 cells were treated with JNJ-64619178 (0.1 μmol/L) or Cpd-2 (10 μmol/L) for either 1 hour (1-hour treatment), 1 hour followed by compound washout and further incubation in compound-free complete media for 72 hours (after washout), or continuously for 72 hours (continuous treatment). Thermal shift (60°C for 3 minutes) was applied, and soluble proteins (supernatant) were separated by WES (ProteinSimple). PRMT5 levels were quantitated, normalized to β-actin and plotted. Dashed red rectangle highlights the washout treatment. Error bars represent mean ± standard deviation (SD). B, PRMT5 downstream signalling in NCI-H1048 cells after compound treatment assessed by immunoblotting against SmD1/3-Me2, SmD3, PRMT5, and MEP50. Cells were either treated with JNJ-64619178 (0.03 and 0.3 μmol/L) or Cpd-2 (0.3 and 3.0 μmol/L) or DMSO (No treatment) as indicated. Dashed red rectangle highlights the washout treatment. C, Inhibition of cell proliferation under intermittent (wash out; y-axis) and continuous (no wash out; x-axis) treatment with JNJ-64619178 (10 pmol/L–1 μmol/L dose response) was assessed over 10 days and AUC (% inhibition/nmol/L) was plotted for 94 cancer cell lines from multiple cancer types (each cancer type represented by different color dots). For washout condition, exponentially growing cells were treated for 3 days with JNJ-64619178, followed by extensive compound washout. Cell growth inhibition was determined after 7 additional days after washout. For the 10-day continuous compound treatment condition (no wash out), cells were continuously treated with JNJ-64619178 at the same concentrations as described above. Linear regression (slope = 0.873) was determined (Pearson's correlation = 0.993).
Furthermore, the SmD1/3-Me2 levels in NCI-H1048 cells were assessed by immunoblotting to explore the effect of JNJ-64619178 on PRMT5 activity (Fig. 3B). In accordance with the data in Fig. 2C, neither JNJ-64619178 nor Cpd-2 influenced SmD1/3-Me2 levels upon a short exposure time (1 hour). However, under continuous treatment (72 hours), SmD1/3-Me2 levels were substantially reduced (Fig. 3B), indicating potent inhibition of SmD1/3 dimethylation. Importantly, only JNJ-64619178 demonstrated reduction of SmD1/3-Me2 levels after compound washout, consistent with its prolonged PRMT5-binding properties (Fig. 3A and Supplementary Fig. S7A). SmD1/3-Me2 levels were unaffected by Cpd-2, even at concentrations 100-fold higher than the lowest active concentration of JNJ-64619178 used (3.0 vs. 0.03 μmol/L, respectively). Similar results were obtained in HCC-78 cells (Supplementary Fig. S7D).
JNJ-64619178′s mechanism of action was further validated by comparing the antiproliferative activity of JNJ-64619178 in a large panel of 94 cancer cell lines originating from 18 different heme and solid cancer types. Cells were treated continuously for 10 days or exposed to a pulsed 3-day treatment followed by compound washout and an additional compound-free growth period of 7 days. The area under the curve (AUC; percentage of inhibition × mol/L) for growth inhibition (% inhibition) was plotted against compound concentration (nmol/L) for each treatment condition (Fig. 3C and Supplementary Fig. S8). The linear regression slope (0.873) suggested that JNJ-64619178 exerted similar antiproliferative activity under continuous or pulsed compound treatment.
In summary, JNJ-64619178 is a potent PRMT5 inhibitor with a long-residence–binding time, leading to sustained inhibition of PRMT5 even after short compound exposure.
JNJ-64619178 demonstrates sustained target engagement associated with potent antitumor activity in tumor-xenograft mouse models
To further confirm the observed JNJ-64619178 activity against cancer cell lines, we expanded our assessment to in vivo settings. First, we determined the pharmacokinetic parameters of JNJ-64619178 on single 2.5 mg/kg intravenous or 10 mg/kg oral doses. Clearance of JNJ-64619178 in vivo was low (6.6 mL/min/kg, corresponding to about 54% of the liver blood flow in mice), and its absolute oral bioavailability was moderate (36%; Fig. 4A). These data highlighted JNJ-64619178 as an orally bioavailable inhibitor with favorable physical properties in mice and supported further evaluation of efficacy against human tumor xenografts.
Figure 4.
![Figure 4. JNJ-64619178 pharmacokinetic properties contribute to inhibition of Sym-Arginine SmD1/3 dimethylation in tumors and efficacy in multiple human lung cancer xenograft models under continuous or intermittent compound dosing. A, Plasma concentrations of JNJ-64619178 over 24 hours following a single intravenous (2.5 mg/kg in PBS) or oral [PO, 10 mg/kg in 20% 2-Hydroxypropyl-β-cyclodextrin solution (20% HP-β-CD)] administration in C57BL/6 mice. Mean ± standard deviation was plotted for 3 animals. B, SmD1/3Me2 levels in tumors from NCI-H1048 lung cancer xenograft-bearing mice (n = 3 mice/group) 24 hours after 6 consecutive days of daily oral administration of JNJ-64619178 (0.1, 1.0, 10 mg/kg). SmD1/3Me2 levels detected by immunoblotting (Supplementary Fig. S9A) were quantified and represented as the percentage (%) of SmD1/3Me2 levels in tumor samples from vehicle (20% HP-β-CD) treated mice. Antitumoral effect of JNJ-64619178 in (C) NCI-H1048, (D) NCI-H441, (E) A427, and (F) NCI-H520 NSCLC xenograft tumors following daily oral treatment for 28, 38, 34, and 27 days, respectively. JNJ-64619178 was orally administered once daily at several doses (0.3, 1, 3, 10 mg/kg) to tumor-bearing nude mice (n = 8–10 mice/group). Mean tumor volume (mm3) ± standard error of the mean (SEM) was determined and plotted on graphs. Tumor growth inhibition (%ΔTGI) values are summarized in Supplementary Table S2. G, Time course of SmD1/3-Me2 levels in NCI-H1048 lung cancer xenograft tumors in mice dosed orally with JNJ-64619178 (10 mg/kg) for 9 consecutive days (gray region), followed by treatment cessation for 11 days. Tumors (n = 3 mice/group) were collected 2 hours after JNJ-64619178 dosing (days 0, 1, 2, 3, and 6) and every day following treatment cessation. SmD1/3-Me2 was detected by immunoblotting and levels normalized to the median levels of β-actin protein detected in tumors from vehicle (20% HP-β-CD)–treated mice. The percentage of SmD1/3-Me2 levels compared with the vehicle-treated group (defined as 100%), are shown as boxplots. Dotted line corresponds to the level of SmD1/3-Me2 in untreated samples. SmD1/3Me2 levels detected by immunoblotting (Supplementary Fig. S9D) were quantified and represented as the percentage (%) of SmD1/3Me2 levels in tumor samples from vehicle. H, Antitumoral effect in NCIH1048 xenograft-bearing mice (n = 8 mice/group) dosed orally once daily with vehicle (20% HP-β-CD) or JNJ-64619178 (10 mg/kg) for either 28 consecutive days or for two cycles of 7 days of drug treatment followed by 7 days dosing holidays (7+/7−, 2 cycles). Tumor growth inhibition (ΔTGI, %) was determined at day 28. In all xenograft studies, the average tumor volume at treatment initiation was about 150 mm3 and mean tumor volumes (mm3) ± SEM of each group were plotted over time.](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=9398174_2317fig4.jpg)
JNJ-64619178 pharmacokinetic properties contribute to inhibition of Sym-Arginine SmD1/3 dimethylation in tumors and efficacy in multiple human lung cancer xenograft models under continuous or intermittent compound dosing. A, Plasma concentrations of JNJ-64619178 over 24 hours following a single intravenous (2.5 mg/kg in PBS) or oral [PO, 10 mg/kg in 20% 2-Hydroxypropyl-β-cyclodextrin solution (20% HP-β-CD)] administration in C57BL/6 mice. Mean ± standard deviation was plotted for 3 animals. B, SmD1/3Me2 levels in tumors from NCI-H1048 lung cancer xenograft-bearing mice (n = 3 mice/group) 24 hours after 6 consecutive days of daily oral administration of JNJ-64619178 (0.1, 1.0, 10 mg/kg). SmD1/3Me2 levels detected by immunoblotting (Supplementary Fig. S9A) were quantified and represented as the percentage (%) of SmD1/3Me2 levels in tumor samples from vehicle (20% HP-β-CD) treated mice. Antitumoral effect of JNJ-64619178 in (C) NCI-H1048, (D) NCI-H441, (E) A427, and (F) NCI-H520 NSCLC xenograft tumors following daily oral treatment for 28, 38, 34, and 27 days, respectively. JNJ-64619178 was orally administered once daily at several doses (0.3, 1, 3, 10 mg/kg) to tumor-bearing nude mice (n = 8–10 mice/group). Mean tumor volume (mm3) ± standard error of the mean (SEM) was determined and plotted on graphs. Tumor growth inhibition (%ΔTGI) values are summarized in Supplementary Table S2. G, Time course of SmD1/3-Me2 levels in NCI-H1048 lung cancer xenograft tumors in mice dosed orally with JNJ-64619178 (10 mg/kg) for 9 consecutive days (gray region), followed by treatment cessation for 11 days. Tumors (n = 3 mice/group) were collected 2 hours after JNJ-64619178 dosing (days 0, 1, 2, 3, and 6) and every day following treatment cessation. SmD1/3-Me2 was detected by immunoblotting and levels normalized to the median levels of β-actin protein detected in tumors from vehicle (20% HP-β-CD)–treated mice. The percentage of SmD1/3-Me2 levels compared with the vehicle-treated group (defined as 100%), are shown as boxplots. Dotted line corresponds to the level of SmD1/3-Me2 in untreated samples. SmD1/3Me2 levels detected by immunoblotting (Supplementary Fig. S9D) were quantified and represented as the percentage (%) of SmD1/3Me2 levels in tumor samples from vehicle. H, Antitumoral effect in NCIH1048 xenograft-bearing mice (n = 8 mice/group) dosed orally once daily with vehicle (20% HP-β-CD) or JNJ-64619178 (10 mg/kg) for either 28 consecutive days or for two cycles of 7 days of drug treatment followed by 7 days dosing holidays (7+/7−, 2 cycles). Tumor growth inhibition (ΔTGI, %) was determined at day 28. In all xenograft studies, the average tumor volume at treatment initiation was about 150 mm3 and mean tumor volumes (mm3) ± SEM of each group were plotted over time.
To explore the functional impact of PRMT5 inhibition in vivo, mice were xenografted with NCI-H1048 and then randomized to vehicle or JNJ-64619178 (0.1–1–10 mg/kg). Because of the delayed effect of PRMT5 inhibition (Fig. 2C), target engagement (assessed as the reduction of the SmD1/3-Me2 levels) was analyzed 24 hours after oral administration of JNJ-64619178 for 6 consecutive days (Fig. 4B and Supplementary Fig. S9A). Normalization of SmD1/3-Me2 levels in tumors from JNJ-64619178-treated mice to vehicle-treated mice revealed dose-dependent inhibition of PRMT5-dependent methylation (25, 65.6, and 93.7% inhibition at 0.1, 1, and 10 mg/kg daily doses, respectively).
Treatment with JNJ-64619178 resulted in a potent dose-dependent tumor growth inhibition (%ΔTGI) in 4 human cell lines derived lung cancer xenograft models tested: NCI-H1048 (Fig. 4C), NCI-H441 (Fig. 4D), A-427 (Fig. 4E), and NCI-H520 (Fig. 4F; %ΔTGI and statistical analysis summarized in Supplementary Table S1). Tumor regression was observed with daily oral doses of 3 mg/kg and 10 mg/kg JNJ-64619178 in all lung cancer models tested, except NCI-H1048, where regression was observed only with the higher dose. All 8 mice bearing NCI-H1048 tumors showed complete tumor regression after 28 days of treatment with 10 mg/kg JNJ-64619178. Approximately 10–14 days after JNJ-64619178 treatment (4 weeks) cessation, 6/8 mice relapsed and succumbed to the disease within 9–12 weeks. Interestingly, 2 mice did not experience tumor regrowth for up to 25 days post-treatment cessation, suggestive of stable remission (Supplementary Fig. S9B). In all studies, no significant body weight loss was observed (Supplementary Fig. S9C).
The extended inhibitory activity of JNJ-64619178 on PRMT5 previously observed in vitro was assessed in vivo. SmD1/3-Me2 in tumors from mice treated with 10 mg/kg JNJ-64619178, which was reduced by >95% after 6 days of treatment, gradually returned to pre-treatment levels by day 16, 6 days after treatment cessation at day 10 (Fig. 4G). These data suggest that an intermittent dosing schedule with JNJ-64619178 would lead to similar efficacy as continuous daily dosing regimens. Hence, we compared efficacy of continuous daily dosing at 10 mg/kg for 28 days with two cycles of 7 days of treatment followed by 7 days without treatment in the NCI-H1048 xenograft model. The continuous and intermittent dosing schedules led to similar efficacy with %ΔTGIs of 77% and 67%, respectively (Fig. 4H).
These data demonstrated that PRMT5 inhibition by JNJ-64619178 is sustained after compound removal, allowing for intermittent dosing, and thereby reducing overall systemic drug exposure, while maintaining antitumor response.
JNJ-64619178 demonstrates broad antiproliferative activity across multiple tumor types
Inhibition of cell proliferation was assessed at multiple JNJ-64619178 concentrations (10−11 to 10−5 mol/L) against 425 cancer cell lines from 8 cancer indications (Supplementary Table S2). GI50 values were determined after 6 or 7 days of continuous compound exposure under 2-dimensional growth conditions (Fig. 5A). The antiproliferative activity of JNJ-64619178 spanned a wide range (∼10,000-fold) in cell lines from most cancer indications tested. However, the cancer types where the proportion of sensitive cell lines (GI50 ≤10−8 mol/L) were highest included pancreatic (49%), hematological (38%), breast (24%), colon (23%), lung (21%), and ovarian (21%) cancer.
Figure 5.
![Figure 5. Broad antiproliferative activity of JNJ-64619178 in cancer cell lines from multiple cancer types leads to greater percentage of change in non-canonical alternative splicing events in a subset of sensitive cell lines. A, Antiproliferative activity of JNJ-64619178, as defined by GI50 (mol/L) values, were obtained after 6 or 7 days of compound treatment (0.01 pmol/L–10 μmol/L dose response), plotted for each cell line and data grouped under specified cancer indications. Cell lines with ≥50% inhibition of proliferation and a calculated AUC [% inhibition of proliferation/JNJ-64619178 concentration (% inhibition/nmol/L)] ≥100, are depicted with round symbols. Red circles, below the red horizontal dashed line (GI50 ≤ 108 mol/L), highlight the most sensitive cell lines. Cell lines in which inhibition did not reach 50% are indicated by green triangles. The assigned GI50 values correspond to the highest concentration tested. GI50 values were determined for cell lines in which proliferation was inhibited by > 50%, and depicted in purple (GI50 > 10−8 mol/L) or red (GI50 ≤ 10−8 mol/L) circles. The Tukey box plot encompasses only the range of calculated GI50 values and the cross bar within the box represents the median value. The total number of cell lines tested (N) and cell lines for which a GI50 value could be determined (n) are listed. The percentage of sensitive cell lines with GI50 values ≤10−8 mol/L, as a fraction of N, is indicated in red. B, In vitro sensitivity parameters [GI50, maximum effect (Ymax; percentage of inhibition of cell proliferation] and AUC (% inhibition of proliferation/drug concentration; % inhibition/mol/L)] were applied to 295 cancer cell lines treated with JNJ-64619178 and 17 cancer cell lines with maximal differential sensitivity/resistance were selected for analysis. Values for the 3 parameters (GI50, Ymax and AUC) are represented for the most (DAN-G, EFM-19, HCC-78, HCC-1937, HUP-T4, LC-2/ad, NCI-H1435, T-47D, and SW-1990) and least (GP5d, HS578T, LS123, LS1034, NCI-H522, NCI-H2030, NCI-H2405, and PANC1005) sensitive cell lines. C, The percentage of change in non-canonical alternatively spliced events identified by RNA-seq of the 17 selected cell lines, 72 hours after JNJ-64619178 (10 nmol/L) treatment compared with control DMSO-treated cells. Two-tailed t test; *, P < 0.01; **, P < 0.05.](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=9398174_2317fig5.jpg)
Broad antiproliferative activity of JNJ-64619178 in cancer cell lines from multiple cancer types leads to greater percentage of change in non-canonical alternative splicing events in a subset of sensitive cell lines. A, Antiproliferative activity of JNJ-64619178, as defined by GI50 (mol/L) values, were obtained after 6 or 7 days of compound treatment (0.01 pmol/L–10 μmol/L dose response), plotted for each cell line and data grouped under specified cancer indications. Cell lines with ≥50% inhibition of proliferation and a calculated AUC [% inhibition of proliferation/JNJ-64619178 concentration (% inhibition/nmol/L)] ≥100, are depicted with round symbols. Red circles, below the red horizontal dashed line (GI50 ≤ 108 mol/L), highlight the most sensitive cell lines. Cell lines in which inhibition did not reach 50% are indicated by green triangles. The assigned GI50 values correspond to the highest concentration tested. GI50 values were determined for cell lines in which proliferation was inhibited by > 50%, and depicted in purple (GI50 > 10−8 mol/L) or red (GI50 ≤ 10−8 mol/L) circles. The Tukey box plot encompasses only the range of calculated GI50 values and the cross bar within the box represents the median value. The total number of cell lines tested (N) and cell lines for which a GI50 value could be determined (n) are listed. The percentage of sensitive cell lines with GI50 values ≤10−8 mol/L, as a fraction of N, is indicated in red. B, In vitro sensitivity parameters [GI50, maximum effect (Ymax; percentage of inhibition of cell proliferation] and AUC (% inhibition of proliferation/drug concentration; % inhibition/mol/L)] were applied to 295 cancer cell lines treated with JNJ-64619178 and 17 cancer cell lines with maximal differential sensitivity/resistance were selected for analysis. Values for the 3 parameters (GI50, Ymax and AUC) are represented for the most (DAN-G, EFM-19, HCC-78, HCC-1937, HUP-T4, LC-2/ad, NCI-H1435, T-47D, and SW-1990) and least (GP5d, HS578T, LS123, LS1034, NCI-H522, NCI-H2030, NCI-H2405, and PANC1005) sensitive cell lines. C, The percentage of change in non-canonical alternatively spliced events identified by RNA-seq of the 17 selected cell lines, 72 hours after JNJ-64619178 (10 nmol/L) treatment compared with control DMSO-treated cells. Two-tailed t test; *, P < 0.01; **, P < 0.05.
JNJ-64619178 modulates PRMT5-mediated spliceosome activity
Although JNJ-64619178 demonstrated a broad antiproliferative activity in cancer cells from various tumor types, an in vitro sensitivity better than 1.0 nmol/L (GI50, 6 days <1.0 nmol/L) was observed in only a fraction of tumor cell lines (Fig. 5A) suggestive of a specific molecular context predictive of JNJ-64619178 sensitivity. Different molecular mechanisms have been proposed to be associated with sensitivity toward PRMT5 inhibition (32, 33); however, none have been extensively validated so far. We explored the sensitivity to JNJ-64619178 of a large panel (>100) of annotated (DNA an RNAseq) cancer cell lines of different tumor types to identify/validate molecular stratification markers of response. Our analyses demonstrated absence of correlation between the sensitivity to JNJ-64619178 and rate of cell proliferation (Supplementary Fig. S10), multiple reported aberrations such as Fanconi anemia pathway member mutation (34), genomic and transcriptional DDR pathway aberrations (9, 10), CLNS1A/RIOK1 expression ratio (35) or MTAP deletion (15). The latter is not surprising based on JNJ-64619178 mode of binding. Although other reported PRMT5 inhibitors bind either in the SAM pocket or the substrate pocket of PRMT5 (36, 37), JNJ-64619178 occupies both the SAM and substrate-binding sites simultaneously with high affinity and will therefore not bind/inhibit the MTAP deletion cancer cell-specific PRMT5–MTA complex. A correlation between the sensitivity to JNJ-64619178 and general protein synthesis rate could also be excluded because no changes, upon treatment with JNJ-64619178, were observed on protein expression of greater than 50 selected proteins, such as, to only name few: PRMT5 interactors MEP50, RIOK1 and pICIn, known PRMT5 substrates SmD1/3, TP53 and E2F1, as well as SRSF1–4 splicing factors.
Several studies have demonstrated the contribution of PRMT5 to aberrant alternative splicing in different human tumors, a well-recognized hallmark of cancer (12, 13). A possible link between splicing and JNJ-64619178-induced cancer cell death was explored by performing comparative RNA-seq analysis of 9 most and 8 least sensitive pancreatic, breast, and lung cancer cell lines to JNJ-64619178 treatment. These cell lines were selected on the basis of their dichotomic response (most and least sensitive) to JNJ-64619178 predicted using 3 key parameters: GI50 (mol/L), cell killing (Ymax %) at 1 μmol/L, and AUC (percentage of inhibition of proliferation/JNJ-64619178 concentration (% inhibition × mol/L) after 6 days of treatment in 2D growth conditions (Fig. 5B). Exposure to JNJ-64619178 increased the percentage of non-canonical alternatively spliced events, predominantly in the sensitive cell lines and independent of cancer type (Fig. 5C). Enrichment in alternative splicing among the sensitive cell lines was most represented by exon skipping, intron retention and novel cassette splicing events. These data further confirmed a role of PRMT5 in the regulation of the splicing machinery and suggest a link between the pharmacological inhibition of PRMT5 and the antiproliferative phenotype associated with changes in alternative splicing events.
Splicing factor mutations and alterations have been demonstrated to be a key event in tumorigenesis and have been associated with oncogenic dependency in hematological malignancies, such as myelodysplastic syndrome (MDS) and AML (24, 38). JNJ-64619178 also showed high antiproliferative activity in the panel of cell lines belonging to the heme malignancies (Fig. 5A). Therefore, we treated 50 primary AML leukapheresis-derived patient leukemic cells ex-vivo with different concentrations of JNJ-64619178 and assessed IC50 values and maximum growth inhibition after 6 days of continuous compound exposure (Fig. 6A and Supplementary Fig. S11). The antiproliferative activity of JNJ-64619178 as measured by IC50 values, ranged from 0.08 nmol/L for the most sensitive to >100 nmol/L for the insensitive cells (∼10,000-fold difference). On the basis of this analysis whole exome sequencing (WES) was performed at baseline on the 10 most sensitive and 10 most resistant patients derived samples. Overall, 7/10 sensitive primary AML cells versus 1/10 resistant primary AML samples showed mutations in splicing factor genes (Fig. 6B) confirming that PRMT5 inhibition leads to more effective cancer cell death in AML with a dysregulated splicing machinery. The most common splicing factor mutations in the 10 sensitive primary AML samples included SRSF2, U2AF1, SF3A1 and SF3B1 mutations in 40%, 20%, 10% and 10% of sensitive cells, respectively. Only one patient derived sample encoding a mutation in the SRSF2 splicing factor showed resistance to PRMT5 inhibition (Fig. 6C). These data were further confirmed using the CRISPR-IN technology to engineer K562 CML cell lines stably expressing mutant SF3B1 K700E splicing factor. The generated lines were expanded by limiting dilution, and the desired substitution and zygosity confirmed using amplicon-based deep sequencing analysis. The SF3B1 locus is diploid in K562 cells, therefore, a mutant allele frequency and expression of ∼50% reflects heterozygous mutations with biallelic expression, as observed in patients (Fig. 6D). As shown in Fig. 6E (and Supplementary Fig. S12), K562 stably carrying the aberrant SF3B1 K700E mutation were more sensitive to a 6-day JNJ-64619178 exposure than parental K562.
Figure 6.

Mutations in splicing factor (SF) genes confer an increased PRMT5 inhibitor sensitivity in ex vivo primary AML samples and in vitro SF mutant engineered cell lines. In vivo, JNJ-64619178 treatment led to increased aberrant splicing events, whereas no correlation between SF mutations and JNJ-64619178 efficacy was observed in solid tumor PDX models. A, Antiproliferative effect measured as IC50 value (nmol/L) of JNJ-64619178 in 20 primary AML samples assessed after 6 days of treatment. B, Day 6 maximal growth inhibition upon JNJ-64619178 treatment compared with baseline in primary AML samples encoding wild-type (WT) or SF mutations (Mutant). C, Heatmap showing SF mutations detected in JNJ-64619178-sensitive and -insensitive primary AML samples (D) K562 cells from WT clones (49, 154, and 389) and SF3B-K700E CRISPR/CAS9 knockin clones (87 and 150) were DNA-seq and RNA-seq to quantitate SF3B1 WT and isogenic K562-K700E mutant allele frequency (%). E, Growth curves normalized to untreated cells are shown for the SF3B1 WT (blue) and SF3B1-K700E mutant (red) following 6 days of JNJ-64619178 exposure at depicted concentrations. F, Alternative splicing burden of solid tumor PDX models before and after daily JNJ-64619178 treatment for 28 days compared with untreated tumors, measured from RNA-seq analysis for the number of novel splicing events per million mapped reads. F, Seventeen PDXs models from 5 different tumor types encoding WT (open circles) or SF mutation (Mutant, closed circles) were assessed for in vivo sensitivity to JNJ-64619178. In this assay, in vivo efficacy of daily-dosed JNJ-64619178 (10 mg/kg) is represented as the percentage of tumor growth inhibition (% TGI) compared with vehicle control on day 28 treatment. Dash bars across the graph represents 80% and 100% TGI.
Since recurrent splicing factor gene mutations have been reported across 33 different tumor types, including solid tumors (39), we assessed the sensitivity to JNJ-64619178 treatment of solid cancers harboring splicing factor mutations. Multiple PDX models with known high prevalence of splicing factor mutations were selected for in vivo sensitivity assessment to JNJ-64619178 treatment. In total, 17 genomically annotated PDX models were analyzed: lung cancer (n = 5), breast cancer (n = 2), pancreatic cancer (n = 4), colorectal cancer (n = 3), and cholangiocarcinoma (n = 3; with splicing factor mutations and WT controls). The PDXs engrafted mice were randomized to vehicle or JNJ-64619178 (10 mg/kg, for 28 days) treatment, TGI determined and RNA-seq analyses performed. Interestingly, JNJ-64619178 treatment increased the percentage of splicing events, independent of the cancer type (Fig. 6F). However, no evident correlation was observed between intrinsic splicing factor mutation and JNJ-64619178 efficacy (Fig. 6G), indicative of additional tumor features driving JNJ-64619178 sensitivity in solid tumors.
Discussion
Aberrant alternative splicing, a hallmark of cancer, is associated with tumor initiation, progression, and therapeutic resistance (40, 41). PRMT5, a key regulator of splicing (12, 42), is an attractive druggable vulnerability in cancers dependent on tumor-specific aberrant splicing events. Here, we describe the discovery and characterization of a highly potent and selective PRMT5 inhibitor, JNJ-64619178, with robust in vitro cancer antiproliferative activity and in vivo efficacy in multiple human xenograft tumor models. We provide evidence that the antitumor effects of JNJ-64619178 are driven by its unique PRMT5 MOI and associated with the modulation of tumor-specific alternative splicing in AML.
In contrast with other classes of oncology small-molecule targets, such as kinases, the optimal pharmacological features required for PRMT5 modulation are less well understood. Of note, the absence of well-documented specific demethylases to counteract PRMT5-dependent symmetric arginine dimethylation renders this substrate modification irreversible. Instead, PRMT5-dependent arginine methylation effects are reversed by turnover of the substrate proteins. These unique features of PRMT5-dependent effects combined with its long protein half-life, make it pharmacologically desirable to develop potent PRMT5 inhibitors with long-residence time. JNJ-64619178 meets the requirements of such an inhibitor, as described previously in this study. Interestingly, other reported inhibitors have been shown to bind either in the PRMT5 SAM pocket (3, 21–25), the PRMT5 substrate pocket (43, 44), or allosterically (45) whereas JNJ-64619178 and its precursor molecule probe we previously described (26) occupy simultaneously both the SAM and substrate-binding sites with high affinity.
The unique biological properties of JNJ-64619178 may be linked to its sustained PRMT5 inhibitory activity. The longer residence time, high binding-affinity, and extended stabilization (Figs. 1 and 3) of PRMT5 in its inactive state makes JNJ-64619178 as a potent inhibitor of PRMT5 with a unique mechanism and highly optimized pharmacological properties that can be leveraged in clinical development. Sustained PRMT5 inhibition was observed following transient exposure of JNJ-64619178 in multiple cellular assays and comparable efficacy was seen with intermittent (7 days on/off) and continuous daily dosing in mice. These observations suggest that intermittent dosing regimens could be applied in clinical trials to limit bone marrow toxicities associated with PRMT5 inhibition (46). It may also improve recovery between dosing cycles and/or may enable combinations with other agents. A potential reduction in toxicity may be attributed to protein half-life differences between cancer and normal tissues, which have been demonstrated for Myc protein (47).
Considering the key role of PRMT5 in spliceosome assembly and pre-mRNA processing (13, 30), tumors displaying splicing instability are thought to be more sensitive to PRMT5 inhibition. Indeed, ex vivo proliferation assays in primary AML samples confirmed a correlation between the presence of splicing factor mutations and sensitivity to JNJ-64619178, supporting clinical assessment of JNJ-64619178 activity in MDS (precursor to AML; linicaltrials.gov Identifier: NCT03573310).
Although a correlation between splicing factor mutations and efficacy of JNJ-64619178 was observed in AML, such correlation was not observed in solid tumor cell lines or xenograft models tested, suggestive of a fundamental difference in splicing targets and/or regulation in hematological malignancies compared with solid tumors. In fact, recurrent somatic mutations in certain components of the spliceosome, mainly SRSF2, U2AF1, SF3B1 and ZRSR2, have been identified in several types of hematological malignancies, including MDS, other myeloid neoplasms, and chronic lymphocytic leukemia; however, very few recurrent mutations in splicing regulators have been detected in solid tumors (48). Although, we demonstrated that PRMT5 inhibition by JNJ-64619178 in solid tumors led to an increased susceptibility to undergo novel alternative splicing events, particularly exon skipping and retention of mutually exclusive exons, in cell lines and in PDX models. These consequences of PRMT5 inhibition can lead to the formation of novel open-reading frames resulting in neoantigens expression (49), which has the potential of increasing immune cell antigen priming and supports the rationale for combining JNJ-64619178 with immunotherapy agents. These observations and the distinctive features of JNJ-64619178 pharmacology support the continued development of this agent in solid tumors and hematological malignancies driven by aberrant PRMT5 activity.
Authors’ Disclosures
D. Brehmer reports a patent for WO2017/032840, WO2020/245365, and WO2018/154104 issued. L. Beke reports a patent for WO2017/032840 and WO2018/154104 pending; and reports employment with Johnson and Johnson. T. Wu reports a patent for WO2017/032840 and a patent for WO2020/245365 pending. C. Moy reports a patent for WO 2017032840 A1 20170302; and C. Moy reports employment with Janssen Research and Development and shareholder in the parent company (Johnson and Johnson). W. Sun reports a patent for WO2017/032840 pending. G. Mannens reports a patent for WO2020/245365 pending; and reports employment with Johnson and Johnson. V. Pande reports a patent for WO2017/032840 pending; and reports employment with Johnson and Johnson. T. Nys reports employment with Johnson and Johnson. E.M. Gustin reports a patent for WO2017/032840 pending to Johnson and Johnson, WO2020/245365 pending to Johnson and Johnson, and WO2018/154104 pending to Johnson and Johnson; and E.M. Gustin reports employment with Janssen Research and Development and shareholder in the parent company (Johnson and Johnson). B. Verbist reports employment with Janssen Research and Development. L. Zhou reports employment with Janssen Research and Development, and shareholder in the parent company (Johnson &Johnson). Y. Fan reports employment with Janssen Research and Development. V. Bhargava reports employment with Janssen Research and Development and shareholder in the parent company (Johnson and Johnson). P. Vinken reports employment with Janssen Research and Development and shareholder in the parent company (Johnson and Johnson). T. Verhulst reports employment with Janssen Research and Development. T.A. Graubert reports other support from Janssen Pharmaceuticals during the conduct of the study. F. Pastore reports employment with Janssen Research and Development. D. Fiore reports employment with Janssen Research and Development. J. Gu reports employment with Janssen Research and Development and shareholder in the parent company (Johnson and Johnson). A. Johnson reports other support from Janssen during the conduct of the study. U. Philippar reports employment with Johnson&Johnson. D. Walker reports employment with Janssen Research and Development. V. Keersmaekers reports employment with Janssen Research and Development. M. Viellevoye reports employment with Research and Development. G. Diels reports a patent for WO2017/032840 pending; and reports employment with Janssen Research and Development. W. Schepens reports a patent for WO2017/032840 pending; and reports employment with Johnson and Johnson. J.W. Thuring reports a patent for WO2017/032840 pending; and reports employment with Janssen Research and Development and shareholder of the parent company (Johnson and Johnson). L. Meerpoel reports a patent for WO2017/032840 pending to Janssen R&D, WO2020/245365 pending to Janssen R&D, and WO2018/154104 pending to Janssen R&D; and reports employment with Janssen Research and Development, and shareholder in the parent company (Johnson and Johnson). M.V. Lorenzi reports employment with and shareholder of Johnson and Johnson. S. Laquerre reports employment with Jonhson and Johnson. No disclosures were reported by the other authors.
Supplementary Material
Supplementary Figures
Materials and Methods
Supplementary Tables
Acknowledgments
Vaibhav Deshpande (SIRO Clinpharm Pvt. Ltd.) provided medical writing assistance and Namit Ghildyal (Janssen Global Services, LLC) provided additional editorial support for this article. We thank Jorge Vialard (Janssen Research and Development) for reviewing this article. The authors also thank the investigators and partner organizations for their leadership in executing the study. In particular, we acknowledge Sebastian K. Wandinger (Evotec, Munich, Germany) to contribute to the cellular target profile process, and David G. Brown, and Colin Robinson (Charles River Laboratories) to provide support for protein sciences and structural biology. This work was sponsored by Janssen Research and Development, LLC.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
This article is featured in Highlights of This Issue, p. 2315
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Therapeutics Online (http://mct.aacrjournals.org/).
Authors' Contributions
D. Brehmer: Conceptualization, resources, data curation, software, formal analysis, supervision, funding acquisition, methodology, writing–original draft, project administration, writing–review and editing. L. Beke: Data curation, formal analysis, methodology, writing–original draft, writing–review and editing. T. Wu: Conceptualization, formal analysis, visualization, writing–review and editing. H.J. Millar: Conceptualization, resources, data curation, formal analysis, funding acquisition, visualization, methodology, project administration, writing–review and editing. C. Moy: Data curation, validation, visualization, writing–review and editing. W. Sun: Data curation, software, writing–review and editing. G. Mannens: Data curation, formal analysis, project administration, writing–review and editing. V. Pande: Formal analysis, investigation, visualization, writing–review and editing. A. Boeckx: Methodology, project administration, writing–review and editing. E. van Heerde: Data curation, software, methodology, project administration, writing–review and editing. T. Nys: Methodology, writing–review and editing. E.M. Gustin: Formal analysis, visualization, writing–review and editing. B. Verbist: Formal analysis, investigation, visualization, writing–review and editing. L. Zhou: Formal analysis, visualization, writing–review and editing. Y. Fan: Conceptualization, formal analysis, writing–original draft. V. Bhargava: Formal analysis, validation, visualization, writing–review and editing. P. Safabakhsh: Formal analysis, visualization, writing–review and editing. P. Vinken: Formal analysis, validation, writing–review and editing. T. Verhulst: Formal analysis, investigation, visualization, writing–review and editing. A. Gilbert: Formal analysis, investigation, methodology, writing–review and editing. S. Rai: Formal analysis, investigation, methodology, writing–review and editing. T.A. Graubert: Formal analysis, investigation, methodology, writing–review and editing. F. Pastore: Formal analysis, investigation, visualization, writing–review and editing. D. Fiore: Formal analysis, visualization, writing–review and editing. J. Gu: Formal analysis, visualization, methodology, writing–review and editing. A. Johnson: Data curation, formal analysis, writing–review and editing. U. Philippar: Data curation, formal analysis, visualization, writing–review and editing. B. Morschhäuser: Formal analysis, visualization, writing–review and editing. D. Walker: Formal analysis, visualization, writing–review and editing. D. De Lange: Data curation, formal analysis, visualization, methodology, writing–review and editing. V. Keersmaekers: Formal analysis, methodology, writing–review and editing. M. Viellevoye: Formal analysis, visualization, writing–review and editing. G. Diels: Formal analysis, writing–review and editing. W. Schepens: Formal analysis, methodology, writing–review and editing. J.W. Thuring: Formal analysis, visualization, writing–review and editing. L. Meerpoel: Formal analysis, visualization, writing–review and editing. K. Packman: Conceptualization, formal analysis, writing–review and editing. M.V. Lorenzi: Conceptualization, formal analysis, supervision, funding acquisition, visualization, writing–review and editing. S. Laquerre: Conceptualization, formal analysis, supervision, funding acquisition, writing–original draft, writing–review and editing.
References
- 1. Blanc RS, Richard S. Arginine methylation: the coming of age. Mol Cell 2017;65:8–24. [DOI] [PubMed] [Google Scholar]
- 2. Hadjikyriacou A, Yang Y, Espejo A, Bedford MT, Clarke SG. Unique features of human protein arginine methyltransferase 9 (PRMT9) and its substrate RNA splicing factor SF3B2. J Biol Chem 2015;290:16723–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Antonysamy S, Bonday Z, Campbell RM, Doyle B, Druzina Z, Gheyi T, et al. Crystal structure of the human PRMT5:MEP50 complex. Proc Natl Acad Sci U S A 2012;109:17960–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burgos ES, Wilczek C, Onikubo T, Bonanno JB, Jansong J, Reimer U, et al. Histone H2A and H4 N-terminal tails are positioned by the MEP50 WD repeat protein for efficient methylation by the PRMT5 arginine methyltransferase. J Biol Chem 2015;290:9674–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kaushik S, Liu F, Veazey KJ, Gao G, Das P, Neves LF, et al. Genetic deletion or small-molecule inhibition of the arginine methyltransferase PRMT5 exhibit antitumoral activity in mouse models of MLL-rearranged AML. Leukemia 2018;32:499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tarighat SS, Santhanam R, Frankhouser D, Radomska HS, Lai H, Anghelina M, et al. The dual epigenetic role of PRMT5 in acute myeloid leukemia: gene activation and repression via histone arginine methylation. Leukemia 2016;30:789–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang S, Ma Y, Hu X, Zheng Y, Chen X. Targeting PRMT5/Akt signalling axis prevents human lung cancer cell growth. J Cell Mol Med 2019;23:1333–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gyorffy B, Surowiak P, Budczies J, Lanczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non–small cell lung cancer. PLoS ONE 2013;8:e82241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Clarke TL, Sanchez-Bailon MP, Chiang K, Reynolds JJ, Herrero-Ruiz J, Bandeiras TM, et al. PRMT5-dependent methylation of the TIP60 coactivator RUVBL1 is a key regulator of homologous recombination. Mol Cell 2017;65:900–16 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hamard PJ, Santiago GE, Liu F, Karl DL, Martinez C, Man N, et al. PRMT5 regulates DNA repair by controlling the alternative splicing of histone-modifying enzymes. Cell Rep 2018;24:2643–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Meister G, Fischer U. Assisted RNP assembly: SMN and PRMT5 complexes cooperate in the formation of spliceosomal UsnRNPs. EMBO J 2002;21:5853–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bezzi M, Teo SX, Muller J, Mok WC, Sahu SK, Vardy LA, et al. Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev 2013;27:1903–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Radzisheuskaya A, Shliaha PV, Grinev V, Lorenzini E, Kovalchuk S, Shlyueva D, et al. PRMT5 methylome profiling uncovers a direct link to splicing regulation in acute myeloid leukemia. Nat Struct Mol Biol 2019;26:999–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jarrold J, Davies CC. PRMTs and arginine methylation: cancer's best-kept secret? Trends Mol Med 2019;25:993–1009. [DOI] [PubMed] [Google Scholar]
- 15. Marjon K, Cameron MJ, Quang P, Clasquin MF, Mandley E, Kunii K, et al. MTAP deletions in cancer create vulnerability to targeting of the MAT2A/PRMT5/RIOK1 Axis. Cell Rep 2016;15:574–87. [DOI] [PubMed] [Google Scholar]
- 16. Smith CR, Kulyk S, Lawson J, Engstrom LD, Aranda R, Briere DM, et al. Fragment based discovery of MRTX9768, a synthetic lethal-based inhibitor designed to bind the PRMT5-MTA complex and selectively target MTAP/CDKN2A-deleted tumors [abstract]. In: Proceedings of the American Association for Cancer Research Annual Meeting 2021; 2021. Apr 10–15 and May 17–21. Philadelphia (PA): AACR; Cancer Res 2021;81(13_Suppl):Abstract No. LB003. [Google Scholar]
- 17. Bottger A, Islam MS, Chowdhury R, Schofield CJ, Wolf A. The oxygenase Jmjd6—a case study in conflicting assignments. Biochem J 2015;468:191–202. [DOI] [PubMed] [Google Scholar]
- 18. Verhoeven J, De Vleeschouwer F, Kong H, Van Hecke K, Pande V, Sun W, et al. Preparation of 4′-spirocyclobutyl nucleoside analogues as novel and versatile adenosine scaffolds. Chemistry 2019;25:15419–23. [DOI] [PubMed] [Google Scholar]
- 19. Jensen AJ, Martinez Molina D, Lundback T. CETSA: a target engagement assay with potential to transform drug discovery. Future Med Chem 2015;7:975–8. [DOI] [PubMed] [Google Scholar]
- 20. Sharma K, Weber C, Bairlein M, Greff Z, Keri G, Cox J, et al. Proteomics strategy for quantitative protein interaction profiling in cell extracts. Nat Methods 2009;6:741–4. [DOI] [PubMed] [Google Scholar]
- 21. Bonday ZQ, Cortez GS, Grogan MJ, Antonysamy S, Weichert K, Bocchinfuso WP, et al. LLY-283, a potent and selective inhibitor of arginine methyltransferase 5, PRMT5, with antitumor activity. ACS Med Chem Lett 2018;9:612–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lin H, Wang M, Zhang YW, Tong S, Leal RA, Shetty R, et al. Discovery of potent and selective covalent protein arginine methyltransferase 5 (PRMT5) inhibitors. ACS Med Chem Lett 2019;10:1033–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Metz PJ, Ching KA, Xie T, Delgado Cuenca P, Niessen S, Tatlock JH, et al. Symmetric arginine dimethylation is selectively required for mRNA splicing and the initiation of type i and type iii interferon signaling. Cell Rep 2020;30:1935–50. [DOI] [PubMed] [Google Scholar]
- 24. Quiroz RV, Reutershan MH, Schneider SE, Sloman D, Lacey BM, Swalm BM, et al. The discovery of two novel classes of 5,5-bicyclic nucleoside-derived prmt5 inhibitors for the treatment of cancer. J Med Chem 2021;64:3911–39. [DOI] [PubMed] [Google Scholar]
- 25. Smil D, Eram MS, Li F, Kennedy S, Szewczyk MM, Brown PJ, et al. Discovery of a dual PRMT5-PRMT7 inhibitor. ACS Med Chem Lett 2015;6:408–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pande V, Sun W, Beke L, Berthelot D, Brehmer D, Brown D, et al. A chemical probe for the methyl transferase PRMT5 with a novel binding mode. ACS Med Chem Lett 2020;11:2227–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guderian G, Peter C, Wiesner J, Sickmann A, Schulze-Osthoff K, Fischer U, et al. RIOK1, a new interactor of protein arginine methyltransferase 5 (PRMT5), competes with pICln for binding and modulates PRMT5 complex composition and substrate specificity. J Biol Chem 2011;286:1976–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bantscheff M, Eberhard D, Abraham Y, Bastuck S, Boesche M, Hobson S, et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat Biotechnol 2007;25:1035–44. [DOI] [PubMed] [Google Scholar]
- 29. Savitski MM, Reinhard FB, Franken H, Werner T, Savitski MF, Eberhard D, et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 2014;346:1255784. [DOI] [PubMed] [Google Scholar]
- 30. Lee SC, Dvinge H, Kim E, Cho H, Micol JB, Chung YR, et al. Erratum: modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med 2016;22:692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang HT, Zeng LF, He QY, Tao WA, Zha ZG, Hu CD. The E3 ubiquitin ligase CHIP mediates ubiquitination and proteasomal degradation of PRMT5. Biochim Biophys Acta 2016;1863:335–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wu Q, Schapira M, Arrowsmith CH, Barsyte-Lovejoy D. Protein arginine methylation: from enigmatic functions to therapeutic targeting. Nat Rev Drug Discov 2021;20:509–530. [DOI] [PubMed] [Google Scholar]
- 33. Kim H, Ronai ZA. PRMT5 function and targeting in cancer. Cell Stress 2020;4:199–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Du W, Amarachintha S, Erden O, Wilson A, Pang Q. The Fanconi anemia pathway controls oncogenic response in hematopoietic stem and progenitor cells by regulating PRMT5-mediated p53 arginine methylation. Oncotarget 2016;7:60005–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Braun CJ, Stanciu M, Boutz PL, Patterson JC, Calligaris D, Higuchi F, et al. Coordinated splicing of regulatory detained introns within oncogenic transcripts creates an exploitable vulnerability in malignant glioma. Cancer Cell 2017;32:411–26 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li X, Wang C, Jiang H, Luo C. A patent review of arginine methyltransferase inhibitors (2010–2018). Expert Opin Ther Pat 2019;29:97–114. [DOI] [PubMed] [Google Scholar]
- 37. Lin H, Luengo JI. Nucleoside protein arginine methyltransferase 5 (PRMT5) inhibitors. Bioorg Med Chem Lett 2019;29:1264–9. [DOI] [PubMed] [Google Scholar]
- 38. Fong JY, Pignata L, Goy PA, Kawabata KC, Lee SC, Koh CM, et al. Therapeutic targeting of RNA splicing catalysis through inhibition of protein arginine methylation. Cancer Cell 2019;36:194–209 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Seiler M, Peng S, Agrawal AA, Palacino J, Teng T, Zhu P, et al. Somatic mutational landscape of splicing factor genes and their functional consequences across 33 cancer types. Cell Rep 2018;23:282–96 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ladomery M. Aberrant alternative splicing is another hallmark of cancer. Int J Cell Biol 2013;2013:463786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang BD, Lee NH. Aberrant RNA splicing in cancer and drug resistance. Cancers 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Deng X, Gu L, Liu C, Lu T, Lu F, Lu Z, et al. Arginine methylation mediated by the Arabidopsis homolog of PRMT5 is essential for proper pre-mRNA splicing. Proc Natl Acad Sci U S A 2010;107:19114–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Duncan KW, Rioux N, Boriack-Sjodin PA, Munchhof MJ, Reiter LA, Majer CR, et al. Structure and property guided design in the identification of PRMT5 tool compound EPZ015666. ACS Med Chem Lett 2016;7:162–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chan-Penebre E, Kuplast KG, Majer CR, Boriack-Sjodin PA, Wigle TJ, Johnston LD, et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol 2015;11:432–7. [DOI] [PubMed] [Google Scholar]
- 45. Palte RL, Schneider SE, Altman MD, Hayes RP, Kawamura S, Lacey BM, et al. Allosteric modulation of protein arginine methyltransferase 5 (PRMT5). ACS Med Chem Lett 2020;11:1688–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu F, Cheng G, Hamard PJ, Greenblatt S, Wang L, Man N, et al. Arginine methyltransferase PRMT5 is essential for sustaining normal adult hematopoiesis. J Clin Invest 2015;125:3532–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Choi SK, Hong SH, Kim HS, Shin CY, Nam SW, Choi WS, et al. JQ1, an inhibitor of the epigenetic reader BRD4, suppresses the bidirectional MYC–AP4 axis via multiple mechanisms. Oncol Rep 2016;35:1186–94. [DOI] [PubMed] [Google Scholar]
- 48. Anczukow O, Krainer AR. Splicing-factor alterations in cancers. RNA 2016;22:1285–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Park J, Chung YJ. Identification of neoantigens derived from alternative splicing and RNA modification. Genomics Inform 2019;17:e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures
Materials and Methods
Supplementary Tables