

Abstract
Research dating back to the 1950s reported an association between the consumption of saturated fatty acids (SFAs) and risk of coronary heart disease. Recent epidemiological evidence, however, challenges these findings. It is well accepted that the consumption of SFAs increases low-density lipoprotein cholesterol (LDL-C), whereas carbohydrates, monounsaturated fatty acids (MUFAs), and polyunsaturated fatty acids (PUFAs) do not. High-density lipoprotein (HDL)-C increases with SFA intake. Among individuals who are insulin resistant, a low-fat, high-carbohydrate diet typically has an adverse effect on lipid profiles (in addition to decreasing HDL-C, it also increases triglyceride and LDL particle concentrations). Consequently, a moderate fat diet in which unsaturated fatty acids replace SFAs and carbohydrates are not augmented is advised to lower LDL-C; compared with a low-fat diet, a moderate-fat diet will lower triglycerides and increase HDL-C. Now, there is some new evidence that is questioning the health benefits of even MUFAs and PUFAs. In addition, in a few recent studies investigators have also failed to demonstrate expected cardiovascular benefits of marine-derived omega-3 fatty acids. To clarify the clinical pros and cons of dietary fats, the National Lipid Association held a fatty acid symposium at the 2011 National Lipid Association Scientific Sessions. During these sessions, the science regarding the effects of different fatty acid classes on coronary heart disease risk was reviewed.
Keywords: Cardiovascular disease, Coronary heart disease, Monounsaturated fatty acids, Polyunsaturated fatty acids, Saturated fat
Fatty acids are biologically active molecules with a wide array of effects. For decades, fatty acids have been a focus of dietary recommendations for heart health. Historically, saturated fatty acids (SFAs) have been a target for reduction. More recently, this dietary restriction has been extended to trans-fatty acids. In contrast, unsaturated fatty acids have been considered to be heart healthy. Thus, in recent years, dietary recommendations have been made to decrease saturated and trans-fatty acids and to emphasize unsaturated fatty acids (both monounsaturated [MUFAs] and polyunsaturated fatty acids [PUFAs]). Since the early 1970s and thereafter, long-chain omega (n)-3 PUFA (notably eicosapentaenoic acid [EPA] and docosahexaenoic acid [DHA]) have been associated with cardiovascular health benefits.
As the science has evolved, new evidence is challenging these time-honored beliefs. Recent epidemiologic research is showing that SFAs and refined carbohydrates are similarly associated with coronary disease risk. There is some evidence from studies in monkeys that suggests similar atherogenic effects of MUFA and SFA, and reanalysis of the n-6 PUFA clinical studies has raised questions about their health benefits, suggesting that they too may have adverse effects. Finally, although the cardiovascular benefits of marine-derived n-3 fatty acids have been demonstrated previously, in new studies investigators have failed to show some of the expected benefits.
The 2011 National Lipid Association’s Fatty Acid Summit provided an opportunity for experts from across the nation to explore confusing, and oftentimes contradictory, aspects of the dietary fatty acid literature and to inform clinicians and scientists about the controversies. The main question addressed by each speaker was, “How does each fatty acid class impact cardiovascular health and disease?” The conference was organized into examinations of SFAs, MUFAs—oleic acid in particular, n-6 fatty acids—linoleic acid (LA) in particular, and n-3 fatty acids—EPA and DHA in particular. The summaries that follow from the program present opposing viewpoints about the cardiovascular health effects of the different fatty acid classes. The intent of the chairs was for the speakers to engage in a dialetic, ie, a forum for discussing opposing views, rather than a debate. Thus, the goal of the summit was to raise awareness of concepts and controversies regarding fatty acid classes and risk of cardiovascular disease (CVD) and to identify topics for future research to answer questions that have been raised by recent research findings.
The fundamentals of fatty acids
Seth J. Baum, MD, FNLA
University of Miami, Miami, FL, USA
Triglycerides (TG), also known as triacylglycerols, are the primary constituents of vegetable oil and animal fats.1 Glycerol, a polyalcohol, is esterified with one, two, or three fatty acids, resulting in monoglycerides, diglycerides, or TG, respectively. During the intestinal absorption of TG, bile acids emulsify fat to very small fat-containing globules.1 The interaction of lipases with TG results in the formation of monoglycerides and free fatty acids (FFAs). These can then be transported into the enterocyte via micelles. Chylomicrons, TG-containing lipoproteins, are formed within the enterocyte. A key aspect of chylomicron synthesis is the formation of TG from monoglycerides, diglycerides, and FFA.
Fatty acids are classified as saturated or unsaturated on the basis of the absence or presence of double bonds. Fatty acids that have no double bonds are “saturated” with hydrogen atoms, hence the name. MUFAs have one double bond; PUFAs have more than one double bond. Oleic acid is the major dietary MUFA. Also important is the cis- versus trans-configuration of the double bonds because it impacts physical and chemical properties. All double bonds in fatty acids are assumed to be in the cis-configuration unless otherwise noted to be trans-. Natural trans fatty acids occur in limited amounts in beef, lamb, and dairy products, which are the predominant dietary sources. Most are produced during chemical hydrogenation of unsaturated fats when some of the cis-isomers are converted to the trans-configuration instead of undergoing complete hydrogenation.
The nomenclature of n-3, n-6, and n-9 PUFA is determined by the location of the first double bond by counting carbons from the terminal (nth or omega) methyl group.2 Alpha-linolenic acid (ALA; 18:3 n-3) is an 18-carbon n-3 essential fatty acid. The 18 refers to the number of carbon atoms, three refers to the number of double bonds, and n-3 refers to the position on the carbon backbone where the first double bond is located. LA (18:2 n-6) is an 18-carbon n-6 essential fatty acid with two double bonds, and oleic acid (18:1 n-9) is an n-9 fatty acid with just one double bond.
The science regarding the biological effects of fatty acids is complex and constantly evolving (Fig. 1). The long-chain PUFAs most often studied include LA, ALA, arachidonic acid (AA; 20:4 n-6), EPA (20:5 n-3), and DHA (22:6 n-3).3 An important point in Figure 1 is that with dietary conditions typical of developed countries, the conversions of LA to AA and ALA to EPA occur to minimal extents in humans. Previously, it was believed that the conversion of LA to AA and ALA to EPA represented processes wherein there was intense competition for the enzymes of elongation and desaturation.4 However, it is now believed that there is minimal forward conversion of these PUFAs to longer and more unsaturated fats. Instead, retroconversion, ie, the small increase in EPA when DHA is fed, may be more consequential. This retroconversion may or may not be attributable to actual enzymatic production of EPA from DHA.5 It is possible that DHA feeding liberates, or otherwise facilitates, the transfer of EPA from noncirculating depots into the blood. Tracer studies are needed to test the retroconversion hypothesis.
Figure 1.
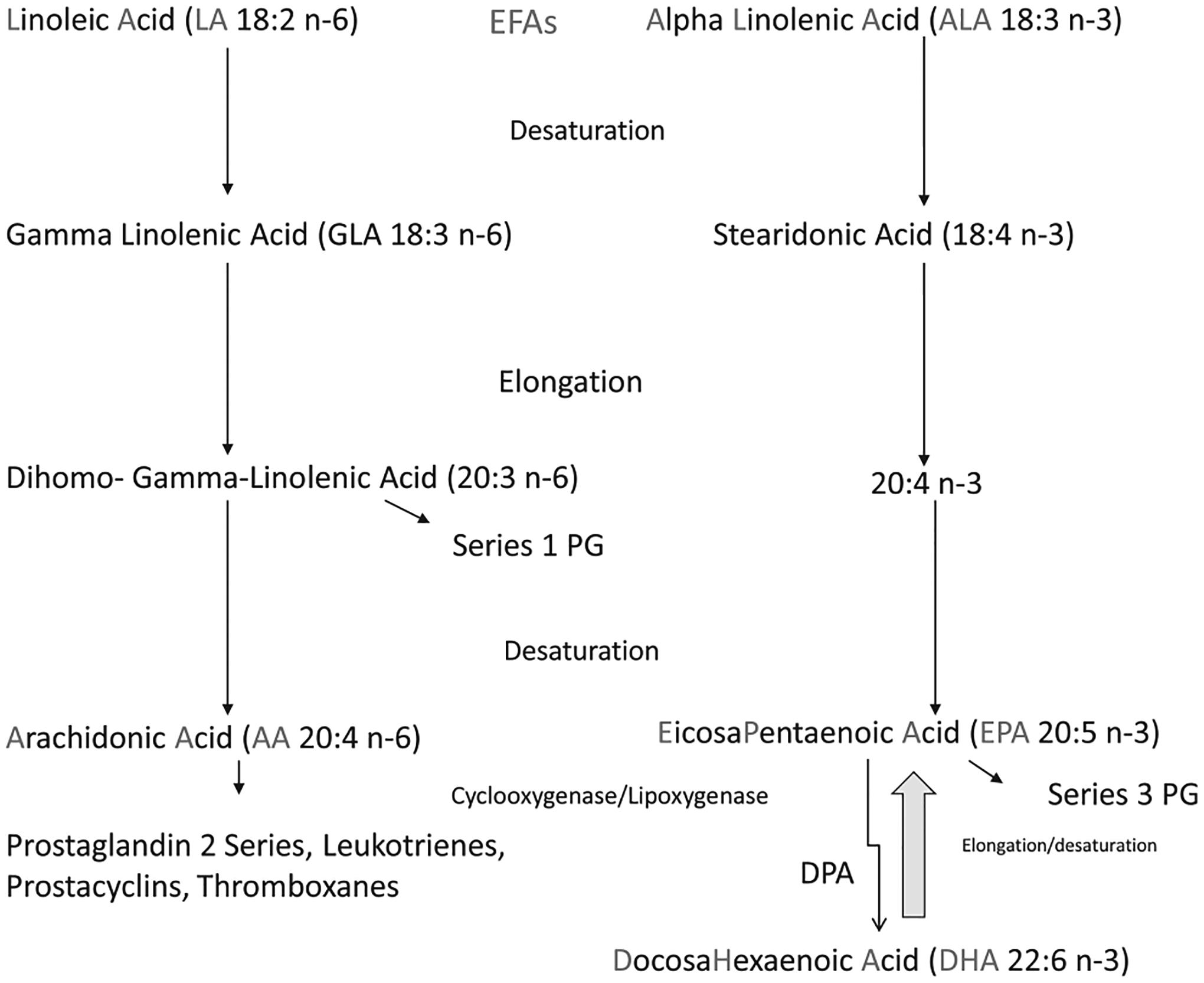
Relationships among the PUFA. EFAs, essential fatty acids; PG, prostaglandin.
AA and EPA are precursors for the eicosanoids, ie, 20-carbon chain compounds, including leukotrienes, prostaglandins, prostacyclins, thromboxanes, and lipoxins.6 DHA and EPA are the predominant long chain n-3 fatty acids in fish oil. They are taken up by cell membranes, where they increase membrane fluidity, regulate gene expression, modulate ion channels, and enhance pinocytosis. EPA is involved in eicosanoid formation, and DHA is involved in the formation of docosanoids.
There are a variety of EPA and DHA pharmaceutical and dietary supplement products, including those in ethyl ester, TG, FFA, and phospholipid forms. The manufacturing of fish oils initially results in an unpurified oil, which must be cleansed and concentrated.7 The maximum quantity of n-3s that can be obtained in unconcentrated oil is ~30% because, at most, only one of the positions on the glycerol backbone is occupied by an n-3 fatty acid. To achieve EPA + DHA concentrations greater than 30%, some manufacturers add alcohol and potassium hydroxide to cleave the fatty acids and form ethyl esters of DHA and EPA, which can then be separated and concentrated. The only pharmaceutical n-3 product currently approved by the Food and Drug Administration contains n-3 acid ethyl esters and is indicated for the treatment of TG ≥500 mg/dL. The TG form of n-3 fatty acids can also be concentrated through a lengthy process that requires re-esterifying the concentrated EPA and DHA ethyl esters back to a glycerol backbone. A FFA form may soon be available as an enteric-coated pharmaceutical product, and the phospholipid form of n-3 fatty acids found in krill oil is also available in dietary supplements.8
The saturated fat and dietary carbohydrate debate: are they “one and the same” relative to cardiovascular risk?
Walter C. Willett, MD, DrPH
Harvard School of Public Health, Boston, MA, USA
This presentation provides an overview of the available evidence on the topic of SFA and carbohydrates in relation to CVD risk. Fifteen years ago, this debate would have been considered impossible because it was believed that SFA intake was the primary determinant of the high rates of CVD in Western countries. However, in recent years, that question has been reexamined, or, more accurately, seriously examined for the first time. In truth, there was not very good epidemiological evidence for this relationship from the beginning.
At a recent meeting in Copenhagen addressing the topic of SFA versus carbohydrate intake in relation to CVD,9 the following issues were discussed: (1) To what is SFA being compared? (2) Do specific fatty acids have different effects on heart disease risk, and should the focus be more on food sources of SFA, recognizing that pure saturated fats or pure unsaturated fats are never consumed? (3) Have the effects of SFA versus carbohydrate changed as obesity has increased, and should the effects of SFA on stroke, cancer, and other health outcomes be considered in recommendations? (4) What types of evidence are sufficient to guide recommendations?
Although randomized controlled trials with clinical end points would be ideal to answer all, or even one, of these questions, such trials are difficult to conduct and often are not feasible. The best-available evidence will likely come from a combination of controlled feeding studies with intermediate end points such as blood lipids, blood pressure, inflammation, and platelet aggregation, in combination with large prospective, observational studies in which investigators examine the relationship between intakes and clinical outcomes.
Several large cohort studies examining diet and health outcomes have been conducted. The Nurses’ Health Study enrolled 121,000 women in 1976 and dietary data collection started in 1980.10 The database on diet is continually updated, now every 4 years, because the food supply and composition are always changing, particularly for manufactured products like margarines and shortenings, and people’s dietary preferences are continually evolving. Intakes of different types of fatty acids were examined in relation to risk of coronary heart disease (CHD), specifically acute myocardial infarction (MI) or fatal CHD. After 14 years, ~1000 such incident events had occurred. When carbohydrates were used for comparison and the changes in risk with increasing percentages of energy from different types of fatty acids were examined, we found that trans-fat was most strongly related to a greater risk of CHD. SFA intake was nonsignificantly related when compared calorie-for-calorie with carbohydrate. MUFAs, and more so, PUFAs (n-6, LA), were associated with a lower risk of CHD. Thus, the tradeoff of SFA versus carbohydrates appeared to be a wash in this study. The best option to reduce risk appears to be the replacement of trans-fat and some SFA with a combination of MUFAs and PUFAs. Again, as far as SFAs are concerned, after 20 years of follow-up in the Nurses’ Health Study, there was a flat dose-response relationship between SFA and CHD risk.11
Results from a recent pooled analysis of large cohort studies indicated that, when compared calorie-for-calorie, there was a statistically significant greater relative risk for CHD with carbohydrate versus SFA (Fig. 2).12 When the comparison was made between n-6 PUFA and SFA, PUFA intake was associated with a statistically significant lower CHD risk. This result was similar to the original Nurses’ Health Study, ie, that the relation of SFA intake to CHD risk depended on the comparator.
Figure 2.
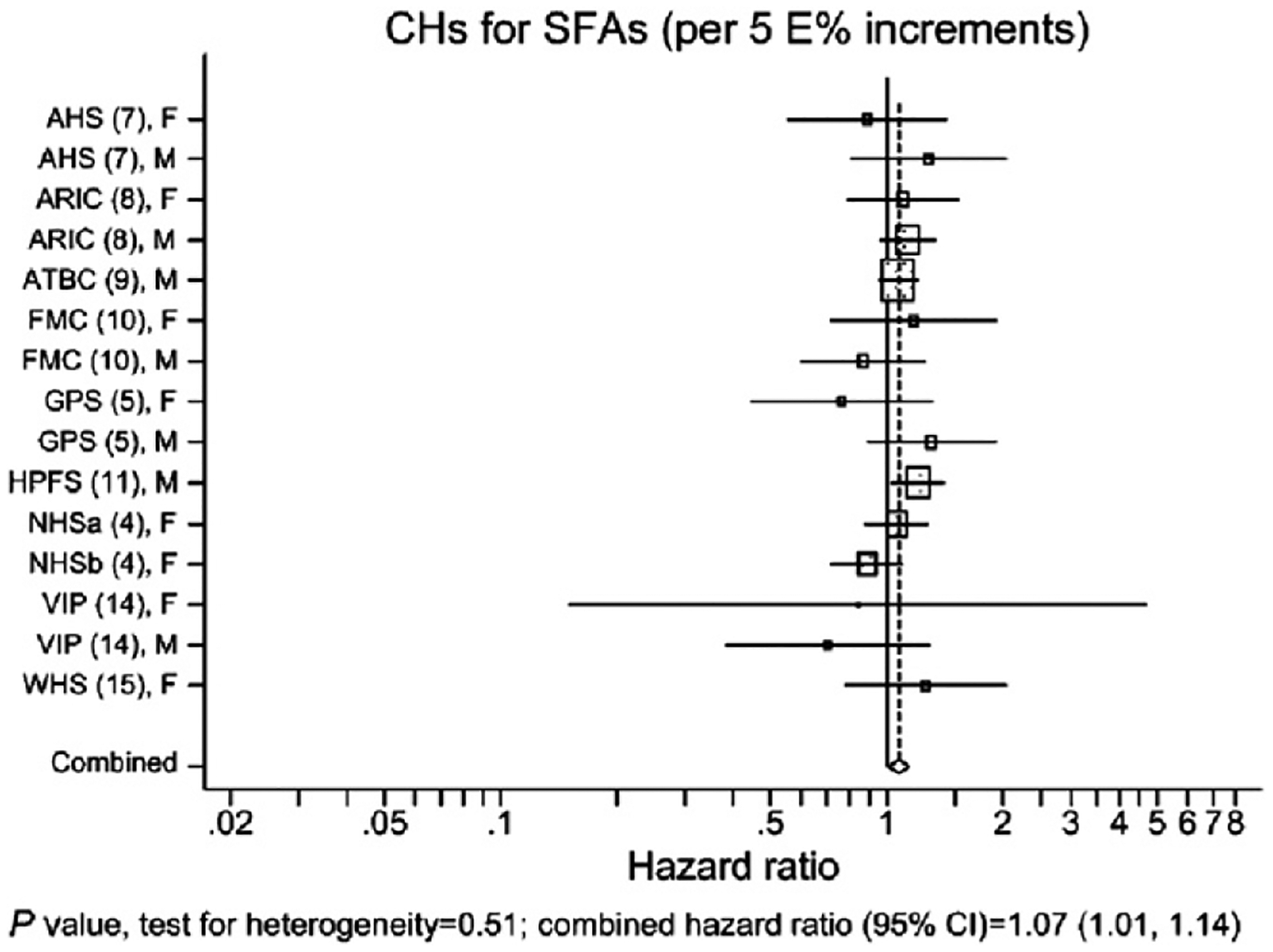
Coronary deaths in the Pooling Project of Cohort Studies on Diet and Coronary Disease. The model included intake of MUFA, PUFA, trans-fatty acids, carbohydrates, and protein expressed as percentages of total energy, fiber, alcohol, and cholesterol intakes; smoking; body mass index; physical activity and educational levels; history of hypertension; and ages at baseline and when questionnaire was returned. Within each study, hazard ratios with 95% CIs for the incidence of coronary events from CHD were calculated by the use of Cox proportional hazards regression with time in study as the time metric. The study-specific logs of hazard ratios were weighted by the inverse of their variances, and a combined estimate of the hazard ratios was computed by using a random effects model. The squares and horizontal lines represent the study-specific hazard ratios and 95% CIs, respectively. The area of the squares reflects the study-specific weight (inverse of the variance). The diamonds represent the combined hazard ratios and 95% CI. AHS, Adventis Health Study; ARIC, Atherosclerosis Risk in Communities Study; ATBC, Alpha-Tocopherol and Beta-Carotene Cancer Prevention Study; CH, carbohydrate; FMC, Finnish Mobile Clinic Health Study; GPS, Glostrup Population Study; HPFS, Health Professionals Follow-Up Study; NHSa, Nurses’ Health Study 1980; NHSb, Nurses’ Health Study 1986; VIP, Västerbotten Intervention Program; WHS, Women’s Health Study. Permission to reuse figure granted by American Society for Nutrition.12
To briefly summarize the metabolic evidence, a seminal study conducted by Mensink and Katan13 showed that reducing SFA and replacing it with carbohydrate resulted in decreased high-density lipoprotein (HDL)-C and increased fasting TG, raising concerns about an increased risk of CHD. Later, they conducted a meta-analysis of controlled feeding studies in which they compared SFAs, MUFAs, and PUFAs versus carbohydrates.14 For HDL-C, all types of fat compared with carbohydrate elevated HDL-C, and, as expected, SFA increased low-density lipoprotein (LDL)-C. However, because SFA intake also increased HDL-C, it had little effect on the LDL-C/HDL-C ratio (a metric that is considered to be a better predictor of CHD risk than LDL-C alone). This finding was consistent with the epidemiologic evidence showing that the exchange of SFA for carbohydrate is a wash in relation to CHD risk. However, replacement of carbohydrate with either MUFA or PUFA, or replacement of SFA with either MUFA or PUFA, substantially improved the LDL-C/HDL-C ratio. For TG, all types of fat, except trans fat, produced about the same response—a reduction compared with carbohydrate.
Because pure SFAs, MUFAs, or PUFAs are never consumed in the real world, Mensink and Katan15 conducted another meta-analysis in which they evaluated the impact on the total C/HDL-C ratio of oils in the forms we typically consume. The best types of fat, in terms of improving the lipid ratio, were canola (rapeseed) oil, soybean oil, and olive oil, whereas the worst types were butter and stick margarine. Not surprisingly, all types of fat were better than pure SFA because even the worst of them do contain some unsaturated fats. There is limited epidemiological evidence on CHD as a function of specific types of SFA, in part, because the different types of SFA are highly correlated and it is difficult to assess independent relationships.
It should be noted that although the aforementioned comparisons were with carbohydrates, not all carbohydrates are the same in terms of composition and relationship with clinical end points. Approximately one-half of the calories in the United States diet are from carbohydrates; the majority in the forms of sugar, refined starch, and starch from potatoes. Therefore, any comparisons with carbohydrates in epidemiologic studies are inherently comparisons with unhealthful choices. In a recent study by Jakobsen et al16 in which they compared different forms of carbohydrates with SFAs, high glycemic index carbohydrates were more strongly associated with risk of CHD than was SFA intake. However, low- or medium-glycemic index carbohydrates, compared with SFAs, were not associated with increased risk.
Thus, the relationship of SFA intake to CVD risk is complex and depends to a great degree on the comparator. The available evidence suggests that replacing SFA with MUFA and PUFA may be beneficial while replacing SFA with carbohydrate may not lower risk or may increase risk if the replacement is with high glycemic index forms of carbohydrate.
An argument for displacing saturated fat from the diet: benefits depend on what replaces it
Alice H. Lichtenstein, DSc
Tufts University, Medford, MA, USA
The objectives of this presentation are to address issues related to the temporal association between dietary fat and CVD rates, dietary fat type and CVD outcomes, and the translation of scientific findings to messages for patients and the general public. Since approximately 1970, the incidence of CVD has decreased dramatically. It is of interest to parse what contributed to this decline, ie, more sophisticated medical modalities, or improved risk factors contributed to by changes in lifestyle. Focusing on the United States, and following the trends in three time frames (1968–1976, 1980–1990, and recent years), one can see that lifestyle modification has had a large effect.17 Regarding lifestyle changes, dietary fat type, and specifically the balance between SFAs and unsaturated fatty acids, is an area that has received substantial attention.
One of the first publications that advised a decrease in SFA intake was published in 1961 by the American Heart Association.18 At that time, there was also a recommendation to decrease total fat intake; the latter recommendation was dropped in the year 2000. Why was there a recommendation to decrease SFA intake 50 years ago? Some of the support came from observational studies, including the iconic Seven-Countries Study of Keys et al, which suggested a positive relationship between SFA intake and CHD death, although there were other differences among the populations.19 One way to examine observational data regarding diet and CHD is to calculate dietary PUFA to SFA (P/S) ratios, thus simultaneously accounting for both dietary components. In the Nurses’ Health Study, a greater P/S ratio was associated with lower relative risk of CHD.20 It is also possible to calculate the expected change in total-C from a change in dietary SFA and PUFA using the predictive equations generated by Keys et al21 and Hegsted et al.22 Both concluded that decreasing SFA intake had about twice the effect to lower total-C as increasing PUFA, and that MUFA was relatively neutral.
A limited number of intervention studies have examined dietary SFA intake and CHD outcomes. It is prohibitively expensive to carry out such studies, and it is unlikely we will see new ones in the near future. Hence, it is important to carefully consider the historical data while acknowledging that some of the methodology is not consistent with current standards. Of note, a common thread among these studies is that not only was SFA intake reduced but PUFA intake was increased. One of the first studies was the Medical Research Council soybean oil study, a secondary CHD prevention study in which SFA were displaced with soybean oil, high in PUFA.23 Among those surviving free of relapse, subjects in the soybean oil group fared better than those in the control (SFA) group. The primary and secondary prevention Los Angeles Veterans Diet Study administered a diet relatively high in total fat (40% of energy) with unsaturated fat, predominately PUFA, tripled at the expense of SFA. Again, fewer events were reported with the intervention diet.24 The secondary prevention Oslo Diet Heart Study also demonstrated that a diet lower in SFA and greater in PUFA resulted in better outcomes.25 However, it should be noted that the increase in PUFA intake was attributable not only to LA but also n-3 fatty acids as described in detail by Dr. Ramsden later in this report.
Investigators from the primary prevention Finnish Mental Hospital Study reported fewer deaths from CHD and lower rates of MI in a hospital that administered dairy products in which SFAs were replaced with PUFA (soybean oil) compared with regular SFA-containing dairy products. With 6 years per treatment phase and the use of a crossover study design, this was probably the longest dietary crossover trial that ever has been conducted.26,27 In the secondary prevention Leiden Intervention Trial, less narrowing in coronary artery vessel diameter was demonstrated in individuals who were compliant with dietary instructions to increase the P/S ratio to greater than two.28
The investigators for the intervention component of the primary prevention Minnesota Coronary Survey reported that increasing the relative amount of PUFA at the expense of SFA did not result in a difference between the control (39% total fat; 18% SFA, 5% PUFA, 16% MUFA, 446 mg cholesterol/d) and intervention groups (38% total fat; 9% SFA, 15% PUFA, 14% MUFA, 166 mg cholesterol/d) with respect to CHD events at 4.5 years follow-up.29 Of note, however, is that 62% of the study subjects were younger than 60 years of age, and the study authors indicated that they suspected a significant reduction might have been evident if the treatment period had been longer in persons in the age range most likely to benefit. The St. Thomas’ Atherosclerosis Regression Study assessed the impact of diet on reversing atherosclerosis, by displacing SFA with PUFA.30 Individuals with the greatest regression were those who reported consuming the least amount of SFA.
Why have questions re-emerged about whether dietary SFA is or is not related to CHD? Starting in 2009, a number of reports have concluded that there is a weak association between either SFA or major foods that contain SFA—meat and milk—and CHD risk.31–33 Strong inverse associations between the incidence of CHD and the Mediterranean diet, a high-quality diet, and a Prudent Heart diet have also been reported, as well as a positive relationship between a Western diet and CHD,31 suggesting that dietary patterns should be the focus rather than individual components of the diet.
As an example of the confusing reports, in one article the authors stated there was no significant association of CHD risk with SFA intake33; however, in a separate article in the same journal issue, the same authors stated “… in summary, although substitution of dietary polyunsaturated fat for saturated fat has been shown to lower CVD risk, there are few epidemiologic or clinical trial data to support the benefit of replacing saturated fat with carbohydrate.”34 In another report, investigators, using pooled data, came to a similar conclusion.12 These findings were corroborated in a separate analysis of the same data (Fig. 3).35 Collectively, these reports emphasize the importance of accounting for not only what is decreased in the diet but also other components of the diet that are altered and even augmented as a consequence of the primary change.
Figure 3.
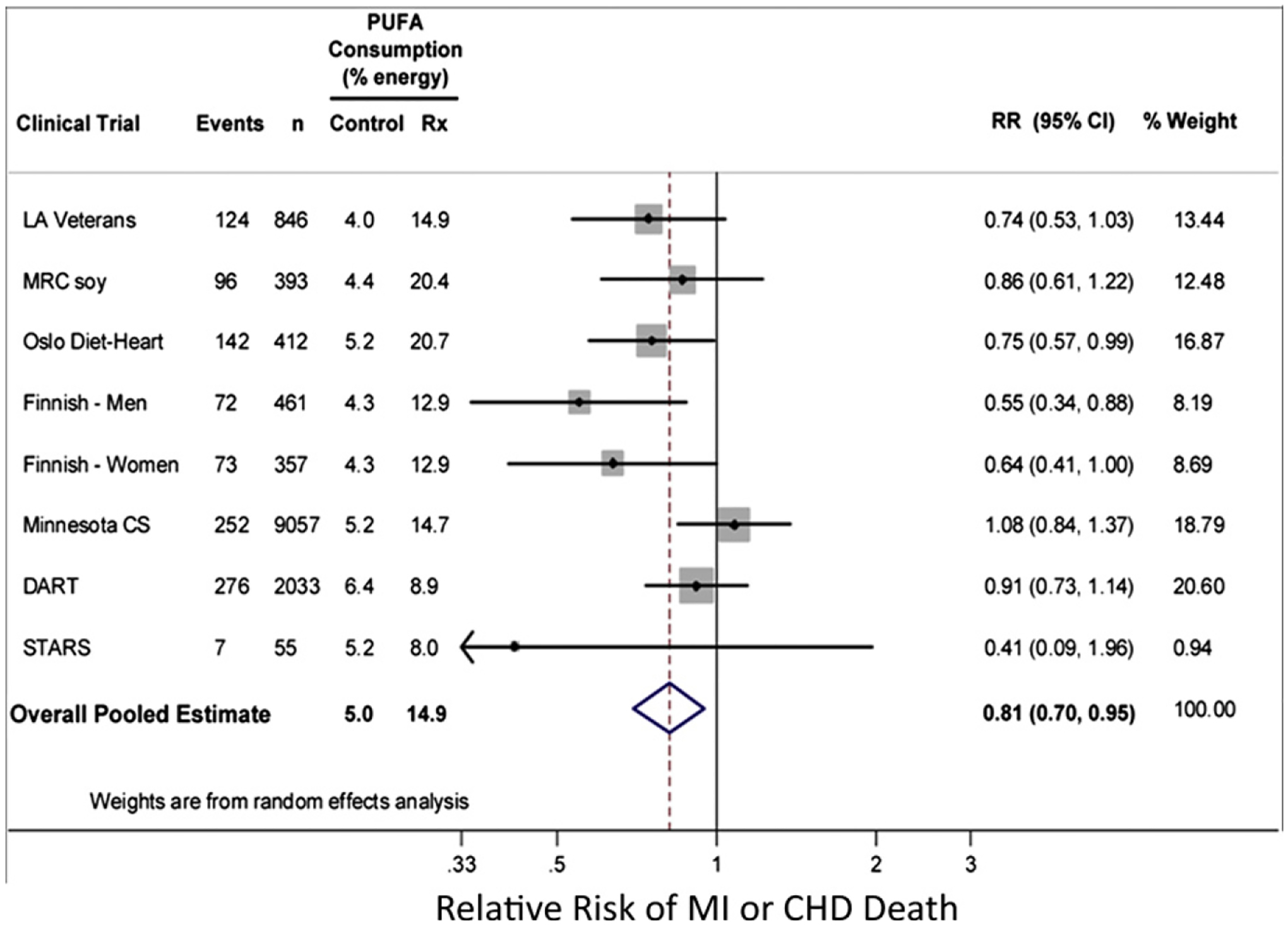
Meta-analysis of randomized controlled trials evaluating effects on CHD events of increasing polyunsaturated fat in place of saturated fat. CS, Coronary Survey; DART, Diet And Reinfarction Trial; LA, Los Angeles; MRC, Medical Research Council; RR, relative risk; Rx, treatment; STARS, St. Thomas’ Atherosclerosis Regression Study. Permission to reuse figure granted by PLoS Medicine.35
This brings the discussion to the last point—message translation. On the basis of available data, the clear message medical professionals should be communicating for both primary and secondary prevention is to displace SFA with unsaturated fats, primarily PUFAs. Although that was the intended message before the 1990s, the message became mutated to “eat a low-fat diet.” This is an appropriate message for major dietary sources of SFA such as meat and milk. Switching from full-fat milk to nonfat milk eliminates the SFA and cuts the calories in half. However, changes such as exchanging butter with sugar in baked products may decrease the SFA, but the price associated with this variation is an increased carbohydrate content of the diet as well as a potentially neutral impact on calories.
A further mutation of the low-fat message is that “low-fat” became synonymous with “low-calorie,” giving the false impression that a product labeled low-fat is automatically also low in calories. As a result, in the United States, there was a proliferation of fat-free and low-fat cookies, brownies, ice creams, and cakes. And even worse, there was a proliferation of products in which PUFA was removed from the food, as often occurs in salad dressings, and replaced with refined carbohydrate, frequently sugar. What was the result of these changes? The carbohydrate content of the diet increased at the expense of the unsaturated fats. Such changes can promote dyslipidemia (low HDL-C, high TG, high LDL particle concentration), especially in overweight and obese individuals. Therefore, it is important to remember that all messages must be embedded within the overarching dictum—to achieve and maintain energy balance.
MUFA—Mediterraneans love olive oil, but is it our friend or foe?
Lawrence L. Rudel, PhD
Wake Forest University School of Medicine, Winston-Salem, NC, USA
According to the lipid hypothesis for atherogenesis, apolipoprotein (apo) B-containing lipoproteins enter the artery wall, accumulate, are oxidized, and become ligands for scavenger receptors on macrophages. They are taken up by and accumulate within macrophages, converting them into foam cells. In this way, the process of atherogenesis begins. HDLs are the cholesterol efflux promoters. When an imbalance between influx and efflux occurs, more foam cells form and atherosclerosis progresses. Not only is the concentration of apo B-containing lipoproteins dramatically affected by the types of fat consumed, but their composition also is affected, and these changes can affect the atherogenicity of the particles.
An old assumption regarding fatty acids and their effects on atherosclerosis was that MUFAs were neutral, SFAs were bad, and PUFAs were good. However, a study conducted by Scott Grundy and Fred Mattson in 1985 turned the “world of MUFA” around.36 They demonstrated that diets rich in SFAs caused a high LDL-C/HDL-C ratio, and that substitution of MUFA for SFA reduced LDL-C but did not reduce HDL-C. Consequently the LDL-C/HDL-C ratio was the lowest with MUFA, given that PUFA reduced HDL-C as well as LDL-C.
Starting with the Seven Countries Study, other authors have shown that in Mediterranean countries versus the United States, CHD mortality rates are appreciably lower, even correcting for plasma cholesterol.37 Many assume that this difference is because of MUFA, specifically olive oil, in the Mediterranean diet. However, the Mediterranean diet differs from the United States diet in many ways.
When diets rich in MUFA versus PUFA are consumed, even for only a few weeks, plasma LDL cholesterol ester fatty acid composition changes dramatically. On a MUFA-rich diet, cholesteryl oleate can become up to 32% of the cholesterol esters, and cholesteryl linoleate comprises roughly 45%. When LA-rich fats are consumed, cholesteryl linoleate increases from 45% to 64%, and cholesteryl oleate decreases from 32% to 13.8%. Why is the effect on cholesterol ester composition important? Data from the Atherosclerosis Risk in Communities study showed that greater carotid artery intimal medial thickness was associated with the highest quartile of the percentage of circulating saturated cholesterol esters.38 The same was also true for cholesteryl oleate; the greatest quartile was associated with greater intimal medial thickness. The inverse was true for cholesteryl linoleate. Data from the Uppsala Longitudinal Study of Adult Men showed a pattern of circulating cholesterol ester percentages similar to those in the aforementioned study: greater mortality was associated with cholesteryl oleate and lower mortality was associated with cholesteryl linoleate (Table 1).39 Collectively, these data suggest that enrichment of plasma lipoproteins with cholesteryl oleate, as promoted by dietary MUFA, may off-set the benefit of an improved LDL-C/HDL-C ratio.
Table 1.
Proportional hazard ratios for fatty acids in serum cholesteryl esters in relation to CVD and total mortality
| Fatty acid and estimated desaturase activities | Scrum proportion* | Cardiovascular disease mortality† (n = 461 events) | Total mortality† (n = 1012 events) | ||
|---|---|---|---|---|---|
| Crude | Adjusted‡ | Crude | Adjusted‡ | ||
| Myristic acid (14:0) | 1.1 ± 0.3 | 1.16 (1.06, 1.27) | 1.12 (1.02, 1.23) | 1.09 (1.03, 1.16) | 1.06 (0.99, 1.13) |
| Palmitic acid (16:0) | 11.7 ± 0.99 | 1.25 (1.14, 1.37) | 1.15 (1.04, 1.26) | 1.16 (1.09, 1.24) | 1.09 (1.02, 1.17) |
| Palmitoleic acid (16:1) | 3.5 (3.0–4.4) | 1.32 (1.21, 1.44) | 1.18 (1.07, 1.30) | 1.28 (1.21, 1.36) | 1.19 (1.11, 1.27) |
| Stearic acid (18:0) | 1.1 (0.97–1.3) | 1.07 (0.97, 1.17) | 1.04 (0.94, 1.15) | 1.01 (0.95, 1.08) | 0.98 (0.91, 1.04) |
| Oleic acid (18:1) | 19.4 ± 2.7 | 1.29 (1.18, 1.41) | 1.18 (1.07, 1.30) | 1.25 (1.18, 1 33) | 1.17 (1.10, 1.25) |
| Linoleic acid (18:2n–6) | 53.9 ± 5.2 | 0.76 (0.70, 0.83) | 0.85 (0.78, 0.94) | 0.80 (0.76, 0.85) | 0.87 (0.81, 0.93) |
| γ-Linolenic acid (18:3n–6) | 0.71 ± 0.30 | 1.15 (1.05, 1.27) | 1.09 (0.98, 1.21) | 1.08 (1.01, 1.15) | 1.04 (0.97, 1.11) |
| α-Linolenic acid (18:3n–3) | 0.66 ± 0.16 | 1.08 (0.99, 1.18) | 1.10 (1.0, 1.21) | 1.03 (0.97, 1.09) | 1.03 (0.97, 1.10) |
| Dihomo-γ-linolenic acid (20:3n–6) | 0.57 ± 0.13 | 1.16 (1.07, 1.27) | 1.06 (0.96, 1.18) | 1.07 (1.01, 1.14) | 1.0 (0.94, 1.08) |
| Arachidonic acid (20:4n–6) | 4.8 ± 0.93 | 1.00 (0.92, 1.10) | 0.95 (0.86, 1.05) | 0.98 (0.92, 1.04) | 0.96 (0.90, 1.02) |
| Eicosapentaenoic acid (20:5n–3) | 1.3 (0.9–1.6) | 1.07 (0.98, 1.17) | 0.99 (0.90, 1.09) | 1.04 (0.98, 1.11) | 1.0 (0.94, 1.08) |
| Docosahexaenoic acid (22:6n–3) | 0.68 (0.56–0.81) | 0.97 (0.89, 1.07) | 0.92 (0.84, 1.02) | 0.96 (0.90, 1.02) | 0.95 (0.89, 1.02) |
| SCD (16:1/16:0) | 0.30 (0.26–0.37) | 1.27 (1.16, 1.39) | 1.15 (1.04, 1.27) | 1.26 (1.18, 1.34) | 1.18 (1.10, 1.26) |
| D6D (18:3n–6 /18:2n–6) | 0.012 (0.009–0.017) | 1.20 (1.10, 1.32) | 1.12 (1.0, 1.24) | 1.12 (1.06, 1.19) | 1.07 (1.0, 1.14) |
| D5D (20:4n–6/20:3n–6) | 8.6 ± 2.0 | 0.84 (0.76, 0.93) | 0.88 (0.80, 0.98) | 0.92 (0.86, 0.98) | 0.96 (0.89, 1.02) |
CVD, cardiovascular disease; FAs, fatty acids; SCD, steroyl-CoA-desaturated; D6D, Δ6-desaturase; D5D, Δ5-desaturase.
Values are means ± SD or means (interquartile ranges).
Values are hazard ratios (95% confidence intervals).
The adjusted model included total-C, body mass index, smoking, physical activity, and hypertension.
Permission to reuse table granted by American Society for Nutrition.39
The following is a discussion of studies conducted in animal models to examine the lipid hypothesis for atherogenesis, with particular focus on fatty acid intakes. The lipid and coronary artery atherosclerosis responses to diets providing 35% of energy as fat (predominantly SFA [palm oil], MUFA [high in oleinate from genetically modified safflower oil], or PUFA [high in LA]) plus cholesterol to boost the amount of atherosclerosis, were examined in male African green monkeys (n = 15 per group) studied for 5 years.40 The lipid effects in the monkeys were the same as those reported by Mattson and Grundy in their study of San Diegoveterans.36 In the monkey studies, average HDL-C was 50 mg/dL in the PUFA group versus 86 and 81 mg/dL in the SFA and MUFA groups, respectively.40 Average plasma LDL-C concentrations in the PUFA and MUFA-fed monkeys were 157 and 167 mg/dL, respectively (no significant difference between them) versus 257 mg/dL in the SFA-fed animals.
The largest effects of these diets were in the LDL cholesterol ester fatty acid composition.40 With PUFA feeding, 70% of cholesterol ester was cholesteryl linoleate; when MUFA oleic acid was fed, 70% of the cholesterol ester was cholesteryl oleate; and the SFA diet resulted in predominantly cholesteryl oleate with a greater percentage of cholesteryl palmitate as well. Because only shorter trials in humans have been conducted to date, it is unknown whether such dramatic changes as those shown in the monkeys would also occur in humans.
Atherosclerotic lesions were circumferential in the SFA- and MUFA-fed monkeys, but were rarely so in the PUFA-fed monkeys.40 The average intimal area for all coronary arteries in each monkey (measured from 16 serial cross sections of heart) showed similar extents of coronary artery atherosclerosis in the SFA- and MUFA-fed animals, whereas monkeys fed PUFA had less atherosclerosis. The cholesterol oleate concentration in the right coronary artery of the MUFA- and SFA-fed monkeys was essentially the same, whereas PUFA-fed monkeys had less accumulation.
Collectively these data showed that despite MUFA-fed monkeys having a lower LDL-C/HDL-C ratio, there was no difference in the amount of atherosclerosis between the SFA- and MUFA-fed monkeys, and only the monkeys fed PUFA were protected against coronary artery atherosclerosis. It was suspected that the high cholesteryl oleate composition of plasma LDL may have influenced this outcome. An examination of the livers of these animals poststudy indicated that the MUFA-fed animals had twice as much cholesteryl ester accumulation compared with monkey livers from the other two groups.41 Isolated liver perfusion yielded a strong correlation (r = 0.8) between the extent of coronary artery atherosclerosis and perfusate cholesterol ester secretion rate.
Acetyl-coenzyme A acetyltransferase 2 (ACAT2) esterifies hepatic cholesterol and is responsible for secretion of cholesterol esters, primarily cholesteryl oleate. In view of the data on hepatic perfusion, it can be hypothesized that dietary MUFA promoted hepatic ACAT2-catalyzed cholesteryl oleate secretion into lipoproteins, leading to enhanced atherogenicity. The aortic cholesterol ester concentration in LDL receptor null, apo-B100–only mice with and without ACAT2 was similar to that seen in the African green monkeys.42 The percentage of cholesterol ester as cholesteryl oleate in mice without ACAT2 was dramatically reduced and replaced by cholesterol esters with PUFA. Furthermore, ACAT2 gene deletion limited the severity of aortic atherosclerosis independent of the type of fat that was fed. These data from two animal models are consistent with the hypothesis that stimulation of hepatic ACAT2-mediated cholesteryl oleate secretion by dietary MUFA can promote atherosclerosis.
In conclusion, the results from the animal studies described herein suggest that the kind of fat consumed impacts the amount of atherosclerosis that develops. Studies in nonhuman primates and mice have shown that dietary MUFA does not protect any better than SFA against the development of coronary artery atherosclerosis. Instead, it is dietary PUFA that offers protection. ACAT2 is the presumed mediator of these fatty acid effects, a point that remains to be proven in humans. Finally, the protection offered by the Mediterranean diet is likely attributable to the many differences in the diet other than olive oil.
Monounsaturated fatty acid intake and atherosclerotic cardiovascular disease risk
Kevin C. Maki, PhD
Biofortis-Provident Clinical Research, Addison, IL, USA
The National Cholesterol Education Program Third Adult Treatment Panel introduced the Therapeutic Lifestyle Changes (TLC) diet, which was more restrictive regarding SFA and cholesterol than the previous Step I diet.43 However, total fat intake was liberalized from <30% to up to 35% of calories, with an emphasis on unsaturated fats, primarily from MUFA (up to 20% total calories) and secondarily PUFA (up to 10% total calories). There are several sources of MUFA in the diet, including nuts and nutlike foods (eg, macadamia, almonds, pistachios, peanuts, and hazelnuts), seeds, and vegetable oils (Table 2).44 Although sometimes underappreciated, foods containing animal fats such as cheddar cheese, whole eggs, and ground beef also contain a high percentage of MUFA (28%–69% of the fatty acids per serving).
Table 2.
Fatty acid content of selected foods and oils44
| Food | Serving size | Total | SFA | MUFA | PUFA |
|---|---|---|---|---|---|
| Fat (g) | |||||
| Nuts and Nutlike Foods | |||||
| Macadamia | 1 oz | 21.6 | 3.4 | 16.8 | 0.4 |
| Almonds | 1 oz | 15.7 | 1.2 | 9.9 | 3.8 |
| Pistachios | 1 oz | 12.9 | 1.6 | 6.8 | 3.9 |
| Peanuts | 1 oz | 14.0 | 1.9 | 6.9 | 4.4 |
| Walnuts | 1 oz | 18.5 | 1.7 | 2.5 | 13.4 |
| Hazelnuts | 1 oz | 17.0 | 1.3 | 12.8 | 2.2 |
| Other foods | |||||
| Avocado | 1 | 19.9 | 2.9 | 13.3 | 2.5 |
| Cheddar cheese | 1 oz | 9.3 | 5.9 | 2.6 | 0.3 |
| Whole egg | 1 medium | 5.0 | 1.6 | 1.9 | 0.7 |
| 95% lean ground beef | 0.25 lb | 5.4 | 2.4 | 2.2 | 0.3 |
| Oils | |||||
| High oleic safflower | 6.2 | 74.6 | 14.4 | ||
| Olive | 13.8 | 73.0 | 10.5 | ||
| Canola | 7.0 | 61.0 | 32.0 | ||
| Peanut | 16.9 | 46.2 | 32.0 | ||
| Corn | 12.9 | 27.6 | 54.7 | ||
MUFA, monounsaturated fatty acids; PUFA, polyunsaturated fatty acids; SFA, saturated fatty acids.
Decades of clinical studies in humans have demonstrated that modification of the fatty acid content of the diet can affect the lipoprotein/lipid profile. For example, LDL-C increases when carbohydrate in the diet is replaced with either trans-fatty acids or SFAs (Fig. 4).45 Conversely, LDL-C is reduced when carbohydrates are replaced with MUFAs or PUFAs, although the effect is more pronounced with PUFA.45 HDL-C is reduced by the addition of trans-fatty acids to the diet, but replacement of carbohydrate with SFAs, MUFAs, or PUFAs will increase HDL-C, with MUFA having an intermediate effect between those of SFA and PUFA. The total-C/HDL-C ratio is raised by trans-fatty acid intake but there is a relatively neutral effect with SFAs because they increase both total-C and HDL-C. Reductions in the total-C/HDL-C ratio occur with both PUFAs and MUFAs.45 It is therefore critical to take into account the nutrient being replaced when considering the lipid effects of fatty acids.
Figure 4.
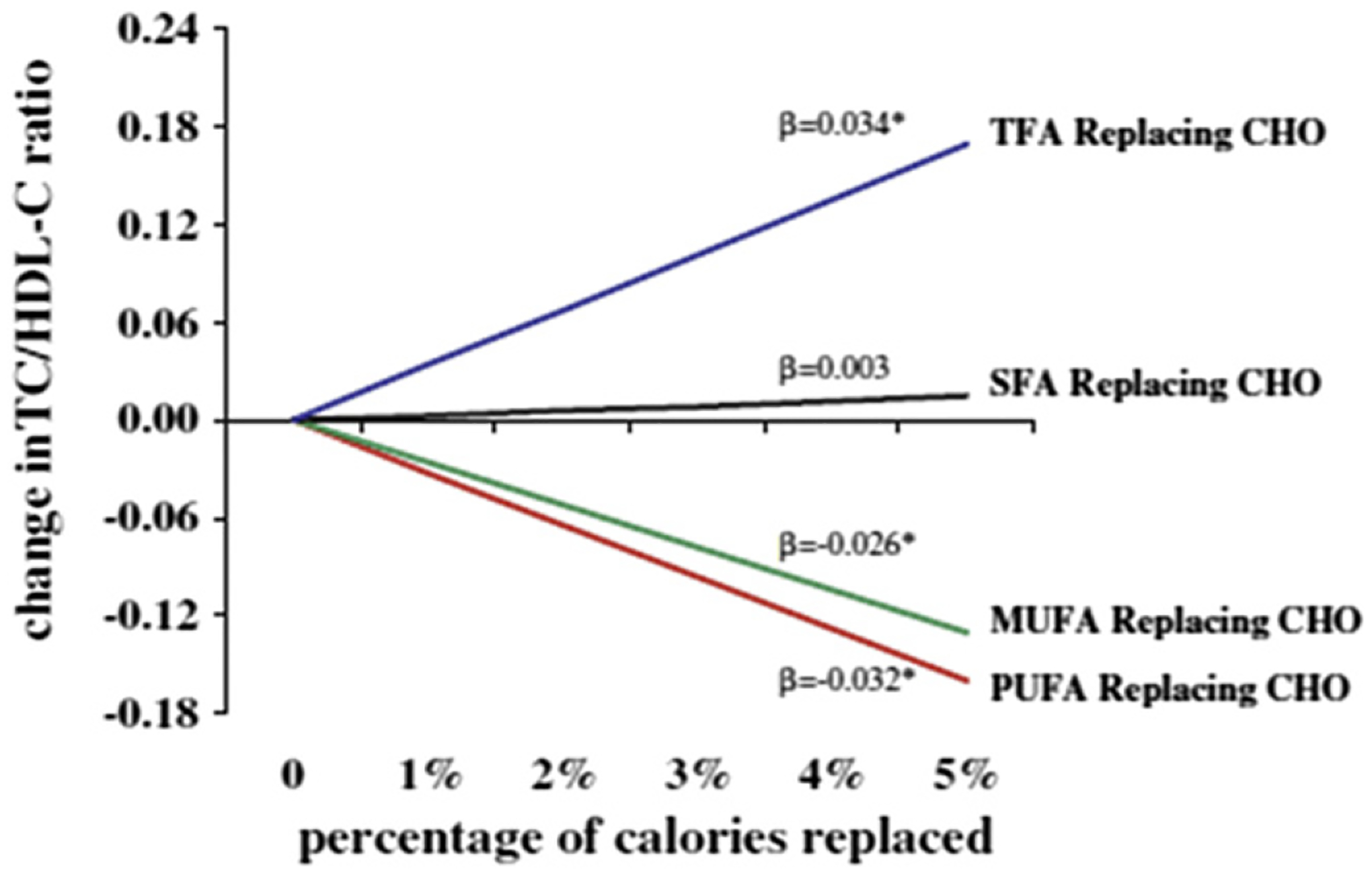
Changes in blood lipid levels following replacement of carbohydrate with various fats. β reflects the change for each 1% energy isocaloric replacement. *P <.05. CHO, carbohydrate; TC, total cholesterol; TFA, trans-saturated fatty acids. Permission to reuse figure granted by Springer.45
Data from diet intervention trials have demonstrated relationships between shifts in dietary fatty acid intakes and changes in cardiovascular risk markers. Jenkins et al46 placed subjects with hypercholesterolemia on a 1-month TLC diet lead-in followed by one of two dietary portfolios for 1 month, both of which were high in soy protein and viscous fiber and designed to be either high (26% energy) or low (13% energy) in MUFA. At the conclusion of the lead-in period, the HDL-C concentrations declined; and when subjects were switched to the high MUFA diet, there was a 12.5% increase in HDL-C that was not present with the low MUFA diet. This was accompanied by an increase in apo A1 concentrations, suggesting a possible increase in the number of HDL particles. LDL-C was reduced following the TLC lead-in diet and continued to decline following both the high- and low-MUFA diets. There were no differences between the two diets in LDL-C or apo B concentrations, indicating no difference in the number of atherogenic particles. Interestingly, high sensitivity C-reactive protein was significantly reduced by the high MUFA diet relative to the lower MUFA diet, a finding that merits additional research.
The results from this and other dietary intervention trials suggest that reductions in LDL-C, non-HDL-C, and apo B occured when MUFAs replaced SFAs in the diet.47–49 When MUFAs replace carbohydrates in the diet, TG, very low-density lipoprotein-C, high-sensitivity C-reactive protein, and blood pressure decrease, while HDL-C and apo A1 increase.48,49 These changes suggest that increased MUFA intake should reduce the risk of cardiovascular events. However, caution is warranted because results from studies in nonhuman primates and mice, as reviewed by Dr. Rudel, have suggested that dietary MUFA compared to SFA intake may not protect against the development of coronary artery atherosclerosis, despite favorable changes in serum lipoprotein lipids.40,42
There is little clinical trial evidence available regarding MUFA intake and CHD outcomes, but in epidemiological studies researchers have examined this relationship. In the Nurses’ Health Study, modeling an isocaloric replacement of 5% of energy from SFA with carbohydrate was associated with a nonsignificant reduction in CHD risk (Fig. 5).10 When carbohydrates were substituted for MUFAs, CHD risk was increased, whereas when MUFAs were substituted for SFAs there was an apparent reduction in risk. However, such results are challenging to interpret because MUFA intake is highly correlated with SFA intake (correlation coefficient of 0.81 in the Nurses’ Health Study data) and is moderately correlated with intakes of PUFA (correlation coefficient 0.30) and trans-fatty acids (correlation coefficient 0.55).
Figure 5.
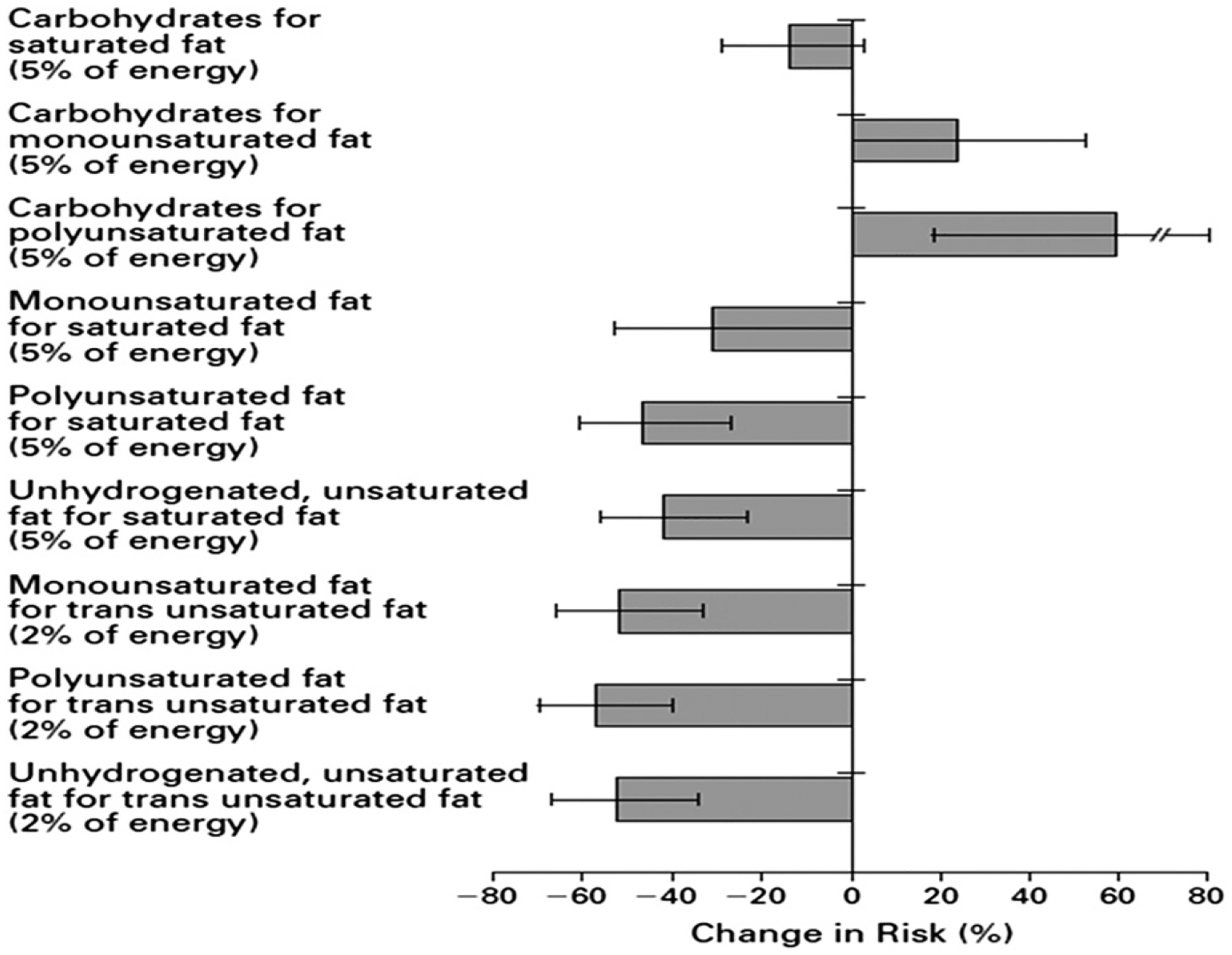
Estimated percent changes in CHD risk with isocaloric substitutions of dietary components: The Nurses’ Health Study. Permission to reuse figure granted by New England Journal of Medicine, Massachusetts Medical Society.10
Tanasescu et al50 evaluated the relationships between specific dietary fatty acids, cholesterol, and CHD risk in 5672 women with type 2 diabetes in the Nurses’ Health Study. The age-adjusted relative risk (RR) between the greatest (>19.9% total energy) and lowest (<13.4% total energy) quintiles of MUFA intakes was 1.22 (95% confidence interval [95% CI] 0.95–1.56). In the multivariate analysis (adjusted for age; smoking; postmenopausal hormone use; parental history of MI before age 60 years; moderate/vigorous activities; body mass index; total caloric, protein, fiber, and alcohol intakes; and multivitamin, vitamin E supplement, and medication use), the association was attenuated (RR 1.10, 95% CI 0.82–1.46), and further adjustments (for intakes of SFA, PUFA, and trans unsaturated fats and cholesterol) led to a reversal in the direction of the association (RR 0.84, 95% CI 0.53–1.34). Although these relationships were not significantly different from unity, the pattern illustrates the difficulties encountered when modeling highly correlated dietary exposures to assess their associations with disease risk.
Mente et al31 evaluated the relationships between various dietary factors and CHD risk and found that a Mediterranean diet was associated with a lower incidence of coronary outcomes, as were diets consisting of a high intake of nuts and MUFA. There are several potential protective components of the Mediterranean diet, and this diet, as well as diets rich in nuts, are typically greater in MUFAs. Other investigations on nut intake have demonstrated benefits on the lipid profile. An analysis of 25 intervention trials including 583 men and women with normal or elevated lipids showed that a mean intake of 67 g/d nuts led to reductions in total-C, LDL-C, and the total-C/HDL-C ratio.51 Pooled data from four cohort studies also reported a significantly lower risk for CHD mortality associated with higher nut intake.51
Mozaffarian et al’s35 review and meta-analysis of randomized controlled trials showed that replacement of SFA with PUFA was associated with lower predicted CHD risk (RR 0.91, 95% CI 0.87–0.95), based on changes in the total-C/HDL-C ratio; (Fig. 6). This finding was consistent with the protective effect observed in a pooled analysis of data from 11 observational cohort studies (RR 0.87, 95% CI 0.77–0.97).35 When MUFAs were substituted for SFAs in clinical trials, there was also a predicted benefit based on changes in the total-C/HDL-C ratio (0.93, 95% CI 0.89–0.96). The pooled analysis of observational cohort data suggested a trend in the opposite direction (RR 1.19, 95% CI 1.00–1.42),35 although, as illustrated by the previous example, the point estimates can be markedly influenced by adjustment for other correlated dietary components, so caution in interpretation of such results is warranted.
Figure 6.
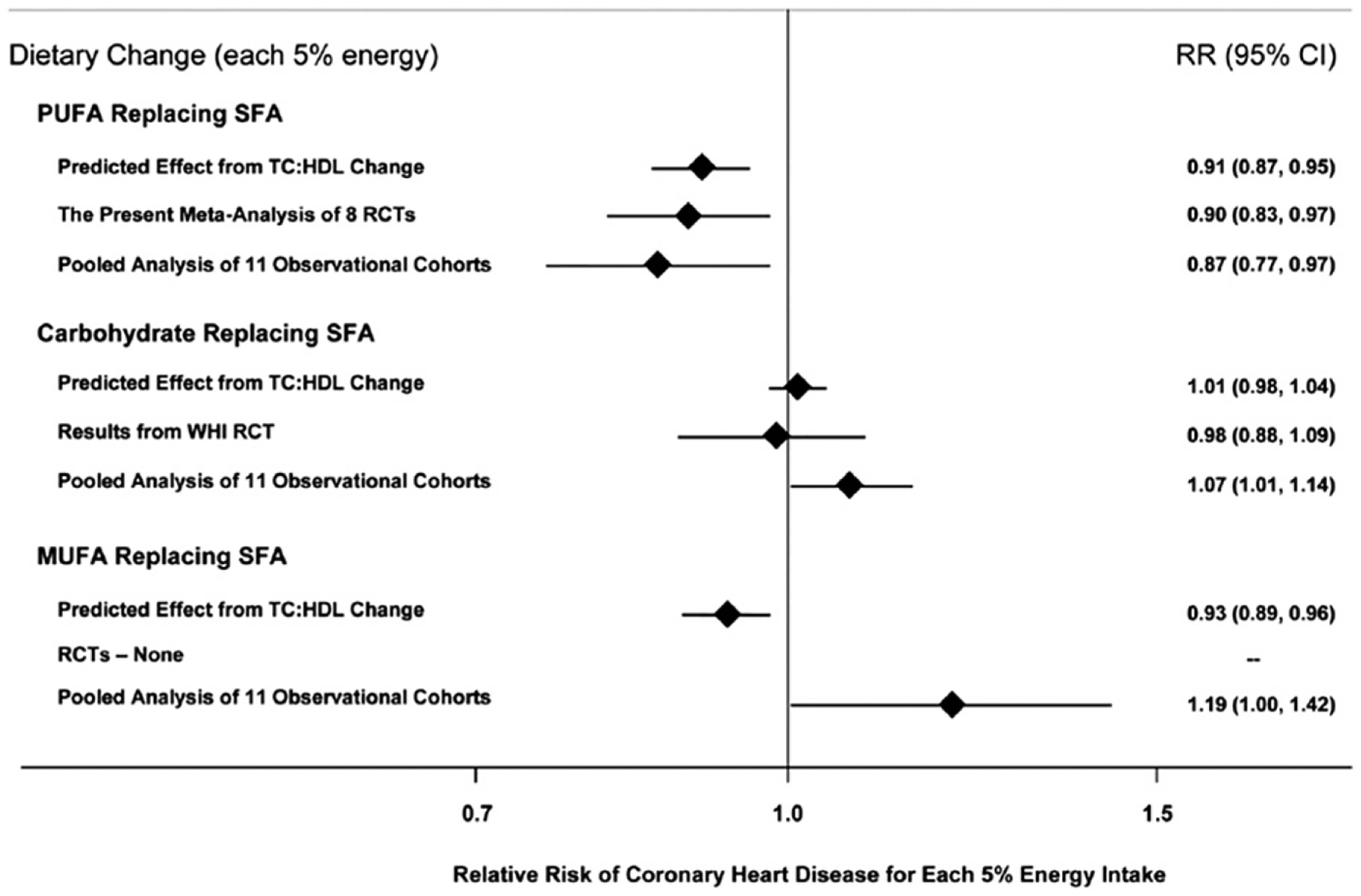
Effects on CHD risk of consuming PUFA, carbohydrate, or MUFA in place of SFA. Predicted effects were determined by changes in the TC/HDL-C ratio in short-term trials (eg, each 5% energy of PUFA replacing SFA lowers TC/HDL-C ratio by 0.16) coupled with observed associations between the TC/HDL-C ratio and CHD outcomes in middle-aged adults. Evidence for effects of dietary changes on actual CHD events came from the meta-analysis of eight randomized controlled trials for PUFA replacing SFA and from the Women’s Health Initiative randomized controlled trial for carbohydrate replacing SFA. Evidence for observed relationships of usual dietary habits with CHD events was from a pooled analysis of 11 prospective cohort studies. RCT, randomized controlled trials; RR, relative risk; TC, total cholesterol; WHI, Women’s Health Initiative. Permission to reuse figure granted by PLoS Medicine.35
Other factors should also be taken into account when considering the relationship between MUFA intake and atherosclerotic CVD risk. For example, MUFAs coexist with SFAs in many foods. In addition, cis- and trans-isomers of MUFAs were sometimes categorized together in epidemiological studies. Studies that specifically assessed trans-fatty acid intake sometimes characterized them poorly, which may have led to residual confounding. Possible effects of the other nutrients in foods being replaced and what is substituted for the displaced foods also have to be considered. Furthermore, the specific food sources of MUFA may influence the results. For example, relatively unrefined olive oil retains several lipophilic components, whereas highly refined olive oil has a low level of some of these potentially bioactive compounds. The net effect of a particular dietary change may also be modified by characteristics of the population being studied. Individuals with insulin resistance, metabolic syndrome, diabetes, or dyslipidemia may respond differently from those who do not have these conditions. Therefore, in light of the uncertainty regarding the relationship between consumption of specific fatty acids and CVD risk, the most prudent recommendation in the author’s view is a dietary pattern that emphasizes whole grains, legumes, nuts and oils, fruits, vegetables, fish, lean meats, and low-fat dairy products, with sparing consumption of refined grains, white rice, potatoes, stick margarines, shortenings, sugar-sweetened sodas, confectionary products, desserts, high-fat meat and high-fat dairy products.52,53
Dietary n-6 PUFA and CVD risk
Jay Whelan, PhD, MPH
University of Tennessee, Knoxville, TN, USA
LA is the parent compound for all n-6 PUFAs. When consumed, LA can be theoretically converted to and enrich tissues with AA via the rate limiting Δ6-desaturase enzyme. AA subsequently can be converted to bioactive eicosanoids, which have been linked to processes involved in the development of cancer, CVD, and inflammation.
Elevated LDL-C and LDL particle concentrations contribute to the atherosclerotic process by injuring the vascular endothelium resulting in lipid deposition and necrosis. It is well established that LDL-C is a risk factor for CVD, and reducing LDL-C decreases CVD risk.
In a study of 1098 United States white, Japanese American, Japanese, and Korean men, greater serum LA concentrations were significantly associated with lower concentrations of LDL particles.54 An inverse relationship between dietary PUFA and CVD was recently confirmed in a review by Astrup et al,9 who summarized the evidence from epidemiologic, clinical, and mechanistic studies in which authors consistently reported that replacement of SFA with PUFA reduced the risk of CHD. In a 15-year study of 1551 white men, energy-adjusted dietary intakes of PUFA and LA in the upper tertile were associated with more than a 50% lower risk for CVD mortality.55 In the 30-year Uppsala Longitudinal Study, the authors reported that CVD mortality was lower for individuals with cholesterol ester LA levels above versus below the median.39 Similarly, investigators from the Nurses’ Health Study reported that increasing intakes of LA corresponded with a gradual reduction in CHD risk, and risk for MI declined with higher levels of LA in serum lipids.10
The American Heart Association Scientific Advisory recommends an n-6 fatty acid or LA intake of 5% to 10% of calories, based primarily on a review of four ecological, two case control, 18 prospective cohort, and 10 randomized controlled trials in which authors concluded that consumption of 5% to 10% of energy from LA or n-6 fatty acids reduced the risk of CHD relative to intakes below 5%.56 In a review of 12 prospective cohorts and 9 randomized controlled trials, Czernichow et al57 concluded that the body of evidence supports the recommendation of n-6 PUFA intake greater than 5%, and ideally near 10% of energy, for reducing risk. Furthermore, consumption below the current lowest values of LA in France, 4% of energy, was not recommended.
At intakes of 12 to 17 g/d, LA is the major PUFA in the United States diet. ALA intake is typically in the range of 1.1 to 1.6 g/d, resulting in an n-6/n-3 ratio of approximately 10:1. When LA consumption increases, consumption of ALA also increases.58 Soybean oil is the main contributor of dietary LA in the United States; 88% of PUFA in soybean oil is LA. The intake of soybean oil (n-6/n-3 ratio of approximately 8:1) dramatically increased beginning around 1970.59 Interestingly, with this huge influx of LA, the mortality rates of CHD precipitously declined.60
One criticism of the aforementioned observational studies is that people do not eat LA in isolation. Typically, n-3 PUFAs, including ALA, accompany LA in the diet. Therefore, a more controlled study to evaluate the effects of LA, separate from n-3, on CHD risk was needed. In an evaluation of previous data, studies were divided into an n-6-only group, and an n-3/n-6 mix in which the LA content of the diet was between 5- and 20-fold greater than the n-3 content.61 There was no significant increase in nonfatal MI risk observed in the n-6-only group, and some reduced risk occurred in the n-3/n-6 group. With regard to relative risk for CHD death, there was no significant risk reduction reported in either of the groups.
A major concern with dietary LA consumption is that it theoretically enriches tissues with AA with subsequent generation of excessive amounts of pro-inflammatory eicosanoids.62–64 To further address the potential adverse mechanism associated with increased LA intake, conversion of LA to AA and subsequent increased eicosanoid production, Rett and Whelan examined 36 papers, identified from more than 4300, and asked: If intakes of LA were increased or decreased from typical dietary intakes, would AA in tissue phospholipids change?65 It was observed that increasing dietary LA levels by up to six-fold, or decreasing them by up to 90%, did not change plasma/serum AA content (Fig. 7).65 These data were consistent with those published by Liou and Innis66 in a randomized crossover study of men consuming a 1% ALA diet containing either 4% or 10% LA. Changing LA content had little or no effect on AA content in plasma phospholipids. In fact, if anything, an inverse relationship was detected; as LA increased, AA levels decreased. In earlier studies, Lands67 also showed little change in AA content between 4% and 10% LA consumption in the United States.
Figure 7.
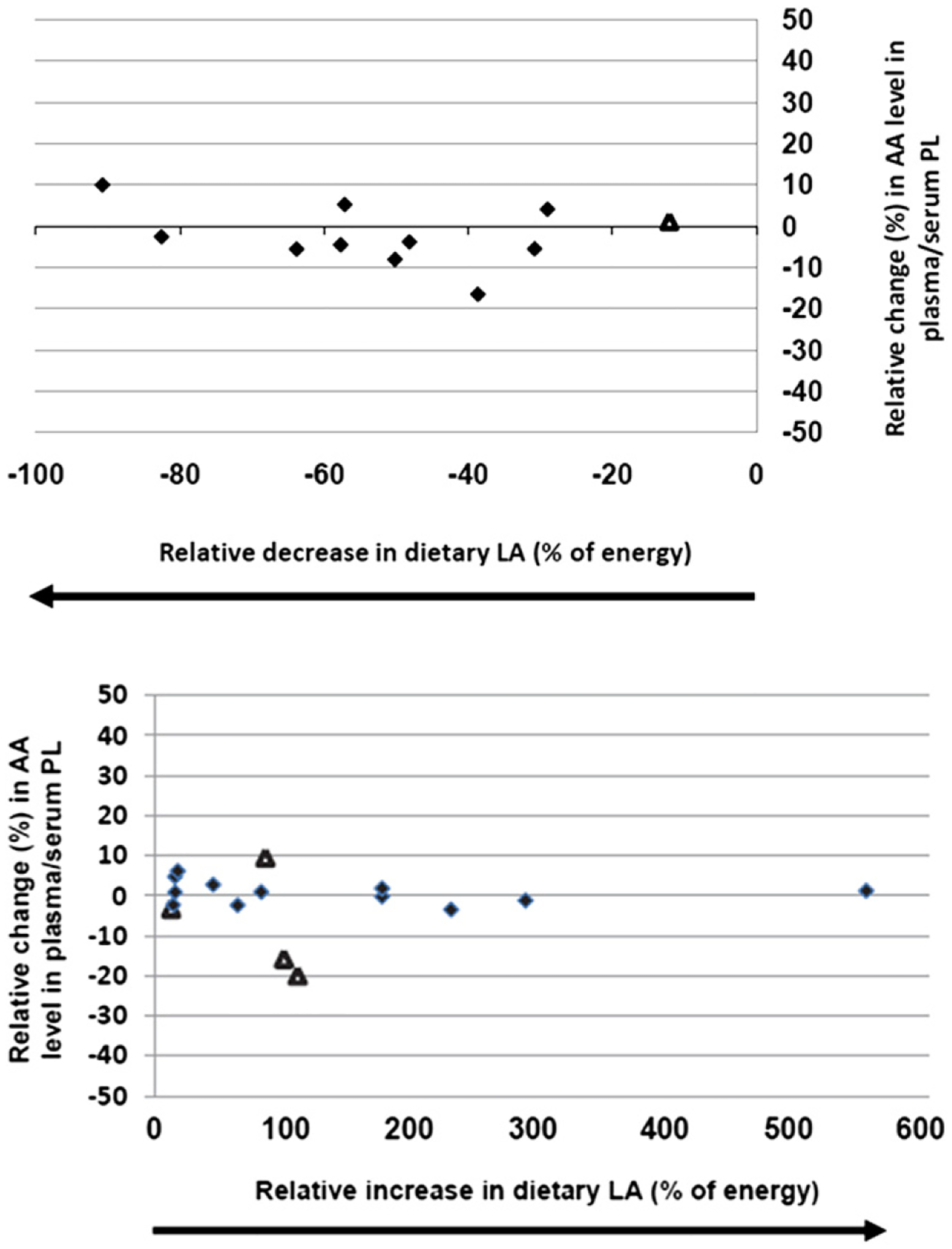
Effects of decreasing and increasing dietary LA intakes on changes in blood phospholipid AA content. Significant (P < .05) changes in AA as reported in the original papers designated as triangles; nonsignificant (P > .05) AA changes as reported in the original papers designated as diamonds. PL, phospholipid. Permission to reuse figure granted by BioMed Central Ltd.65
Despite these findings, it has been suggested that LA intakes should be reduced to less than 2% of energy to change the tissue AA levels. However, if LA were reduced or eliminated, there is no guarantee that dietary gammalinolenic acid or AA would not replenish tissue AA levels and maintain those levels in tissues. Because LA is a major constituent in most common foods consumed, it is difficult to determine how to decrease LA intake from 14.5 g/d (6% of energy) to 4.5 g/d (2% of energy). Selective elimination from the diet of foods with high LA content could also be undesirable. For example, as discussed previously, the advent of increasing soybean oil (88% of PUFA is LA) in the food supply was associated with reductions in the risk of CVD.59,60 Even so, the elimination of soybean oil, a major source of LA in the diet, would require changing the entire food supply infrastructure in the United States, which is a difficult proposition. Similarly, targeting foods that contribute to a balanced diet, such as walnuts, which have an LA content of 81% of PUFA, would also be undesirable because walnut consumption decreases total-C and LDL-C and has been associated with lower CVD risk.68 Thus, reducing LA does not appear to be the best way to decrease AA in tissue phospholipids. If it were desirable to do so, the best strategy for reducing AA would be to increase the intake of long chain n-3 PUFA. The American Heart Association recommends an EPA + DHA intake of 500 mg/d in patients without coronary artery disease (ie, 400 mg/d in addition to the ~100 mg/d that is typically consumed) which would result in a 20% to 25% reduction in tissue AA content, and has been associated with a significantly reduced risk of sudden death compared with lower n-3 blood levels.69 The 2010 Dietary Guidelines for Americans recommends 250 mg/day, which is twice current intake.70
In conclusion, available evidence suggests that increasing intake of LA correlates with reduced CHD risk. However, because of the theoretical potential for LA to be converted to AA leading to putative increased production of potentially damaging eicosanoids, additional research that supports current dietary recommendations for PUFA would resolve the lingering controversy about their health benefits.
Are All PUFA created equal? LA-specific PUFA interventions show no benefit and a signal toward harm in randomized controlled trials
Christopher E. Ramsden, MD, USPHS
NIH, Bethesda, MD, USA
Are all PUFAs created equal? The main objective of this presentation is to evaluate whether n-6 LA is cardioprotective, on the basis of the best-available evidence from clinical trials that selectively increased LA in place of SFA and trans-fatty acids. Key questions addressed are: (1) Are all PUFAs equivalent? Use of the general term PUFA implies that all PUFAs are a single molecular species with shared metabolic and health effects when, in reality, the situation is more complex; and (2) Is the specific PUFA composition of a given dietary intervention a critical determinant of its effect on CHD risk?
The pooled analysis of 11 prospective cohorts by Jakobsen et al12 demonstrated that for every 5% increase in energy as PUFA, in place of SFA, there was a 13% reduction in CHD risk. It should be noted that although the most abundant PUFA was LA, all PUFAs, including n-3 and n-6 fatty acids, defined the exposure variable(s). It is not necessarily a valid assumption to attribute effects of PUFA in general to LA. Similarly, it is not a valid assumption that because 88% of PUFA content of soybean oil is LA (ratio of LA to ALA of 8:1), that, soybean oil is equivalent to LA.
A pooled analysis of randomized controlled trials of dietary PUFA and clinical CHD outcomes reported a 10% CHD risk reduction for every 5% of energy substitution of PUFA in place of SFA.35 However, again, the independent variable was total PUFA including not only n-6 LA, but also n-3 ALA, EPA, and DHA. In the American Heart Association advisory, the reviewed data similarly evaluated the CHD effects of PUFA in general but the advice was more specific, in advising Americans to maintain or increase consumption of n-6 PUFA.56
One extreme example of confounding resulting from a lack of clarity in defining the independent variable(s) is from the Oslo Diet Heart Study.71 According to Leren,71 men from Oslo had a 7-fold increased incidence of first MI in the two decades before the trial, indicating that the normal diet was likely atherogenic, perhaps as the result of the high trans-fatty acid content. The control diet contained massive amounts of partially hydrogenated fish oil and vegetable oil margarines (~65 g/d), with ~9% to 10% of calories from trans fatty acids, including some 20 and 22-carbon trans-PUFA. The control diet was very low in LA and ALA, as well as EPA and DHA. In the intervention group, margarines were replaced with a large amount of soybean oil, which markedly increased LA to nearly 16% of calories, but, importantly, also substantially increased ALA to about 4.5 times average United States intake. Perhaps most importantly, the investigators provided Norwegian sardines canned in cod liver oil and advised the substitution of fish, shellfish, and whale in place of meats and eggs. The intervention group consumed ~5 g/d of EPA and DHA, which is equivalent to approximately 16 fish oil capsules per day. Despite this dominant confounder, the Oslo trial has previously been considered strong evidence for the benefits of n-6 LA.
Interestingly, ALA and/or EPA and DHA were also increased substantially in all but one of the randomized controlled trials pooled by Mozaffarian et al.35 The only trial included which selectively increased LA (in place of SFAs and trans fatty acids) showed a slight signal in the opposite direction.
Controlled trials that substituted LA for SFAs and trans-fatty acids are arguably the best available evidence for evaluating the effects of LA, without the potentially confounding role of n-3 EPA + DHA and/or ALA. In an effort to more specifically evaluate the effects of LA separate from other PUFAs, a detailed evaluation of the dietary interventions and oils provided in studies described in cited randomized controlled trial manuscripts, other obscure manuscripts from these trials, R01 and other grant materials, and data from interim analyses were included.61 From the database constructed using this information, it was apparent that there were three trials (four datasets) with 9569 participants, that increased LA selectively. Four other datasets with 1706 participants substantially increased n-3 ALA and/or EPA + DHA in addition to n-6 LA, thereby preventing a specific evaluation of the effects of LA. These datasets are described later.
The Rose Corn Oil Trial, a study with 54 participants, was the first LA-selective PUFA trial.72 Corn oil was substituted for typical fat sources, including hydrogenated oils, and was also taken as a supplement. There was more than a four-fold increased risk of CHD death and death from all causes in the corn oil group. Rose concluded that “corn oil cannot be recommended in the treatment of ischemic heart disease” because it is “most unlikely to be beneficial and possibly harmful.”
The next LA selective PUFA trial, the Sydney Diet Heart Study (n = 458), has received little attention.73 An obscure publication identified the oil provided as ~2 tablespoons/d of liquid safflower oil as well as a safflower polyunsaturated margarine (Miracle Margarine, Marrickville Margarine Party Limited). After intervention, there was a 49% increased risk of death from all causes in the LA selective PUFA group, which approached statistical significance despite the replacement of animal fats and common margarines. Because most of these deaths were caused by CHD (91% of total deaths were CHD deaths; 96% were CVD deaths in the two groups combined), omission of the Sydney data from previous analyses likely led to an overestimation of the apparent benefits of PUFA on CHD risk reduction in general and an underestimation of possible harm of n-6 LA.
The final n-6 LA-selective PUFA randomized controlled trial was the Minnesota Coronary Survey, in which corn oil and corn oil polyunsaturated margarine, providing ~14.5% of calories from LA, were the intervention oils provided for an average of slightly longer than 1 year.29 This is the only randomized controlled trial that included women (>4600). There was no alteration in the risk for CHD or death associated with the LA intervention in the 4500 male participants. However, in women in the experimental group there were statistically insignificant increased risks for nonfatal MI (+47%), composite CHD and CVD events (+30%), and death (+16%).
Analyses of individual trials and a meta-analysis of pooled data from the aforementioned three studies (four datasets) resulted in a relatively consistent increased risk associated with LA consumption (Fig. 8).61 When the Sydney Study was included in the pooled analysis, with the assumption that 91% of all deaths in each group were caused by CHD, it increased the risk for CHD death (+28%), CHD events (+25%), and death from all causes (+16%) associated with LA. Exclusion of the Sydney data from the meta-analysis resulted in a statistically insignificant risk ratio for CHD events of 1.13. Thus, there was no indication of benefit and a nonsignificant signal toward harm from LA-specific PUFA interventions as a replacement for SFAs and trans-fatty acids. Important limitations of this analysis included the small number of datasets, moderate number of participants with LA-specific interventions (fewer than 10,000), limited generalizability to other patient populations, and high LA dose (14%–15% of calories as LA, compared with the average American consumption of 6%–7%).
Figure 8.
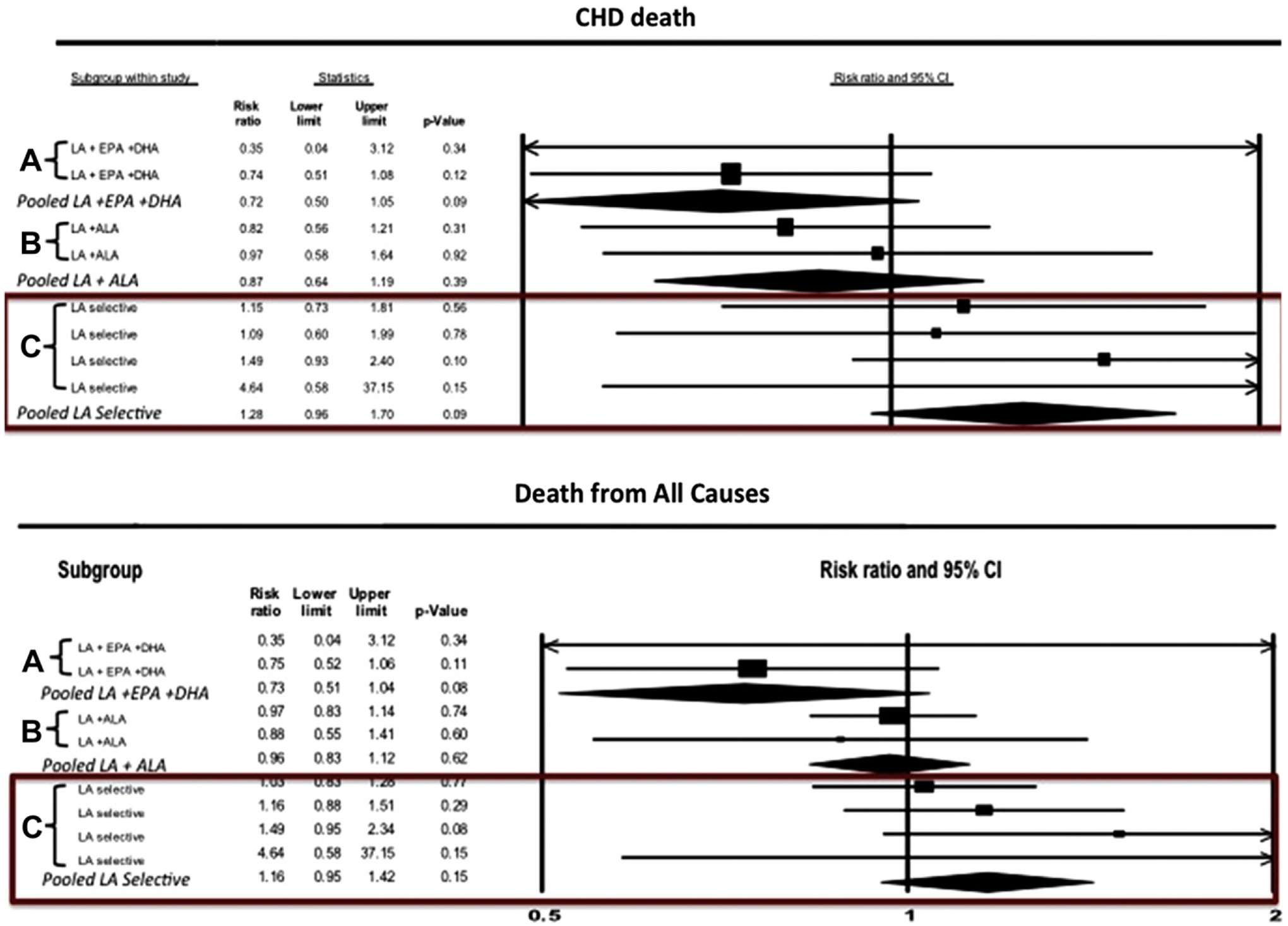
Forest plots of CHD death and death from all causes from a meta-analysis of randomized controlled trials in which dietary interventions increased n-6 PUFA specifically, or increased both n-3 and n-6 PUFA. (A) indicates mixed n-3/n-6 randomized controlled trial datasets providing substantial quantities of n-3 EPA + DHA and n-6 LA; (B) indicates mixed n-3/n-6 datasets providing substantial quantities of n-3 ALA and n-6 LA; (C) indicates n-6 specific datasets selectively increasing n-6 LA. Permission to reuse figure granted by Cambridge University Press.61
Evaluation of the studies that substantially increased n-3 and n-6 intakes indicated a different result. When categorized into three groups: (1) LA + EPA + DHA interventions showed a significant benefit; (2) LA + ALA without EPA + DHA showed a signal toward benefit; and (3) LA-selective PUFA interventions showed a signal in the opposite direction (Fig. 8).61 As is apparent, the results for LA were not in the right direction. Mechanisms through which EPA and DHA may have cardioprotective effects are well-documented. Some have also speculated that ALA may reduce risk for arrhythmias, enhance arterial compliance, and lower levels of inflammatory markers.74 One might therefore speculate that n-3s in general, and especially EPA + DHA, were cardioprotective in these trials, and that these benefits were attenuated by the concomitant increase in LA.
There are two major gaps in the critical evidence. First, there are no randomized controlled trial data evaluating effects on CHD of diets with LA intakes between 2% and 5% of calories, the advised cutoff. To put this into context, it is estimated that the range of LA intake in historical (evolutionary) diets was ~2–3%. At the beginning of the 20th century, the LA intake in the United States was ~2% of calories. Because it is difficult to consume more than 5% of calories from LA without added seed oils, a reduction in LA to 2% to 3% of calories would not create an atypical scenario. In fact, that is the amount of LA provided by virtually any whole food-based diet, whether it is meat-based, or it is predominantly vegetable-based. The second gap in the critical evidence is that there have been no studies assessing the effect on CHD of selectively increasing n-3 ALA, without increasing LA.
n-3 PUFA: Does new research debunk old truths? The omega-3 debate
Robert C. Block, MD, MPH
University of Rochester Medical Center, Rochester, NY, USA
This presentation reviews recent marine-derived n-3 fatty acid clinical trials that evaluated potential benefits for reducing CVD. Also considered are evidence-based dietary guidelines for n-3 fatty acids, as well as the potential safety, and multi-organ effects of EPA and DHA.
The Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto (GISSI) Miocardico Prevenzione Trial randomized ~11,000 subjects (85% men) soon after they had an MI (within 3 months) to 1 g/d fish oil concentrate, which provided ~840 mg combined EPA and DHA, similar to the prescription fish oil product available in the United States.75 The authors found that there was a significant reduction in sudden cardiac death and cardiovascular events that was evident after approximately 3 months of treatment. A subsequent randomized double-blind controlled trial by the same group studied patients with congestive heart failure. Participants were given either 1 g/d fish oil concentrate (840 mg of EPA + DHA) or placebo and followed for 3.9 years.76 The investigators reported a significant difference between fish oil and placebo treatments in those who died from any cause (27% and 29%, respectively) and those who died from or were admitted to hospital for cardiovascular reasons (57% and 59%, respectively). The authors estimated that 56 patients needed to be treated for a median duration of ~4 years to avoid one death, and in the same timeframe 44 patients needed to be treated to avoid one event of admission to hospital for cardiovascular reasons. In both groups, gastrointestinal disorders, although infrequent, were the most common adverse reactions, which has been the case in nearly every n-3 fatty acid study.77 The rates of other adverse events tended not to differ significantly from placebo.
In a more recent randomized controlled, prospective double-blind parallel study, 663 United States outpatients with symptomatic paroxysmal or persistent atrial fibrillation received 6720 mg/d of EPA + DHA for 7 days and then 3360 mg/d of EPA + DHA for 24 weeks in the FDA-approved prescription fish oil product.78 There was no benefit of prescription n-3 in reducing the first episode of recurrent symptomatic atrial fibrillation among all subjects, nor were there any significant effects detected in subgroups on the basis of age, sex, race, smoking status, or diabetes. Limitations of this study included that the population was primarily persons with paroxysmal atrial fibrillation, without other significant cardiac issues, such as reduced left ventricular ejection fraction or other mechanical problems. Furthermore, this trial was not powered to investigate episodes of stroke or CVD, so it was not possible to evaluate other clinically important events such as mortality, and it was not designed to evaluate a dose effect. Another weakness of this trial could be that fish intake was not determined; however, it is not practical to obtain 3.6 g/d of EPA + DHA by consuming fish, so this design flaw probably had little impact on the findings.
In the Alpha Omega trial, a randomized double-blind placebo-controlled multicenter study, investigators assessed whether EPA and DHA, given for 40 months to individuals ~3.5 to 4 years after MI, would improve cardiovascular health.79 N-3 fatty acids were provided as one of four trial margarines supplemented with: 400 mg of combined EPA + DHA, 2 g of ALA, 400 mg of EPA + DHA + 2 g of ALA, or placebo. The dose of ALA was significantly more than what is typically consumed in the United States, and the dose of EPA + DHA was relatively low. However, if the amount of EPA + DHA already present in the diet (~130 mg/d) was considered, subjects in the EPA and DHA treatment group consumed ~500 mg/d, which meets current recommendations from various organizations.
In the Alpha Omega trial, there were no significant differences between groups in the primary outcome, major cardiovascular events, or other end points (Table 3).79 Although the EPA + DHA arm had fewer fatal cardiovascular events early in the study, Kaplan-Meier curves showed no significant benefit with regard to either fatal CHD or major cardiovascular events. This study was considered to show a neutral effect; however, subgroup analyses conducted post hoc showed that patients with diabetes receiving EPA + DHA had significantly reduced risk of incident CVD and death from CHD.80
Table 3.
Primary and secondary outcomes according to n-3 supplementation in the Alpha Omega trial*
| EPA-DHA (N = 2404) | Placebo or ALA Only (N = 2433) | |||||
|---|---|---|---|---|---|---|
| Outcome | no. (%) | rate/1000 patient-yr | no. (%) | rate/1000 patient-yr | Hazard Ratio (95% Cl)† | P Value |
| Primary outcome: major cardiovascular events‡ | 336 (14.0) | 46.0 | 335 (13.8) | 45.7 | 1.01 (0.87–1.17) | .93 |
| Secondary outcomes | ||||||
| Incident cardiovascular disease | 170 (7.1) | 22.4 | 185 (7.6) | 24.3 | 0.92 (0.75–1.13) | .43 |
| Death from cardiovascular disease | 80 (3.3) | 10.3 | 82 (3.4) | 10.5 | 0.98 (0.72–1.33) | .89 |
| Death from coronary heart disease | 67 (2.8) | 8.7 | 71 (2.9) | 9.1 | 0.95 (0.68–1.32) | .75 |
| Ventricular–arrhythmia–related events§ | 67 (2.8) | 8.7 | 74 (3.0) | 9.6 | 0.90 (0.65–1.26) | .55 |
| Death from any cause | 186 (7.7) | 24.0 | 184 (7.6) | 23.7 | 1.01 (0.82–1.24) | .92 |
| ALA (N = 2409) | Placebo or EPA–DHA Only (N = 2428) | |||||
| no. (%) | rate/1000 patient-yr | no. (%) | rate/1000 patient-yr | Hazard Ratio (95% Cl)† | P Value | |
| Primary outcome: major cardiovascular events | 319 (13.2) | 43.6 | 352 (14.5) | 48.1 | 0.91 (0.78–1.05) | .20 |
| Secondary outcomes | ||||||
| Incident cardiovascular disease | 168 (7.0) | 22.1 | 187 (7.7) | 24.5 | 0.90 (0.73–1.11) | .34 |
| Death from cardiovascular disease | 78 (3.2) | 10.1 | 84 (3.5) | 10.8 | 0.94 (0.69–127) | .67 |
| Death from coronary heart disease | 66 (2.7) | 8.6 | 72 (3.0) | 9.2 | 0.92 (0.66–1.29) | .64 |
| Ventricular–arrhythmia–related events§ | 62 (2.6) | 8.1 | 79 (3.3) | 10.2 | 0.79 (0.57–1.10) | .16 |
| Death from any cause | 182 (7.6) | 23.5 | 188 (7.7) | 24.1 | 0.97 (0.79–1.19) | .80 |
ALA, alpha-linolenic acid; CI, confidence interval; DHA, docosahexaenoic acid; EPA, eicosapentaenoic acid.
The two groups that received EPA + DHA were combined and compared with the two groups that did not receive EPA + DHA (ie, the groups that received either placebo or only ALA). Similarly, the two groups that received ALA were combined and compared with the two groups that did not receive ALA (ie, the groups that received either placebo or only EPA + DHA).
The hazard ratios and 95% confidence intervals were calculated with the use of Cox proportional-hazards models.
Major cardiovascular events comprised fatal and nonfatal cardiovascular events and the cardiac interventions percutaneous coronary intervention and coronary-artery bypass grafting.
Ventricular-arrhythmia-related events comprised sudden death, fatal and nonfatal cardiac arrest, and placement of implantable cardioverter defibrillators.
Permission to reuse table granted by New England Journal of Medicine, Massachusetts Medical Society.79
Differences between the Alpha Omega trial79 and the GISSI heart failure study76 were that patients in Alpha Omega had received optimal treatment for a period of time, and the study intervention occurred significantly past their incident MI. Also, in the Alpha Omega trial, subjects received state-of-the-art antihypertensive, lipid-lowering (up to 90% for statins), and antithrombotic drugs at much greater proportions than subjects in the GISSI study. Limitations of the Alpha Omega trial included low doses of EPA and DHA, and the fact that subjects were enrolled years after their MI, thereby missing opportunities for evaluating effects of EPA and DHA on myocardial healing, arrhythmias, and ischemia in the acute and post-MI setting. Furthermore, the number of subjects with diabetes was relatively low, hindering statistical power for evaluations in this high-risk population.
The OMEGA trial was a randomized double-blind multicenter trial that evaluated 840 mg of EPA + DHA—the same dose that was used in the GISSI study—versus an olive oil control.81 Subjects included 3851 survivors of an acute MI (ST elevation or non-ST elevation) who were randomized soon after the MI, similar to the GISSI study. The primary outcome of sudden cardiac death, which was reduced in the GISSI study, was not benefitted with treatment in the OMEGA trial (Fig. 9).81 Furthermore, there were no reductions in cardiovascular events, or statistically significant differences in total mortality or revascularization. Essentially, this was a neutral study, although the guidelines for treatment of acute cardiovascular events have improved since the time of the GISSI study, which may explain why the percentage of patients with sudden cardiac death was significantly lower in the OMEGA trial compared with the GISSI trial.
Figure 9.
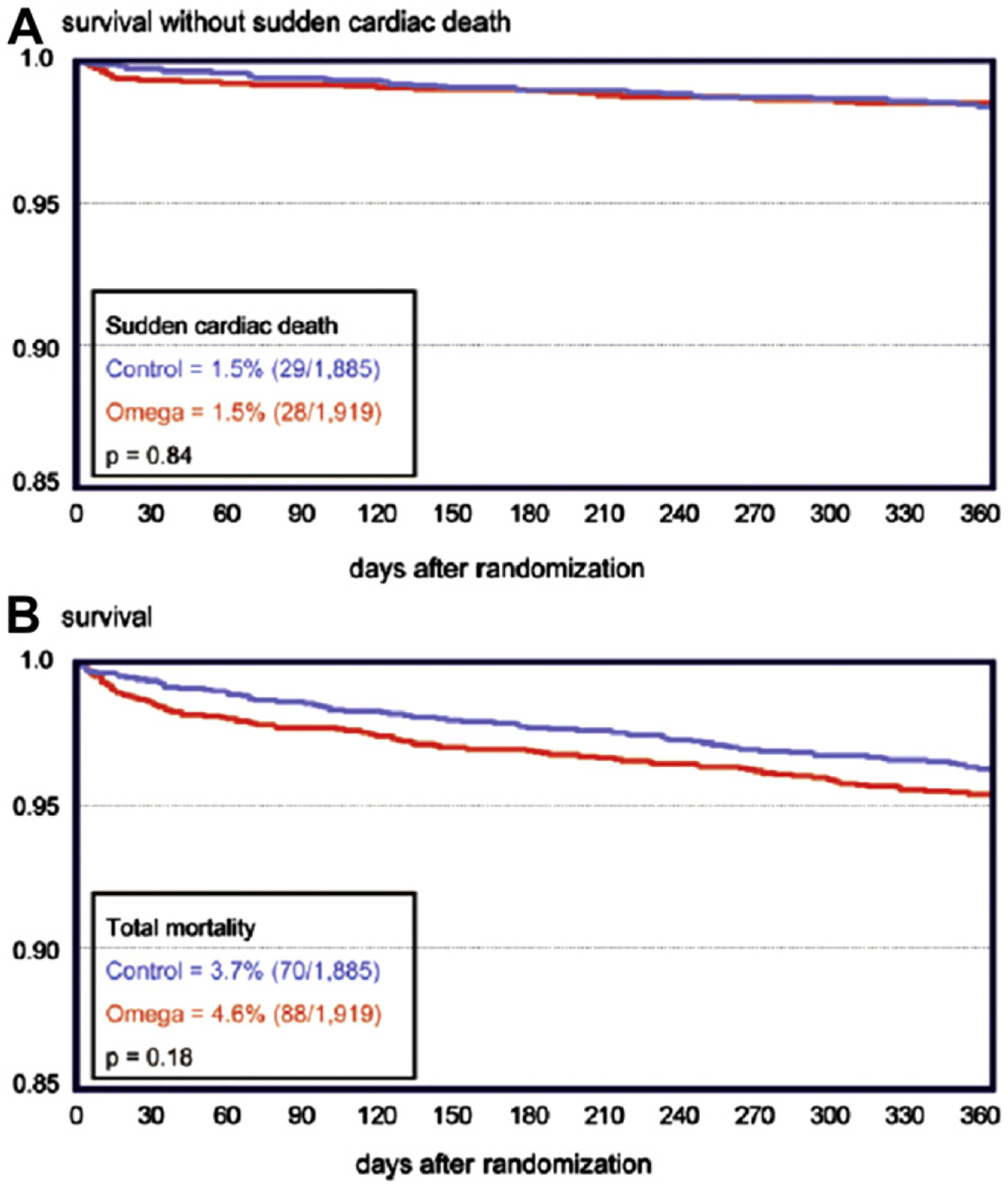
Kaplan-Meier diagrams from the OMEGA study in which 1 g/d n-3-acid ethyl esters were administered after acute MI. P-values are from the univariate analysis. A, Survival without sudden cardiac death during 1-year follow-up (red line, omega group; blue line, olive oil control group). B, total survival during one-year follow-up (red line, omega group; blue line, olive oil control group). Permission to reuse figure granted by the American Heart Association.81
A major problem with the OMEGA trial was that the investigators changed the inclusion criteria after the trial had started because they realized that the study lacked statistical power. In a statistical power calculation performed before the study had been completed, it was determined that ~20,000 patients would have been required to demonstrate significant differences in the outcomes. Other limitations are that fish consumption increased by nearly a factor of two in both groups, and the OMEGA follow up period of ~1 year is quite short.
According to the 2010 Dietary Guidelines for Americans, the amount and variety of seafood consumed should be increased by choosing seafood in place of some meat and poultry.70 This is the first time that such a recommendation has been issued in the Dietary Guidelines. The median intake of seafood in the United States is approximately 3.5 oz/week. Seafood varieties commonly consumed in the United States that are greater in EPA and DHA and tend to be lower in mercury, include salmon, anchovies, and herring. The Dietary Guidelines also suggest that women who are pregnant or breastfeeding consume 8 to 12 oz of seafood/week from a variety of seafood types. This recommendation is based on improved infant visual and cognitive development, even in the presence of greater mercury levels. Because of its greater methyl mercury content, white albacore tuna should be limited to 6 oz per week, and fish that accumulate mercury, such as tilefish, shark, swordfish, and king mackerel, should be avoided, especially by pregnant and breast feeding women.
Beyond the effects of EPA and DHA on CVD events, these fatty acids have pleiotropic effects and very few side effects, as well as few interactions with other drugs.82 Areas of ongoing EPA and DHA research include the fields of macular degeneration, corneal healing, neural health and regeneration, anti-inflammation, inflammation-resolving, antiapoptotic, attention deficit hyperactivity disorder, and other psychiatric disorders. When patients with major depressive disorder and bipolar disorder were given n-3, they had reduced symptoms of depression,83 which suggests that EPA and DHA may prove to be a viable option for treating MI patients who have a high risk of depression after being hospitalized.84 However, other evidence has not been supportive of this benefit.85 Results from some studies suggest that n-3 fatty acids benefit inflammation/autoimmune diseases, such as asthma and rheumatoid arthritis, and lower the need to use anti-inflammatory drugs.86 There is also interest in the potential use of intravenous EPA and DHA infusions for people who are injured badly on the battlefield, due to the purported benefits on inflammation and neural protection. Resolvins and protectins are metabolites of EPA and DHA with anti-inflammatory and neuroprotective effects up to about 1000 times greater than EPA and DHA.87 There could be great interest in the clinical benefits of these EPA and DHA metabolites in the future.
In conclusion, the benefits of EPA + DHA supplementation for CVD prevention via supplements are supported if fish intake is low, particularly in individuals with elevated TG. Nonetheless, pleiotropic effects and possible benefits for depression and inflammatory diseases, issues that affect many patients, are supportive of the use of these n-3 fatty acids. Additional research is needed to more clearly define the groups that would benefit from supplemental n-3 fatty acid intake and the mechanisms responsible for the benefits observed in observational studies and some clinical trials.
Conclusion
The National Lipid Association 2011 Fatty Acid Summit included presentations that summarized current controversies in fatty acid science relative to CVD risk. Food is extraordinarily complex; thus, it is unlikely that randomized controlled trials assessing dietary interventions will be able to determine definitively the effects of altering intakes of various fatty acids on CVD risk. To make dietary recommendations, we will have to rely on epidemiologic evidence coupled with controlled clinical trials on surrogate markers, along with an evolving understanding of the pathophysiology of CVD. Messages conveyed at the summit were that SFAs have unfavorable effects on LDL-C and are probably equivalent to refined and high glycemic index carbohydrates with respect to CVD risk (based on epidemiological studies).
Furthermore, the presentations underscored that, when evaluating diet studies one must carefully consider which foods and nutrients are substituted for those that are displaced. With regard to MUFA, specifically oleic acid—one element of a Mediterranean diet, substantial evidence was presented for favorable effects on CVD risk markers and a suggestion of benefit from some observational studies. However, a MUFA-rich diet has also been shown to increase the percentage of cholesteryl oleate in cholesterol esters, which has been found to correlate with increased arterial intima-media thickness, greater mortality in observational studies, and with coronary artery atherosclerosis in intervention studies conducted in mice and non-human primates. More information on the relationship between oleic acid intake and CVD risk is needed. Although cardioprotective effects of n-6 PUFA, specifically LA, have been promoted based, in part, on results from several clinical trials, a detailed evaluation of the dietary interventions provided in those studies suggested possible confounding in some by inclusion of significant amounts of ALA, EPA, and DHA. Thus, it was concluded that LA is essential and may be beneficial for cardiovascular health, although there are limitations in the available literature. Finally, with respect to n-3 fatty acids and CVD risk, the message conveyed at this summit was a reaffirmation of current recommendations regarding consumption of EPA and DHA for CVD prevention, and particularly for secondary prevention.
References
- 1.Mu H, Hoy CE. The digestion of dietary triacylglycerols. Prog Lipid Res. 2004;43:105–133. [DOI] [PubMed] [Google Scholar]
- 2.IUPAC-IUB Commission on Biochemical Nomenclature (CBN). The nomenclature of lipids (recommendations 1976). Eur J Biochem. 1977;79:11–21. [Google Scholar]
- 3.Russo GL. Dietary n-6 and n-3 polyunsaturated fatty acids: from biochemistry to clinical implications in cardiovascular prevention. Biochem Pharmacol. 2009;77:937–946. [DOI] [PubMed] [Google Scholar]
- 4.Gregory MK, Gibson RA, Cook-Johnson RJ, et al. Elongase reactions as control points in long-chain polyunsaturated fatty acid synthesis. PLoS One. 2011;6:e29662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brenna JT, Salem N Jr., Sinclair AJ, Cunnane SC. International Society for the Study of Fatty Acids and Lipids (ISSFAL). Alpha-linolenic acid supplementation and conversion to n-3 long-chain polyunsaturated fatty acids in humans. Prostaglandins Leukot Essent Fatty Acids. 2009;80:85–91. [DOI] [PubMed] [Google Scholar]
- 6.Calder PC. Polyunsaturated fatty acids, inflammatory processes and inflammatory bowel diseases. Mol Nutr Food Res. 2008;52:885–897. [DOI] [PubMed] [Google Scholar]
- 7.McKenney JM, Sica D. Prescription omega-3 fatty acids for the treatment of hypertriglyceridemia. Am J Health Syst Pharm. 2007;64: 595–605. [DOI] [PubMed] [Google Scholar]
- 8.Schuchardt JP, Schneider I, Meyer H, et al. Incorporation of EPA and DHA into plasma phospholipids in response to different omega-3 fatty acid formulations—a comparative bioavailability study of fish oil vs. krill oil. Lipids Health Dis. 2011;10:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Astrup A, Dyerberg J, Elwood P, et al. The role of reducing intakes of saturated fat in the prevention of cardiovascular disease: where does the evidence stand in 2010? Am J Clin Nutr. 2011;93:684–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu FB, Stampfer MJ, Manson JE, et al. Dietary fat intake and the risk of coronary heart disease in women. N Engl J Med. 1997;337: 1491–1499. [DOI] [PubMed] [Google Scholar]
- 11.Oh K, Hu FB, Manson JE, et al. Dietary fat intake and risk of coronary heart disease in women: 20 years of follow-up of the nurses’ health study. Am J Epidemiol. 2005;161:672–679. [DOI] [PubMed] [Google Scholar]
- 12.Jakobsen MU, O’Reilly EJ, Heitmann BL, et al. Major types of dietary fat and risk of coronary heart disease: a pooled analysis of 11 cohort studies. Am J Clin Nutr. 2009;89:1425–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mensink RP, Katan MB. Effect of monounsaturated fatty acids versus complex carbohydrates on high-density lipoproteins in healthy men and women. Lancet. 1987;1:122–125. [DOI] [PubMed] [Google Scholar]
- 14.Mensink RP, Katan MB. Effect of dietary fatty acids on serum lipids and lipoproteins. A meta-analysis of 27 trials. Arterioscler Thromb. 1992;12:911–919. [DOI] [PubMed] [Google Scholar]
- 15.Mensink RP, Zock PL, Kester AD, et al. Effects of dietary fatty acids and carbohydrates on the ratio of serum total to HDL cholesterol and on serum lipids and apolipoproteins: a meta-analysis of 60 controlled trials. Am J Clin Nutr. 2003;77:1146–1155. [DOI] [PubMed] [Google Scholar]
- 16.Jakobsen MU, Dethlefsen C, Joensen AM, et al. Intake of carbohydrates compared with intake of saturated fatty acids and risk of myocardial infarction: importance of the glycemic index. Am J Clin Nutr. 2010;91:1764–1768. [DOI] [PubMed] [Google Scholar]
- 17.Ford ES, Ajani UA, Croft JB, et al. Explaining the decrease in U.S. deaths from coronary disease, 1980–2000. N Engl J Med. 2007;356: 2388–2398. [DOI] [PubMed] [Google Scholar]
- 18.American Heart Association. Dietary fat and its relation to heart attacks and strokes. JAMA. 1961;175:135–137. [PubMed] [Google Scholar]
- 19.Keys A. Coronary heart disease in seven countries. 1970. Nutrition. 1997;13:250–252. [DOI] [PubMed] [Google Scholar]
- 20.Hu FB, Stampfer MJ, Manson JE, et al. Dietary intake of alpha-linolenic acid and risk of fatal ischemic heart disease among women. Am J Clin Nutr. 1999;69:890–897. [DOI] [PubMed] [Google Scholar]
- 21.Keys A, Anderson JT, Grande F. Serum-cholesterol response to dietary fat. Lancet. 1957;1:787–795. [Google Scholar]
- 22.Hegsted DM, McGandy RB, Myers ML, et al. Quantitative effects of dietary fat on serum cholesterol in man. Am J Clin Nutr. 1965;17: 281–295. [DOI] [PubMed] [Google Scholar]
- 23.Medical Research Council. Controlled trial of soya-bean oil in myocardial infarction. Lancet. 1968;2:693–699. [PubMed] [Google Scholar]
- 24.Dayton S, Pearce ML. Prevention of coronary heart disease and other complications of arteriosclerosis by modified diet. Am J Med. 1969; 46:751–762. [DOI] [PubMed] [Google Scholar]
- 25.Leren P The Oslo Diet-Heart Study: eleven-year report. Circulation. 1970;42:935–942. [DOI] [PubMed] [Google Scholar]
- 26.Miettinen M, Turpeinen O, Karvonen MJ, et al. Cholesterol-lowering diet and mortality from coronary heart-disease. Lancet. 1972;2: 1418–1419. [DOI] [PubMed] [Google Scholar]
- 27.Turpeinen O Effect of cholesterol-lowering diet on mortality from coronary heart disease and other causes. Circulation. 1979;59:1–7. [DOI] [PubMed] [Google Scholar]
- 28.Arntzenius AC, Kromhout D, Barth JD, et al. Diet, lipoproteins, and the progression of coronary atherosclerosis. The Leiden Intervention Trial. N Engl J Med. 1985;312:805–811. [DOI] [PubMed] [Google Scholar]
- 29.Frantz ID Jr., Dawson EA, Ashman PL, et al. Test of effect of lipid lowering by diet on cardiovascular risk: The Minnesota Coronary Survey. Arteriosclerosis. 1989;9:129–135. [DOI] [PubMed] [Google Scholar]
- 30.Watts GF, Jackson P, Mandalia S, et al. Nutrient intake and progression of coronary artery disease. Am J Cardiol. 1994;73:328–332. [DOI] [PubMed] [Google Scholar]
- 31.Mente A, de Koning L, Shannon HS, et al. A systematic review of the evidence supporting a causal link between dietary factors and coronary heart disease. Arch Intern Med. 2009;169:659–669. [DOI] [PubMed] [Google Scholar]
- 32.Skeaff CM, Miller J. Dietary fat and coronary heart disease: a summary of evidence from prospective cohort and randomised controlled trials. Ann Nutr Metab. 2009;55:173–201. [DOI] [PubMed] [Google Scholar]
- 33.Siri-Tarino PW, Sun Q, Hu F, et al. Meta-analysis of prospective cohort studies evaluating the association of saturated fat with cardiovascular disease. Am J Clin Nutr. 2010;91:535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Siri-Tarino PW, Sun Q, Hu FB, et al. Saturated fat, carbohydrate, and cardiovascular disease. Am J Clin Nutr. 2010;91:502–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mozaffarian D, Micha R, Wallace S. Effects on coronary heart disease of increasing polyunsaturated fat in place of saturated fat: a systematic review and meta-analysis of randomized controlled trials. PLoS Med. 2010;7:e1000252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mattson FH, Grundy SM. Comparison of effects of dietary saturated, monounsaturated, and polyunsaturated fatty acids on plasma lipids and lipoproteins in man. J Lipid Res. 1985;26:194–202. [PubMed] [Google Scholar]
- 37.Keys A, Menotti A, Karvonen MJ, et al. The diet and 15-year death rate in the seven countries study. Am J Epidemiol. 1986;124: 903–915. [DOI] [PubMed] [Google Scholar]
- 38.Ma J, Folsom AR, Lewis L, et al. Relation of plasma phospholipid and cholesterol ester fatty acid composition to carotid artery intima-media thickness: the Atherosclerosis Risk in Communities (ARIC) Study. Am J Clin Nutr. 1997;65:551–559. [DOI] [PubMed] [Google Scholar]
- 39.Warensjö E, Sundström J, Vessby B, et al. Markers of dietary fat quality and fatty acid desaturation as predictors of total and cardiovascular mortality: a population-based prospective study. Am J Clin Nutr. 2008; 88:203–209. [DOI] [PubMed] [Google Scholar]
- 40.Rudel LL, Parks KS, Sawyer JK. Compared with dietary monounsaturated and saturated fat, polyunsaturated fat protects African green monkeys from coronary artery atherosclerosis. Arterioscler Thromb Vasc Biol. 1995;15:2101–2110. [DOI] [PubMed] [Google Scholar]
- 41.Rudel LL, Haines J, Sawyer JK, et al. Hepatic origin of cholesteryl oleate in coronary artery atherosclerosis in African green monkeys. Enrichment by dietary monounsaturated fat. J Clin Invest. 1997;100: 74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rudel LL, Kelley K, Sawyer JK, et al. Dietary monounsaturated fatty acids promote aortic atherosclerosis in LDL receptor-null, human Apo B100-overexpressing transgenic mice. Arterioscler Thromb Vasc Biol. 1998;18:1818–1827. [DOI] [PubMed] [Google Scholar]
- 43.Expert Panel on Detection, Evaluation and Treatment of High Blood Cholesterol in Adults. Executive summary of the third report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA. 2001;285:2486–2497. [DOI] [PubMed] [Google Scholar]
- 44.U.S. Department of Agriculture. What’s in the foods you eat. Search Tool, 4.1 Available at: http://www.ars.usda.gov/Services/docs.htm?docid=17032. Accessed April 16, 2012.
- 45.Micha R, Mozaffarian D. Saturated fat and cardiometabolic risk factors, coronary heart disease, stroke, and diabetes: a fresh look at the evidence. Lipids. 2010;45:893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jenkins DJA, Chiavaroli L, Wong JMW, et al. Adding monounsaturated fatty acids to a dietary portfolio of cholesterol lowering foods in hypercholesterolemia. CMAJ. 2010;182:1961–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Allman-Farinelli MA, Gomes K, Favaloro EJ, et al. A diet rich in high-oleic sunflower oil favorably alters low-density lipoprotein cholesterol, triglycerides, and factor VII coagulant activity. J Am Diet Assoc. 2005;105:1071–1079. [DOI] [PubMed] [Google Scholar]
- 48.Appel LJ, Sacks FM, Carey VJ, et al. Effects of protein, monounsaturated fat, and carbohydrate on blood pressure and serum lipids. JAMA. 2005;294:2455–2464. [DOI] [PubMed] [Google Scholar]
- 49.Berglund L, Lefevre M, Ginsberg HN, et al. , DELTA Investigators. Comparison of monounsaturated fat with carbohydrates as a replacement for saturated fat in subjects with a high metabolic risk profile: studies in fasting and postprandial states. Am J Clin Nutr. 2007;86:1611–1620. [DOI] [PubMed] [Google Scholar]
- 50.Tanasescu M, Cho E, Manson JE, et al. Dietary fat and cholesterol and the risk of cardiovascular disease among women with type 2 diabetes. Am J Clin Nutr. 2004;79:999–1005. [DOI] [PubMed] [Google Scholar]
- 51.Sabaté J, Oda K, Ros E. Nut consumption and blood lipid levels. Arch Intern Med. 2010;170:821–827. [DOI] [PubMed] [Google Scholar]
- 52.Hu FB. Dietary pattern analysis: a new direction in nutritional epidemiology. Curr Opin Lipidol. 2002;13:3–9. [DOI] [PubMed] [Google Scholar]
- 53.Maki KC. Dietary factors in the prevention of diabetes mellitus and coronary artery disease associated with the metabolic syndrome. Am J Cardiol. 2004;93(suppl):12C–7C. [DOI] [PubMed] [Google Scholar]
- 54.Choo J, Ueshima H, Curb JD, et al. , ERA-JUMP Study Group. Serum n-6 fatty acids and lipoprotein subclasses in middle-aged men: the population-based cross-sectional ERA-JUMP study. Am J Clin Nutr. 2010;91:1195–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Laaksonen DE, Nyyssönen K, Niskanen L, et al. Prediction of cardiovascular mortality in middle-aged men by dietary and serum linoleic and polyunsaturated fatty acids. Arch Intern Med. 2005;165:193–199. [DOI] [PubMed] [Google Scholar]
- 56.Harris WS, Mozaffarian D, Rimm E, et al. Omega-6 fatty acids and risk for cardiovascular disease: a science advisory from the American Heart AssociationNutrition Subcommittee of the Council onNutrition, Physical Activity, and Metabolism; Council on Cardiovascular Nursing; and Council on Epidemiology and Prevention. Circulation. 2009;119:902–907. [DOI] [PubMed] [Google Scholar]
- 57.Czernichow S, Thomas D, Bruckert E. n-6 fatty acids and cardiovascular health: a review of the evidence for dietary intake recommendations. Br J Nutr. 2010;104:788–796. [DOI] [PubMed] [Google Scholar]
- 58.Friesen RW, Innis SM. Linoleic acid is associated with lower long-chain n-6 and n-3 fatty acids in red blood cell lipids of Canadian pregnant women. Am J Clin Nutr. 2010;91:23–31. [DOI] [PubMed] [Google Scholar]
- 59.Blasbalg TL, Hibbeln JR, Ramsden CE, et al. Changes in consumption of omega-3 and omega-6 fatty acids in the United States during the 20th century. Am J Clin Nutr. 2011;93:950–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Roberts DC. Dietary factors in the fall in coronary heart disease mortality. Prostaglandins Leukot Essent Fatty Acids. 1991;44:97–101. [DOI] [PubMed] [Google Scholar]
- 61.Ramsden CE, Hibbeln JR, Majchrzak SF, et al. N-6 fatty acid-specific and mixed polyunsaturated dietary interventions have different effects on CHD risk: a meta-analysis of randomised controlled trials. Br J Nutr. 2010;104:1586–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Simopoulos AP. The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp Biol Med (Maywood). 2008;233:674–688. [DOI] [PubMed] [Google Scholar]
- 63.Lagarde M, Chen P, Véricel E, et al. Fatty acid-derived lipid mediators and blood platelet aggregation. Prostaglandins Leukot Essent Fatty Acids. 2010;82:227–230. [DOI] [PubMed] [Google Scholar]
- 64.Wall R, Ross RP, Fitzgerald GF, et al. Fatty acids from fish: the anti-inflammatory potential of long-chain omega-3 fatty acids. Nutr Rev. 2010;68:280–289. [DOI] [PubMed] [Google Scholar]
- 65.Rett BS, Whelan J. Increasing dietary linoleic acid does not increase tissue arachidonic acid content in adults consuming Western-type diets: a systematic review. Nutr Metab (Lond). 2011;8:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liou YA, Innis SM. Dietary linoleic acid has no effect on arachidonic acid, but increases n-6 eicosadienoic acid, and lowers dihomo-gamma-linolenic and eicosapentaenoic acid in plasma of adult men. Prostaglandins Leukot Essent Fatty Acids. 2009;80:201–206. [DOI] [PubMed] [Google Scholar]
- 67.Lands WE. Biochemistry and physiology of n-3 fatty acids. FASEB J. 1992;6:2530–2536. [DOI] [PubMed] [Google Scholar]
- 68.Mukuddem-Petersen J, Oosthuizen W, Jerling JC. A systematic review of the effects of nuts on blood lipid profiles in humans. J Nutr. 2005; 135:2082–2089. [DOI] [PubMed] [Google Scholar]
- 69.Von Schacky C, Harris WS. Cardiovascular benefits of omega-3 fatty acids. Cardiovasc Res. 2007;73:310–315. [DOI] [PubMed] [Google Scholar]
- 70.U.S. Department of Agriculture. Dietary Guidelines for Americans, 2010. Available at: http://www.cnpp.usda.gov/dietaryguidelines.htm. Accessed April 16, 2012.
- 71.Leren P The effect of plasma cholesterol lowering diet in male survivors of myocardial infarction. A controlled clinical trial. Acta Med Scand Suppl. 1966;466:1–92. [PubMed] [Google Scholar]
- 72.Rose GA, Thompson WB, Williams RT. Corn oil in treatment of ischaemic heart disease. Br Med J. 1965;1:1531–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Woodhill JM, Palmer AJ, Blacket RB. Dietary habits and their modification in a coronary prevention programme for Australians. Food Technol Aust. 1969;21:264–271. [Google Scholar]
- 74.Anderson BM, Ma DL. Are all n-3 polyunsaturated fatty acids created equal? Lipids Health Dis. 2009;8:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction results of the GISSI-Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto miocardico. Lancet. 1999;354:447–455. [PubMed] [Google Scholar]
- 76.GISSI-HF Investigators; Tavazzi L, Maggioni AP, Marchioli R, et al. Effects of n-3 polyunsaturated fatty acids in patients with chronic heart failure (the GISSI-HF trial): a randomised, double-blind, placebo-controlled trial. Lancet. 2008;372:1223–1230. [DOI] [PubMed] [Google Scholar]
- 77.Harris WS, Ginsberg HN, Arunkakul N, et al. Safety and efficacy of Omacor in severe hypertriglyceridemia. Cardiovasc Risk. 1997;4: 385–391. [PubMed] [Google Scholar]
- 78.Kowey PR, Reiffel JA, Ellenbogen KA, et al. Efficacy and safety of prescription omega-3 fatty acids for the prevention of recurrent symptomatic atrial fibrillation: a randomized controlled trial. JAMA. 2010; 304:2363–2372. [DOI] [PubMed] [Google Scholar]
- 79.Kromhout D, Giltay EJ, Geleijnse JM; Alpha Omega Trial Group. N-3 fatty acids and cardiovascular events after myocardial infarction. N Engl J Med. 2010;363:2015–2026. [DOI] [PubMed] [Google Scholar]
- 80.Kromhout D, Geleijnse JM, de Goede J, et al. N-3 fatty acids, ventricular arrhythmia-related events, and fatal myocardial infarction in post-myocardial infarction patients with diabetes. Diabetes Care. 2011;34: 2515–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rauch B, Schiele R, Schneider S, et al. , OMEGA Study Group. OMEGA, a randomized, placebo-controlled trial to test the effect of highly purified omega-3 fatty acids on top of modern guideline-adjusted therapy after myocardial infarction. Circulation. 2010;122:2152–2159. [DOI] [PubMed] [Google Scholar]
- 82.Kar S. Omacor and omega-3 fatty acids for treatment of coronary artery disease and the pleiotropic effects. Am J Ther. 2011;Oct 4 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 83.Ross BM, Seguin J, Sieswerda LE. Omega-3 fatty acids as treatments for mental illness: which disorder and which fatty acid? Lipids Health Dis. 2007;18:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Thombs BD, Bass EB, Ford DE, et al. Prevalence of depression in survivors of acute myocardial infarction. J Gen Intern Med. 2006;21: 30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Carney RM, Freedland KE, Rubin EH, et al. Omega-3 augmentation of sertraline in treatment of depression in patients with coronary heart disease: a randomized controlled trial. JAMA. 2009;301:1651–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Simopoulos AP. Omega-3 fatty acids in inflammation and autoimmune diseases. J Am Coll Nutr. 2002;21:495–505. [DOI] [PubMed] [Google Scholar]
- 87.Serhan CN, Petasis NA. Resolvins and protectins in inflammation resolution. Chem Rev. 2011;111:5922–5943. [DOI] [PMC free article] [PubMed] [Google Scholar]