

Abstract
While effective in treating abdominal pain, opioids have significant side effects. Recent legalization of cannabis will likely promote use of cannabinoids as an adjunct or alternative to opioids, despite a lack of evidence. We aimed to investigate whether cannabinoids inhibit mouse colonic nociception, alone or in combination with opioids at low doses. Experiments were performed on C57BL/6 male and female mice. Visceral nociception was evaluated by measuring visceromotor responses (VMR), afferent nerve mechanosensitivity in flat-sheet colon preparations, and excitability of isolated DRG neurons. Blood oxygen saturation, locomotion, and defecation were measured to evaluate side effects. An agonist of cannabinoid 1 receptor (CB1R), arachidonyl-2′-chloroethylamide (ACEA), dose-dependently decreased VMR. ACEA and HU-210 (another CB1R agonist) also attenuated colonic afferent nerve mechanosensitivity. Additionally, HU-210 concentration-dependently decreased DRG neuron excitability, which was reversed by the CB1R antagonist AM-251. Conversely, cannabinoid 2 receptor (CB2R) agonists did not attenuate VMR, afferent nerve mechanosensitivity, or DRG neuron excitability. Combination of subanalgesic doses of CB1R and µ-opioid receptor agonists decreased VMR; importantly, this analgesic effect was preserved after 6 d of twice daily treatment. This combination also attenuated afferent nerve mechanosensitivity and DRG neuron excitability, which was inhibited by neuronal nitric oxide synthase and guanylate cyclase inhibitors. This combination avoided side effects (decreased oxygen saturation and colonic transit) caused by analgesic dose of morphine. Activation of CB1R, but not CB2R, decreased colonic nociception both alone and in synergy with µ-opioid receptor. Thus, CB1R agonists may enable opioid dose reduction and avoid opioid-related side effects.
SIGNIFICANCE STATEMENT One of the most cited needs for patients with abdominal pain are safe and effective treatment options. The effectiveness of opioids in the management of abdominal pain is undermined by severe adverse side effects. Therefore, strategies to replace opioids or reduce the doses of opioids to suppress abdominal pain is needed. This study in mice demonstrates that cannabinoid 1 receptor (CB1R) agonists inhibit visceral sensation. Furthermore, a combination of subanalgesic doses of µ-opioid receptor agonist and CB1R agonist markedly reduce abdominal pain without causing the side effects of high-dose opioids. Thus, CB1R agonists, alone or in combination with low-dose opioids, may be a novel and safe treatment strategy for abdominal pain.
Keywords: afferent nerve, cannabinoid, colon, opioid, visceral pain
Introduction
Visceral pain is a debilitating symptom associated with a variety of gastrointestinal (GI) disorders. The consultation prevalence for abdominal pain in primary care is 2.8%; and in about one-third of these patients, the cause cannot be specified (Viniol et al., 2014). Currently, opioids are the mainstay for treating severe pain, despite many adverse side effects (Elikkottil et al., 2009). Moreover, the opioid epidemic has become a global health crisis (Wiese and Wilson-Poe, 2018). The estimated overdose deaths from opioids in the United States was 75,673 in the 12 month period ending in April 2021 (U.S. National Center for Health Statistics, https://www.cdc.gov/nchs/pressroom/nchs_press_releases), and there was a 95% increase in opioid toxicity deaths in Canada in 2021 compared with the year prior (Public Health Agency of Canada, https://health-infobase.canada.ca/substance-related-harms/opioids-stimulants/). This highlights the need for developing new treatments for visceral pain. While the analgesic actions of cannabinoids have been reported for centuries (Elikkottil et al., 2009), the recent legalization of cannabis in many countries will likely promote cannabinoid use. However, there are few mechanistic studies to support their use to treat visceral pain, either alone or in combination with opioids.
The actions of cannabinoids are largely mediated by two subtypes of cannabinoid receptors. Cannabinoid 1 receptors (CB1Rs) are widely distributed in many cell types in the GI tract, including enteric neurons, extrinsic vagal and spinal primary afferent nerves, and enteroendocrine cells, whereas cannabinoid 2 receptors (CB2Rs) are mainly expressed by immune cells (Sharkey and Wiley, 2016). The U.S. FDA has approved one cannabis-derived cannabinoid (cannabidiol) for treatment of seizures and three synthetic cannabinoids for nausea and anorexia. Although studies have suggested analgesic effects of cannabinoids in both somatic pain models (Clayton et al., 2002; Liang et al., 2007) and in visceral sensation models (Sanson et al., 2006; Brusberg et al., 2009; Hong et al., 2009), the use of nonselective agonists and use of in vivo techniques leave a critical knowledge gap regarding whether the analgesic effect of cannabinoids involves activation of CB1R and/or CB2R on afferent nerves innervating the GI tract.
The synergy of cannabinoids and opioids has been reported in somatic pain models. For example, coapplication potentiates their analgesic effects compared with either drug alone (Reche et al., 1996; Smith et al., 1998; Cichewicz et al., 1999; Chen et al., 2019). However, a recent systematic review examining 92 preclinical and clinical studies on opioid sparing effects of cannabinoids reveals inconsistent outcomes (Nielsen et al., 2022). Moreover, the majority of these studies investigated somatic pain, and there is little known whether this synergy influences visceral pain signaling and, if so, what underlying molecular mechanisms are involved. A number of mechanisms have been proposed, including convergence in their common intracellular signaling pathways (Shapira et al., 1998; Maguire and France, 2014), or involvement of endogenous cannabinoids and opioids (Pacheco et al., 2009). Additionally, both opioids and cannabinoids can inhibit nociception via nitric oxide (NO) signaling (Reis et al., 2009; Hervera et al., 2011), which may also be a mechanism of synergy. Studies have revealed that anandamide stimulates NO release from immunocytes, microglia, and macrophages via activation of CB1R (Stefano et al., 1996), while morphine stimulates NO release from endothelial cells by activating µ opioid receptor (MOR) (Stefano et al., 1995). Since NO plays an inhibitory modulatory role in primary sensory afferent nerves of the GI tract (Page et al., 2009; Yu et al., 2019a), release of NO on activation of both cannabinoid and opioid receptors may potentiate the inhibitory actions on nociceptive nerves.
This study aimed to determine whether CB1R or CB2R agonists alone reduce visceral pain signaling in peripheral intestinal nerves. We also investigated whether cannabinoid and opioid receptor agonists have a synergistic action on these nerves but with reduced side effects compared with higher doses of either agonist alone. Additionally, the role for NO signaling was examined as a first step toward understanding the mechanisms of this interaction.
Materials and Methods
All authors had access to the study data and reviewed and approved the final manuscript.
Animals and ethical approval
All experiments were approved by Queen's University Animal Care Committee, in accordance with the guideline of the Canadian Council for Animal Care. Male and female C57BL/6 mice (body weight 20-25 g, 8-10 weeks old) were purchased from Charles River Laboratories. They were housed individually under a standard light-dark cycle (lights on: 7:00 A.M.; lights off: 7:00 P.M.) with free access to food (Lab Diet, 5015) and water. Mice were given 1 week to acclimate the new housing before any procedures. Mice were killed by isoflurane inhalation followed by cervical dislocation.
Visceromotor responses (VMRs) to colorectal distention
Mice were implanted with a PhysioTel ETA-F10 telemetric transmitter (Data Science International) to measure EMG activity in response to colorectal distention (CRD) (Mondelaers et al., 2016). Mice were anesthetized via isoflurane inhalation (1.5%-2.5% isoflurane; oxygen flow: 1 L/min), placed on a heating pad, and given bupivacaine (2 mg/kg intradermally) and tramadol (20 mg/kg subcutaneously) before surgery as an analgesic. The telemetric transmitter was inserted within the abdominal cavity, with the noninsulated tips of the electrodes sutured onto the external oblique muscle (∼5-10 mm apart). Mice were allowed to recover for 10 d, and tramadol (20 mg/kg) was given subcutaneously daily for 3 d as a postoperative analgesic. Following recovery, mice were acclimatized in a restrainer (Kent Scientific) for 30 min daily for 2 d before VMR recording. On the day of recording, mice were lightly anesthetized with isoflurane, placed in a restrainer, and a 4F arterial embolectomy catheter (Fogerty 120804FF, Edwards Lifesciences) was inserted into the colorectum ∼0.5 cm from the anus and secured to the tail with tape. Mice were allowed to recover from anesthesia for 20 min before the catheter was distended with volumes of 20-80 µl (in 20 µl increments) in duplicate, with each distention lasting 10 s and a 3 min rest period in between. VMR was measured 20 min (Sanson et al., 2006) after intraperitoneal (i.p.) injection of vehicle (3% methyl acetate or 7% DMSO in sterile saline), arachidonyl-2′-chloroethylamide (ACEA; 0.3, 1, 3 mg/kg) (Izzo et al., 2003), HU-308 (0.3, 1, 3 mg/kg) (Ofek et al., 2006), morphine (0.3, 3 mg/kg) (Ingram et al., 2007), or a mixture of ACEA and morphine (0.3 mg/kg of each). Each group of experiments (the same drug at different doses) was performed on the same group of mice with one treatment per day. For chronic treatment with ACEA-morphine mix (0.3 mg/kg each), mice received i.p. injections twice daily for 6 d. VMR in response to 80 µl distention was measured 30 min after the first injection of the mix on day 1, 3, and 6, and normalized to the vehicle response on the same day. VMR was analyzed as the mean EMG activity during distention (10 s) subtracted by the mean value of the basal activity using the Ponemah version 6.5 software (Data Science International). Results were presented as %VMR relative to the maximal VMR following vehicle injection for a given mouse.
Perforated patch-clamp recording
DRG (T13-L2) neurons were dissociated as previously described (Sessenwein et al., 2017). Following overnight culture, coverslips containing isolated neurons were placed in a recording chamber on an inverted microscope and superfused with external solution containing the following (in mm): NaCl 140, KCl 5, MgCl2 1, CaCl2 2, HEPES 10, and glucose 10, pH 7.4 with NaOH. Only small-diameter DRG neurons (≤30 pF) were selected, as they are putative nociceptors (Yu et al., 2019b). Patch electrodes were pulled from premium custom 8520 patch glass (Warner Instruments) and filled with an internal solution containing the following (in mm): K-gluconate 110, KCl 30, MgCl2 1, CaCl2 2, HEPES 10, pH 7.25 with KOH. Amphotericin B (240 µg/ml) was added to the pipette solution. Neuronal excitability was assessed by determining rheobase, the minimum amount of current required to elicit an action potential. Neurons were incubated with CB1R, CB2R, MOR agonists or a combination for 30 min at 37°C before making a recording (no agonist was present during the recording). Neurons were also incubated in corresponding vehicle controls (3.8% methanol for HU-210, 0.1% DMSO for HU-308, 0.03% methyl acetate for JWH-133). All antagonists or inhibitors were applied 15 min before the addition of the agonists.
Retrograde labeling
To enable identification of colon-projecting DRG neurons, surgeries were performed in a subset of mice to allow injection of a retrograde neuronal tracer Fast Blue (17 740-1, Polysciences). Mice were anesthetized, prepared for surgery, and received postsurgery care as described above. The colon was carefully exposed, and Fast Blue (1.7% w/v in sterile water) was injected in small volumes (1-2 µl) into multiple sites on the colon wall. Mice were killed 7-14 d after surgery (10-13 weeks old) and DRGs (T10-L2) were acutely dissociated for patch-clamp recordings.
Extracellular afferent nerve recording
Afferent nerve activity of the lumbar splanchnic nerve innervating the isolated distal colon was recorded as previously described (Yu et al., 2019b). The distal colon (∼3 cm) with attached inferior mesenteric artery was collected and placed in an organ bath continuously superfused with gassed (5% CO2 and 95% O2) Krebs buffer (composition, in mm as follows: NaCl, 118.4; NaHCO3, 24.9; MgSO4, 1.2; KH2PO4, 1.2; glucose, 11.7; CaCl2, 1.9) at 34°C. The colon was cut open along the mesentery border and pinned flat. The lumbar splanchnic nerves were isolated proximal to the inferior mesenteric ganglion and drawn into a glass suction electrode attached to a Neurolog headstage (NL100, Digitimer). Afferent nerve signals were amplified (NL104), filtered (NL125 band pass filter), and recorded on a computer via a Micro 1401 interface and Spike 2 software (version 7, Cambridge Electronic Design). Krebs buffer contained the L-type calcium channel blocker nifedipine (3 µm) and the mAChR antagonist atropine (5 µm) to suppress smooth muscle activity, as well as the cyclooxygenase inhibitor indomethacin (3 µm) to suppress potential inhibitory actions of endogenous prostaglandins (Yu et al., 2019b). Receptive fields were identified by systematically stroking the mucosal surface and mesenteric attachment of the colon with a fine brush. Once identified, receptive fields were tested with three distinct mechanical stimuli to allow classification: probing (1 × g von Frey filament, Remington Medical), mucosal stroking (0.4 × g) and circular stretch (assessed by lightly pulling on the colonic tissue with forceps). Only vascular afferent nerves (i.e., those that only respond to probing of the gut wall or the mesenteric attachment) (Brookes et al., 2013) were included. After a 30 min equilibration period, mechanosensitivity was evaluated by probing of the receptive field using a von Frey filament (1 × g for 3 s, a total of three probes separated by 10 s intervals). Afferent nerve responses to probing were reevaluated after 15 min superfusion of cannabinoid and/or opioid receptor agonists as well as 15 and 30 min after washout. Single-unit analysis was performed offline to discriminate individual afferent nerve activity. Each probing response was calculated as the mean firing frequency over a 3 s period using a custom-made script. Average of three repeated probing responses was used for comparisons.
Pulse oximetry and heart rate (HR) test
Mice were lightly anesthetized with isoflurane (1.5%) on a heating blanket. A pulse oximeter and HR monitor with a paw sensor (MouseSTAT Jr., Kent Scientific) was used to measure oxygen saturation (SpO2) and HR. After a 10 min baseline recording, mice were randomized to receive one of the three treatments (i.p.): morphine (3 mg/kg), ACEA (3 mg/kg), or morphine plus ACEA (both 0.3 mg/kg). Measurements were collected every 5 min until 50 min after injection. The same group of mice received all three treatments (one treatment at the same time of each day) in random order.
Open field test and pelleting test
These two tests were performed simultaneously. Mice were randomized to receive one of the four treatments (i.p.): vehicle (3% methyl acetate), morphine (3 mg/kg), ACEA (3 mg/kg), or morphine plus ACEA (both 0.3 mg/kg). Fecal pellets were counted for 90 min after injection. Thirty minutes after injection, locomotion was recorded for 10 min in an open top box (45 × 45 cm) and processed using the Smart Video Tracking System version 3.0 (Panlab) software. The same group of mice received all treatments (one treatment at the same time each day) in random order.
Drugs and compounds
ACEA (91054, 10 mg/ml in methyl acetate), AM-251 (71670, dissolved in DMSO to 1 mm stock), JWH-133 (10005428, 10 mg/ml in methyl acetate), Nω-propyl-L-arginine (NPL, 80587, dissolved in 1:10 mix of ethanol and DMSO to 1 mm stock), L-NIL (80310, dissolved in distilled water to 10 mm stock), and 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ (81 410, dissolved in DMSO to 10 mm stock) were purchased from Cayman Chemical. HU-210 (H673500, 0.1 mg/ml in methanol) was obtained from Toronto Research Chemical. HU-308 (3088, dissolved in DMSO to 10 mm stock) was from Tocris Bioscience. Morphine sulfate injection USP (5530) was from Sandoz Canada. DAMGO (E7384, dissolved in distilled water to 5 mm stock) was from Millipore-Sigma. All stock solution was kept frozen at −20°C, as per the suppliers' specifications, and diluted to their final concentration before application. Choice of different combinations of CB1R and MOR agonists in in vitro and in vivo experiments was made based on legal restrictions (drug availability), solvent toxicity (methanol for HU-210), agonist specificity, potency, and clinical relevance.
Data analysis and statistics
All data are expressed as mean ± SEM unless otherwise stated. Significant differences were determined by Student's t test (two-tailed) and/or one- or two-way ANOVA with Bonferroni test as appropriate using GraphPad Prism 9. Normality was checked with D'Agostino & Pearson test. When a normality test failed, the Kruskal–Wallis test was used. N refers to number of animals, and n indicates number of cells or afferent units. p < 0.05 was considered significant. p values of the post hoc multiple comparisons (if applicable) are listed in figure legends. Experiments and analysis were not performed blindly.
Results
CB1R activation decreased colonic nociceptive signaling in healthy mice
To examine the analgesic effect of CB1R activation on visceral pain in vivo, a highly specific CB1R agonist ACEA (Hillard et al., 1999) was injected intraperitoneally into conscious mice 20 min before assessment of the VMR to CRD. ACEA dose-dependently decreased VMR (Fig. 1A,B; F(3,18) = 14.55, p < 0.0001, two-way repeated-measures ANOVA with Bonferroni test, N = 7 mice) with significant differences at 80 µl distention at 1 mg/kg (p = 0.0068) and 3 mg/kg (p < 0.0001). In ex vivo extracellular recordings, superfusion of ACEA (1 and 10 µm) significantly reduced afferent nerve mechanosensitivity compared with superfusion of vehicle (Fig. 1D; 1 µm: F(3,24) = 3.317, p = 0.0369, n = 9 units, N = 5 mice; 10 µm: F(3,21) = 5.972, p = 0.0041, n = 8 units, N = 5 mice, respectively, one-way repeated-measures ANOVA with Bonferroni test for both). A lower concentration of ACEA (100 nm) had no effect on mechanosensitivity (F(3,18) = 0.2470, p = 0.8624, n = 7 units, N = 5 mice, one-way repeated-measures ANOVA). Superfusion of another CB1R agonist HU-210 (1 µm) caused significant inhibition of mechanosensitivity compared with vehicle (Fig. 1E; F(3,24) = 4.799, p = 0.0093, one-way repeated-measures ANOVA with Bonferroni test, n = 10 units, N = 6 mice); lower concentrations (10 and 100 nm) had no effect (F(3,30) = 0.1908, p = 0.9018, n = 11 units, N = 5 mice; F(3,28) = 1.865, p = 0.1584, n = 10 units, N = 7 mice, respectively, one-way repeated-measures ANOVA). In agreement with the afferent nerve recordings, HU-210 concentration-dependently decreased the excitability of DRG neurons (Fig. 1F,G; F(3,45) = 4.584, p = 0.0070, n = 10-15 neurons, N = 6 mice, one-way ANOVA with Bonferroni test), with a significantly increased rheobase observed after a 30 min incubation with HU-210 1 µm (p = 0.0440) and 10 µm (p = 0.0047). This inhibitory effect of HU-210 (10 µm) was blocked by a specific CB1R antagonist, AM-251 (1 µm) (Trang et al., 2007) (Fig. 1H; F(2,33) = 6.801, p = 0.0034, one-way ANOVA with Bonferroni test, n = 11-13 neurons, N = 5 mice).
Figure 1.

CB1R agonists ACEA and HU-210 dose-dependently reduced VMR, colonic afferent nerve mechanosensitivity, and DRG neuronal excitability. A, A representative recording showing the effect of ACEA (3 mg/kg) on EMG activity to CRD (10 s, 80 µl). B, ACEA dose-dependently reduced VMR to CRD (N = 7). C, A representative recording showing the effect of ACEA (10 μm) on colonic afferent nerve responses to probing with a 1 × g von Frey filament. D, ACEA (1 and 10 μm) significantly reduced colonic mechanosensitivity to probing (1 μm: p = 0.0494, 10 μm: p = 0.0202), while a lower concentration (100 nm) had no effect (n = 8-10). E, HU-210 (1 μm) significantly reduced colonic mechanosensitivity to probing with von Frey filament (p = 0.0046), while lower concentrations (10 and 100 nm) had no effect (p > 0.9999 for both). F, Representative recordings showing the effect of HU210 on DRG neuron excitability. HU-210 (1 and 10 μm) increased the rheobase (i.e., decrease excitability) of DRG neurons (1 μm: p = 0.044, 10 μm: p = 0.0047) (G), and this effect was blocked by AM-251 (p = 0.0058) (H). *p < 0.05. **p < 0.01. ****p < 0.0001.
CB2R activation did not decrease colonic nociceptive signaling
In contrast to the CB1R agonist, the CB2R agonist HU-308 did not reduce VMR to CRD (Fig. 2A; F(3,21) = 1.255, p = 0.3151, two-way ANOVA, N = 8), with 3 mg/kg HU-308 having a tendency to increase VMR. Superfusion of the CB2R agonist HU-308 (1 and 10 µm) did not alter colonic afferent nerve mechanosensitivity compared with vehicle (Fig. 2B). This effect was mirrored by another specific CB2R agonist, JWH-133 (Huffman et al., 1999), where 1 and 10 µm concentrations had no effect on colonic afferent nerve mechanosensitivity compared with vehicle (Fig. 2C). In patch-clamp recordings, neither HU-308, nor JWH-133, attenuated DRG neuron excitability; a higher concentration of HU-308 (10 µm) increased DRG neuron excitability (Fig. 2D; F(3,38) = 5.941, p = 0.0020, one-way ANOVA with Bonferroni test, n = 10 or 11 neurons, N = 5 mice). JWH-133 had no significant effect on rheobase (Fig. 2E; F(3,40) = 0.7058, p = 0.5542, one-way ANOVA with Bonferroni test, n = 10-13 neurons, N = 5 mice). Together, CB2R agonists did not have an analgesic effect on colonic nociceptive signaling.
Figure 2.

CB2R agonists had no analgesic effect in mice. A, HU-308 did not reduce VMR to CRD. B, HU-308 did not alter colonic afferent nerve mechanosensitivity to probing (1 μm: F(3,24) = 0.0908, p = 0.9644, 10 μm: F(3,28) = 0.1098, p = 0.9537, one-way repeated-measures ANOVA with Bonferroni test, N = 5 or 6 mice for each). C, Similarly, JWH-133 did not alter colonic mechanosensitivity to probing (1 μm: F(3,24) = 1.617, p = 0.2117, 10 μm: F(3,27) = 0.3189, p = 0.8116, one-way repeated-measures ANOVA with Bonferroni test, N = 5 or 6 mice for each). D, Incubation with HU-308 (100 nm and 1 μm) did not significantly alter excitability of DRG neurons (100 nm: p > 0.9999, 1 μm: p > 0.9999), but 10 μm HU-308 decreased the rheobase (i.e., increased excitability; p = 0.0021). E, JWH-133 did not change rheobase of DRG neurons (100 nm: p > 0.9999, 1 μm: p > 0.9999, 10 μm: p = 0.4666). **p < 0.01.
Combination of subanalgesic doses of CB1R and MOR agonists inhibited colonic nociceptive signaling
The next series of experiments examined whether combining a CB1R agonist with a MOR agonist at subanalgesic doses (i.e., a dose of the individual agonist does not inhibit VMR) could reduce visceral pain. First, we examined the effects of morphine alone on visceral pain by performing VMR recording 20 min after injection of vehicle or morphine. Morphine (0.3 mg/kg) did not reduce VMR compared with vehicle, while a higher dose (3 mg/kg) attenuated VMR at both 60 µl (p = 0.0014) and 80 µl (p < 0.0001) distention (Fig. 3A; F(2,8) = 12.03, p = 0.0039, two-way repeated-measures ANOVA with Bonferroni test, N = 5 mice). We then examined the effect of simultaneously administering a combination of the subanalgesic dose of ACEA (0.3 mg/kg, Fig. 1B) with the subanalgesic dose of morphine (0.3 mg/kg) (“ACEA-morphine mix”) on VMR. Interestingly, the ACEA-morphine mix significantly reduced VMR to CRD compared with vehicle (Fig. 3B; F(1,7) = 68.06, p < 0.0001, two-way repeated-measures ANOVA with Bonferroni test, N = 8 mice). To examine whether chronic treatment with a twice daily injection of this mixture resulted in tolerance, mice received the ACEA-morphine mix twice daily for 6 d. VMR was measured 30 min after the first injection on day 1, 3, and 6. The inhibitory effect of the ACEA-morphine mix on VMR was not changed over this time course (Fig. 3C; F(2,8) = 0.4277, p = 0.6661, one-way repeated-measures ANOVA with Bonferroni test, N = 5 mice). We then investigated whether the mixture of subanalgesic concentrations of CB1R and MOR agonists reduced nociceptive signaling at the level of colonic afferent nerves. In ex vivo afferent nerve recordings, superfusion of HU-210 (100 nm) or the MOR agonist DAMGO (1 nm) alone in the same recording had no effect. However, when both agonists were superfused together, there was a significant reduction in mechanosensitivity (Fig. 3D; F(4,36) = 6.046, p = 0.0008, one-way repeated-measures ANOVA with Bonferroni test, n = 10 units, N = 8 mice). Similarly, a combination of subanalgesic concentrations of the two agonists also reduced DRG neuronal excitability. While incubation with HU-210 (100 nm) or DAMGO (1 nm) alone had no effect on DRG neuron excitability (p > 0.9999), a combination of HU-210 and DAMGO, both at subanalgesic concentrations, significantly decreased DRG neuron excitability (Fig. 3E; F(3,39) = 3.629, p = 0.0211, one-way ANOVA with Bonferroni test, n = 10 or 11 neurons, N = 8 mice). Furthermore, while incubation with DAMGO alone at a higher concentration (100 nm) significantly decreased excitability of DRG neurons (116.4 ± 12.6 vs 70.9 ± 9.4 pA, p = 0.0334), we did not observe a greater reduction when combining it with a subanalgesic concentration of HU-210 (100 nm) (116.4 ± 12.6 vs 122.0 ± 13.2 pA, p > 0.9999), suggesting that this synergistic effect may have reached a ceiling. Collectively, these data suggest that a combination of subanalgesic doses of CB1R and MOR agonists reduces visceral pain at the level of nociceptive nerves in the GI tract.
Figure 3.

Combination of subanalgesic dose of CB1R and MOR agonists reduced nociception. A, Morphine (0.3 mg/kg) did not reduce VMR, while a higher dose (3 mg/kg) significantly reduced VMR to CRD (60 µl: p = 0.0014; 80 µl: p < 0.0001). B, A combination of ACEA and morphine at subanalgesic doses (0.3 mg/kg each) significantly reduced VMR to CRD (40 µl: p = 0.0166, 60 µl: p = 0.0023, 80 µl: p < 0.0001). C, A chronic treatment with this ACEA-morphine mix for 6 d given twice daily did not change its inhibitory effect on VMR. D, In male mice, when superfused alone, HU-210 (100 nm) and the MOR agonist DAMGO (1 nm) had no effect on colonic mechanosensitivity. When superfused together, they significantly inhibited colonic mechanosensitivity to probing (p = 0.0002). E, Similarly, when HU-210 (100 nm) and DAMGO (1 nm) were applied alone, there was no change in DRG neuronal excitability. However, the combination of the two agonists significantly decreased DRG neuronal excitability (p = 0.0389). F, In female mice, independent superfusion of ACEA (100 nm) and DAMGO (1 nm) had no effect on colonic mechanosensitivity in afferent nerve recordings. When superfused together, they significantly inhibited colonic mechanosensitivity to probing (p<0.0001). G, In retrogradely labeled colon-projecting DRG neurons from female mice, when low concentrations of HU-210 (100 nm) and DAMGO (1 nm) were applied alone, there was no change in neuronal excitability. However, the combination of the two agonists significantly decreased DRG neuronal excitability (p = 0.0009). *p < 0.05. **p<0.01. ***p < 0.001. ****p < 0.0001.
To enhance the impact of this study, we verified some of the key findings on colon preparations and colon-projecting DRG neurons from female mice. In afferent nerve recordings, superfusion of ACEA (100 nm) or DAMGO (1 nm) alone had no effect. However, when both agonists were superfused together, there was a significant reduction in mechanosensitivity (Fig. 3F; F(4,41) = 11.31, p < 0.0001, one-way ANOVA with Bonferroni test, n = 16 units, N = 8 mice). In patch-clamp recordings from retrogradely labeled colon-projecting DRG neurons, 10 µm HU-210 significantly decreased neuron excitability (111.7 ± 14.1 vs 56.7 ± 5.8 pA, p < 0.01, one-way ANOVA with Bonferroni test, n = 12 neurons, N = 8 mice). While neither 100 nm HU-210 (p > 0.9999, n = 12) nor 1 nm DAMGO alone (p = 0.2378, n = 12 neurons, N = 8 mice) significantly changed the rheobase, combining them significantly decreased DRG neuron excitability (Fig. 3G; F(3,45) = 5.995, p = 0.0016, one-way ANOVA with Bonferroni test, n = 13 neurons, N = 8 mice). Collectively, these data are consistent with our observations on colon afferent nerves and nonlabeled DRG neurons from male mice, suggesting that the synergistic effects of CB1R and MOR agonists on visceral pain signaling are present in both sexes.
In contrast to the combination of CB1R and MOR agonists, coapplication of the CB2R agonist HU-308 (1 µm) and DAMGO (1 nm) had no effect on colonic mechanosensitivity (Fig. 4A; F(3,15) = 0.4751, p = 0.7043, one-way repeated-measures ANOVA, n = 6 units, N = 5 mice) or on DRG neuron excitability (Fig. 4B; F(2,29) = 0.9201, p = 0.4098, one-way ANOVA with Bonferroni test, n = 10-12 neurons, N = 5 mice). Similarly, superfusion of a combination of JWH-133 (1 µm) and DAMGO (1 nm) had no effect on colonic mechanosensitivity (Fig. 4C; F(3,24) = 0.6472, p = 0.5923, one-way repeated-measures ANOVA with Bonferroni test, n = 9 units, N = 6 mice) or DRG neuron excitability (Fig. 4D; F(2,33) = 0.1723, p = 0.8424, one-way ANOVA with Bonferroni test, n = 11-13 neurons, N = 5 mice).
Figure 4.

Combination of CB2R and MOR agonists had no effect on nociception. A, A combination of HU-308 (1 μm) and DAMGO (1 nm) did not reduce colonic mechanosensitivity (p = 0.7043), or DRG neuron excitability (B, p = 0.4098). C, Similarly, combining JWH-133 (1 μm) and DAMGO (1 nm) had no effect on colonic mechanosensitivity (p = 0.5923), nor DRG neuron excitability (D, p = 0.8424).
Role of NO in the inhibition of nociceptive signaling by subanalgesic concentrations of CB1R and MOR agonists in combination
In somatic pain models, combining cannabinoids and opioids reduced pain (Reche et al., 1996; Smith et al., 1998; Cichewicz et al., 1999), but the mechanism of this synergy is unknown. Given that we found ∼80% of colonic afferent nerves were inhibited (>20% inhibition) by the combination of CB1R and MOR agonists at subanalgesic concentrations, while only 30%-40% of DRG neurons express CB1R immunoreactivity (Binzen et al., 2006; Veress et al., 2013), we hypothesized that this synergy may also involve release of a mediator that inhibits neuronal activity. Therefore, we examined the role of NO as both cannabinoids and opioids can signal via NO (Stefano et al., 1995, 1996; Reis et al., 2009; Hervera et al., 2011). While ODQ (10 µm), a selective inhibitor of NO-sensitive guanylyl cyclase, alone did not change the excitability of DRG neurons, it inhibited the synergistic effect of mix of HU-210 (100 nm) and DAMGO (1 nm) (Fig. 5A; F(3) = 11.83, p = 0.0080, Kruskal–Wallis test, n = 10-15 neurons, N = 6 mice). To identify the origin of NO, DRG neurons were preincubated with either NPL, a neuronal NO synthase (nNOS) inhibitor, or L-NIL, an inducible NOS (iNOS) inhibitor. NPL (100 nm) blocked the inhibitory effect of combined HU-210 and DAMGO on DRG neuron excitability (Fig. 5B; F(5,65) = 3.653, p = 0.0056, one-way ANOVA with Bonferroni test, n = 10-14 neurons, N = 6 mice). Although pretreatment with L-NIL (10 µm) reduced the effect of the mix compared with the mix alone (105.8 ± 9.9 vs 80.7 ± 5.5 pA p = 0.1231, n = 11-14 neurons, N = 6 mice), this was not statistically significant. Similarly, NPL (100 nm) significantly reduced the inhibitory effect of the mix of HU-210 and DAMGO on colonic afferent nerve mechanosensitivity (Fig. 5C; F(2,26) = 10.55, p = 0.0004, one-way ANOVA with Bonferroni test, n = 8-11 units, N = 5-9 mice), whereas L-NIL (10 µm) did not have a significant effect .
Figure 5.
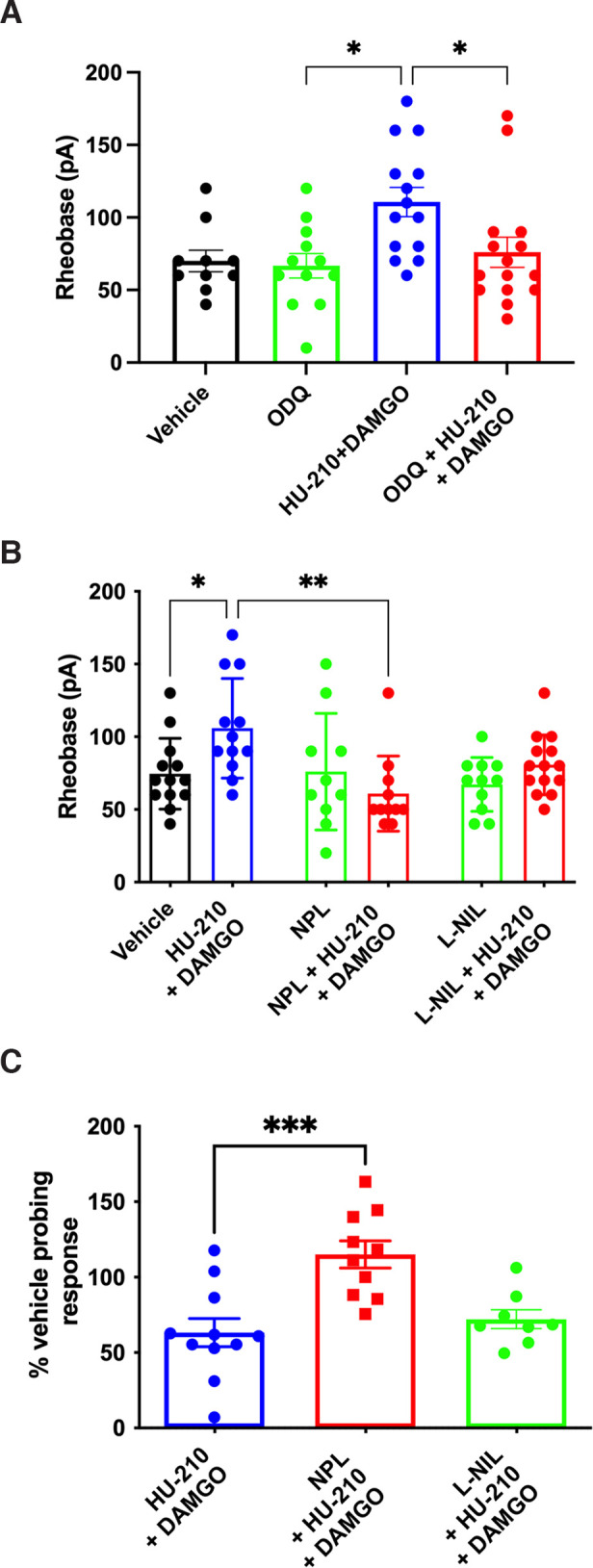
The synergy of CB1R and MOR was mediated via NO. A, The inhibitory effect of the mix of HU-210 and DAMGO on DRG neuron excitability was reversed by ODQ (p = 0.0155). B, The inhibitory effect of the mix on DRG neuron excitability was attenuated by preincubation with NPL (p = 0.0012). The effect of L-NIL was not significant (p = 0.1231). C, In colonic afferent nerve recordings, the inhibitory effect of the mix was reduced by NPL (p = 0.0003) but not L-NIL (p = 0.9695). *p < 0.05. **p < 0.01. ***p < 0.001.
Combination of subanalgesic doses of CB1R and MOR agonists lacked side effects
SpO2 and HR
Readings of SpO2 and HR were taken every 5 min beginning 10 min before administration of CB1R and/or MOR agonists until 50 min after their administration. At analgesic doses, morphine (3 mg/kg) significantly decreased SpO2, whereas ACEA (3 mg/kg) and the ACEA-morphine mix (0.3 mg/kg each) had no effect (Fig. 6A; F(12,36) = 5.869, p < 0.0001, two-way repeated-measures ANOVA with Bonferroni test, N = 5 mice each). Interestingly, ACEA increased HR while morphine decreased HR, but the ACEA-morphine mix had no effect (p > 0.9999 for all time points) (Fig. 6B; F(2,6) = 19.75, p = 0.0023, two-way repeated-measures ANOVA with Bonferroni test, N = 5 mice each).
Figure 6.

Evaluation of side effects of analgesic doses of CB1R agonist, MOR agonist, and subanalgesic doses of CB1R-MOR agonists in combination. A, Morphine (3 mg/kg) significantly reduced oxygen saturation from 10 to 50 min after injection, while ACEA (0.3 mg/kg) or the ACEA-morphine mix (0.3 mg/kg each) had no effect (N = 5). B, ACEA significantly increased HR, while morphine significantly decreased HR, after injection; the ACEA-morphine mix had no effect (N = 5). A, B, Asterisks indicate significant differences compared with time 0. C, D, E, Morphine, ACEA, or the ACEA-morphine mix had no effect on locomotion after injection. F, Morphine significantly reduced pelleting in mice, compared with vehicle (p = 0.0104), while neither ACEA nor the ACEA-morphine mix altered pelleting over 90 min (N = 5). G, The movement patterns of mice did not differ between vehicle or treatment groups (N = 5). *p < 0.05. **p < 0.01. ***p < 0.001. ****p < 0.0001.
Locomotion
Mice were placed in the open-field box for 10 min to assess their locomotion. At analgesic doses, morphine (3 mg/kg), ACEA (3 mg/kg), and the ACEA-morphine mix (0.3 mg/kg each) had no effect on distance traveled (F(3,12) = 2.022, p = 0.1646), mean speed with resting (F(3,12) = 2.032, p = 0.1631), and mean speed without resting (F(3,12) = 0.6936, p = 0.5734) (Fig. 6C–E,G; one-way repeated-measures ANOVA with Bonferroni test, N = 5 mice for all).
Pelleting
Mouse fecal pellets expelled over 90 min were counted in a clean cage, without bedding, to assess GI motility following administration of analgesic doses of morphine (3 mg/kg), ACEA (3 mg/kg), the ACEA-morphine mix (0.3 mg/kg each), or a vehicle control. Morphine significantly reduced pelleting (p = 0.0104), while ACEA (p = 0.5243) and the ACEA-morphine mix (p > 0.9999) had no effect compared with vehicle (Fig. 6F; F(3,12) = 9.053, p = 0.0021, one-way repeated-measures ANOVA with Bonferroni test, N = 5 mice each).
Discussion
Opioid-associated side effects and mortality highlight the need for better pain management strategies. The recent legalization of cannabis in some countries will likely increase the number of patients using cannabis as an alternative or adjunct to opioids, despite a lack of evidence, particularly for visceral pain. This study evaluated the effect of cannabinoids alone or in combination with opioids on colonic nociceptive signaling using both in vivo as well as in vitro techniques. We demonstrated that CB1R, but not CB2R, activation in mice decreased VMR to colorectal distention. Our findings that CB1R agonists reduce colonic afferent nerve mechanosensitivity and DRG neuronal excitability suggest that the reduction in visceral pain in vivo by activation of CB1R occurs, at least in part, at the level of the nerves innervating the colon. Importantly, combining CB1R and MOR agonists at subanalgesic doses/concentrations attenuated colonic nociceptive signaling both acutely and after chronic treatment, while avoiding side effects caused by a higher, analgesic dose of morphine. We further explored the mechanisms underlying the synergy of cannabinoids and opioids, and our findings suggest that NO signaling may be involved.
There is a critical knowledge gap regarding the peripheral actions of cannabinoids on extrinsic afferent nerves innervating the GI tract, as most previous studies used in vivo techniques, where actions may be contaminated by central behavioral effects, or nonselective agonists. We therefore examined the effects of specific CB1R and CB2R agonists on colonic nociceptive signaling using three complementary approaches: in vivo measure of visceral pain, recordings of afferent nerves with terminals in the gut wall, and recordings from the cell bodies of these neurons that reside in the DRG. We found that CB1R, but not CB2R, agonists attenuated VMR, colonic afferent nerve mechanosensitivity, and DRG neuronal excitability in healthy mice. This finding provides strong evidence that peripheral CB1R activation reduces visceral pain. Some peripherally restricted CB1R agonists have been developed to produce analgesic effects on neuropathic pain conditions with reduced CNS-mediated side effects (Hossain et al., 2020), and these agonists may have potential to manage visceral pain. A recent study demonstrated that olorinab, a selective CB2R agonist, did not alter VMR and colonic afferent mechanosensitivity in healthy mice, but significantly reduced nociception in mice with trinitrobenzene sulfonic acid colitis and post-trinitrobenzene sulfonic acid chronic visceral hypersensitivity (Castro et al., 2022). Since trinitrobenzene sulfonic acid colitis did not change CB2R expression, this study suggests that inflammation upregulates the function of CB2R in visceral pain pathways. Interestingly, we found the CB2R agonist HU-308 had a tendency to increase the VMR and had an excitatory effect in DRG neuron excitability at a higher concentration, while it had no effect on colonic afferent nerve mechanosensitivity. Another study showed that the endocannabinoid anandamide had concentration-dependent biphasic effects on tension-sensitive vagal afferents: low concentrations had an inhibitory effect mediated by Gαi/o and protein kinase A, whereas high concentrations displayed an excitatory effect mediated by Gαq and protein kinase C (Christie et al., 2020). However, this study only investigated the involvement of CB1R. We did not observe an excitatory effect on DRG neurons with the other CB2R agonist JWH-133. Taken with the findings of the lack of effect of olorinab on VMR in healthy mice (Castro et al., 2022), this may suggest a nonspecific excitatory effect of HU-308 rather than increased neuronal excitability via CB2R activation. Together, this suggests that activation of CB1R, but not CB2R, has analgesic effects in the noninflamed gut, but this may be altered in the presence of inflammation.
Although several studies have documented synergistic interactions of cannabinoids and opioids in somatic pain models, it was unknown whether this synergy applies to visceral pain and whether it occurs in the periphery (i.e., GI tract), independent of actions of either agonist in the CNS. Our data demonstrated that combined CB1R and MOR agonists at subanalgesic doses/concentrations inhibited visceral pain in vivo and decreased colonic afferent nerve mechanosensitivity and DRG neuron excitability in vitro. Although we cannot completely exclude any central actions of the low-dose combination in vivo, our data suggest that their synergistic actions reduce visceral nociception at peripheral nerve terminals. Importantly, we found that a chronic treatment with this combination did not result in tolerance during the given treatment period, although it may be necessary to assess longer treatment periods in future studies. Thus, this synergy may have great clinical significance to reduce the opioid dose required for analgesia and associated side effects and tolerance. Some clinical studies in chronic pain have suggested that combining cannabinoids and opioids may enhance analgesia. A small-scale clinical trial suggested that combined treatment with opioids and vaporized cannabis enhanced the relief of chronic pain more than the opioid alone (Abrams et al., 2011). Additionally, δ-9-tetrahydrocannabinol (Δ9-THC) resulted in additional analgesia among patients taking opioids for chronic noncancer pain (Narang et al., 2008). A recent Phase II clinical trial evaluated the analgesic effect of oral hydromorphone combined with several oral doses of Δ9-THC; the analgesic effect of hydromorphone was enhanced by the lowest dose of Δ9-THC, although a consistent dose–effect relationship across different pain tests was not observed (Dunn et al., 2021). In contrast, another study found that Δ9-THC did not reduce the need for the opioid piritramide during the 2 d after surgery (Seeling et al., 2006). To our knowledge, there are no clinical studies that have examined combining subanalgesic doses of cannabinoids and opioids to reduce visceral pain. Based on our findings, future clinical studies should examine the ability of cannabinoids to reduce opioid doses used to treat visceral pain.
The mechanisms underlying the synergistic actions of subanalgesic cannabinoids and opioids are unclear, but there is evidence suggesting both paracrine and intracellular signaling pathways. In our study, we examined the potential paracrine actions of NO signaling as a first step, given the evidence that both cannabinoids and opioids inhibit nociception via NO signaling (Stefano et al., 1995, 1996; Reis et al., 2009; Hervera et al., 2011). In our study, we demonstrated that inhibition of NO-sensitive guanylyl cyclase and nNOS, and to a minor degree iNOS, inhibited the synergy of CB1R and MOR, suggesting that nNOS is an important player in this synergy. nNOS is expressed by 20%-50% of DRG neurons in the lower thoracic and upper lumbar levels (Kolesar et al., 2017); and in addition, glial cells may be another source of nNOS (Anneser et al., 2001). In the colon, nNOS immunoactivity can be detected in nerve fibers, enteric neurons, and smooth muscle (Spencer, 2013; Jo et al., 2014). Although future studies should identify the source of nNOS-derived NO, these findings provide novel insights into a mechanism underlying the synergy of cannabinoids and opioids. In addition to NO, endogenous opioids and cannabinoids also act in a neuromodulatory and paracrine manner to mediate analgesic functions. For example, exogenous cannabinoids induce release of endogenous opioids to inhibit nociception and vice versa (Welch, 1993; Houser et al., 2000; Pacheco et al., 2009; Negrete et al., 2011). Furthermore, we cannot rule out the contributions of intracellular signaling convergence, although its contribution is less clear given the dependence on receptor colocalization within the same DRG neurons. We observed that 80% of colonic afferent nerves were inhibited by the subanalgesic combination, which we estimated to be much higher than receptor colocalization on the same neuron as expression of either receptor alone on DRG neurons is only as high as ∼30%-40% (Binzen et al., 2006; Veress et al., 2013; Guerrero-Alba et al., 2018), although this could be an underrepresentation depending on sensitivity of the antibody. Thus, future studies should investigate both intracellular mechanisms of this synergy and the involvement of endogenous cannabinoids and opioids.
Opioids produce a variety of side effects, including sedation, nausea, constipation, and respiratory depression (Elikkottil et al., 2009). Cannabinoids also cause a range of side effects, including disruption of motor control, catalepsy, immobility, and cognitive impairment (Kazantzis et al., 2016). We tested indicators of three common side effects of opioids (blood oxygen saturation, locomotion, and defecation) after acute treatment of analgesic doses of morphine or ACEA alone or the combination of the two at subanalgesic doses. Morphine alone significantly decreased oxygen saturation, HR, and defecation rate. While the analgesic dose of ACEA only increased the HR, other studies suggest that cannabinoids either unalter or decrease oxygen saturation and HR (Abdallah et al., 2018). Some mice also appeared to exhibit reduced locomotion after morphine treatment, but there was no significant change overall. This may be because of the biphasic effect of morphine on locomotion, with high doses increasing and low doses decreasing locomotion (Heidari et al., 2006; Zarrindast et al., 2007). Importantly, we demonstrated that subanalgesic doses of morphine and ACEA in combination achieved a comparable analgesic effect to morphine alone at a higher dose without inducing any of the side effects tested.
In conclusion, the current study has not only shown a peripheral analgesic action of CB1R agonists in the GI tract but importantly that combined subanalgesic doses of CB1R agonists and opioids have similar effects to analgesic doses of opioids. Moreover, the combination appears to lack the side effects commonly seen with opioid analgesics. These studies could set the stage for clinical trials to examine this combination in patients suffering from abdominal pain as a strategy for opioid dose minimization.
Footnotes
D.E.R. was supported by Natural Sciences and Engineering Research Council, Canadian Institutes of Health Research, and Department of Medicine, Queen's University. S.J.V. was supported by Canadian Institutes of Health Research and Crohn's and Colitis Canada operating grants. All the relevant data generated and analyzed during the current study are available upon reasonable request to the corresponding author. We thank Ayssar Tashtush, Dr. Cintya Lopez-Lopez, and Dr. Nestor Jimenez-Vargas for technical assistance.
The authors declare no competing financial interests.
References
- Abdallah SJ, Smith BM, Ware MA, Moore M, Li PZ, Bourbeau J, Jensen D (2018) Effect of vaporized cannabis on exertional breathlessness and exercise endurance in advanced chronic obstructive pulmonary disease: a randomized controlled trial. Ann Am Thorac Soc 15:1146–1158. 10.1513/AnnalsATS.201803-198OC [DOI] [PubMed] [Google Scholar]
- Abrams DI, Couey P, Shade SB, Kelly ME, Benowitz NL (2011) Cannabinoid-opioid interaction in chronic pain. Clin Pharmacol Ther 90:844–851. 10.1038/clpt.2011.188 [DOI] [PubMed] [Google Scholar]
- Anneser JM, Cookson MR, Ince PG, Shaw PJ, Borasio GD (2001) Glial cells of the spinal cord and subcortical white matter up-regulate neuronal nitric oxide synthase in sporadic amyotrophic lateral sclerosis. Exp Neurol 171:418–421. 10.1006/exnr.2001.7756 [DOI] [PubMed] [Google Scholar]
- Binzen U, Greffrath W, Hennessy S, Bausen M, Saaler-Reinhardt S, Treede RD (2006) Co-expression of the voltage-gated potassium channel Kv1.4 with transient receptor potential channels (TRPV1 and TRPV2) and the cannabinoid receptor CB1 in rat dorsal root ganglion neurons. Neuroscience 142:527–539. 10.1016/j.neuroscience.2006.06.020 [DOI] [PubMed] [Google Scholar]
- Brookes SJ, Spencer NJ, Costa M, Zagorodnyuk VP (2013) Extrinsic primary afferent signalling in the gut. Nat Rev Gastroenterol Hepatol 10:286–296. 10.1038/nrgastro.2013.29 [DOI] [PubMed] [Google Scholar]
- Brusberg M, Arvidsson S, Kang D, Larsson H, Lindstrom E, Martinez V (2009) CB1 receptors mediate the analgesic effects of cannabinoids on colorectal distension-induced visceral pain in rodents. J Neurosci 29:1554–1564. 10.1523/JNEUROSCI.5166-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro J, Garcia-Caraballo S, Maddern J, Schober G, Lumsden A, Harrington A, Schmiel S, Lindstrom B, Adams J, Brierley SM (2022) Olorinab (APD371), a peripherally acting, highly selective, full agonist of the cannabinoid receptor 2, reduces colitis-induced acute and chronic visceral hypersensitivity in rodents. Pain 163:e72–e86. 10.1097/j.pain.0000000000002314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Cowan A, Inan S, Geller EB, Meissler JJ, Rawls SM, Tallarida RJ, Tallarida CS, Watson MN, Adler MW, Eisenstein TK (2019) Opioid-sparing effects of cannabinoids on morphine analgesia: participation of CB1 and CB2 receptors. Br J Pharmacol 176:3378–3389. 10.1111/bph.14769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie S, O'Rielly R, Li H, Wittert GA, Page AJ (2020) Biphasic effects of methanandamide on murine gastric vagal afferent mechanosensitivity. J Physiol 598:139–150. 10.1113/JP278696 [DOI] [PubMed] [Google Scholar]
- Cichewicz DL, Martin ZL, Smith FL, Welch SP (1999) Enhancement mu opioid antinociception by oral delta9-tetrahydrocannabinol: dose-response analysis and receptor identification. J Pharmacol Exp Ther 289:859–867. [PubMed] [Google Scholar]
- Clayton N, Marshall FH, Bountra C, O'Shaughnessy CT (2002) CB1 and CB2 cannabinoid receptors are implicated in inflammatory pain. Pain 96:253–260. 10.1016/S0304-3959(01)00454-7 [DOI] [PubMed] [Google Scholar]
- Dunn KE, Bergeria CL, Huhn AS, Speed TJ, Mun CJ, Vandrey R, Campbell CM (2021) Within-subject, double-blinded, randomized, and placebo-controlled evaluation of the combined effects of the cannabinoid dronabinol and the opioid hydromorphone in a human laboratory pain model. Neuropsychopharmacology 46:1451–1459. 10.1038/s41386-021-01007-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elikkottil J, Elikottil J, Gupta P, Gupta K (2009) The analgesic potential of cannabinoids. J Opioid Manag 5:341–357. [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Alba R, Valdez-Morales EE, Jimenez-Vargas NN, Bron R, Poole D, Reed D, Castro J, Campaniello M, Hughes PA, Brierley SM, Bunnett N, Lomax AE, Vanner S (2018) Co-expression of mu and delta opioid receptors by mouse colonic nociceptors. Br J Pharmacol 175:2622–2634. 10.1111/bph.14222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidari P, Sahebgharani M, Riazi G, Zarrindast MR (2006) Influence of morphine and dopamine receptor sensitization on locomotor activity in mice. Pharmacology 78:185–192. 10.1159/000096428 [DOI] [PubMed] [Google Scholar]
- Hervera A, Negrete R, Leanez S, Martin-Campos JM, Pol O (2011) Peripheral effects of morphine and expression of mu-opioid receptors in the dorsal root ganglia during neuropathic pain: nitric oxide signaling. Mol Pain 7:25. 10.1186/1744-8069-7-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillard CJ, Manna S, Greenberg MJ, DiCamelli R, Ross RA, Stevenson LA, Murphy V, Pertwee RG, Campbell WB (1999) Synthesis and characterization of potent and selective agonists of the neuronal cannabinoid receptor (CB1). J Pharmacol Exp Ther 289:1427–1433. [PubMed] [Google Scholar]
- Hong S, Fan J, Kemmerer ES, Evans S, Li Y, Wiley JW (2009) Reciprocal changes in vanilloid (TRPV1) and endocannabinoid (CB1) receptors contribute to visceral hyperalgesia in the water avoidance stressed rat. Gut 58:202–210. 10.1136/gut.2008.157594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain MZ, Ando H, Unno S, Kitagawa J (2020) Targeting peripherally restricted cannabinoid receptor 1, cannabinoid receptor 2, and endocannabinoid-degrading enzymes for the treatment of neuropathic pain including neuropathic orofacial pain. Int J Mol Sci 21:1423. 10.3390/ijms21041423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houser SJ, Eads M, Embrey JP, Welch SP (2000) Dynorphin B and spinal analgesia: induction of antinociception by the cannabinoids CP55,940, Delta (9)-THC and anandamide. Brain Res 857:337–342. 10.1016/s0006-8993(00)01981-8 [DOI] [PubMed] [Google Scholar]
- Huffman JW, Liddle J, Yu S, Aung MM, Abood ME, Wiley JL, Martin BR (1999) 3- (1′,1′-Dimethylbutyl)-1-deoxy-Δ8-THC and related compounds: synthesis of selective ligands for the CB2 receptor. Bioorg Med Chem 7:2905–2914. 10.1016/S0968-0896(99)00219-9 [DOI] [PubMed] [Google Scholar]
- Ingram SL, Fossum EN, Morgan MM (2007) Behavioral and electrophysiological evidence for opioid tolerance in adolescent rats. Neuropsychopharmacology 32:600–606. 10.1038/sj.npp.1301139 [DOI] [PubMed] [Google Scholar]
- Izzo AA, Capasso F, Costagliola A, Bisogno T, Marsicano G, Ligresti A, Matias I, Capasso R, Pinto L, Borrelli F, Cecio A, Lutz B, Mascolo N, Di Marzo V (2003) An endogenous cannabinoid tone attenuates cholera toxin-induced fluid accumulation in mice. Gastroenterology 125:765–774. 10.1016/s0016-5085(03)00892-8 [DOI] [PubMed] [Google Scholar]
- Jo HJ, Kim N, Nam RH, Kang JM, Kim JH, Choe G, Lee HS, Park JH, Chang H, Kim H, Lee MY, Kim YS, Kim JS, Jung HC (2014) Fat deposition in the tunica muscularis and decrease of interstitial cells of Cajal and nNOS-positive neuronal cells in the aged rat colon. Am J Physiol Gastrointest Liver Physiol 306:G659–G669. 10.1152/ajpgi.00304.2012 [DOI] [PubMed] [Google Scholar]
- Kazantzis NP, Casey SL, Seow PW, Mitchell VA, Vaughan CW (2016) Opioid and cannabinoid synergy in a mouse neuropathic pain model. Br J Pharmacol 173:2521–2531. 10.1111/bph.13534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolesar D, Kolesarova M, Kyselovic J (2017) Distribution pattern of dorsal root ganglion neurons synthesizing nitric oxide synthase in different animal species. Can J Physiol Pharmacol 95:328–332. [DOI] [PubMed] [Google Scholar]
- Liang YC, Huang CC, Hsu KS (2007) The synthetic cannabinoids attenuate allodynia and hyperalgesia in a rat model of trigeminal neuropathic pain. Neuropharmacology 53:169–177. 10.1016/j.neuropharm.2007.04.019 [DOI] [PubMed] [Google Scholar]
- Maguire DR, France CP (2014) Impact of efficacy at the µ-opioid receptor on antinociceptive effects of combinations of µ-opioid receptor agonists and cannabinoid receptor agonists. J Pharmacol Exp Ther 351:383–389. 10.1124/jpet.114.216648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondelaers SU, Theofanous SA, Florens MV, Perna E, Aguilera-Lizarraga J, Boeckxstaens GE, Wouters MM (2016) Effect of genetic background and postinfectious stress on visceral sensitivity in Citrobacter rodentium-infected mice. Neurogastroenterol Motil 28:647–658. 10.1111/nmo.12759 [DOI] [PubMed] [Google Scholar]
- Narang S, Gibson D, Wasan AD, Ross EL, Michna E, Nedelikovic SS, Jamison RN (2008) Efficacy of dronabinol as an adjuvant treatment for chronic pain patients on opioid therapy. J Pain 9:254–264. 10.1016/j.jpain.2007.10.018 [DOI] [PubMed] [Google Scholar]
- Negrete R, Hervera A, Leanez S, Martin-Campos JM, Pol O (2011) The antinociceptive effects of JWH-015 in chronic inflammatory pain are produced by nitric oxide-cGMP-PKG-KATP pathway activation mediated by opioids. PLoS One 6:e26688. 10.1371/journal.pone.0026688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen S, Picco L, Murnion B, Winters B, Matheson J, Graham M, Campbell G, Parvaresh L, Khor KE, Betz-Stablein B, Farrell M, Lintzeris N, Le Foll B (2022) Opioid-sparing effect of cannabinoids for analgesia: an updated systematic review and meta-analysis of preclinical and clinical studies. Neuropsychopharmacology 47:1315–1330. 10.1038/s41386-022-01322-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofek O, Karsak M, Leclerc N, Fogel M, Frenkel B, Wright K, Tam J, Attar-Namdar M, Kram V, Shohami E, Mechoulam R, Zimmer A, Bab I (2006) Peripheral cannabinoid receptor, CB2, regulates bone mass. Proc Natl Acad Sci USA 103:696–701. 10.1073/pnas.0504187103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco DD, Klein A, Perez AC, Pacheco CM, de Francischi JN, Reis GM, Duarte ID (2009) Central antinociception induced by mu-opioid receptor agonist morphine, but not delta- or kappa-, is mediated by cannabinoid CB1 receptor. Br J Pharmacol 158:225–231. 10.1111/j.1476-5381.2009.00310.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page AJ, O'Donnell TA, Cooper NJ, Young RL, Blackshaw LA (2009) Nitric oxide as an endogenous peripheral modulator of visceral sensory neuronal function. J Neurosci 29:7246–7255. 10.1523/JNEUROSCI.6099-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reche I, Fuentes JA, Ruiz-Gayo M (1996) Potentiation of delta (9)-tetrahydrocannabinol-induced analgesia by morphine in mice: involvement of mu- and kappa-opioid receptors. Eur J Pharmacol 318:11–16. 10.1016/S0014-2999(96)00752-2 [DOI] [PubMed] [Google Scholar]
- Reis GM, Pacheco D, Perez AC, Klein A, Ramos MA, Duarte ID (2009) Opioid receptor and NO/cGMP pathway as a mechanism of peripheral antinociceptive action of the cannabinoid receptor agonist anandamide. Life Sci 85:351–356. 10.1016/j.lfs.2009.06.012 [DOI] [PubMed] [Google Scholar]
- Sanson M, Bueno L, Fioramonti J (2006) Involvement of cannabinoid receptors in inflammatory hypersensitivity to colonic distension in rats. Neurogastroenterol Motil 18:949–956. 10.1111/j.1365-2982.2006.00819.x [DOI] [PubMed] [Google Scholar]
- Seeling W, Kneer L, Büchele B, Gschwend JE, Maier L, Nett C, Simmet T, Steffen P, Schneider M, Rockemann M (2006) Delta (9)-tetrahydrocannabinol and the opioid receptor agonist piritramide do not act synergistically in postoperative pain. Anaesthesist 55:391–400. 10.1007/s00101-005-0963-6 [DOI] [PubMed] [Google Scholar]
- Sessenwein JL, Baker CC, Pradhananga S, Maitland ME, Petrof EO, Allen-Vercoe E, Noordhof C, Reed DE, Vanner SJ, Lomax AE (2017) Protease-mediated suppression of DRG neuron excitability by commensal bacteria. J Neurosci 37:11758–11768. 10.1523/JNEUROSCI.1672-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira M, Gafni M, Sarne Y (1998) Independence of, and interactions between, cannabinoid and opioid signal transduction pathways in N18TG2 cells. Brain Res 806:26–35. 10.1016/s0006-8993(98)00697-0 [DOI] [PubMed] [Google Scholar]
- Sharkey KA, Wiley JW (2016) The role of the endocannabinoid system in the brain-gut axis. Gastroenterology 151:252–266. 10.1053/j.gastro.2016.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith FL, Cichewicz D, Martin ZL, Welch SP (1998) The enhancement of morphine antinociception in mice by delta9-tetrahydrocannabinol. Pharmacol Biochem Behav 60:559–566. 10.1016/s0091-3057(98)00012-4 [DOI] [PubMed] [Google Scholar]
- Spencer NJ (2013) Characteristics of colonic migrating motor complexes in neuronal NOS (nNOS) knockout mice. Front Neurosci 7:184. 10.3389/fnins.2013.00184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefano GB, Hartman A, Bilfinger TV, Magazine HI, Liu Y, Casares F, Goligorsky MS (1995) Presence of the mu3 opiate receptor in endothelial cells: coupling to nitric oxide production and vasodilation. J Biol Chem 270:30290–30293. 10.1074/jbc.270.51.30290 [DOI] [PubMed] [Google Scholar]
- Stefano GB, Liu Y, Goligorsky MS (1996) Cannabinoid receptors are coupled to nitric oxide release in invertebrate immunocytes, microglia, and human monocytes. J Biol Chem 271:19238–19242. 10.1074/jbc.271.32.19238 [DOI] [PubMed] [Google Scholar]
- Trang T, Sutak M, Jhamandas K (2007) Involvement of cannabinoid (CB1)-receptors in the development and maintenance of opioid tolerance. Neuroscience 146:1275–1288. 10.1016/j.neuroscience.2007.02.031 [DOI] [PubMed] [Google Scholar]
- Veress G, Meszar Z, Muszil D, Avelino A, Matesz K, Mackie K, Nagy I (2013) Characterisation of cannabinoid 1 receptor expression in the perikarya, and peripheral and spinal processes of primary sensory neurons. Brain Struct Funct 218:733–750. 10.1007/s00429-012-0425-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viniol A, Keunecke C, Biroga T, Stadje R, Dornieden K, Bosner S, Donner-Banzhoff N, Haasenritter J, Becker A (2014) Studies of the symptom abdominal pain: a systematic review and meta-analysis. Fam Pract 31:517–529. 10.1093/fampra/cmu036 [DOI] [PubMed] [Google Scholar]
- Welch SP (1993) Blockade of cannabinoid-induced antinociception by norbinaltorphimine, but not N,N-diallyl-tyrosine-Aib-phenylalanine-leucine, ICI 174,864 or naloxone in mice. J Pharmacol Exp Ther 265:633–640. [PubMed] [Google Scholar]
- Wiese B, Wilson-Poe AR (2018) Emerging evidence for cannabis' role in opioid use disorder. Cannabis Cannabinoid Res 3:179–189. 10.1089/can.2018.0022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Park SJ, Beyak MJ (2019a) Inducible nitric oxide synthase-derived nitric oxide reduces vagal satiety signalling in obese mice. J Physiol 597:1487–1502. 10.1113/JP276894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Villalobos-Hernandez EC, Pradhananga S, Baker CC, Keating C, Grundy D, Lomax AE, Reed DE (2019b) Deoxycholic acid activates colonic afferent nerves via 5-HT3 receptor-dependent and -independent mechanisms. Am J Physiol Gastrointest Liver Physiol 317:G275–G284. 10.1152/ajpgi.00016.2019 [DOI] [PubMed] [Google Scholar]
- Zarrindast MR, Heidari-Darvishani A, Rezayof A, Fathi-Azarbaijani F, Jafari-Sabet M, Hajizadeh-Moghaddam A (2007) Morphine-induced sensitization in mice: changes in locomotor activity by prior scheduled exposure to GABAA receptor agents. Behav Pharmacol 18:303–310. 10.1097/FBP.0b013e3282186baa [DOI] [PubMed] [Google Scholar]