

Abstract
Individuals who have Down syndrome (DS) frequently develop early onset Alzheimer's disease (AD), a neurodegenerative condition caused by the buildup of aggregated amyloid-β (Aβ) and tau proteins in the brain. Aβ is produced by amyloid precursor protein (APP), a gene located on chromosome 21. People who have DS have three copies of chromosome 21 and thus also an additional copy of APP; this genetic change drives the early development of AD in these individuals. Here we use a combination of next-generation mouse models of DS (Tc1, Dp3Tyb, Dp(10)2Yey and Dp(17)3Yey) and a knockin mouse model of Aβ accumulation (AppNL-F) to determine how chromosome 21 genes, other than APP, modulate APP/Aβ in the brain when in three copies. Using both male and female mice, we demonstrate that three copies of other chromosome 21 genes are sufficient to partially ameliorate Aβ accumulation in the brain. We go on to identify a subregion of chromosome 21 that contains the gene(s) causing this decrease in Aβ accumulation and investigate the role of two lead candidate genes, Dyrk1a and Bace2. Thus, an additional copy of chromosome 21 genes, other than APP, can modulate APP/Aβ in the brain under physiological conditions. This work provides critical mechanistic insight into the development of disease and an explanation for the typically later age of onset of dementia in people who have AD in DS, compared with those who have familial AD caused by triplication of APP.
SIGNIFICANCE STATEMENT Trisomy of chromosome 21 is a commonly occurring genetic risk factor for early-onset Alzheimer's disease (AD), which has been previously attributed to people with Down syndrome having three copies of the amyloid precursor protein (APP) gene, which is encoded on chromosome 21. However, we have shown that an extra copy of other chromosome 21 genes modifies AD-like phenotypes independently of APP copy number (Wiseman et al., 2018; Tosh et al., 2021). Here, we use a mapping approach to narrow down the genetic cause of the modulation of pathology, demonstrating that gene(s) on chromosome 21 decrease Aβ accumulation in the brain, independently of alterations to full-length APP or C-terminal fragment abundance and that just 38 genes are sufficient to cause this.
Keywords: amyloid precursor protein, amyloid-β, BACE2, Down syndrome, DYRK1A
Introduction
Down syndrome (DS) is caused by trisomy of human chromosome 21 (Hsa21), and occurs in ∼1/1000 live births in Europe (de Graaf et al., 2021). Most individuals who have DS develop the neuropathological features of Alzheimer's disease (AD); amyloid-β (Aβ) plaques and tau neurofibrillary tangles by the age of 50 (Davidson et al., 2018), and 80% of individuals will have developed dementia by age 65 (McCarron et al., 2017). The high prevalence of AD in DS is in part because of the gene encoding amyloid precursor protein (APP) being located on Hsa21, thereby raising APP and Aβ protein levels (Glenner and Wong, 1984; Cheon et al., 2008; Doran et al., 2017). Recent studies in preclinical systems have demonstrated that an extra copy of other genes on Hsa21 can modulate APP biology (Garcia-Cerro et al., 2017; Wiseman et al., 2018, Alic et al., 2020, Tosh et al., 2021) and thus may alter the earliest stages of AD in individuals who have DS. These extra genes may act to promote or to reduce Aβ accumulation; the mechanism that predominates is currently unclear. Notably, the age of clinical dementia diagnosis occurs slightly later in individuals who have DS, compared with those who have early-onset familial AD caused by duplication of APP (i.e., with three copies of WT APP) (Wiseman et al., 2015). However, a direct comparison between these two causes of AD is confounded by the different diagnostic criteria used (as necessitated by the underlying intellectual disability that occurs in people who have DS) (Benejam et al., 2020). Understanding these processes is crucial to the appropriate selection of treatments for AD-primary prevention trials in people who have DS.
Previous in vivo studies have either examined the processing of endogenous mouse APP or used APP transgenic models to address this biology, but both of these approaches have limitations (Garcia-Cerro et al., 2017; Sasaguri et al., 2017; Wiseman et al., 2018, Tosh et al., 2021). Mouse APP differs in sequence from the human protein. In the Aβ region, these differences reduce both cleavage of the protein by β-secretase and the tendency of the Aβ generated to aggregate (Serneels et al., 2020), thus limiting our ability to determine how changes to biology affect accumulation of Aβ, a key early aspect of AD. The over- and mis-expression of APP in transgenic mouse models may cause artifactual phenotypes, masking the modulatory effect of the extra copy of Hsa21 genes and causing elevated mortality, which may confound data interpretation (Saito et al., 2014; Sasaguri et al., 2017).
Here we take a combinatorial approach: assessing the effect of an additional copy of Hsa21 genes, using a series of DS mouse models (O'Doherty et al., 2005; Yu et al., 2010; Lana-Elola et al., 2016) on the biology of endogenous mouse APP and on APP generated from a partially humanized App knock-in allele that also carries AD-causal Swedish (NL) and Iberian (F) point mutations, which does not cause elevated mortality (AppNL-F; Fig. 1) (Saito et al., 2014). These data indicate that trisomy of genes on Hsa21 reduces Aβ accumulation and that people who have DS are partly protected from their raised APP gene dose by the additional copy of other genes on the chromosome. We go on to show that one of the gene or genes that cause this change in biology is located on mouse chromosome 16 between Mir802 and Zbtb21. This region contains 38 genes, and we specifically test whether mechanisms linked to two lead candidate genes in this region, Dyrk1a and Bace2, occur in our novel in vivo AD in DS (AD-DS) model system.
Figure 1.
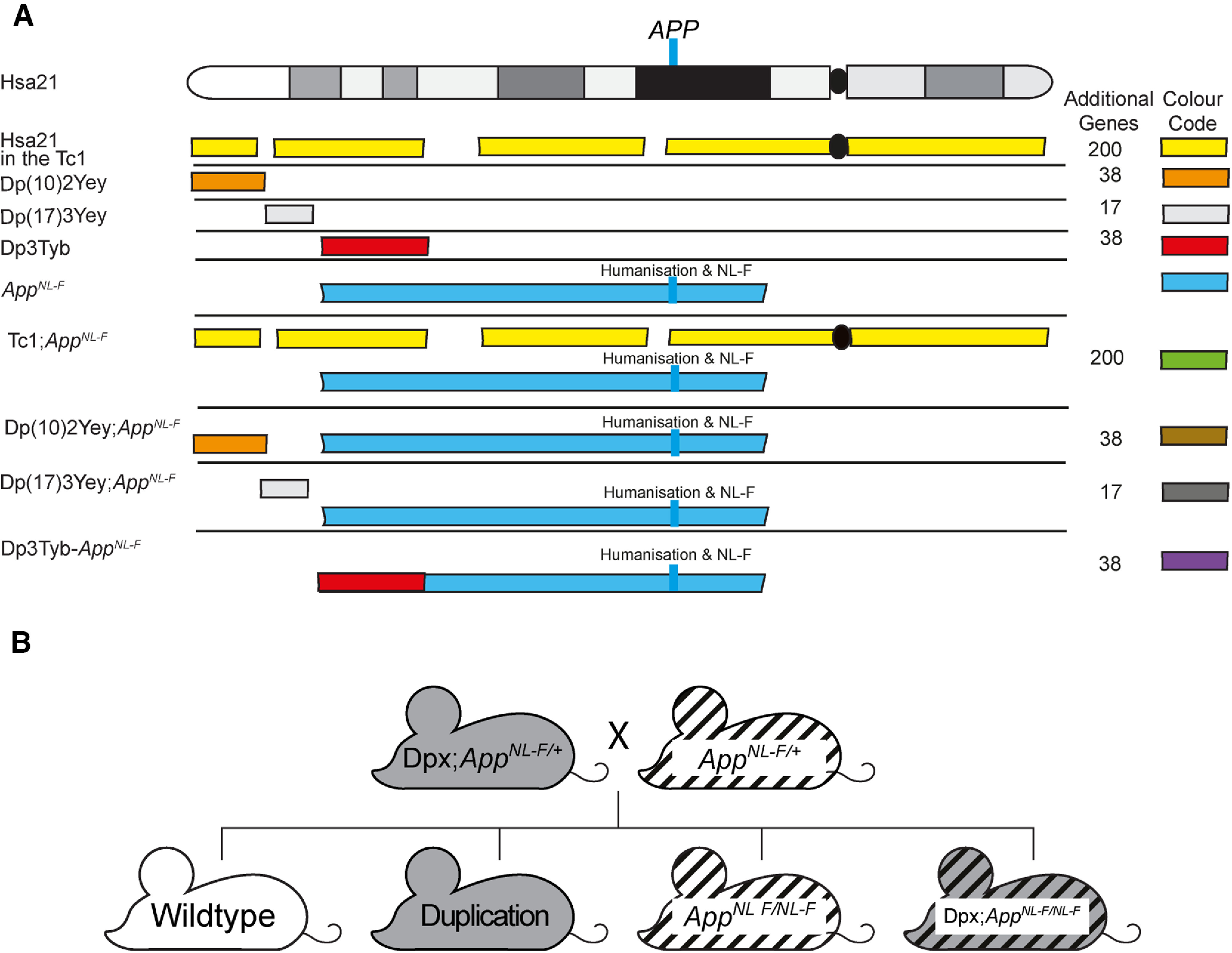
A schematic of Hsa21 indicating the major karyotypic bands, and the regions of Hsa21 or homologous regions of mouse chromosomes that are in three copies in the mouse models used. A, The additional Hsa21 gene content in Tc1 (yellow), in the Dp(10)2Yey (orange), Dp(17)3Yey (light gray), and Dp3Tyb (red) segmental duplication models of DS as shown. The number of additional human genes in the Tc1, and the additional number of mouse genes in the Dp(10)2Yey, Dp(17)3Yey, and Dp3Tyb as listed. These lines were crossed with the AppNL-F mouse model of Aβ accumulation (blue), to generate Tc1;AppNL-F(green), Dp(10)2Yey;AppNL-F (brown), Dp(17)3Yey;AppNL-F(dark gray), and Dp3Tyb-AppNL-F (purple) offspring, homozygous for the AppNL-F allele and carrying the additional gene content. Blue line indicates the location of APP/App. This locus is altered in the AppNL-F model such that the Aβ sequence is humanized and the model carries the AD-causal Swedish (NL) and Iberian (F) mutations. This color scheme is used in subsequent figures to code for each of the mouse models. B, Schematic of the generation of experimental cohorts for the duplication models (Dp(10)2Yey, Dp(17)3Yey, and Dp3Tyb) used in the studies. Experimental cohorts were produced by crossing Dpx;AppNL-F/+ mice with AppNL-F/+ mice.
Materials and Methods
Animal welfare and husbandry
All experiments were undertaken in accordance with the Animals (Scientific Procedures) Act 1986 (United Kingdom), after local institutional ethical review by the Medical Research Council, University College London and in accordance with ARRIVE2 guidelines (Percie du Sert et al., 2020). Mice were housed in individually ventilated cages (Techniplast) with Grade 5, autoclaved dust-free wood bedding, paper bedding, and a translucent red “mouse house” at a specific pathogen-free facility. Free access to food (Picolab Rodent Diet 20 Labdiet) and water was provided. The animal facility was maintained at a constant temperature of 19°C-23°C with 55 ± 10% humidity in a 12 h light/dark cycle. Pups were weaned at 21 d and moved to standardized same-sex group housing with a maximum of 5 mice per cage.
The following mouse strains were used in this paper, showing abbreviated name and then the official name and unique Mouse Genome Informatics (MGI) identifier: AppNL-F (Apptm2.1Tcs, MGI:5637816), Tc1 (Tc(HSA21)1TybEmcf, MGI:3814712), Dp3Tyb (Dp(16Mir802-Zbtb21)3TybEmcf, MGI:5703802), Dp(10)2Yey (Dp(10Prmt2-Pdxk)2Yey, MGI:4461400), and Dp(17)3Yey (Dp(17Abcg1-Rrp1b)3Yey, MGI:4461398).
Tc1 mice were maintained by mating Tc1 females to F1 (129S8 × C57BL/6) males. All other mouse strains were maintained by backcrossing males and females to C57BL/6J mice (imported from The Jackson Laboratory). Experimental cohorts for Tc1, Dp(10)2Yey, and Dp(17)3Yey studies were produced by crossing mice carrying the additional Hsa21 or Hsa21 orthologous duplications with AppNL-F/+ animals in a two-generation cross to generate all required genotypes from the second generation (WT, Tc1/Dpx, AppNL-F/NL-F, Tc1/Dpx;AppNL-F/NL-F; Fig. 1; Table 1). As both the Dp3Tyb segmental duplication and the AppNL-F gene are located on mouse chromosome 16 (Mmu16), for this cross we first generated a Dp3Tyb-AppNL-F recombinant Mmu16, by crossing the two lines together and then back-crossing to C57BL/6J mice to identify recombined Mmu16, carrying both genetic changes on the same chromosome. Mice with the recombined Mmu16 were then crossed with AppNL-F/+ animals to generate Dp3Tyb;AppNL-F/NL-F progeny. For this cross, AppNL-F/NL-F controls were generated from AppNL-F/+ × AppNL-F/+ matings, in addition to rare re-recombinations, resulting in offspring without the Dp3Tyb segmental duplication but two copies of the App knock-in allele. Dp3Tyb controls were generated from Dp3Tyb × C57BL/6J matings generated at the same time as the Dp3Tyb;AppNL-F/NL-F mice. WT controls were taken from all three matings.
Table 1.
Summary of mouse cohortsa
| Cohort | Crosses | Age (mo) | Outcomes | Figure | Female 'n' |
Male 'n' |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | Tc1/Dpx | AppNL-F/NL-F | Tc1/Dpx; AppNL-F/NL-F | WT | Tc1/Dpx | AppNL-F/NL-F | Tc1/Dpx; AppNL-F/NL-F | |||||
| 1 | Tc1;AppNL-F/+ and AppNL-F/+ | 3 | APP and APP-CTF abundance | 3 | 4 | 3 | 2 | 4 | 4 | 5 | 2 | 4 |
| 2 | Dp(10)2Yey;AppNL-F/+ and AppNL-F/+ | 3 | APP and APP-CTF abundance | 8 | 1 | 0 | 0 | 2 | 4 | 8 | 9 | 4 |
| 3 | Dp3Tyb-AppNL-F/+ and AppNL-F/+ Dp3Tyb and C57BL/6J AppNL-F/+ and AppNL-F/+ | 3 | APP, APP-CTF, DYRK1A, and BACE2 abundance | 7, 10, 11 | 6 | 5 | 8 | 5 | 11 | 7 | 7 | 8 |
| Human Aβ fragments (only AppNL-F carriers analyzed) | ||||||||||||
| 4 | Dp3Tyb and C57BL/6J | 3 | Mouse Aβ abundance | 9 | 5 | 7 | NA | NA | 11 | 12 | NA | NA |
| 5 | Tc1;AppNL-F/+ and AppNL-F/+ | 8 | 82E1 plaque counts | 2 | 5 | 5 | 15 | 8 | 0 | 3 | 8 | 10 |
| Human Aβ abundance | ||||||||||||
| 6 | Dp3Tyb-AppNL-F/+ and AppNL-F/+ AppNL-F/+ and AppNL-F/+ | 8 | 82E1 plaque counts | 4–6 | 2 | NA because of breed scheme | 2 | 9 | 2 | NA because of breed scheme | 7 | 5 |
| Human Aβ abundance | ||||||||||||
| 7 | Dp(10)2Yey;AppNL-F/+ and AppNL-F/+ | 8 | 82E1 plaque counts | 4–6 | 2 | 2 | 4 | 6 | 4 | 1 | 5 | 3 |
| Human Aβ abundance | ||||||||||||
| 8 | Dp(17)3Yey;AppNL-F/+ and AppNL-F/+ | 8 | 82E1 plaque counts | 4–6 | 3 | 3 | 7 | 7 | 3 | 3 | 5 | 7 |
| Human Aβ abundance | ||||||||||||
aSummary of the cohorts of mice, the cross(es) used to generate them, the age at which the cohort was killed, the outcome measured, and the figure in which the data are presented.
Animals were killed by exposure to rising carbon dioxide, followed by confirmation of death by dislocation of the neck in accordance with the Animals (Scientific Procedures) Act 1986 (United Kingdom).
Tissue preparation and western blotting
For analysis of protein abundance in the hippocampus and cortex, tissues were dissected under ice-cold PBS before snap freezing. Samples were then homogenized in RIPA buffer (150 mm sodium chloride, 50 mm Tris, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) plus complete protease inhibitors (Calbiochem) by mechanical disruption. Total protein content was determined by Bradford assay or Pierce 660 nm assay (Thermo Fisher Scientific). Samples from individual animals were analyzed separately and were not pooled.
Equal amounts of total brain proteins were then denatured in LDS denaturing buffer (Invitrogen) and β-mercaptoethanol, before separation by SDS-PAGE gel electrophoresis using precast 4%-12% Bis-Tris gels (Invitrogen). Proteins were transferred to nitrocellulose or PVDF membranes before blocking in 5% milk/PBST (0.05% Tween 20) or 5%-10% BSA/PBST. Primary antibodies were diluted in 1% BSA/PBST; HRP-conjugated secondary anti-rabbit, anti-mouse, and anti-goat antibodies (Dako) were diluted 1:10,000 in 1% BSA/PBST. Linearity of antibody binding was confirmed by a twofold dilution series of cortical protein samples. Band density was analyzed using ImageJ. Relative signal of the antibody of interest compared with the internal loading control was then calculated, and the relative signal was then normalized to the mean relative signal of control samples electrophoresed on the same gel. Means of technical replicates were calculated and used for ANOVA, such that biological replicates were used as the experimental unit.
Primary antibodies against C-terminal APP (Sigma A8717, 1:10,000), β-actin (Sigma A5441, 1:60,000), DYRK1A (7D10, Abnova, 1:500), and BACE2 (Abcam ab5670 1:1000) were used.
Biochemical fractionation of mouse brain tissues for the analysis of human Aβ
Cortical proteins were fractionated as described by Shankar et al. (2009). A half cortex was weighed on a microscale and homogenized in 4 volumes of ice-cold TBS (50 mm Tris-HCl, pH 8.0) containing a cocktail of protease and phosphatase inhibitors (Calbiochem) using a handheld mechanical homogenizer and disposable pestles (Anachem). Samples were then transferred to 1.5 ml Microfuge tubes (Beckman Coulter, #357448), balanced by adding more TBS and centrifuged at 175,000 × g with an RC-M120EX ultracentrifuge (Sorvall) fitted with rotor S100AT5 at 4°C for 30 min. Supernatant (the Tris- fraction) was removed and stored at −80°C. The remaining pellet was homogenized in 5 volumes of ice-cold 1% Triton X-100 (Sigma-Aldrich) in TBS (50 mm Tris-HCl, pH 8.0), balanced and centrifuged at 175,000 × g for 30 min at 4°C. The resultant supernatant (the Triton-soluble fraction) was removed and stored at −80°C. The pellet was then resuspended in 8 volumes (by original cortical weight) of TBS (50 mm Tris-HCl, pH 8.0), containing 5 m guanidine HCl and left overnight at 4°C on a rocker to ensure full resuspension, and subsequently stored at −80°C. A Bradford assay or Pierce 660 nm protein assay (Thermo Fisher Scientific) was performed to determine protein concentration.
Biochemical preparation of mouse brain tissues for the analysis of mouse Aβ
A half cortex was weighed on a microscale and homogenized in 3 volumes of ice-cold TBS (50 mm Tris-HCl, pH 8.0) containing a cocktail of protease and phosphatase inhibitors (Calbiochem) using a handheld mechanical homogenizer and disposable pestles (Anachem) based on the method in Holtta et al. (2013). Homogenates were centrifuged at 21,130 × g at 4°C for 1 h, and the resultant supernatant was stored at −80°C for onward analysis.
Quantification of Aβ abundance by meso scale discovery (MSD) assay
Aβ38, Aβ40, and Aβ42 levels were quantified on Multi-Spot 96-well plates precoated with anti-Aβ38, Aβ40, and Aβ42 antibodies using multiplex MSD technology, as described by Wiseman et al. (2018). A 6E10 detection antibody was used to quantify human Aβ and 4G8 detection antibody for the quantification of mouse Aβ. Amounts of Aβ38, Aβ40, and Aβ42 were normalized to the original starting weight of cortical material.
Immunohistochemistry
Half brains were immersion fixed in 10% buffered formal saline (Pioneer Research Chemicals) for a minimum of 48 h before being processed to wax (Leica ASP300S tissue processor). The blocks were trimmed laterally from the midline by ∼0.9-1.4 mm to give a sagittal section of the hippocampal formation. Two 4 µm sections at least 40 µm apart were analyzed. The sections were pretreated with 98% formic acid (FA) for 8 min, followed by washing. The slides were wet loaded onto a Ventana XT for staining (Ventana Medical Systems). The protocol included the following steps: heat-induced epitope retrieval (mCC1) for 30 min in Tris boric acid EDTA buffer, pH 9.0, superblock (8 min) and manual application of 100 µl directly biotinylated mouse monoclonal IgG1 antibody against the N-terminus of Aβ (82E1, IBL, 0.2 µg/ml) for 8 h. Staining was visualized using a ChromoMap DAB kit followed by counterstaining with hematoxylin. The sections were dehydrated, cleared, and mounted in DPX before scanning (Leica SCN400F slide scanner). All images were analyzed using ImageJ and by manual plaque counting by two independent researchers.
Biochemical preparation of mouse brain tissues for mass spectrometry (MS) of Aβ
A half cortex was weighed on a microscale and was homogenized in 5 volumes of tris(hydroxymethyl)aminomethane TBS, pH 7.6, containing cOmplete Protease Inhibitor Cocktail (Roche, catalog #04693116001). For the homogenization, one 5 mm bead per sample was used in a TissueLyser (QIAGEN) for 4 min at 30 Hz. After homogenization, additional TBS with protease inhibitor cocktail was added up to 1 ml and transferred to a new tube to be centrifuged at 31,000 × g for 1 h at 4°C. The pellet was resuspended in 1 ml of 70% FA (v/v), followed by further homogenization in the TissueLyser for 2 min at 30 Hz and subsequent sonication for 30 s. The homogenate was centrifuged again at 31,000 × g for 1 h at 4°C, and the supernatant (FA fraction) was dried down in a vacuum centrifuge.
Initially, 400 µl of 70% FA (v/v) was added to the dried FA fractions, shaken for 30 min at 21°C, and centrifuged at 31,000 × g for 1 h at 4°C. After the removal of the supernatant, neutralization with 8 ml 0.5 m Tris was performed. Immunoprecipitation was performed as previously described with some modifications (Gkanatsiou et al., 2019). Briefly, 50 µl of sheep anti-mouse magnetic beads (Thermo Fisher Scientific) that had previously been linked with 4 µg each of mouse monoclonal 6E10 and 4G8 antibodies (Biolegend) was added to the neutralized FA fraction. This complex was incubated overnight at 4°C in 0.2% Triton X-100 in PBS (v/v). By using an automated magnetic-particle KingFisher ml system (Thermo Fisher Scientific), the samples were then washed with PBS Triton X-100, PBS, and 50 mm ammonium bicarbonate separately before elution in 100 µl 0.5% FA. Eluates were dried down in a vacuum centrifuge and stored at −80°C pending MS analysis.
MS
Liquid chromatography-MS was conducted in a similar manner as described previously (Portelius et al., 2007). Briefly, a nanoflow liquid chromatograph was coupled to an electrospray ionization hybrid quadrupole–orbitrap tandem MS (Dionex Ultimate 3000 system and Q Exactive, both Thermo Fisher Scientific). Samples were reconstituted in 7 µl 8% FA/8% acetonitrile in water (v/v/v) and loaded onto an Acclaim PepMap 100 C18 trap column (length 20 mm; inner diameter 75 µm; particle size 3 µm; pore size 100 Å) for online desalting, and thereafter separated on a reversed-phase Acclaim PepMap RSLC column (length 150 mm, inner diameter 75 µm; particle size 2 µm; pore size 100 Å) (both Thermo Fisher Scientific). Mobile phases were as follows: A, 0.1% FA in water (v/v); and B, 0.1% FA/84% acetonitrile in water (v/v/v). The flow rate was 300 nl/min, and a linear gradient of 3%-40% B for 50 min was applied. The temperature of the column oven was 60°C. Mass spectrometer settings were as follows: positive ion mode; mass-to-charge (m/z) interval 350-1800 m/z units; data-dependent acquisition with 1 precursor ion acquisition (MS) followed by up to 5 fragment ion acquisitions (MS/MS); resolution setting 70,000 (for both MS and MS/MS); number of microscans 1 (MS and MS/MS); target values 106 (MS and MS/MS); maximum injection time 250 ms (MS and MS/MS); fragmentation type was higher-energy collisional dissociation fragmentation; normalized collision energy setting 25; singly charged ions and ions with unassigned charge were excluded for MS/MS selection. Database search (including isotope and charge deconvolution) and label-free quantification were performed with PEAKS Studio version 8.5 (Bioinformatics Solutions) against a custom-made APP database. All suggested fragment mass spectra were evaluated manually. MS signal was normalized to starting weight of the cortex before data analysis.
Statistical analysis and experimental design
All experiments and analyses were undertaken blind to genotype and sex. A 6-digit identification number was allocated to each animal before genotyping, which was used to blind samples. Experimental groups for all experiments were pseudo-randomized using Mendelian inheritance. Mice not carrying the correct combination of alleles (i.e., AppNL-F/+ heterozygous mice) were excluded from the analysis; no other animals were excluded. Some samples were lost from the study because of technical issues during tissue processing to wax or during fractionation. Data were analyzed as indicated in the figure legends by univariate ANOVA with between-subject factors being genetic status (App+/+/AppNL-F/NL-F and/or WT/Hsa21 or WT/Dpx) and sex. Fractionation batch was included as a between-subject factor for analysis of Aβ by MSD assays. The subject means of technical replicates were calculated and used in the ANOVA for western blot and MSD assays, as the number of replicates for which data were available varied between samples. Repeated-measures ANOVA was used for manual plaque counts (combining the data of two independent researchers).
Results
Trisomy of chromosome 21 decreases accumulation of Aβ in the cortex of the AppNL-F mouse model
Following on from our previous studies, which indicated that an additional copy of Hsa21 alters APP biology and the accumulation of Aβ in vivo in an APP transgenic model (Wiseman et al., 2018; Tosh et al., 2021), here we determined whether an additional copy of Hsa21 modulated APP/Aβ biology in the AppNL-F knock-in mouse model. We undertook a two-generation cross of the Tc1 mouse model of DS (O'Doherty et al., 2005), which contains a freely segregating copy of Hsa21 (but not a functional additional copy of APP) (Gribble et al., 2013), with the AppNL-F model to generate four genotypes of mice (WT, Tc1, AppNL-F/NL-F, and Tc1;AppNL-F/NL-F). We quantified the number of 82E1+ Aβ deposits in the cortex of these four genotypes of mice at 8 months of age. 82E1+ deposits were not observed in WT or Tc1 mice, consistent with our previous study (Wiseman et al., 2018). We found a significant decrease in the number of deposits in Tc1;AppNL-F/NL-F compared with AppNL-F/NL-F controls (Fig. 2A,G).
Figure 2.

Trisomy of Hsa21 results in a decrease in Aβ deposition in the AppNL-F/NL-F model. A, G, Aβ deposition (82E1) in the cortex was quantified at 8 months of age in male and female mice by manual plaque counting. A, Representative image of 82E1-stained Aβ deposits (brown) in WT, Tc1, AppNL-F/NL-F, and Tc1;AppNL-F/NL-F mice. G, Significantly fewer Aβ deposits in the cortex were observed in Tc1;AppNL-F/NL-F compared with AppNL-F/NL-F controls (F(1,23) = 24.997, p < 0.001). AppNL-F/NL-F female n = 11, male n = 7, Tc1; AppNL-F/NL-F female n = 4, male n = 6. No Aβ deposits were observed in WT (female n = 5) or Tc1 (female n = 5, male n = 3) age-matched littermate controls (data not shown). B–F, Total cortical proteins were biochemically fractionated and amyloid abundance analyzed by MSD assay. B, No statistically significant difference in the abundance of 5 m guanidine hydrochloride-soluble Aβ42 was observed between Tc1;AppNL-F/NL-F compared with AppNL-F/NL-F controls (F(1,13) = 2.262, p = 0.156). AppNL-F/NL-F female n = 15, male n = 8; Tc1;AppNL-F/NL-F female n = 8, male n = 10. WT and Tc1 samples that do not produce human Aβ were included as negative controls. WT n = 4, of which 1 was below limit of detection (mean = 0.543 pg/mg, SEM = 0.433); Tc1 n = 6, of which 4 samples were below the limit of detection (mean = 2.175 pg/mg, SEM 1.437). C, No difference in the abundance of 1% Triton X-100-soluble Aβ42 was observed between Tc1;AppNL-F/NL-F compared with AppNL-F/NL-F controls (F(1,5) = 0.015, p = 0.907). AppNL-F/NL-F female n = 7, male n = 2 (n = 8 below limit of detection); Tc1;AppNL-F/NL-F female n = 5, male n = 2 (n = 11 below limit of detection). WT and Tc1 samples that do not produce human Aβ were included as negative controls. WT n = 4, of which 2 were below the limit of detection (mean = 0.021 pg/mg, SEM = 0.007); Tc1 n = 6, of which all samples were below the limit of detection. D, Significantly more Tris-soluble Aβ42 was observed in Tc1; AppNL-F/NL-F compared with AppNL-F/NL-F controls (F(1,5) = 10.697, p = 0.022). AppNL-F/NL-F female n = 7, male n = 2 (n = 8 below limit of detection) Tc1;AppNL-F/NL-F female n = 5, male n = 2 (n = 11 below limit of detection). WT and Tc1 samples that do not produce human Aβ were included as negative controls. WT n = 4, of which all were below the limit of detection; Tc1 n = 6, of which all samples were below the limit of detection. E, No difference in the abundance of 5 m guanidine hydrochloride-soluble Aβ40 was observed between Tc1;AppNL-F/NL-F compared with AppNL-F/NL-F controls (F(1,13) = 0.005, p = 0.946). AppNL-F/NL-F female n = 11, male n = 8, n = 4 below the limit of detection; Tc1;AppNL-F/NL-F female n = 7, male n = 5, n = 6 below the limit of detection. WT and Tc1 samples that do not produce human Aβ were included as negative controls. WT n = 4, of which 3 were below the limit of detection (1.870 pg/mg); Tc1 n = 6, of which 5 samples were below the limit of detection (1.150 pg/mg). F, No difference in the abundance of 1% Triton X-100-soluble Aβ40 was observed between Tc1;AppNL-F/NL-F compared with AppNL-F/NL-F control (F(1,3) = 0.000, p = 0.991). AppNL-F/NL-F female n = 4, male n = 0 (n = 17 below the limit of detection); Tc1;AppNL-F/NL-F female n = 4, male n = 2 (n = 14 below the limit of detection). Tris-soluble Aβ40 was not detected in these samples. WT and Tc1 samples that do not produce human Aβ were included as negative controls (Table 2). WT n = 4, of which 2 samples were below limit of detection (mean = 0.090 pg/mg, SEM = 0.005); Tc1 n = 6, of which all samples were below the limit of detection. Error bars indicate SEM, *p < 0.05 and ***p < 0.001. Data points represent independent mice.
We also determined whether trisomy of Hsa21 modulated the biochemical aggregation of Aβ40 and Aβ42 in the cortex at 8 months of age, using biochemical protein fractionation by stepwise homogenization and ultracentrifugation in sequentially more disruptive solutions (Tris-HCl, Tris-HCl 1% Triton X-100, and finally 5 m guanidine hydrochloride). We then quantified human Aβ40 and Aβ42 in each fraction normalized to starting brain weight (6E10 MSD triplex assay). Samples from mice without a humanized App allele, that do not produce human Aβ, were used as negative controls. Aβ42 in the guanidine hydrochloride fraction was not significantly reduced in the presence of the extra copy of Hsa21 (Fig. 2B). A significant increase in Tris-soluble Aβ42 was seen in the cortex of Tc1;AppNL-F/NL-F compared with AppNL-F/NL-F controls (Fig. 2D). However, we note that this effect is being driven by one outlier, and significance is lost if this animal is excluded from the analysis; moreover, this analyte could not be detected in a significant proportion of the samples analyzed. Thus, a replication study is required to determine the validity of this result. No significant difference in Triton-soluble Aβ42 abundance was observed (Fig. 2C). The amount of human Aβ40 in the AppNL-F/NL-F model is very low because of the Iberian mutation in the modified App allele, and this analyte was below the limit of detection in the Tris-soluble fraction and did not significantly differ between genotypes in the Triton and 5 m guanidine hydrochloride fractions (Fig. 2E,F). These data indicate that an additional copy of a Hsa21 gene or genes is sufficient to reduce the deposition of Aβ in the cortex; this may occur via an effect on Aβ formation, clearance, or aggregation.
Decreased Aβ accumulation in the Tc1-AppNL-F/NL-F model does not occur because of a reduction of APP or CTF-β abundance
The significant increase in Tris-soluble Aβ42 in the Tc1;AppNL-F/NL-F cortex suggests that the decrease in Aβ accumulation observed is likely to be caused by a change in peptide aggregation. However, previous studies have suggested that genes on Hsa21, other than APP, can increase APP protein level in vivo and modulate the abundance of the Aβ precursor APP-C-terminal fragment-β (CTF-β) (Garcia-Cerro et al., 2017; Naert et al., 2018; Wiseman et al., 2018). Thus, we used western blotting to determine whether the additional Hsa21 genes altered APP or CTF-β abundance in our new model system. Here, we found no evidence of decreased mouse or human full-length APP (FL-APP) in the cortex of the Tc1 and Tc1;AppNL-F/NL-F compared with WT and AppNL-F/NL-F mice (Fig. 3A,D). We note that significantly less FL-APP is detected in humanized models compared with controls (using antibody A8717); this may reflect a reduction in antibody binding rather than a biological reduction in protein level.
Figure 3.
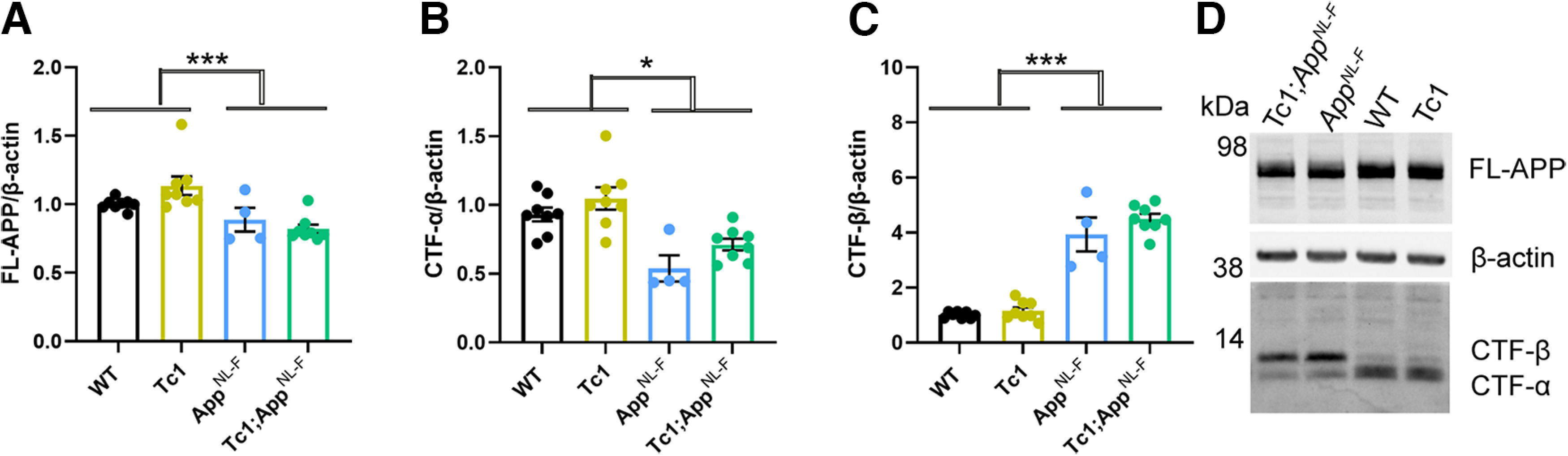
The abundance of FL-APP and CTF is not altered by trisomy of Hsa21. A–D, The relative abundance of FL-APP, APP β-C-terminal fragment (β-CTF), and APP α-C-terminal fragment (α-CTF) compared with β-actin was measured by western blot using A8717 primary antibody in the cortex at 3 months of age in female and male mice. A, Significantly less FL-APP was observed in mice in which App was humanized and mutated (F(1,19) = 23.837, p < 0.001). An additional copy of Hsa21 did not alter FL-APP abundance (F(1,19) = 0.599, p = 0.449). B, Significantly less CTF-α was observed in mice in which App was humanized and mutated (F(1,19) = 5.950, p = 0.025) but an additional copy of Hsa21 did not alter α-CTF abundance (F(1,19) = 3.012, p = 0.099). C, Significantly more β-CTF was observed in mice in which App was humanized and mutated (F(1,19) = 868.431, p < 0.001). By ANOVA, a significant effect of Hsa21 on CTF-β abundance was detected (F(1,19) = 23.462, p < 0.001); however, WT and Tc1 (Bonferroni pairwise comparison p = 1.000) and AppNL-F/NL-F and Tc1;AppNL-F/NL-F (Bonferroni pairwise comparison p = 0.118) were not statistically significant. WT female n = 4, male n = 4: Tc1 female n = 3, male n = 5; AppNL-F/NL-F female n = 2, male n = 2; Tc1;AppNL-F/NL-F female n = 4, male n = 4. D, Representative image of western blot in WT, Tc1, AppNL-F/NL-F, and Tc1;AppNL-F/NL-F mice. Error bars indicate SEM, *p < 0.05 and ***p < 0.001. Data points represent independent mice.
We found a significant increase in CTF-β and a significant decrease in CTF-α in the cortex of AppNL-F/NL-F mice consistent with the reported effects of the introduced mutations on APP processing (Fig. 3B–D) (Saito et al., 2014). An additional copy of Hsa21 did not significantly alter WT APP CTF-β or CTF-α levels in the cortex of the Tc1 compared with WT mice or in the Tc1;AppNL-F/NL-F compared with AppNL-F/NL-F. This finding contrasted with the large increase in CTF-β in male Tc1;APP transgenic model that we previously reported (Wiseman et al., 2018). These data suggest that the reduction in deposition of Aβ in the Tc1;AppNL-F/NL-F model is likely mediated by an enhancement of Aβ clearance or an impairment of peptide aggregation rather than a decrease in APP or CTF-β abundance.
Decreased accumulation of Aβ is caused by an additional copy of 38 Hsa21 orthologous genes
Many DS-associated phenotypes are multigenic, caused by the combined effect of multiple Hsa21 genes acting together on one biological pathway. To understand the mechanisms underlying the decrease in Aβ accumulation in the Tc1;AppNL-F/NL-F model further, we used a series of mouse models of DS that carry an extra copy of subregions of mouse chromosomes that are orthologous with Hsa21, to identify the combination of regions/genes responsible for the changes (Fig. 1) (Yu et al., 2010; Lana-Elola et al., 2016). We used three mouse lines that carry similar gene content to the Tc1 mouse model. However, because of the limitations of available models, we were unable to explore the Hsa21 genes closest to App on Mmu16, which are in three copies in the Tc1 model, as using recombination to generate the required combination of alleles in the available Dp9Tyb model was not feasible.
An extra copy of the genes between Mir802 and Zbtb21 (Dp3Tyb) was sufficient to decrease the accumulation of Aβ in the cortex, as quantified by 82E1 plaque counts (Fig. 4A). However, an extra copy of the genes between Abcg1 and Rrp1b (Dp(10)2Yey) and between Prmt2 and Pdxk (Dp(17)3Yey) was not sufficient to significantly alter the accumulation of Aβ in the cortex, as quantified by 82E1 plaque counts in 8-month-old mice (Fig. 4B,C).
Figure 4.
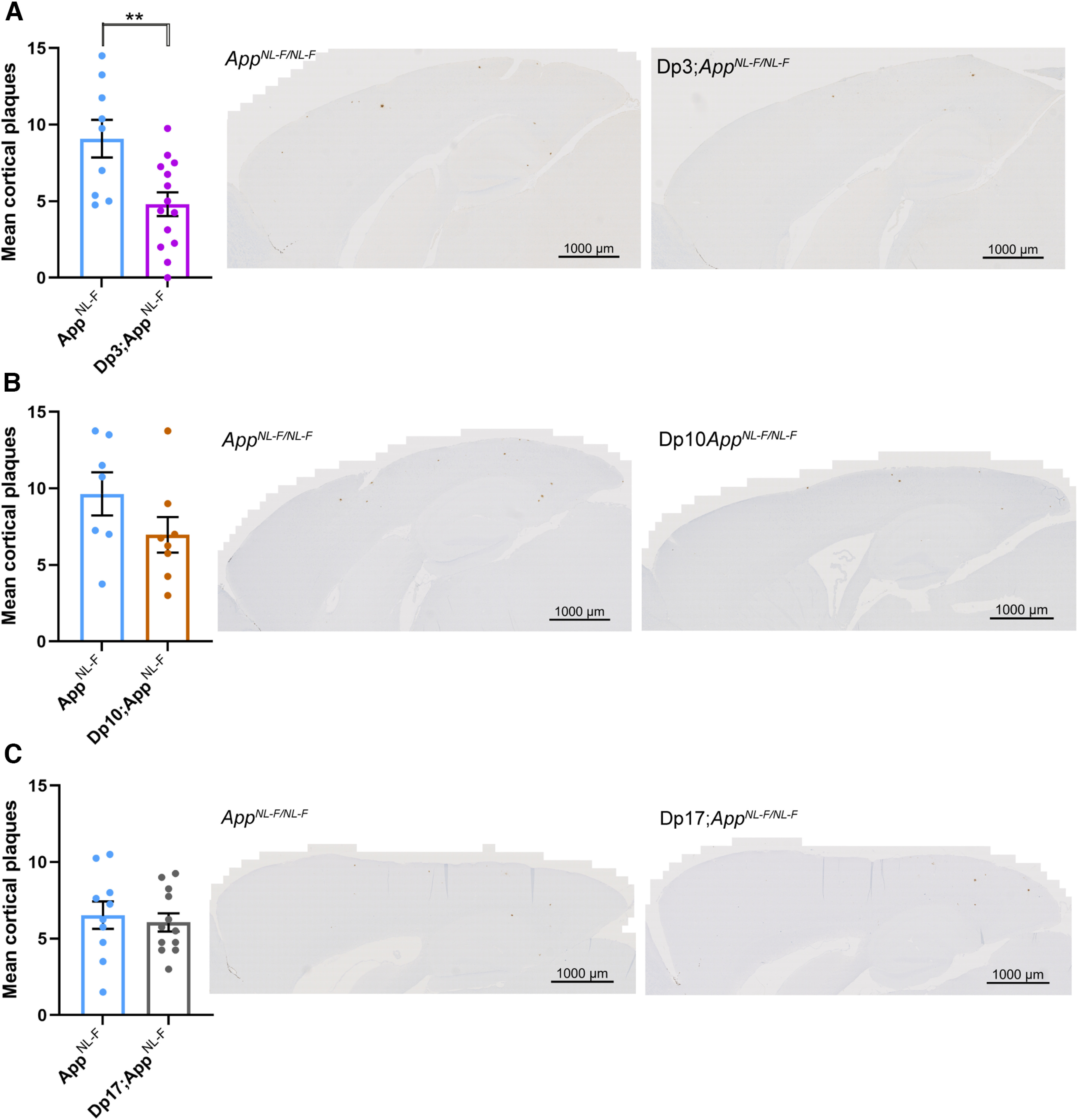
Duplication of the Dp3Tyb region of mouse chromosome 21 orthologous with Hsa21 is sufficient to decrease Aβ accumulation in the AppNL-F/NL-F model. A–C, Aβ deposition (82E1) in the cortex was quantified at 8 months of age in male and female mice by manual plaque counting. A, Significantly fewer Aβ deposits were observed in the cortex of Dp3Tyb;AppNL-F/NL-F compared with AppNL-F/NL-F controls (F(1,18) = 12.359, p = 0.002). AppNL-F/NL-F female n = 2, male n = 7; Dp3Tyb;AppNL-F/NL-F female n = 9, male n = 5. No amyloid deposits were observed in n = 4 WT controls. B, A nonsignificant trend for reduced Aβ deposits in the cortex was observed in Dp(10)2Yey;AppNL-F/NL-F compared with AppNL-F/NL-F controls (F(1,11) = 12.359, p = 0.077). AppNL-F/NL-F female n = 4, male n = 3; Dp(10)2Yey;AppNL-F/NL-F female n = 6, male n = 2. No Aβ deposits were observed in WT n = 6 and Dp(10)2Yey n = 3 controls. C, No difference in Aβ deposits was observed in the cortex of Dp(17)3Yey;AppNL-F/NL-F compared with AppNL-F/NL-F controls (F(1,18) = 0.021, p = 0.885). AppNL-F/NL-F female n = 6, male n = 4; Dp(17)3Yey AppNL-F/NL-F female n = 6, male n = 6. No Aβ deposits were observed in WT n = 6 and Dp(17)3Yey n = 6 controls. For clarity: Dp3, Dp3Tyb; Dp10, Dp(10)2Yey; Dp17, Dp(17)3Yey. Error bars indicate SEM, **p < 0.01. Data points represent independent mice.
The abundance of Aβ42 in the guanidine hydrochloride fraction was not significantly altered in the Dp3Tyb;AppNL-F/NL-F, Dp(10)2Yey;AppNL-F/NL-F, or Dp(17)3Yey;AppNL-F/NL-F mice compared with AppNL-F/NL-F controls at 8 months of age (Fig. 5A–C; Table 2). No significant difference in the abundance of Tris- or Triton-soluble Aβ42 was observed in Dp3Tyb;AppNL-F/NL-F, Dp(10)2Yey;AppNL-F/NL-F, or Dp(17)3Yey;AppNL-F/NL-F compared with controls (Fig. 5D–I). No difference in the abundance of guanidine hydrochloride or Triton-soluble Aβ40 was observed between Dp(10)2Yey;AppNL-F/NL-F or Dp(17)3Yey;AppNL-F/NL-F compared with controls (Fig. 6A–D). These data indicate that a gene or genes in 3 copies in the Dp3Tyb model is sufficient to decrease the deposition of Aβ in the brain.
Figure 5.

Biochemical solubility of Aβ42 is not altered by an additional copy of the Dp3Tyb, Dp(10)2Yey, or Dp(17)3Yey Hsa21 orthologous regions. Total cortical proteins were biochemically fractionated from 8-month-old mice and Aβ abundance analyzed by MSD assay (6E10). No difference in the abundance of (A) 5 m guanidine hydrochloride-soluble Aβ42 (F(1,12) = 0.128, p = 0.726), (D) 1% Triton X-100-soluble Aβ42 (F(1,12) = 2.863, p = 0.116), or (G) Tris-soluble Aβ42 (F(1,10) = 0.281, p = 0.608) was observed between Dp3Tyb;AppNL-F/NL-F compared with AppNL-F/NL-F controls. AppNL-F/NL-F female n = 2, male n = 6; Dp3Tyb;AppNL-F/NL-F female n = 7, male n = 3. No difference in the abundance of (B) 5 m guanidine hydrochloride-soluble (F(1,11) = 2.337, p = 0.115), (E) 1% Triton X-100-soluble Aβ42 (F(1,11) = 0.145, p = 0.711), or (H) Tris-soluble Aβ42 (F(1,11) = 0.001, p = 0.978) was observed between Dp(10)2Yey;AppNL-F/NL-F compared with AppNL-F/NL-F controls. AppNL-F/NL-F female n = 4, male n = 5; Dp(10)2Yey;AppNL-F/NL-F female n = 6, male n = 3. No difference in the abundance of (C) 5 m guanidine hydrochloride-soluble (F(1,20) = 4.079, p = 0.057), (F) 1% Triton X-100-soluble Aβ42 (F(1,20) = 0.124, p = 0.728), or (I) Tris-soluble (F(1,20) = 0.521, p = 0.479) Aβ42 was observed between Dp(17)3Yey;AppNL-F/NL-F compared with AppNL-F/NL-F controls. AppNL-F/NL-F female n = 7, male n = 5; Dp(17)3Yey;AppNL-F/NL-F female n = 7, male n = 7. Details of negative controls, which do not carry an AppNL-F allele in Table 2. For clarity: Dp3, Dp3Tyb; Dp10, Dp(10)2Yey; Dp17, Dp(17)3Yey. Error bars indicate SEM. Data points represent independent mice.
Table 2.
Summary of negative control samples, which do not carry an AppNL-F allele and do not produce human Aβ, used in MSD assaysa
| Figure | Measurement | Genotype | Total 'n' | Below the limit of detection | Above limit of detection |
||
|---|---|---|---|---|---|---|---|
| n | Mean (pg/mg) | SD | |||||
| 5 A | Guanidine HCl Aβ42/ total brain weight | WT | 4 | 4 | 0 | NA | NA |
| 5 A | Guanidine HCl Aβ42/ total brain weight | Dp3Tyb | NA | NA | NA | NA | NA |
| 5 B | Guanidine HCl Aβ42/total brain weight | WT | 6 | 0 | 6 | 0.505 | 0.444 |
| 5 B | Guanidine HCl Aβ42/ total brain weight | Dp(10)2Tyb | 3 | 0 | 3 | 0.59 | 0.411 |
| 5 C | Guanidine HCl Aβ42/ total brain weight | WT | 5 | 4 | 1 | 6.154 | NA |
| 5 C | Guanidine HCl Aβ42/ total brain weight | Dp(17)3Tyb | 5 | 5 | 0 | NA | NA |
| 5 D | 1% Triton Aβ42/total brain weight | WT | 4 | 3 | 1 | 0.002 | NA |
| 5 D | 1% Triton Aβ42/ total brain weight | Dp3Tyb | NA | NA | NA | NA | NA |
| 5 E | 1% Triton Aβ42/ total brain weight | WT | 6 | 5 | 1 | 0.009 | NA |
| 5 E | 1% Triton Aβ42/ total brain weight | Dp(10)2Tyb | 3 | 2 | 1 | 0.007 | NA |
| 5 F | 1% Triton Aβ42/ total brain weight | WT | 5 | 2 | 3 | 0.475 | 0.814 |
| 5 F | 1% Triton Aβ42/ total brain weight | Dp(17)3Tyb | 5 | 3 | 2 | 0.074 | NA |
| 5 G | Tris Aβ42/ total brain weight | WT | 4 | 3 | 1 | 0.015 | NA |
| 5 G | Tris Aβ42/ total brain weight | Dp3Tyb | NA | NA | NA | NA | NA |
| 5 H | Tris Aβ42/total brain weight | WT | 6 | 5 | 1 | 0.006 | NA |
| 5 H | Tris Aβ42/ total brain weight | Dp(10)2Tyb | 3 | 2 | 1 | 0.007 | NA |
| 5 I | Tris Aβ42/ total brain weight | WT | 5 | 2 | 3 | 0.304 | 0.517 |
| 5 I | Tris Aβ42/ total brain weight | Dp(17)3Tyb | 5 | 2 | 3 | 0.011 | 0.009 |
| 6 A | Guanidine HCl Aβ40/total brain weight | WT | 6 | 0 | 6 | 2.097 | 1.246 |
| 6 A | Guanidine HCl Aβ40/ total brain weight | Dp(10)2Tyb | 3 | 0 | 3 | 2.255 | 1.796 |
| 6 B | Guanidine HCl Aβ40/ total brain weight | WT | 5 | 5 | 0 | NA | NA |
| 6 B | Guanidine HCl Aβ40/ total brain weight | Dp(17)3Tyb | 5 | 5 | 0 | NA | NA |
| 6 C | 1% Triton Aβ42/ total brain weight | WT | 6 | 4 | 2 | 0.058 | 0.004 |
| 6 C | 1% Triton Aβ42/ total brain weight | Dp(10)2Tyb | 3 | 1 | 2 | 0.035 | 0.023 |
| 6 D | 1% Triton Aβ42/ total brain weight | WT | 5 | 4 | 1 | 3.241 | NA |
| 6 D | 1% Triton Aβ42/ total brain weight | Dp(17)3Tyb | 5 | 5 | 5 | NA | NA |
aNegative control samples from mice that do not carry the AppNL-F allele and do not produce human Aβ were assayed by 6E10 MSD assay alongside samples homozygous for the AppNL-F allele and which do produce human Aβ. The epitope for 6E10 lies within the region of sequence difference between human and mouse Aβ; the antibody is against the human sequence. In some cases, as indicated, all samples without an AppNL-F allele were below the limit of detection for the assay; thus, mean and SDs are not available (NA).
Figure 6.

Biochemical solubility of Aβ40 is not altered by an additional copy of the Dp(10)2Yey or Dp(17)3Yey Hsa21 orthologous regions. Total cortical proteins were biochemically fractionated from 8-month-old mice and Aβ abundance analyzed by MSD assay (6E10). A, No difference in the abundance of 5 m guanidine hydrochloride-soluble Aβ40 was observed between Dp(10)2Yey;AppNL-F/NL-F compared with AppNL-F/NL-F controls (F(1,12) = 2.137, p = 0.169). AppNL-F/NL-F female n = 6, male n = 3; Dp(10)2Yey;AppNL-F/NL-F female n = 4, male n = 5. B, No difference in the abundance of 5 m guanidine hydrochloride-soluble Aβ40 was observed between Dp(17)3Yey;AppNL-F/NL-F compared with AppNL-F/NL-F controls (F(1,1) = 0.782, p = 0.539). AppNL-F/NL-F female n = 1, male n = 2, n = 9 below limit of detection; Dp(17)3Yey;AppNL-F/NL-F female n = 2, male n = 0, n = 12 below limit of detection. C, No difference in the abundance of 1% Triton X-100-soluble Aβ40 was observed between Dp(10)2Yey;AppNL-F/NL-F compared with AppNL-F/NL-F controls (F(1,11) = 0.540, p = 0.478). AppNL-F/NL-F female n = 6, male n = 3; Dp(10)2Yey;AppNL-F/NL-F female n = 4, male n = 4. D, No difference in the abundance of 1% Triton X-100-soluble Aβ40 was observed between Dp(17)3Yey;AppNL-F/NL-F compared with AppNL-F/NL-F controls (F(1,19) = 0.007, p = 0.982). AppNL-F/NL-F female n = 7, male n = 5; Dp(17)3Yey;AppNL-F/NL-F female n = 7, male n = 6, n = 1 below limit of detection. For details of negative controls, which do not carry an AppNL-F allele, see Table 2. Error bars indicate SEM. Data points represent independent mice. For clarity: Dp10, Dp(10)2Yey; Dp17, Dp(17)3Yey.
Increased DYRK1A does not lead to increased APP or Aβ in the Dp3Tyb model of DS
The Dp3Tyb model contains an additional copy of 38 genes, including Dyrk1a, a kinase that phosphorylates APP (Ryoo et al., 2008), increasing the abundance of the protein in vivo, contributing to raised soluble Aβ abundance in the Ts65Dn mouse model of DS (Garcia-Cerro et al., 2017). We wanted to determine whether we could observe this previously reported biology in our new model system. We found that an extra copy of the Dp3Tyb region raises the abundance of DYRK1A in the cortex of 3-month-old animals, including in the context of AppNL-F knock-in mutations (Fig. 7A,B). This is consistent with numerous previous reports of dosage sensitivity of DYRK1A in the mouse throughout the lifespan (Sheppard et al., 2012; Garcia-Cerro et al., 2017; Yin et al., 2017). We also note that, consistent with previous reports in other mouse model systems, 3 copies of Dyrk1a in the Dp3Tyb;AppNL-F/NL-F model were associated with an increase in cortical weight (Guedj et al., 2012) (see Fig. 10A).
Figure 7.
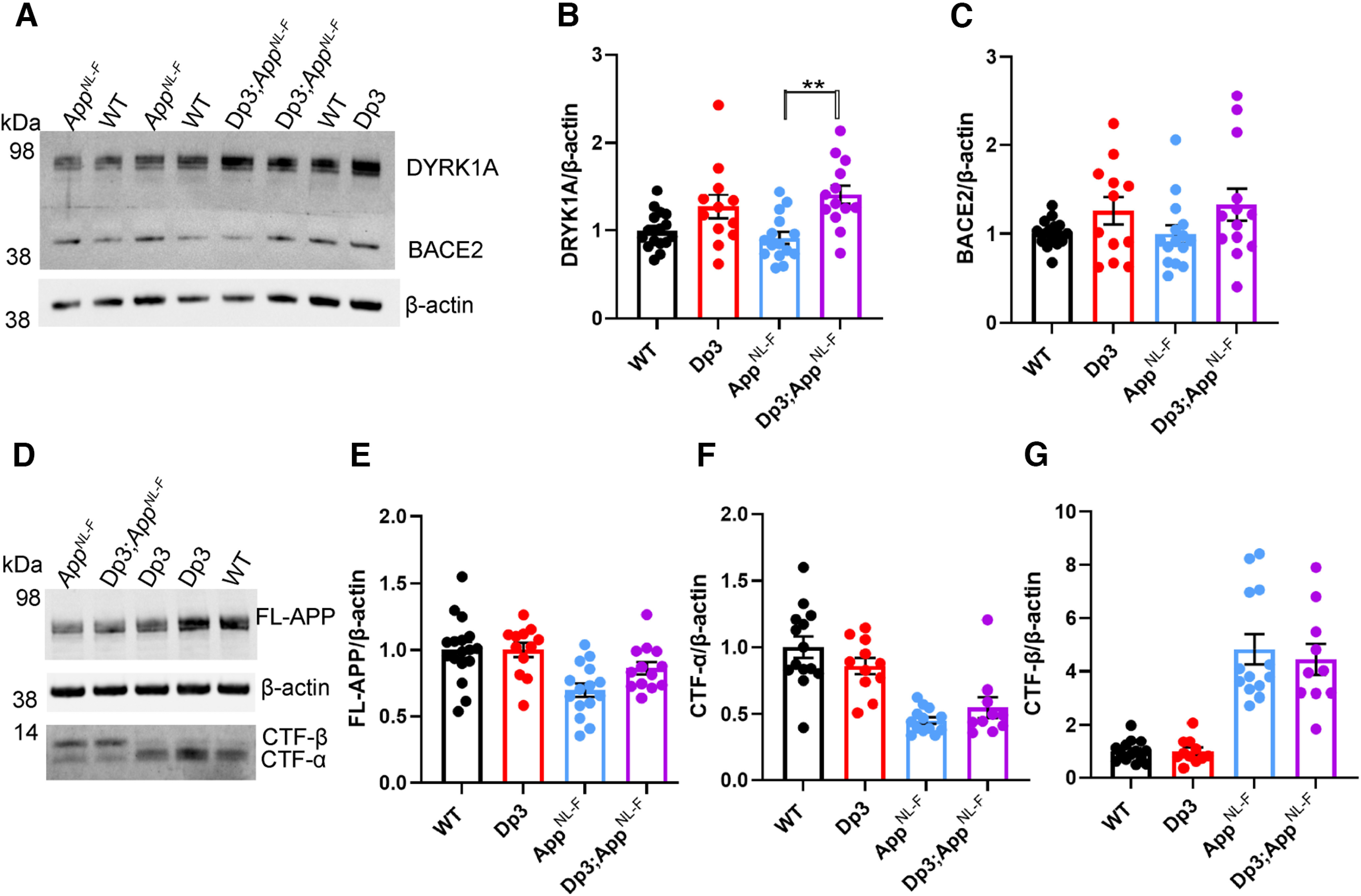
Duplication of the Dp3Tyb region is sufficient to raise the protein abundance of DYRK1A in the cortex but does not significantly alter BACE2, FL-APP, or CTF abundance. The abundance of (A,B) DYRK1A, (A,C) BACE2, (D,E) FL-APP, (D,F) C-terminal fragment-α (CTF-α), and (D,G) CTF-β relative to β-actin loading control was measured by western blot in the cortex at 3 months of age in male and female mice. ANOVA indicated that an additional copy of the Dp3Tyb region increased the abundance of (B) DYRK1A (F(1,49) = 16.511, p < 0.001) and (C) BACE2 F(1,49) = 4.444, p = 0.040). Post hoc pairwise comparison with Bonferroni correction for multiple comparison, demonstrated that significantly higher levels of DYRK1A were observed in Dp3Tyb;AppNL-F/NL-F compared with AppNL-F/NL-F cortex (p = 0.002) but that BACE2 levels did not differ between these two genotypes of mice (p = 0.359). There was no effect of an extra copy of the Dp3Tyb region on the abundance of (E) FL-APP level (F(1,49) = 2.183, p = 0.126), (F) CTF-α (F(1,40) = 0.040, p = 0.843), or (G) CTF-β (F(1,40) = 0.008, p = 0.929). As previously reported (Saito et al., 2014), mice homozygous for the AppNL-F allele had lower abundance of (E) FL-APP (F(1,49) = 16.790, p < 0.001), (F) CTF-α (F(1,40) = 15.739, p < 0.001), and a higher abundance of (G) CTF-β (F(1,40) = 147.440, p < 0.001). WT (female n = 6, male n = 11), Dp3Tyb (female n = 5, male n = 7), AppNL-F (female n = 8, male n = 7), and Dp3Tyb;AppNL-F/NL-F (female n = 5, male n = 8). CTF were below the limit of detection in WT (n = 3), Dp3Tyb (n = 1), AppNL-F (n = 2), and Dp3Tyb;AppNL-F/NL-F (n = 3) samples. Error bars indicate SEM, ** = p < 0.01. Data points represent independent mice. For clarity: Dp3, Dp3Tyb.
Figure 10.
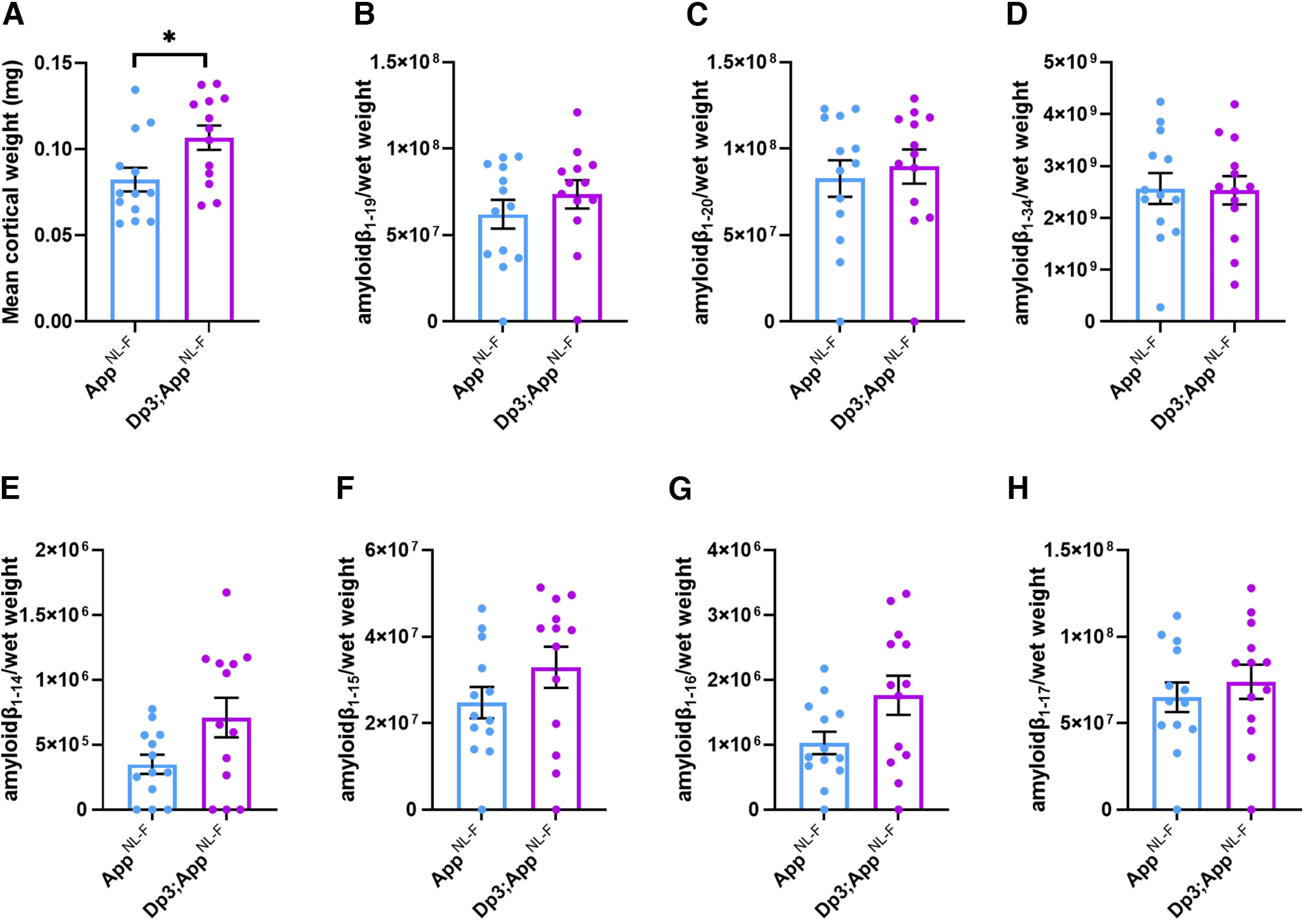
BACE2 Aβ cleaved fragments do not have increased abundance in the young cortex of the Dp3Tyb;AppNL-F/NL-F model. Liquid chromatography-MS analysis of immunoprecipitated cortical Aβ from FA fraction normalized to weight of cortical tissue was used to determine whether the Dp3Tyb region was sufficient to alter the abundance of putative BACE2 cleavage products at 3 months of age. A, Dp3Tyb;AppNL-F/NL-F cortex weighs more than AppNL-F/NL-F cortex at 3 months of age (F(1,22) = 7.772, p = 0.011). AppNL-F/NL-F (female n = 8, male n = 5), Dp3Tyb;AppNL-F/NL-F (female n = 4, male n = 9). No significant increase in the abundance of (B) Aβ1-19, (F(1,20) = 0.166 p = 0.688), (C) Aβ1-20 (F(1,20) = 0.274 p = 0.607), or (D) Aβ1-34 (F(1,20) = 0.005 p = 0.942) was observed. No difference in the abundance of (E) Aβ1-14, (F(1,15) = 1.622, p = 0.222), (F) Aβ1-15, (F(1,19) = 0.496, p = 0.490), (G) Aβ1-16, (F(1,19) = 2.274, p = 0.148), or (H) Aβ1-17, (F(1,19) = 0.079, p = 0.781) was detected in the cortex of Dp3Tyb;AppNL-F/NL-F compared with AppNL-F/NL-F mice. AppNL-F/NL-F (female n = 8, male n = 5), Dp3Tyb;AppNL-F/NL-F (female n = 4, male n = 9). For clarity: Dp3, Dp3Tyb. Error bars indicate SEM, *p < 0.05. Data points represent independent mice. For Aβ1-19 and Aβ1-20, n = 1 sample was below the limit of detection per genotype. Aβ1-14, n = 3 samples were below the limit of detection per genotype, Aβ1-15, 1-16, 1-17, n = 1 samples were below the limit of detection per genotype. These samples are shown on the graphs but were excluded from ANOVA.
However, we found no evidence of changes to the abundance of humanized-APP or mouse-APP in Dp3Tyb;AppNL-F/NL-F or Dp3Tyb models compared with AppNL-F/NL-F controls in the cortex at 3 months of age (Fig. 7D,E). Similarly no change in human-CTF-β or mouse-CTFβ was observed in the Dp3Tyb model (Fig. 7F,G). We also found no change in human/mouse APP or CTF-β abundance in the Dp(10)2Yey;AppNL-F/NL-F mouse (Fig. 8). We found no evidence of changes to total mouse Aβ40 or Aβ42 in young Dp3Tyb compared with WT controls (3 months of age) or insoluble human Aβ40 or Aβ42 in young (3 months of age) Dp3Tyb;AppNL-F/NL-F compared with AppNL-F/NL-F controls (Fig. 9; see Fig. 11). These data suggest that the decreased accumulation of human Aβ in the Dp3Tyb model is not likely to be the result of changed abundance of APP, CTF-β, or Aβ in the young brain; and a mechanism that decreases aggregation or enhances clearance of Aβ may be causal, consistent with the data from the Tc1;AppNL-F/NL-F model.
Figure 8.
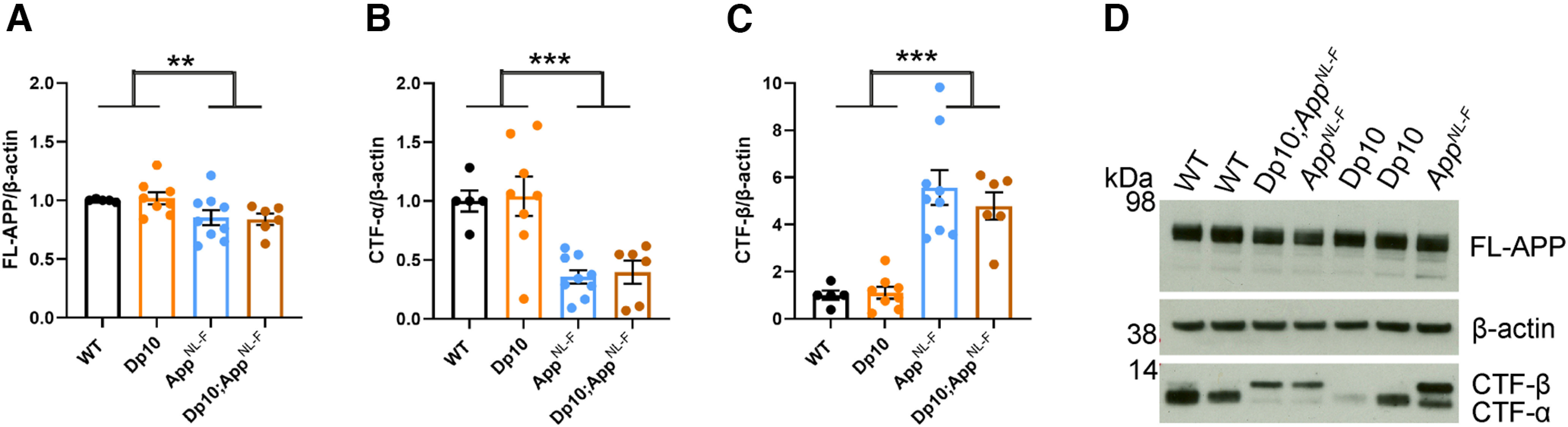
The abundance of FL-APP and CTF is not altered by duplication of the Dp10Yey Hsa21 orthologous region. The abundance of (A,D) FL-APP, (B,D) C-terminal fragment-α (CTF-α) and (C,D) CTF-β relative to β-actin loading control was measured by western blot in the cortex at 3 months of age in male and female mice. There was no effect of an extra copy of the Dp(10)2Yey region on the abundance of (A) FL-APP level (F(1,22) = 0.828, p = 0.372), (B) CTF-α (F(1,22) = 0.054, p = 0.819), or (C) CTF-β abundance (F(1,22) = 0.829, p = 0.372). As previously observed, mice homozygous for the AppNL-F allele had lower abundance of (A) FL-APP (F(1,22) = 8.168, p = 0.009), (B) CTF-α (F(1,22) = 72.150, p < 0.001), and a higher abundance of (C) CTF-β (F(1,22) = 15.300, p < 0.001). WT (female n = 1, male n = 4), Dp(10)2Yey (male n = 8), AppNL-F (male n = 9), and Dp(10)2Yey;AppNL-F/NL-F (female n = 2, male n = 4). Error bars indicate SEM, **p < 0.01 and ***p < 0.001. Data points represent independent mice. For clarity: Dp10, Dp(10)2Yey.
Figure 9.

An additional copy of the Dp3Tyb region does not alter the abundance of mouse Aβ in the young cortex. Total cortical proteins were homogenized in TBS buffer, from mice of 3 months of age, and Aβ abundance was analyzed by MSD assay (4G8). An additional copy of the Dp3Tyb region did not alter the abundance of (A) Aβ40 (F(1,14) = 0.693, p = 0.419) or (B) Aβ42 (F(1,7) = 0.410, p = 0.718). WT (female n = 5, male n = 11), Dp3Tyb (female n = 7, male n = 12). Aβ40 (WT n = 4 and Dp3Tyb n = 5) and Aβ42 (WT n = 8 and Dp3Tyb n = 8) were below the limit of detection. Error bars indicate SEM. Data points represent independent mice. For clarity: Dp3, Dp3Tyb.
Figure 11.

Duplication of the Dp3Tyb region of mouse chromosome 21 orthologous with Hsa21 is not sufficient to decrease Aβ biochemical aggregation in the cortex of the AppNL-F/NL-F model at 3 months of age. Total cortical proteins of 3-month-old mice were prepared for MS analysis and Aβ abundance analyzed in the FA-soluble fraction by MSD assay (6E10). No difference in the abundance of FA-soluble (A) Aβ38 (F(1,21) = 1.001, p = 0.328), (B) Aβ40 (F(1,21) = 0.032 p = 0.860), or (C) Aβ42 (F(1,21) = 0.306, p = 0.443) was detected. AppNL-F/NL-F (female n = 8, male n = 5), Dp3Tyb;AppNL-F/NL-F (female n = 4, male n = 9). For clarity: Dp3, Dp3Tyb. Error bars indicate SEM. Data points represent independent mice.
In Dp3Tyb mice, three copies of Bace2 do not raise BACE2 abundance or decrease Aβ abundance in the young adult brain
The Dp3Tyb model carries an extra copy of Bace2, which encodes a secretase that has been previously reported to cleave APP at the θ site, resulting in the production of Aβ1-19 (Sun et al., 2006; Alic et al., 2020). BACE2 has also been suggested to clear Aβ, leading to reduced accumulation and production of Aβ1-20 and Aβ1-34 in an organoid model of DS (Sun et al., 2006; Alic et al., 2020). Thus, we wanted to investigate BACE2 in our AD-DS model. Using western blotting, we found that an extra copy of the Dp3Tyb region did not cause BACE2 abundance to be significantly higher in the Dp3Tyb;AppNL-F/NL-F compared with AppNL-F/NL-F cortex (Fig. 7A,C), likely because of the underlying high variability in the abundance of this protein in the cortex. We went on to determine whether the putative BACE2 Aβ degradation products (human-Aβ1-20 and human-Aβ1-34) or the APP-θ cleavage product (human-Aβ1-19) were altered by the Dp3Tyb region. No difference in these analytes and human-Aβ1-14, human-Aβ1-15, human-Aβ1-16 or human-Aβ1-17 was observed between Dp3Tyb;AppNL-F/NL-F and AppNL-F/NL-F cortex at 3 months of age (Fig. 10).
As we had observed significant variability in the abundance of BACE2 in the cortex, we investigated whether BACE2 protein levels within individual mice predicted the abundance of Aβ1-19, Aβ1-20, and Aβ1-34 in the same animals. No relationships between the level of BACE2 protein and the analytes were observed (Aβ1-19 R2 = 0.0001, Aβ1-20 R2 = 0.0079, Aβ1-34 R2 = 0.0004). These data suggest that differences in BACE2 protein abundance in the young adult mouse brain are not sufficient to cause a detectable alteration in the clearance of Aβ or enhanced θ cleavage. One caveat is that, at 3 months of age, we cannot yet detect an alteration of the aggregation of human Aβ in the Dp3Tyb;AppNL-F/NL-F, as detected by MSD assay after biochemical isolation in FA (Fig. 11). Thus, our data could be consistent with a BACE2 aging-dependent mechanism leading to the observed decrease in Aβ in the Dp3Tyb;AppNL-F/NL-F model at 8 months of age, but further analysis is required to support this.
Discussion
Here we use a series of mouse models of DS to identify which combination of Hsa21 genes (other than APP) are sufficient to modulate APP/Aβ. Our systematic approach identified a region of Hsa21 that contains 38 genes, which is sufficient to decrease the deposition of Aβ in vivo at an early time point. This region contains two lead candidate genes Bace2 and Dyrk1A.
DYRK1A is widely expressed in both the developing and adult mouse brain (Marti et al., 2003; Hammerle et al., 2008) and is found in all major cell types in both the human and mouse brain (Zhang et al., 2014, 2016). It is a primer kinase that can phosphorylate a large number of proteins, including APP, which has been suggested to increase the protein's abundance (Ryoo et al., 2008; Branca et al., 2017). Inhibition of the kinase in a transgenic mouse model of AD decreased Aβ accumulation (Branca et al., 2017; Velazquez et al., 2019). Consistent with this, a decrease in APP and Aβ abundance is observed in a mouse model of DS with a normalized Dyrk1a gene dose (Garcia-Cerro et al., 2017). In the Dp3Tyb model, which contains an additional copy of Dyrk1a, we observe no increase in APP abundance (endogenous mouse APP and partially humanized APPSW) or evidence of enhanced Aβ accumulation. This suggests that, in our knock-in model system, 3 copies of Dyrk1a are not sufficient to modulate APP protein abundance or promote Aβ accumulation. Notably, inhibition of DYRK1A has been suggested as both a possible therapeutic to enhance cognition in people who have DS and as a possible AD therapeutic strategy (De la Torre et al., 2014; Yin et al., 2017; Neumann et al., 2018; Nguyen et al., 2018). Our data do not support the use of DYRK1A inhibitors as a treatment strategy to decrease early Aβ accumulation in people who have DS. However, inhibiting the kinase may have other beneficial effects, such as to slow or prevent the formation of tau neurofibrillary tangles or to alter the response of cells within the brain to accumulating pathology. Further research is needed, in next-generation AD-DS mouse models, to address these key questions.
BACE2 has been reported to be expressed in a subset of astrocytes and neurons in the human brain (Holler et al., 2012). In the mouse, the gene is expressed in a subset of neurons with the highest level in the CA3 and subiculum, and some expression is also reported in oligodendrocytes and astrocytes lining the lateral ventricles (Voytyuk et al., 2018). The protein may function as a θ-secretase (decreasing CTF-β), a β-secretase (increasing CTF-β), and as an Aβ degrading protease (decreasing Aβ) (Sun et al., 2006; Abdul-Hay et al., 2012); which mechanism predominates in the human brain is not well understood. A previous study in which BACE2 was overexpressed in a WT APP overexpressing mouse model reported no evidence of altered Aβ40 or Aβ42 abundance in the brain (Azkona et al., 2010). Conversely, knocking-down Bace2 in the AppNL-G-F mouse model of Aβ accumulation has been reported to decrease CTF-β and soluble Aβ (Wang et al., 2019). In contrast, reducing BACE2 copy number in human organoids produced from individuals with DS or APP duplication increased Aβ and triggered the formation of deposits within the model system. The authors propose that this occurs because of the amyloid clearance function of BACE2, as evidenced by the raised levels of Aβ degradation products in trisomic compared with isogenic disomic control (Alic et al., 2020).
In the Dp3Tyb model that contains an additional copy of Bace2, we observe no significant change in CTF-β (endogenous mouse APP and partially humanized APPSW), soluble Aβ (endogenous mouse and partially humanized APPSW), or Aβ degradation products in the young adult brain. However, we observe a significant decrease in Aβ deposition in older mice (8 months of age) consistent with the role of BACE2 as an Aβ degrading protease (Alic et al., 2020). Further studies are required in aged mice to determine whether enhanced amyloid clearance can be detected, or whether BACE2 gene-dose correction is sufficient to reverse the reduction in amyloid accumulation caused by the Dp3Tyb model.
Genes other than Dyrk1a or Bace2 may be responsible for the decreased Aβ accumulation observed in our model systems, perhaps via enhancing Aβ clearance pathways. However, we previously studied the rate of extracellular clearance of human Aβ in an alternative model system (cross of the Tc1 with the J20 APP transgenic model) and found no evidence that the Aβ clearance rate was modulated by the extra copy of Hsa21 genes (Wiseman et al., 2018). In addition, in the same model system, we found no evidence of increased abundance of the key clearance enzymes neprilysin or insulin degrading enzyme in the young adult brain. Changes to intracellular clearance/formation of Aβ may contribute to the alterations in accumulation of the peptide reported here. Notably DS is associated with significant alterations to endosomal biology (Botte and Potier, 2020); neuronal endosomes have a key role in Aβ formation and astrocyte and microglia endosomes in Aβ clearance. Further studies focusing on cell type-specific effects of extra chromosome 21 genes are required to understand this complex and important biology.
DS is caused by the increase in the abundance of Hsa21 gene products; however, not all genes on the chromosome are dosage-sensitive in all contexts. For example, APP dosage sensitivity has been reported to vary over the lifespan (Choi et al., 2009), and BACE2 has been reported to be both dosage-sensitive (Barbiero et al., 2003) and insensitive in the human brain (Cheon et al., 2008; Holler et al., 2012). In contrast, the abundance of DYRK1A is highly sensitive to gene dose in the vast majority of reported studies (Arbones et al., 2019), including previously in the adult brain of the Tc1 mouse model (Sheppard et al., 2012; Ahmed et al., 2013; Naert et al., 2018). Consistent with this, here we report a significant increase in DYRK1A in the adult brain of the new Dp3Tyb model. However, we could not detect a significant increase in BACE2; this lack of statistical significance is likely caused by the higher intra-animal variation in abundance of this protein in the mouse brain. Similar overlapping levels of Bace2 gene expression have previously been reported at RNA level in the brain of an alternative mouse model of DS compared with euploid controls (Sultan et al., 2007).
The relative abundance of proteins in AD-DS model systems is particularly important, as DS is caused by an imbalance in the relative abundance of gene products. Previous research using APP transgenic models, which overexpress APP protein, may mask the subtle but physiologically relevant effects of the 50% increase in the abundance of other Hsa21 gene products. This study addresses this limitation; here we systematically investigate the effect of additional copies of Hsa21 gene orthologs, other than App, on APP biology in a knock-in mouse model system. However, because of technical limitations, we were not able to study the effect of an extra copy of genes located close to App, including the role of key genes, such as Adamts1 (Kunkle et al., 2019) and Usp25 (Zheng et al., 2021). We were also unable to determine whether genes located far apart on the chromosome act synergistically to cause a modulation of APP biology. Thus, our data do not preclude a multigenic role for the genes in the Dp(10)2Yey or Dp(17)3Yey regions when combined with other Hsa21 genes. A further limitation is the use of AD-associated mutations in APP to drive pathology as these mutations alter the subcellular localization and processing of APP (Sasaguri et al., 2017), which may modulate the effect of trisomy of Hsa21 on these processes. We have partially addressed this issue by undertaking a side-by-side comparison of mouse APP/Aβ and partially humanized APP/Aβ.
We studied two time points in our model systems: (1) the young adult brain at 3 months of age, before the accumulation of human Aβ in the AppNL-F to understand the modulation of early upstream processes by the additional copies of Hsa21 orthologs; and (2) the middle-aged adult brain at 8 months of age, which, in the AppNL-F mouse, models the initiation of Aβ accumulation (Saito et al., 2014). Thus, in this study, we focus on how additional copies of Hsa21 gene orthologs modulate the earliest stages of AD pathology, the formation and early accumulation of Aβ in the brain. We note that the effect of the additional genes may differ later in disease, including during the maturation of Aβ plaques, the formation of tau neurofibrillary tangles, and the response of astrocytes, microglia and neurons to AD pathology. We studied both male and female mice, to ensure the generalizability of our findings, as sex is known to modulate APP/Aβ in mouse models (Jankowsky and Zheng, 2017). We did not find evidence that sex modulated the effect of additional Hsa21 genes on amyloid accumulation in either the Tc1 or Dp3 models.
Our results indicate that an additional copy of genes on Hsa21 in people who have DS can modulate key AD biology. The extent of this effect is likely to differ between individuals both because of genetic variation on Hsa21 and differences in lifestyle factors that modulate dementia risk. This may contribute to the variation in age of onset of both pathology and dementia observed in people who have DS.
In conclusion, DS is a complex condition that alters multiple aspects of neurobiology and physiology. Here we demonstrate using physiological mouse models that an additional copy of Hsa21 genes reduces the accumulation of Aβ within the brain, one of the earliest steps in the AD pathogenic process. Thus, trisomy of Hsa21 may partially protect individuals who have DS from the accumulation of Aβ, resultant from their extra copy of APP. Moreover, we predict that individuals who have DS will accumulate Aβ more slowly than other groups who develop other genetic forms of early-onset AD, including that caused by duplication of APP. However, multiple studies of adults who have DS demonstrate that these individuals do accumulate substantial Aβ within their brains by mid-life; thus, the effect of an additional copy of other genes on the chromosome is not sufficient to delay pathology development, and AD primary prevention therapies targeting Aβ are likely to significantly benefit individuals who have DS (Fortea et al., 2021). Treatments for AD-DS and other medical conditions associated with DS must take into account the differences in biology of individuals with the syndrome to ensure that interventions do not reverse the beneficial effect of the additional genes.
Footnotes
F.K.W., H.Z. and P.M. were supported by the UK Dementia Research Institute, which receives its funding from DRI Ltd, funded by the UK Medical Research Council, Alzheimer's Society and Alzheimer's Research UK UKDRI-1014, and by Alzheimer's Research UK Senior Research Fellowship ARUK-SRF2018A-001. F.K.W. also received funding that contributed to the work in this paper from the MRC via CoEN award MR/S005145/1. J.T. was supported by an Alzheimer's Society PhD studentship awarded to F.K.W. and E.M.C.F. This work was also supported by Wellcome Trust Strategic Award Grant 098330/Z/12/Z awarded to the London Down Syndrome (LonDownS) Consortium (V.T., and E.M.C.F); Wellcome Trust Joint Senior Investigators Award Grants 098328 and 098327 to V.T. and E.M.C.F.; and Medical Research Council Program U117527252 to V.T. V.T. was also supported by Francis Crick Institute, which receives its core funding from Medical Research Council FC001194, Cancer Research UK FC001194, and Wellcome Trust FC001194. H.Z. is a Wallenberg Scholar supported by Swedish Research Council Grant 2018-02532, European Research Council Grant 681712, Swedish State Support for Clinical Research Grant ALFGBG-720931, Alzheimer Drug Discovery Foundation Grant 201809-2016862, AD Strategic Fund and Alzheimer's Association Grants DSF-21-831376-C, ADSF-21-831381-C, and ADSF-21-831377-C, the Olav Thon Foundation, the Erling-Persson Family Foundation, Stiftelsen för Gamla Tjänarinnor, Hjärnfonden, Sweden FO2019-0228, the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie Grant Agreement 860197 (MIRIADE), and the UK Dementia Research Institute at UCL. K.B. was supported by Swedish Research Council 2017-00915, Swedish Alzheimer Foundation AF-742881, Hjärnfonden, Sweden FO2017-0243, Swedish state under the agreement between the Swedish government and the County Councils, ALF Agreement ALFGBG-715986, and Alzheimer's Association 2021 Zenith Award ZEN-21-848495. For the purpose of open access, the author has applied a CC-BY public copyright license to any Author Accepted Manuscript version arising from this submission. All authors read and approved the final manuscript. We thank Dr. Amanda Heslegrave (UCL) for assistance with this project.
H.Z. has served at scientific advisory boards and/or as a consultant for Abbvie, Alector, Annexon, Artery Therapeutics, AZTherapies, CogRx, Denali, Eisai, Nervgen, Pinteon Therapeutics, Red Abbey Labs, Passage Bio, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics, and Wave; has given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure, and Biogen; and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program (outside submitted work). K.B. has served as a consultant, at advisory boards, or at data monitoring committees for Abcam, Axon, Biogen, JOMDD/Shimadzu, Julius Clinical, Lilly, MagQu, Novartis, Prothena, Roche Diagnostics, and Siemens Healthineers; and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program, all unrelated to the work presented in this paper. The remaining authors declare no competing financial interests.
References
- Abdul-Hay SO, Sahara T, McBride M, Kang D, Leissring MA (2012) Identification of BACE2 as an avid ss-amyloid-degrading protease. Mol Neurodegener 7:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed MM, Dhanasekaran AR, Tong S, Wiseman FK, Fisher EM, Tybulewicz VL, Gardiner KJ (2013) Protein profiles in Tc1 mice implicate novel pathway perturbations in the Down syndrome brain. Hum Mol Genet 22:1709–1724. 10.1093/hmg/ddt017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alic I, et al. (2020) Patient-specific Alzheimer-like pathology in trisomy 21 cerebral organoids reveals BACE2 as a gene dose-sensitive AD suppressor in human brain. Mol Psychiatry 26:5766–5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbones ML, Thomazeau A, Nakano-Kobayashi A, Hagiwara M, Delabar JM (2019) DYRK1A and cognition: a lifelong relationship. Pharmacol Ther 194:199–221. 10.1016/j.pharmthera.2018.09.010 [DOI] [PubMed] [Google Scholar]
- Azkona G, Levannon D, Groner Y, Dierssen M (2010) In vivo effects of APP are not exacerbated by BACE2 co-overexpression: behavioural characterization of a double transgenic mouse model. Amino Acids 39:1571–1580. 10.1007/s00726-010-0662-8 [DOI] [PubMed] [Google Scholar]
- Barbiero L, Benussi L, Ghidoni R, Alberici A, Russo C, Schettini G, Pagano SF, Parati EA, Mazzoli F, Nicosia F, Signorini S, Feudatari E, Binetti G (2003) BACE-2 is overexpressed in Down's syndrome. Exp Neurol 182:335–345. 10.1016/S0014-4886(03)00049-9 [DOI] [PubMed] [Google Scholar]
- Benejam B, Videla L, Vilaplana E, Barroeta I, Carmona-Iragui M, Altuna M, Valldeneu S, Fernandez S, Gimenez S, Iulita F, Garzon D, Bejanin A, Bartres-Faz D, Videla S, Alcolea D, Blesa R, Lleo A, Fortea J (2020) Diagnosis of prodromal and Alzheimer's disease dementia in adults with Down syndrome using neuropsychological tests. Alzheimers Dement (Amst) 12:e12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botte A, Potier MC (2020) Focusing on cellular biomarkers: the endo-lysosomal pathway in Down syndrome. Prog Brain Res 251:209–243. [DOI] [PubMed] [Google Scholar]
- Branca C, Shaw DM, Belfiore R, Gokhale V, Shaw AY, Foley C, Smith B, Hulme C, Dunckley T, Meechoovet B, Caccamo A, Oddo S (2017) Dyrk1 inhibition improves Alzheimer's disease-like pathology. Aging Cell 16:1146–1154. 10.1111/acel.12648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheon MS, Dierssen M, Kim SH, Lubec G (2008) Protein expression of BACE1, BACE2 and APP in Down syndrome brains. Amino Acids 35:339–343. 10.1007/s00726-007-0618-9 [DOI] [PubMed] [Google Scholar]
- Choi JH, Berger JD, Mazzella MJ, Morales-Corraliza J, Cataldo AM, Nixon RA, Ginsberg SD, Levy E, Mathews PM (2009) Age-dependent dysregulation of brain amyloid precursor protein in the Ts65Dn Down syndrome mouse model. J Neurochem 110:1818–1827. 10.1111/j.1471-4159.2009.06277.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson YS, Robinson A, Prasher VP, Mann DM (2018) The age of onset and evolution of Braak tangle stage and Thal amyloid pathology of Alzheimer's disease in individuals with Down syndrome. Acta Neuropathol Commun 6:56. 10.1186/s40478-018-0559-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf G, Buckley F, Skotko BG (2021) Estimation of the number of people with Down syndrome in Europe. Eur J Hum Genet 29:402–410. 10.1038/s41431-020-00748-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- De la Torre R, et al. (2014) Epigallocatechin-3-gallate, a DYRK1A inhibitor, rescues cognitive deficits in Down syndrome mouse models and in humans. Mol Nutr Food Res 58:278–288. 10.1002/mnfr.201300325 [DOI] [PubMed] [Google Scholar]
- Doran E, Keator D, Head E, Phelan MJ, Kim R, Totoiu M, Barrio JR, Small GW, Potkin SG, Lott IT (2017) Down syndrome, partial trisomy 21, and absence of Alzheimer's disease: the role of APP. J Alzheimers Dis 56:459–470. 10.3233/JAD-160836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortea J, Zaman SH, Hartley S, Rafii MS, Head E, Carmona-Iragui M (2021) Alzheimer's disease associated with Down syndrome: a genetic form of dementia. Lancet Neurol 20:930–942. 10.1016/S1474-4422(21)00245-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Cerro S, Rueda N, Vidal V, Lantigua S, Martinez-Cue C (2017) Normalizing the gene dosage of Dyrk1A in a mouse model of Down syndrome rescues several Alzheimer's disease phenotypes. Neurobiol Dis 106:76–88. 10.1016/j.nbd.2017.06.010 [DOI] [PubMed] [Google Scholar]
- Gkanatsiou E, Portelius E, Toomey CE, Blennow K, Zetterberg H, Lashley T, Brinkmalm G (2019) A distinct brain beta amyloid signature in cerebral amyloid angiopathy compared to Alzheimer's disease. Neurosci Lett 701:125–131. 10.1016/j.neulet.2019.02.033 [DOI] [PubMed] [Google Scholar]
- Glenner GG, Wong CW (1984) Alzheimer's disease and Down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun 122:1131–1135. 10.1016/0006-291X(84)91209-9 [DOI] [PubMed] [Google Scholar]
- Gribble SM, et al. (2013) Massively parallel sequencing reveals the complex structure of an irradiated human chromosome on a mouse background in the Tc1 model of Down syndrome. PLoS One 8:e60482. 10.1371/journal.pone.0060482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedj F, Pereira PL, Najas S, Barallobre MJ, Chabert C, Souchet B, Sebrie C, Verney C, Herault Y, Arbones M, Delabar JM (2012) DYRK1A: a master regulatory protein controlling brain growth. Neurobiol Dis 46:190–203. 10.1016/j.nbd.2012.01.007 [DOI] [PubMed] [Google Scholar]
- Hammerle B, Elizalde C, Tejedor FJ (2008) The spatio-temporal and subcellular expression of the candidate Down syndrome gene Mnb/Dyrk1A in the developing mouse brain suggests distinct sequential roles in neuronal development. Eur J Neurosci 27:1061–1074. 10.1111/j.1460-9568.2008.06092.x [DOI] [PubMed] [Google Scholar]
- Holler CJ, Webb RL, Laux AL, Beckett TL, Niedowicz DM, Ahmed RR, Liu Y, Simmons CR, Dowling AL, Spinelli A, Khurgel M, Estus S, Head E, Hersh LB, Murphy MP (2012) BACE2 expression increases in human neurodegenerative disease. Am J Pathol 180:337–350. 10.1016/j.ajpath.2011.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtta M, Hansson O, Andreasson U, Hertze J, Minthon L, Nagga K, Andreasen N, Zetterberg H, Blennow K (2013) Evaluating amyloid-beta oligomers in cerebrospinal fluid as a biomarker for Alzheimer's disease. PLoS One 8:e66381. 10.1371/journal.pone.0066381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Zheng H (2017) Practical considerations for choosing a mouse model of Alzheimer's disease. Mol Neurodegener 12:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkle BW, et al. (2019) Genetic meta-analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet 51:414–430. 10.1038/s41588-019-0358-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lana-Elola E, Watson-Scales S, Slender A, Gibbins D, Martineau A, Douglas C, Mohun T, Fisher EM, Tybulewicz V (2016) Genetic dissection of Down syndrome-associated congenital heart defects using a new mouse mapping panel. Elife 5:e11614. 10.7554/eLife.11614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti E, Altafaj X, Dierssen M, de la Luna S, Fotaki V, Alvarez M, Perez-Riba M, Ferrer I, Estivill X (2003) Dyrk1A expression pattern supports specific roles of this kinase in the adult central nervous system. Brain Res 964:250–263. 10.1016/S0006-8993(02)04069-6 [DOI] [PubMed] [Google Scholar]
- McCarron M, McCallion P, Reilly E, Dunne P, Carroll R, Mulryan N (2017) A prospective 20-year longitudinal follow-up of dementia in persons with Down syndrome. J Intellect Disabil Res 61:843–852. 10.1111/jir.12390 [DOI] [PubMed] [Google Scholar]
- Naert G, Ferre V, Keller E, Slender A, Gibbins D, Fisher EM, Tybulewicz VL, Maurice T (2018) In vivo and ex vivo analyses of amyloid toxicity in the Tc1 mouse model of Down syndrome. J Psychopharmacol 32:174–190. 10.1177/0269881117743484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann F, Gourdain S, Albac C, Dekker AD, Bui LC, Dairou J, Schmitz-Afonso I, Hue N, Rodrigues-Lima F, Delabar JM, Potier MC, Le Caer JP, Touboul D, Delatour B, Cariou K, Dodd RH (2018) DYRK1A inhibition and cognitive rescue in a Down syndrome mouse model are induced by new fluoro-DANDY derivatives. Sci Rep 8:2859. 10.1038/s41598-018-20984-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TL, Duchon A, Manousopoulou A, Loaec N, Villiers B, Pani G, Karatas M, Mechling AE, Harsan LA, Limanton E, Bazureau JP, Carreaux F, Garbis SD, Meijer L, Herault Y (2018) Correction of cognitive deficits in mouse models of Down syndrome by a pharmacological inhibitor of DYRK1A. Dis Model Mech 11:dmm035634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Doherty A, Ruf S, Mulligan C, Hildreth V, Errington ML, Cooke S, Sesay A, Modino S, Vanes L, Hernandez D, Linehan JM, Sharpe PT, Brandner S, Bliss TV, Henderson DJ, Nizetic D, Tybulewicz VL, Fisher EM (2005) An aneuploid mouse strain carrying human chromosome 21 with Down syndrome phenotypes. Science 309:2033–2037. 10.1126/science.1114535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percie du Sert N, et al. (2020) The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. PLoS Biol 18:e3000410. 10.1371/journal.pbio.3000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portelius E, Tran AJ, Andreasson U, Persson R, Brinkmalm G, Zetterberg H, Blennow K, Westman-Brinkmalm A (2007) Characterization of amyloid beta peptides in cerebrospinal fluid by an automated immunoprecipitation procedure followed by mass spectrometry. J Proteome Res 6:4433–4439. 10.1021/pr0703627 [DOI] [PubMed] [Google Scholar]
- Ryoo SR, Cho HJ, Lee HW, Jeong HK, Radnaabazar C, Kim YS, Kim MJ, Son MY, Seo H, Chung SH, Song WJ (2008) Dual-specificity tyrosine(Y)-phosphorylation regulated kinase 1A-mediated phosphorylation of amyloid precursor protein: evidence for a functional link between Down syndrome and Alzheimer's disease. J Neurochem 104:1333–1344. 10.1111/j.1471-4159.2007.05075.x [DOI] [PubMed] [Google Scholar]
- Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, Iwata N, Saido TC (2014) Single App knock-in mouse models of Alzheimer's disease. Nat Neurosci 17:661–663. 10.1038/nn.3697 [DOI] [PubMed] [Google Scholar]
- Sasaguri H, Nilsson P, Hashimoto S, Nagata K, Saito T, De Strooper B, Hardy J, Vassar R, Winblad B, Saido TC (2017) APP mouse models for Alzheimer's disease preclinical studies. EMBO J 36:2473–2487. 10.15252/embj.201797397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serneels L, T'Syen D, Perez-Benito L, Theys T, Holt MG, Strooper BD (2020) Modeling the beta-secretase cleavage site and humanizing amyloid-beta precursor protein in rat and mouse to study Alzheimer's disease. Mol Neurodegener 15:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Leissring MA, Adame A, Sun X, Spooner E, Masliah E, Selkoe DJ, Lemere CA, Walsh DM (2009) Biochemical and immunohistochemical analysis of an Alzheimer's disease mouse model reveals the presence of multiple cerebral Abeta assembly forms throughout life. Neurobiol Dis 36:293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard O, Plattner F, Rubin A, Slender A, Linehan JM, Brandner S, Tybulewicz VL, Fisher EM, Wiseman FK (2012) Altered regulation of tau phosphorylation in a mouse model of down syndrome aging. Neurobiol Aging 33:e831–e844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan M, Piccini I, Balzereit D, Herwig R, Saran NG, Lehrach H, Reeves RH, Yaspo ML (2007) Gene expression variation in Down's syndrome mice allows prioritization of candidate genes. Genome Biol 8:R91. 10.1186/gb-2007-8-5-r91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, He G, Song W (2006) BACE2, as a novel APP theta-secretase, is not responsible for the pathogenesis of Alzheimer's disease in Down syndrome. FASEB J 20:1369–1376. 10.1096/fj.05-5632com [DOI] [PubMed] [Google Scholar]
- Tosh JL, Rhymes ER, Mumford P, Whittaker HT, Pulford LJ, Noy SJ, Cleverley K, LonDown SC, Walker MC, Tybulewicz VL, Wykes RC, Fisher EM, Wiseman FK (2021) Genetic dissection of down syndrome-associated alterations in APP/amyloid-beta biology using mouse models. Sci Rep 11:5736. 10.1038/s41598-021-85062-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velazquez R, Meechoovet B, Ow A, Foley C, Shaw A, Smith B, Oddo S, Hulme C, Dunckley T (2019) Chronic Dyrk1 inhibition delays the onset of AD-like pathology in 3xTg-AD mice. Mol Neurobiol 56:8364–8375. 10.1007/s12035-019-01684-9 [DOI] [PubMed] [Google Scholar]
- Voytyuk I, Mueller SA, Herber J, Snellinx A, Moechars D, van Loo G, Lichtenthaler SF, Strooper BD (2018) BACE2 distribution in major brain cell types and identification of novel substrates. Life Sci Alliance 1:e201800026. 10.26508/lsa.201800026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Xu Q, Cai F, Liu X, Wu Y, Song W (2019) BACE2, a conditional beta-secretase, contributes to Alzheimer's disease pathogenesis. JCI Insight 4:e123431. 10.1172/jci.insight.123431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman FK, Al-Janabi T, Hardy J, Karmiloff-Smith A, Nizetic D, Tybulewicz VL, Fisher EM, Strydom A (2015) A genetic cause of Alzheimer disease: mechanistic insights from Down syndrome. Nat Rev Neurosci 16:564–574. 10.1038/nrn3983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman FK, et al. (2018) Trisomy of human chromosome 21 enhances amyloid-beta deposition independently of an extra copy of APP. Brain 141:2457–2474. 10.1093/brain/awy159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Jin N, Shi J, Zhang Y, Wu Y, Gong CX, Iqbal K, Liu F (2017) Dyrk1A overexpression leads to increase of 3R-tau expression and cognitive deficits in Ts65Dn Down syndrome mice. Sci Rep 7:619. 10.1038/s41598-017-00682-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, et al. (2010) A mouse model of Down syndrome trisomic for all human chromosome 21 syntenic regions. Hum Mol Genet 19:2780–2791. 10.1093/hmg/ddq179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ (2014) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34:11929–11947. 10.1523/JNEUROSCI.1860-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MS, Li G, Duncan JA 3rd, Cheshier SH, Shuer LM, Chang EF, Grant GA, Gephart MG, Barres BA (2016) Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89:37–53. 10.1016/j.neuron.2015.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng QG, Li S, Wang Y, Zhou K, Liu Y, Gao Y, Zhou L, Zheng L, Zhu Q, Deng M, Wu A, Di L, Zhang Y, Zhao H, Zhang H, Sun C, Dong H, Xu X, Wang (2021) Trisomy 21-induced dysregulation of microglial homeostasis in Alzheimer's brains is mediated by USP25. Sci Adv 7:eabe1340. [DOI] [PMC free article] [PubMed] [Google Scholar]