

Abstract
Cancer is a heterogeneous disease, and its treatment requires the identification of new ways to thwart tumor cells. Amongst such emerging targets are glucose transporters (GLUTs, SLC2 family), which are overexpressed by almost all types of cancer cells; their inhibition provides a strategy to disrupt tumor metabolism selectively, leading to antitumor effects. Here, novel Thiazolidinedione (TZD) derivatives were designed, synthesized, characterized, and evaluated for their GLUT1, GLUT4, and GLUT5 inhibitory potential, followed by in vitro cytotoxicity determination in leukemic cell lines. Compounds G5, G16, and G17 inhibited GLUT1, with IC50 values of 5.4 ± 1.3, 26.6 ± 1.8, and 12.6 ± 1.2 μM, respectively. G17 was specific for GLUT1, G16 inhibited GLUT4 (IC50=21.6 ± 4.5 μM) comparably but did not affect GLUT5. The most active compound, G5, inhibited all three GLUT types, with GLUT4 IC50 = 9.5 ± 2.8 μM, and GLUT5 IC50 = 34.5 ± 2.4 μM. Docking G5, G16, and G17 to the inward- and outward-facing structural models of GLUT1 predicted ligand binding affinities consistent with the kinetic inhibition data and implicated E380 and W388 of GLUT1 vs. their substitutions in GLUT5 (A388 and A396, respectively) in inhibitor preference for GLUT1. G5 inhibited the proliferation of leukemia CEM cells at low micromolar range (IC50 = 13.4 μM) while being safer for normal blood cells. Investigation of CEM cell cycle progression after treatment with G5 showed that cells accumulated in the G2/M phase. Flow cytometric apoptosis studies revealed that compound G5 induced both early and late-stage apoptosis in CEM cells.
Keywords: Thiazolidinedione (TZD), GLUT1, GLUT4, GLUT5, Leukemia, Apoptosis, Ligand Specificity
Graphical Abstract
1. Introduction
Recently glucose transporter (GLUT, SLC2 gene family) inhibitors are being explored as anticancer agents. PET diagnostic technique, which uses radiolabeled glucose to identify tumors in the body, relies on the fact that the transformed cells require more energy and consume more glucose. Due to rapid growth, cancer cells demand an increased supply of glucose, which serves both as a source of energy and building blocks for macromolecule synthesis. The direct consequence of this is the reprogramming of several metabolic pathways to meet the glucose demand of proliferating cancer cells. One such change is the overexpression of GLUTs, transporters responsible for the cellular uptake of glucose, which is the first rate-limiting step in the glycolysis. For example, GLUTs are up-regulated in solid and hematological malignancies (Amann and Hellerbrand, 2009; Guo et al., 2019; Siebeneicher et al., 2016a). Thus, inhibition of glucose transporters is a promising strategy for antitumor therapy, and these transporters have been targeted with natural products and small molecules (Granchi et al., 2016; Reckzeh and Waldmann, 2019; Zambrano et al., 2019). Among GLUTs, GLUT1 has been studied extensively, and its overexpression has been reported in many types of cancers, including prostate (Effert et al., 2004), ovarian (Zambrano et al., 2019), lung (Suzuki et al., 2018), kidney (Chan et al., 2011), brain (Nishioka et al., 1992), breast (Wellberg et al., 2016), colon (Merigo et al., 2018), and various types of leukemias (Coskun and Sutton, 2005; Laister et al., 2015; Pontes et al., 2013). Several GLUT1 inhibitors have already been described, including resveratrol, naringenin (Harmon and Patel, 2003), phloretin, WZB 117(Liu et al., 2012), BAY 786 (Siebeneicher et al., 2016b), thiazolidinediones (Arafa et al., 2014; Chen et al., 2015; Wei et al., 2010), and STF-31 (Chan et al., 2011). With most GLUT inhibitors, while they exhibit excellent GLUT inhibition, their overall anticancer effects are either scant or observed at very high concentrations. Though a few GLUT inhibitors are in the early phase of clinical trials, none of them have made it up to clinical use. Thus, there is still a high demand to discover GLUT inhibitors with the potential to reach clinical use.
2. RESULTS AND DISCUSSION
2.1. Designing of the Molecules
In several types of cancer cells, the TZD-scaffold containing agents such as Troglitazone (TG) and ciglitazone (CG), PPARγ agonists used to treat diabetes, have been shown in-vitro and in-vivo to elicit cellular responses that are characterized by energy restriction, but at relatively higher doses. Δ2TG and Δ2CG, which are PPARγ inactive analogs of troglitazone and ciglitazone, respectively, with double bond adjoining to the terminal TZD ring, have been reported to possess higher antitumor potency compared to their parent compounds (Omar et al., 2013). Interestingly, these PPARγ inactive analogs retain their parent compounds’ capability to target many key signaling pathways of the cancer cell cycle regulation and apoptosis. (Huang et al., 2005; Wei et al., 2010). The pleiotropic mode of action of these compounds comes from their ability to mimic the state of glucose deficiency by inhibiting GLUTs (glucose transporters) (Arafa et al., 2014; Chen et al., 2015; Wei et al., 2010). Some of the cellular responses induced by TZDs as a result of energy restriction are decreased glycolysis, induction of the Sirt1 gene (silent information regulator 1), activation of AMPK, and induction of ER (endoplasmic reticulum) stress (Wei et al., 2010).
These findings offer a mechanistic basis for using Δ2CG and Δ2TG, derivatives of CG and TG, respectively, as a scaffold to develop a new and more potent class of GLUT inhibitors. Reported modifications of Δ2CG involve permutations of Δ2CG (1) along with alterations of terminal phenolic and hydrophobic moieties, leading to the discovery of GLUT inhibitor compounds (3) (Chen et al., 2015). We observed the structural similarity of our previously reported TZD derivative, (4) (with excellent anti-proliferative effects in myeloid leukemic K562 cells), with the permutated Δ2CG (1), and the commercially available GLUT1 inhibitor Fasentin (2) (Wood et al., 2008) and the compound reported by Chen et al. (3) (Fig. 1). Thus, we modified our series of molecules to inhibit GLUTs. The terminal lipophilic part of our reported TZD (4) has structural similarity to Fasentin (2), while the benzylidene TZD portion of (4) is common to Δ2CG (1) and compound (3). With all these considerations, we designed our novel series of GLUT1 inhibitors (Fig. 1). Interestingly, a few TZD scaffold-containing compounds activated the protein expression (mRNA level) of both PPARγ and GLUT4, showing therapeutic potential as multitarget anti-diabetic agents (Colín-Lozano et al., 2018; Gutierréz-Hernández et al., 2019). Examining their structures reveals that the location of the TZD ring within the molecule may determine the action of the compound as an activator of GLUT expression or GLUT inhibitor. Thus, we hypothesized that when the TZD ring is at the terminus, the compounds will exhibit GLUT upregulation, whereas when it is centrally located (Fig. 1., Compound 3), the molecules will have GLUT inhibitory potential. Hence, we kept the TZD ring in the middle of the molecule, as in the structures of GLUT inhibitors reported by Chen. et al., to avoid potential GLUT upregulation.
Figure 1. Designing a novel series of GLUT1 inhibitors G1-G17. G1-G17.

compounds were designed to incorporate features of two potent GLUT1 inhibitors, compound (3) reported by Chen et al. and Fasentin (2), shared with our previously reported TZD-based antiproliferative series (Patil et al., 2010)(4): the aryl acetyl amido moiety (blue) and the 5-benzylidene-2,4-TZD scaffold (red).
2.2. Chemistry
The synthetic route for the synthesis of G1-G17 is outlined in Scheme 1 and involves a sequence of four steps. In step one heteroaryl/aryl amines 1a-1d reacted with chloroacetyl chloride to give chloroacetylated aryl amides 2a-2d. In step two, thiazolinediones 4 was condensed with various aldehydes 3a-3g in sodium acetate and acetic acid to give knoevenagel products 5a-5g. In the third step, compounds 5a-5g were refluxed with ethanolic potassium hydroxide to obtain the salt form of compounds 6a-6g. In the final step, compounds 2a-2d and salts 6a-6g were refluxed in acetone to yield the final derivatives G1-G17. All final products were characterized by IR, 1H NMR, 13C NMR, and mass spectrometry; HPLC established the purity of compounds. FT-IR spectra of G1-G17 displayed characteristic absorption bands corresponding to the amide group NH(s) vibration at 3390–3200 cm−1 and that of C=O(s) at 1700–1680 cm−1. Characteristic absorption peaks for C-Cl, C-F, and C-Br were observed in the respective compounds. Proton NMR data of compounds G1-G17 displayed a peculiar singlet of -NH of amide linkage in a region of δ=10.1–10.4 ppm. A characteristic singlet of benzylidene C=CH- was detected in the 7.3–8.1 ppm region for intermediates 5a-5g, salts 6a-6g, and final compounds G1-G17. A Singlet of two protons corresponding to methylene -CH2 was also observed at δ= 4.5–4.7 ppm. The presence of this singlet confirmed the formation of target compounds by condensation of 2a-2d and 6a-6g. The spectra also showed various peaks in the aromatic and aliphatic regions corresponding to the protons of variously substituted aromatic and heteroaromatic groups attached to the N atom of an amide linkage. Peaks of a furan ring appeared in the aromatic area of δ= 6.50–8.00 ppm. The formation of salts 6a-6g from 5a-5g was confirmed by the disappearance of a singlet of -NH in the region of 12.0–13.0 ppm (which corresponds to -NH of TZD moiety where salt formation occurs) from spectra of salts 6a-6g. In 13C-NMR, C=O of amide showed a peak at 165 δ ppm, and C=O of TZD showed a peak at 164–170 δ ppm. The aromatic carbons had peaks at δ= 110–140 ppm. Methylene carbons showed a peak at δ= 40–50 ppm in 13C-NMR. The mass spectrum was recorded for both positive and negative ionization. There was extensive ionization in positive scanning, but in the negative scan, the characteristic [M-H+] peak was observed at 100% intensity for almost all the compounds. The details of synthesis and spectral observations are in the experimental section.
Scheme 1. Synthetic route for the intermediates and final derivatives G1-G17.
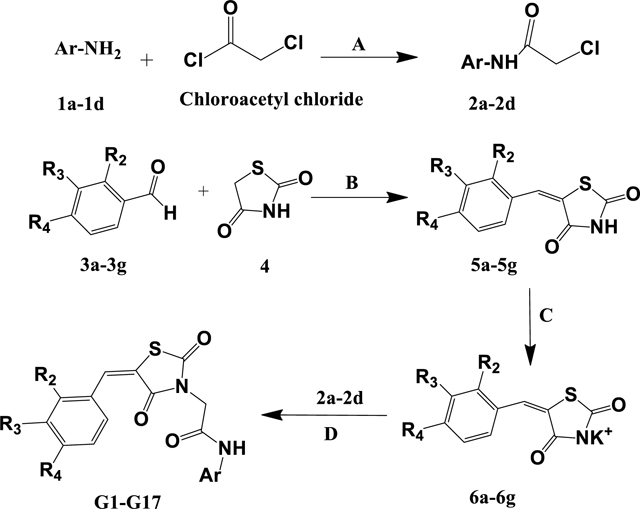
a) K2CO3, DCM, Stir at RT for 24 h. B) Acetic acid, Sodium acetate, reflux 3–6 h; C) EtOH, KOH, reflux 1–2h. d) Acetone, reflux 6–10h. The R2, R3, R4, and Ar are the substituents, and they are defined for each specific product and mentioned in Table 4.
2.3. SwissADME Prediction of physicochemical, pharmacokinetic, and ADME properties
The modern process of drug discovery utilizes in-vitro testing methods and in silico property predictions to optimize lead compounds. SwissADME is a free webtool for predicting pharmacokinetic and physicochemical properties of the newly synthesized small molecules, including molecular weight, solubility, partition coefficient, topological surface area, and pharmacokinetic properties, such as absorption, distribution, metabolism, and elimination of molecules, which are key parameters for lead molecules to proceed to in-vivo preclinical and clinical stage (Daina et al., 2017). Based on these predictions, lead molecules can be modified. For example, if lead molecules are predicted to have poor solubility, then its actual solubility can be experimentally determined and improved.
The 2D structures of G1-G17 were drawn using Chemdraw ultra version 12.0 software and were imported in the swissADME environment (http://www.swissadme.ch/), where SMILES of the structures were generated to run the swissADME tool. The values for various physicochemical and pharmacokinetic parameters were obtained by importing the CSV file. The bioavailability radar plots generated by the swissADME tool were directly used for the interpretation.
Results (Table 1) showed that the molecular weights of the compounds G1-G17 were 388.39 – 519.72 g/mol, ilogP values (Daina et al., 2014) were 2.32 – 3.43, topological polar surface area (TPSA) was 91.78 Å2 for G1-G16 and 150.74 Å2 for G17. The number of hydrogen bond acceptors (HBA) was <10, and that of hydrogen bond donors (HBD) was <5. All compounds were predicted to be moderately to poorly soluble, which can be problematic in dosage formulation and absorption. The solubility enhancement methods could help to tackle this issue. 70% of the compounds displayed high gastrointestinal (GI) absorption. All the compounds were predicted to be non-penetrating to BBA (blood-brain barrier), reducing the chances of CNS adverse effects. Also, the compounds were non- substrates for P- glycoprotein (P- gp), which is located in various organs and acts as efflux transporter pumping out xenobiotics from the cells, leading to enhanced clearance of the drugs. The bioavailability score of 0.55 fulfills the drug-likeness as indicted by Lipinski’s rule of five (Lipinski et al., 2001), Ghose filter (Ghose et al., 1999), Egan rule (Egan et al., 2000), Veber’s rule (Veber et al., 2002), and Muegge’s filter (Muegge et al., 2001), with the violation of molecular weight in some cases.
Table 1.
SwissADME predictions for G1-G17.
| Code | MWa | Fraction Csp3 | RBsb | HBAc | HBDd | TPSAe | iLOGPf | ESOL Classg | GIAh | BBBI permeant | P-gpj substrate |
|---|---|---|---|---|---|---|---|---|---|---|---|
| G1 | 519.72 | 0.11 | 6 | 6 | 1 | 91.78 | 3.32 | Poorly soluble | Low | No | No |
| G2 | 453.26 | 0.06 | 5 | 5 | 1 | 91.78 | 3.43 | Moderately soluble | High | No | No |
| G3 | 468.88 | 0.19 | 6 | 6 | 1 | 91.78 | 3.03 | Poorly soluble | Low | No | No |
| G4 | 402.41 | 0.15 | 5 | 5 | 1 | 91.78 | 2.78 | Moderately soluble | High | No | No |
| G5 | 475.27 | 0.11 | 6 | 6 | 1 | 91.78 | 2.98 | Poorly soluble | Low | No | No |
| G6 | 408.81 | 0.06 | 5 | 5 | 1 | 91.78 | 3.05 | Moderately soluble | High | No | No |
| G7 | 476.8 | 0.11 | 6 | 8 | 1 | 91.78 | 3.14 | Moderately soluble | Low | No | No |
| G8 | 410.34 | 0.06 | 5 | 7 | 1 | 91.78 | 2.99 | Moderately soluble | High | No | No |
| G9 | 458.81 | 0.11 | 6 | 7 | 1 | 91.78 | 2.63 | Moderately soluble | High | No | No |
| G10 | 392.35 | 0.06 | 5 | 6 | 1 | 91.78 | 2.41 | Moderately soluble | High | No | No |
| G11 | 454.85 | 0.15 | 6 | 6 | 1 | 91.78 | 3.36 | Moderately soluble | High | No | No |
| G12 | 388.39 | 0.11 | 5 | 5 | 1 | 91.78 | 2.57 | Moderately soluble | High | No | No |
| G13 | 440.82 | 0.11 | 6 | 6 | 1 | 91.78 | 2.68 | Moderately soluble | High | No | No |
| G14 | 374.36 | 0.06 | 5 | 5 | 1 | 91.78 | 2.83 | Moderately soluble | High | No | No |
| G15 | 442.36 | 0.11 | 6 | 8 | 1 | 91.78 | 2.79 | Moderately soluble | High | No | No |
| G16 | 424.37 | 0.11 | 6 | 7 | 1 | 91.78 | 2.32 | Moderately soluble | High | No | No |
| G17 | 429.9 | 0.05 | 5 | 4 | 1 | 132.91 | 2.74 | Moderately | Low | No | No |
Molecular weight
Rotatable bonds
Hydrogen bond acceptors
Hydrogen bond donors
Topological surface area
octanol–water partition coefficient
Solubility class
Gastro-intestinal absorption
Blood brain barrier
P- glycoprotein
The swissADME also displayed a bioavailability (BA) radar plot, which gives the probable profile of oral bioavailability of the compounds; the pink-colored area of the radar plot is the supreme zone indicating lead-like molecules. The six physicochemical properties considered to generate this supreme radar zone, are size (SIZE), polarity (POLAR), lipophilicity (LIPO), solubility (INSOLU), saturation (INSATU), and flexibility (FLEX). Fig. 2 shows the bioavailability radar plot of compounds G5 and G17. The compounds G1-G17 were in the pink area, with the degree of saturation deviated slightly out of the supreme area, which can be attributed to the fraction of SP3 hybridized carbons of < 0.25. Thus, prediction data indicates that these series of compounds may display favorable pharmacokinetic and physicochemical profiles to be drug-like molecules.
Figure 2.

Bioavailability radar plot of compound G5 (A) and G17 (B) generated on SwissADME platform.
2.4. Screening for transport activity inhibition of GLUT1, GLUT4, and GLUT5 and Docking analysis at active site of GLUTs.
2.4.1. Screening for transport activity inhibition of GLUT1, GLUT4, and GLUT5
To validate the correlation between designing considerations and GLUT inhibitory activity, we screened all the derivatives for GLUT inhibition. Based on the sequence homology, GLUTs are divided into three classes: class I (GLUT1–4), class II (GLUTs 5, 7, 9, and 11), and class III (GLUTs 6, 8, 10, 12, and HMIT) (Mueckler and Thorens, 2013). The most studied GLUTs are those from class I, followed by those from class II. Previously described GLUT inhibitors have different degrees of selectivity for the various GLUT isoforms. For instance, natural products such as rubusoside (George Thompson et al., 2015), (−)-epicatechin-gallate, and (−)-epigallocatechin-gallate (Slavic et al., 2009) affect both class I and II GLUTs; cytochalasin B and phloretin distinguish between class I and class II GLUTs (Li et al., 2004), while Astragalin-6-glucoside (George Thompson et al., 2015) and N-[4-(methylsulfonyl)-2-nitrophenyl]-1,3-benzodioxol-5-amine (MSNBA) (George Thompson et al., 2016) only inhibit GLUT5. To establish the selectivity of the compounds, besides GLUT1, we included in the transport activity check another class I member, GLUT4, and a class II member, GLUT5.
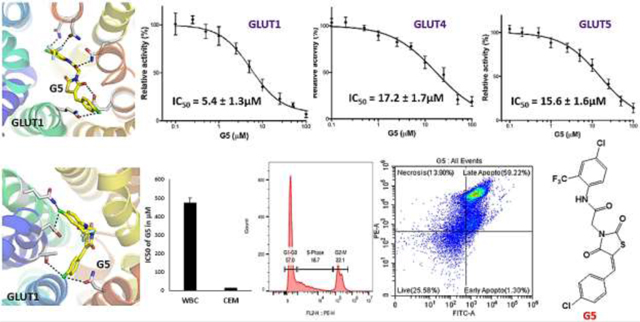
We checked the effect of compounds G1-G17 on the transport activity of an individual GLUT by using a GLUT-specific assay method, which utilizes hexose transporter deficient yeast cells (hxt0) expressing a particular human GLUT (Tripp et al., 2017; Wieczorke et al., 2003). In hxt0 strains, yeast cell growth in hexose-based media relies solely on the transport activity of the expressing human GLUT for bringing the carbon source into cells. Thus, GLUT transport activity is determined as the C14-hexose uptake into whole cells (Tripp et al., 2017; Wieczorke et al., 2003). The primary inhibition screening was carried out at 100 μM compound concentration on three glucose transporters: GLUT1, GLUT4, and GLUT5 (Fig. 3- A, E, H).
Figure 3. Screening of G1-G17 for inhibition of the transport activity by GLUT1, GLUT4, and GLUT5.

Relative transport activity of GLUT1 (A), GLUT4 (E), and GLUT5 (H) in the presence of 100 μM concentration of G1-G17. The dose response curve of GLUT1 for G5 (B), G16 (C), and G17 (D). The dose response curve of GLUT4 for G5 (F) and G16 (G). The dose response curve of GLUT5 for G5 (I). Error bars represent the standard deviation from three independent measurements. Transport activity was determined in hxt0 yeast cells expressing a single GLUT. Transport assay was initiated by the addition of 5 mM C14- glucose for GLUT1 or GLUT4 or 10 mM C14-fructose for GLUT5 to whole cells and stopped after 10 minutes (see Materials and Methods for details).
In the primary screen, three compounds (G5, G16, and G17) out of 17 compounds decreased the relative transport activity >50% of at least one GLUT among GLUT1, GLUT4, and GLUT5. G5 decreased the activities of all three GLUTs, G16 those of GLUT1 and GLUT4, and G17 that of GLUT1. For these three compounds, the concentration was varied in the assay from 0.1 to 100 μM to determine inhibitor IC50. G5 inhibited all three GLUTs, and the IC50 values for GLUT1, GLUT4, and GLUT5 were 5.4 ± 1.3 μM, 9.5 ± 2.8 μM, and 34.5 ± 2.4 μM, respectively (Fig. 3- B, F, I). Thus, G5 was a PAN inhibitor of GLUTs, with slightly better specificity for class I GLUTs (GLUT1 and GLUT4) than GLUT5 (class II GLUTs). G16 was an equipotent dual inhibitor of GLUT1 and GLUT4 with IC50 values of 26.8 ± 1.8 μM, and 21.6 ± 4.5 μM, respectively (Fig. 3- C, G). G17 was a selective inhibitor of GLUT1, with an IC50 value of 12.6 ± 1.2 μM (Fig. 3- I). Surveying the structures of all three compounds revealed that the compound G5 with 4-chlorobenzylidene ring at one side of the centrally located TZD ring and two substituents, CF3 and Cl groups, on the phenyl ring at the other side of the compound exhibited pan inhibition of all three types of GLUTs; G16 with the 4-halobenzylidene ring and only the CF3 group substituted on the phenyl ring lead to GLUT1 and GLUT4 inhibition, whereas G17 with unsubstituted benzylidene ring and Cl substituted benzothiazole ring lead to specific inhibition of GLUT1. All other derivatives with di-halo, di-methyl or methyl benzylidene ring were found to be inactive, indicating that monohalosubstituted or unsubstituted benzylidene ring is preferred for retention of GLUT inhibitory effects. The GLUT1-specific inhibitor G17 has Cl substituted on the benzothiazole ring. In our previously reported pyridyl TZDs (Upadhyay et al., 2020), compound P19 with a benzothiazole ring was a GLUT1 inhibitor. Therefore, it seems that the benzothiazole ring’s presence may be important for selective targeting of GLUT1.
2.4.2. Docking at active site of GLUTs
Ligand docking and structural model generation were done with the software package Molecular Operating Environment (MOE) (Chemical Computing Group Inc, Montreal, Quebec, Canada). The structural model for the inward-facing conformation of GLUT1 was the crystal structure of GLUT1, PDB ID 4PYP. The outward-facing conformation of GLUT1 was generated with the ‘Homology Model’ function, using chain A of PDB ID 4ZWC (the crystal structure for GLUT3) as the template. The generated models were scored with GB/VI, and the model with the lowest free energy was used further. Before ligand docking, the structural models were optimized by protonation with ‘Protonate 3D’, and energy minimization using Amber10:EHT force field and default parameters. Ligands G5, G16, and G17 were put in a ligand database, and the conformations were generated with ‘Conformational Search’, using LowModeMD method and default parameters, with Amber10:EHT force field, resulting in 33, 92, and 42 conformations for G5, G16, and G17, respectively. For the molecular docking simulation, the docking sites in the prepared protein structural models were determined with ‘Site Finder’; the first site in the Site Finder list was selected, and dummy atoms were created at the alpha centers. The ligand conformational database was docked on the dummy atoms, with the function ‘Dock’, using as a placement method ‘Triangle Matcher’. The top 30 poses, according to London dG scoring, were further refined, and scored with GBVI/WSA dG (Corbeil et al., 2012); the best five poses for each ligand were kept and examined. The best ligand-transporter complexes were selected based on its free binding energy value, and ligand-protein interactions.
GLUTs belong to the major facilitator superfamily (MFS), one of the largest and most ubiquitous secondary transporter family (Marger and Saier, 1993; Saier et al., 1999). They transport their substrate by alternately opening the substrate cavity to each side of the membrane, cycling through the so-called outward-facing (substrate cavity opened toward periplasm) or inward-facing (substrate cavity opened toward lumen) conformations (Fig. 4-A & E) (Abramson et al., 2003; Yan, 2015). Cytochalasin B, a well-known inhibitor of class I GLUTs, binds to the inward-facing conformation of the transporter, as shown by biochemical studies and confirmed by the crystal structure of GLUT1 complexed with the inhibitor (Carruthers and Helgerson, 1991; Kapoor et al., 2016). A GLUT inhibitor may bind to one or both conformations of the transporter. It can either block substrate access to the active site or lock the transporter in a particular state, preventing the cycling between the inward- and outward-facing conformations. For example, cytochalasin B stabilizes the inward-facing conformation, as evidenced by the fact that GLUT1 crystallized in the inward-facing conformation in the presence of the inhibitor, and also binds at the active site impeding glucose binding (Kapoor et al., 2016).
Figure 4. Ligand docking of G5, G16, and G17 to the structural models for the inward- and outward-facing conformations of GLUT1.

Global views of the inward-facing (A) and outward-facing (E) conformations of GLUT1 structural models, based on the crystal structures PDB IDs 4PYP and 4ZWC, with docked G5. Close-up views of the ligand binding sites for docked G5 (B, F), G16 (C, G), and G17 (D, H) to the inward-facing (B, C, D) or outward-facing (F, G, H) conformations of GLUT1. Black-dotted lines indicate interactions of the docked ligand with the protein residues. H1-H12 denote the twelve transmembrane helices of the transporter. GLUT1 residues T30, E380, W388, and N411 are not conserved in GLUT5; only T30 is not conserved in GLUT4 (Table 3).
To examine the molecular basis of the inhibitor selectivity and potency, we carried out ligand docking studies for G5, G16, and G17 to the inward- and outward-facing conformations of GLUT1 (Fig. 4). Each ligand docked to both states of the transporter but adopted different conformations and interactions with the protein residues (Fig. 4) from the substrate cavity. Thus, unlike cytochalasin B, the compounds did not seem to prefer one of the transporter conformations over the other (Table 2). The extent of ligand-protein interactions (Fig. 4) and associated ligand affinity (Table 2) correlated with the ligand inhibition potency (IC50, Fig. 3), with G5 and G17 having more interactions than G16, in both transporter states.
Table 2.
Calculated ligand affinity for G5, G16, and G17 docked to the GLUT1 structural models.
| GLUT1 conformation | Computed affinity energy for docked ligand to GLUT1 (kcal/mol) |
||
|---|---|---|---|
| G5 | G16 | G17 | |
| Inward-facing | −8.1148 | −7.7447 | −7.8908 |
| Outward-facing | −7.9021 | −7.4839 | −7.5917 |
Analysis of the interactions between GLUT1 and G5 in the two states of the transporter (Fig. 4B & F, Table 3) reveals just one difference between GLUT1 and GLUT4: the hydroxyl group of T30 in GLUT1 outward-facing conformation interacts with the benzyl Chlorine of G5, interaction that lacks in GLUT4 (which has I42) and GLUT5 (which has V36). Collectively GLUT1 and GLUT4 differ from GLUT5 in two other residues: N411 and E380 of GLUT1 are H419 and A388 of GLUT5, respectively (Table 3). T30 and N411 interact with glucose both in the inward- and outward-facing conformations of GLUT1, with N411 having more protracted hydrogen bonds with the substrate than T30, whereas E380 interacts with glucose in the inward-facing conformation of GLUT1(Galochkina et al., 2019). The degree of differences in these residues among GLUT1, GLUT4, and GLUT5 may explain why G5 inhibits GLUT1 most potently and GLUT5 the least (Fig. 3B, F, I).
Table 3.
Sequence conservation in GLUT4 and GLUT5 for GLUT1 residues predicted to interact with G5, G16, or G17(Fig.4). Bolded are residues that vary among GLUT1, GLUT4, and GLUT5.
| GLUT | Protein sequence position | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GLUT1 | T30 | N34 | S80 | Q161 | Q172 | Q282 | N288 | N317 | E380 | W388 | N411 | N415 |
| GLUT4 | I42 | N46 | S96 | Q177 | Q188 | Q298 | N304 | N333 | E396 | W404 | N427 | N431 |
| GLUT5 | V36 | N40 | S86 | Q167 | Q178 | Q288 | N294 | N325 | A388 | A396 | H419 | N423 |
G16 and G17 interact with GLUT1 residues conserved in GLUT4 (Fig. 4C, D, G, H, Table 3), consistent with the comparable inhibition of GLUT1 and GLUT4 by G16 (Fig. 3C, G), but inconsistent with the differing effect of G17 on GLUT1 and GLUT4. W388 interacts with both G16 and G17 in the inward-facing conformation of GLUT1. It is conserved in GLUT4, but in GLUT5, it is A396, a drastic substitution that is likely to significantly weaken the ligand binding in GLUT5, consistent with the lack of GLUT5 inhibition by G16 and G17. Another notable difference is the afore-mentioned E380 of GLUT1, which is the A396 of GLUT5 (Table 3). This change would impact the protein interactions of G17 in the outward-facing conformation (Fig. 4H), consistent with the insensitivity of GLUT5 to G17. Thus, the ligand docking analysis suggests that the key to G16 and G17 inhibition selectivity in GLUT1 over GLUT5 are residues E380 and W388 of GLUT1. A396, which was previously shown to be critical for substrate specificity, with GLUT5A396W mutant being able to transport glucose besides fructose; wild-type GLUT5 only transports fructose (George Thompson et al., 2015). The larger space in GLUT5 substrate cavity created by the substitution of a bulky side chain (Trp) with a small side-chain (A396) seems crucial in GLUT5 ligand selectivity, including inhibitors. As the residues recognizing the inhibitors are known to interact with glucose, overall, the inhibitory action of G5, G16, and G17 in GLUT1 seems to rely on hindering glucose access to its active site in either conformation of the transporter.
We had designed the molecules G1-G17 to target GLUT1, but found that G5 inhibited all three GLUTs; G16 and G17 inhibited GLUT1 and GLUT4, though with different affinities and specificities. Nevertheless, neither G16 nor G17 altered the transport activity of GLUT5. While both G16 and G17 seem to discriminate between class I and class II GLUTs, G17 also distinguishes between GLUT1 and GLUT4, making G17 a promising starting point for the design of future GLUT1-specific probes.
2.5. In-vitro cytotoxicity assay
GLUT inhibitors show moderate to potent in-vitro antiproliferative effects in solid as well as hematological malignancies. (+)-Cryptocaryone, glucose uptake inhibitor exhibited antiproliferative effects in human colon cancer HT-29 cells (IC50 = 0.32 μM), murine leukemia P-388 cells (IC50 = 0.04 μM), and in human prostate cancer PC-3 cells (IC50 = 1.6 μM)(Bonnet et al., 2014). A series of methyl-ketoxime derivatives discovered as GLUT1 inhibitors had antiproliferative effects in a human non-small cell lung cancer cell line with IC50 values in the range 10–18 μM (Granchi et al., 2016). A group of polyphenolic esters with GLUT1 inhibition potential was found to inhibit the growth of H1299 lung cancer cells (IC50 = 10 μM)(Liu et al., 2010; Zhang et al., 2010). Naringenin, a natural flavanone, has also shown mild antiproliferative effects in MCF-7 breast cancer cells (Harmon and Patel, 2004; So et al., 1996). Another GLUT inhibitor, STF-31, displayed antiproliferative effects in VHL-deficient RCC4 cells with an IC50 value of 7.9 μM (Chan et al., 2011). Some GLUT4 inhibitors have shown promising antiproliferative effects in myeloid leukemia cells (Laister et al., 2015).
As GLUT inhibitors have been found effective in hematological malignancies (Bonnet et al., 2014; Laister et al., 2015), in-vitro cytotoxicity of the compounds was determined by MTT assay with two hematological cancer cell types, myeloid leukemia cell line - K562 and T-cell lymphoma cell line - CEM. For the MTT assay, each cell line was exposed to the test compounds for 48 h. To compare the cytotoxic effects, Paclitaxel, a clinically used anticancer drug, and Pioglitazone, a TZD-scaffold containing compound, were the standard references. Untreated cells were the negative control. The IC50 values are in Table 4, and the dose response curves (DRCs) are in supplementary data, (Supplementary Fig. S1-S3). In K562 cells, the compounds had moderate activity compared to Paclitaxel, but compounds G1, G3, G5, G8, and G17 showed better or equal activity to that of pioglitazone. All the compounds exhibited antiproliferative effects in CEM cells, ranging from sub-micromolar to mid-micromolar concentrations, with the most active compounds being G1, G3, G5, G8, G10, G12, and G16 (IC50 values 4.7 – 30 μM).
Table 4.
Inhibitory concentration 50% (IC50) values of the active G-series compounds in the leukemic cell lines, CEM and K-562.
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Compound | R2 | R3 | R4 | Ar | K562- IC50 (μM) | CEM- IC50 (μM) |
|
| ||||||
| G1 | H | H | Br | 4-Cl, 2-CF3 phenyl | 23.97±0.58 | 25.13±1.84 |
| G2 | H | H | Br | 2,4-diF phenyl | >50 | 47.71±2.12 |
| G3 | H | CH3 | CH3 | 4-Cl, 2-CF3 phenyl | 26.36±3.07 | 25.93±2.95 |
| G4 | H | CH3 | CH3 | 2,4-diF phenyl | >50 | 31.92±2.27 |
| G5 | H | H | Cl | 4-Cl, 2-CF3 phenyl | 34.94±1.07 | 13.49±3.88 |
| G6 | H | H | Cl | 2,4-diF phenyl | 48.23±1.23 | 39.33±2.68 |
| G7 | F | H | F | 4-Cl, 2-CF3 phenyl | 36.8±1.77 | 40.38±2.94 |
| G8 | F | H | F | 2,4-diF phenyl | 34.5±3.01 | 4.77±1.51 |
| G9 | H | H | F | 4-Cl, 2-CF3 phenyl | 43.6±2.23 | 38.02±3.34 |
| G10 | H | H | F | 2,4-diF phenyl | 45.4±1.13 | 27.39±3.06 |
| G11 | H | H | CH3 | 4-Cl, 2-CF3 phenyl | 34.60±3.57 | 31.77±3.42 |
| G12 | H | H | CH3 | 2,4-diF phenyl | >50 | 9.93±2.03 |
| G13 | H | H | H | 4-Cl, 2-CF3 phenyl | 41.68±3.90 | 47.51±2.75 |
| G14 | H | H | H | 2,4-diF phenyl | >50 | 32.27±1.75 |
| G15 | F | H | F | 3-CF3 phenyl | >50 | 17.26±1.16 |
| G16 | H | H | F | 3- CF3 phenyl | 23.50±1.94 | 30.12±2.32 |
| G17 | H | H | H | 4-Cl benzothiazol-2-yl | 78.97±3.66 | 35.150±1.77 |
| Paclitaxel | - | - | - | - | 0.29±0.15 | 0.31±0.12 |
| Pioglitazone | - | - | - | - | 31.97±3.66 | 17.91±2.45 |
IC50 values determined in triplicates are presented as Mean±SD, n=3. Data analysis and IC50 determination were performed with GraphPad Prism, Ver 5.1.
The activity of G5, G8, G12, and G15 was better than that of pioglitazone in CEM cells. Thus, our newly synthesized compounds, which are permutated TZD derivatives of glitazones, had better antiproliferative profiles than the parent non-permutated glitazones. Our results agree with Omar et al.’s observations that the Δ2 analogs of glitazones possess higher antitumor potency than their parent compounds. The derivatives with Ar = 2-CF3, 2-Cl phenyl show higher antiproliferative activity than that with 2,4-DiF, except for G8. Also, R2, R3, or R4 substituted with halogens (F, Cl or Br) have enhanced antiproliferative effects compared to the unsubstituted derivatives. Removal of Cl substitution from G9 and G7 gives G16 and G15, respectively, both of which displayed better antiproliferative effects in CEM cells than their unsubstituted counterparts. Hence, a monosubstituted benzylidene ring and a monosubstituted phenyl ring (Ar) around the centrally located TZD ring seem important for the antiproliferative activity.
2.6. Determination of apoptosis induction by flow cytometry
GLUT inhibitors have been reported to cause apoptotic cell death in several cancerous cell lines. GLUT1 inhibitor, (+)-Cryptocaryone, is a natural compound found to induce apoptosis by activating caspase-8 and −3 (Chen et al., 2007). Fasentin, a GLUT1/GLUT4 inhibitor, induces apoptosis by sensitizing cells to FAS, a death receptor belonging to the TNF family (Granchi et al., 2016). WZB117, a GLUT1 inhibitor, decreased the levels of GLUT1, intracellular ATP, and glycolytic enzymes, leading to cell-cycle arrest, senescence, and necrosis through the increase in expression of AMPK, an ATP sensing enzyme, and decrease in cyclin E2(Liu et al., 2012). OSU-CG5, a thiazolidinedione derivative reported as a GLUT1 inhibitor, induced the apoptosis through induction of ER stresses (Arafa et al., 2014). All these observations indicate that the antiproliferative effects of GLUT inhibition include: induction of apoptosis, necrosis, and cell cycle arrest. In agreement with these observations, our GLUT1/GLUT4 inhibitor, G5, when tested for apoptosis induction by flow cytometric analysis, exhibited apoptotic and necrotic cell death in lymphoid leukemic CEM cells.
Treatment of CEM cells with G5 at its IC50 concentration for 24 h. increased the population of cells in late apoptotic and necrotic phase (Fig. 5A) compared with the control (Supplementary Fig. S4). The percentage of cells in early and late apoptotic phase was 1.30% and 59.22%, respectively. The percentage of live cells was only 25.58%, as compared with 92.15% in control. The percentage of necrotic cell population increased to 13.90% in G5-treated cells. A sizable number of cells (13.90%) were in the necrotic region, in agreement with observations by Liu et al., who have reported that WZB117, a GLUT1 inhibitor, causes cell-cycle arrest and necrosis as a consequence of GLUT1 inhibition(Liu et al., 2012). Thus, our results indicate that compound G5 has the potential to bring apoptotic cell death in the CEM cells through the induction of apoptosis and necrosis. These results indicate that G5 dramatically stimulated cell apoptosis in CEM cells, consistent with the inhibition of cell proliferation.
Figure 5. Effect of G5 on the cell cycle, cell viability, and HDAC-8 activity. (A).

Graphical depiction of apoptotic events of untreated and G5 treated CEM cells (L-Live cells, EA-Early apoptosis, LA-Late apoptosis, N-Necrosis), n=3, p=<0.0001. (B) Flow cytometry cell cycle analysis of CEM cells treated with G5 in comparison to untreated cells as control. n=3, *p=<0.01, ***p=<0.0001. (C) Comparison of IC50 values of G5 for white blood cells (WBC) and CEM cells. G5 inhibits cell proliferation in the lymphoid CEM cells significantly more than it does in normal WBCs after 48 hours of treatment. (D) Dose response curves of G3 (circle), G6 (square), G9 (up triangle), G11 (down triangle), and G13 (diamond) against HDAC-8.
2.7. Effect of G5 on the cell cycle in CEM leukemic cells
After observing encouraging results from GLUT inhibition assay, antiproliferative effects in sub-micromolar range, and apoptotic cell death induction, we further investigated the effect of G5 on cell cycle phases of leukemic lymphoid CEM cells. Treatment of CEM cells with IC50 concentration of G5 lead to subtle changes in the cell cycle profile of treated CEM cells (Fig. 5B) compared to untreated control cells (Supplementary Fig. S5). We observed an increase in the G2-M population with a subsequent decrease in the G0-G1 phase cell population. The percentage of cells arrested in the G2-M phase was 22.1%, and that of control was 3.58%, and the percentage of cells arrested in G0-G1 phase was 52.0% and that of control was 73.4%. There were slight changes in the level of cells in the S phase. Thus, results suggest that compound G5 can cause cell cycle arrest in the G2-M phase in the CEM cell line and characteristic apoptotic behavior for the DNA content, which is influenced by G0/G1, S, and G2/M phases of the cell cycle. Energy restriction mimetic TZDs with a similar structural framework as G5, have been found to exert the anticancer effects through downregulation of β-TrCP (β-transducin repeat-containing protein) pathway (Arafa et al., 2014; Wei et al., 2010). β-TrCP facilitates the proteolysis of various proteins involved in the regulation of cell cycle and apoptosis. Thus, the results showing the effect of G5 on the cell cycle and apoptosis, suggest the downregulation of β-TrCP as a result of GLUT inhibition as a possible mechanism for cell cycle arrest and induction of apoptosis.
2.8. Effect on viability of non-transformed WBCs
To establish the cytotoxicity of G5 for normal cells, we performed the MTT assay with white blood cells (WBCs), similarly to the described antiproliferative assay. The IC50 value was determined by treating WBCs with increasing concentrations of G5 for 48 h. The IC50 value of G5 on WBCs was 475.80 μM, which is 35 times that on CEM cell line (Fig. 5C). More than 66% of the WBC cells were viable after 48 h treatment with 100 μM of G5. Thus, these findings indicate that the G5 could be safer to noncancerous normal cells.
2.9. Screening for inhibition of HDAC-4 and HDAC-8 and ligand docking analysis at the active site of HDAC8.
2.9.1. HDAC enzyme activity assay.
We have been exploring the TZD scaffold in targeted anticancer drug discovery, and we have reported the N-Substituted TZDs with HDAC inhibition potential (Mohan et al., 2012). G1-G17 are also N-substituted TZD derivatives, though with entirely different substitution pattern than our previously reported N-substituted TZD analogs. To establish and compare their selectivity for GLUTs over HDACs or vice a versa, we screened G1-G17 on two subtypes of HDAC, HDAC-4, and HDAC-8. These two isoforms were selected as they represent two different classes of HDAC, HDAC-8 (Class I) and HDA-4 (class II). The primary screening was done at 50 μM of G1-G17. The observed relative activity is shown in Supplementary Fig. S6. The relative enzyme activity for HDAC-4 was >1, indicating that G1-G17 do not inhibit HDAC-4 up to 50 μM.
For HDAC-8 six compounds G1, G3, G6, G9, G11, and G13 decreased the relative enzyme activity (< 50 %), and were further examined to determine their IC50. The IC50 values for G1, G3, G6, G9, G11, and G13 are in Table 5, and the corresponding dose response curves are in Fig. 5D. G1, G3, G11, and G13 were further screened against HDAC-1 and HDAC-6. G11 is a pan HDAC inhibitor, inhibiting HDAC-1, HDAC-6, and HDAC-8 equally.
Table 5:
IC50-values of indicated compounds of the G-series against a panel of HDAC isoenzymes. HDAC-1, HDAC-4, and HDAC-6 are typical representatives of classes I, IIa, and IIb, respectively. HDAC-8 is formally a member of class I but stands between class I and class II in terms of substrate selectivity and sequence similarity.
| IC50/μM | ||||
|---|---|---|---|---|
| Code | HDAC1 | HDAC4 | HDAC6 | HDAC8 |
| G1 | > 50 | > 50 | 16 ± 2 | 20 ± 3 |
| G3 | > 50 | > 50 | 17 ± 2 | 16 ± 2 |
| G6 | nd* | > 50 | nd* | 49 ± 5 |
| G9 | nd* | > 50 | nd* | 45 ± 5 |
| G11 | 31±1.1 | > 50 | 14 ± 3 | 18 ± 2 |
| G13 | > 50 | > 50 | 42 | 11 ± 1 |
| PCI-34051 | 3.0* | 10* | 18* | 0.024* |
IC50 values were determined in triplicates and presented as Means ±SD, n=3.
Kleinschek, A., et al. (2016). “Potent and Selective Non-hydroxamate Histone Deacetylase 8 Inhibitors.” ChemMedChem 11(23): 2598–2606. nd*- not determined.
Furthermore, G13 is not only most potent against HDAC-8, but also the compound from the G-series TZDs with the best selectivity against HDAC-1 (a typical representative of class I HDACs), HDAC-4 (class IIa HDAC), and HDAC-6 (class IIb HDAC) (Table 5). Surprisingly none of these five compounds exhibited GLUT1/4/5 inhibition even up-to 100 μM (Fig. 3).
2.9.2. Docking analysis at active site of HDAC-8
To rationalize the inhibitory activity of the active substances against human HDAC-8, we performed docking studies using the crystal structures of representative HDAC8 conformations in complex with various inhibitors (closed: PDB-ID 1T69, open: PDB-ID 1VKG and PDB-ID 3SFF with widened lower selectivity pocket).These crystal structures were prepared for docking using MOE 2019 software (Chemical Computing Group ULC, Canada). Docking, scoring, and energy minimization were performed with the same software package and AMBER 14 forcefield according to previously described procedures (Tilekar et al., 2020). Best binding poses were energy minimized within a radius of 10 Å around the respective ligand.
In agreement with the moderate activity of the G-series compounds, the docking scores are less favorable than that of the ligand in the corresponding crystal structure of HDAC-8, which has a higher binding affinity (Table 6). Best docking scores were obtained for PDB-ID 3SFF (Table 6). All six active compounds (G1, G3, G6, G9, G11, and G13) docked into very similar binding poses at the bottom of the active site (Supplementary data Fig. S7.). The energetically minimized docking pose of G13 is shown in Fig. 6. The carbonyl oxygen coordinates the catalytic zinc ion, and the substituted phenylalanine ring protrudes into the acetate release channel (Fig. 6- A, B). There is a hydrogen bond between the amide nitrogen of G13 and the hydroxyl oxygen of Y306. The complex between G13 and HDAC8 includes Pi-stacking between the phenyl ring connected with the thiazolidine ring and the active site flanking aromatic amino acids F152 and F208. Also, G13 undergoes Pi-interactions through the thiazolidine ring (to H180) and the phenylalanine (to W141). Interestingly, G13 shows an almost perfect superposition with the α-amino-peptide inhibitor in the crystal structure of its complex with HDAC-8 (PDB-ID: 3SFF, Fig. 6- C). Most recently, Marek et al. identified a selectivity pocket of HDAC-8 at the surface between the L1- and L6-loop (Marek et al., 2018). Some inhibitors, such as the well-known HDAC8-selective inhibitor PCI-34051, have a constrained L-shape that allows for perfect binding into this selectivity pocket. However, G13 shows an entirely different binding mode (Fig. 6- D). The aromatic capping groups of PCI-34051 and G13 point into different grooves of the protein surface, and PCI-34051 does not occupy the acetate release channel. The higher binding affinity of PCI-34051 can be explained by its hydroxamic acid warhead that chelates the catalytic zinc ion. Nevertheless, the G-series compounds appear to be promising pharmacophores because they do not contain a problematic zinc chelating group. Moreover, these compounds show selectivity over class II HDAC isoenzymes, most probably due to their binding into the unique acetate release pocket in HDAC-8.
Table 6.
Docking scores of active substances against representative crystal structures of HDAC-8.
| Compound | PDB-ID 3SFF | PDB-ID 1T69 | PDB-ID 1VKG |
|---|---|---|---|
|
| |||
| G1 | −8.7 | −6.3 | −6.9 |
| G3 | −8.5 | −7.0 | −6.9 |
| G6 | −8.3 | −7.4 | −6.6 |
| G9 | −8.7 | −7.5 | −6.7 |
| G11 | −8.3 | −7.5 | −6.7 |
| G13 | −8.8 | −8.0 | −6.6 |
| Ligand* | −9.4 | −12.1 | −12.4 |
Ligand in respective crystal structure as reference.
Figure 6. Energy minimized docking pose of G13 in HDAC-8 (PDB-ID: 3SFF).

A) 3D-docking scheme showing major interactions as dotted lines: Pi-stacking (pink), hydrogen bond, metal complexation (grey). B) 2D-interactions demonstrating multiple interactions with adjacent amino acids. C) Overlay of the α-amino-peptide ligand (orange) in PDB-ID 3SFF and G13 (green) docked into this protein structure filling the lower pocket in the transition area between active site and acetate release channel. D) Overlay of G13 (green) docked into PDB-ID 3SFF and the crystal structure of smHDAC8 from Schistosoma mansoni and PCI-34051 inhibitor (blue, PDB-ID: 6HQY). The capping groups of both compounds bind to different surface areas.
3. CONCLUSION.
N-substituted benzylidene thiazolidinedione (TZD) derivatives were designed as GLUT1 inhibitors wherein we discovered three compounds, G5, G16, and G17, as inhibitors of GLUTs, with G5 being the most potent of all on GLUT1 (IC50 = 5.4 ± 1.3 μM). Thus, with experimental evidence of GLUT1 and GLUT4 inhibition, our hypothesis of distinguishing TZD derivatives as activators of GLUT protein level or GLUT transport activity inhibitors, based on placing the TZD ring terminally or centrally within this kind of molecules, may serve as a critical milestone in the design of TZD analogs as GLUT modulating agents and their subsequent application as antidiabetic and anticancer agents. Docking of G5, G16, and G17 to the inward- and outward-facing structural models of GLUT1 suggested that the compounds block glucose access to the active site in both transporter conformations. Antiproliferative assay in leukemic CEM and K562 cells indicated good cytotoxic potential for this series of compounds; it was better than that of the unpermuted TZD analog, Pioglitazone, indicating that permuted Δ2 analogs (benzylidene) exerts better antiproliferative effects compared to parent compounds. G5, as the most potent GLUT1 inhibitor, was also found to induce apoptosis and disrupt the cell cycle leading to growth arrest in the G2-M phase for CEM cells, but was safer to noncancerous blood cells, WBCs, making it a promising candidate for further development. Possibly, the higher cytotoxic action of G5 relative to G16 and G17 in CEM cells may stem from the ability of G5 to also inhibit GLUT4 and GLUT5, thus restricting more efficiently the cell energy source. Altogether, G5 and its analogs open the door to novel chemotherapeutics against leukemia acting through the inhibition of GLUTs. The moderate activity of some compounds against HDAC8 holds the potential for the development of dual acting GLUT-/HDAC-inhibitors for an improved synergistic combat of leukemic cells.
4. MATERIALS AND METHODS
4.1. Chemistry
Commercial reagents from S D Fine, Research Lab, or Sigma Aldrich were procured from suppliers in India. Thin layer chromatography (TLC) was performed on Merck pre-coated Silica Gel 60 F254. Melting points were determined by the open capillary method on a VEEGO melting point apparatus and are uncorrected. Infrared spectra were recorded on a Schimadzu FT/IR-8400S by direct sampling technique. 1H and 13C NMR spectra were recorded at 400 MHz on a Bruker instrument using TMS as internal standard, and chemical shifts (δ) are reported in ppm. J values for coupling constants are reported in hertz (Hz). Abbreviations used in NMR are as follows: s - singlet, d - doublet, t – triplet, and m - multiplet. Mass spectra were recorded using LC-MS Agilent Technologies 1260 Infinity instrument. The purity of final derivatives (≥95 %) was confirmed with HPLC (high-performance liquid chromatography) Agilent 1100 system. The conditions used for chromatography are: column- Hemochrome C18, 15 cm, detection wavelength – 300 nm, detector - UV visible detector, flow rate −1mL/min, oven temperature, 30 °C; gradient elution with a run time of 15 min using mobile phase-Methanol: (0.1%) Formic acid (70:30). The synthesis consists of four steps, and the synthetic route followed to arrive at G1-G17 is depicted in Scheme 1.
4.1.1. Synthesis of amides 2a-2d by chloroacetylation of amines 1a-1d
Amides 2a-2d were prepared as previously reported (Scheme 1)(Kabir et al., 2019; Patil et al., 2010; Tilekar et al., 2020). Briefly, Chloroacetyl chloride was added dropwise to the solution of amines 1a-1d and potassium carbonate in DCM and stirred overnight. The crude product was collected by vacuum evaporation of the solvent and recrystallized from ethanol.
4.1.2. Synthesis of Knoevenagel intermediates 5a-5g, by condensation of aldehydes 2a-2g with thiazolidine-2, 4-dione.
Knoevenagel compounds 6a-6g were synthesized by refluxing thiazolidine-2, 4-dione, 4 (5 gm, 0.04 moles) with aromatic aldehydes 3a-3g (5 gm, 0.07 moles) in the presence of sodium acetate (3 gm, 0.003 moles), using acetic acid (10 ml) as solvent (Scheme 1). The reaction mixture was refluxed for 3–6 hours with intermittent stirring. The reaction was monitored by TLC using hexane and ethyl acetate as the mobile phase in the ratio of 4:1. On cooling the reaction mixture, a crystalline product was obtained, which was then pump-filtered and washed with water, dried, and taken further for the next step. Initially, the reaction was tried with toluene as solvent and piperidinium benzoate as a catalyst. Though the condensation reaction was proceeding, it was slow, and the yield was too low. The crude product thus formed was impure and needed recrystallization from alcohol, which further impacted the yield. The method with sodium acetate in acetic acid was more effective in yield, and the condensed product obtained was almost pure and in crystalline form, which was taken to the next step without any purification.
5-(4-bromobenzylidene)thiazolidine-2,4-dione (5a)
Yield: 8.3 g (83 %); M.P.: 241.5–242.5°C; Creamish white shiny crystals; IR (cm−1) : 3088.14, 3051.49, 1712.85, 1606.76, 1481.38, 896.93; 1H NMR (400 MHz, DMSO-d6, δ ppm) 7.27 (s, 1H), 7.38–7.42(m, 2H), 7.49–7.51 (m, 2H), 12.57 (s, 1H).
5-(3,4-dimethylbenzylidene)thiazolidine-2,4-dione (5b)
Yield: 9.1 g (91 %); M.P.: 219.4–219.9 °C; White shiny crystals; IR (cm−1) : 3117.07, 3045.70, 1737.92, 1674.27, 1595.18, 1564.32; 1H NMR (400 MHz, DMSO-d6, δ ppm) 7.25–7.32 (m, 3H), 7.67 (s, 1H), 12.51 (s, 1H), 2.27 (s, 6H).
5-(4-chlorobenzylidene)thiazolidine-2,4-dione (5c)
Yield: 8.6 g (86 %); M.P.: 228.7–229.3 °C; White shiny crystals; IR (cm−1) : 3151.79, 3001.34, 1670.41, 1602.90, 1535.39, 815.92; 1H NMR (400 MHz, DMSO-d6, δ ppm) 7.56–7.62 (m, 4H), 7.77(s, 1H), 12.63 (s, 1H).
5-(2,4-difluorobenzylidene)thiazolidine-2,4-dione (5d)
Yield: 8.8 g (88 %); M.P.: 223.9–224.1 °C; Light yellow shiny crystals; IR (cm−1) : 3037.99, 1662.69, 1624.12, 1614.47, 1585.54, 1537.32, 1012.66; 1H NMR (400 MHz, DMSO-d6, δ ppm) 7.24–7.29(m, 1H), 7.42–7.48 (m, 1H), 7.55–7.71 (m, 1H), 7.70 (s, 1H), 12.71 (s, 1H).
5-(4-fluorobenzylidene)thiazolidine-2,4-dione (5e)
Yield: 7.5 g ( 75 %); M.P.: 239.1–239.6 °C; Light yellow shiny crystals; IR (cm−1) : 3142.15, 3045.70, 1728.28, 1680.05, 1583.61, 1502.60; 1H NMR (400 MHz, DMSO-d6, δ ppm) 7.34–7.40 (m, 2H), 7.63–7.68 (m, 2H), 7.79 (s, 1H), 12.60 (s, 1H).
5-(4-methylbenzylidene)thiazolidine-2,4-dione (5f)
Yield: 7.7 g (77 %); M.P.: 228.9–229.2 °C; Creamish shiny crystals; IR (cm−1) : 3153.72, 3039.91, 1680.05, 1595.18, 1585.54; 1H NMR (400 MHz, DMSO-d6, δ ppm) 7.32–7.34 (m, 2H), 7.47–7.49 (m, 2H), 7.74 (s, 1H), 12.54 (s, 1H), 2.34 (s, 3H).
5-benzylidenethiazolidine-2,4-dione (5g)
Yield: 9.0 g (90 %); M.P.: 248.0–248.6 °C; White shiny crystals; IR (cm−1) : 3039.91, 1666.55, 1612.54, 1585.54, 1537.32; 1H NMR (400 MHz, DMSO-d6, δ ppm) 7.43–7.58(m, 5H), 7.76 (s, 1H), 12.58 (s, 1H).
4.1.3. Synthesis of potassium salts 6a-6g of Knoevenagel intermediates 3a-3g.
Potassium hydroxide (2.5 gm, 0.044 moles) in ethanol was added to 5a-5g (5.0 gm, 0,025 moles) and refluxed for 1–3 h (Scheme 1). White fine solid was obtained after cooling the reaction mixture, which was filtered at pump, washed with cold ethanol, and dried to obtain 6a-6g. The salts were readily soluble in water.
potassium-5-(4-bromobenzylidene)-2,4-dioxothiazolidin-3-ide (6a)
Yield: 5.2 g (69.3 %); M.P.: 289.7–290.6 °C; White powder; IR (cm−1) : 3082.35, 1666.55, 1618.33, 1537.32, 1519.96, 914.29; 1H NMR (400 MHz, DMSO-d6, δ ppm) 7.24 (s, 1H), 7.44–7.46(m, 2H), 7.58–7.61 (m, 2H).
potassium-5-(3,4-dimethylbenzylidene)-2,4-dioxothiazolidin-3-ide (6b)
Yield: 4.8 g (64 %); M.P.: 284.1–284.7 °C; White powder; IR (cm−1): 3012.91, 1739.85, 1672.34, 1602.90, 1537.32; 1H NMR (400 MHz, DMSO-d6, δ ppm) 7.15–7.23 (m, 3H), 7.26 (s, 1H), 2.17–2.22 (d, J = 3.6 Hz, 6H).
potassium-5-(4-chlorobenzylidene)-2,4-dioxothiazolidin-3-ide (6c)
Yield: 5.3 g (70.66 %); M.P.: 245.7–246.2 °C; White powder; IR (cm−1) : 3014.84, 1670.41, 1600.97, 1535.39, 817.85; 1H NMR (400 MHz, DMSO-d6, δ ppm) 7.26 (s, 1H), 7.44–7.48(m, 2H), 7.50–7.54 (m, 2H).
potassium-5-(2,4-difluorobenzylidene)-2,4-dioxothiazolidin-3-ide (6d)
Yield: 5.0 g (66.66 %); M.P.: 255.9–256.5 °C; White powder; IR (cm−1) : 3045.70, 1664.62, 1614.47, 1585.54, 1012.66; 1H NMR (400 MHz, DMSO-d6, δ ppm) 7.17–7.22 (m, 1H), 7.28–7.33 (m, 1H), 7.33 (s, 1H), 7.58–7.64 (m, 1H).
potassium-5-(4-fluorobenzylidene)-2,4-dioxothiazolidin-3-ide (6e)
Yield: 4.4 g ( 58.66 %); M.P.: >300 °C; White powder; IR (cm−1) : 3014.84, 1670.41, 1616.40, 1600.97, 1537.32, 891.14; 1H NMR (400 MHz, DMSO-d6, δ ppm) 7.22–7.27 (m, 3H), 7.53–7.57 (m, 2H).
potassium-5-(4-methylbenzylidene)-2,4-dioxothiazolidin-3-ide (6f)
Yield: 5.5 g (73.33 %); M.P.: 286.4–287.0 °C; Creamish shiny crystals; IR (cm−1) : 3018.70, 2914.54, 1668.48, 1616.40, 1537.32; 1H NMR (400 MHz, DMSO-d6, δ ppm) 7.21–7.23(m, 2H), 7.26 (s, 1H), 7.38–7.40 (m, 2H), 2.30 (s, 3H).
potassium-5-benzylidene-2,4-dioxothiazolidin-3-ide (6g)
Yield: 6.0 g (80 %); M.P.: 280.0–281.1 °C; White powder; IR (cm−1) : 3037.99, 1668.48, 1616.40, 1535.39; 1H NMR (400 MHz, DMSO-d6, δ ppm) 7.25–7.29 (m, 2H), 7.38–7.42(m, 2H), 7.49–7.52 (m, 2H).
4.1.4. Synthesis of final products (G1-G17)
The final products were obtained by refluxing amides 2a-2d (0.044 moles) and potassium salts 6a-6g (0.044 moles) in an equimolar ration, for 6–10 hours, with acetone as the solvent (Scheme 1). The reaction was monitored by TLC using hexane and ethyl acetate as the mobile phase. On completion of the reaction, the reaction mixture was poured over crushed ice. The precipitated crude product was filtered and purified by column or flash chromatography with ethyl acetate: hexane as a solvent in the ration of 10:90 to 40:60.
2-[5-(4-Bromobenzylidene)-2,4-dioxo-thiazolidin-3-yl]-N-(4-chloro-2-trifluromethylphenyl) acetamide (G1)
Yield: 1.6 g (65%); M.P.: charred at 263.0–264.1°C; Cream amorphous powder; IR (cm−1): 3271, 1734, 1666, 1581; 1H NMR (400 MHz, DMSO-d6, δ ppm): 4.57 (s, 2H), 7.58–7.53 (m, 3H), 7.74–7.70 (m, 4H), 7.95 (s, 1H), 10.15 (s, 1H); 13C NMR (400 MHz, DMSO-d6): 43.59, 133.39, 132.97, 132.31, 132, 131.85, 131.63, 126.28, 124.32, 121.73, 166.9, 165.22, 165; Theoretical mass: 519.72, LC-MS (m/z, I%): 520.6 [(M+H)+, 33%]; UV- Spectrum (10 ppm): λmax 331.4nm; HPLC Purity: % Area 97.40, RT 4.04 mins.
2-[5-(4-Bromobenzylidene)-2,4-dioxo-thiazolidin-3-yl]-N-(2,4-diflurophenyl)acetamide(G2)
Yield: 1.2 g (54%); M.P.: charred at 277.7–278.1°C; White amorphous powder; IR (cm−1): 3395, 1747, 1687, 1597; 1H NMR (400 MHz, DMSO-d6, δ ppm): 4.60 (s, 2H), 7.02–6.98 (m, 1H), 7.24–7.18 (m, 1H), 7.57 (d, J = 8Hz, 2H), 7.72 (d, J = 8Hz, 2H), 7.91–7.85 (m, 1H), 7.95 (s, 1H), 10.27 (s, 1H); 13C NMR (400 MHz, DMSO-d6): 43.74, 132.34, 132.27, 131.97, 131.80, 124.94, 124.83, 121.94, 121.72, 114.14, 110.89, 104.03, 166.65, 165.05, 164.21; UV Spectrum (10 ppm): λmax 331.4nm; Theoretical mass: 451.96, LC-MS (m/z, I %): 452.8 [(M+H) +, 78%]; HPLC Purity: % Area 98.65, RT 5.83 mins.
N-(4-Chloro-2-trifluoromethylphenyl)-2-[5-(3,4-dimethylbenzylidene)-2,4-dioxothiazolidin-3-yl] acetamide (G3)
Yield: 1.8 g (54%); M.P.: charred at 286.7–287.0°C; White amorphous powder; IR (cm−1): 3288, 1735, 1670, 1591; 1H NMR (400 MHz, DMSO-d6, δ ppm): 3.06 (s, 6H), 4.54 (s, 2H), 7.79 (d, J = 8Hz, 1H), 7.44 (d, J = 8Hz, 2H), 7.55 (d, J = 8Hz, 1H), 7.71–7.66 (m, 2H), 7.81 (s, 1H), 10.06 (s, 1H); 13C NMR (400 MHz, DMSO-d6): 11.88, 132.30, 134.44, 139.58, 143.94, 167.33, 165.35; Theoretical mass: 468.05, LC-MS (m/z, I%): 470.6 [(M+2H)+, 100%]; HPLC Purity: % Area 97.11, RT 5.96 mins.
N-[2,4-Difluorophenyl)-2-[5-(3,4-dimethylbenzylidene)-2,4-dioxothiazolidin-3-yl]acetamide (G4)
Yield: 0.8 g (45%); M.P.: charred at 299.1–200.8°C; White amorphous powder; IR (cm−1): 3279, 1681, 1660, 1589; 1H NMR (400 MHz, DMSO-d6, δ ppm): 3.05 (s, 6H), 4.56 (s, 2H), 6.8 (d, J = 8Hz, 2H), 7.23–7.18 (m, 1H), 7.46 (d, J = 8Hz, 2H), 7.82 (s, 1H), 7.91–7.85 (m, 1H), 10.24 (s, 1H); 13C NMR (400 MHz, DMSO-d6): 43.50, 134.51, 132.37, 125.01, 124.89, 122.10, 119.63, 112.85, 119.96, 111.15, 110.96, 104.33, 104.06, 103.82 167.28, 165.40, 164.54; Theoretical mass: 402.08, LC-MS (m/z, I %): 403.0 [(M+H)+, 58%]; HPLC Purity: % Area 96.14, RT 4.81mins.
2-[5-(4-Chlorobenzylidene)-2,4-dioxothiazolidin-3-yl]-N-(4-chloro-2-trifluromethylphenyl) acetamide (G5)
Yield: 0.4 g (62%); M.P.: 257.7–258.3°C; White amorphous powder; IR (cm−1): 3323, 1732, 1672, 1593; 1H NMR (400 MHz, DMSO-d6, δ ppm): 4.58 (s, 2H), 7.56–7.53 (m, 3H), 7.62 (d, J = 8Hz, 2H), 7.69–7.63 (m, 1H), 7.71 (s, 1H), 7.95 (s, 1H), 10.10 (s, 1H); 13C NMR (400 MHz, DMSO-d6): 43.60, 135.44, 133.42, 132.24, 131.71, 131.67,131.29, 129.88, 126.23, 121.64, 121.07, 166.61, 165.23, 165; UV Spectrum (10 ppm): λmax 330.3 nm; Theoretical mass: 473.98, LC-MS (m/z, I%): 475.0 [(M+H) +, 30%]; HPLC Purity: % Area 99.57, RT 7.20 mins.
2-[5-(4-Chlorobenzylidene)-2,4-dioxothiazolidin-3-yl]-N-(2,4-diflurophenyl)acetamide (G6)
Yield: 0.8 g (47%); M.P.: charred at 254.8–255.1°C; White amorphous powder; IR (cm−1): 3389, 1691, 1591; 1H NMR (400 MHz, DMSO-d6, δ ppm): 4.59 (s, 2H), 7.03–6.98 (m, 1H), 7.25–7.20 (m, 1H), 7.59 (d, J = 8Hz, 2H), 7.66 (d, J = 8Hz, 1H), 7.90–7.84 (m, 1H), 7.98 (s, 1H), 10.27 (s, 1H); 13C NMR (400 MHz, DMSO-d6): 43.74, 135.48, 132.25, 131.60, 131.64, 130.72, 129.35, 124.99, 124.90, 122.02, 110.95, 104.04, 103.80, 166.68, 165.06, 164.24; UV Spectrum (10 ppm): λmax 330.3 nm; Theoretical mass: 408.01, LC-MS (m/z, I%): 409.1 [(M+H) +, 72%]; HPLC Purity: % Area 96.66, RT 4.08 mins.
N-(4-Chloro-2-trifluoromethylphenyl)-2-[5-(2,4-difluorobenzylidene)-2,4-dioxothiazolidin-3-yl] acetamide (G7)
Yield: 1.2 g (64%); M.P.: charred at 243.9–244.6°C; White amorphous powder; IR (cm−1): 3352, 1737, 1672, 1595; 1H NMR (400 MHz, DMSO-d6, δ ppm): 4.59 (s, 2H), 7.08 (t, J = 8Hz, 1H), 7.43 (s, 1H), 7.56 (d, J = 8Hz, 1H), 7.70–7.61 (m, 2H), 7.9 (s, 1H), 10.10 (s, 1H); 13C NMR (400 MHz, DMSO-d6): 43.20, 137.50, 129.46, 127.94, 127.46, 127.43, 125.26, 125.25, 124.12, 118.92, 118.69, 115.86, 115.77, 170.88, 165.45, 164.58; UV Spectrum (10 ppm): λmax 327.4 nm; Theoretical mass: 476.0, LC-MS (m/z, I%): 476.8 [(M+H) +, 46%]; HPLC Purity: % Area 97.1, RT 5.28 mins.
2-[5-(2,4-Difluorobenzylidene)-2,4-dioxothiazolidin-3-yl]-N-(2,4-diflurophenyl)acetamide (G8)
Yield: 0.9 g (53%); M.P.: charred at 230.6–231.2°C; White amorphous powder; IR (cm−1): 3279, 1734, 1672, 1591; 1H NMR (400 MHz, DMSO-d6, δ ppm): 4.46 (s, 2H), 6.93 (t, J = 8Hz, 1H), 7.11–7.06 (m, 1H), 7.26–7.18 (m, 2H), 7.64– 7.60 (dd, J = 16Hz, 1H), 7.95–7.89 (m, 2H), 10.22 (s, 1H); 13C NMR (400 MHz, DMSO-d6): 43.38, 140.10, 139.99, 137.56, 130.51, 130.41, 129.52, 128, 125.21, 124.19, 114.22, 110.01, 106.14, 105.86, 170.92, 165.49, 164.28; UV Spectrum (10 ppm): λmax 327.6 nm; Theoretical mass: 410.03 LC-MS (m/z, I%): 410.9 [(M+H)+, 67%]; HPLC Purity: % Area 97.50, RT 4.41mins.
N-(4-Chloro-2-trifluoromethylphenyl)-2-[5-(4-fluorobenzylidene)-2,4-dioxothiazolidin-3-yl]acetamide (G9)
Yield: 1.0 g (68%); M.P.: charred at 265.0–266.1°C; White amorphous powder; IR (cm−1): 3302, 1707, 1579; 1H NMR (400 MHz, DMSO-d6, δ ppm): 4.57 (s, 2H), 7.54 (d, J = 8Hz, 1H), 7.84–7.73 (m, 4H), 7.91 (d, J = 8Hz, 1H), 8.01 (s, 1H), 8.11 (s, 1H), 10.19 (s, 1H); 13C NMR (400 MHz, DMSO-d6): 43.20, 133.95, 133.36, 132.76, 132.07, 131.91, 131.70, 130.46, 123.13, 165.21; UV Spectrum (10 ppm): λmax 326.30 nm; Theoretical mass: 458.01, LC-MS (m/z, I%): 459.0 [(M+H) +, 80%]; HPLC Purity: % Area 98.38, RT 5.29 mins.
N-(2,4-Difluoropheyl)-2-[5-(4-fluoro-benzylidene)-2,4-dioxothiazolidin-3-yl]acetamide (G10)
Yield: 1.2 g (38%); M.P.: charred at 260.8–261.2°C; White amorphous powder; IR (cm−1): 3076, 1734, 1701, 1581; 1H NMR (400 MHz, DMSO-d6, δ ppm): 4.57 (s, 2H), 7.33 (t, J = 8Hz, 2H), 7.55 (d, J = 8Hz, 1H), 7.73–7.67 (m, 4H), 7.91 (s, 1H), 10.11 (s, 1H); 13C NMR (400 MHz, DMSO-d6): 43.37, 138.36, 137.54, 131.15, 130.69, 129.53, 127.98, 125.22, 124.17, 120.41, 119.13, 170.89, 165.45, 164.44; UV Spectrum (10 ppm): λmax 326.40 nm; Theoretical mass: 392.35, LC-MS (m/z, I%): 393.7 [(M+H)+, 100%]; HPLC Purity: % Area 98.17, RT 4.09 mins.
N-(4-Chloro-2-trifluoromethylphenyl)-2-[5-(4-methylbenzylidene)-2,4-dioxothiazolidin-3-yl]acetamide (G11)
Yield: 0.5 g (39%); M.P.: 171.5–272.4°C; White amorphous powder; IR (cm−1): 3383, 1716, 1681, 1604, 1539; 1H NMR (400 MHz, DMSO-d6, δ ppm): 2.50 (s, 3H), 4.54 (s, 3H), 7.38 (t, J = 8Hz, 2H), 7.56–7.51 (m, 3H), 7.78 (d, J = 8Hz, 1H), 7.84 (s, 1H), 7.96 (s, 1H), 10.20 (s, 1H); 13C NMR (400 MHz, DMSO-d6): 43.12, 137.64, 129.84, 128.03, 125.30, 124.26, 122.85, 121.93, 111.35, 111.13, 104.50, 104.26, 104, 170.92, 165.49, 164.57; UV Spectrum (10 ppm): λmax 332.6 nm; Theoretical mass: 454.04 LC-MS (m/z, I%): 455.1 [(M+H)+, 66%]; HPLC Purity: % Area 98.94, RT 6.73 mins.
N-(2,4-Difluoropheyl)-2-[5-(4-methylbenzylidene)-2,4-dioxothiazolidin-3-yl]acetamide (G12)
Yield: 0.6 g (48%); M.P.: 268.5–269.3°C; White amorphous powder; IR (cm−1): 3381, 1790, 1649, 1622, 1595; 1H NMR (400 MHz, DMSO-d6, δ ppm): 2.28 (s, 3H), 4.48 (s, 2H), 7.01–6.96 (m, 1H), 7.30–7.24 (m, 3H), 7.47 (d, J = 8Hz, 2H), 7.70–7.56 (m, 1H), 7.87 (s, 1H), 10.20 (s, 1H); 13C NMR (400 MHz, DMSO-d6): 21.13, 43.79, 141.04, 133.61, 130.18, 130.05, 129.91, 119.65, 166.98, 165.32, 164.32; UV Spectrum (10 ppm): λmax 332.8 nm; Theoretical mass: 388.07, LC-MS (m/z, I %): 389.0 [(M+H)+, 100%]; HPLC Purity: % Area 96.41, RT 5.07 mins.
2-[5-Benzylidene-2,4-dioxothiazolidin-3-yl]-N-(4-chloro-2-trifluoromethylphenyl)acetamide (G13)
Yield: 1.3 g (66%); M.P.: 265.0–265.6°C; White amorphous powder; IR (cm−1): 3317, 1660, 1604, 1512; 1H NMR (400 MHz, DMSO-d6, δ ppm): 4.55 (s, 1H), 7.58–7.49 (m, 4H), 7.66 (d, J = 8Hz, 2H), 7.79–7.76 (m, 1H), 7.83 (d, J = 4Hz, 1H), 7.99 (s, 1H), 10.21 (s, 1H); 13C NMR (400 MHz, DMSO-d6): 43.59, 133.57, 133.46, 133.13, 132.80, 131.83, 131.36, 130.79, 130.17, 129.40, 126.42, 126.36, 123.84, 121.12, 120.92, 166.97, 166.39, 166.15; UV Spectrum (10 ppm): λmax 325.7 nm; Theoretical mass: 440.02, LC-MS (m/z, I %): 441.6 [(M+H)+, 100%]; HPLC Purity: % Area 97.02, RT 5.27 mins.
2-[5-Benzylidene-2,4-dioxothiazolidin-3-yl]-N-(2,4-difluorophenyl)acetamide (G14)
Yield: 1.3 g (67%); M.P.: 231.7–232.6°C; White amorphous powder; IR (cm−1): 3333, 1699, 1658, 1579; 1H NMR (400 MHz, DMSO-d6, δ ppm): 4.59 (s, 1H), 7.07 (m, 1H), 7.37–7.32 (m, 1H), 7.54–7.49 (m, 3H), 7.65 (d, J = 8Hz, 2H), 7.86–7.80 (m, 1H), 7.99 (s, 1H), 10.21 (s, 1H); 13C NMR (400 MHz, DMSO-d6): 43.75, 159.89, 159.78, 157.47, 155.06, 154. 94, 152.59, 133.60, 132.79, 130.71, 129.87, 129.21, 125.28, 125.19, 122.0, 121.91, 121.88, 120.91, 111.35, 111.32, 111.13, 167.08, 165.23, 164.44; UV- Spectrum (10 ppm): λmax 326.2 nm; Theoretical mass: 374.09, LC-MS (m/z, I %): 374.8 [(M+H)+, 73%]; HPLC Purity: % Area 99.25, RT 4.00 mins.
2-(5-(2,4-difluorobenzylidene)-2,4-dioxothiazolidin-3-yl)-N-(3-(trifluoromethyl)phenyl) acetamide ( G15)
Yield: 1.2g (57%); M.P.: 255.2–256.1°C; White color solid; IR (cm−1): 3319.60, 1676.20, 1608.69, 1550.82, 1251.84; 1H NMR (400 MHz, DMSO-d6, δ ppm): 4.57 (s, 2H), 7.29–7.33 (t, J = 7.8Hz, 1H), 7.43–7.50 (m, 2H), 7.56–7.60 (t, J = 7.8Hz, 1H), 7.65–7.74 (m, 2H), 7.90 (s, 1H), 8.04 (s, 1H), 10.78 (s, 1H); 13C NMR (400 MHz, DMSO-d6): 44.121, 105.112, 112.868, 113.121, 115.214, 117.457, 117.578, 120.151, 122.748, 123.401, 124.099, 124.157, 125.302, 129.721, 130.173, 130.713, 130.815, 138.996, 164.377, 164.919, 166.706; UV Spectrum (10 ppm): λmax 327.7nm; Theoretical mass: 442.04, LC-MS (m/z, I %): 441.0 [(M-H)+, 100%]; HPLC Purity: % Area 96.18, RT 6.22 mins.
2-(5-(4-fluorobenzylidene)-2,4-dioxothiazolidin-3-yl)-N-(3-(trifluoromethyl)phenyl)acetamide (G16)
Yield: 0.9g (60%); M.P.: charred at 245.9–246.9°C; White color solid; IR (cm−1): 3367.82, 1670.41, 1614.47, 1595.18, 1552.75; 1H NMR (400 MHz, DMSO-d6, δ ppm): 4.56 (s, 2H), 7.39–7.46 (m, 3H), 7.57–7.60 (m, 1H), 7.73–7.76 (m, 3H), 8.01–8.04 (m, 2H), 10.78 (s, 1H); 13C NMR (400 MHz, DMSO-d6): 44.038, 116.494, 116.712, 120.604, 122.766, 129.478, 130.207, 132.672, 132.761, 139.018, 164.484, 165.197, 167.006; UV Spectrum (10 ppm) λmax 326.50 nm; Theoretical mass: 424.05, LC-MS (m/z, I%): 423.1 [(M-H)+, 100%]; HPLC Purity: % Area 99.34, RT 5.55 mins.
2-(5-benzylidene-2,4-dioxothiazolidin-3-yl)-N-(4-chlorobenzo[d]thiazol-2-yl)acetamide (G17)
Yield: 0.85 g (55%); M.P.: charred at 278.3–279.5°C; White color solid; IR (cm−1): 3211.59, 3082.35, 3032.20, 1745.64, 1691.63, 1666.55, 1595.18; 1H NMR (400 MHz, DMSO-d6, δ ppm): 4.69 (s, 2H), 7.30–7.34 (m, 1H), 7.50–7.59 (m, 4H), 7.66–7.68 (m, 2H), 7.97–7.99 (m, 1H) 8.00 (s, 1H), 13.24 (s, 1H); 13C NMR (400 MHz, DMSO-d6): 39.718, 39.926, 40.135, 43.548, 120.968, 124.545, 124.689, 126.315, 129.400, 130.173, 130.831, 132.789, 133.041, 133.791, 145.313, 158.450, 165.098, 165.743, 167.043; UV Spectrum (10 ppm): λmax 327.60nm; Theoretical mass: 429.90, LC-MS (m/z, I%): 431.0[(M+H)+, 100%]; HPLC Purity: % Area 96.63, RT 8.57 mins.
4.2. In-vitro GLUT transport assay
Compounds G1-G17 were analyzed for their effect on the transport activity of GLUT1 (Wieczorke et al., 2003), GLUT4 (Wieczorke et al., 2003), and GLUT5 (Tripp et al., 2017) expressed in hexose transporter null yeast cells (hxt0). The assay was carried out as described previously (Upadhyay et al., 2020). The culturing of yeast cells was done at 30 °C with shaking (180 rpm). VW4000fgy1 yeast cells expressing GLUT1 were grown for 2–3 days in synthetic media without uracil (SC-uracil) with 2% (w/v) maltose. Cells were washed once in SC-uracil, 2% (w/v) glucose media, transferred in the same media so that OD600nm~0.5 and grown further for 1 to 2 days. VW4000fgy1erg4 yeast cells expressing GLUT4 (Wieczorke et al., 2003) were cultured like GLUT1, however in media with a lower percentage of maltose and glucose: SC-uracil, 1% (w/v) maltose media for the initial cell culture, and SC-uracil, 0.2% (w/v) glucose media for the last cell culture. VW4000 yeast cells expressing GLUT5 (Tripp et al., 2017) were grown for one day in YEP [1% (w/v) yeast and 2% (w/v) peptone], 2% (w/v) maltose media, 100 μg/ml geneticin G418. Cells were washed once in YEP, 2% (w/v) fructose media, transferred in the same media at initial OD600nm~0.5, and grown further for 1–2 days. For transport activity assay, cells in the hexose media were centrifuged (1000 xg, 5 minutes), washed once with PBS solution (10 mM Na2HPO4, 1.8 mM KH2PO4, 2.7 mM KCl, 137 mM NaCl, pH 7.4), and resuspended in PBS buffer at an OD600nm ~ 10; each assay determination contained 100 μl of this cell solution.
Transport activity assay was started by adding C14-hexose (5 mM glucose for GLUT1 or GLUT4, and 10 mM fructose for GLUT5). Transport activity assay was halted after 10 minutes by adding 3-ml ice-chilled Quench buffer (0.1 M KPi, 0.1 M LiCl, pH 5.5), followed by filtration through a glass fiber channel (GC50; Advantec, Tokyo, Japan) under vacuum, and another wash with 3-ml Quench buffer and filtration. The filtration membranes were transferred into scintillation vials with 10 ml of Scintillation Solution (BioSafeII; Research Products International, Mount Prospect, IL, USA), and vortexed briefly. The radioactivity was determined with a scintillation counter (Tri-carb 2900TR, Perkin Elmer, USA). As all compounds were solubilized in DMSO, controls for determining the relative transport activity included 1% (v/v) DMSO to represent DMSO used as vehicle control, and known inhibitors as positive controls, 200 μM phloretin for GLUT1 and GLUT4 (Kasahara and Kasahara, 1997), and 100 μM N-[4-(methylsulfonyl)- 2-nitrophenyl]-1,3-benzodioxol-5-amine (MSNBA) for GLUT5(George Thompson et al., 2016). Primary screening was done at 100 μM compound concentration, and the IC50 values were further calculated for the compounds that diminished the relative transport activity by half or more (>50%). Data were analyzed with GraphPad Prism (San Diego, CA, USA).
4.3. In-vitro HDAC screening
HDAC8 was produced by the recombinant technique, as described recently (Jänsch et al., 2019). Recombinant cHDAC4 was expressed using a pET14b vector (Novagen, EMD Millipore) containing the codon-optimized catalytic domain of human HDAC4. A sequential dilution of inhibitor in a buffer (25 mM Tris-HCl, 75 mM KCl, pH 8.0, 0.001 % Pluronic F-127) was made. Various inhibitor concentrations were incubated for an hour with HDAC isoforms in a 96-well black microtiter half-area plate (Greiner) at 30 °C. Then the reaction was started by adding 20 μM of Boc-Lys(trifluoroacetyl)-AMC (Bachem) as a substrate for HDAC4 and HDAC8. After incubation for 60 min at 30 °C, the reaction was terminated by adding 1.7 μM SATFMK for both HDAC4 and 8. The deacetylated substrate was transformed into a fluorescent product by adding 0.4 mg/ml trypsin (Applichem). All experiments were performed in triplicate, and the IC50 values were determined as mean±SD. The release of AMC was tracked in a microplate reader (PheraStar Plus, BMG Labtech) at 450 nm (λEx = 350 nm) and was correlated to the activity of enzyme. IC50 values were derived from the dose-response curves (DRCs).
4.4. Cell Culture
K562 cells (Chronic myeloid leukemia) were obtained from National Center for Cell Sciences (NCCS), Pune, and CEM cells (acute T-cell lymphoblastic leukemia) were acquired from the Department of Biochemistry Indian Institute of Science, Bangalore. K562 and CEM cells were cultured in suspension using RPMI 1640 medium (Thermo fisher scientific) supplied with 10% (v/v) FBS (fetal bovine serum, Gibco, Invitrogen) and 1% anti-microbial antimycotic 100X solution (100 units/mL of penicillin, 100 μg/mL of streptomycin, and 0.25 μg/mL of Gibco Amphotericin B). Cells were kept up in a humidified 5% CO2 air environment at 37°C.
4.5. MTT assay
MTT test was utilized to decide the cytotoxic capability of G1-G17 on leukemic/lymphoid cell lines, CEM and K562. The assay was conducted as depicted before (Tilekar et al., 2020). Quickly, the cells were seeded at a thickness of around 5×103 cells/well in a flat 96-well plate and kept overnight at 37 °C in 95% humidity supplied with 5% CO2. Various concentrations of compounds were made with DMSO and added to the cells. The maximum DMSO concentration in the investigations was <1.5% and demonstrated no cell lethality. The cells were incubated with the compounds for 48 h. Next, 96-well plates were centrifuged at 300 g, 4 °C for 5 minutes and cells in each well were washed with phosphate buffer twice. Then, 20 μL of the MTT staining reagent (5 mg/ml in phosphate cradle arrangement) was added to each well, and the plates were incubated at 37 °C. After 4 h, 100 μL of dimethyl sulfoxide (DMSO) was added to each well to solubilize the formazan crystals, and absorbance was recorded at 570 nm with a microplate reader. All experiments were performed in triplicates, and IC50 values are presented as mean±SD.
4.6. Cell cycle analysis
Cells were seeded in a 24-well microplate (flat bottom) and incubated overnight at 37 °C in a CO2 incubator for 24 h. The media was replaced with new media, and the cells were treated with the IC50 concentration of G5 for 24 h. Untreated cells were the negative control. After incubation, the cells were washed with PBS, centrifuged for 5 minutes at 300 xg and 4 °C, and the supernatant was discarded. Cells were re-suspended in 0.5 ml of PBS, then incubated on ice in 4.5 ml of super cold 70 % ethanol for 2 h, and centrifuged for 5 min at 200 xg and 4 °C. The ethanol was discarded. The resulting cell pellets were suspended in 5 ml PBS for one minute, and centrifuged for 5 min at 200 xg and 4 °C. They were further treated with 1 ml PI (propidium iodide) staining reagent for 15 mins at 37 °C; within 30 mins, cells were analyzed (BD AccuriTM C5 stream cytometer, BD Biosciences, CA, USA). All experiments were performed in triplicate. Data was analyzed with FlowJo software (adaptation 10.1, Ashland, OR, USA).
4.7. Apoptosis
The induction of apoptosis by compound G5 was established by flow cytometry, as described previously (Tilekar et al., 2020). Briefly, cells were seeded in a 24-well microplate and incubated overnight at 37 °C in a CO2 incubator for 24 h. The media was exchanged with fresh media, and the cells were treated with G5 for 24 h. Untreated cells were utilized as the negative control. After incubation, cells were washed with PBS, centrifuged for 5 minutes at 500 xg and 4 °C, and the supernatant was discarded. The cell pellets were resuspended in ice-cold 1X Binding Buffer; 1 μL of annexin V-FITC solution and 5 μL PI (propidium iodide) were added and mixed. Tubes were kept on ice and incubated for 15 minutes in the dark, then combined with 400 μL of ice-cold 1X binding buffer. The cell preparations were analyzed by flow cytometry (BD Accuri C5 flow cytometer, BD Biosciences, CA, USA). All experiments were performed in triplicates. Cytometry data was analyzed with FlowJo software (version 10.1, Ashland, OR, USA).
4.8. Isolation of WBC and assessment of Effect on viability of non-transformed WBCs.
Whole blood was collected from a healthy adult volunteer after obtaining ethical clearance from institutional EC (ethical committee) of Maratha Mandal’s NGH Institute of Dental Sciences, Belagavi. WBCs were isolated as described previously (Dagur and McCoy, 2015), with slight modifications. Briefly, 7.5 ml of whole blood was centrifuged along with 2.5 ml of HiSep at 400 xg for 30 mins. The plasma layer was discarded, and the remaining cell pack volume was diluted with RBC lysis buffer and centrifuged again. This was repeated till the RBCs were visible on the cell pack. Finally, the cell pellets were suspended in PBS, washed to remove the RBC lysis buffer, centrifuged at 400 x g for 10 minutes, resuspended in PBS, and maintained in a CO2 incubator at 37 °C until further use for MTT assay. To find out the cytotoxicity, isolated WBC’s were seeded in a 96-well flat-bottom microplate and maintained at 37 °C in 95% humidity and 5% CO2 overnight. Then the cells were treated with increasing concentrations of G5 ranging from 2.5 to 100 μM for 48 hours. The wells were washed twice with PBS, and 20 μL of the MTT staining solution was added to each well, and the plate was incubated at 37 °C. After 4 h, 100 μL of DMSO was added to each well to dissolve the formazan crystals, and absorbance was recorded at 570 nm with a microplate reader.
Supplementary Material
Highlights.
A series of novel permuted thiazolidinedione derivatives were designed and synthesized as GLUT1 inhibitors.
The representative Compound G5 demonstrated GLUT1, GLUT4 and GLUT5 inhibitory activity.
Compound G5 exhibited excellent antiproliferative activity against CEM cells.
Compound G5 could block the cell cycle in the sub G0/G1 phase and induce apoptosis.
Few compounds of the series were also found as HDAC8 inhibitor.
Acknowledgements
This study was supported by Indo-Poland joint research program grant from Department of Science and Technology (DST) with reference number DST/INT/Pol/P-27/2016 (to CSR), the United State NIH grant R01GM123103 (to JC).
Abbreviations
- TZD
Thiazolidinedione
- GLUT1/4/5
Glucose transporter 1/4/5
- HDAC
Histone deacetylase
- DMF
Dimethyl Formamide
- DCM
Dichloromethane
- NMR
Nuclear magnetic resonance spectroscopy
- IR
Infrared Spectroscopy
- DMEM
Dulbecco’s Modified Eagle Medium
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- FBS
fetal calf serum
- RPMI
Roswell Park Memorial Institute media
- PBS
Phosphate-buffered saline, SD-Standard deviation, RT-Room temperature
Footnotes
Competing Interests. The authors declare no competing interests.
Credit author statement
Kalpana Tilekar: Writing - Original Draft, Investigation, Formal analysis, Investigation, Methodology; Neha Upadhyay: Investigation, Formal analysis, Methodology; Markus Schweipert: Investigation, Formal analysis, Methodology; Jessica D. Hess: Investigation, Formal analysis, Methodology; Lucasantiago Henze Macias: Investigation, Formal analysis; Piotr Mrowka: Investigation, Formal analysis; Franz-Josef Meyer-Almes: Methodology, Investigation, Formal analysis, Software, Supervision; Renato J. Aguilera: Supervision, Resources, Project administration; Cristina V. Iancu: Investigation, Resources, Formal analysis; Jun-yong Choe: Methodology, Supervision, Funding acquisition, Software, Formal analysis, Project administration; C. S. Ramaa: Conceptualization, Supervision, Funding acquisition, Project administration.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S, 2003. Structure and mechanism of the lactose permease of Escherichia coli. Science 301, 610–615. 10.1126/science.1088196 [DOI] [PubMed] [Google Scholar]
- Amann T, Hellerbrand C, 2009. GLUT1 as a therapeutic target in hepatocellular carcinoma. Expert Opin. Ther. Targets 13, 1411–1427. 10.1517/14728220903307509 [DOI] [PubMed] [Google Scholar]
- Arafa EA, Abdelazeem AH, Arab HH, Omar HA, 2014. OSU-CG5, a novel energy restriction mimetic agent, targets human colorectal cancer cells in vitro. Acta Pharmacol. Sin 35, 394–400. 10.1038/aps.2013.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet M, Flanagan JU, Chan DA, Giaccia AJ, Hay MP, 2014. Identifying novel targets in renal cell carcinoma: design and synthesis of affinity chromatography reagents. Bioorg. Med. Chem 22, 711–720. 10.1016/j.bmc.2013.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carruthers A, Helgerson AL, 1991. Inhibitions of sugar transport produced by ligands binding at opposite sides of the membrane. Evidence for simultaneous occupation of the carrier by maltose and cytochalasin B. Biochemistry 30, 3907–3915. 10.1021/bi00230a015 [DOI] [PubMed] [Google Scholar]
- Chan DA, Sutphin PD, Nguyen P, Turcotte S, Lai EW, Banh A, Reynolds GE, Chi J-T, Wu J, Solow-Cordero DE, Bonnet M, Flanagan JU, Bouley DM, Graves EE, Denny WA, Hay MP, Giaccia AJ, 2011. Targeting GLUT1 and the Warburg Effect in Renal Cell Carcinoma by Chemical Synthetic Lethality. Sci. Transl. Med. 3, 94ra70–94ra70 10.1126/scitranslmed.3002394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C-S, Wang D, Kulp SK, 2015. Glucose transporter inhibitors. US9174951B2. [Google Scholar]
- Chen Y-C, Chen I-S, Guh J-H, 2007. Cryptocaryone, isolated from Cryptocarya infectoria, induces apoptosis through extrinsic pathways: The involvement of death receptor clustering and FADD/caspase-8 activation cascades. Clin. Cancer Res 13, C42–C42. [Google Scholar]
- Colín-Lozano B, Estrada-Soto S, Chávez-Silva F, Gutiérrez-Hernández A, Cerón-Romero L, Giacoman-Martínez A, Almanza-Pérez J, Hernández-Núñez E, Wang Z, Xie X, Cappiello M, Balestri F, Mura U, Navarrete-Vazquez G, 2018. Design, Synthesis and in Combo Antidiabetic Bioevaluation of Multitarget Phenylpropanoic Acids. Molecules 23, 340. 10.3390/molecules23020340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbeil CR, Williams CI, Labute P, 2012. Variability in docking success rates due to dataset preparation. J. Comput. Aided Mol. Des 26, 775–786. 10.1007/s10822-012-9570-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun AK, Sutton RE, 2005. Expression of Glucose Transporter 1 Confers Susceptibility to Human T-Cell Leukemia Virus Envelope-Mediated Fusion. J. Virol 79, 4150–4158. 10.1128/JVI.79.7.4150-4158.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagur PK, McCoy JP, 2015. Collection, Storage, and Preparation of Human Blood Cells. Curr. Protoc. Cytom 73. 10.1002/0471142956.cy0501s73 [DOI] [PMC free article] [PubMed]
- Daina A, Michielin O, Zoete V, 2017. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep 7, 42717. 10.1038/srep42717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina A, Michielin O, Zoete V, 2014. iLOGP: A Simple, Robust, and Efficient Description of n -Octanol/Water Partition Coefficient for Drug Design Using the GB/SA Approach. J. Chem. Inf. Model 54, 3284–3301. 10.1021/ci500467k [DOI] [PubMed] [Google Scholar]
- Effert P, Beniers AJ, Tamimi Y, Handt S, Jakse G, 2004. Expression of glucose transporter 1 (Glut-1) in cell lines and clinical specimens from human prostate adenocarcinoma. Anticancer Res. 24, 3057–3063. [PubMed] [Google Scholar]
- Egan WJ, Merz KM, Baldwin JJ, 2000. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem 43, 3867–3877. 10.1021/jm000292e [DOI] [PubMed] [Google Scholar]
- Galochkina T, Ng Fuk Chong M, Challali L, Abbar S, Etchebest C, 2019. New insights into GluT1 mechanics during glucose transfer. Sci. Rep 9, 998. 10.1038/s41598-018-37367-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- George Thompson AM, Iancu CV, Nguyen TTH, Kim D, Choe J, 2015. Inhibition of human GLUT1 and GLUT5 by plant carbohydrate products; insights into transport specificity. Sci. Rep 5, 12804. 10.1038/srep12804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George Thompson AM, Ursu O, Babkin P, Iancu CV, Whang A, Oprea TI, Choe J, 2016. Discovery of a specific inhibitor of human GLUT5 by virtual screening and in vitro transport evaluation. Sci. Rep 6, 24240. 10.1038/srep24240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghose AK, Viswanadhan VN, Wendoloski JJ, 1999. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem 1, 55–68. 10.1021/cc9800071 [DOI] [PubMed] [Google Scholar]
- Granchi C, Fortunato S, Minutolo F, 2016. Anticancer agents interacting with membrane glucose transporters. MedChemComm 7, 1716–1729. 10.1039/C6MD00287K [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Cheng Z, Wang J, Liu W, Peng H, Wang Y, Rao AVS, Li R, Ying X, Korangath P, Liberti MV, Li Y, Xie Y, Hong SY, Schiene-Fischer C, Fischer G, Locasale JW, Sukumar S, Zhu H, Liu JO, 2019. Discovery of a Potent GLUT Inhibitor from a Library of Rapafucins by Using 3D Microarrays. Angew. Chem. Int. Ed 58, 17158–17162. 10.1002/anie.201905578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierréz-Hernández A, Galván-Ciprés Y, Domínguez-Mendoza EA, Aguirre-Vidal Y, Estrada-Soto S, Almanza-Pérez JC, Navarrete-Vázquez G, 2019. Design, Synthesis, Antihyperglycemic Studies, and Docking Simulations of Benzimidazole-Thiazolidinedione Hybrids. J. Chem 2019, 1–8. 10.1155/2019/1650145 [DOI] [Google Scholar]
- Harmon AW, Patel YM, 2004. Naringenin inhibits glucose uptake in MCF-7 breast cancer cells: a mechanism for impaired cellular proliferation. Breast Cancer Res. Treat 85, 103–110. 10.1023/B:BREA.0000025397.56192.e2 [DOI] [PubMed] [Google Scholar]
- Harmon AW, Patel YM, 2003. Naringenin inhibits phosphoinositide 3-kinase activity and glucose uptake in 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun 305, 229–234. 10.1016/S0006-291X(03)00720-4 [DOI] [PubMed] [Google Scholar]
- Huang J-W, Shiau C-W, Yang Y-T, Kulp SK, Chen K-F, Brueggemeier RW, Shapiro CL, Chen C-S, 2005. Peroxisome proliferator-activated receptor gamma-independent ablation of cyclin D1 by thiazolidinediones and their derivatives in breast cancer cells. Mol. Pharmacol 67, 1342–1348. 10.1124/mol.104.007732 [DOI] [PubMed] [Google Scholar]
- Jänsch N, Meyners C, Muth M, Kopranovic A, Witt O, Oehme I, Meyer-Almes F-J, 2019. The enzyme activity of histone deacetylase 8 is modulated by a redox-switch. Redox Biol. 20, 60–67. 10.1016/j.redox.2018.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabir A, Tilekar K, Upadhyay N, Ramaa CS, 2019. Novel Anthraquinone Derivatives as Dual Inhibitors of Topoisomerase 2 and Casein Kinase 2: In Silico Studies, Synthesis and Biological Evaluation on Leukemic Cell Lines. Anticancer Agents Med. Chem 18, 1551–1562. 10.2174/1871520618666180423111309 [DOI] [PubMed] [Google Scholar]
- Kapoor K, Finer-Moore JS, Pedersen BP, Caboni L, Waight A, Hillig RC, Bringmann P, Heisler I, Müller T, Siebeneicher H, Stroud RM, 2016. Mechanism of inhibition of human glucose transporter GLUT1 is conserved between cytochalasin B and phenylalanine amides. Proc. Natl. Acad. Sci. U. S. A 113, 4711–4716. 10.1073/pnas.1603735113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara T, Kasahara M, 1997. Characterization of rat Glut4 glucose transporter expressed in the yeast Saccharomyces cerevisiae: comparison with Glut1 glucose transporter. Biochim. Biophys. Acta 1324, 111–119. 10.1016/s0005-2736(96)00217-9 [DOI] [PubMed] [Google Scholar]
- Laister RC, Minden MD, Mak TW, 2015. Inhibition of Glycolysis as a Therapeutic Strategy in Acute Myeloid Leukemias. Target. Ther. Acute Myeloid Leuk. 709–723 10.1007/978-1-4939-1393-0_38 [DOI] [Google Scholar]
- Li Q, Manolescu A, Ritzel M, Yao S, Slugoski M, Young JD, Chen X-Z, Cheeseman CI, 2004. Cloning and functional characterization of the human GLUT7 isoform SLC2A7 from the small intestine. Am. J. Physiol. Gastrointest. Liver Physiol 287, G236–242. 10.1152/ajpgi.00396.2003 [DOI] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ, 2001. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev 46, 3–26. 10.1016/s0169409x(00)00129-0 [DOI] [PubMed] [Google Scholar]
- Liu Y, Cao Y, Zhang W, Bergmeier S, Qian Y, Akbar H, Colvin R, Ding J, Tong L, Wu S, Hines J, Chen X, 2012. A Small-Molecule Inhibitor of Glucose Transporter 1 Downregulates Glycolysis, Induces Cell-Cycle Arrest, and Inhibits Cancer Cell Growth In Vitro and In Vivo. Mol. Cancer Ther 11, 1672–1682. 10.1158/1535-7163.MCT-12-0131 [DOI] [PubMed] [Google Scholar]
- Liu Yi, Zhang W, Cao Y, Liu Yan, Bergmeier S, Chen X, 2010. Small compound inhibitors of basal glucose transport inhibit cell proliferation and induce apoptosis in cancer cells via glucose-deprivation-like mechanisms. Cancer Lett. 298, 176–185. 10.1016/j.canlet.2010.07.002 [DOI] [PubMed] [Google Scholar]
- Marek M, Shaik TB, Heimburg T, Chakrabarti A, Lancelot J, Ramos-Morales E, Da Veiga C, Kalinin D, Melesina J, Robaa D, Schmidtkunz K, Suzuki T, Holl R, Ennifar E, Pierce RJ, Jung M, Sippl W, Romier C, 2018. Characterization of Histone Deacetylase 8 (HDAC8) Selective Inhibition Reveals Specific Active Site Structural and Functional Determinants. J. Med. Chem 61, 10000–10016. 10.1021/acs.jmedchem.8b01087 [DOI] [PubMed] [Google Scholar]
- Marger MD, Saier MH, 1993. A major superfamily of transmembrane facilitators that catalyse uniport, symport and antiport. Trends Biochem. Sci 18, 13–20. 10.1016/0968-0004(93)90081-w [DOI] [PubMed] [Google Scholar]
- Merigo F, Brandolese A, Facchin S, Missaggia S, Bernardi P, Boschi F, D’Incà R, Savarino EV, Sbarbati A, Sturniolo GC, 2018. Glucose transporter expression in the human colon. World J. Gastroenterol 24, 775–793. 10.3748/wjg.v24.i7.775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan R, Sharma AK, Gupta S, Ramaa CS, 2012. Design, synthesis, and biological evaluation of novel 2,4-thiazolidinedione derivatives as histone deacetylase inhibitors targeting liver cancer cell line. Med. Chem. Res 21, 1156–1165. 10.1007/s00044-011-9623-3 [DOI] [Google Scholar]
- Mueckler M, Thorens B, 2013. The SLC2 (GLUT) family of membrane transporters. Mol. Aspects Med 34, 121–138. 10.1016/j.mam.2012.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muegge I, Heald SL, Brittelli D, 2001. Simple selection criteria for drug-like chemical matter. J. Med. Chem 44, 1841–1846. 10.1021/jm015507e [DOI] [PubMed] [Google Scholar]
- Nishioka T, Oda Y, Seino Y, Yamamoto T, Inagaki N, Yano H, Imura H, Shigemoto R, Kikuchi H, 1992. Distribution of the glucose transporters in human brain tumors. Cancer Res. 52, 3972–3979. [PubMed] [Google Scholar]
- Omar HA, Salama SA, Arafa E-SA, Weng J-R, 2013. Antitumor effects of energy restriction-mimetic agents: thiazolidinediones. Biol. Chem 394, 865–870. 10.1515/hsz-2013-0139 [DOI] [PubMed] [Google Scholar]
- Patil V, Tilekar K, Mehendale-Munj S, Mohan R, Ramaa CS, 2010. Synthesis and primary cytotoxicity evaluation of new 5-benzylidene-2,4-thiazolidinedione derivatives. Eur. J. Med. Chem 45, 4539–4544. 10.1016/j.ejmech.2010.07.014 [DOI] [PubMed] [Google Scholar]
- Pontes HAR, Pontes FSC, Silva BS de F, Fonseca FP, Andrade B.A.B. de, Rizo VHT, Romanach MJ, Leon JE, Almeida O.P. de, 2013. Extranodal Nasal NK/T-Cell Lymphoma: A Rare Oral Presentation and FASN, CD44 and GLUT-1 Expression. Braz. Dent. J 24, 284–288. 10.1590/0103-6440201302202 [DOI] [PubMed] [Google Scholar]
- Reckzeh ES, Waldmann H, 2019. Development of Glucose Transporter (GLUT) Inhibitors: Development of Glucose Transporter (GLUT) Inhibitors. Eur. J. Org. Chem 10.1002/ejoc.201901353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saier MH, Beatty JT, Goffeau A, Harley KT, Heijne WH, Huang SC, Jack DL, Jähn PS, Lew K, Liu J, Pao SS, Paulsen IT, Tseng TT, Virk PS, 1999. The major facilitator superfamily. J. Mol. Microbiol. Biotechnol 1, 257–279. [PubMed] [Google Scholar]
- Siebeneicher H, Bauser M, Buchmann B, Heisler I, Müller T, Neuhaus R, Rehwinkel H, Telser J, Zorn L, 2016a. Identification of novel GLUT inhibitors. Bioorg. Med. Chem. Lett 26, 1732–1737. 10.1016/j.bmcl.2016.02.050 [DOI] [PubMed] [Google Scholar]
- Siebeneicher H, Cleve A, Rehwinkel H, Neuhaus R, Heisler I, Müller T, Bauser M, Buchmann B, 2016b. Identification and Optimization of the First Highly Selective GLUT1 Inhibitor BAY-876. ChemMedChem 11, 2261–2271. 10.1002/cmdc.201600276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavic K, Derbyshire ET, Naftalin RJ, Krishna S, Staines HM, 2009. Comparison of effects of green tea catechins on apicomplexan hexose transporters and mammalian orthologues. Mol. Biochem. Parasitol 168, 113–116. 10.1016/j.molbiopara.2009.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- So FV, Guthrie N, Chambers AF, Moussa M, Carroll KK, 1996. Inhibition of human breast cancer cell proliferation and delay of mammary tumorigenesis by flavonoids and citrus juices. Nutr. Cancer 26, 167–181. 10.1080/01635589609514473 [DOI] [PubMed] [Google Scholar]
- Suzuki S, Okada M, Takeda H, Kuramoto K, Sanomachi T, Togashi K, Seino S, Yamamoto M, Yoshioka T, Kitanaka C, 2018. Involvement of GLUT1-mediated glucose transport and metabolism in gefitinib resistance of non-small-cell lung cancer cells. Oncotarget 9, 32667–32679. 10.18632/oncotarget.25994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilekar K, Upadhyay N, Jänsch N, Schweipert M, Mrowka P, Meyer-Almes FJ, Ramaa CS, 2020. Discovery of 5-naphthylidene-2,4-thiazolidinedione derivatives as selective HDAC8 inhibitors and evaluation of their cytotoxic effects in leukemic cell lines. Bioorganic Chem. 95, 103522. 10.1016/j.bioorg.2019.103522 [DOI] [PubMed] [Google Scholar]
- Tripp J, Essl C, Iancu CV, Boles E, Choe J-Y, Oreb M, 2017. Establishing a yeast-based screening system for discovery of human GLUT5 inhibitors and activators. Sci. Rep 7, 6197. 10.1038/s41598-017-06262-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyay N, Tilekar K, Jänsch N, Schweipert M, Hess JD, Henze Macias L, Mrowka P, Aguilera RJ, Choe J, Meyer-Almes F-J, Ramaa CS, 2020. Discovery of novel N-substituted thiazolidinediones (TZDs) as HDAC8 inhibitors: in-silico studies, synthesis, and biological evaluation. Bioorganic Chem. 100, 103934. 10.1016/j.bioorg.2020.103934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veber DF, Johnson SR, Cheng H-Y, Smith BR, Ward KW, Kopple KD, 2002. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem 45, 2615–2623. 10.1021/jm020017n [DOI] [PubMed] [Google Scholar]
- Wei S, Kulp SK, Chen C-S, 2010. Energy restriction as an antitumor target of thiazolidinediones. J. Biol. Chem 285, 9780–9791. 10.1074/jbc.M109.065466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellberg EA, Johnson S, Finlay-Schultz J, Lewis AS, Terrell KL, Sartorius CA, Abel ED, Muller WJ, Anderson SM, 2016. The glucose transporter GLUT1 is required for ErbB2-induced mammary tumorigenesis. Breast Cancer Res. 18, 131. 10.1186/s13058-016-0795-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieczorke R, Dlugai S, Krampe S, Boles E, 2003. Characterisation of mammalian GLUT glucose transporters in a heterologous yeast expression system. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol 13, 123–134. 10.1159/000071863 [DOI] [PubMed] [Google Scholar]
- Wood TE, Dalili S, Simpson CD, Hurren R, Mao X, Saiz FS, Gronda M, Eberhard Y, Minden MD, Bilan PJ, Klip A, Batey RA, Schimmer AD, 2008. A novel inhibitor of glucose uptake sensitizes cells to FAS-induced cell death. Mol. Cancer Ther 7, 3546–3555. 10.1158/1535-7163.MCT-08-0569 [DOI] [PubMed] [Google Scholar]
- Yan N, 2015. Structural Biology of the Major Facilitator Superfamily Transporters. Annu. Rev. Biophys 44, 257–283. 10.1146/annurev-biophys-060414-033901 [DOI] [PubMed] [Google Scholar]
- Zambrano A, Molt M, Uribe E, Salas M, 2019. Glut 1 in Cancer Cells and the Inhibitory Action of Resveratrol as A Potential Therapeutic Strategy. Int. J. Mol. Sci 20, 3374. 10.3390/ijms20133374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Liu Y, Chen X, Bergmeier SC, 2010. Novel inhibitors of basal glucose transport as potential anticancer agents. Bioorg. Med. Chem. Lett 20, 2191–2194. 10.1016/j.bmcl.2010.02.027 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.