

Abstract
Objective
Emerging evidence highlights the clinical implications of N6-methyladenosine (m6A) modification in HCC. Yet, the roles of m6A modification in modulating cancer immunity and shaping tumor microenvironment (TME) are undefined in hepatocellular carcinoma (HCC).
Methods
Here, m6A modification classification was determined for HCC through 23 m6A modifier levels by employing consensus clustering approach. Prognosis analysis was presented for comparing the differences in survival outcomes. The ssGSEA and ESTIMATE approaches were adopted for evaluating the abundances of tumor-infiltrating immune cell populations. The m6A scoring system was computed for reflecting m6A modification classification via PCA algorithm.
Results
Three m6A modifier-mediated modification patterns were established among HCC specimens, which were characterized by different prognosis, signaling pathways, and TME features. After extracting m6A phenotype-associated DEGs, we determined m6A scores in individual HCC and stratified patients into high- and low-score groups. Patients with low m6A score displayed the survival advantage and higher sensitivity to gemcitabine. Moreover, those with low m6A score possessed the better anti-PD-1/PD-L1 therapeutic response in the IMvigor210 immunotherapy cohort.
Conclusion
Our findings highlighted that m6A modification exerted a nonnegligible role in remodeling diverse and complex TME. Quantification of the m6A modification patterns of individual HCC may enhance the comprehension of TME features and facilitate immunotherapeutic plans.
1. Introduction
Hepatocellular carcinoma (HCC) represents a complex neoplasm with multiple etiologies, comprising 75% to 85% of liver cancer cases [1]. Over 1 million HCC patients will die from HCC in 2030, as estimated by the World Health Organization [2]. Early-stage HCC patients suitably receive curative therapy including resection, ablation, and transplantation, with expected 5-year survival rate up to 60% to 80% [3]. Nevertheless, less than 20% patients are eligible for curative therapy [4]. Intermediate-stage patients usually experience locoregional therapy [5]. Meanwhile, systemic therapy is reserved for advanced patients. For instance, sorafenib is the first systemic agent with efficacy for advanced HCC [6]. In recent years, immunotherapy like antiprogrammed cell death-1 (anti-PD-1), anti-PD-1 ligand (anti-PD-L1), and anti-cytotoxic T-lymphocyte antigen-4 (anti-CTLA-4) that may activate the host's natural defense system, and identify and eliminate the tumor cells [7], have emerged as a prospective alternative treatment strategy against advanced HCC with durable responses [8]. Despite this, only a minority of patients benefit from the immunotherapy [9]. Hence, it urgently demands novel therapeutic predictors for identifying the ideal HCC subgroups for immunotherapy.
N6-methyladenosine (m6A) is the most abundant messenger RNA (mRNA) modification that occurs in humans, occupying 0.1% to 0.4% total adenosine residues [10]. The m6A modification represents a dynamic reversible process in humans [11]. Hence, exploring the regulatory genes may assist to uncover the roles and mechanisms of m6A modification at posttranscriptional levels. Emerging evidences have confirmed that deregulation and genetic alterations of m6A modifiers contribute to HCC initiation and progression [12]. For instance, m6A reader YTHDF1 accelerates HCC progression via inducing FZD5 mRNA translation with an m6A-dependent manner [13]. M6A eraser ALKBH5 inhibits malignancy of HCC through m6A-dependent epigenetic suppression of LYPD1 [14]. M6A writer KIAA1429 facilitates migration and invasion of HCC through elevating m6A-mediated ID2 level [15]. HCC progression represents a multistep event, comprising the genetic and epigenetic alterations within tumor cells and the surrounding tumor microenvironment (TME) [16]. Cancer cells elicit various biological behavior alterations via the direct or indirect interplay with TME [17]. The in-depth comprehending of the diverse and complex TME may reveal its key roles in tumor development, immune escape, and immunotherapeutic responsiveness [18]. Emerging evidence suggests that TME is specially correlated to m6A modification [19]. For instance, m6A eraser ALKBH5 may enhance the effects of anti-PD-1 agent through modulating lactate accumulation along with immunosuppressive cell populations in the TME [20]. Inhibiting m6A writers METTL3/14 may increase the responsiveness to anti-PD-1 therapy [21]. Nevertheless, above findings are limited to one or two m6A modifiers due to limited technology. Hence, comprehensively discerning the TME traits modulated by different m6A modifiers may enhance the cognition of antitumor immunity.
Herein, we presented an overall evaluation concerning the interactions of m6A modification with TME traits through integration of the HCC transcriptomic and genomic profiles from public data sets. We established three m6A modification patterns with diverse outcomes and TME. Also, an m6A scoring system was proposed for quantifying the m6A modification patterns, which could predict survival outcomes and immunotherapy responses. Thus, m6A machinery exerts a nonnegligible function in shaping diverse TME and regulating cancer immunity in HCC.
2. Materials and Methods
2.1. Acquisition of HCC Cohorts and Preprocessing
Transcriptome profiling and clinicopathological annotation of HCC were gathered from The Cancer Genome Atlas (TCGA) together with Gene-Expression Omnibus (GEO) repositories. Specimens with incomplete follow-up data were removed. For TCGA data set, RNA-seq profiling (FPKM value) of 373 HCC samples and 50 normal samples was gained from the Genomic Data Commons (GDC, https://portal.gdc.cancer.gov/) with TCGAbiolinks package [22]. Afterwards, FPKM was converted to TPM form. Somatic mutations and copy number variations (CNVs) were also retrieved from TCGA. For microarray data from the GSE14520 data set, the raw “CEL” file of 242 HCC samples was retrieved, which was corrected by background and normalized by quantile with robust multiarray averaging algorithm [23]. Through Rcircos package [24], the locations of 23 m6A modifiers in chromosome were drawn. The clinical information of TCGA and GSE14520 data sets was shown in Supplementary Tables 1 and 2.
2.2. Unsupervised Clustering for 23 m6A Regulators
Totally, this study extracted 23 m6A modifiers comprising 8 writers (CBLL1, KIAA1429, METTL3/14, RBM15/15B, WTAP, and ZC3H13), 2 erasers (ALKBH5 and FTO), and 13 readers (ELAVL1, FMR1, HNRNPA2B1, HNRNPC, IGF2BP/2/3, LRPPRC, YTHDC1/2, and YTHDF1/2/3) from the obtained data sets. Distinct m6A regulator-mediated modification patterns were classified for HCC through unsupervised clustering analyses in the light of the level of aforementioned modifiers. By employing consensus clustering approach, the number and consistency of clustering were determined via ConsensuClusterPlus package with 1000 times repetitions [25].
2.3. Clinical Specimens
Three fresh HCC and matched adjacent normal liver tissues were harvested in the Affiliated Traditional Chinese Medicine Hospital of Southwest Medical University from February 2021 to May 2021. No patients experienced preoperative chemo- or radiotherapy before operation. Each subject provided written informed consent following the guideline of the Declaration of Helsinki. The study was approved by the Ethics Committee of The Affiliated Traditional Chinese Medicine Hospital of Southwest Medical University (approval id: 2021017).
2.4. Western Blot
Tissue and cell specimens were lysed by RIPA lysis reagent (Beyotime, China) on the ice for half hour and sonicated in an ice bath for 3 min. Following centrifugation at 4°C at 12,000 r/min for 10 min, the supernatant was harvested. The protein concentrations were calculated with BCA kit (Beyotime, China). Then, lysate was boiled with 5 × SDS loading buffer at 100°C for 5 min. Then, protein was separated by SDS-PAGE electrophoresis as well as transferred onto PVDF membranes (Millipore, Germany). Following being washed with TBST, the membranes were blocked by 5% milk/TBST lasting 1 h. Then, the membranes were probed with primary antibodies against METTL3 (1 : 1000; 15073-1-AP; Proteintech, Wuhan, China), ZC3H13 (1 : 1000; DF4623; AFFINITY, USA), YTHDF2 (1 : 1000; 24744-1-AP; Proteintech, Wuhan, China), or GAPDH (1 : 5000; ATA00013Rb; AtaGenix, Wuhan, China) at 4°C overnight. The membranes were washed by PBST for three times. Afterwards, the membranes were exposed to HRP-labeled goat antirabbit secondary antibody (SA00001-2; Proteintech, Wuhan, China) at room temperature for 1 h. The membranes were developed with luminescent buffer and investigated by ChemiDoc™ XRS + gel imaging system (Bio-Rad, Shanghai, China).
2.5. Immunofluorescence
Immunofluorescence was performed for detecting METTL3, ZC3H13, and YTHDF2 expression in HCC and normal tissues. In brief, paraformaldehyde-fixed and paraffin-embedded tissue slices were cut into 5 μm thickness. The slices were incubated by anti-METTL3 (1 : 50; 15073-1-AP; Proteintech, Wuhan, China), anti-ZC3H13 (1 : 50; DF4623; AFFINITY, USA), and anti-YTHDF2 (1 : 50; 24744-1-AP; Proteintech, Wuhan, China) antibodies lasting 2 h. Following being washed, the slices were probed with ALexa Fluor 488-conjugated Affinipure goat antirabbit IgG (H + L) (SA00006-2; Proteintech, Wuhan, China) and DAPI (D9542; Sigma, USA). Then, the sections were mounted with glycerol and investigated under a fluorescence microscope.
2.6. Functional Annotation Analyses
The activity of biological processes and pathways between different clusters was compared through gene set variation analysis (GSVA) [26] that represents a nonparametric and unsupervised gene set enrichment method. The Hallmark gene set was retrieved from the Molecular Signatures Database as a reference. Functional annotation analyses of m6A regulators or m6A-related genes were carried out through clusterProfiler package [27].
2.7. Cell Culture and Transfection
HCC cell lines (Hep3B, HUH-7; Chinese Academy of Sciences; Shanghai, China) were grown in DMEM (Thermo Fisher Scientific, USA) plus 10% FBS (Thermo Fisher Scientific, USA), 100 units/mL ampicillin together with 100 μg/mL streptomycin at 37°C with 5% CO2. ZC3H13 plasmid was gained from GenePharma company (USA), which was transfected into Hep3B and HUH-7 cells via Lipofectamine™ 2000 transfection reagent (Thermo Fisher Scientific, USA).
2.8. 5-Ethynyl-2′-deoxyuridine (EdU) Assay
Cellular proliferation was determined utilizing BeyoClick™ EdU-594 cell proliferation detection kit (C0078S; Beyotime, Shanghai, China). The transfected cells were inoculated onto 24-well plates (8 × 105 cells/well). All operations were carried out following the instructions. Under a fluorescence microscopy, the images were acquired and analyzed.
2.9. Transwell Assay
Migration and invasion were examined utilizing Transwell chambers (Corning, Shanghai, China). For invasion test, the chambers were coated by Matrigel (BD, USA), without Matrigel for migration test. HCC cells were inoculated onto the upper chambers plus serum-free media (3 × 104 cells/well). DMEM media with 10% FBS were added to the lower chambers. Following 24 h, migrated or invasive cells were fixed by 4% PFA (Beyotime, China), and dyed utilizing crystal violet. The number of migrated or invaded cells was counted at ×100 magnification utilizing an inverted light microscope.
2.10. Assessment of the TME
Single-sample gene set enrichment analyses (ssGSEA) were adopted to infer the relative infiltrations of 28 immune cell populations in the TME. The enrichment scores ranging from 0 to 1 were used to denote the relative infiltrations of each immune population based on the markers of each immune population [28, 29].
2.11. Quantifying Immune Response Predictive Factors
The Estimation of Stromal and Immune Cells in Malignant Tumors using Expression Data (ESTIMATE) method was adopted for computing immune/stromal score that could be predictive immune/stromal cell abundance [30]. The Tumor Immune Dysfunction and Exclusion (TIDE) score may infer cancer immunotherapy response [31]. This score was based on two major tumor immune evasion mechanisms: dysfunctional tumor-infiltrating cytotoxic T lymphocytes (CTLs) as well as CTL exclusion via immunosuppressors. Immunophenoscore (IPS) that was developed by four types of immune-related genes: MHC, checkpoint or immunomodulator, effector/suppressor cell population was used to estimate anti-CTLA-4/PD-1 therapeutic response [29].
2.12. Dimension Reduction and Ferroptosis Score
Differentially expressed genes (DEGs) with adjusted p < 0.05 and |fold-change| > 1.5 were screened between m6A methylation patterns with limma package, called as m6A [32]. The overlapped DEGs between distinct m6A methylation patterns were chosen, called as m6A phenotype-associated DEGs. Univariate Cox regression analysis was conducted for screening prognostic m6A phenotype-associated DEGs with p < 0.05. According to the expression profiling of prognostic m6A phenotype-relevant DEGs, HCC subjects were clustered into distinct m6A genomic phenotypes. The expression profiles of the prognostic m6A phenotype-associated DEGs were utilized for performing PCA, followed by extraction of principal components 1 and 2 as m6A score. The approach mostly depended upon the scores on the gene sets with the most favorable association (or inverse association) genes blocks, and down-weighted contribution of genes that cannot be tracked with other set members. The formula [33, 34] was adopted for defining the m6A score: m6A score = ∑PC1i + ∑PC1i, in which i denoted m6A phenotype-associated DEG level.
2.13. Prediction of Chemotherapy and Immunotherapy Response
The response to two commonly chemotherapeutic agents (gemcitabine and cisplatin) was inferred through the Genomics of Drug Sensitivity in Cancer (GDSC; https://www.cancerrxgene.org/) [35]. The half maximal inhibitory concentration (IC50) values were determined through pRRophetic package [36]. The available data of immunotherapy were obtained from IMvigor210 data set in the light of Creative Commons 3.0 License [37]. The immunotherapeutic efficacy was estimated based on subclass mapping (SubMap) analyses [38].
2.14. Statistical Analysis
All the computational and statistical analyses were carried out with R programming and GraphPad Prism. Pearson test was applied for assessing the interactions between variables. T-distributed stochastic neighbor embedding (t-SNE) was implemented for the differences between HCC and controls in the light of the mRNA levels of m6A modifiers. Kaplan–Meier curves of overall survival (OS), disease-free survival (DFS), disease-specific survival (DSS) together with progression-free interval (PFI) were constructed and the survival differences were computed utilizing log-rank tests. Univariate and multivariate analyses were utilized for assessing the independency of variables in predicting prognosis. Comparisons between two groups were presented with student's t or Wilcoxon tests, with one-way analysis of variance or Kruskal–Wallis test among more than two groups. p values < 0.05 were regarded as statistical significance.
3. Results
3.1. Gene Mutations and Expression of m6A Regulators in HCC
Here, the present research observed the roles of 23 m6A modifiers across HCC comprising 8 writers, 2 erasers, and 13 readers (Figure 1(a)). The prevalence of somatic variations of above regulators in HCC was summarized in Figure 1(b). Totally, 39 of the 364 (10.71%) HCC specimens displayed somatic variations of m6A regulators, mainly containing missense mutation, nonsense mutation, splice site, in frame deletion, and frame shift deletion. HNRNPC, LRPPRC, ZC3H13, ELAVL1, FMR1, RNPA2B1, YTHDC1, YTHDC2, WTAP, and IGF2BP3 occurred somatic variations in HCC. Further analysis revealed the prevalent CNVs in m6A regulators and gain was the most frequent CNV type (Figure 1(c)). We further ascertained whether the aforementioned gene mutations affected the mRNA level of m6A modifiers across HCC. Compared to control specimens, most exhibited higher expression in HCC specimens (Figure 1(d)). The m6A regulators with gain CNVs significantly had increased expression in HCC. This indicated that CNVs might prominently contribute to the perturbation on the m6A modifier level. In accordance with 23 m6A modifiers, HCC specimens were distinctly distinguished from control specimens (Figure 1(e)). Pearson correlation analyses indicated the significantly mutual regulation between regulators, as shown in Figure 1(f). Among 23 m6A regulators, we selected three regulators METTL3, ZC3H13, and YTHDF2 to verify their expression in three paired HCC and normal tissues. Our Western blot (Figures 1(g) and 1(h)) and immunofluorescence assays (Figures 1(i) and 1(j)) confirmed that METTL3 and YTHDF2 were significantly up-regulated, while ZC3H13 possessed distinct down-regulation in HCC versus normal specimens. These data unveiled the heterogeneity in mutations and levels of m6A modifiers between HCC and controls together with their important implications in liver tumorigenesis.
Figure 1.
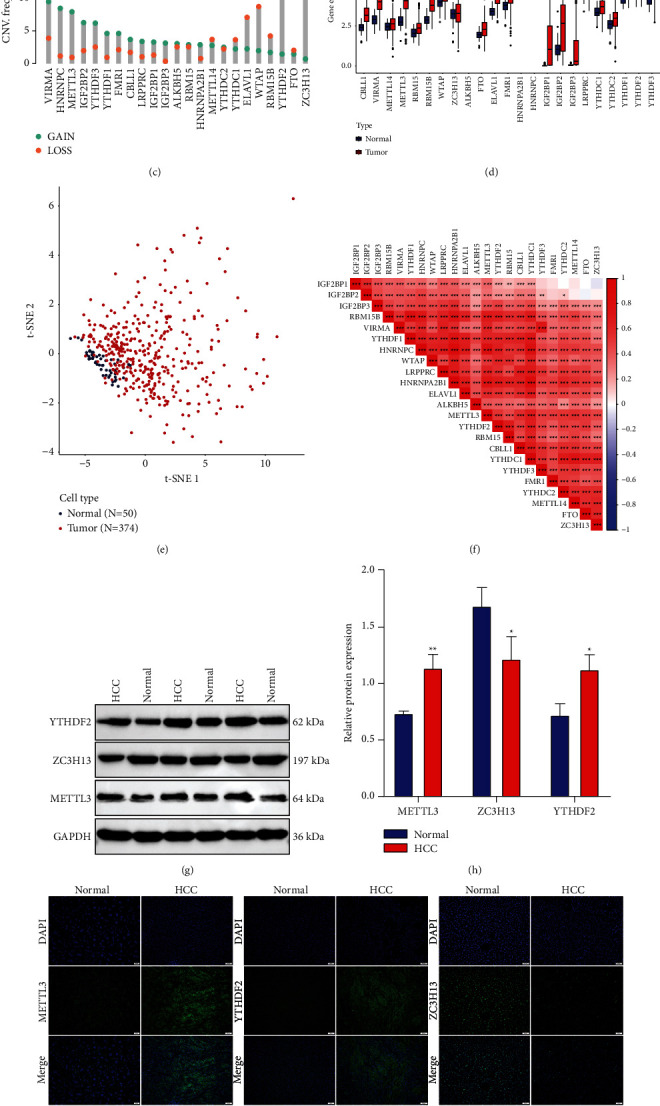
Overall mutations and expression of m6A regulatory genes across HCC: (a) the locations of CNV alterations of 23 m6A modifiers on human chromosome. (b) The frequency of somatic mutations of 23 m6A modifiers across HCC specimens from TCGA cohort. The number on the right denoted the variation frequencies of modifies and the right bars displayed the proportions of mutation types. Each column represented individual samples. The stacked bars below indicated the fractions of transformation across specimens. (c) The CNV frequencies of m6A modifiers across HCC specimens from TCGA cohort. The column height indicated the mutation frequencies. Blue represented the frequency of gain and orange represented the frequency of loss. (d) The differences in mRNA levels of 23 m6A modifiers between HCC and normal tissues in TCGA cohort. (e) The t-SNE for distinguishing HCC (red dot) from control (blue dot) specimens based on the regulators. (f) Pearson analyses for the mutual regulation among regulators at the mRNA expression. The darker the red, the stronger the correlation. (g, h) Western blot of the expression of three regulators METTL3, ZC3H13, YTHDF2 in 3 paired HCC and normal tissues. (i, j) Immunofluorescence for the levels of above regulators. Bar = 50 μm. Ns: no significance; ∗p < 005; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
3.2. Prognostic Implications and Biological Functions of m6A Regulators in HCC
Functional annotation analyses confirmed the influence of 23 m6A regulators on mRNA methylation, RNA modification, RNA stability, and the like (Figure 2(a)). As depicted in Figure 2(b), there were close correlations between 23 writer, reader, and eraser modifiers. This indicated the functions of distinct m6A modifiers on HCC pathogenesis. Univariate and multivariate analyses showed the survival implication of above regulators (Figures 2(c) and 2(d)). Among them, METTL3, ZC3H13, and YTHDF2 could act as independent prognostic indicators. Previous research has reported that METTL3 and YTHDF2 may facilitate HCC progression [39]. Here, we observed the biological functions of ZC3H13 in HCC. In HepG2 and HUH-7 cells, ZC3H13 was successfully overexpressed following transfection with ZC3H13 plasmid (Figures 2(e) and 2(f)). EdU staining showed that ZC3H13 overexpression markedly weakened proliferation of HepG2 and HUH-7 cells (Figures 2(g) and 2(h)). Also, the migrated (Figures 2(i) and 2(j)) and invasive capacities (Figures 2(k) and 2(l)) were distinctly suppressed in HCC cells after overexpressing ZC3H13. Figure 2(m) showed the top-30 genes associated with ZC3H13 in HCC. Collectively, m6A regulators possessed the important prognostic implications and biological functions in HCC.
Figure 2.
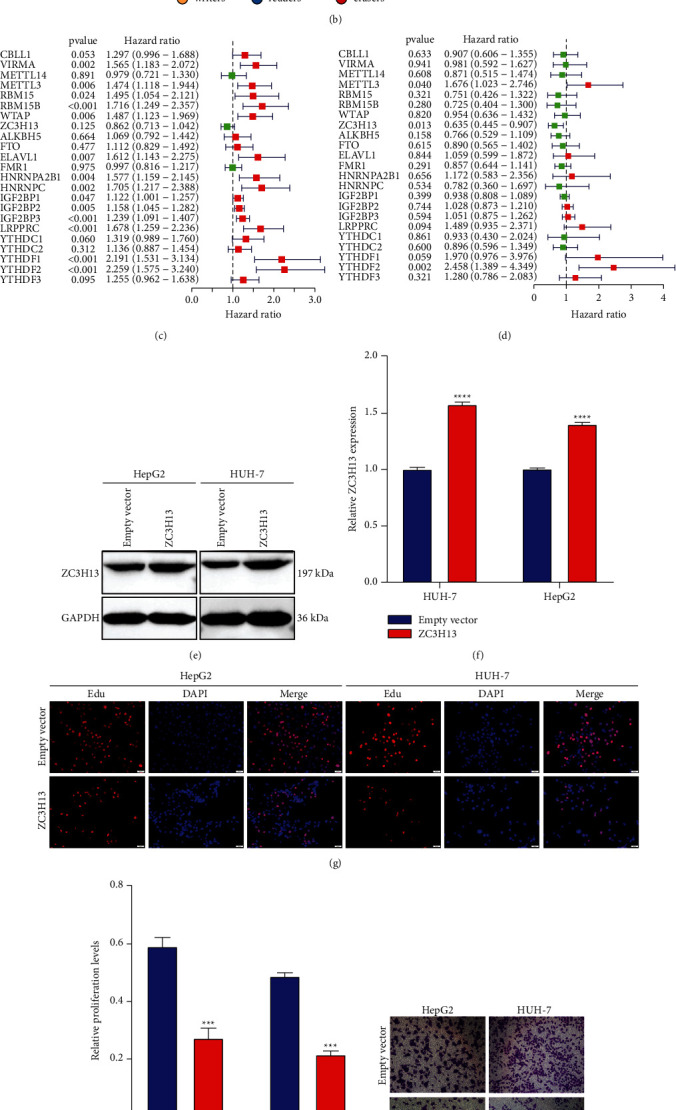
Characterization of prognostic implications and biological functions of 23 m6A regulators across HCC: (a) functional annotations of the regulators with GO enrichment analyses. The bar length represented the number of enriched regulators. (b) The interaction of expression on 23 m6A regulators (writers, yellow; readers, blue; erasers, red) in HCC. The lines that connected modifiers indicated their interactions, and the circle size indicated the survival value of regulators and scaled by p values. (c) Uni- and (d) multivariate cox regression analyses of 23 m6A regulators in HCC prognosis. (e, f) Western blot of the expression of ZC3H13 in HeG2 and HUH-7 cells transfected with empty vector and ZC3H13 up-regulation. (g, h) EdU staining for cell proliferation of HeG2 and HUH-7 cells with empty vector and ZC3H13 up-regulation. (i–l) Transwell assays for the migration and invasion of HeG2 and HUH-7 cell lines following transfection with empty vector and ZC3H13 overexpression. (m) Heatmap showing the top 30 genes associated with ZC3H13 in HCC. ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
3.3. Characterization of m6A Regulator Expression Patterns with Distinct Prognosis and TME Landscape
Our aforementioned data indicated that the interplay between m6A regulators exerted a key function in taking shape diverse m6A modification patterns of HCC. Through ConsensusClusterPlus package, we classified HCC subjects in TCGA cohort as diverse m6A modification patterns in the light of the mRNA levels of 23 m6A regulatory genes. As a result, three distinct modification patterns were characterized with unsupervised clustering analyses (Supplementary Figures 1(a)–1(d)). Our data showed the distinct discrepancy in m6A regulator levels among three m6A modification patterns (Figure 3(a)). For most m6A regulators, cluster B displayed the highest expression, with the moderate expression in cluster A and the lowest expression in cluster C. For exploring the biological molecular alterations underlying three clusters, GSVA was carried out based on the Hallmark gene set (Figure 3(b)). We found that cluster A was markedly enriched in metabolism-related processes (fatty acid/bile acid/xenobiotic metabolisms, etc.). Meanwhile, cluster B displayed prominently enriched pathways that were in relation to carcinogenic activation, stromal and immune pathways like PI3K-Akt-mTOR, P53, Hedgehog, Notch, Wnt-β-catenin, TGF-β, complement, IL2-STAT5, and IL6-JAK-STAT3 pathways. Nevertheless, most pathways were down-regulated in cluster C. Prognosis analysis showed that cluster A presented a distinct survival superiority, while cluster B possessed the poorest outcomes in TCGA data set (Figure 3(c)). Further analyses confirmed that stromal activation was distinctly strengthened in cluster B, as displayed by EMT and WNT-target processes (Figure 3(d)). Genetic mutation pathways were also markedly activated in cluster B including DNA damage repair, DNA replication, nucleotide excision repair, homologous recombination, and mismatch repair, with cell cycle-related processes in cluster B. The ssGSEA approaches were adopted for estimating the immune population abundance across HCC. Pearson correlation analysis indicated the significantly positive or negative interactions between 23 m6A modifiers and infiltrating immune populations (Figure 3(e)). Heatmap was depicted for visualizing the differences in immune population abundance among patterns. Antitumor lymphocyte populations like effector memory CD4+ T, activated CD4+ T, and natural killer cells exhibited primary activation in cluster B (Figure 3(f)). By ESTIMATE approach, this study quantified the overall infiltrations of immune together with stromal cell populations. Cluster A was characterized by enhanced stromal score, cluster C displayed relatively high immune score and cluster B had the highest CD274 (PD-L1) expression (Figure 3(g)).
Figure 3.
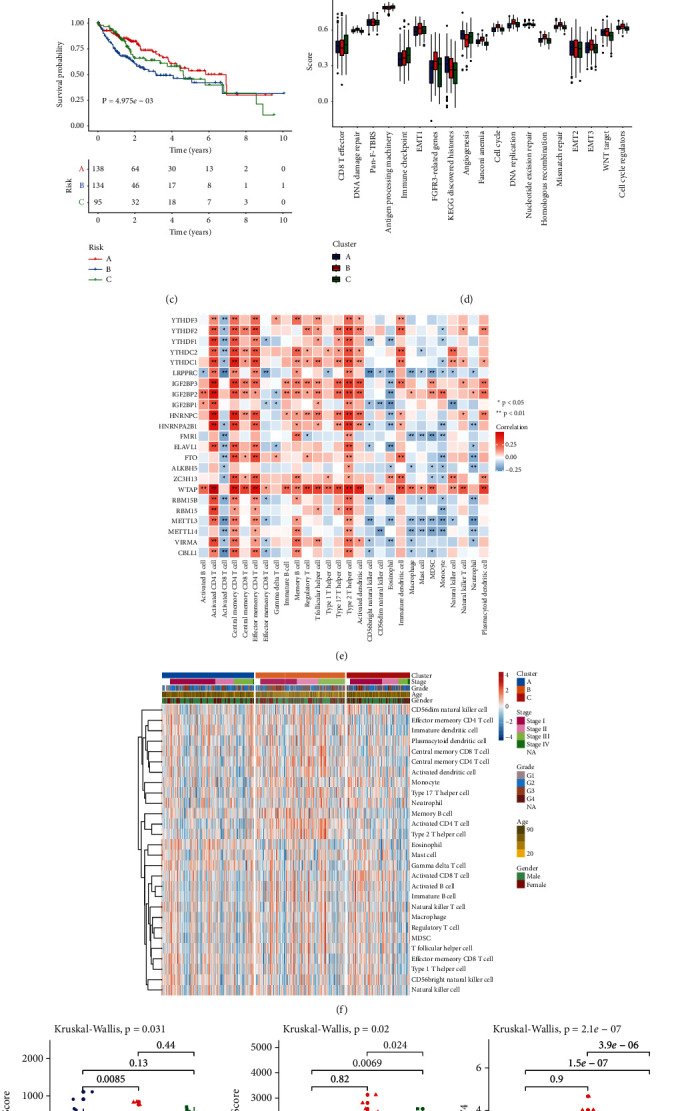
Characterization of m6A methylation patterns with diverse prognosis and TME landscape in TCGA cohort: (a) Heatmaps for the mRNA levels of 23 m6A modifiers among three m6A machinery phenotypes. (b) Heatmap of the GSVA scores of Hallmark pathways in distinct m6A modification patterns. (c) Kaplan–Meier OS curves for HCC patients from TCGA data set with diverse patterns (log-rank tests). (d) The differences in the activation of several pathways among three patterns. Kruskal-Wallis H test was applied for comparing three clusters. Ns: no significance; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. (e) Heatmap of the correlations of 23 m6A modifiers with infiltrating immune cells based on ssGSEA method. Positive correlation, red; negative correlation, blue. (f) Heatmap for the differences in 28 immune population abundance among three patterns. (g) The discrepancy in stromal/immune score and CD274 expression among three patterns.
3.4. Construction of m6A Genomic Phenotypes in HCC
For more deeply investigating the underlying biological behaviors in each m6A cluster, 331 overlapping genes with deregulation were selected among distinct m6A modification patterns with limma approach, called as m6A phenotype-associated DEGs (Figure 4(a); Supplementary Table 3). Functional annotation analyses showed the distinct enrichment of the m6A phenotype-associated DEGs in metabolic processes (Figure 4(b)) and pathways (Figure 4(c)), confirming the critical roles of m6A methylation in HCC progression. For validating the regulation mechanisms, unsupervised clustering analysis was carried out according to above 331 m6A phenotype-associated DEGs. Consistent with the m6A modification patterns, patients were classified as three genomic phenotypes (Supplementary Figures 2(a)–2(d)). Prognosis analysis showed that gene cluster B displayed the worst survival outcomes among three genomic phenotypes (Figure 4(d)). Heatmap visualized the expression of the m6A phenotype-associated DEGs among three gene clusters (Figure 4(e)). Consistent with m6A modification patterns, gene cluster B displayed the largest stromal score, with the highest immune score in cluster C and the activated CD274 expression in cluster B (Figure 4(f)).
Figure 4.
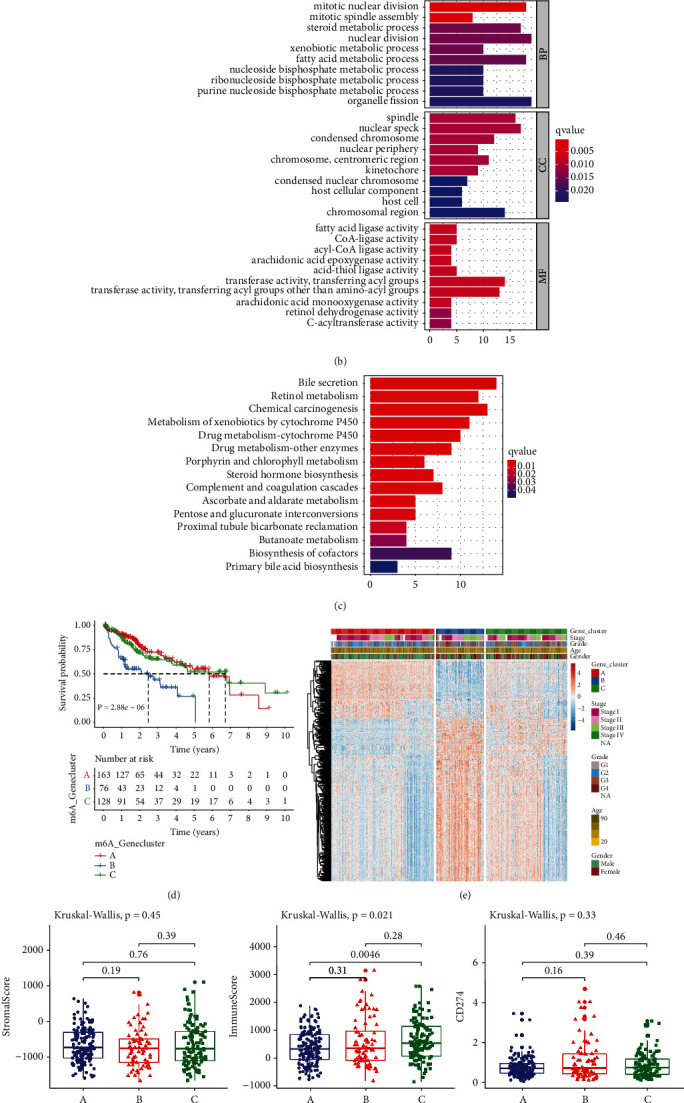
Construction of m6A genomic phenotypes in HCC from TCGA cohort: (a) Venn diagram for the 331 m6A phenotype-associated DEGs between three genomic phenotypes. (b) GO and (c) KEGG enrichment results of the m6A phenotype-associated DEGs. (d) Kaplan–Meier curves of OS for three gene clusters (log-rank tests). (e) Heatmap for the levels of the m6A phenotype-associated DEGs among clusters. (f) The discrepancy in stromal score, immune score and CD274 expression among clusters.
3.5. Establishment of an m6A Scoring System and Characterization of Its Clinical Implications
For accurately predicting m6A machinery classification across each HCC sample, this study generated the m6A scoring system according to the prognostic m6A phenotype-associated DEGs (Figure 5(a)). We compared m6A score among distinct m6A genomic phenotypes. Consequently, genomic cluster B exhibited the largest m6A score, whereas cluster A possessed the lowest m6A score (Figure 5(b)). Prognosis analysis showed that high m6A score indicated poorer OS than low m6A score (Figure 5(c)). Following univariate and multivariate analyses, m6A score acted as an independent survival indicator (Figures 5(d) and 5(e)). After stratifying HCC cases into diverse subgroups in the light of clinicopathological traits (age, sex, grade, and stage), patients with high m6A score also displayed unfavorable outcomes than those with low m6A score (Supplementary Figure 3(a)–3(h)). No notable differences in stromal score were measured between high and low m6A scores (Figure 5(f)). Nevertheless, low m6A score displayed the increased immune score compared to high m6A score (Figure 5(g)). CD274 expression was also compared between groups. In Figure 5(h), high m6A score was characterized by elevated CD274 expression. We also found that m6A score was positively associated with stromal pathways (including EMT and WNT-target) and genetic mutation-related pathways (Figure 5(i)). Furthermore, high m6A score indicated the poorer DFI (Figure 5(j)), DFS (Figure 5(k)), DSS (Figure 5(l)), and PFI (Figure 5(m)) compared to low m6A score, indicating that m6A score could be utilized for predicting HCC recurrence and progression.
Figure 5.
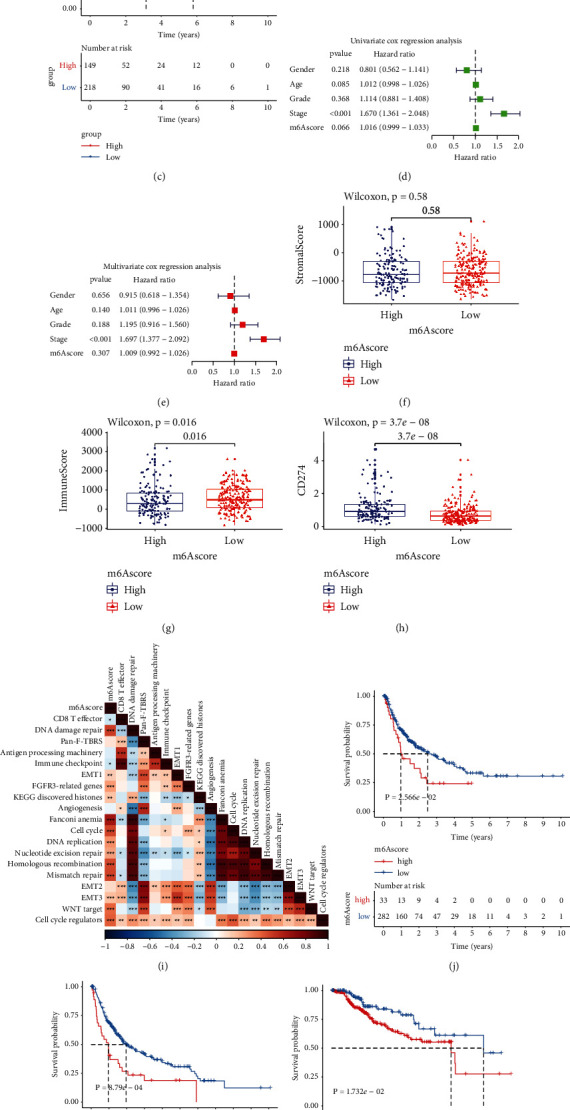
Establishment of an m6A scoring system along with characterization of its clinical implications in TCGA cohort: (a) alluvial chart of m6A modification, gene cluster, m6A score along with survival status. (b) The discrepancy in m6A score between three m6A-modified gene clusters. P values were calculated with Kruskal-Wallis tests. (c) Kaplan–Meier OS curves for patients high or low m6A score (log-rank test). (d) Uni- and (e) multivariate analyses of the independency of the m6A score in predicting HCC outcomes. (f–h) The discrepancy in (f) stromal score, (g) immune score and (h) CD274 expression between groups. P values were determined using Wilcoxon tests. (i) Associations between m6A score and several pathways in HCC specimens. (j–m) Kaplan–Meier curves of (j) DFI, (k) DFS, (l) DSS and (m) PFI for cases with high or low m6A scores (log-rank tests). ∗p < 005; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
3.6. The m6A Score Can Predict Chemotherapeutic and Immunotherapeutic Benefits
Tumor somatic mutations were compared between m6A score subgroups in TCGA cohort. As depicted in Figures 6(a) and 6(b), more prevalent somatic mutations occurred in high m6A group compared to low m6A group. The responses to two commonly chemotherapeutic drugs were assessed by GDSC database. After comparison, the estimated IC50 value of gemcitabine was markedly lower in high m6A score population (Figure 6(c)). This demonstrated that patients possessing high m6A score were more likely to benefit from gemcitabine. Nevertheless, no significant difference in cisplatin was found between groups (Figure 6(d)). TIDE score, an emerging predictor of immunotherapy, was calculated in each HCC specimen. Compared to high m6A score group, decreased TIDE score was found in low m6A score population (Figure 6(e)), indicating the more benefits from immunotherapy. Figure 6(f) visualized the proportions of patients that responded to anti-PD-L1 immunotherapy in the IMvigor210 data set. The responser/nonresponser was 15%/85% in high m6A score population and 32%/68% in another population. This confirmed that cases with low m6A score exhibited the distinct therapeutic advantage in anti-PD-L1 therapy. In addition, we compared the survival differences between populations in this cohort. In Figure 6(g), cases with low m6A score possessed the significant prognostic advantage. SubMap analyses also indicated that low m6A score cases possessed the greater possibility of responding to anti-PD-1 therapy (Figure 6(h)). The IPS score was determined for evaluating the immune response, which was markedly increased in low m6A score population (Figure 6(i)). Thus, m6A modification patterns could be involved in mediating the tumor immune response.
Figure 6.
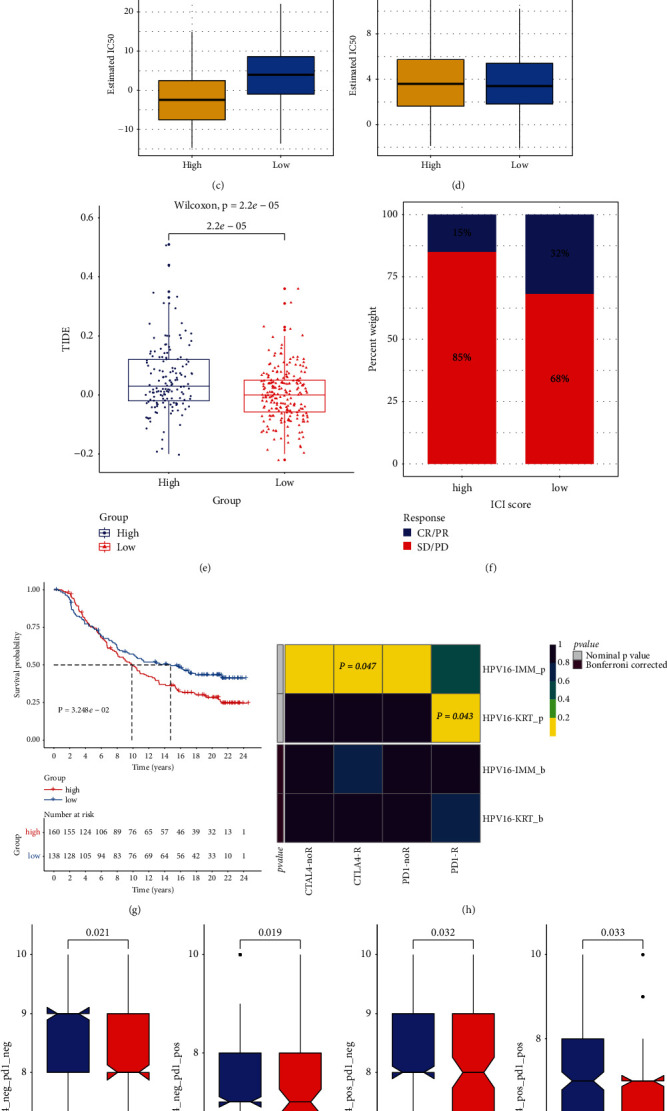
The roles of m6A score in prediction of chemo- and immunotherapeutic benefits of HCC patients in TCGA cohort: (a, b) Somatic mutation landscape of significantly mutated genes across (a) high or (b) low m6A score populations. (c, d) The differences in estimated IC50 values between populations. (e) The comparisons in TIDE score between populations. P values were determined with Wilcoxon tests. (f) The proportions of subjects responding to anti-PD-L1therapy in two populations in the IMvigor210 cohort. (g) Kaplan–Meier OS curves for cases with high and low m6A scores (log-rank test). (h) The response of cases with high and low m6A scores to anti-CTLA-4/PD-1 therapies through SubMap analyses. (i) The comparisons of IPS scores in two populations.
3.7. External Validation of m6A Methylation Patterns and m6A Score
The m6A regulator-based m6A methylation patterns were validated among 242 HCC patients in the GSE14520 cohort. Consistent with the TCGA cohort, HCC cases were classified as three m6A methylation patterns (Figure 7(a)). Heatmap visualized the notable discrepancy in 23 m6A modifier levels among three m6A methylation patterns (Figure 7(b)). Similarly, cluster B exhibited the poorest OS outcomes (Figure 7(c)). Based on the m6A-related DEGs, we also calculated m6A score of each HCC case in the GSE14520 data set. Data demonstrated that cases with low m6A score exhibited the significant survival advantage (Figure 7(d)). These data confirmed the accurate classifications of HCC based on 23 m6A regulators.
Figure 7.
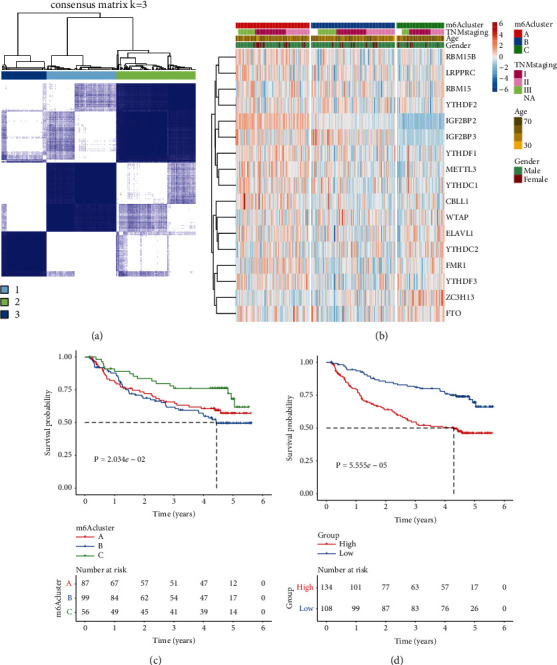
Verification of the m6A methylation patterns and m6A score for HCC in the GSE14520 data set: (a) Consensus clustering analysis for classifying 242 HCC patients into three m6A machinery phenotypes in the light of the expression profiles of 23 m6A modifiers. (b) Heatmap for the expression of 23 m6A regulators among three patterns. (c) Kaplan–Meier OS curves among three phenotypes. (d) Kaplan-Meier OS curves for cases with high or low m6A score. P values were calculated with log-rank tests.
4. Discussion
Mounting evidences suggest that m6A machinery is closely in relation to innate immunity, inflammatory response along with anticancer effects by interaction with different m6A regulatory genes [40]. Here, we established three m6A regulator-based m6A modification phenotypes with diverse prognostic outcomes, biological processes along with TME traits across HCC according to the expression profiling of 8 writers, 2 erasers, and 13 readers. Based on m6A-associated DEGs, we developed an m6A scoring system for each HCC specimen. The m6A score enabled to infer immunotherapeutic response. Hence, the current research highlighted the roles of m6A machinery in shaping TME along with immunity modulation, which might promote precision immunotherapeutic strategies.
Herein, we comprehensively uncovered the somatic mutations, CNVs and expression patterns of 23 m6A regulators. Most displayed the gain CNVs and high expression in HCC, and their expression profiling could distinguish HCC from normal liver tissues. Also, most were distinctly correlated to HCC prognosis. These data suggested that m6A regulators participated in HCC initiation and progress. We verified METTL3, ZC3H13, and YTHDF2 levels across HCC and normal specimens via Western blot and immunofluorescence. Our data confirmed the up-regulation of METTL3 and YTHDF2 as well as the down-regulation of ZC3H13 in HCC. So far, there is no study about the implications of ZC3H13 across HCC. However, previous research has found that ZC3H13 acts as a tumor suppressor gene in breast carcinoma [41]. It mitigates growth along with invasive capacity of colorectal cancer via inactivation of Ras-ERK signaling [42]. Herein, our experiments showed that ZC3H13 up-regulation exerted an inhibitory effect on proliferation and invasion of HCC cells, confirming the anti-HCC effects of ZC3H13.
At the mRNA levels, 23 m6A regulators exhibited significant synergistic effects in HCC. Hence, the current research established three m6A machinery patterns in the light of their expression. In cluster A, metabolism-related processes were distinctly activated while cluster B displayed the activation of carcinogenic, stromal, and immune pathways. This explained why cluster B had the worst survival outcomes. The three m6A methylation patterns were characterized by distinct TME. For instance, anti-tumor lymphocyte cells were mainly activated in cluster B. Totally, 331 DEGs were identified among patterns, which were mainly in relation to metabolic processes. The evidence suggests the biological implications of metabolic process in HCC outcomes [2]. On the basis of m6A-associated DEGs, three m6A gene phenotypes were clustered. Consistent with the m6A methylation patterns, three gene phenotypes were characterized by distinct prognosis and TME. For defining m6A modification patterns, we developed an m6A scoring system, which might guide treatment plans for each subject. Our prognosis analyses unveiled that the m6A score acted as a credible independent survival predictor for HCC. Elevated m6A score was markedly correlated to undesirable OS, recurrence, and progression of HCC patients. Moreover, m6A score exhibited the strong correlations to immune predictors such as TIDE and IPS. Low m6A score was more likely to be responsive to anti-PD-1/PD-L1 therapy in the IMvigor210 cohort. Our data confirmed that the m6A machinery patterns can be adopted in clinical application as well as immunotherapy plans for HCC.
There are advantages in our study. First, several experimental studies have verified the roles of single m6A regulators in HCC [43, 44]. For instance, RBM15 is highly expressed in HCC, and its up-regulation is indicative of undesirable survival outcomes as well as triggers HCC progression by modulating m6A-modified YES1 depending upon IGF2BP1 [45]. However, the function of ZC3H13 in HCC progression remains unclear. Herein, this research for the first time confirmed the low level of ZC3H13 in HCC tissue and ZC3H13 up-regulation inhibited the proliferation, migration along with invasion of HCC cells through experiments. Second, although a recent study constructed distinct m6A regulator-based m6A modification patterns for HCC in TCGA cohort, this classification was not externally verified in independent cohorts. Herein, we determined three m6A regulator-mediated modification patterns for HCC, and confirmed the classification accuracy in the GSE14520 cohort. Third, we developed an m6A score that could predict survival outcomes and therapeutic responses for HCC patients. Despite this, several limitations should be pointed out. First, although the current research reviewed the literature and collected 23 known m6A modifiers, new identified regulators should be included for optimizing the accuracy of the m6A machinery phenotypes. Second, the m6A machinery phenotypes and m6A score were proposed based on retrospective cohorts. Therefore, more prospective cohorts of HCC cases are needed to verify our conclusion.
5. Conclusion
Collectively, the current research synthetically evaluated the m6A regulator-based m6A machinery phenotypes for HCC that were characterized by different prognosis, TME features, and activation of biological processes. Moreover, quantification of the m6A modification patterns by m6A score may enhance the cognition of TME features as well as offer key insights into immunotherapy responses.
Acknowledgments
This research was supported by the Sichuan Provincial Administration of Traditional Chinese Medicine Special Project of Traditional Chinese Medicine (grant nos. 2020LC0227 and 2020LC0228).
Abbreviations
- HCC:
Hepatocellular carcinoma
- m6A:
N6-methyladenosine
- mRNA:
Messenger RNA
- TME:
Tumor microenvironment
- TCGA:
The cancer genome atlas
- GEO:
Gene-expression omnibus
- CNV:
Copy number variation
- GSVA:
Gene set variation analysis
- EdU:
5-Ethynyl-2′-deoxyuridine
- ssGSEA:
Single sample gene set enrichment analyses
- TIDE:
Tumor immune dysfunction and exclusion
- DEGs:
Differentially expressed genes
- GDSC:
Genomics of drug sensitivity in cancer
- IC50:
Half maximal inhibitory concentration
- t-SNE:
T-distributed stochastic neighbor embedding
- OS:
Overall survival
- DFS:
Disease-free survival
- DSS:
Disease-specific survival
- PFI:
Progression-free interval.
Contributor Information
Zongchao Liu, Email: 565409672@qq.com.
Chenyi Huang, Email: xnykdfszyyhcy@swmu.edu.cn.
Data Availability
The data used to support the findings of this study are included within the supplementary materials.
Conflicts of Interest
The authors declare no conflicts of interest.
Authors' Contributions
Fei Liu and Xinyue Zhang contributed equally.
Supplementary Materials
Supplementary Figure 1. Consensus clustering analyses of stratifying HCC cases in TCGA cohort into three m6A methylation patterns according to 23 m6A regulators. (A) Heatmap for the consensus matrix k = 3. (B) Cumulative distribution function (CDF) under diverse k values. (C) Delta area diagram for relative alterations in area under CDF curves. (D) The tracking plot for HCC samples under different k values. Supplementary Figure 2. Consensus clustering analyses for clustering three m6A genomic phenotypes in the light of the expression profiling of m6A-associated genes in TCGA cohort. (A) Heatmap for the consensus matrix k = 3. (B) CDF under diverse k values. (C) Delta area diagram for relative alterations in area under CDF curves. (D) The tracking plot for HCC samples under different k values. Supplementary Figure 3. Subgroup analysis of the prognosis value of m6A score among HCC patients in TCGA data set. Kaplan-Meir curves of cases with high or low m6A score in each subgroup: (A) age ≥ 65; (B) age < 65; (C) female; (D) male; (E) G1-2; (F) G3-4; (G) stage I-II; (H) stage III-IV. P values were determined through log-rank tests. Supplementary Table 1. The clinical information of HCC samples in the TCGA data set. Supplementary Table 2. The clinical information of HCC samples in the GSE14520 data set. Supplementary Table 3. The list of 331 m6A phenotype-associated DEGs.
References
- 1.Calvaruso V., Bruix J. Towards personalized screening for hepatocellular carcinoma: still not there. Journal of Hepatology . 2020;73:1319–1321. doi: 10.1016/j.jhep.2020.06.032. [DOI] [PubMed] [Google Scholar]
- 2.Yang C., Huang X., Liu Z., Qin W., Wang C. Metabolism-associated molecular classification of hepatocellular carcinoma. Molecular Oncology . 2020;14:896–913. doi: 10.1002/1878-0261.12639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang B. H., Yang J., Jiang L., et al. Development and validation of a 14-gene signature for prognosis prediction in hepatocellular carcinoma. Genomics . 2020;112(4):2763–2771. doi: 10.1016/j.ygeno.2020.03.013. [DOI] [PubMed] [Google Scholar]
- 4.Lee Y. H., Tai D., Yip C., Choo S. P., Chew V. Combinational immunotherapy for hepatocellular carcinoma: radiotherapy, immune checkpoint blockade and beyond. Frontiers in Immunology . 2020;11 doi: 10.3389/fimmu.2020.568759.568759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang J. D., Heimbach J. K. New advances in the diagnosis and management of hepatocellular carcinoma. BMJ . 2020;371 doi: 10.1136/bmj.m3544.m3544 [DOI] [PubMed] [Google Scholar]
- 6.Vogel A., Saborowski A. Current strategies for the treatment of intermediate and advanced hepatocellular carcinoma. Cancer Treatment Reviews . 2020;82 doi: 10.1016/j.ctrv.2019.101946.101946 [DOI] [PubMed] [Google Scholar]
- 7.Kong X., Fu M., Niu X., Jiang H. Comprehensive analysis of the expression, relationship to immune infiltration and prognosis of TIM-1 in cancer. Frontiers in Oncology . 2020;10:p. 1086. doi: 10.3389/fonc.2020.01086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang L., Ding J., Li H. Y., Wang Z. H., Wu J. Immunotherapy for advanced hepatocellular carcinoma, where are we? Biochimica et Biophysica Acta (BBA)—Reviews on Cancer . 2020;1874(2) doi: 10.1016/j.bbcan.2020.188441.188441 [DOI] [PubMed] [Google Scholar]
- 9.Brown Z. J., Greten T. F., Heinrich B. Adjuvant treatment of hepatocellular carcinoma: prospect of immunotherapy. Hepatology . 2019;70(4):1437–1442. doi: 10.1002/hep.30633. [DOI] [PubMed] [Google Scholar]
- 10.He L., Li H., Wu A., Peng Y., Shu G., Yin G. Functions of N6-methyladenosine and its role in cancer. Molecular Cancer . 2019;18(1):p. 176. doi: 10.1186/s12943-019-1109-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Niu X., Xu J., Liu J., Chen L., Qiao X., Zhong M. Landscape of N (6)-methyladenosine modification patterns in human ameloblastoma. Frontiers in Oncology . 2020;10 doi: 10.3389/fonc.2020.556497.556497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang T., Kong S., Tao M., Ju S. The potential role of RNA N6-methyladenosine in Cancer progression. Molecular Cancer . 2020;19(1):p. 88. doi: 10.1186/s12943-020-01204-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu X., Qin J., Gao T., et al. RETRACTED: YTHDF1 facilitates the progression of hepatocellular carcinoma by promoting FZD5 mRNA translation in an m6A-dependent manner. Molecular Therapy—Nucleic Acids . 2020;22:750–765. doi: 10.1016/j.omtn.2020.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Chen Y., Zhao Y., Chen J., et al. ALKBH5 suppresses malignancy of hepatocellular carcinoma via m (6)A-guided epigenetic inhibition of LYPD1. Molecular Cancer . 2020;19(1):p. 123. doi: 10.1186/s12943-020-01239-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng X., Li M., Rao X., et al. KIAA1429 regulates the migration and invasion of hepatocellular carcinoma by altering m6A modification of ID2 mRNA. OncoTargets and Therapy . 2019;12:3421–3428. doi: 10.2147/ott.s180954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Q., He Y., Luo N., et al. Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell . 2019;179(4):829–845. doi: 10.1016/j.cell.2019.10.003. [DOI] [PubMed] [Google Scholar]
- 17.Chen L., Niu X., Qiao X., et al. Characterization of interplay between autophagy and ferroptosis and their synergistical roles on manipulating immunological tumor microenvironment in squamous cell carcinomas. Frontiers in Immunology . 2021;12 doi: 10.3389/fimmu.2021.739039.739039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu Q., Zhou L., Lv D., Zhu X., Tang H. Exosome-mediated communication in the tumor microenvironment contributes to hepatocellular carcinoma development and progression. Journal of Hematology & Oncology . 2019;12(1):p. 53. doi: 10.1186/s13045-019-0739-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y., Gu J., Xu F., et al. Molecular characterization, biological function, tumor microenvironment association and clinical significance of m6a regulators in lung adenocarcinoma. Briefings in Bioinformatics . 2020;22 doi: 10.1093/bib/bbaa225. [DOI] [PubMed] [Google Scholar]
- 20.Li N., Kang Y., Wang L., et al. ALKBH5 regulates anti-PD-1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proceedings of the National Academy of Sciences of the United States of America . 2020;117(33):20159–20170. doi: 10.1073/pnas.1918986117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang L., Hui H., Agrawal K., et al. m (6 A RNA methyltransferases METTL3/14 regulate immune responses to anti-PD-1 therapy. EMBO Journal . 2020;39(20) doi: 10.15252/embj.2020104514.e104514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Colaprico A., Silva T. C., Olsen C., et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Research . 2016;44(8):p. e71. doi: 10.1093/nar/gkv1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Irizarry R. A., Hobbs B., Collin F., et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics . 2003;4(2):249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 24.Zhang H., Meltzer P., Davis S. RCircos: an R package for Circos 2D track plots. BMC Bioinformatics . 2013;14(1):p. 244. doi: 10.1186/1471-2105-14-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilkerson M. D., Hayes D. N. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics . 2010;26(12):1572–1573. doi: 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hänzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics . 2013;14(1):p. 7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu G., Wang L. G., Han Y., He Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS: A Journal of Integrative Biology . 2012;16(5):284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jia Q., Wu W., Wang Y., et al. Local mutational diversity drives intratumoral immune heterogeneity in non-small cell lung cancer. Nature Communications . 2018;9(1):p. 5361. doi: 10.1038/s41467-018-07767-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Charoentong P., Finotello F., Angelova M., et al. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Reports . 2017;18(1):248–262. doi: 10.1016/j.celrep.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 30.Yoshihara K., Shahmoradgoli M., Martínez E., et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nature Communications . 2013;4(1):p. 2612. doi: 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang P., Gu S., Pan D., et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nature Medicine . 2018;24(10):1550–1558. doi: 10.1038/s41591-018-0136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ritchie M. E., Phipson B., Wu D., et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Research . 2015;43(7):p. e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chong W., Shang L., Liu J., et al. m(6 A regulator-based methylation modification patterns characterized by distinct tumor microenvironment immune profiles in colon cancer. Theranostics . 2021;11(5):2201–2217. doi: 10.7150/thno.52717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang B., Wu Q., Li B., Wang D., Wang L., Zhou Y. L. m(6 A regulator-mediated methylation modification patterns and tumor microenvironment infiltration characterization in gastric cancer. Molecular Cancer . 2020;19(1):p. 53. doi: 10.1186/s12943-020-01170-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang W., Soares J., Greninger P., et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Research . 2012;41(D1):D955–D961. doi: 10.1093/nar/gks1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geeleher P., Cox N., Huang R. S. pRRophetic: an R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS One . 2014;9 doi: 10.1371/journal.pone.0107468.e107468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mariathasan S., Turley S. J., Nickles D., et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature . 2018;554(7693):544–548. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoshida Y., Brunet J. P., Tamayo P., Golub T. R., Mesirov J. P. Subclass mapping: identifying common subtypes in independent disease data sets. PLoS One . 2007;2(11) doi: 10.1371/journal.pone.0001195.e1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen M., Wei L., Law C. T., et al. RNA N6-methyladenosinemethyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology . 2018;67(6):2254–2270. doi: 10.1002/hep.29683. [DOI] [PubMed] [Google Scholar]
- 40.Su R., Dong L., Li Y., et al. Targeting FTO suppresses cancer stem cell maintenance and immune evasion. Cancer Cell . 2020;38(1):79–96. doi: 10.1016/j.ccell.2020.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gong P. J., Shao Y. C., Yang Y., et al. Analysis of N6-methyladenosine methyltransferase reveals METTL14 and ZC3H13 as tumor suppressor genes in breast cancer. Frontiers in Oncology . 2020;10 doi: 10.3389/fonc.2020.578963.578963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu D., Zhou J., Zhao J., et al. ZC3H13 suppresses colorectal cancer proliferation and invasion via inactivating Ras-ERK signaling. Journal of Cellular Physiology . 2019;234(6):8899–8907. doi: 10.1002/jcp.27551. [DOI] [PubMed] [Google Scholar]
- 43.Li J., Zhu L., Shi Y., Liu J., Lin L., Chen X. m6A demethylase FTO promotes hepatocellular carcinoma tumorigenesis via mediating PKM2 demethylation. American Journal of Translational Research . 2019;11(9):6084–6092. [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang C., Huang S., Zhuang H., et al. YTHDF2 promotes the liver cancer stem cell phenotype and cancer metastasis by regulating OCT4 expression via m6A RNA methylation. Oncogene . 2020;39(23):4507–4518. doi: 10.1038/s41388-020-1303-7. [DOI] [PubMed] [Google Scholar]
- 45.Cai X., Chen Y., Man D., et al. RBM15 promotes hepatocellular carcinoma progression by regulating N6-methyladenosine modification of YES1 mRNA in an IGF2BP1-dependent manner. Cell Death & Disease . 2021;7(1):p. 315. doi: 10.1038/s41420-021-00703-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Consensus clustering analyses of stratifying HCC cases in TCGA cohort into three m6A methylation patterns according to 23 m6A regulators. (A) Heatmap for the consensus matrix k = 3. (B) Cumulative distribution function (CDF) under diverse k values. (C) Delta area diagram for relative alterations in area under CDF curves. (D) The tracking plot for HCC samples under different k values. Supplementary Figure 2. Consensus clustering analyses for clustering three m6A genomic phenotypes in the light of the expression profiling of m6A-associated genes in TCGA cohort. (A) Heatmap for the consensus matrix k = 3. (B) CDF under diverse k values. (C) Delta area diagram for relative alterations in area under CDF curves. (D) The tracking plot for HCC samples under different k values. Supplementary Figure 3. Subgroup analysis of the prognosis value of m6A score among HCC patients in TCGA data set. Kaplan-Meir curves of cases with high or low m6A score in each subgroup: (A) age ≥ 65; (B) age < 65; (C) female; (D) male; (E) G1-2; (F) G3-4; (G) stage I-II; (H) stage III-IV. P values were determined through log-rank tests. Supplementary Table 1. The clinical information of HCC samples in the TCGA data set. Supplementary Table 2. The clinical information of HCC samples in the GSE14520 data set. Supplementary Table 3. The list of 331 m6A phenotype-associated DEGs.
Data Availability Statement
The data used to support the findings of this study are included within the supplementary materials.