

Abstract
Calcineurin (CaN) is a calcium/calmodulin-dependent serine/threonine phosphatase with a crucial role in cellular homeostasis. It is also the target of the Food and Drug Administration (FDA) approved immunosuppressant drugs FK506 and cyclosporine A. Recent work from our group and others indicated that an uncontrolled increase in CaN activity causes synaptic dysfunction and neuronal death in various models of neurodegenerative diseases associated with calcium dysregulation. Furthermore, pharmacological normalization of CaN activity can prevent disease progression in animal models. However, none of the FDA-approved CaN inhibitors bind CaN directly, leading to adverse side effects. The development of direct CaN inhibitors is required to reduce off-target effects, but its highly conserved active site and similar mechanism of action with other protein serine/threonine phosphatases impose a significant challenge. In this work, we developed a novel pharmacophore model to screen for CaN-specific inhibitors. Then, we performed a virtual screen for molecules having the pharmacophore model. We also show that the molecules identified in this screen can inhibit CaN with a low micromolar IC50. Interestingly, the inhibitors identified from the screen do not inhibit phosphoprotein phosphatase 2A, a member of the serine/threonine phosphatase family that shares 43% sequence identity with the CaN active site. The pharmacophore model that we developed and validated in this work may help to accelerate the development of specific CaN inhibitors.
Highlights
-
•
In this work, we developed a novel pharmacophore model to screen for CaN-specific inhibitors.
-
•
We show that the molecules identified in this screen can inhibit CaN with a low micromolar IC50.
-
•
Importantly, the inhibitors identified from the screen do not inhibit phosphoprotein phosphatase 2A, a member of the serine/threonine phosphatase family that shares 43% sequence identity with the CaN active site.
1. Introduction
Calcineurin (CaN), a serine/threonine phosphatase of type 2B (PP2B), plays a key role in orchestrating the function of different transcription factors, ion channels and other cellular targets under oscillating bivalent calcium (Ca2+) concentration [1,2]. It is a heterodimeric enzyme composed of a 60 kDa catalytic subunit (calcineurin A; CnA) and an 18 kDa regulatory subunit (calcineurin B; CnB) [3]. In resting conditions, an auto-inhibitory domain (CnAI) blocks the active site of the enzyme, rendering it almost inactive [4]. Ca2+-dependent binding of calmodulin (CaM) to the CnA-CnB complex removes CnAI from the catalytic site and activates the enzyme in a Ca2+ concentration-dependent manner [3,5].
Optimum CaN activity is vital in various biological processes, including immune responses, muscle development, cardiac hypertrophy, neurodevelopment and memory formation [1,6]. Being the only Ca2+-dependent and most abundant phosphatase present in neurons, CaN plays a crucial role in synaptic plasticity and neuronal survival [2,7]. However, uncontrolled CaN activity alters the balance of the phosphorylation states for its targets, thus leading to the dysregulation of neuronal homeostasis [7,8]. Chronic CaN activity up-regulation is associated with synaptic impairment and neuronal death in Alzheimer's disease (AD), Parkinson's disease (PD) and prion diseases [[8], [9], [10], [11], [12], [13], [14], [15], [16], [17]]. CaN is an ideal candidate for therapeutic intervention because of its well-established crystal structure and dual activity on synaptic alteration and neuronal death.
Pharmacological normalization of chronically upregulated CaN has exhibited beneficial effects for neurodegenerative diseases in both experimental and clinical settings [[9], [10], [11], [12], [13], [14], [15], [16], [17], [18]]. However, FDA-approved CaN inhibitors, FK506 and cyclosporine (CyA), are currently used to prevent post-transplant immune response [3,19]. Furthermore, instead of binding CaN directly, FK506 and CyA form complexes with their respective cognate immunophilins, FKBP12 and cyclophilin, which inhibit substrate entry into the CaN active site [19,20]. Unfortunately, long-term use of FK506 or CyA produces many different side effects, including nephrotoxicity, hypertension, hypercholesterolemia, diabetes and/or tremors. Some of these effects are attributed to the drug binding to immunophilins [21,22].
In the search for a direct CaN inhibitor, we recently developed a high-throughput screening assay to unbiasedly screen for CaN inhibitors [23]. However, CaN shares a highly conserved active site and similar catalytic mechanisms with other serine-threonine protein phosphatases (PSPs) from the phosphoprotein phosphatase (PPP) family, which imposes a serious challenge in CaN-specific inhibitor development [[24], [25], [26], [27]]. Although several approaches are under investigation, CaN-specific inhibitor development continues to remain an unmet need [[28], [29], [30], [31], [32]].
Virtual screening is an in-silico method used to filter and select compounds from large chemical spaces. Although there are limitations [33], it has been widely and successfully applied to identify biologically active compounds [34]. In this work, we describe a novel pharmacophore model that mimics the blockage of the active site by the auto-inhibitory domain, a unique domain for CaN compared to other phosphatases in the PPP family. After performing virtual screening, the small molecules selected were tested in vitro and shown to specifically inhibit CaN. These results suggest that our pharmacophore model could serve as a powerful tool to accelerate the discovery of novel CaN-specific inhibitors.
2. Results
Pharmacophore design: The CnAI is known to block CaN phosphatase activity by interacting with the CaN active site during the absence of Ca2+/calmodulin [20]. Therefore, we designed pharmacophore models to mimic this interaction based on the crystallized three-dimensional structure of the human CaN heterodimer (PDB: 1AUI) using a ligand (CnAI) – receptor (active site) based methodology [35]. We identified the following amino acids as part of the active site: Asp90, His92, Asp118, Arg122, Asn150, His151, Leu156, Phe160, His199, Trp232, Asp234, Asn251, Thr252, Val253, Arg254, Phe259, His281, Glu282, Ala283, Phe306, Tyr311, Leu312 and Tyr315. The following amino acids were identified as part of the CnAI: Ser469, Phe470, Glu471, Glu472, Ala473, Lys474, Glu475, Leu476, Asp477, Asp477, Arg478, Ile479, Asn 480, Glu481, Arg482, Pro484 and Pro485. We identified the exact intramolecular interactions (aromatic centers, hydrophobicity, hydrogen bonding, salt bridges and the van der Waals surface) of the active site with the CnAI domain (the van der Waals surfaces are shown in Fig. 1A). We calculated the van der Waals surface at 4.5 Å of the active site. Then, we analyzed its shape and composition. All calculations were performed using the Chemical Computing Group's Molecular Operating Environment software (MOE 2011). As described in the LigandScout user manual, the following distance constraints were used to define the interactions: aromatic 2.8–4.5 Å, hydrophobic 1.0–5.9 Å, salt bridge 1.5–5.5 Å and hydrogen bond 2.2–3.8 Å. A total of three-dimensional pharmacophore models were generated based on different combinations of the observed interactions. Pharmacophore selection was conducted through a retrospective virtual screening using a conformational database, which was able to distinguish the CnAI domain from a subset of unrelated compounds with similar molecular properties. The selected pharmacophore model is shown in Fig. 1B. In this model, the amino acids in the CaN active site that were determined to be critical for binding CnAI were Arg122, Tyr315, Glu282 and Thr252 (Table 1). Importantly, all of these amino acids were specific for the CaN active site except Arg122, which is shared with other phosphatases (PP1 and PP2A).
Fig. 1.

Pharmacophore design and in silico analysis. A) Van der Waals surface at 4.5 Å of the active site. Color code: magenta, H-bonding regions; green, hydrophobic regions; blue, mild polar regions. The alpha-helix of the auto-inhibitory domain is colored in red. B) Selected pharmacophore model designed on the basis of the CnAI interactions with the active site. 1 Cationic, 2 Anionic, 3 Hydrophobic/Aromatic, 4 Anionic and Hydrogen bond acceptor, 5 Hydrogen bond donor, 6 Anionic and Hydrogen bond acceptor, 7 Aromatic.
Table 1.
Observed intramolecular interactions between calcineurin autoinhibitory domain (CnAI) and calcineurin.
| CnAI | Calcineurin | Interaction | Distance (Å) |
|---|---|---|---|
| Phe470 | Tyr315 | Aromatic | 3.28 |
| Ala473 | Tyr315 | Hydrophobic | 3.66 |
| Lys474 | Glu282 | Salt bridge | 5.52 |
| Asp477 | Tyr315 | Hydrogen bond | 2.71 |
| Asp477 | Arg122 | Salt bridge | 2.79 |
| Glu481 | HOH | Hydrogen bond | 2.60 |
| Arg482 | Thr252 | Hydrogen bond | 2.50 |
Virtual screening and validation of candidates: To perform the prospective virtual screening, we used the Molecular Operating Environment database (MOE), which contains 653,233 Oprea lead molecules. After the virtual screening, our pharmacophore model selected 33 candidates. Since eight of these candidates were commercially available, we tested their inhibitory potential with a 12-point dose-response assay against CaN. To perform the inhibition assays, we first determined the Km value of CaN for the well-established substrate RIIP (a 19 amino acid peptide fragment [81-99aa], phosphorylated at the serine residue, from regulatory subunit type II of the CaN substrate cAMP-dependent protein kinase [PKA]) using similar conditions as published previously [23]. All of the inhibition assays were performed at the Km concentration of the substrate. One out of eight molecules (PMD0011) inhibited CaN with an IC50 of 56.62 ± 1.22 μM (Fig. 2). The structure and physicochemical properties of the active compound is presented in Table 2.
Fig. 2.

Inhibitory potential of selected molecules against CaN. Inhibition potentials for the two out of eight candidates with the best dose responses in the CaN phosphatase inhibition assay: PMD0011, IC50 = 56.62 ± 1.22 μM. Each data point represents the mean ± SEM of duplicates.
Table 2.
Structure and molecular properties of the prospective virtual screening hits that were used to perform the first set of in vitro assays.
| Code | Structure | Drug like properties |
|---|---|---|
| PMD0011 |  |
MW 414 |
| LogP 1.51 | ||
| RB 7 | ||
| Hdon 3 | ||
| Hacc 6 |
MW = molecular weight, LogP = octanol/water partition coefficient, RB = number of rotatable bonds, Hdon = number of H-bond donors, Hacc = number of H-bond acceptors.
Structure-Activity Relationship studies: We attempted to improve the inhibition potential of PMD0011 by using its scaffold to produce derivatives with elevated potency and solubility. For this purpose, we chose modifications expected to significantly alter its structure and physicochemical properties (Table 3). To determine the requirement of the fluoro group, a few commercially available compounds were selected, in which the fluoro group was replaced by 2-Nitro (PMD0033), 3,4-Dihydroxy (PMD0022) or 4-N-dimethyl (PMD0026). All replacements exhibited decreased potency, as respectively indicated by the high IC50 values of 63, >200 and > 500 μM (Table 3). However, substituting the fluoro group with a heterocyclic thiophene ring (PMD0024), containing an electron-rich S atom in the aromatic ring, increased the potency 7 times to an IC50 of 8.56 ± 1.41 μM (Fig. 3). To confirm the requirement of carboxylic group in the PDM0024 molecule, the carboxylic group was replaced with a 3,4-Dichloro group (PDM0023). This single replacement led to a loss of activity with an IC50 greater than 200 μM. Our results clearly indicate that the most potent structure requires a carboxylic group at the para position of the left-hand phenyl ring and a thiophene group on the right-hand side. Although we were able to significantly improve the potency of the CaN inhibitor by rationally modifying the structure, it, unfortunately, did not improve the solubility.
Table 3.
Seven different derivatives of the lead compound were tested to study the effect of different functional groups in the core structure..
| Code | R | R′ | R’’ | IC50 (μM) |
|---|---|---|---|---|
| PMD0033 | 2-Nitro | -COOH | H | 63 |
| PMD0022 | 3,4-Dihydroxy | -COOH | H | >200 |
| PMD0026 | 4-N-(CH3)2 | -COOH | H | >500 |
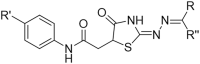 | ||||
| Code |
R |
R′ |
R’’ |
IC50 (μM) |
| PMD0024 | 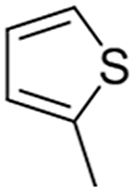 |
-COOH | CH3 | 8.6 |
| PMD0023 | 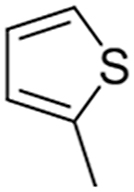 |
3,4-Dichloro | CH3 | >200 |

Fig. 3.

Improved potency of derivatives of PMD0011 inhibitor. We generated several derivatives of PMD0011 for improved inhibitory potential and solubility (also see Table 3). PMD0024 showed a nearly 7 times increase in inhibition potential with IC50 = 8.56 ± 1.41 μM. Each data point represents the mean ± SEM of duplicates.
Specificity of the inhibitors: PMD0011 and PMD0024 should be specific CaN inhibitors based on the filters used to generate our pharmacophore model. In order to confirm CaN specificity, we tested the inhibition potential of these two compounds against PP2A, a PPP family member possessing 43% sequence identity with the CaN active site [26,27]. First, we determined the Km value for PP2A and the RIIP substrate. Next, the inhibitory potential for PMD0011 and PMD0024 against PP2A were tested using the 12-point dose-response assay. Both PMD0011 and PMD0024 failed to inhibit PP2A activity (Fig. 4A and B), even at the highest concentrations tested. Importantly, these concentrations were approximately 6 and 30 times higher than the IC50 observed with CaN, respectively.
Fig. 4.

CaN inhibitors do not inhibit PP2A. We tested the inhibition potential of both A) PMD0011 and its derivative B) PMD0024 against PP2A. Both of them failed to inhibit PP2A. Each data point represents the mean ± SEM of duplicates.
3. Discussion
Unregulated increase in cytoplasmic Ca2+ leads to sustained activation of CaN. Chronically active CaN results in synaptic abnormalities and neuronal apoptosis observed in a spectrum of neurodegenerative diseases [7,8]. Although a potential therapeutic target, CaN has a remarkably conserved active site with other members of the PPP family, which imposes a challenge in developing a CaN-specific inhibitors. Attempts to inhibit CaN with small molecules targeting the active site will likely inhibit other phosphatases of the PPP family, thus leading to deleterious consequences in cells. In this work, we developed and validated a unique strategy to identify CaN-specific inhibitors. We and others have demonstrated that chronic upregulation of CaN leads to synaptic impairment and neuronal death in AD, PD, and prion diseases [[8], [9], [10], [11], [12], [13], [14], [15], [16], [17]]. We hope that our work will facilitate the development of a novel class of anti-CaN therapeutics for these devasting disorders of the brain.
The functional diversity and specificity of CaN originates from its unique regulatory subunits, domains and binding partners [24,25]. In contrast to the other PPP members, the binding of CaN to Ca2+/CaM results in the displacement of CnAI resulting in enzyme activation. Targeting CaN-CaM binding as a mechanism to inhibit CaN activity may produce off-target effects by inhibiting CaM binding to its other known partners. Moreover, it has been shown that in AD, proteolytic truncation of the CnAI domain makes CaN constitutively active, without the requirement of Ca2+/CaM binding [36]. Since CnAI occlusion of the active site is unique to CaN, we developed a novel pharmacophore model to screen for specific CaN inhibitors by mimicking the blockage of the CnA active site with CnAI. The pharmacophore models were generated using a ligand (CnAI)–receptor (active site) based methodology, where the intermolecular interactions were evaluated and determinant pharmacophore features were included in the filters. The combination of both approaches allows a detailed and precise description of the binding mode between the CnAI and the active site of the protein, therefore leading to specific screening for new inhibitors [33,35].
An in-silico screen with our pharmacophore model identified 33 candidate molecules, 8 of which were commercially available. Two out of eight candidate molecules produced a dose-dependent inhibition of CaN in an enzyme assay. Moreover, the potency of the inhibitors was significantly improved by modifying the active compound. Importantly, our newly identified CaN inhibitors (PMDAM0011 and PMDAM0024) did not inhibit the activity of PP2A, another closely related member of the PPP family. Taken together, these results strongly indicate the specificity of these inhibitors for CaN, validating our pharmacophore model.
Development of specific serine/threonine phosphatase inhibitors for therapeutic purposes has traditionally been difficult [37]. The similarity in the active site imposed a risk that even the low-affinity nonspecific inhibition of the off-target phosphates may produce devastating effects. Further work is needed to validate the general specificity of the novel CaN inhibitors developed in this work against other PPP family members. However, our work provided proof of concept demonstration of specificity that these novel CaN inhibitors do not inhibit PP2A activity. In addition, targeting protein-protein interactions with small molecules is considered to be a challenging strategy with limited success [38]. The pharmacophore model we developed and validated in this work may help to overcome both of these obstructions to accelerate the discovery of specific CaN inhibitors.
4. Methods
Retrospective and prospective virtual screening: To construct the retrospective VS database, the AI of CaN was taken as positive control and a set of assumed inactive compounds with similar 1D and 2D molecular properties to Endothall (Molecular weight: 184.147) and Norcantharidin (Molecular weight: 168.148) (known CaN inhibitors) were selected as negative controls from the PubChem Database (see Table below). Such controls are listed here: CID_641395, CID_600929 CID_598359, CID_598966, CID_597215, CID_597130, CID_596714, CID_596646 and CID_595632. A set of different conformations was generated for each molecule using MMFF94x forcefield. A limit of 4.5 kcal/mol strain energy was imposed with a limit of 500 conformations per molecule. Duplicate conformations were removed with a heavy atom RMSD tolerance of 0.6 Å. Prospective database included 653, 533 (Oprea) lead-like chemical structures (MW: max. 450, logP: −3,5 to +4,5, H-bond acceptors: max. 8, H-bond donors: max. 5) from the MOE molecular database. The structures come from the following sources: A-Synthese-Biotech, ASDI, Akos, Art-Chem, Asinex, Aurora, Biofocus, Bionet, Biotech Corp. Of America, Cerep, Chem T&I, Chembridge, Chemdiv, Chemical Block, Chemstar, Comgenex, EMC Microcollection, Enamine, Exclusive Chemistry, FCHC, InnovaPharm, InterBioScreen, Labotest, Life Chemicals, Lithuania, MDD World Molecules, MDPI, Maybridge, Menai, Moscow MedChem Labs, Nanosyn, Otava, Peakdale, Pharmeks, Princeton Biomolecular, Pyxis Discovery, Scientific Exchange, Sigma-Aldrich, SPECS, Spectrum Info, TimTec, Toslab, Tripos and Vitas-M Lab. Both retrospective and prospective three-dimensional virtual screenings were performed. MMFF94x forcefield was used to generate the conformations of the prospective database. A limit of 4.5 kcal/mol strain energy was imposed with a limit of 500 conformations per molecule. Duplicate conformations were removed with a heavy atom RMSD tolerance of 0.6 Å (0.75 Å for conformations with strain greater than 3.5 kcal/mol). Selection of the best pharmacophore filter was conducted through retrospective virtual screening, using a conformational test database. The selection of the pharmacophore model was based on the ability to effectively identify positive and negative controls from the training database. Once the best filter was selected, prospective virtual screening was applied to the MOE database, which contains 653,233 lead-like chemical structures (Oprea).
Enzyme assay: The CaN assay was performed as previously described [23]. In brief, an enzyme master mix was prepared in 2X assay buffer containing 100 mM HEPES (pH 7.0), 1 mM DTT,1 mM CaCl2, 2 mM MnCl2, 16 nM CaN (R&D; Cat # 3160-CA-020) and 32 nM CM (EMD Millipore; Cat # 208694). As a substrate we used a 19 amino acid peptide fragment (81-99aa), phosphorylated at the serine residue, from regulatory subunit type II of the CaN substrate cAMP-dependent protein kinase (PKA). This peptide fragment is called RIIP. The master mix was incubated for 30 min at 37 °C. 25 μl of master mix and 25 μl RIIP substrate (in deionized water; American Peptide; Product No. 310258) were mixed to initiate the reaction. The reaction was stopped by adding 50 μl of malachite green (BioAssay System; Cat # POMG-25H) reagent. The absorbance of the phospho-malachite green complex was measured at 620 nm using Molecular Devices' Gemini Spectromax spectrophotometer. The enzyme assays with PP2A (Cayman chemicals; item # 10011237) were similar and also used RIIP as substrate [31].
Enzyme inhibition assay: Inhibitor stock solution was prepared in DMSO (Fisher; Cat #D128-500) at a 50 mM concentration. 10 different concentrations of inhibitor were made by diluting DMSO stock in assay buffer (1.5 mM - 1.4 μm). 10 μl of each concentration was then incubated with 20 μL of the activated master mix for another 30 min at 37 °C. The final DMSO concentration in the assay was 0.6%. The positive controls were prepared in the same way and had only 0.6% DMSO without any inhibitor. The enzyme reaction was started by adding 20 μL of 625 μM RIIP substrate. After a 30 min incubation, substrate hydrolysis was stopped by adding 50 μL of the stop solution. The absorbance value was recorded in a colorimeter at 620 nm. Absorbance values were converted to amount of free phosphate released from the phosphate standard curve. Percentage inhibition was calculated using equation (1). IC50 values were determined by fitting the data points to log (inhibitor) vs response-variable slope (four parameters) using equation (2).
| % Inhibition = (Positive control –test well)*100/ Positive control | equation 1 |
| Y=Bottom + (Top-Bottom)/(1 + 10∧((LogIC50-X)*HillSlope)) | equation 2 |
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors consented for publication.
Availability of data and material
All the data generated in this work is available from the corresponding authors upon request.
Funding
The work is funded by a generous gift from The Cynthia and George Mitchell Foundation awarded to CS.
Authors' contributions
AM and CS conceived the idea. AM and CS supervised the work.
KC-C designed the pharmacophore model and performed the in silico screening. AM and AS performed the enzyme inhibition and specificity assays. AM, CS and KC-C analyzed the in silico screening data. AM, CS and AS analyzed the enzyme inhibition and specificity data. AM prepared the manuscript. CS, KC-C and AS edited the manuscript.
Declaration of competing interest
I would like to confirm that none of the authors have any conflict of interest.
Acknowledgments
We would like to convey our sincere thanks to the The Cynthia and George Mitchell Foundation for generously providing the funding. We would also like to thank Dr. Rabab Al-Lahham for reviewing the manuscript.
Contributor Information
Abhisek Mukherjee, Email: Abhisek.Mukherjee@uth.tmc.edu.
Claudio Soto, Email: Claudio.Soto@uth.tmc.edu.
Data availability
Data will be made available on request.
References
- 1.Rusnak F., Mertz P. Calcineurin: form and function. Physiol. Rev. 2000 Oct;80(4):1483–1521. doi: 10.1152/physrev.2000.80.4.1483. [DOI] [PubMed] [Google Scholar]
- 2.Mansuy I.M. Calcineurin in memory and bidirectional plasticity. Biochem. Biophys. Res. Commun. 2003 Nov 28;311(4):1195–1208. doi: 10.1016/j.bbrc.2003.10.046. [DOI] [PubMed] [Google Scholar]
- 3.Kissinger C.R., Parge H.E., Knighton D.R., Lewis C.T., Pelletier L.A., Tempczyk A., Kalish V.J., Tucker K.D., Showalter R.E., Moomaw E.W., et al. Crystal structures of human calcineurin and the human FKBP12-FK506-calcineurin complex. Nature. 1995 Dec 7;378(6557):641–644. doi: 10.1038/378641a0. [DOI] [PubMed] [Google Scholar]
- 4.Hashimoto Y., Perrino B.A., Soderling T.R. Identification of an autoinhibitory domain in calcineurin. J. Biol. Chem. 1990 Feb 5;265(4):1924–1927. [PubMed] [Google Scholar]
- 5.Li H.C., Chan W.W. Activation of brain calcineurin towards proteins containing Thr(P) and Ser(P) by Ca2+, calmodulin, Mg2+ and transition metal ions. Eur. J. Biochem. 1984 Nov 2;144(3):447–452. doi: 10.1111/j.1432-1033.1984.tb08486.x. [DOI] [PubMed] [Google Scholar]
- 6.Lim H.W., Molkentin J.D. Calcineurin and human heart failure. Nat. Med. 1999 Mar;5(3):246–247. doi: 10.1038/6430. [DOI] [PubMed] [Google Scholar]
- 7.Mukherjee A., Soto C. Role of calcineurin in neurodegeneration produced by misfolded proteins and endoplasmic reticulum stress. Curr. Opin. Cell Biol. 2011 Apr;23(2):223–230. doi: 10.1016/j.ceb.2010.12.006. PMCID:PMC3078182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reese L.C., Taglialatela G. A role for calcineurin in Alzheimer's disease. Curr. Neuropharmacol. 2011 Dec;9(4):685–692. doi: 10.2174/157015911798376316. PMCID:PMC3263462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu H.Y., Hudry E., Hashimoto T., Kuchibhotla K., Rozkalne A., Fan Z., Spires-Jones T., Xie H., Arbel-Ornath M., Grosskreutz C.L., et al. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J. Neurosci. 2010 Feb 17;30(7):2636–2649. doi: 10.1523/JNEUROSCI.4456-09.2010. PMCID:PMC2841957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mukherjee A., Morales-Scheihing D., Gonzalez-Romero D., Green K., Taglialatela G., Soto C. Calcineurin inhibition at the clinical phase of prion disease reduces neurodegeneration, improves behavioral alterations and increases animal survival. PLoS Pathog. 2010 Oct 7;6(10) doi: 10.1371/journal.ppat.1001138. PMCID:PMC2951383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dineley K.T., Hogan D., Zhang W.R., Taglialatela G. Acute inhibition of calcineurin restores associative learning and memory in Tg2576 APP transgenic mice. Neurobiol. Learn. Mem. 2007 Sep;88(2):217–224. doi: 10.1016/j.nlm.2007.03.010. PMCID:PMC2031869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin Z.S., Neugebauer V., Dineley K.T., Kayed R., Zhang W., Reese L.C., Taglialatela G. alpha-Synuclein oligomers oppose long-term potentiation and impair memory through a calcineurin-dependent mechanism: relevance to human synucleopathic diseases. J. Neurochem. 2012 Feb;120(3):440–452. doi: 10.1111/j.1471-4159.2011.07576.x. PMCID:PMC3253169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reese L.C., Zhang W., Dineley K.T., Kayed R., Taglialatela G. Selective induction of calcineurin activity and signaling by oligomeric amyloid beta. Aging Cell. 2008 Dec;7(6):824–835. doi: 10.1111/j.1474-9726.2008.00434.x. PMCID:PMC2954114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taglialatela G., Hogan D., Zhang W.R., Dineley K.T. Intermediate- and long-term recognition memory deficits in Tg2576 mice are reversed with acute calcineurin inhibition. Behav. Brain Res. 2009 Jun 8;200(1):95–99. doi: 10.1016/j.bbr.2008.12.034. PMCID:PMC2663011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hong J.-M., Moon J.-H., Park S.-Y. Human prion protein-mediated calcineurin activation induces neuron cell death via AMPK and autophagy pathway. Int. J. Biochem. Cell Biol. 2020 Feb;119 doi: 10.1016/j.biocel.2019.105680. PMID: 31866508. [DOI] [PubMed] [Google Scholar]
- 16.Kuchibhotla K.V., Goldman S.T., Lattarulo C.R., Wu H.Y., Hyman B.T., Bacskai B.J. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008 Jul 31;59(2):214–225. doi: 10.1016/j.neuron.2008.06.008. PMCID:PMC2578820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rozkalne A., Hyman B.T., Spires-Jones T.L. Calcineurin inhibition with FK506 ameliorates dendritic spine density deficits in plaque-bearing Alzheimer model mice. Neurobiol. Dis. 2011 Mar;41(3):650–654. doi: 10.1016/j.nbd.2010.11.014. PMCID:PMC3031746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taglialatela G., Rastellini C., Cicalese L. Reduced incidence of dementia in solid organ transplant patients treated with calcineurin inhibitors. J.Alzheimers.Dis. 2015;47(2):329–333. doi: 10.3233/JAD-150065. PMCID:PMC4923720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin L., Harrison S.C. Crystal structure of human calcineurin complexed with cyclosporin A and human cyclophilin. Proc. Natl. Acad. Sci. U.S.A. 2002 Oct 15;99(21):13522–13526. doi: 10.1073/pnas.212504399. PMCID:PMC129706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kissinger C.R., Parge H.E., Knighton D.R., Lewis C.T., Pelletier L.A., Tempczyk A., Kalish V.J., Tucker K.D., Showalter R.E., Moomaw E.W., et al. Crystal structures of human calcineurin and the human FKBP12-FK506-calcineurin complex. Nature. 1995 Dec 7;378(6557):641–644. doi: 10.1038/378641a0. [DOI] [PubMed] [Google Scholar]
- 21.Liu E.H., Siegel R.M., Harlan D.M., O'Shea J.J. T cell-directed therapies: lessons learned and future prospects. Nat. Immunol. 2007 Jan;8(1):25–30. doi: 10.1038/ni1429. [DOI] [PubMed] [Google Scholar]
- 22.Naesens M., Kuypers D.R., Sarwal M. Calcineurin inhibitor nephrotoxicity. Clin. J. Am. Soc. Nephrol. 2009 Feb;4(2):481–508. doi: 10.2215/CJN.04800908. [DOI] [PubMed] [Google Scholar]
- 23.Mukherjee A., Syeb K., Concannon J., Callegari K., Soto C., Glicksman M.A. Development of a fluorescent quenching based high throughput assay to screen for calcineurin inhibitors. PLoS One. 2015;10(7) doi: 10.1371/journal.pone.0131297. PMCID:PMC4503349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McConnell J.L., Wadzinski B.E. Targeting protein serine/threonine phosphatases for drug development. Mol. Pharmacol. 2009 Jun;75(6):1249–1261. doi: 10.1124/mol.108.053140. PMCID:PMC2684880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCluskey A., Sim A.T., Sakoff J.A. Serine-threonine protein phosphatase inhibitors: development of potential therapeutic strategies. J. Med. Chem. 2002 Mar 14;45(6):1151–1175. doi: 10.1021/jm010066k. [DOI] [PubMed] [Google Scholar]
- 26.Barton G.J., Cohen P.T., Barford D. Conservation analysis and structure prediction of the protein serine/threonine phosphatases. Sequence similarity with diadenosine tetraphosphatase from Escherichia coli suggests homology to the protein phosphatases. Eur. J. Biochem. 1994 Feb 15;220(1):225–237. doi: 10.1111/j.1432-1033.1994.tb18618.x. [DOI] [PubMed] [Google Scholar]
- 27.Cohen P., Cohen P.T. Protein phosphatases come of age. J. Biol. Chem. 1989 Dec 25;264(36):21435–21438. [PubMed] [Google Scholar]
- 28.Matsoukas M.T., Aranguren-Ibanez A., Lozano T., Nunes V., Lasarte J.J., Pardo L., Perez-Riba M. Identification of small-molecule inhibitors of calcineurin-NFATc signaling that mimic the PxIxIT motif of calcineurin binding partners. Sci. Signal. 2015 Jun 23;8(382):ra63. doi: 10.1126/scisignal.2005918. [DOI] [PubMed] [Google Scholar]
- 29.Sieber M., Baumgrass R. Novel inhibitors of the calcineurin/NFATc hub - alternatives to CsA and FK506? Cell Commun. Signal. 2009 Oct 27:7–25. doi: 10.1186/1478-811X-7-25. PMCID:PMC2774854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sieber M., Karanik M., Brandt C., Blex C., Podtschaske M., Erdmann F., Rost R., Serfling E., Liebscher J., Patzel M., et al. Inhibition of calcineurin-NFAT signaling by the pyrazolopyrimidine compound NCI3. Eur. J. Immunol. 2007 Sep;37(9):2617–2626. doi: 10.1002/eji.200737087. [DOI] [PubMed] [Google Scholar]
- 31.Erdmann F., Weiwad M., Kilka S., Karanik M., Patzel M., Baumgrass R., Liebscher J., Fischer G. The novel calcineurin inhibitor CN585 has potent immunosuppressive properties in stimulated human T cells. J. Biol. Chem. 2010 Jan 15;285(3):1888–1898. doi: 10.1074/jbc.M109.024844. PMCID:PMC2804347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Erdmann F., Weiwad M. Calcineurin inhibitors: status quo and perspectives. Biomol. Concepts. 2011 Apr 1;2(1–2):65–78. doi: 10.1515/bmc.2011.011. [DOI] [PubMed] [Google Scholar]
- 33.Scior T., Bender A., Tresadern G., Medina-Franco J.L., Martinez-Mayorga K., Langer T., Cuanalo-Contreras K., Agrafiotis D.K. Recognizing pitfalls in virtual screening: a critical review. J. Chem. Inf. Model. 2012 Apr 23;52(4):867–881. doi: 10.1021/ci200528d. [DOI] [PubMed] [Google Scholar]
- 34.Matter H., Sotriffer C. Virtual Screening. Wiley-VCH Verlag GmbH & Co. KGaA; 2011. Applications and success stories in virtual screening; pp. 319–358. [Google Scholar]
- 35.Drwal M.N., Griffith R. Combination of ligand- and structure-based methods in virtual screening. Drug Discov. Today Technol. 2013 Sep;10(3):e395–e401. doi: 10.1016/j.ddtec.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 36.Liu F., Grundke-Iqbal I., Iqbal K., Oda Y., Tomizawa K., Gong C.X. Truncation and activation of calcineurin A by calpain I in Alzheimer disease brain. J. Biol. Chem. 2005 Nov 11;280(45):37755–37762. doi: 10.1074/jbc.M507475200. [DOI] [PubMed] [Google Scholar]
- 37.Hopkins A.L., Groom C.R. The druggable genome. Nat. Rev. Drug Discov. 2002 Sep;1(9):727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 38.Scott D.E., Bayly A.R., Abell C., Skidmore J. Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug Discov. 2016 Aug;15(8):533–550. doi: 10.1038/nrd.2016.29. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All the data generated in this work is available from the corresponding authors upon request.
Data will be made available on request.