

Abstract
Chronic kidney diseases can progress to kidney failure and require dialysis or transplantation, while early diagnosis can alter the course of disease and lead to better outcomes in both pediatric and adult patients. Significant CKD comorbidities include the manifestation of cardiovascular disease, heart failure, coronary disease and hypertension. The pathogenesis of chronic kidney diseases can present as subtle and especially difficult to distinguish between different glomerular pathologies. Early detection of adult and pediatric CKD and detailed mechanistic understanding of the kidney damage can be helpful in delaying or curtailing disease progression via precise intervention toward diagnosis and prognosis. Clinically, serum creatinine and albumin levels can be indicative of CKD, but often are a lagging indicator only significantly affected once kidney function has severely diminished. The evolution of proteomics and mass spectrometry technologies has begun to provide a powerful research tool in defining these mechanisms and identifying novel biomarkers of CKD. Many of the same challenges and advances in proteomics apply to adult and pediatric patient populations. Additionally, proteomics analysis of adult CKD patients can be transferred directly toward advancing our knowledge of pediatric CKD as well. In this review, we highlight applications of proteomics that have yielded such biomarkers as PLA2R, SEMA3B and other markers of membranous nephropathy as well as KIM-1, MCP-1 and NGAL in lupus nephritis among other potential diagnostic and prognostic markers. The potential for improving the clinical toolkit toward better treatment of pediatric kidney diseases is significantly aided by current and future development of proteomics applications.
Keywords: biomarker, proteomics, mass spectrometry, pediatric glomerular disease, chronic kidney disease
Introduction
Chronic kidney disease is one of the most burdensome medical issues facing western medicine. Currently, dialysis or transplantation are the only viable long-term treatments for kidney failure. Kidney disease treatment costs are the largest yearly expenditure by Medicare for a single medical issue, typically totaling more than $35.9 billion annually [1]. Kidney disease manifests subtly in pediatric patients with accumulating deleterious effects throughout childhood and can progress to more chronic and difficult to manage disease states. Pediatric patients with glomerular diseases are typically treated with glucocorticosteroids at initial diagnosis or inpatient visit for kidney failure symptoms. Some pediatric patients will respond to therapy, while others have steroid resistant characteristics that may require more focused and intensive clinical management. Clinical presentation and manifestation of disease can be highly variable and presents special challenges in pediatric populations as the urgency to blunt disease progress is most important to prevent longer-term damage. The mechanism of disease pathology can be highly important in determination of the course of treatment. Typically, proteinuria, urine blood, serum creatinine and other biomarkers are key clinical parameters used to determine treatment approaches in pediatric patients, yet again, can be highly variable in prediction of treatment response and these markers can lag disease progression for unknown reasons. Kidney biopsy is the gold standard for diagnosis and follow-up prognostics for determination of therapeutic responsiveness along with measuring blood and urine chemistry (creatinine, micro- and macro-albuminuria). Kidney biopsy is invasive and therefore not a primary approach for the pediatric nephrologist, leaving secondary biomarkers as the most significant means for proper diagnosis and selection of the optimal therapeutic course.
Proteomics, metabolomics and transcriptomics offer the potential to analytically probe the molecular mechanisms of pediatric kidney diseases and offer solutions to the clinician not available until recently. Omics is a relatively new field that utilizes systems biology approaches toward a more global understanding of biological systems. Proteomics has been a powerful Omics tool in the realm of nephrology in recent decades. Biomarker research has expanded markedly within the past 20 years as indicated in the growth in publications, although much work is required to fully implement these findings into the clinic. This review will illustrate the history of proteomics and some applications in kidney diseases and highlight applications in both pediatric and adult populations with particular emphasis on recent breakthroughs and what the future holds for pediatric kidney patients regarding this field of basic and clinical kidney research.
Brief History: Application of Proteomics
Mass spectrometry and proteomics have become a mainstay approach and more readily accessible in clinical biomedical research over the previous two decades. The development and refinement of tools such as electrospray ionization (ESI), the orbitrap and hybrid orbitraps, TOF (time of flight), bioinformatics algorithms and search databases and the expansion of computational power to manage such data-intensive methods has led to a significant leap forward in capability and applications of proteomics, particularly in kidney research.
Coupling different mass spectrometry platforms (i.e. ion trap, orbitrap and TOF instruments) with low or nano-flow rate liquid chromatography systems has allowed for the advancement of proteomics into the realm of complex and low-abundance protein and peptide detection and quantification [2, 3]. These improvements have coincided with the popular application of so-called “shotgun” proteomics. Similar to shotgun genome sequencing of the same era, shotgun proteomics applications are employed to identify as many proteins as possible from a complex sample set without the tedium of physical purification of every protein to be analyzed [4, 5]. Shotgun proteomics is inherently useful in clinical applications because of the broad array of identifications and label-free quantitative data acquired. Shotgun proteomics offers a high-throughput analysis platform that can be implemented in virtually any sample context and can identify thousands of proteins from virtually any sample source. As would be expected, pitfalls of this approach include lack of reproducibility and data variance. Often, shotgun proteomics analysis can serve as the impetus for more in-depth specific analysis of a subset or sub-proteome sample.
Improvements in technical aspects of mass spectrometry and bioinformatics have led to exponentially improved identification rates and throughput. These improvements have made proteomics approaches more amenable to clinical research goals. Liquid handling systems and chromatography methods now allow for deep resolution of complex biological samples by spatio-temporal separation of proteins and peptides, which leads to improved coverage and overall proteome identification and quantification. A greater availability of materials such as highly standardized and pre-packed columns tailored to a variety of targets offer highly reproducible performance, while powerful software packages with intuitive user interfaces have taken mass spectrometry from the subbasement rooms of universities to medical clinics and laboratories.
Quantitative analysis in proteomics has made great strides recently. As described above, initial proteomics experiments implemented fluorescent dye labeling and sample multiplexing in-gel platforms followed by downstream quantitation and mass spectrometry analysis. This approach is very low-throughput and highly irreproducible. Recent improvements in chemical tagging of peptides and isobaric reporter ion technology (TMT-tandem mass tagging) has increased multiplexing capacity from 6- to 16- or even 27-plex in a single MS analysis [6, 7]. Clinical studies often require hundreds of samples for statistical power; multiplexing offers the ability to robustly analyze these types of samples in a manageable timeframe. Simultaneous analysis decreases variability in the data and allows the use of quality control samples to be injected with every run to offer a quantitative standard to compare unique results from one run to the next.
As mentioned above, direct pathogenic diagnosis in pediatric kidney disease is a challenge at best due to the invasive nature of kidney biopsies. Therefore, identification and characterization of complex sample types such as serum, plasma and urine could provide needed tools for both early and serialized clinical assessment of early-onset pediatric kidney diseases. Early diagnosis would allow initiation of therapy prior to the accumulation of massive damage required for more crude diagnostic methods. Serialized collection of non-invasive samples will allow constant assessment of the therapeutic response so that therapies can be modified or dropped completely if at any time they are found to be less than optimal. With a fully realized proteomic toolset, modern medical practice may finally begin to offer the personalized medicine initially promised with the human genome project, improving outcomes by providing a precision-based medicine decision-making process with proteomics integration in the field of kidney disease management, which has largely been left out of the push toward the personalization of medicine due to the difficulty and invasive nature of obtaining specimens which directly show disease pathogenesis. Much progress has already been made in identifying candidate molecular indicators of disease initiation, progression and prognostics. However, for these studies to be fruitful, proper study design and validation are especially important to reveal bona fide biomarkers of kidney diseases. Figure 1 illustrates the typical flow and design of a biomarker experiment that implements a discovery, verification and finally a validation phase using independent discovery and validation cohorts for a rigorous investigative platform. Each method has pros and cons. Gel-based imaging allows for individual spot/band identification based on isoelectric point and molecular weight, and allows for potential quantification, though gels are very labor intensive and low throughput and not very reproducible. LC-MS approaches such as DDA allow for a broad array of identifications across multiple molecular weight ranges and physicochemical parameters (hydrophobicity, charge, etc.). Data-dependent analysis (DDA) is ideal for shotgun proteomics and requires genome sequences for mapping spectra to an in-silico digest of the cognate peptides and proteins. Confirmation of biomarkers can be conducted in a variety of alternative approaches that include antibody-based approaches as well as parallel or selected ion monitoring by mass spectrometry. Ideally, mass spectrometry-based approaches can be robustly implemented in clinical diagnostics laboratories within most major hospitals.
Figure 1. Biomarker study design.
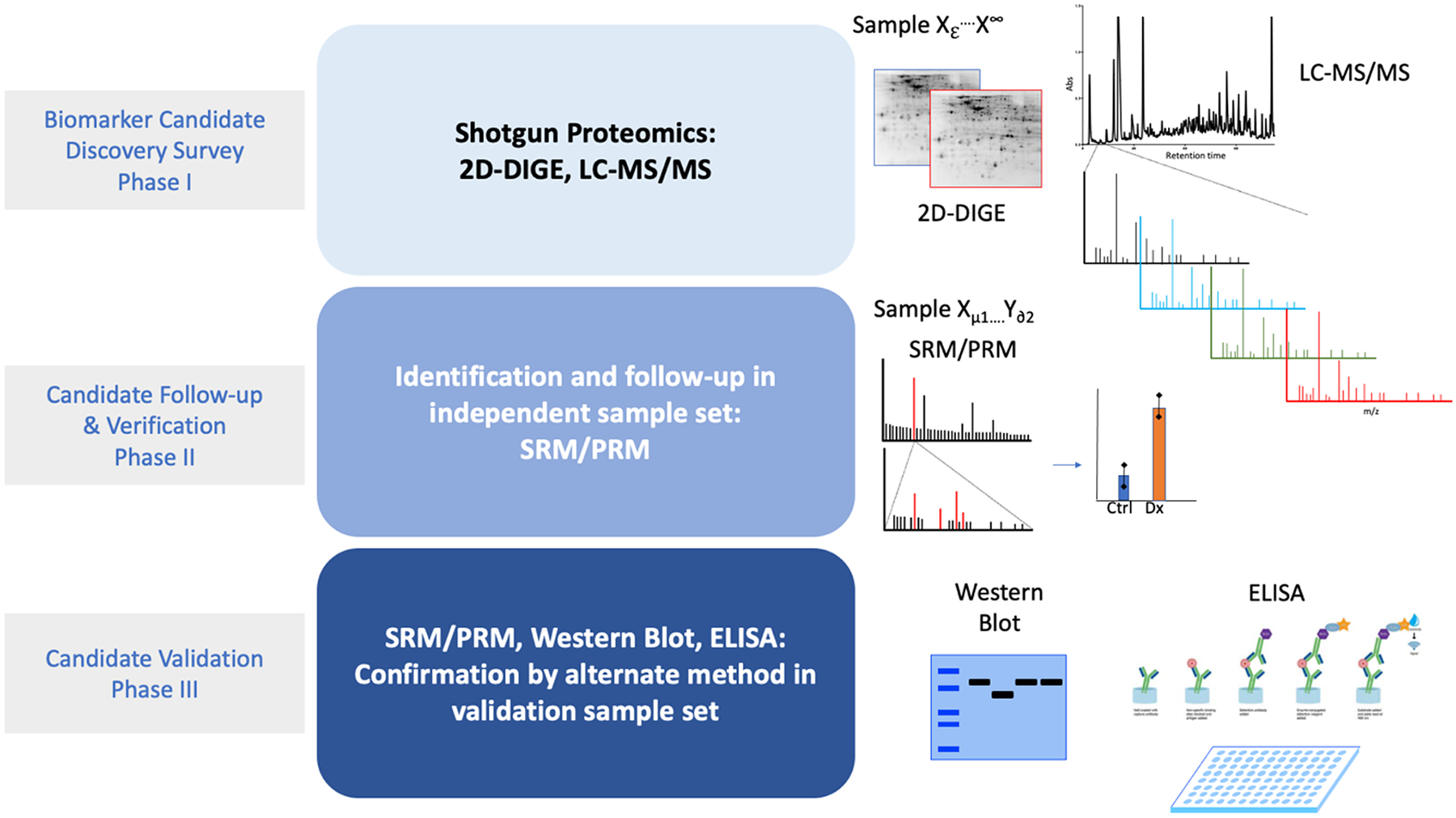
Candidate biomarker discovery is performed by shotgun proteomics implementing a resolving or separation technique such as gel electrophoresis (2-dimensional differential gel electrophoresis; 2D-DIGE), LC-MS/MS directly into the mass spectrometry platform. Candidate verification can be performed in a different focused absolute quantitative platform such as selected-reaction monitoring or parallel reaction monitoring into a triple-quadrupole mass spectrometer to follow specific signature ions/peptides from known candidates in an independent sample set. Ideally, validation of candidate biomarkers can be performed using specific biochemical assays such as western blot, ELISA or bead-based ligand-antibody assays such as Luminex.
Application of Proteomics in Chronic Kidney Disease
Focal segmental glomerulosclerosis (FSGS)
Biofluid proteins and metabolites can provide initial indications of kidney injury in pediatric and adult patients. Early studies of pediatric kidney diseases have focused on identification of urine biomarkers of glucocorticoid responsiveness. Biomarkers of effective steroid treatment for patient subpopulations allows for more targeted treatment and prevents unnecessary administration of ineffective medications. In a 2006 study of steroid-resistant and steroid-sensitive pediatric nephrotic syndrome, Khurana and colleagues used surface-enhanced laser desorption-ionization time of flight mass spectrometry to identify five spectral peaks that correlated with the steroid resistance [8]. The authors focused on isolation and identification of proteins from the spectra by employing repeated multidimensional chromatographic fractionation of the urine until the peak of interest was sufficiently isolated and could then be identified by an alternate mass spectrometry approach. The isolated fraction was trypsin digested and submitted to MALDI-TOF/TOF MS (matrix assisted laser desorption ionization time of flight/time of flight mass spectrometry) and identified as β2-microglobulin, a potential biomarker of steroid-resistant nephrotic syndrome in pediatric patients. β2-microglobulin has been shown to be implicated in several kidney pathologies and can be utilized in calculating the estimated glomerular filtration rate (GFR) [9–11]. Other studies of steroid resistance in pediatric kidney disease have utilized targeted cytokine array panels for proteomics analysis. Arrays are a biased or focused approach toward implicating specific cytokines in the disease process that have defined roles in inflammation, cytoskeletal regulation and other important biological processes. Specific cytokine antibodies are covalently bound to a surface to allow for binding of soluble urine proteins for estimation of the abundance of proteins. The authors employed a high throughput array to measure TGF-β1/2/3, IFN-γ, ICAM-1, TNF-α/β and interleukin isoforms among other cytokines in urine from pediatric patients with idiopathic nephrotic syndrome [12]. Although this study did not distinguish between steroid responsive and resistant nephrotic syndrome, TGF-β1 abundance was useful in distinguishing FSGS (focal segmental glomerulosclerosis) and MCD (minimal change disease). These studies begin to lay the groundwork for more in-depth exploration of the mechanisms and biomarkers of pediatric nephrotic syndrome by applying different proteomics approaches. More recent follow-up studies from our group on the plasma of pediatric patients with steroid resistance reveal a panel of proteins correlated with steroid resistance [13]. Proteomics analysis was conducted on 15 paired plasma samples in patients pre- and post-steroid treatment. This study design allows for the identification of protein biomarkers of steroid resistance where patients did not respond to treatment. Candidate biomarkers can be used to potentially distinguish steroid responsive patients. Candidate biomarkers were identified from label-free spectral abundance estimation and validated by biochemical analysis using western blotting. Vitamin D binding protein (VDB) and APOL1 were associated with steroid resistance and hemopexin. Adiponectin and sex hormone-binding globulin were predictive of differences between steroid resistance and sensitivity.
FSGS is a prevalent glomerular disease in pediatric kidney patients. The complexity of this kidney disease cannot be overstated, as it presents as a spectrum of pathology that can often be difficult to distinguish from other glomerulopathies. Proteomics studies are beginning to help distinguish the mechanisms of FSGS in comparison to other kidney diseases and may soon offer important clues about differences in mechanism.
Membranous Nephropathy (MN)
Advances in proteomics applications have also played a key role in the identification of biomarkers of idiopathic membranous nephropathy (iMN) in the past decade. Our laboratory has identified three primary antigens in iMN: PLA2R, THSD7A and HTRA1 [14–16]. Identification of PLA2R antigen in ~70% of primary adult iMN cases was an important contribution in the diagnosis and prognosis of iMN. In this work we were able to utilize a well-curated biorepository of serially collected iMN serum samples to probe for antigen interaction in normal human glomerular extracts. Previous work had suggested the primary antigen in iMN is glycosylated, therefore, lectin purification of the target antigen(s) was utilized to resolve the suspect protein(s) from a complex mixture of glomerular proteins. MN patient serum was then used as a probe for reactive bands which were excised and proteolytically digested. Multi-dimensional HPLC separated peptides were then eluted into a linear ion trap for identification by MS/MS. This approach facilitated identification of PLA2R as the putative target antigen in membranous nephropathy.
New antigens involved in MN have been identified by employing rigorous proteomics experimentation including at least one in pediatric patient samples. In 2020, Sethi et al reported that semaphorin 3B is associated with MN, specifically in pediatric populations, and appears to be a distinct form of the disease in younger patients [17]. This study employed proteomics to identify new antigens from 70 biopsy-confirmed PLA2R negative cases (Mayo cohort). The discovery cohort yielded 2 cases where MS/MS was used to identify and quantify semaphorin 3B in patient sera. Spectral counting was applied to estimate the amount of semaphorin 3B abundance. Validation was performed in 3 further cohorts of 59 biopsy-confirmed MN cases (French cohort 1), 43 MN cases confirmed by immunofluorescence (Italian cohort pediatric only), and a third cohort of 16 IF confirmed cases (French cohort 2, pediatric only). These validations showed a total of 9 semaphorin 3B-associated pediatric cases only or in one adult who had semaphorin 3B in adolescence from longitudinal samples collected at age 1 and 19. These findings emphasize the combined importance of proteomics applications and patient biopsy and sample collection at different ages to confirm the presence of antigens at various stages of pathology and development. Utilizing longitudinal samples allowed for the extrapolation of semaphorin 3B association in an adult patient who showed pediatric onset of MN.
Minimal Change Disease (MCD)
Minimal change disease and FSGS are two common glomerular diseases that often present with similar pathological sequelae and can be difficult to distinguish. Typically, MCD and FSGS present with proteinuria and other altered clinical parameters and require tissue biopsy to confirm the pathology and disease status. Proteomics offers the ability to perform protein level analysis of biofluids to separate and characterize these distinct glomerular pathologies and provide potential biomarkers.
In a 2017 comparative analysis of biopsy-confirmed MCD versus FSGS patient cohorts, Perez and colleagues performed 2D-DIGE analysis of the adult urine proteome to quantify and identify proteome differences in these pathologies [18]. Spots were extracted and identified by MALDI-TOF mass spectrometry as the discovery phase analysis. From the discovery analysis of 10 MCD and 11 FSGS samples they found 11 proteins “up-regulated” in MCD and 5 in FSGS. Validation from 14 MCD and 14 FSGS samples was performed by ELISA-based quantitative assays and showed AAT (SERPINA1), TF (transferrin), and MRPL17 (mitochondrial ribosomal protein L17) were statistically significantly increased in MCD as compared to FSGS. DIGE spot volume estimation translated into ELISA validation, though not at a 1:1 mathematical relationship, and was an indicator of the robustness of proteomics analysis. SERPINA1 has been reported in the literature to be affected by nephrotic syndrome [19]. In this study the authors compared MCD, FSGS and iMN urine proteomes to suggest major differences in urine protein composition across the pathologies. Interestingly, this work partially validates Perez and colleagues in regard to SERPINA1 in MCD. Unfortunately, the parallel work challenges some aspects of the results in regard to albumin levels in MCD compared to FSGS. Choi et al found albumin to be increased in MCD while Perez found some spots corresponding to albumin to be both increased and decreased, creating a degree of confusion about whether MCD leads to more urine albumin than FSGS or if different isoforms of albumin could be responsible for the confounding results. Importantly, Choi and colleagues normalized protein abundance estimates to urine creatinine to correct for differences in urine protein concentration due to the dilution or concentration of urine from collection at random times, which was not performed in the Perez study. Normalization to creatinine appears to significantly affect results from urine proteomics studies and must be considered in primary study designs [20].
One caveat to both studies is the use of gels as the primary fractionation approach. Gel band extraction and proteolytic digestion can affect quantitative reproducibility and may affect the comparison of these results. Additionally, both studies suffer from underpowered group analysis. Ideally for clinical inference, multiple dozens of patient samples should be employed in the study design to improve robustness and provide for more reproducible findings. Studies implementing in-solution proteolysis and on-line peptide fractionation will remove experimental effects of peptide extraction from gels.
Suresh et al conducted a urine proteomics study on MCD and FSGS pediatric patients with steroid sensitivity, frequent relapse steroid-dependence or relapse free patients (n = 3–20/group) [21]. This study design integrated isobaric tag labeling (iTRAQ) of proteins for relative quantitative comparison across multiple groups in a multiplex manner to allow high throughput. Each urine sample was depleted of IgG and albumin, then labelled with distinct reporters and combined for chromatographic fractionation (strong-cation exchange) to improve proteome coverage by spatially separating un-depleted high abundance components to allow for deep mass spectrometry analysis of lower abundance signal peptide ions. Quantitative outputs were normalized to urine creatinine to correct for urine dilution effects on protein concentration. The reported results show that APOA1, A2M, ORM2, RBP4 and A2GP were found to be differentially regulated across multiple groups. APOA1 appears to be correlated with steroid resistance, A2M, RBP4, PRM2 and LRG1 were FSGS dependent markers with A2M having the strongest indication of a true biomarker. Interestingly, A2M mRNA levels have been shown to be correlated with FSGS disease progression in a different study [22].
Minimal change disease and FSGS share enough common pathological features for which distinguishing biomarkers could provide great improvement in clinical decision-making in pediatric patients showing initial signs of CKD. The use and application of quantitative proteomics will continue to provide new insights into the CKD-related urine proteome, as study designs improve and more well-curated patient samples become available. These experimental approaches will become ever more important in deconvoluting the mechanisms of disease progression and treatment responsivity in patient sub-populations.
Immunoglobulin-A Nephropathy (IgAN)
IgAN is the most common glomerular disease and a leading cause of kidney failure. IgAN is characterized by immunoglobulin and complement deposit in the glomerulus that leads to immune overactivation and eventual pathological effects from kidney decline and eventual failure. IgAN may present with subtle clinical characteristics yet can be diagnosed from measuring a variety of clinical parameters. Hematuria and minimal changes of tissue features can be a strong clinical indicator of IgAN, though conclusive diagnosis can require staining of tissue for immunohistochemical analysis or further electron microscopy (EM). This type of analysis is required to distinguish from lupus nephritis, thin basement membrane nephropathy and Alport syndrome [23]. Proteomic analysis of urine or serum could provide a clearer and more expedient analysis by quantifying and characterizing the protein content in the biofluids of the patient.
Mechanistically, little is known about the exact processes driving IgAN disease pathology. Post-translational modification of proteins involved in IgAN pathology could be important in gaining better insight into the progression and initiation of this form of kidney disease. Proteomics is well-suited to help begin to understand the role of post-translational modification of proteins integral to IgAN and other kidney pathologies. Glycosylation of proteins can affect a multitude of biological functions including localization and enzyme activity. Serum contains different types of IgA with differentially glycosylated forms of immunoglobulin, with IgA1 O-glycosylation being most prominent in the hinge region. Dotz el al conducted high resolution mass spectrometry analysis of 83 IgAN patient serum samples and 244 healthy age- and sex-matched controls [24]. This approach was used to characterize the O- and N-glycosylation of proteins in IgAN serum in comparison with controls. Interestingly, this study identified glycopeptides that predicted glomerular function better than a lectin-based ELISA for IgA1 levels.
An early analysis of the IgAN urinary proteome was reported in a 2007 case report of IgAN associated with Hodgkin’s disease [25]. This study applied 2-dimensional gel electrophoresis on the urine proteome of a 14-year-old patient before and during chemotherapy treatment to determine protein quantification in response to IgAN and therapy. Gel spots were quantified and extracted for identification by MS, revealing 14 differentially abundant proteins in response to therapy.
More recently, Fang and colleagues performed a data-independent MS/MS analysis on pediatric IgAN and HSPN (Henoch-Schonlein Purpura Nephritis) patients to determine differences in the urine proteome in these overlapping and comorbid pathologies [26]. In this work the authors applied technical mass spectrometry analysis by performing the MS/MS method of DIA (data-independent-analysis) which collects all m/z ion transitions possible within the duty cycle limits of the instrument (30 for this analysis). Typically, conventional MS experiments performed in a DDA mode (data-dependent analysis) generate MS/MS fragment ions based on intensity of the parent ion and can exclude ions once measurement has occurred to minimize fragmentation of already analyzed species, usually 12–20 per measurement cycle. Inherently, the DDA mode is biased toward the most highly enriched ion species and can lead to fewer low abundance protein identifications. DIA attempts to capture all ion species within an analysis time-frame are limited only by instrument speed, or what is known as cycle times. DIA can be useful in the detection of lower abundance species.
Here the authors compared 19 urinary proteomes of IgAN versus HSPN where patients exhibited proteinuria, mesangial expansion and IgA deposition. Additionally, healthy patient urine samples were collected and compared to the pathological samples. In this patient cohort it would be difficult to distinguish IgAN versus HSPN using traditional clinical parameters because of the overlap in clinical features without invasive biopsy and deep clinical characterization. Urine proteome analysis using DIA-MS/MS across all patient groups yielded ~1,800 protein identifications where 276 were differentially abundant in IgAN and 125 in HSPN.
Hierarchical clustering analysis illustrated major differences between the urine proteome of IgAN compared to HSPN. Further bioinformatics analysis using GO (Gene Ontology) and KEGG (Kyoto Encyclopedia of Genes and Genomes) was utilized to delve into the enriched pathways and suggest biological mechanisms involved in the pathogenesis of the two overlapping diagnoses as compared to normal urine proteome. Ontology analysis suggested pathways involved in the extracellular matrix, PI3K signaling, and cell-adhesion. Complement and coagulation cascades were among some of the most enriched components in the urine proteomes.
Finally, validation of a small subset of proteins was performed for A1BG and AFAMIN by performing ELISAs to quantify differences in the IgAN and HSPN proteome as compared to normal healthy urine. Though differences were validated between healthy urine and IgAN and HSPN, no validations were performed to compare differences between IgAN and HSPN. This work begins to show the versatility in applications of a variety of technical aspects of mass spectrometry and proteomics. Detailed follow-up to this type of proteomics analysis will hopefully yield more in-depth understanding of the differences in IgAN and HSPN and the methodology that can be applied to characterize other pathologically affected urine proteomes.
Systemic Lupus Erythematous (SLE) and Lupus nephritis (LN)
Lupus is an autoimmune disease that affects the kidney through elevated inflammation and infiltration of the organ with complement, antibodies and autoantibodies. Sustained inflammatory damage to the glomerular basement membrane and specifically podocytes negatively affect glomerular function. In pediatric lupus populations, ~80% of patients will develop lupus nephritis and in some countries the 5-year survival rate can approach 45%. In most developed medical systems an 80–95% 5-year survival rate can be expected with targeted and early clinical intervention [27–29]. Urine biomarkers are an attractive alternative to repeated and invasive core tissue biopsies in pediatric patients, which are usually not ordered by the pediatric nephrologist. Urine biomarkers could offer the ability to track and predict disease flare and progression, and monitor treatment efficacy from longitudinal sampling of this biofluid for diagnostic/prognostic biomarkers. Several novel urine biomarkers of LN have been elucidated and further investigated but fail to be useful in the clinic. Robustness of study design is often affected by lack of patient samples to effectively analyze in a statistically rigorous manner, likely due to variability in heterogeneity of disease progression in LN. Some novel biomarkers of LN include NGAL, MCP-1, VCAM-1 and a panel of cytokines or other inflammatory associated markers including IL-6 and IP10 [30]. Recent proteomic and mass spectrometry studies of LN urine biomarkers have reported orosomucoid (ORM), transferrin (TF), α−2-macroglobulin (A2M), haptoglobin (HPT), afamin (AFM), vimentin, ICAM-1, α−1-antitrypsin (A1AT), ceruloplasmin (CP) and PTGDS as potentially useful in diagnosis and prognosis of the disease [30, 31].
Investigations by Anania and colleagues undertook an impressive 3-pronged proteomics approach toward uncovering biomarkers of LN. Here the authors conducted 1) gel-based proteomics, 2) TMT-isobaric reporter tagging LC-MS/MS, and 3) DIA (data-independent analysis) followed by targeted candidate quantitation by parallel reaction monitoring (PRM) of specific peptides from 7 proteins identified in the 3-pronged discovery phase [31]. Methodologically, this work revealed that TMT identified significantly more overall proteins than DIA which is useful information for the mass spectrometrist interested in studying urine proteomes. Interestingly, the design of these experiments called for a smaller discovery phase patient enrollment (3 healthy controls versus 3 LN disease active) followed by validation in 20 independent healthy controls and 20 LN disease active samples using targeted peptide level monitoring by quantitative PRM methods. This design allows for more technical replicates in the discovery phase and deep quantitative profiling in validation cohorts. Combining 3 shotgun approaches shows the overlap in mass spectrometry methods and some of the gains from using alternate methods. In this application, DIA clearly measures lower abundance proteins while TMT has the advantage of broader proteome coverage. Inherent in each of these MS-coupled approaches are technical differences that should be considered if only one approach is available.
Advanced proteomics technologies can provide novel insights into the process of LN biology. Study design coupled with robust quantitative proteomics are particularly important in gaining helpful insights into biomarkers of LN that could be useful in clinical settings. The ability to implement a panel of biomarkers to accurately and precisely determine disease flare, remission and therapeutic efficacy will be a welcome advance for the clinician toolbox in pediatric nephrology.
Integration of Metabolomics and Proteomics technology
Diagnostic procedures in pediatric populations present with a degree of difficulty in misdiagnosis from routine blood sampling and biopsy extraction [32]. Overcoming these challenges is the need of the hour, therefore, omics-based technologies offers unparalleled solutions to these limitations toward discovery of novel and reliable biomarkers. New biomarker discovery will allude to mechanistic drivers of idiopathic forms of kidney disease from minimal blood or urine sampling. Moreover, discovery-based approaches open new avenues of research. For example, podocyte-focused research was jumpstarted in the case of iMN after the proteomics-based discovery of autoantibodies to PLA2R, HTRA1 and THSD7A [14–16]. Interestingly, this surge in focused podocyte research has led to the finding that elevated fumarate in urine of patients with PLA2R-associated MN leads to alterations in phenotypic profile of cultured podocytes [33]. Whereas, in the case of LC/MS based metabolomics, a study performed on urine from mice with acute kidney injury (AKI) revealed niacinamide levels as an indicator of developing AKI risk with loss- and gain-of-function of peroxisome proliferator-activated receptor γ coactivator 1-α (PGC1 α) pointing toward the importance of oxidative stress-based research [34]. Although single omics-based approaches can reveal novel information on disease pathogenicity, integration of multiple omics-based platforms provides an extra layer of information with the potential for development of more effective treatment strategies – for example novel gene-protein-metabolite linkages can offer insights into disease modifier mechanisms [35–37]. In the context of kidney diseases, only a handful of current studies exist that support multi-omics analysis as a promising strategy. For instance, integration of serum proteomic and metabolomics profiles from a rat model of kidney fibrosis revealed complement and coagulation cascade and regulation of actin cytoskeleton as altered pathways [38]. Another study integrated RNA-Seq, proteomics and metabolomics from kidney biopsies of diabetic nephropathy (DN) patients [39]. These findings reveal enzymes and metabolites linked to linoleic acid metabolism and ß-oxidation as potentially targetable pathways/molecules for the treatment of DN.
As anticipated, multi-omics integration will improve our understanding of pediatric kidney diseases at different levels, but complex and large omics dataset analysis is often the bottleneck in novel discovery. Often, this is because many analytical tools traditionally used for single omics platforms are not well-suited for intelligent integration [40]. However, emerging trends in bioinformatics have allowed improved data handling such as MetaboAnalyst 5.0 [41, 42], similarity network fusion (SNF) [43], mixOmics [44], and multi-omics factor analysis (MOFA) [45]. Utility of these approaches depends on joint pathway analysis, matrix factorization, network fusion, canonical correlation and factor analysis. The latest version of MetaboAnalyst has narrowed the gap between high resolution mass spectrometry (HRMS) and functional insights by adding different features, especially weighted joint pathway analysis modules for multi-omics integration, and a new function for data-driven network analysis that uses a well-established debiased sparse partial correlation (DSPC) algorithm which can differentiate between direct and indirect associations between metabolites [42, 46]. SNF integrates diverse types of data by constructing networks of samples (e.g., patients) for each available data type and then efficiently fusing this into one network that represents the full spectrum of available data (for example, [43]), to create a comprehensive view of a disease in a cohort of patients. MixOmics offers a wide range of multivariate methods where the number of variables (e.g., genes, proteins, metabolites) is much larger than number of samples (e.g., patients, cells, mice) to integrate the large data sets into a graphical output that enable better understanding of relationships and correlation structures [47, 48]. MOFA is a statistical method for integrating multiple modalities of omic data in an unsupervised fashion. MOFA infers (hidden) factors that could be driving sources of variation across data modalities, thus facilitating the identification of molecular states or subgroups of samples [45]. In all, these tools are organized based on ability to address biological questions that are broadly categorized into: 1) Disease subtyping and classification based on multi-omics profiles, 2) Predication of biomarkers including diagnostics and driver genes for diseases, and 3) Insights into disease biology [49].
Since these tools provide in-depth insight into biological states or processes, at the same time they are also evolving to circumvent the challenges associated with multi-omics integration, which are essentially the underlying heterogeneity in individual omics data, large size of data sets, different formats of data sets and data preprocessing [49]. As these limitations significantly impact the integrative analysis, the right choice of tool and an appropriate know-how of handling the data are prerequisites, and important to address the biological question-of-interest.
Interestingly, deep learning (DL) and machine learning (ML) approaches combined with integrative analysis can dramatically improve accuracy in disease prediction, patient stratification decision-making and delivery of precision medicine [50]. For instance, in a neuroblastoma data integration study, integrative network fusion framework was combined with the ML classifier to extract features which can discriminate between different outcomes of patients [51]. The major AI tools applied to date are the supervised learning-based support vector machine (SVM) algorithm, a widely-used approach for analysis of multi-omics data for better task performance [50]. Unsupervised learning-based autoencoders consist of an encoder and a decoder. The encoder extracts important features from large input data, and the decoder tries to construct an output very similar to the input using only the extracted features. This approach excludes the redundant data [50]. AI-based methodologies described above are mainly applied to cancer classification and prognosis, but application of these tools coupled with metabolomics and proteomics will provide new insights and understanding of pediatric CKD [52–54]. However, in order to increase utility of these AI-based platforms, the availability of large data sets from kidney consortia are crucial for development of sophisticated and validated platforms to enable accurate discovery of reliable biomarkers, novel drivers of kidney disease and algorithms for patient stratification, all contributing to development of effective personalized medicine in pediatric CKD.
Current Applications for Future Directions in Pediatric Nephrology
In addition to patient recruitment/cohort design and consistent methods for collection of patient samples (blood, serum, plasma or urine), use of remnant, archived patient kidney biopsy material for targeted proteomic analyses increases successful identification of surrogate biomarkers for kidney disease. Laser capture microdissection (LCMD) allows isolation of different kidney nephron segments to understand normal development of specific kidney structures (68), response to injury, and disease progression. Coupled with proteomics, LCMD isolation of glomeruli identified neural cell adhesion molecule 1 as a novel autoantigen in lupus nephritis [56]. This same technical approach determined the prevalence of semaphorin 3B-associated membranous nephropathy in pediatric patients [17]. Another study with LCMD of glomeruli in membranous nephropathy associated increased expression of complement proteins in glomeruli with increased complement activation products in urine, highlighting the role of complement in disease progression [57]. Additional approaches with immunofluorescent labeling of cells and structures for isolation by LCMD [58] allow coupling protein expression to regional transcriptomics. Our group subjected LCMD collected glomerular sections to sequential protein extraction protocols separating the cellular compartment from the extracellular matrix (ECM), for mass spectrometry analysis [59], and expanded the number of previously-reported glomerular ECM proteins [60]. We utilized the same approaches to define significant differences in basement membrane, structural ECM proteins, and ECM-affiliated and regulating proteins, in glomeruli of patients with collapsing FSGS compared to other FSGS variants [61]. Similar approaches have been utilized in glomerular and tubule compartments of kidney allografts with antibody-mediated rejection (77), and in glomeruli with IgA nephropathy (78). Defining quantitative and qualitative glomerular ECM composition from limited human biopsy material allows analysis of ECM, which may indicate early disease transitional remodeling processes, or better define and associate with histologic classification and disease progression [62, 63]. Utilization of approaches combining LCMD of renal structures with proteomics can increase understanding of pediatric renal disease pathogenesis and progression.
Proteomic Array Technology
Mass spectrometry-based approaches offer unmatched analytical measurement range on a wide array of targets, while antibody-based methods boast sensitives even below the picogram per milliliter range on single targets. Many of the biomarkers of CKD mentioned thus far are strong standalone predictors for diagnosis, prognosis, and/or response to therapy. That said, the kidneys are complex organs, and the combination of multiple biomarkers may produce diagnostic algorithms capable of earlier detection or more accurate estimation of therapeutic response when compared to the use of a single biomarker. It is likely that as datasets grow for specific conditions, newly defined biomarkers with lower predictive value may show increased diagnostic power as panels rather than as independent results. As with many other disease conditions (RAIL – Kidney Activity Score for Lupus), a diagnostic score could be derived from the combination of several independent factors such as creatinine clearance or urine protein concentration combined with multiple inflammatory cytokines or other markers of CKD.
Luminex is a bead-based array platform that has been in the proteomic toolset for decades due multiplexing capacity. Luminex assays can measure up to 500 targets in a single run with sensitivity in the pg/mL range [64]. Custom panels can be tailored to specific needs of a patient set to include the necessary inflammatory cytokines, chemokines, and cellular injury markers specific to disease state. Recently, Cody et al produced a convenient method for simultaneous detection of 4 RAIL biomarkers (NGAL, KIM-1, MCP-1, and adiponectin) with sufficient power to produce a RAIL score, albeit with 2 targets failing to be detected [65].
Somalogic platforms utilize single stranded DNA-based protein-like structures to capture corresponding targets, with over 7,000 possible targets measured on a single chip. This technology boasts a 10-log dynamic range, far more than ELISA and even dwarfing LC-MS/MS. Chips are expensive, yet stable, and effective for quantifying lower abundance cytokines and chemokines. In this paper, Niewczas and colleagues identified a panel of biomarkers for diagnosing DN and demonstrate that this panel can be effectively used to gauge response to therapy [66]. As we overlap technologies with the proteomic world, the potential exists for these methods to be combined and address challenges in pediatric CKD diagnosis and prognosis.
Final Perspectives
Although this review has covered the topic of proteomics applications in pediatric kidney diseases such as LN, FSGS, MN, MCD, IgAN and others, the basic methodology described within can be applied in a variety of fields and should be considered applicable in a variety of kidney contexts as well as other organ systems. Other areas of promise include applications of peptidomics and metabolomics to identify molecular biomarkers of diabetic kidney disease, congenital anomalies of the kidney and urinary tract, and the above-mentioned glomerular pathologies. Discovering novel biomarkers of pediatric kidney disease holds a great deal of promise with improving technology and focused development of biorepository capacity and diversity. Although much work remains to be conducted to continue the trajectory toward clinically viable findings, the fundamental improvements in mass spectrometry, bioinformatics and sample availability are beginning to pay dividends. Improvements in mass spectrometry platforms are beginning to help identify and develop new leads in adult kidney diseases and can be transferred directly into the pediatric realm. The burgeoning fields of proteomics, genomics and metabolomics are primed for application in pediatric diseases and should be particularly helpful in pediatric chronic kidney diseases. Integration of each component of omics technology into a seamless data pipeline will be a step toward actuation of the goal of precision medicine in pediatric nephrology.
Funding
This work was supported by NIDDK-NIH (DK110077 to JBK, WES), and clinical research funding from the University of Louisville Kidney Disease Program.
Footnotes
Conflict of interest statement
The authors have no financial interests or conflicts of interest to declare.
References
- 1.United States Renal Data System (2019) 2019 USRDS Annual Data Report: Epidemiology of kidney disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD. https://www.usrds.org/annual-data-report/. Accessed 16 Aug 2021 [Google Scholar]
- 2.Davis MT, Beierle J, Bures ET et al. (2001) Automated LC-LC-MS-MS platform using binary ion-exchange and gradient reversed-phase chromatography for improved proteomic analyses. J Chromatogr B Biomed Sci App 752:281–291. 10.1016/s0378-4347(00)00547-8 [DOI] [PubMed] [Google Scholar]
- 3.Vissers JPC, Blackburn RK, Moseley MA (2002) A novel interface for variable flow nanoscale LC/MS/MS for improved proteome coverage. J Am Soc Mass Spectrom 13:760–771. 10.1016/S1044-0305(02)00418-X [DOI] [PubMed] [Google Scholar]
- 4.Wolters DA, Washburn MP, Yates JR (2001) An automated multidimensional protein identification technology for shotgun proteomics. Anal Chem 73:5683–5690. 10.1021/ac010617e [DOI] [PubMed] [Google Scholar]
- 5.McDonald WH, Yates JR (2002) Shotgun proteomics and biomarker discovery. Dis Markers 18:99–105. 10.1155/2002/505397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dayon L, Hainard A, Licker V et al. (2008) Relative quantification of proteins in human cerebrospinal fluids by MS/MS using 6-plex isobaric tags. Anal Chem 80:2921–2931. 10.1021/ac702422x [DOI] [PubMed] [Google Scholar]
- 7.Wang Z, Yu K, Tan H et al. (2020) 27-Plex Tandem Mass Tag Mass Spectrometry for Profiling Brain Proteome in Alzheimer’s Disease. Anal Chem 92:7162–7170. 10.1021/acs.analchem.0c00655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khurana M, Traum AZ, Aivado M et al. (2006) Urine proteomic profiling of pediatric nephrotic syndrome. Pediatr Nephrol 21:1257–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Juraschek SP, Coresh J, Inker LA et al. (2013) Comparison of serum concentrations of β-trace protein, β2-microglobulin, cystatin C, and creatinine in the US population. Clin J Am Soc Nephrol 8:584–592. 10.2215/CJN.08700812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inker LA, Tighiouart H, Coresh J et al. (2016) GFR Estimation Using β-Trace Protein and β2-Microglobulin in CKD. Am J Kidney Dis 67:40–48. 10.1053/j.ajkd.2015.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Argyropoulos CP, Chen SS, Ng Y-H et al. (2017) Rediscovering Beta-2 Microglobulin As a Biomarker across the Spectrum of Kidney Diseases. Front Med 4:73. 10.3389/fmed.2017.00073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woroniecki RP, Shatat IF, Supe K et al. (2008) Urinary cytokines and steroid responsiveness in idiopathic nephrotic syndrome of childhood. Am J Nephrol 28:83–90. 10.1159/000109396 [DOI] [PubMed] [Google Scholar]
- 13.Agrawal S, Merchant ML, Kino J et al. (2020) Predicting and Defining Steroid Resistance in Pediatric Nephrotic Syndrome Using Plasma Proteomics. Kidney Int Rep 5:66–80. 10.1016/j.ekir.2019.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Al-Rabadi LF, Caza T, Trivin-Avillach C et al. (2021) Serine Protease HTRA1 as a Novel Target Antigen in Primary Membranous Nephropathy. J Am Soc Nephrol 32:1666–1681. 10.1681/ASN.2020101395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tomas NM, Beck LH, Meyer-Schwesinger C et al. (2014) Thrombospondin type-1 domain-containing 7A in idiopathic membranous nephropathy. N Engl J Med 371:2277–2287. 10.1056/NEJMoa1409354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beck LH, Bonegio RGB, Lambeau G et al. (2009) M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med 361:11–21. 10.1056/NEJMoa0810457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sethi S, Debiec H, Madden B et al. (2020) Semaphorin 3B-associated membranous nephropathy is a distinct type of disease predominantly present in pediatric patients. Kidney Int 98:1253–1264. 10.1016/j.kint.2020.05.030 [DOI] [PubMed] [Google Scholar]
- 18.Pérez V, López D, Boixadera E et al. (2017) Comparative differential proteomic analysis of minimal change disease and focal segmental glomerulosclerosis. BMC Nephrol 18:49. 10.1186/s12882-017-0452-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi YW, Kim YG, Song M-Y et al. (2017) Potential urine proteomics biomarkers for primary nephrotic syndrome. Clin Proteomics 14:18. 10.1186/s12014-017-9153-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Waikar SS, Sabbisetti VS, Bonventre JV (2010) Normalization of urinary biomarkers to creatinine during changes in glomerular filtration rate. Kidney Int 78:486–494. 10.1038/ki.2010.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suresh CP, Saha A, Kaur M et al. (2016) Differentially expressed urinary biomarkers in children with idiopathic nephrotic syndrome. Clin Exp Nephrol 20:273–283. 10.1007/s10157-015-1162-7 [DOI] [PubMed] [Google Scholar]
- 22.Menon R, Otto EA, Hoover P et al. (2020) Single cell transcriptomics identifies focal segmental glomerulosclerosis remission endothelial biomarker. JCI Insight 5:e133267. 10.1172/jci.insight.133267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wenderfer SE, Gaut JP (2017) Glomerular Diseases in Children. Adv Chronic Kidney Dis 24:364–371. 10.1053/j.ackd.2017.09.005 [DOI] [PubMed] [Google Scholar]
- 24.Dotz V, Visconti A, Lomax-Browne H et al. (2021) O- and N-Glycosylation of Serum Immunoglobulin A is Associated with IgA Nephropathy and Glomerular Function. J Am Soc Nephrol 32:2455–2465. 10.1681/ASN.2020081208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khositseth S, Kanitsap N, Warnnissorn N, Thongboonkerd V (2007) IgA nephropathy associated with Hodgkin’s disease in children: a case report, literature review and urinary proteome analysis. Pediatr Nephrol 22:541–546. 10.1007/s00467-006-0382-1 [DOI] [PubMed] [Google Scholar]
- 26.Fang X, Lu M, Xia Z et al. (2021) Use of liquid chromatography-tandem mass spectrometry to perform urinary proteomic analysis of children with IgA nephropathy and Henoch-Schönlein purpura nephritis. J Proteomics 230:103979. 10.1016/j.jprot.2020.103979 [DOI] [PubMed] [Google Scholar]
- 27.Boneparth A, Wenderfer SE, Moorthy LN et al. (2017) Clinical characteristics of children with membranous lupus nephritis: the Childhood Arthritis and Rheumatology Research Alliance Legacy Registry. Lupus 26:299–306. 10.1177/0961203316662720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boneparth A, Ilowite NT; CARRA Registry Investigators (2014) Comparison of renal response parameters for juvenile membranous plus proliferative lupus nephritis versus isolated proliferative lupus nephritis: a cross-sectional analysis of the CARRA Registry. Lupus 23:898–904. 10.1177/0961203314531841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pereira M, Muscal E, Eldin K et al. (2017) Clinical presentation and outcomes of childhood-onset membranous lupus nephritis. Pediatr Nephrol 32:2283–2291. 10.1007/s00467-017-3743-z [DOI] [PubMed] [Google Scholar]
- 30.Suzuki M, Wiers K, Brooks EB et al. (2009) Initial validation of a novel protein biomarker panel for active pediatric lupus nephritis. Pediatr Res 65:530–536. 10.1203/PDR.0b013e31819e4305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anania VG, Yu K, Pingitore F et al. (2019) Discovery and Qualification of Candidate Urinary Biomarkers of Disease Activity in Lupus Nephritis. J Proteome Res 18:1264–1277. 10.1021/acs.jproteome.8b00874 [DOI] [PubMed] [Google Scholar]
- 32.Traum AZ, Schachter AD (2007) Proteomic analysis in pediatric renal disease. Semin Nephrol 27:652–657. 10.1016/j.semnephrol.2007.09.009 [DOI] [PubMed] [Google Scholar]
- 33.Jo HA, Hyeon JS, Yang SH et al. (2021) Fumarate modulates phospholipase A2 receptor autoimmunity-induced podocyte injury in membranous nephropathy. Kidney Int 99:443–455. 10.1016/j.kint.2020.06.031 [DOI] [PubMed] [Google Scholar]
- 34.Poyan Mehr A, Tran MT, Ralto KM et al. (2018) De novo NAD+ biosynthetic impairment in acute kidney injury in humans. Nat Med 24:1351–1359. 10.1038/s41591-018-0138-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feng J, Zhang Q, Zhou Y et al. (2018) Integration of Proteomics and Metabolomics Revealed Metabolite-Protein Networks in ACTH-Secreting Pituitary Adenoma. Front Endocrinol 9:678. 10.3389/fendo.2018.00678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bahado-Singh R, Poon LC, Yilmaz A et al. (2017) Integrated Proteomic and Metabolomic prediction of Term Preeclampsia. Sci Rep 7:16189. 10.1038/s41598-017-15882-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Costanzo M, Zacchia M, Bruno G et al. (2017) Integration of Proteomics and Metabolomics in Exploring Genetic and Rare Metabolic Diseases. Kidney Dis (Basel) 3:66–77. 10.1159/000477493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cao H, Zhang A, Sun H et al. (2015) Metabolomics-proteomics profiles delineate metabolic changes in kidney fibrosis disease. Proteomics 15:3699–3710. 10.1002/pmic.201500062 [DOI] [PubMed] [Google Scholar]
- 39.Sha Q, Lyu J, Zhao M et al. (2020) Multi-Omics Analysis of Diabetic Nephropathy Reveals Potential New Mechanisms and Drug Targets. Front Genet 11:616435. 10.3389/fgene.2020.616435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pinu FR, Beale DJ, Paten AM et al. (2019) Systems Biology and Multi-Omics Integration: Viewpoints from the Metabolomics Research Community. Metabolites 9:E76. 10.3390/metabo9040076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chong J, Wishart DS, Xia J (2019) Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr Protoc Bioinforma 68:e86. 10.1002/cpbi.86 [DOI] [PubMed] [Google Scholar]
- 42.Pang Z, Chong J, Zhou G et al. (2021) MetaboAnalyst 5.0: narrowing the gap between raw spectra and functional insights. Nucleic Acids Res 49:W388–W396. 10.1093/nar/gkab382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang B, Mezlini AM, Demir F et al. (2014) Similarity network fusion for aggregating data types on a genomic scale. Nat Methods 11:333–337. 10.1038/nmeth.2810 [DOI] [PubMed] [Google Scholar]
- 44.Rohart F, Gautier B, Singh A, Lê Cao KA (2017) mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput Biol 13:e1005752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Argelaguet R, Velten B, Arnol D et al. (2018) Multi-Omics Factor Analysis-a framework for unsupervised integration of multi-omics data sets. Mol Syst Biol 14:e8124. 10.15252/msb.20178124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Basu S, Duren W, Evans CR et al. (2017) Sparse network modeling and metscape-based visualization methods for the analysis of large-scale metabolomics data. Bioinforma Oxf Engl 33:1545–1553. 10.1093/bioinformatics/btx012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Amezquita RA, Lun ATL, Becht E et al. (2020) Orchestrating single-cell analysis with Bioconductor. Nat Methods 17:137–145. 10.1038/s41592-019-0654-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bioconductor. Open Source Software for Bioinformatics. https://bioconductor.org/. Accessed 17 Aug 2021
- 49.Subramanian I, Verma S, Kumar S et al. (2020) Multi-omics Data Integration, Interpretation, and Its Application. Bioinforma Biol Insights 14:1177932219899051. 10.1177/1177932219899051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reel PS, Reel S, Pearson E et al. (2021) Using machine learning approaches for multi-omics data analysis: A review. Biotechnol Adv 49:107739. 10.1016/j.biotechadv.2021.107739 [DOI] [PubMed] [Google Scholar]
- 51.Francescatto M, Chierici M, Rezvan Dezfooli S et al. (2018) Multi-omics integration for neuroblastoma clinical endpoint prediction. Biol Direct 13:5. 10.1186/s13062-018-0207-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sealfon RSG, Mariani LH, Kretzler M, Troyanskaya OG (2020) Machine learning, the kidney, and genotype-phenotype analysis. Kidney Int 97:1141–1149. 10.1016/j.kint.2020.02.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saez-Rodriguez J, Rinschen MM, Floege J, Kramann R (2019) Big science and big data in nephrology. Kidney Int 95:1326–1337. 10.1016/j.kint.2018.11.048 [DOI] [PubMed] [Google Scholar]
- 54.Bülow RD, Dimitrov D, Boor P, Saez-Rodriguez J (2021) How will artificial intelligence and bioinformatics change our understanding of IgA Nephropathy in the next decade? Semin Immunopathol 43:739–752. 10.1007/s00281-021-00847-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Potter SS, Brunskill EW (2014) Building an atlas of gene expression driving kidney development: pushing the limits of resolution. Pediatr Nephrol 29:581–588. 10.1007/s00467-013-2602-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Caza TN, Hassen SI, Kuperman M et al. (2021) Neural cell adhesion molecule 1 is a novel autoantigen in membranous lupus nephritis. Kidney Int 100:171–181. 10.1016/j.kint.2020.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ayoub I, Shapiro JP, Song H et al. (2021) Establishing a Case for Anti-complement Therapy in Membranous Nephropathy. Kidney Int Rep 6:484–492. 10.1016/j.ekir.2020.11.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barwinska D, Ferkowicz MJ, Cheng YH, Winfree S, Dunn KW, Kelly KJ, Sutton TA, Rovin BH, Parikh SV, Phillips CL, Dagher PC, El-Achkar TM, Eadon MT; Kidney Precision Medicine Project (2020) Application of Laser Microdissection to Uncover Regional Transcriptomics in Human Kidney Tissue. J Vis Exp 160:10.3791/61371. 10.3791/61371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hobeika L, Barati MT, Caster DJ et al. (2017) Characterization of glomerular extracellular matrix by proteomic analysis of laser-captured microdissected glomeruli. Kidney Int 91:501–511. 10.1016/j.kint.2016.09.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lennon R, Byron A, Humphries JD et al. (2014) Global analysis reveals the complexity of the human glomerular extracellular matrix. J Am Soc Nephrol 25:939–951. 10.1681/ASN.2013030233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Merchant ML, Barati MT, Caster DJ et al. (2020) Proteomic Analysis Identifies Distinct Glomerular Extracellular Matrix in Collapsing Focal Segmental Glomerulosclerosis. J Am Soc Nephrol 31:1883–1904. 10.1681/ASN.2019070696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Clotet-Freixas S, McEvoy CM, Batruch I,et al. (2020) Extracellular Matrix Injury of Kidney Allografts in Antibody-Mediated Rejection: A Proteomics Study. J Am Soc Nephrol 31:2705–2724. 10.1681/ASN.2020030286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Paunas FTI, Finne K, Leh S et al. (2019) Characterization of glomerular extracellular matrix in IgA nephropathy by proteomic analysis of laser-captured microdissected glomeruli. BMC Nephrol 20:410. 10.1186/s12882-019-1598-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Solier C, Langen H (2014) Antibody-based proteomics and biomarker research - current status and limitations. Proteomics 14:774–783. 10.1002/pmic.201300334 [DOI] [PubMed] [Google Scholar]
- 65.Cody EM, Bennett MR, Gulati G et al. (2021) Successful Urine Multiplex Bead Assay to Measure Lupus Nephritis Activity. Kidney Int Rep 6:1949–1960. 10.1016/j.ekir.2021.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Niewczas MA, Pavkov ME, Skupien J et al. (2019) A signature of circulating inflammatory proteins and development of end-stage renal disease in diabetes. Nat Med 25:805–813. 10.1038/s41591-019-0415-5 [DOI] [PMC free article] [PubMed] [Google Scholar]