

Abstract
Here, we discuss the principles of allosteric activating mutations, propagation downstream of the signals that they prompt, and allosteric drugs, with examples from the Ras signaling network. We focus on Abl kinase where mutations shift the landscape toward the active, imatinib binding-incompetent conformation, likely resulting in the high affinity ATP outcompeting drug binding. Recent pharmacological innovation extends to allosteric inhibitor (GNF-5)-linked PROTAC, targeting Bcr-Abl1 myristoylation site, and broadly, allosteric heterobifunctional degraders that destroy targets, rather than inhibiting them. Designed chemical linkers in bifunctional degraders can connect the allosteric ligand that binds the target protein and the E3 ubiquitin ligase warhead anchor. The physical properties and favored conformational state of the engineered linker can precisely coordinate the distance and orientation between the target and the recruited E3. Allosteric PROTACs, noncompetitive molecular glues, and bitopic ligands, with covalent links of allosteric ligands and orthosteric warheads, increase the effective local concentration of productively oriented and placed ligands. Through covalent chemical or peptide linkers, allosteric drugs can collaborate with competitive drugs, degrader anchors, or other molecules of choice, driving innovative drug discovery.
Keywords: ensembles, allosteric PROTAC, allosteric molecular glues, autoinhibition, kinase drug discovery, enzyme catalysis, cellular network
Graphical Abstract
Introduction
Classically, throughout past decades, the life sciences aimed at morphological descriptions of organisms, their classification, and their habitats. The emergence of the structure – function paradigm was inspired by the recognition that even living things must conform to the laws of physics, including the laws of motion and structural chemistry [1]. The gradual conceptual change merged with technological advances that were able to capture the conformational heterogeneity [1–5]. Allostery, which involves dynamic shifts of the distributions of the conformational ensembles prompted by some triggering events, is at the center of regulated molecular behavior [6–15]. Changes in the conformational distributions provoked by allosteric PTMs (post-translational modifications), interactions with ions, cofactors, mutations, allosteric drugs, changes in concentrations, and more [16–30], result in conformational and dynamic changes [12,13,31–41] and can be captured by the dynamic free energy landscapes [37,42]. The resulting energetic frustration, or local energetic conflicts in the structure [43–46] reinforced by the allosteric conformational changes, propagate and are the key to protein function [47–52]. Allostery signals at short ranges. If the domains are separated by long disordered linkers, as in the case of Raf kinase [53], the relatively similar free energies of the conformational states (the depths of the basins in the landscape) characteristic of disordered ensembles, support multiple routes through which the allosteric signals can travel [54]. Funnels of folding, binding and function [55–58] showed that folding progresses via multiple routes, with some dominantly populated, which may not be the case for allosteric propagation or folding of disordered states.
Catalysis and regulation are the beating heart of biology [59]. In catalysis, ensembles of activated conformations can catalyze multiple reaction steps [60]. The transition-state ensemble [61] can also employ multiple paths from the rugged saddle region as shown for the yeast chorismate mutase [62,63]. Thus, conformational ensemble concepts [64] extend from folding to binding to enzymatic catalysis, and below we apply them to allosteric cancer drivers and innovative allosteric drug concepts. Considering that to have a functional impact allostery-prompted signals of activating mutations need to propagate downstream, we further take up the vital and formidable question of measurements of signal thresholds and outline some principles [65].
Allosteric driver mutations in cancer
Here, we consider diseases that are the outcome of allosteric driver mutations [33,66–79]. Not all drivers are allosteric [80]; mutations at the functional (or active) site are not, as exemplified by oncogenic Ras drivers at G12, or Q61. More on this below. Allosteric drivers work by destabilizing the inactive protein state, stabilizing the active state, or both [33]. In repressors, allosteric mutations stabilize the inactive state [70]. The differences in energy between the inactive and active states are typically relatively small. To get stabilized, that is, to become the favorable state, the protein needs to undergo a conformational change. Mutations can do this by breaking the interactions that stabilize it, and/or forming alternative stabilizing interactions, such as salt bridges or hydrophobic cores. By definition, these are allosteric events [6] that can lead to significant increase in protein activity over the basal state. Driver mutations promote cancer since they bestow a growth edge [81]. Current therapeutic regimens seek knowledge of the patient’s driver mutations to guide treatment [82–85]. Driver mutations are commonly identified by their high frequencies of occurrence, making detection of rare allosteric driver mutations that may be equally potent a daunting task [86]. Thus, in addition to the statistics of occurrence, strategies to identify driver mutations include structural location and organization, and functional consequences [81]. Clustering of mutations not only in protein sequences [87] but especially in structures [88–93] is one such strategy. Allosteric drivers often cluster since clustering intensifies their effect. A classic example of clustered residue hotspots is the “hot regions” [94,95]. The residues are structurally highly conserved residing within locally tightly packed regions, where they form a network of interactions making their contributions to the stability cooperative. Identification of drivers can also be helped by molecular dynamics (MD) simulations, which can observe conformational changes. Residue interaction networks [96] can further reveal whether they lie on the same propagation pathway of the intramolecular allosteric signal, which can strengthen the signal outcome. Other approaches include (i) machine learning, which can identify dynamic signatures (e.g., [40,97–100]), and whose vast contributions to the field will be reviewed elsewhere, (ii) dynamic residue networks and (iii) perturbation response scanning which were used to identify allosteric hot spots of human Hsp90 as cancer drug target, and it was shown that both approaches are in agreement [101]. A recent review summarized the approaches to identify allosteric regions/hotspots. It includes residue interaction networks and other approaches. It also extensively discusses allosteric drugs [102]. Additional approaches have also been devised [103–109]. From the conformational standpoint, the hallmark of allosteric activity is the behavior of the ensemble. If it is shifted toward an active (or inactive in repressors) state, then the mutations are activating. Ensemble shifts can be captured through, e.g., schematic, funnel-like “function diagrams” [110], nuclear magnetic resonance (NMR), and MD simulations [111–114]. Recently, we proposed that a key mechanism of allosteric driver mutations is relieving autoinhibition and provided a few mechanistic examples, such as protein kinase B (AKT) and mammalian target of rapamycin (mTOR) protein kinases, phosphoinositide 3-kinase α (PI3Kα) lipid kinase, SH2 domain-containing phosphatase 2 (SHP2) phosphatase, NIMA-related kinase 7 (NEK7) and 9 (NEK9) protein kinases, and engineered mutations in MAP/microtubule affinity-regulating kinase 1 (MARK1) kinase-associated-1 (KA1) domain [66]. Mutations that relieve autoinhibition are allosteric drivers, whether they are frequent or statistically rare [86]. Below, we go deeper into PI3Kα with recent data, which now also include double/multiple mutations, Raf, and phosphatase and tensin homolog (PTEN) phosphatase for comparison and discuss Ras non-allosteric mutations.
PI3Kα lipid kinase
PI3Kα is a lipid kinase. It is a key component in the PI3K/AKT/mTOR signaling pathway, phosphorylating signaling lipid phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-trisphosphate (PIP3) at the membrane [115,116]. Phosphorylation events play a key role in PI3Kα autoinhibition and activation [117]. PI3Kα has two domains, p85α and p110α. Single, double, and multiple cancer driver mutations have been identified in PI3Kα (Figure 1). They can be strong, functionally weak, or relatively rare. When co-occurring they can collaborate to more potently transform cells [118–120]. Strong drivers include E542K, E545K in the p110α helical domain and H1047R in the kinase domain. E726K and M1043V/I are weak drivers in the kinase domain. The other weak mutations are N345K, C420R, E453K/Q in the C2 domain and R38H/C, R88Q, R93Q, R108H, and G118D in the adaptor binding domain (ABD). Their actions can be additive or cooperative. The driver mutations activate PI3Kα by relieving the autoinhibition exerted by the nSH2 domain of the p85α subunit, which covers the active site and blocks access to membrane, and by positioning the kinase domain appropriately at the membrane. Under physiological conditions, the favorable interaction of the nSH2 with a phosphorylated tyrosine motif at the C-terminal of an insulin receptor, a receptor tyrosine kinase (RTK), promotes conformational changes that lead to exposure to the active site. The E542K and E545K driver mutations in p110α disrupt the interfacial salt bridges, relieving the autoinhibition and leading to a reorganization at the active site. In the absence of an incoming physiological signal from the RTK and Ras activation, the H1047R strong driver promotes the interaction with the membrane. The weak drivers (e.g., E453K/Q and E726K on the surface of the N-lobe, and M1043V/I in the interior of the C-lobe of the kinase domain) can couple with the driver hotspots [118,121]. They too help promote population shift toward the active state by enhancing the activation mechanisms, all of which mimic the physiological activation of PI3K, which involves two components: release of autoinhibition by the nSH2 domain and attaching favorably to the membrane. Some mutations (e.g., M1043V/I) enhance the population shift by stabilizing the hydrophobic core. For further details and the roles played by other driver mutations see [115].
Figure 1.
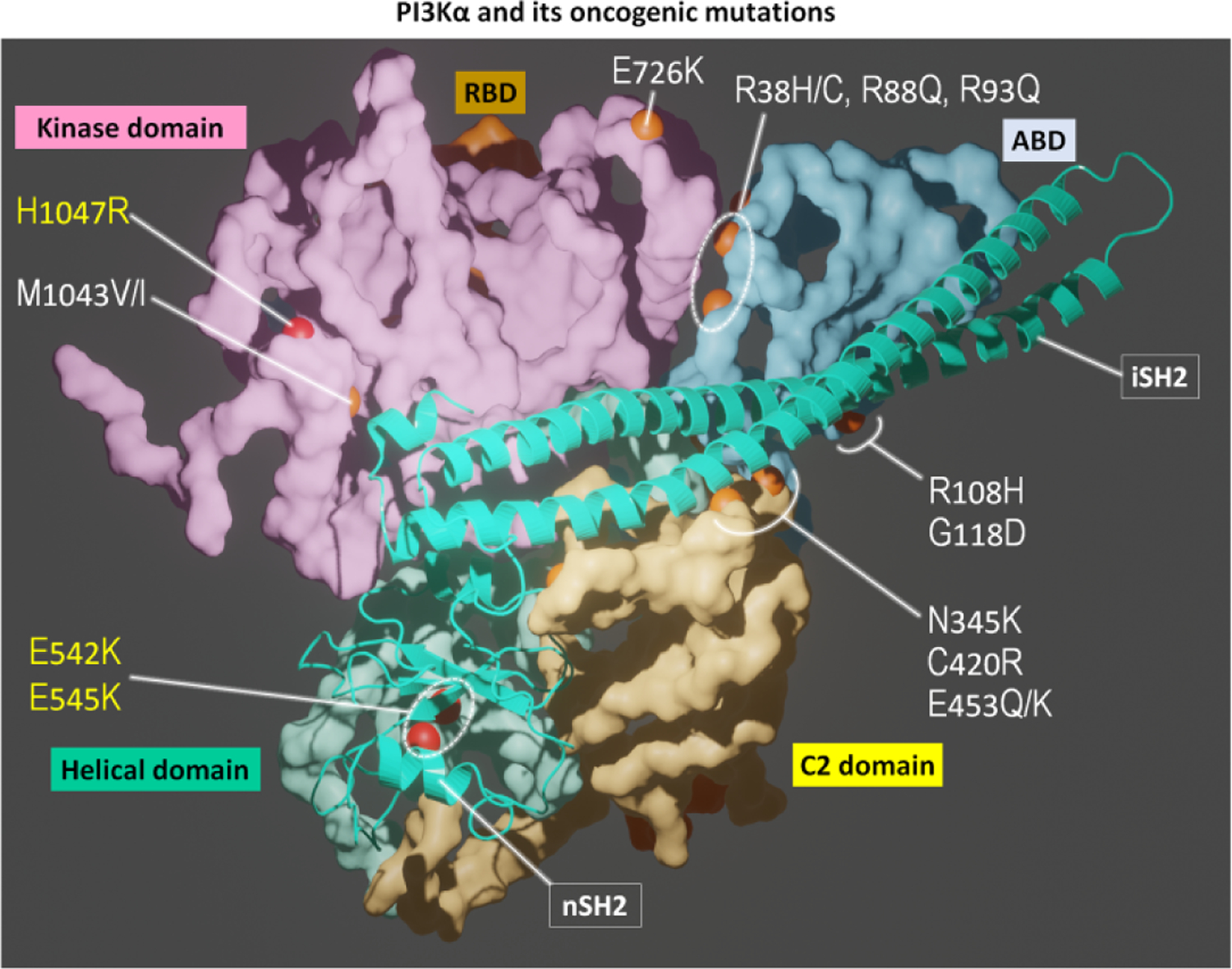
PI3Kα and its oncogenic mutations. PI3Kα is an obligate dimer with catalytic p110α (surface representation) and regulatory p85α (cartoon representation) subunits. p110α contains ABD (cyan), RBD (orange), C2 domain (yellow), helical domain (light green), and kinase domain (pink). The iSH2 and nSH2 domains in p85α interact with p110α subunit. PI3Kα contains hotspot mutations (H1047R in the kinase domain; E542K and E545K in the helical domain, yellow letter) and weak mutations (M1043V/I and E726K in the kinase domain; R38H/C, R88Q, R93Q, R108H, G118D in the ABD; N345K, C420R, E453Q/K in the C2 domain, white letter). These mutations can collaborate to transform cells more potently.
Raf kinase
Raf is a key protein kinase in the mitogen-activated protein kinase (MAPK) signaling cascade [122–126]. It consists of three conserved regions (CRs). CR1 contains the Ras binding domain (RBD), which binds activated Ras at the membrane [53,127,128], and the cysteine rich domain (CRD), which anchors at the membrane [129,130]. Wild-type active Raf is a dimer [131]. In the inactive autoinhibited state, it is a monomer [132–134]. Both RBD and CRD are involved in Raf’s autoinhibition by binding CR3, which results in occluding the dimerization surface of the kinase domain (Figure 2). Binding to Ras and the membrane exposes the catalytic kinase domain surface for dimerization and activation. CR2 has a flexible linker between CR1 and CR3 which contains a serine/threonine rich region. In the autoinhibited state, it is phosphorylated and binds the 14-3-3 protein. The binding stabilizes the ternary autoinhibited state [135–137]. CR3 is the kinase domain, which consists of two lobes connected by a hinge. When activated, it phosphorylates mitogen-activated protein kinase kinase 1/2 (MEK1/2), a key action in MAPK signal propagation. The activation loop (A-loop) runs from the conserved DFG motif to the APE motif in the C-terminal lobe. Phosphorylated Thr599 and Ser602 in the A-loop [138–144] of active, dimeric B-Raf destabilize the inactive and stabilize the active state. The switching from the inactive to the active state involves allosteric structural changes [145].
Figure 2.
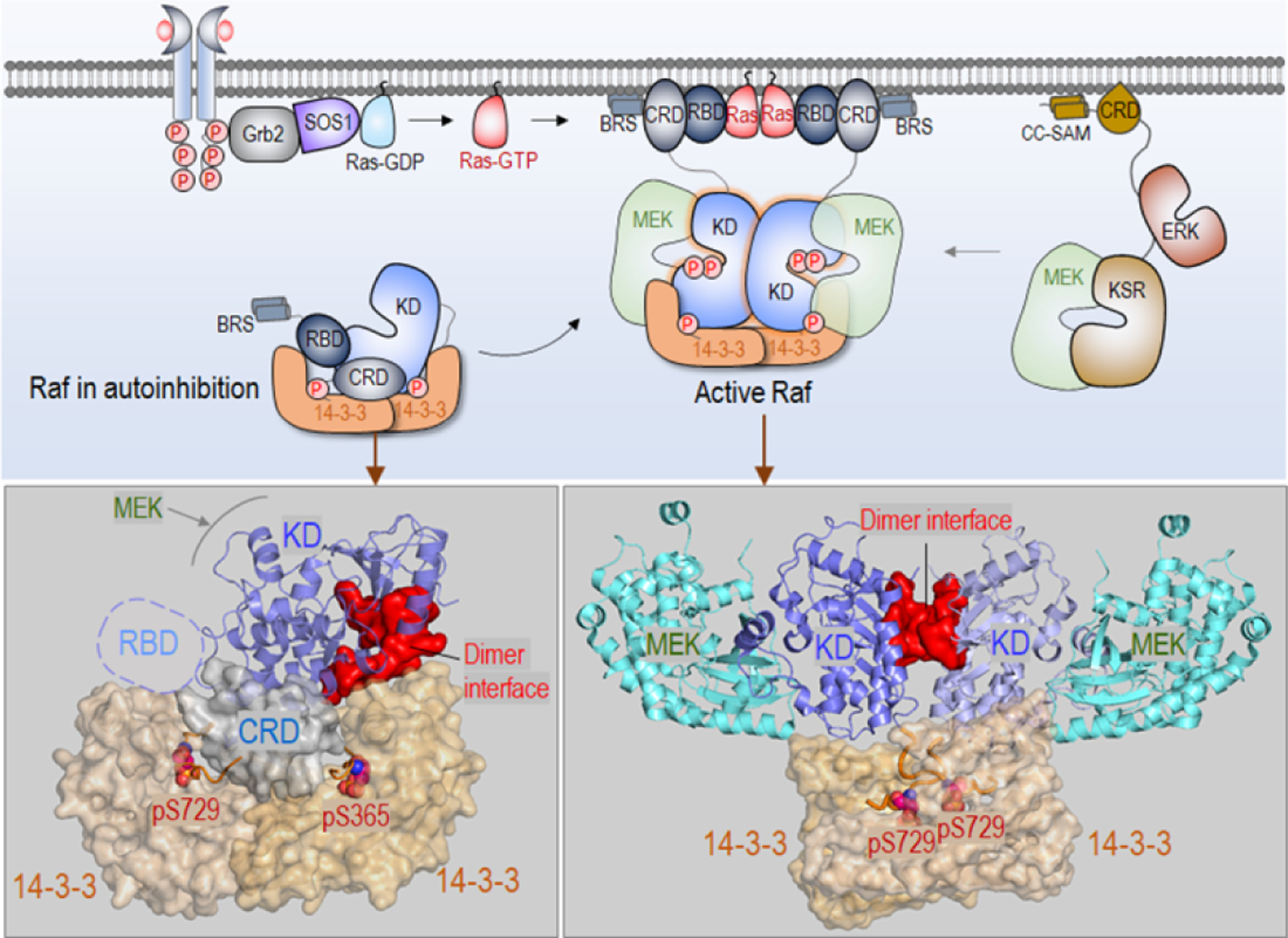
Autoinhibited and activated Raf in the Ras/Raf/MEK/ERK pathway. Here, examples are shown for B-Raf (top panel). In the cytosol, Raf monomer is autoinhibited via the interaction with 14-3-3 proteins. Activated Ras by Son of sevenless 1 (SOS1) recruits Raf to the membrane, releasing autoinhibition. Raf is activated through side-by-side dimerization of the kinase domain. Active Raf dimer phosphorylates and activates MEK, and subsequently phosphorylates and activates ERK, leading to cell proliferation. Kinase suppressor of Ras (KSR) can act as a scaffolding protein, promoting the signaling. In Raf, KD denotes the kinase domain and BRS domain denotes B-Raf specific domain. In KSR, CC-SAM denotes coiled coil sterile α motif. The crystal structure of autoinhibited B-Raf interacting with 14-3-3 proteins (PDB: 6NYB) (bottom left). Two phosphorylated sites, pS729 and pS365, have strong interactions with 14-3-3 proteins. The dimer interface of the kinase domain (red surface) is blocked by 14–3-3 protein. The crystal structure of active Raf dimer in complex with 14-3-3 proteins (PDB ID: 6Q0J) (bottom right). pS729 interacts with 14-3-3 protein, and the MEK proteins are loaded to the kinase domain for phosphorylation.
V600E, the most frequent cancer mutation in B-Raf, is a potent driver mutation [146,147]. MD simulations provided mechanistic details as to how it allosterically shifts B-Raf from the inactive to the active state by mimicking the mechanism of activation of the wild-type [120,123]. The A-loop is extended, and when the DFG motif orients such that Phe595 rotates away from the αC-helix, the helix moves inward (Figure 3). This allows formation of a salt bridge between Lys483 and Glu501. The mutation further acts by destabilizing the inactive state. It breaks the hydrophobic interactions and stabilizes the active state a salt bridge between Glu600 and Lys507.
Figure 3.
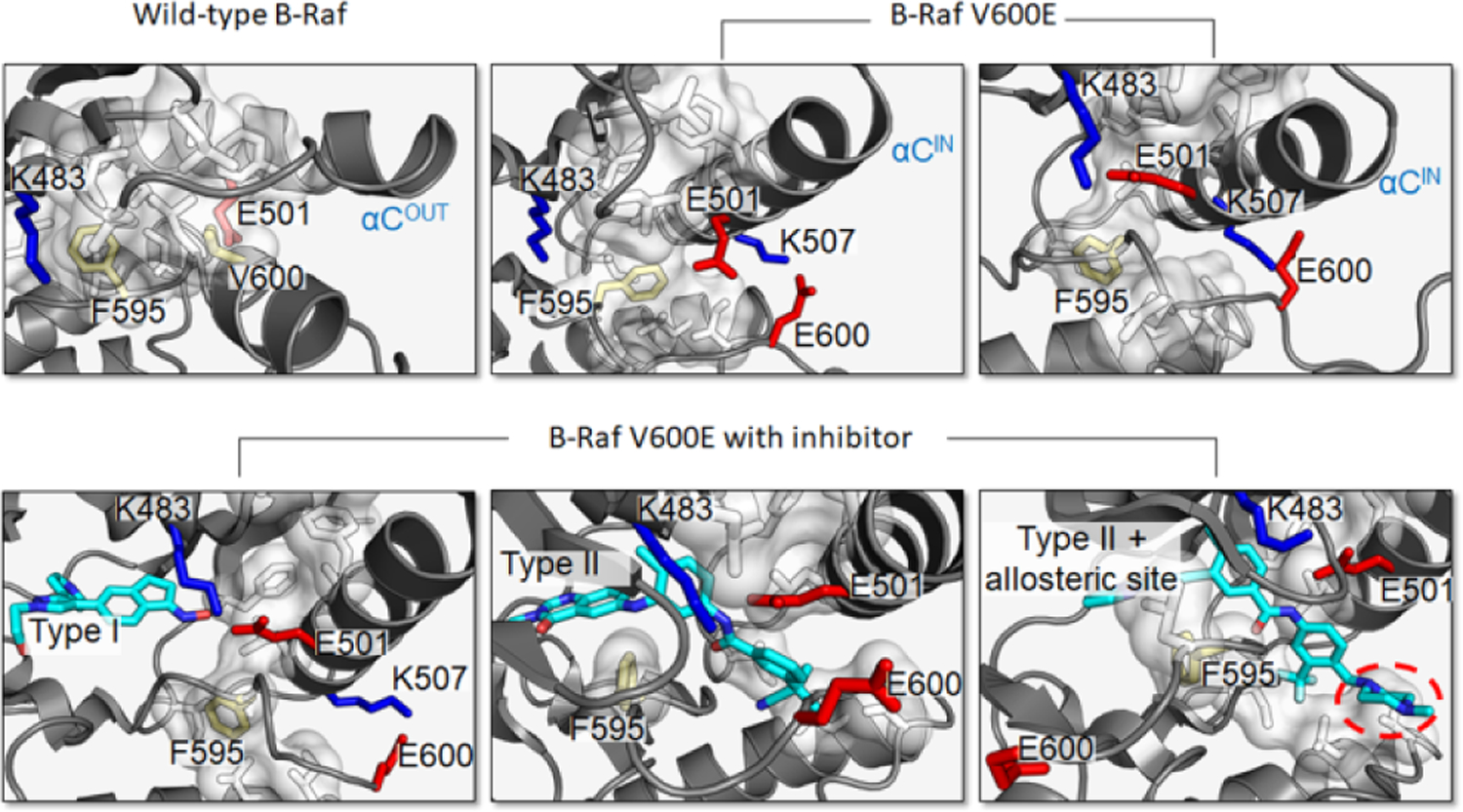
B-Raf V600E destabilizes the inactive state and stabilizes the active state. Inactive wild-type B-Raf contains a large hydrophobic pocket (white) that includes Phe595 and Val600 (yellow), which prevents the formation of the Lys483–Glu501 salt bridge and maintains an outward αC-helix (top left). The V600E mutation disrupts the hydrophobic pocket and causes the activation loop of B-Raf to extend and forms the Lys507–GluE600 salt bridge. However, in a low population state, Phe595 can orient to prevent the Lys483–Glu501 salt bridge formation, which keeps the αC-helix from moving fully inward (top middle). B-Raf V600E with proper orientation of Phe595 to allow both Lys507–GluE600 and Lys483–Glu501 salt bridge formation, stabilizing the active state (top right). B-Raf V600E maintains an active configuration in the presence of Type I inhibitor GDC0879 (bottom left). Type II inhibitor derived from diarylthiazole allosterically inhibits B-Raf V600E by stabilizing the “DFG out” orientation (bottom middle). Type II inhibitor Ponatinib extends to an allosteric site (red circle), stabilizing the “DFG out” orientation and displacing the activation loop and E600 from its typical conformation (bottom right). Inhibitors are colored cyan. Cartoons depict the crystal structures of B-Raf V600E with inhibitor (PDB: 4MNF, 4CQE, 6P3D).
PTEN tumor suppressor
PTEN is a tumor suppressor phosphatase [117]. It dephosphorylates signaling lipid PIP3 at the membrane to PIP2, opposing PI3K phosphorylation of PIP2, thus suspending signaling. Its activation and catalytic reaction have been described [148]. PTEN consists of the PIP2-binding motif (PBM, residues 1–15), the phosphatase domain (residues 16–185), the C2 domain which anchors at the membrane (residues 190–350), the C-terminal tail (CTT), and the PDZ binding motif (Figure 4). In the autoinhibited state, the phosphorylated residues in CTT interact with the arginine loop in the phosphatase domain, and the CBR3 and Cα2 loops in the C2 domain. Being a tumor suppressor, its driver mutations reduce the vital membrane interactions (S10N, K13E, G20E, L42R, and F90S) [149], obstruct the catalysis (R130Q/G), obstruct the essential phosphatase/C2 interface (S170N/G/I/R and R173C/H/L) [150], and relieve the autoinhibition. Its pathway is among the most highly mutated in cancer, particularly when involving mutations in PTEN, and to lesser extent, mutations in PI3Kα and AKT1 [151]. PTEN deletion mutations are also common in cancer [152] as are S170N/G/I/R and R173C/H/L [153]. Some of the mutations are allosteric and our on-going work aims to reveal their detailed mechanisms. Notably, some of PTEN’s mutations have been observed to promote autism spectrum disorder (ASD) [154]. Neurodevelopmental disorders have also been associated with mutations in PI3K, Raf, Ras, and more [155,156]. Recently, we resolved the tantalizing question of how to understand same gene, and even same mutations, promoting both cancer and neurodevelopmental disorder [157].
Figure 4.
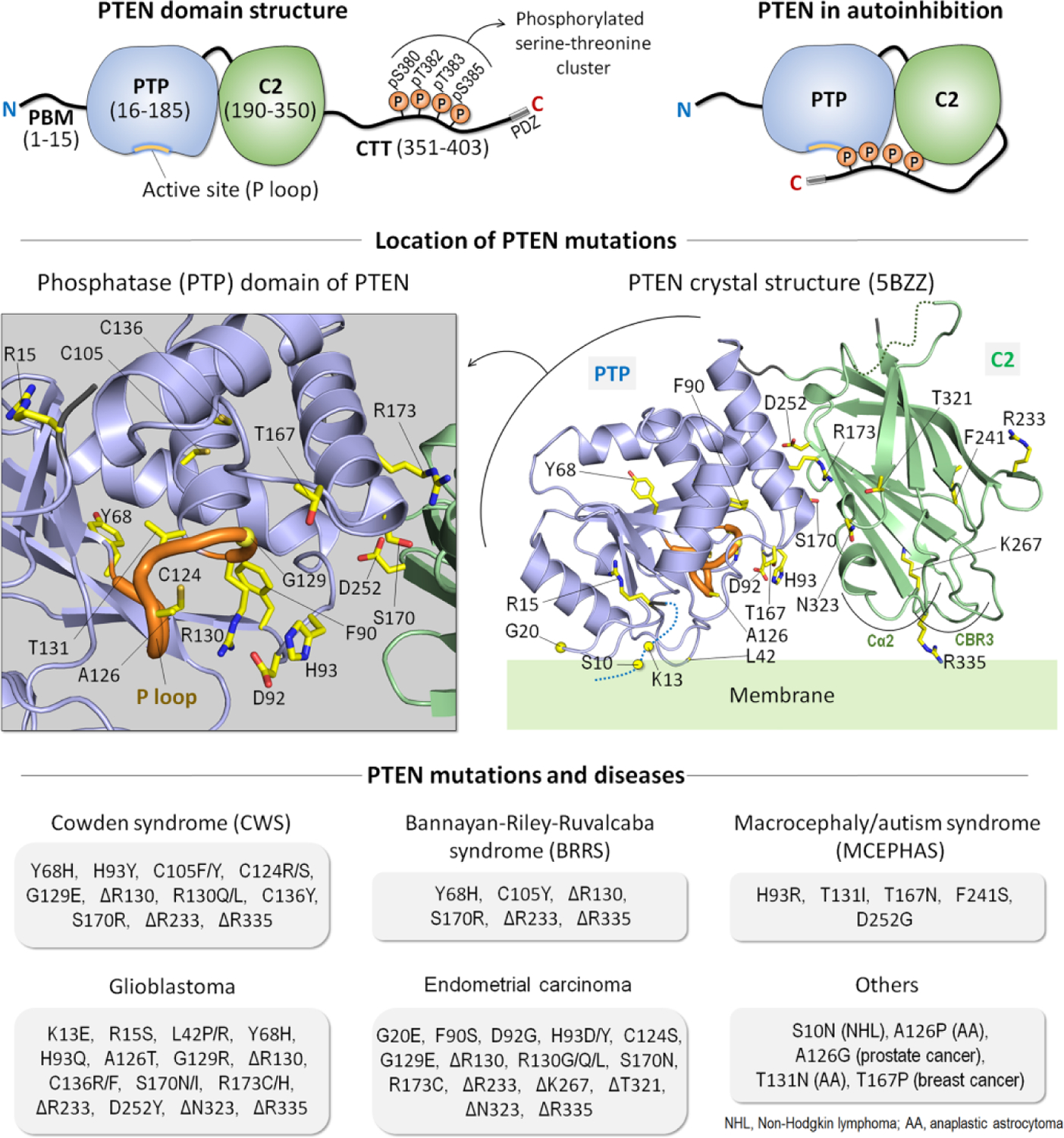
PTEN structure and mutations. Domain structure of phosphatase and tensin homolog (PTEN) (top left). It contains the N-terminal PIP2-binding motif (PBM), the phosphatase (PTP) domain, the C2 domain, and the C-terminal tail (CTT). The three residues, 401TKV403, at the C-terminus serve as the PDZ binding motif at the membrane. In the cytosol, PTEN is autoinhibited by its CTT with phosphorylation on a serine-threonine cluster (pS380, pT382, pT383, and pS385) yielding a closed conformation (top right). Dephosphorylation on the CTT relieves the autoinhibition, promoting membrane localization. The locations of mutations mapped on the crystal structure of PTEN (PDB: 5BZZ) (middle right) and a highlight of the active site (P loop) in the PTP domain (middle left). Types of the mutations and their associated deceases are summarized (bottom). These include germline mutations of the neurodevelopmental disorders (e.g., Cowden syndrome (CWS), Bannayan-Riley-Ruvalcaba syndrome (BRRS), and macrocephaly/autism syndrome (MCEPHAS)) and somatic mutations of the tumors (e.g., glioblastoma, endometrial carcinoma, prostate cancer, breast cancer, non-Hodgkin lymphoma, and anaplastic astrocytoma).
Not all driver mutations are allosteric: The Ras example.
Wild-type Ras regulates cell growth and division [158–165]. It binds multiple effectors and signals through multiple pathways. Among these, MAPK and PI3K/AKT/mTOR stand out as the major, and most consequential in cell life, including development, and disease. Above we have already discussed its two main effectors, PI3K and B-Raf. These, along with Ras activators, regulators and its other effectors, and signaling cascades (e.g., PTEN), including events such as mutations, have been studied and reviewed in multiple publications (e.g., [54,128,130,136,137,166–189]). The MAPK signal enters the G1 phase of the cell cycle, where cyclin-dependent kinases promote passage to the S (synthesis) phase. Together with the Ras/PI3K/AKT/mTOR pathway signal [190,191], they promote cell growth and division, accelerating proliferation [192–194]. Ras inactivation is mediated by GTPase-activating proteins (GAPs) that hydrolyze the GTP to GDP [195]. Driver mutations such as those at G12 hinder GAP-assisted hydrolysis, keeping Ras in the active GTP-bound state [196–199]. Weaker drivers enhance the exchange of GDP by GTP, e.g., A146T in KRas4B [200] as well as influence the intrinsic hydrolysis. In the steady state of the cell, about seventy five percent of KRasG12C is GTP bound, reflecting its high, millimolar-range concentration and picomolar affinity. G12C shows relatively higher rate of intrinsic hydrolysis as compared to other mutations such as G12D, G12V, G13D, and Q61H [201].
Different than kinases, in Ras activating mutations work by blocking deactivation, not by stabilizing the active and (or) destabilizing the inactive state; that is, not by shifting the equilibrium which results in increasing the number of the active molecules [158]. In contrast, kinases switch from the inactive αC-helix-out to the active αC-helix-in, movements that involve rotation and shift. In epidermal growth factor receptor (EGFR), the driver L858R mutation in the A-loop destabilizes the inactive αC-helix-out conformation with Arg breaking the hydrophobic interactions in the αC-helix-out, and the driver T790M mutation stabilizes the active αC-helix-in conformation by stabilizing the hydrophobic R-spine. T315I in Bcr-Abl, T334I in c-Abl, T341I in Src, T670I in Kit, and T674I in platelet-derived growth factor receptor α (PDGFRα), all also stabilize the hydrophobic R-spine [33].
Allosteric drugs: Bcr-Abl, allosteric molecular glues and allosteric PROTACs
Targeting Bcr-Abl
Allosteric drugs have been extensively reviewed by us and others (e.g., [12,31,33,34,202–210]). Below following a brief overview, we touch on some recent examples focusing on Bcr-Abl, a fusion of BCR and ABL genes present in most patients with different phenotypes of leukemia, including acute lymphoblastic leukemia (ALL), chronic myeloid leukemia (CML), and neutrophilic-chronic myeloid leukemia (CML-N) (Figure 5). This fusion results from the reciprocal translocation of chromosomes 9 and 22. CML has been treated by tyrosine kinase inhibitors (TKIs), especially imatinib that binds to Abl inactive state [211]. Drug resistance eventually emerges, either by mutations at the active site or elsewhere in the kinase domain, allosterically altering the active site. Overcoming such mutational events requires a drug with higher affinity than the substrate or cofactor, which can be challenging to achieve. In the quintessential kinases example, this requires higher affinity than the low micromolar ATP [212]. It also requires sufficiently high dosage. High dosage leads to binding to related kinases with conserved active sites, thus toxic side-effects. Allosteric drugs bind at sites other than the active, or functional site, which are not conserved. Their resulting high specificity has low chance of side effects. Covalent allosteric drugs combine the pharmacological merits of covalent drugs and the high specificity of allosteric drugs [202,212]. Cooperativity between the orthosteric and allosteric ligand binding sites has been proposed [6] and recently observed for RAR-related orphan receptor γ isoform 2 (RORγt, a.k.a. RORγ2) [213] and discussed further in a number of works (e.g., [214] and references therein). How to quantify design parameters [18,215,216] was also overviewed.
Figure 5.
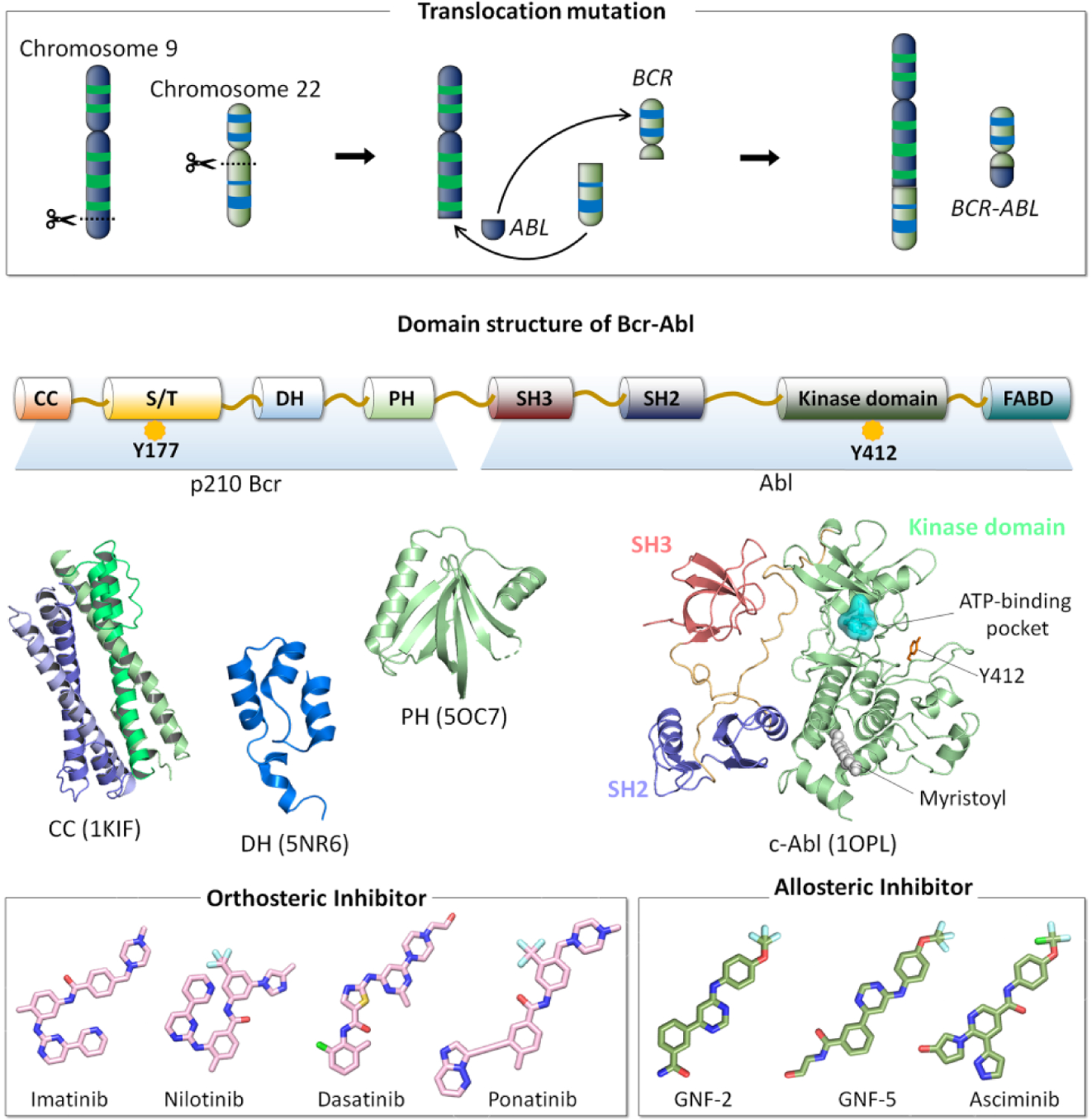
Chromosome 9 consists of the ABL gene and chromosome 22 contains the BCR gene. In BCR-ABL-induced leukemia oncogenesis, the translocation mutation between chromosome 9 and chromosome 22 leads to the generation of the fusion BCR-ABL gene, which encodes the Bcr-Abl oncoprotein. p210 Bcr-Abl protein is the hallmark of chronic myeloid leukemia (CML). p210 Bcr region possesses the coiled coil (CC) domain, the serine/threonine (S/T) domain, the Dbl homology (DH) domain, and the pleckstrin homology (PH) domain. Phosphorylation of Tyr177 in the S/T domain enables to Bcr-Abl recruitment of Grb2 through its SH2 interaction, which activate Ras/MAPK pathway. Abl region contains the SH3 domain, the SH2 domain, the kinase domain, and the FABD (F-actin-binding domain). Phosphorylation of Tyr412 (numbered by Abl 1b isoform) in the activation loop activates Abl kinase. In Abl kinase domain, orthosteric inhibitors (e.g., imatinib, nilotinib, dasatinib, and ponatinib) bind to the ATP-binding pocket in the N-lobe of the kinase domain, while allosteric inhibitors (e.g., GNF-2, GNF-5, and asciminib) prefer to occupy the myristoyl-binding pocket in the C-lobe of the kinase domain.
A recent study reported that multiple patient-derived imatinib-resistant Abl kinase domain mutants still bound imatinib [217]. Kinetic analyses suggested that the allosteric drug resistance mutations resulted in considerably faster drug dissociation from the mutant as compared to the wild type. These observations can be explained by considering that the difference in energy between the active and inactive states of the kinase is small. The conformational changes promoted by the allosteric oncogenic mutations shift the free energy landscape from the inactive kinase to populate the now more stable active state, which is not imatinib binding competent. These changes destabilize imatinib binding [218–220], lowering its affinity which is outcompeted by the high affinity of ATP. Gene duplication, a frequent event in cancer, will increase Bcr-Abl expression, which will aggravate the plight. On a related note, resistance may also be augmented by upregulation of a redundant kinase [217,219,221–228].
In kinases, the A-loop, the DFG motif, the regulatory spine, and the gatekeeper residue are all key elements in identification of the active/inactive states. Recently two inactive states have been detected by NMR for the Abl kinase domain [218]. The studies revealed that the kinase domain interconverts between one active, populated 90% of the time, and two discrete inactive states, each transiently populated 5% of the time. One of these is imatinib binding competent. Resistance mutations shift the ensemble toward the active state facilitated by the small difference in energy. As to the two inactive states, in principle both can be used for drug design if new features are captured in the second. Additional Abl allosteric strategies have been discussed as well (e.g., see [223,229]).
Innovative allosteric drugs: from heterobifunctional PROTACs to molecular glues
Allosteric drugs have recently taken an innovative turn [230,231] through the powerful concept of molecular glues [232] and heterobifunctional PROteolysis TArgeting Chimeras (PROTACs) [233]. Molecular glues are small molecules that bind at the interface between two proteins and induce their interactions [234]. Induction of proximity is a groundbreaking concept in drug discovery, with molecular glues promising to broaden the therapeutic landscape. Their potential has been validated by natural products (e.g., rapamycin, tacrolimus (FK506), and sanglifehrin A), synthetic small molecules (e.g., IMiD, immunomodulatory imide drug) including thalidomide and its analogues, lenalidomide and pomalidomide), and anticancer sulfonamides (e.g., tasisulam, indisulam, and chloroquinoxaline sulfonamide (CQS)) (Figure 6). Uncompetitive molecular glues acting as active state stabilizers were shown to expand protein-protein modulation drugs for peptide hormone receptors [235]. Interfacial binding of the IMiD ligands induces a protein-protein interaction e.g., between cereblon (CRBN) and the target proteins [236]. The ‘molecular glue’ connotation was attributed not only to heteromeric protein-protein interactions but also to homomeric sigma-1 receptor (Sig1R) interactions to explain how allosteric modulators could increase the number of Sig1R in the agonist state conformation, thus activity [237]. Molecular glue compounds can stabilize weak protein interactions as shown in the case of the Cdc34A (a.k.a. UBE2R1, ubiquitin-conjugating enzyme E2 R1) where molecular glue compounds inhibit a noncovalent E2 enzyme–ubiquitin complex [238], and promote cyclin-dependent kinase 12 (CDK12)–DDB1 (CUL4 adaptor protein) interaction to trigger cyclin K degradation [239].
Figure 6.
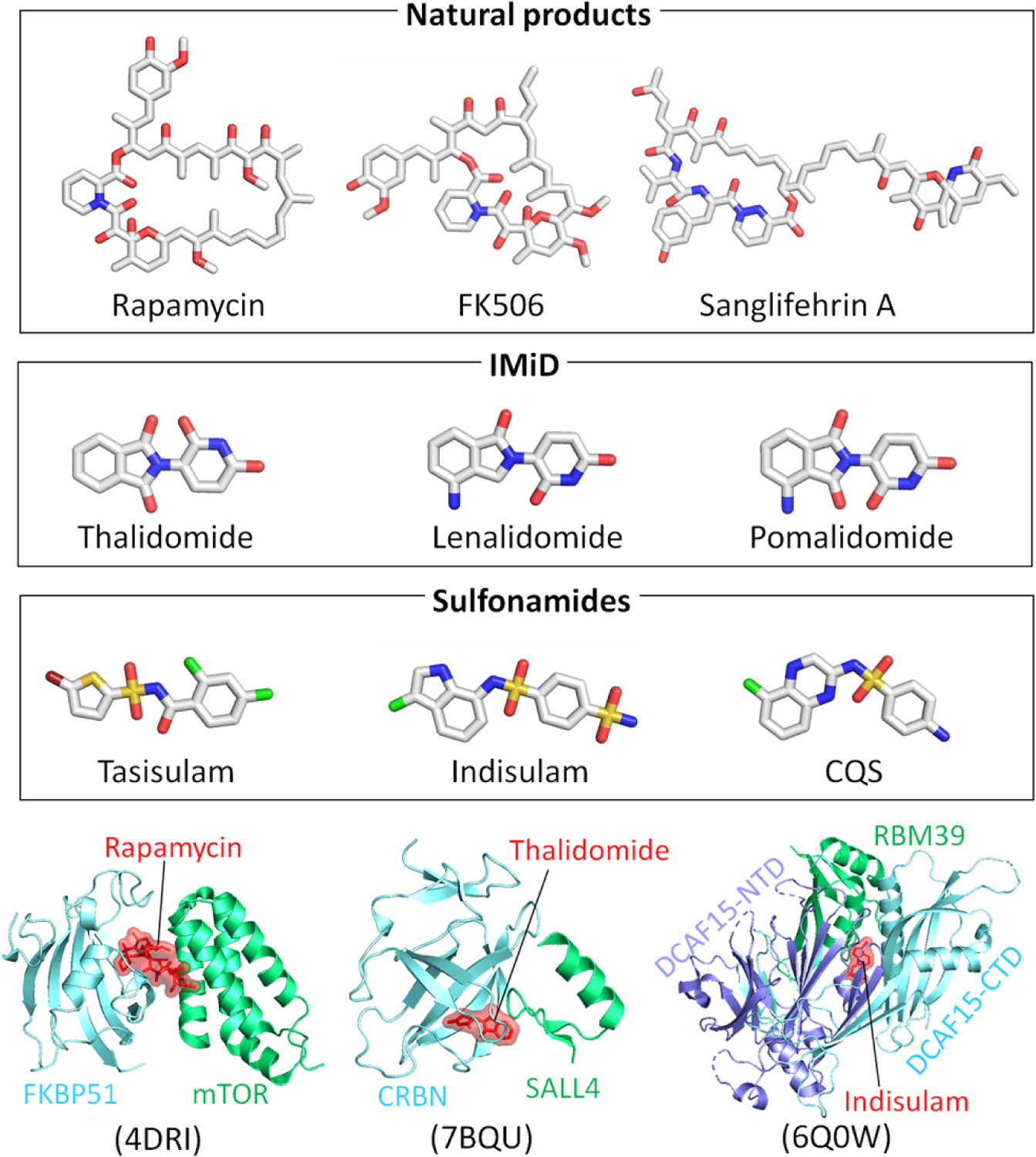
Structures of different types of molecular glues, including natural products (rapamycin, FK506, and sanglifehrin A), IMiD (thalidomide, lenalidomide, and pomalidomide), and sulfonamides (tasisulam, indisulam, and CQS). Examples of the molecular glues in the intermolecular interface of protein complexes for rapamycin (PDB: 4DRI), thalidomide (PDB: 7BQU), and indisulam (PDB: 6Q0W).
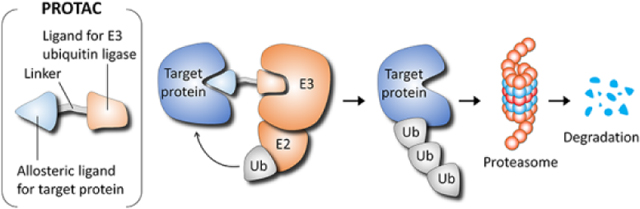
The PROTACs do not inhibit proteins [230,240–242]. They destroy them. While conceptually resembling molecular glues [243], PROTACS also differ. Unlike the molecular glues which consist of single small molecule, a PROTAC is a linkage of two. PROTACs are heterobifunctional degraders that enter cells and reduce their targeted proteins through the ubiquitination system. Allosteric PROTAC consists of a molecule (the warhead) that recruits an E3 ligase. The molecule is also linked to a high affinity allosteric inhibitor of the target protein. The inhibitor and the warhead molecules are covalently joined by a chemically suitable linker such that the target protein and the ubiquitination complex are brought into proximity. The IMiD molecular glue ligands above provide a good example [236], with the drug property downside of the construct. PROTACs have been exploited in mediated ternary complex formation (e.g., [244–247]) and their cooperativity have been delineated as well [248,249]. The challenge in designing them is in tuning the affinity and cooperativity of binding at both ends [248]. Especially challenging is the linker [250]. Its length and flexibility, essential to induce a ternary complex with the two proteins at the appropriate distance and orientation, require an optimal conformational bias. Differential allosteric PROTAC substrate specificity has been shown to be dictated by orientation of recruited E3 ligase [251], and the impact of linker length on the activity of PROTACs has also been investigated [252,253]. A PROTAC database has also been developed [254]. A list of selected degraders in, and approaching, the clinic has been compiled [233].
Recently, the first allosteric PROTACs were constructed. Abl1 contains an allosteric myristoyl binding site [255]. Multiple allosteric drugs have been designed to bind in this pocket [256,257]. The PROTACs were designed to covalently link to the allosteric inhibitor (GNF-5, an analog of GNF-2 [256]) (Figure 7) to degrade the Bcr-Abl1 mutant protein (with the T315I mutation). Since GNF-5 acts together with competitive inhibitors, such as imatinib, the allosteric PROTACs can collaborate with the competitive inhibitors trampling the oncogenic recalcitrant Bcr-Abl mutant, permitting a reduced drug dose and lesser side effects. GNF-5–PROTAC also acts on Bcr-Abl1 lacking mutations, permitting broad therapeutic stem cell applications [258].
Figure 7.
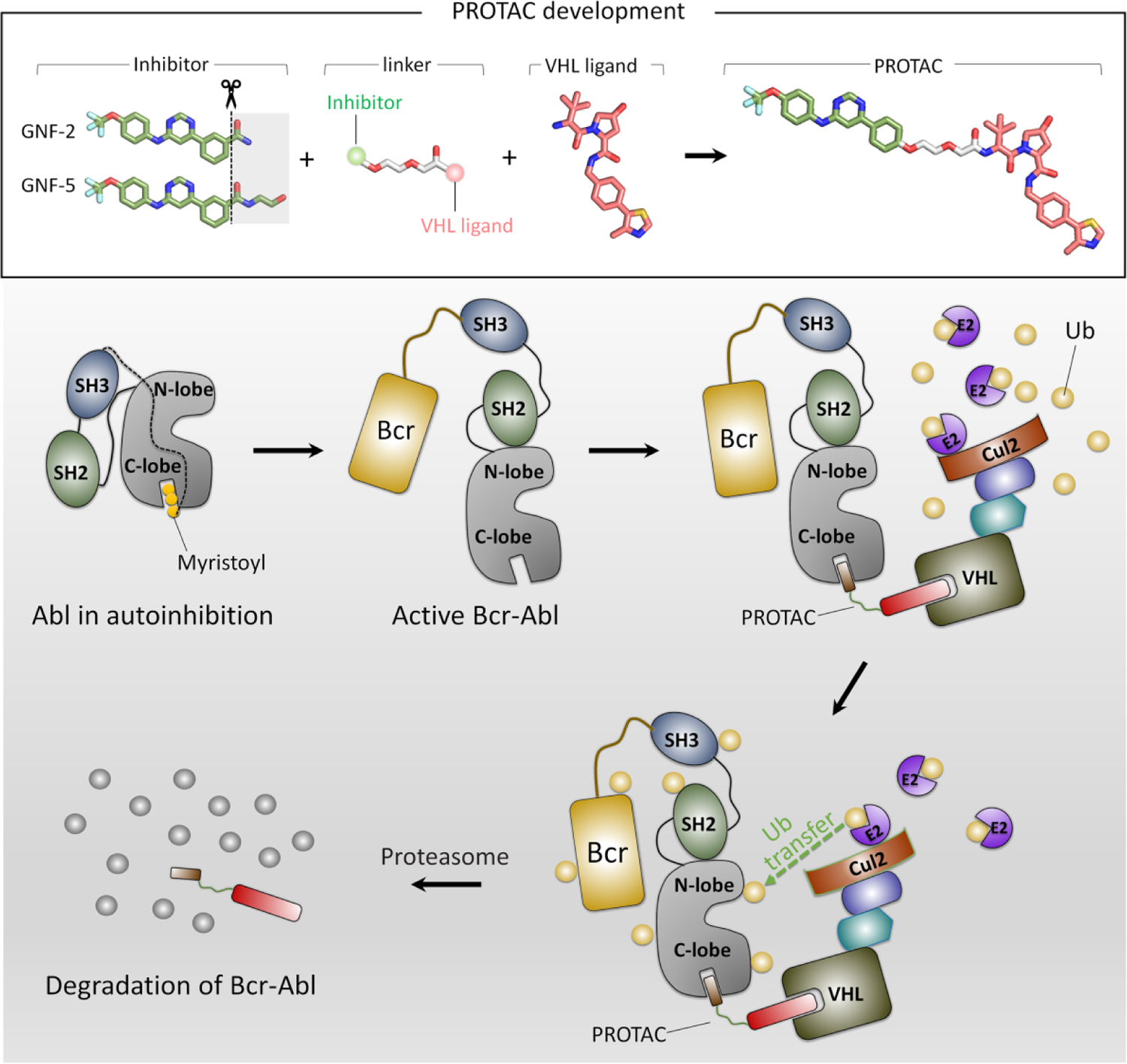
GNF-5-PROTAC is formed by a GNF-5 inhibitor and a warhead molecule which are connected by a linker. Release of the myristoyl group from the C-lobe of Abl kinase domain activates Abl protein, leading to a vacant myristoyl-binding pocket. GNF-5, an allosteric inhibitor, can bind to the myristoyl-binding pocket of Abl. Of the heterobifunctional GNF-5-PROTAC molecule, GNF5 is responsible for the binding of Abl, while the other end recruits E3 ligase. This forms a Abl/GNF-5-PROTAC/E3 ternary complex. Then, this complex is brought into the ubiquitin system. The E3 ligase binds to E2 enzyme, regulating the transfer of ubiquitin protein to Abl from E2 enzyme. The Abl protein is degraded by proteasome accompanied with the dissociation of Abl/GNF-5-PROTAC/E3 ternary complex.
Bifunctional molecules developed for targeted protein degradation through ubiquitination [243] are new potential allosteric drugs. In principle, this technology can bring any two proteins together through small molecules that target each partner. Details, outlines, and examples of the development of small molecule degraders that work by stabilizing and increasing the affinity of the ternary complexes and cases that have been shown to work have recently been reviewed in-depth, including an induced cooperativity in the catalytic degradation profiles [259]. The potential and innovation of such applications is only beginning to be apparent. One recent example has been discussed [238]. In another, G protein-coupled receptor (GPCR) example [235], it has been employed to promote the association of protein complexes, an emerging therapeutic strategy. The authors discovered a GPCR ligand that stabilizes an active state conformation by cooperatively binding both the receptor and orthosteric ligand. Drug resistance could arise through mutations in the degradation pathway, suggesting combination strategies to slow it. Additional innovative allosteric drug approaches have also been reviewed (e.g., [18,260]).
In another innovative and promising feat [261], a chemical biology approach developed a bitopic ligand for the RORγt nuclear receptor (NR), which concomitantly exploited two binding pockets. Three candidates were obtained, yielding an orthosteric and allosteric RORγt pharmacophore covalently linked via a polyethylene glycol (PEG) linker. Covalent occlusion of the RORγt ligand binding pocket was further shown to permit targeting of an allosteric site [262]. Here too, the linker’s length influences the RORγt binding mode. Bitopic ligands can powerfully improve the affinity and (or) selectivity profiles [263]. Apart from identification of the allosteric pocket, the challenge is in the properties of the linker. Currently, linkers are mostly synthetic polymers such as polyethylene glycols, making an engineered conformational bias a formidable task.
Signal transduction
Molecular events, such as activation, are governed by the conformational behavior of the protein [1,2,6,8,10,32,92,264–276]. To be effective, the allosteric signal – physiologic or oncogenic – needs to propagate downstream. For that it needs to be sufficiently strong but not too strong. We dubbed productive signal transduction “signaling by-the-numbers”. Signaling by the numbers does not imply a stronger functional effect [277], but rather, as setting the threshold for the signal for propagating downstream through the pathway to the cell cycle to activate (repress) transcription, or exit via physiologic senescence, premature developmental senescence, or oncogene induced senescence (OIS). Activity of a single protein (node) in the pathway cannot serve as the threshold for passing from one node to the other. To calculate the threshold, three quantities are needed: the expression level of the protein, how much is located (recruited) to the right place, and the population activated by the upstream signaling node. Cell type [278], cell state [279], timing window [280–283] in cell development, and chromatin modeling, all play a role [284–289].
Conclusions: future of studies of allostery
Where will studies of allostery go? If we can venture to predict, we see them focusing on translation, including the mechanisms of activating allosteric mutations, identification of druggable allosteric sites, and innovative and productive allosteric drugs. The advantages of allosteric drugs are well-established, especially, the higher specificity which is coupled with reduced side effects. Besides increasing the drug repertoire, efforts will focus on higher affinity, and potency. Exploiting the already available repertoire, which has already undergone clinical trials and is in use, will save development time and cost. Covalent linkage of such drugs via appropriate linkers to warheads that recruit degraders, as in the case of Bcr-Abl1’s PROTAC, or to orthosteric drugs, can accomplish such a goal. In a way, this is analogous to repurposing drugs that are already in the clinic with documented safety profiles. The challenge is in the construction of linkers (or spacers) with appropriate length and conformational bias such that the orthosteric and allosteric pharmacophores yield bitopic ligands that achieve improved affinity and selectivity. The search is also on for allosteric drugs at newly discovered pockets or covalently linked to residues other than Cys, such as Tyr [290]. Innovative allosteric drugs may also be engineered to mimic allosteric rescue mutations [194].
Over 55 years have elapsed since Monod, Wyman, and Changeux have proposed the transformational two-state concerted model (MWC model) [291]. In the late 1990’s, we proposed the conformational selection and population shift versus induced fit model to explain how biological functions are achieved through allostery [6,55,292]. Going forward, the challenge is in identifying rare allosteric activating mutations, their mechanisms, and innovative allosteric pharmacology.
In addition, we foresee conceiving ways of defining and measuring signal transduction, initiated by allosteric events, and propagating downstream, a formidable but achievable aim [65]. As we emphasized recently in “allostery, and how to define and measure signal transduction”, such measurements are vastly important in setting thresholds for ‘actionable’ signals, which would permit assessing and predicting oncogenic signaling in tumor development and drug resistance. On their own, activating mutations may or may not influence the cell cycle and the level of expression. We have been trying to contribute our share toward these aims [65,157].
We overview the principles of allosteric activating mutations and allosteric drugs
Examples of activating mutations include the Ras signaling network and Abl kinase
We overview innovative allosteric drug concepts, underscoring the challenge
The review links allostery on the molecular level and productive cell signaling
From the cellular standpoint, we propose a signaling by-the-numbers lens
Acknowledgements
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261201500003I. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This Research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Abbreviations
- MD
molecular dynamics
- NMR
nuclear magnetic resonance
- AKT
protein kinase B
- mTOR
mammalian target of rapamycin
- PI3K
phosphoinositide 3-kinase
- SHP2
SH2 domain-containing phosphatase 2
- NEK
NIMA-related kinase
- MARK
MAP/microtubule affinity-regulating kinase
- KA1
kinase-associated-1
- PTEN
phosphatase and tensin homolog
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PIP3
phosphatidylinositol 3,4,5-trisphosphate
- ABD
adaptor binding domain
- RTK
receptor tyrosine kinase
- MAPK
mitogen-activated protein kinase
- CR
conserved region
- RBD
Ras binding domain
- CRD
cysteine rich domain
- MEK
mitogen-activated protein kinase kinase
- A-loop
activation loop
- PBM
PIP2-binding motif
- CTT
C-terminal tail
- ASD
autism spectrum disorder
- GAP
GTPase-activating protein
- EGFR
epidermal growth factor receptor
- PDGFR
platelet-derived growth factor receptor
- ALL
acute lymphoblastic leukemia
- CML
chronic myeloid leukemia
- CML-N
neutrophilic-chronic myeloid leukemia
- TKI
tyrosine kinase inhibitor
- RORγ
RAR-related orphan receptor γ
- PROTAC
proteolysis targeting chimera
- IMiD
immunomodulatory imide drug
- CRBN
cereblon
- Sig1R
sigma-1 receptor
- CDK
cyclin-dependent kinase
- GPCR
G protein-coupled receptor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CRediT author statement
Ruth Nussinov: Conceptualization, Investigation, Writing - Original Draft, Supervision, Project administration. Mingzhen Zhang: Resources, Visualization. Ryan Maloney: Resources, Visualization. Yonglan Liu: Resources, Visualization. Chung-Jung Tsai: Resources, Visualization. Hyunbum Jang: Conceptualization, Investigation, Writing - Review & Editing, Supervision.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Nussinov R & Wolynes PG (2014). A second molecular biology revolution? The energy landscapes of biomolecular function. Phys Chem Chem Phys 16, 6321–2. [DOI] [PubMed] [Google Scholar]
- 2.Frauenfelder H, Sligar SG & Wolynes PG (1991). The energy landscapes and motions of proteins. Science 254, 1598–603. [DOI] [PubMed] [Google Scholar]
- 3.Tsai CJ & Nussinov R (2014). The free energy landscape in translational science: how can somatic mutations result in constitutive oncogenic activation? Phys Chem Chem Phys 16, 6332–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wei G, Xi W, Nussinov R & Ma B (2016). Protein Ensembles: How Does Nature Harness Thermodynamic Fluctuations for Life? The Diverse Functional Roles of Conformational Ensembles in the Cell. Chem Rev 116, 6516–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nussinov R, Tsai CJ & Jang H (2019). Dynamic Protein Allosteric Regulation and Disease. Adv Exp Med Biol 1163, 25–43. [DOI] [PubMed] [Google Scholar]
- 6.Tsai CJ & Nussinov R (2014). A unified view of “how allostery works”. PLoS Comput Biol 10, e1003394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.del Sol A, Tsai CJ, Ma B & Nussinov R (2009). The origin of allosteric functional modulation: multiple pre-existing pathways. Structure 17, 1042–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cui Q & Karplus M (2008). Allostery and cooperativity revisited. Protein Sci 17, 1295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Motlagh HN, Wrabl JO, Li J & Hilser VJ (2014). The ensemble nature of allostery. Nature 508, 331–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gunasekaran K, Ma B & Nussinov R (2004). Is allostery an intrinsic property of all dynamic proteins? Proteins 57, 433–43. [DOI] [PubMed] [Google Scholar]
- 11.Changeux JP & Christopoulos A (2016). Allosteric Modulation as a Unifying Mechanism for Receptor Function and Regulation. Cell 166, 1084–1102. [DOI] [PubMed] [Google Scholar]
- 12.Chatzigoulas A & Cournia Z (2021). Rational design of allosteric modulators: Challenges and successes. Wiley Interdisciplinary Reviews-Computational Molecular Science 11, e1529. [Google Scholar]
- 13.Kar G, Keskin O, Gursoy A & Nussinov R (2010). Allostery and population shift in drug discovery. Curr Opin Pharmacol 10, 715–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hilser VJ, Wrabl JO & Motlagh HN (2012). Structural and energetic basis of allostery. Annu Rev Biophys 41, 585–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Changeux JP (2012). Allostery and the Monod-Wyman-Changeux model after 50 years. Annu Rev Biophys 41, 103–33. [DOI] [PubMed] [Google Scholar]
- 16.Schann S, Mayer S, Franchet C, Frauli M, Steinberg E, Thomas M, Baron L & Neuville P (2010). Chemical switch of a metabotropic glutamate receptor 2 silent allosteric modulator into dual metabotropic glutamate receptor 2/3 negative/positive allosteric modulators. J Med Chem 53, 8775–9. [DOI] [PubMed] [Google Scholar]
- 17.Schwartz TW & Holst B (2007). Allosteric enhancers, allosteric agonists and agoallosteric modulators: where do they bind and how do they act? Trends Pharmacol Sci 28, 366–73. [DOI] [PubMed] [Google Scholar]
- 18.Wenthur CJ, Gentry PR, Mathews TP & Lindsley CW (2014). Drugs for allosteric sites on receptors. Annu Rev Pharmacol Toxicol 54, 165–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fang Z, Grutter C & Rauh D (2013). Strategies for the selective regulation of kinases with allosteric modulators: exploiting exclusive structural features. ACS Chem Biol 8, 58–70. [DOI] [PubMed] [Google Scholar]
- 20.Quaglia F, Lazar T, Hatos A, Tompa P, Piovesan D & Tosatto SCE (2021). Exploring Curated Conformational Ensembles of Intrinsically Disordered Proteins in the Protein Ensemble Database. Curr Protoc 1, e192. [DOI] [PubMed] [Google Scholar]
- 21.Bah A & Forman-Kay JD (2016). Modulation of Intrinsically Disordered Protein Function by Post-translational Modifications. J Biol Chem 291, 6696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jin F & Grater F (2021). How multisite phosphorylation impacts the conformations of intrinsically disordered proteins. PLoS Comput Biol 17, e1008939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nussinov R, Tsai CJ, Xin F & Radivojac P (2012). Allosteric post-translational modification codes. Trends Biochem Sci 37, 447–55. [DOI] [PubMed] [Google Scholar]
- 24.Stetz G, Tse A & Verkhivker GM (2018). Dissecting Structure-Encoded Determinants of Allosteric Cross-Talk between Post-Translational Modification Sites in the Hsp90 Chaperones. Sci Rep 8, 6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Venne AS, Kollipara L & Zahedi RP (2014). The next level of complexity: crosstalk of posttranslational modifications. Proteomics 14, 513–24. [DOI] [PubMed] [Google Scholar]
- 26.Lechtenberg BC, Gehring MP, Light TP, Horne CR, Matsumoto MW, Hristova K & Pasquale EB (2021). Regulation of the EphA2 receptor intracellular region by phosphomimetic negative charges in the kinase-SAM linker. Nat Commun 12, 7047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alaalm L, Crunden JL, Butcher M, Obst U, Whealy R, Williamson CE, O’Brien HE, Schaffitzel C, Ramage G, Spencer J & Diezmann S (2021). Identification and Phenotypic Characterization of Hsp90 Phosphorylation Sites That Modulate Virulence Traits in the Major Human Fungal Pathogen Candida albicans. Front Cell Infect Microbiol 11, 637836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sostaric N & van Noort V (2021). Molecular dynamics shows complex interplay and long-range effects of post-translational modifications in yeast protein interactions. PLoS Comput Biol 17, e1008988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang H, He J, Hu G, Zhu F, Jiang H, Gao J, Zhou H, Lin H, Wang Y, Chen K, Meng F, Hao M, Zhao K, Luo C & Liang Z (2021). Dynamics of Post-Translational Modification Inspires Drug Design in the Kinase Family. J Med Chem 64, 15111–15125. [DOI] [PubMed] [Google Scholar]
- 30.Liu HF & Liu R (2020). Structure-based prediction of post-translational modification cross-talk within proteins using complementary residue- and residue pair-based features. Brief Bioinform 21, 609–620. [DOI] [PubMed] [Google Scholar]
- 31.Nussinov R & Tsai CJ (2012). The different ways through which specificity works in orthosteric and allosteric drugs. Curr Pharm Des 18, 1311–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Byun JA, VanSchouwen B, Akimoto M & Melacini G (2020). Allosteric inhibition explained through conformational ensembles sampling distinct “mixed” states. Comput Struct Biotechnol J 18, 3803–3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nussinov R & Tsai CJ (2013). Allostery in disease and in drug discovery. Cell 153, 293–305. [DOI] [PubMed] [Google Scholar]
- 34.Wagner JR, Lee CT, Durrant JD, Malmstrom RD, Feher VA & Amaro RE (2016). Emerging Computational Methods for the Rational Discovery of Allosteric Drugs. Chem Rev 116, 6370–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang L, Li M & Liu Z (2018). A comprehensive ensemble model for comparing the allosteric effect of ordered and disordered proteins. PLoS Comput Biol 14, e1006393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gupta AK, Wang X, Pagba CV, Prakash P, Sarkar-Banerjee S, Putkey J & Gorfe AA (2019). Multi-target, ensemble-based virtual screening yields novel allosteric KRAS inhibitors at high success rate. Chem Biol Drug Des 94, 1441–1456. [DOI] [PubMed] [Google Scholar]
- 37.Biddle JW, Martinez-Corral R, Wong F & Gunawardena J (2021). Allosteric conformational ensembles have unlimited capacity for integrating information. Elife 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Acuner Ozbabacan SE, Gursoy A, Keskin O & Nussinov R (2010). Conformational ensembles, signal transduction and residue hot spots: application to drug discovery. Curr Opin Drug Discov Devel 13, 527–37. [PubMed] [Google Scholar]
- 39.Musafia B & Senderowitz H (2010). Biasing conformational ensembles towards bioactive-like conformers for ligand-based drug design. Expert Opin Drug Discov 5, 943–59. [DOI] [PubMed] [Google Scholar]
- 40.Marchetti F, Moroni E, Pandini A & Colombo G (2021). Machine Learning Prediction of Allosteric Drug Activity from Molecular Dynamics. J Phys Chem Lett 12, 3724–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greives N & Zhou HX (2014). Both protein dynamics and ligand concentration can shift the binding mechanism between conformational selection and induced fit. Proc Natl Acad Sci U S A 111, 10197–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feher VA, Durrant JD, Van Wart AT & Amaro RE (2014). Computational approaches to mapping allosteric pathways. Curr Opin Struct Biol 25, 98–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guzovsky AB, Schafer NP, Wolynes PG & Ferreiro DU (2022). Localization of Energetic Frustration in Proteins. Methods Mol Biol 2376, 387–398. [DOI] [PubMed] [Google Scholar]
- 44.Jenik M, Parra RG, Radusky LG, Turjanski A, Wolynes PG & Ferreiro DU (2012). Protein frustratometer: a tool to localize energetic frustration in protein molecules. Nucleic Acids Res 40, W348–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gianni S, Freiberger MI, Jemth P, Ferreiro DU, Wolynes PG & Fuxreiter M (2021). Fuzziness and Frustration in the Energy Landscape of Protein Folding, Function, and Assembly. Acc Chem Res 54, 1251–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Freiberger MI, Wolynes PG, Ferreiro DU & Fuxreiter M (2021). Frustration in Fuzzy Protein Complexes Leads to Interaction Versatility. J Phys Chem B 125, 2513–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liang Z, Verkhivker GM & Hu G (2020). Integration of network models and evolutionary analysis into high-throughput modeling of protein dynamics and allosteric regulation: theory, tools and applications. Brief Bioinform 21, 815–835. [DOI] [PubMed] [Google Scholar]
- 48.Astl L & Verkhivker GM (2019). Data-driven computational analysis of allosteric proteins by exploring protein dynamics, residue coevolution and residue interaction networks. Biochim Biophys Acta Gen Subj. [DOI] [PubMed] [Google Scholar]
- 49.Ponzoni L & Bahar I (2018). Structural dynamics is a determinant of the functional significance of missense variants. Proc Natl Acad Sci U S A 115, 4164–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Csermely P, Korcsmaros T, Kiss HJ, London G & Nussinov R (2013). Structure and dynamics of molecular networks: a novel paradigm of drug discovery: a comprehensive review. Pharmacol Ther 138, 333–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ma X, Meng H & Lai L (2016). Motions of Allosteric and Orthosteric Ligand-Binding Sites in Proteins are Highly Correlated. J Chem Inf Model 56, 1725–33. [DOI] [PubMed] [Google Scholar]
- 52.Astl L & Verkhivker GM (2020). Dynamic View of Allosteric Regulation in the Hsp70 Chaperones by J-Domain Cochaperone and Post-Translational Modifications: Computational Analysis of Hsp70 Mechanisms by Exploring Conformational Landscapes and Residue Interaction Networks. J Chem Inf Model 60, 1614–1631. [DOI] [PubMed] [Google Scholar]
- 53.Nussinov R, Tsai CJ & Jang H (2019). Does Ras Activate Raf and PI3K Allosterically? Front Oncol 9, 1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nussinov R, Tsai CJ & Jang H (2020). Ras assemblies and signaling at the membrane. Curr Opin Struct Biol 62, 140–148. [DOI] [PubMed] [Google Scholar]
- 55.Ma B, Kumar S, Tsai CJ & Nussinov R (1999). Folding funnels and binding mechanisms. Protein Eng 12, 713–20. [DOI] [PubMed] [Google Scholar]
- 56.Onuchic JN, Luthey-Schulten Z & Wolynes PG (1997). Theory of protein folding: the energy landscape perspective. Annu Rev Phys Chem 48, 545–600. [DOI] [PubMed] [Google Scholar]
- 57.Tsai CJ, Ma B & Nussinov R (1999). Folding and binding cascades: shifts in energy landscapes. Proc Natl Acad Sci U S A 96, 9970–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Frauenfelder H, Parak F & Young RD (1988). Conformational substates in proteins. Annu Rev Biophys Biophys Chem 17, 451–79. [DOI] [PubMed] [Google Scholar]
- 59.Nussinov R & Shirouzu M (2021). Editorial overview: Catalysis and regulation: The beating heart of biology. Curr Opin Struct Biol 71, iii–v. [DOI] [PubMed] [Google Scholar]
- 60.Ma B, Kumar S, Tsai CJ, Hu Z & Nussinov R (2000). Transition-state ensemble in enzyme catalysis: possibility, reality, or necessity? J Theor Biol 203, 383–97. [DOI] [PubMed] [Google Scholar]
- 61.Onuchic JN, Socci ND, Luthey-Schulten Z & Wolynes PG (1996). Protein folding funnels: the nature of the transition state ensemble. Fold Des 1, 441–50. [DOI] [PubMed] [Google Scholar]
- 62.Strater N, Schnappauf G, Braus G & Lipscomb WN (1997). Mechanisms of catalysis and allosteric regulation of yeast chorismate mutase from crystal structures. Structure 5, 1437–52. [DOI] [PubMed] [Google Scholar]
- 63.Lin SL, Xu D, Li A & Nussinov R (1998). Electrostatics, allostery, and activity of the yeast chorismate mutase. Proteins 31, 445–52. [DOI] [PubMed] [Google Scholar]
- 64.Dill KA & Chan HS (1997). From Levinthal to pathways to funnels. Nat Struct Biol 4, 10–9. [DOI] [PubMed] [Google Scholar]
- 65.Nussinov R, Tsai CJ & Jang H (2022). Allostery, and how to define and measure signal transduction. Biophys Chem 283, 106766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nussinov R, Tsai CJ & Jang H (2020). Autoinhibition can identify rare driver mutations and advise pharmacology. FASEB J 34, 16–29. [DOI] [PubMed] [Google Scholar]
- 67.Nussinov R, Jang H, Tsai CJ & Cheng F (2019). Precision medicine review: rare driver mutations and their biophysical classification. Biophys Rev 11, 5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Song K, Li Q, Gao W, Lu S, Shen Q, Liu X, Wu Y, Wang B, Lin H, Chen G & Zhang J (2019). AlloDriver: a method for the identification and analysis of cancer driver targets. Nucleic Acids Res 47, W315–W321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rochman ND, Wolf YI & Koonin EV (2020). Deep phylogeny of cancer drivers and compensatory mutations. Commun Biol 3, 551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Padua RAP, Sun Y, Marko I, Pitsawong W, Stiller JB, Otten R & Kern D (2018). Mechanism of activating mutations and allosteric drug inhibition of the phosphatase SHP2. Nat Commun 9, 4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Walker C, Wang Y, Olivieri C, Karamafrooz A, Casby J, Bathon K, Calebiro D, Gao J, Bernlohr DA, Taylor SS & Veglia G (2019). Cushing’s syndrome driver mutation disrupts protein kinase A allosteric network, altering both regulation and substrate specificity. Sci Adv 5, eaaw9298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morjaria S (2021). Driver mutations in oncogenesis. Int J Mol Immuno Oncol 6, 100–2. [Google Scholar]
- 73.Tee WV, Tan ZW, Lee K, Guarnera E & Berezovsky IN (2021). Exploring the Allosteric Territory of Protein Function. J Phys Chem B 125, 3763–3780. [DOI] [PubMed] [Google Scholar]
- 74.Zhou Y, Zhao J, Fang J, Martin W, Li L, Nussinov R, Chan TA, Eng C & Cheng F (2021). My personal mutanome: a computational genomic medicine platform for searching network perturbing alleles linking genotype to phenotype. Genome Biol 22, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lu S, Qiu Y, Ni D, He X, Pu J & Zhang J (2020). Emergence of allosteric drug-resistance mutations: new challenges for allosteric drug discovery. Drug Discov Today 25, 177–184. [DOI] [PubMed] [Google Scholar]
- 76.Klein MI, Cannataro VL, Townsend JP, Newman S, Stern DF & Zhao H (2021). Identifying modules of cooperating cancer drivers. Mol Syst Biol 17, e9810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guarnera E & Berezovsky IN (2020). Allosteric drugs and mutations: chances, challenges, and necessity. Curr Opin Struct Biol 62, 149–157. [DOI] [PubMed] [Google Scholar]
- 78.Tan ZW, Tee WV, Guarnera E, Booth L & Berezovsky IN (2019). AlloMAPS: allosteric mutation analysis and polymorphism of signaling database. Nucleic Acids Res 47, D265–D270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tan ZW, Guarnera E, Tee WV & Berezovsky IN (2020). AlloSigMA 2: paving the way to designing allosteric effectors and to exploring allosteric effects of mutations. Nucleic Acids Res 48, W116–W124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tee WV, Guarnera E & Berezovsky IN (2019). On the Allosteric Effect of nsSNPs and the Emerging Importance of Allosteric Polymorphism. J Mol Biol 431, 3933–3942. [DOI] [PubMed] [Google Scholar]
- 81.Pon JR & Marra MA (2015). Driver and passenger mutations in cancer. Annu Rev Pathol 10, 25–50. [DOI] [PubMed] [Google Scholar]
- 82.Nussinov R, Jang H, Tsai CJ & Cheng F (2019). Review: Precision medicine and driver mutations: Computational methods, functional assays and conformational principles for interpreting cancer drivers. PLoS Comput Biol 15, e1006658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nussinov R, Jang H & Tsai CJ (2014). The structural basis for cancer treatment decisions. Oncotarget 5, 7285–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Raphael BJ, Dobson JR, Oesper L & Vandin F (2014). Identifying driver mutations in sequenced cancer genomes: computational approaches to enable precision medicine. Genome Med 6, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Barros EP, Demir O, Soto J, Cocco MJ & Amaro RE (2020). Markov state models and NMR uncover an overlooked allosteric loop in p53. Chem Sci 12, 1891–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nussinov R, Tsai CJ & Jang H (2019). Why Are Some Driver Mutations Rare? Trends Pharmacol Sci 40, 919–929. [DOI] [PubMed] [Google Scholar]
- 87.Getz G, Hofling H, Mesirov JP, Golub TR, Meyerson M, Tibshirani R & Lander ES (2007). Comment on “The consensus coding sequences of human breast and colorectal cancers”. Science 317, 1500. [DOI] [PubMed] [Google Scholar]
- 88.Davoli T, Xu AW, Mengwasser KE, Sack LM, Yoon JC, Park PJ & Elledge SJ (2013). Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 155, 948–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Das JK, Thakuri B, MohanKumar K, Roy S, Sljoka A, Sun GQ & Chakraborty A (2021). Mutation-Induced Long-Range Allosteric Interactions in the Spike Protein Determine the Infectivity of SARS-CoV-2 Emerging Variants. ACS Omega 6, 31312–31327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.McCormick JW, Russo MA, Thompson S, Blevins A & Reynolds KA (2021). Structurally distributed surface sites tune allosteric regulation. Elife 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.de Regt AK, Kim S, Sohn J, Grant RA, Baker TA & Sauer RT (2015). A conserved activation cluster is required for allosteric communication in HtrA-family proteases. Structure 23, 517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Leander M, Yuan Y, Meger A, Cui Q & Raman S (2020). Functional plasticity and evolutionary adaptation of allosteric regulation. Proc Natl Acad Sci U S A 117, 25445–25454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xu J, Pham CG, Albanese SK, Dong Y, Oyama T, Lee CH, Rodrik-Outmezguine V, Yao Z, Han S, Chen D, Parton DL, Chodera JD, Rosen N, Cheng EH & Hsieh JJ (2016). Mechanistically distinct cancer-associated mTOR activation clusters predict sensitivity to rapamycin. J Clin Invest 126, 3526–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Keskin O, Ma B & Nussinov R (2005). Hot regions in protein--protein interactions: the organization and contribution of structurally conserved hot spot residues. J Mol Biol 345, 1281–94. [DOI] [PubMed] [Google Scholar]
- 95.Keskin O, Gursoy A, Ma B & Nussinov R (2008). Principles of protein-protein interactions: what are the preferred ways for proteins to interact? Chem Rev 108, 1225–44. [DOI] [PubMed] [Google Scholar]
- 96.Verkhivker GM (2019). Biophysical simulations and structure-based modeling of residue interaction networks in the tumor suppressor proteins reveal functional role of cancer mutation hotspots in molecular communication. Biochim Biophys Acta Gen Subj 1863, 210–225. [DOI] [PubMed] [Google Scholar]
- 97.Agajanian S, Odeyemi O, Bischoff N, Ratra S & Verkhivker GM (2018). Machine Learning Classification and Structure-Functional Analysis of Cancer Mutations Reveal Unique Dynamic and Network Signatures of Driver Sites in Oncogenes and Tumor Suppressor Genes. J Chem Inf Model 58, 2131–2150. [DOI] [PubMed] [Google Scholar]
- 98.Rodrigues CH, Ascher DB & Pires DE (2018). Kinact: a computational approach for predicting activating missense mutations in protein kinases. Nucleic Acids Res 46, W127–W132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ferraro M, Moroni E, Ippoliti E, Rinaldi S, Sanchez-Martin C, Rasola A, Pavarino LF & Colombo G (2021). Machine Learning of Allosteric Effects: The Analysis of Ligand-Induced Dynamics to Predict Functional Effects in TRAP1. J Phys Chem B 125, 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhou H, Dong Z & Tao P (2018). Recognition of protein allosteric states and residues: Machine learning approaches. J Comput Chem 39, 1481–1490. [DOI] [PubMed] [Google Scholar]
- 101.Penkler DL, Atilgan C & Tastan Bishop O (2018). Allosteric Modulation of Human Hsp90alpha Conformational Dynamics. J Chem Inf Model 58, 383–404. [DOI] [PubMed] [Google Scholar]
- 102.Sheik Amamuddy O, Veldman W, Manyumwa C, Khairallah A, Agajanian S, Oluyemi O, Verkhivker G & Tastan Bishop O (2020). Integrated Computational Approaches and Tools forAllosteric Drug Discovery. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ye J, Pavlicek A, Lunney EA, Rejto PA & Teng CH (2010). Statistical method on nonrandom clustering with application to somatic mutations in cancer. BMC Bioinformatics 11, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ryslik GA, Cheng Y, Cheung KH, Modis Y & Zhao H (2013). Utilizing protein structure to identify non-random somatic mutations. BMC Bioinformatics 14, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Carter H, Chen S, Isik L, Tyekucheva S, Velculescu VE, Kinzler KW, Vogelstein B & Karchin R (2009). Cancer-specific high-throughput annotation of somatic mutations: computational prediction of driver missense mutations. Cancer Res 69, 6660–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rodrigues T, Werner M, Roth J, da Cruz EHG, Marques MC, Akkapeddi P, Lobo SA, Koeberle A, Corzana F, da Silva EN Junior, Werz O & Bernardes GJL (2018). Machine intelligence decrypts beta-lapachone as an allosteric 5-lipoxygenase inhibitor. Chem Sci 9, 6899–6903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Greener JG & Sternberg MJ (2018). Structure-based prediction of protein allostery. Curr Opin Struct Biol 50, 1–8. [DOI] [PubMed] [Google Scholar]
- 108.Malik G & Kloczkowski A (2018). Biophysical Society Annual Meeting, San Francisco, CA, USA. [Google Scholar]
- 109.Fleetwood O, Kasimova MA, Westerlund AM & Delemotte L (2020). Molecular Insights from Conformational Ensembles via Machine Learning. Biophys J 118, 765–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nussinov R & Tsai CJ (2014). Free energy diagrams for protein function. Chem Biol 21, 311–8. [DOI] [PubMed] [Google Scholar]
- 111.Angyan AF, Szappanos B, Perczel A & Gaspari Z (2010). CoNSEnsX: an ensemble view of protein structures and NMR-derived experimental data. BMC Struct Biol 10, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jamroz M, Kolinski A & Kmiecik S (2014). CABS-flex predictions of protein flexibility compared with NMR ensembles. Bioinformatics 30, 2150–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gaalswyk K, Liu Z, Vogel HJ & MacCallum JL (2021). An Integrative Approach to Determine 3D Protein Structures Using Sparse Paramagnetic NMR Data and Physical Modeling. Front Mol Biosci 8, 676268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Torchia DA (2015). NMR studies of dynamic biomolecular conformational ensembles. Prog Nucl Magn Reson Spectrosc 84–85, 14–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nussinov R, Zhang M, Maloney R & Jang H (2021). Drugging multiple same-allele driver mutations in cancer. Expert Opin Drug Discov 16, 823–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tangye SG (2016). CIS Annual Meeting: Immune Deficiency & Dysregulation North American Conference, Boston, MA, USA. [DOI] [PubMed] [Google Scholar]
- 117.Nussinov R, Zhang M, Tsai CJ & Jang H (2021). Phosphorylation and Driver Mutations in PI3Kalpha and PTEN Autoinhibition. Mol Cancer Res 19, 543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Vasan N, Razavi P, Johnson JL, Shao H, Shah H, Antoine A, Ladewig E, Gorelick A, Lin TY, Toska E, Xu G, Kazmi A, Chang MT, Taylor BS, Dickler MN, Jhaveri K, Chandarlapaty S, Rabadan R, Reznik E, Smith ML, Sebra R, Schimmoller F, Wilson TR, Friedman LS, Cantley LC, Scaltriti M & Baselga J (2019). Double PIK3CA mutations in cis increase oncogenicity and sensitivity to PI3Kalpha inhibitors. Science 366, 714–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Saito Y, Koya J, Araki M, Kogure Y, Shingaki S, Tabata M, McClure MB, Yoshifuji K, Matsumoto S, Isaka Y, Tanaka H, Kanai T, Miyano S, Shiraishi Y, Okuno Y & Kataoka K (2020). Landscape and function of multiple mutations within individual oncogenes. Nature 582, 95–99. [DOI] [PubMed] [Google Scholar]
- 120.Zhang M, Jang H & Nussinov R (2021). PI3K Driver Mutations: A Biophysical Membrane-Centric Perspective. Cancer Res 81, 237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sun M, Hillmann P, Hofmann BT, Hart JR & Vogt PK (2010). Cancer-derived mutations in the regulatory subunit p85alpha of phosphoinositide 3-kinase function through the catalytic subunit p110alpha. Proc Natl Acad Sci U S A 107, 15547–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Barbosa R, Acevedo LA & Marmorstein R (2021). The MEK/ERK Network as a Therapeutic Target in Human Cancer. Mol Cancer Res 19, 361–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Maloney RC, Zhang M, Jang H & Nussinov R (2021). The mechanism of activation of monomeric B-Raf V600E. Comput Struct Biotechnol J 19, 3349–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pfister S, Janzarik WG, Remke M, Ernst A, Werft W, Becker N, Toedt G, Wittmann A, Kratz C, Olbrich H, Ahmadi R, Thieme B, Joos S, Radlwimmer B, Kulozik A, Pietsch T, Herold-Mende C, Gnekow A, Reifenberger G, Korshunov A, Scheurlen W, Omran H & Lichter P (2008). BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest 118, 1739–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lavoie H & Therrien M (2015). Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol 16, 281–98. [DOI] [PubMed] [Google Scholar]
- 126.Tsai CJ & Nussinov R (2018). Allosteric activation of RAF in the MAPK signaling pathway. Curr Opin Struct Biol 53, 100–106. [DOI] [PubMed] [Google Scholar]
- 127.Lu S, Jang H, Gu S, Zhang J & Nussinov R (2016). Drugging Ras GTPase: a comprehensive mechanistic and signaling structural view. Chem Soc Rev 45, 4929–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Travers T, Lopez CA, Van QN, Neale C, Tonelli M, Stephen AG & Gnanakaran S (2018). Molecular recognition of RAS/RAF complex at the membrane: Role of RAF cysteine-rich domain. Sci Rep 8, 8461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Improta-Brears T, Ghosh S & Bell RM (1999). Mutational analysis of Raf-1 cysteine rich domain: requirement for a cluster of basic aminoacids for interaction with phosphatidylserine. Mol Cell Biochem 198, 171–8. [DOI] [PubMed] [Google Scholar]
- 130.Li S, Jang H, Zhang J & Nussinov R (2018). Raf-1 Cysteine-Rich Domain Increases the Affinity of K-Ras/Raf at the Membrane, Promoting MAPK Signaling. Structure 26, 513–525 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nan X, Collisson EA, Lewis S, Huang J, Tamguney TM, Liphardt JT, McCormick F, Gray JW & Chu S (2013). Single-molecule superresolution imaging allows quantitative analysis of RAF multimer formation and signaling. Proc Natl Acad Sci U S A 110, 18519–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cutler RE Jr., Stephens RM, Saracino MR & Morrison DK (1998). Autoregulation of the Raf-1 serine/threonine kinase. Proc Natl Acad Sci U S A 95, 9214–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Terai K & Matsuda M (2006). The amino-terminal B-Raf-specific region mediates calcium-dependent homo- and hetero-dimerization of Raf. EMBO J 25, 3556–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Freed E, Symons M, Macdonald SG, McCormick F & Ruggieri R (1994). Binding of 14-3-3 proteins to the protein kinase Raf and effects on its activation. Science 265, 1713–6. [DOI] [PubMed] [Google Scholar]
- 135.Zhang M, Jang H, Li Z, Sacks DB & Nussinov R (2021). B-Raf autoinhibition in the presence and absence of 14-3-3. Structure 29, 768–777 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Park E, Rawson S, Li K, Kim BW, Ficarro SB, Pino GG, Sharif H, Marto JA, Jeon H & Eck MJ (2019). Architecture of autoinhibited and active BRAF-MEK1–14-3-3 complexes. Nature 575, 545–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kondo Y, Ognjenovic J, Banerjee S, Karandur D, Merk A, Kulhanek K, Wong K, Roose JP, Subramaniam S & Kuriyan J (2019). Cryo-EM structure of a dimeric B-Raf:14-3-3 complex reveals asymmetry in the active sites of B-Raf kinases. Science 366, 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Terrell EM, Durrant DE, Ritt DA, Sealover NE, Sheffels E, Spencer-Smith R, Esposito D, Zhou Y, Hancock JF, Kortum RL & Morrison DK (2019). Distinct Binding Preferences between Ras and Raf Family Members and the Impact on Oncogenic Ras Signaling. Mol Cell 76, 872–884 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Weber CK, Slupsky JR, Kalmes HA & Rapp UR (2001). Active Ras induces heterodimerization of cRaf and BRaf. Cancer Res 61, 3595–8. [PubMed] [Google Scholar]
- 140.Hu J, Stites EC, Yu H, Germino EA, Meharena HS, Stork PJS, Kornev AP, Taylor SS & Shaw AS (2013). Allosteric activation of functionally asymmetric RAF kinase dimers. Cell 154, 1036–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Morrison DK, Heidecker G, Rapp UR & Copeland TD (1993). Identification of the major phosphorylation sites of the Raf-1 kinase. J Biol Chem 268, 17309–16. [PubMed] [Google Scholar]
- 142.Kolch W (2000). Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem J 351 Pt 2, 289–305. [PMC free article] [PubMed] [Google Scholar]
- 143.Michaud NR, Therrien M, Cacace A, Edsall LC, Spiegel S, Rubin GM & Morrison DK (1997). KSR stimulates Raf-1 activity in a kinase-independent manner. Proc Natl Acad Sci U S A 94, 12792–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rajakulendran T, Sahmi M, Lefrancois M, Sicheri F & Therrien M (2009). A dimerization-dependent mechanism drives RAF catalytic activation. Nature 461, 542–5. [DOI] [PubMed] [Google Scholar]
- 145.Shaw AS, Kornev AP, Hu J, Ahuja LG & Taylor SS (2014). Kinases and pseudokinases: lessons from RAF. Mol Cell Biol 34, 1538–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Armstrong SA, Malley R & Weinberg BA (2020). Glimmers of Hope—New Strategies for Overcoming Treatment Resistance in Patients with BRAF V600E-mutated Metastatic Colorectal Cancer. touchREVIEWS Oncol Hematol 16, 31–5. [Google Scholar]
- 147.Nikanjam M, Tinajero J, Barkauskas DA & Kurzrock R (2021). BRAF V600E/V600K Mutations versus Nonstandard Alterations: Prognostic Implications and Therapeutic Outcomes. Mol Cancer Ther 20, 1072–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Jang H, Smith IN, Eng C & Nussinov R (2021). The mechanism of full activation of tumor suppressor PTEN at the phosphoinositide-enriched membrane. iScience 24, 102438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Masson GR & Williams RL (2020). Structural Mechanisms of PTEN Regulation. Cold Spring Harb Perspect Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Smith IN, Thacker S, Jaini R & Eng C (2019). Dynamics and structural stability effects of germline PTEN mutations associated with cancer versus autism phenotypes. J Biomol Struct Dyn 37, 1766–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Degan SE & Gelman IH (2021). Emerging Roles for AKT Isoform Preference in Cancer Progression Pathways. Mol Cancer Res 19, 1251–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kotelevets L, Trifault B, Chastre E & Scott MGH (2020). Posttranslational Regulation and Conformational Plasticity of PTEN. Cold Spring Harb Perspect Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, Dixon JE, Pandolfi P & Pavletich NP (1999). Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell 99, 323–34. [DOI] [PubMed] [Google Scholar]
- 154.Ngeow J & Eng C (2020). PTEN in Hereditary and Sporadic Cancer. Cold Spring Harb Perspect Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Maeda Y, Tidyman WE, Ander BP, Pritchard CA & Rauen KA (2021). Ras/MAPK dysregulation in development causes a skeletal myopathy in an activating Braf(L597V) mouse model for cardio-facio-cutaneous syndrome. Dev Dyn 250, 1074–1095. [DOI] [PubMed] [Google Scholar]
- 156.Castel P, Rauen KA & McCormick F (2020). The duality of human oncoproteins: drivers of cancer and congenital disorders. Nat Rev Cancer 20, 383–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Nussinov R, Tsai CJ & Jang H (2022). How can same-gene mutations promote both cancer and developmental disorders? Sci Adv 8, eabm2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Nussinov R, Jang H, Gursoy A, Keskin O & Gaponenko V (2021). Inhibition of Nonfunctional Ras. Cell Chem Biol 28, 121–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bryant KL, Mancias JD, Kimmelman AC & Der CJ (2014). KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci 39, 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Cox AD & Der CJ (2010). Ras history: The saga continues. Small GTPases 1, 2–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Crespo P & Leon J (2000). Ras proteins in the control of the cell cycle and cell differentiation. Cell Mol Life Sci 57, 1613–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Drosten M, Dhawahir A, Sum EY, Urosevic J, Lechuga CG, Esteban LM, Castellano E, Guerra C, Santos E & Barbacid M (2010). Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J 29, 1091–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lu S, Jang H, Muratcioglu S, Gursoy A, Keskin O, Nussinov R & Zhang J (2016). Ras Conformational Ensembles, Allostery, and Signaling. Chem Rev 116, 6607–65. [DOI] [PubMed] [Google Scholar]
- 164.Pylayeva-Gupta Y, Grabocka E & Bar-Sagi D (2011). RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer 11, 761–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wiesmuller L & Wittinghofer F (1994). Signal transduction pathways involving Ras. Mini review. Cell Signal 6, 247–67. [DOI] [PubMed] [Google Scholar]
- 166.Moran MF, Polakis P, McCormick F, Pawson T & Ellis C (1991). Protein-tyrosine kinases regulate the phosphorylation, protein interactions, subcellular distribution, and activity of p21ras GTPase-activating protein. Mol Cell Biol 11, 1804–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bandaru P, Kondo Y & Kuriyan J (2019). The Interdependent Activation of Son-of-Sevenless and Ras. Cold Spring Harb Perspect Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Huang WYC, Alvarez S, Kondo Y, Lee YK, Chung JK, Lam HYM, Biswas KH, Kuriyan J & Groves JT (2019). A molecular assembly phase transition and kinetic proofreading modulate Ras activation by SOS. Science 363, 1098–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Liao TJ, Jang H, Fushman D & Nussinov R (2018). Allosteric KRas4B Can Modulate SOS1 Fast and Slow Ras Activation Cycles. Biophys J 115, 629–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Liao TJ, Jang H, Nussinov R & Fushman D (2020). High-Affinity Interactions of the nSH3/cSH3 Domains of Grb2 with the C-Terminal Proline-Rich Domain of SOS1. J Am Chem Soc 142, 3401–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Nussinov R, Tsai CJ & Jang H (2019). Is Nanoclustering essential for all oncogenic KRas pathways? Can it explain why wild-type KRas can inhibit its oncogenic variant? Semin Cancer Biol 54, 114–120. [DOI] [PubMed] [Google Scholar]
- 172.Nussinov R, Tsai CJ & Jang H (2019). Oncogenic KRas mobility in the membrane and signaling response. Semin Cancer Biol 54, 109–113. [DOI] [PubMed] [Google Scholar]
- 173.Solman M, Ligabue A, Blazevits O, Jaiswal A, Zhou Y, Liang H, Lectez B, Kopra K, Guzman C, Harma H, Hancock JF, Aittokallio T & Abankwa D (2015). Specific cancer-associated mutations in the switch III region of Ras increase tumorigenicity by nanocluster augmentation. Elife 4, e08905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sutton MN, Lu Z, Li YC, Zhou Y, Huang T, Reger AS, Hurwitz AM, Palzkill T, Logsdon C, Liang X, Gray JW, Nan X, Hancock J, Wahl GM & Bast RC Jr. (2019). DIRAS3 (ARHI) Blocks RAS/MAPK Signaling by Binding Directly to RAS and Disrupting RAS Clusters. Cell Rep 29, 3448–3459 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhou Y, Prakash P, Gorfe AA & Hancock JF (2018). Ras and the Plasma Membrane: A Complicated Relationship. Cold Spring Harb Perspect Med 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Nussinov R, Zhang M, Tsai CJ, Liao TJ, Fushman D & Jang H (2018). Autoinhibition in Ras effectors Raf, PI3Kalpha, and RASSF5: a comprehensive review underscoring the challenges in pharmacological intervention. Biophys Rev 10, 1263–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Rezaei Adariani S, Buchholzer M, Akbarzadeh M, Nakhaei-Rad S, Dvorsky R & Ahmadian MR (2018). Structural snapshots of RAF kinase interactions. Biochem Soc Trans 46, 1393–1406. [DOI] [PubMed] [Google Scholar]
- 178.Fang Z, Lee KY, Huo KG, Gasmi-Seabrook G, Zheng L, Moghal N, Tsao MS, Ikura M & Marshall CB (2020). Multivalent assembly of KRAS with the RAS-binding and cysteine-rich domains of CRAF on the membrane. Proc Natl Acad Sci U S A 117, 12101–12108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Li ZL, Prakash P & Buck M (2018). A “Tug of War” Maintains a Dynamic Protein-Membrane Complex: Molecular Dynamics Simulations of C-Raf RBD-CRD Bound to KRas4B at an Anionic Membrane. ACS Cent Sci 4, 298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Okada T, Hu CD, Jin TG, Kariya K, Yamawaki-Kataoka Y & Kataoka T (1999). The strength of interaction at the Raf cysteine-rich domain is a critical determinant of response of Raf to Ras family small GTPases. Mol Cell Biol 19, 6057–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sarkar S & Garcia AE (2020). Presence or Absence of Ras Dimerization Shows Distinct Kinetic Signature in Ras-Raf Interaction. Biophys J 118, 1799–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Holderfield M, Deuker MM, McCormick F & McMahon M (2014). Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer 14, 455–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Lavoie H, Thevakumaran N, Gavory G, Li JJ, Padeganeh A, Guiral S, Duchaine J, Mao DY, Bouvier M, Sicheri F & Therrien M (2013). Inhibitors that stabilize a closed RAF kinase domain conformation induce dimerization. Nat Chem Biol 9, 428–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lee Y, Phelps C, Huang T, Mostofian B, Wu L, Zhang Y, Tao K, Chang YH, Stork PJ, Gray JW, Zuckerman DM & Nan X (2019). High-throughput, single-particle tracking reveals nested membrane domains that dictate KRas(G12D) diffusion and trafficking. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Muratcioglu S, Aydin C, Odabasi E, Ozdemir ES, Firat-Karalar EN, Jang H, Tsai CJ, Nussinov R, Kavakli IH, Gursoy A & Keskin O (2020). Oncogenic KRas4B Dimerization Enhances Downstream Mitogen-activated Protein Kinase Signaling. J Mol Biol 432, 1199–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Muratcioglu S, Chavan TS, Freed BC, Jang H, Khavrutskii L, Freed RN, Dyba MA, Stefanisko K, Tarasov SG, Gursoy A, Keskin O, Tarasova NI, Gaponenko V & Nussinov R (2015). GTP-Dependent K-Ras Dimerization. Structure 23, 1325–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.De Luca A, Maiello MR, D’Alessio A, Pergameno M & Normanno N (2012). The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin Ther Targets 16 Suppl 2, S17–27. [DOI] [PubMed] [Google Scholar]
- 188.Young A, Lou D & McCormick F (2013). Oncogenic and wild-type Ras play divergent roles in the regulation of mitogen-activated protein kinase signaling. Cancer Discov 3, 112–23. [DOI] [PubMed] [Google Scholar]
- 189.Gao Y, Zhou J, Xie Z, Wang J, Ho CK, Zhang Y & Li Q (2019). Mechanical strain promotes skin fibrosis through LRG-1 induction mediated by ELK1 and ERK signalling. Commun Biol 2, 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Downward J (2008). Targeting RAS and PI3K in lung cancer. Nat Med 14, 1315–6. [DOI] [PubMed] [Google Scholar]
- 191.Sheridan C & Downward J (2013). Inhibiting the RAS-PI3K pathway in cancer therapy. Enzymes 34 Pt. B, 107–36. [DOI] [PubMed] [Google Scholar]
- 192.Gul A, Leyland-Jones B, Dey N & De P (2018). A combination of the PI3K pathway inhibitor plus cell cycle pathway inhibitor to combat endocrine resistance in hormone receptor-positive breast cancer: a genomic algorithm-based treatment approach. Am J Cancer Res 8, 2359–2376. [PMC free article] [PubMed] [Google Scholar]
- 193.Pokrass MJ, Ryan KA, Xin T, Pielstick B, Timp W, Greco V & Regot S (2020). Cell-Cycle-Dependent ERK Signaling Dynamics Direct Fate Specification in the Mammalian Preimplantation Embryo. Dev Cell 55, 328–340 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zhang M, Jang H & Nussinov R (2020). PI3K inhibitors: review and new strategies. Chem Sci 11, 5855–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Wittinghofer A, Scheffzek K & Ahmadian MR (1997). The interaction of Ras with GTPase-activating proteins. FEBS Lett 410, 63–7. [DOI] [PubMed] [Google Scholar]
- 196.Adjei AA (2001). Blocking oncogenic Ras signaling for cancer therapy. J Natl Cancer Inst 93, 1062–74. [DOI] [PubMed] [Google Scholar]
- 197.Schubbert S, Shannon K & Bollag G (2007). Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer 7, 295–308. [DOI] [PubMed] [Google Scholar]
- 198.Smith MJ, Neel BG & Ikura M (2013). NMR-based functional profiling of RASopathies and oncogenic RAS mutations. Proc Natl Acad Sci U S A 110, 4574–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Vigil D, Cherfils J, Rossman KL & Der CJ (2010). Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer 10, 842–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Poulin EJ, Bera AK, Lu J, Lin YJ, Strasser SD, Paulo JA, Huang TQ, Morales C, Yan W, Cook J, Nowak JA, Brubaker DK, Joughin BA, Johnson CW, DeStefanis RA, Ghazi PC, Gondi S, Wales TE, Iacob RE, Bogdanova L, Gierut JJ, Li Y, Engen JR, Perez-Mancera PA, Braun BS, Gygi SP, Lauffenburger DA, Westover KD & Haigis KM (2019). Tissue-Specific Oncogenic Activity of KRAS(A146T). Cancer Discov 9, 738–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S & Westover KD (2015). Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol Cancer Res 13, 1325–35. [DOI] [PubMed] [Google Scholar]
- 202.Nussinov R & Tsai CJ (2015). The design of covalent allosteric drugs. Annu Rev Pharmacol Toxicol 55, 249–67. [DOI] [PubMed] [Google Scholar]
- 203.Nussinov R & Tsai CJ (2014). Unraveling structural mechanisms of allosteric drug action. Trends Pharmacol Sci 35, 256–64. [DOI] [PubMed] [Google Scholar]
- 204.Abdel-Magid AF (2015). Allosteric modulators: an emerging concept in drug discovery. ACS Med Chem Lett 6, 104–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Conn PJ, Christopoulos A & Lindsley CW (2009). Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov 8, 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Conn PJ, Lindsley CW, Meiler J & Niswender CM (2014). Opportunities and challenges in the discovery of allosteric modulators of GPCRs for treating CNS disorders. Nat Rev Drug Discov 13, 692–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Reyes-Alcaraz A, Lucero Garcia-Rojas EY, Bond RA & McConnell BK (2020). Allosteric Modulators for GPCRs as a Therapeutic Alternative with High Potential in Drug Discovery. In Molecular Pharmacology (Catala A & Ahmad U, eds.). IntechOpen. [Google Scholar]
- 208.Ni D, Li Y, Qiu Y, Pu J, Lu S & Zhang J (2020). Combining Allosteric and Orthosteric Drugs to Overcome Drug Resistance. Trends Pharmacol Sci 41, 336–348. [DOI] [PubMed] [Google Scholar]
- 209.Grover AK (2013). Use of allosteric targets in the discovery of safer drugs. Med Princ Pract 22, 418–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Shaffer C (2019). Importance of Allostery in Pharmacology. In News Medical. [Google Scholar]
- 211.Moisoiu V, Teodorescu P, Parajdi L, Pasca S, Zdrenghea M, Dima D, Precup R, Tomuleasa C & Soverini S (2019). Assessing Measurable Residual Disease in Chronic Myeloid Leukemia. BCR-ABL1 IS in the Avant-Garde of Molecular Hematology. Front Oncol 9, 863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Becher I, Savitski MM, Savitski MF, Hopf C, Bantscheff M & Drewes G (2013). Affinity profiling of the cellular kinome for the nucleotide cofactors ATP, ADP, and GTP. ACS Chem Biol 8, 599–607. [DOI] [PubMed] [Google Scholar]
- 213.de Vries R, Meijer FA, Doveston RG, Leijten-van de Gevel IA & Brunsveld L (2021). Cooperativity between the orthosteric and allosteric ligand binding sites of RORgammat. Proc Natl Acad Sci U S A 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Wei S, Zhao T, Wang J & Zhai X (2021). Approach in Improving Potency and Selectivity of Kinase Inhibitors: Allosteric Kinase Inhibitors. Mini Rev Med Chem 21, 991–1003. [DOI] [PubMed] [Google Scholar]
- 215.Kenakin T (2013). Analytical pharmacology and allosterism: the importance of quantifying drug parameters in drug discovery. Drug Discov Today Technol 10, e229–35. [DOI] [PubMed] [Google Scholar]
- 216.Gingell JJ, Simms J, Barwell J, Poyner DR, Watkins HA, Pioszak AA, Sexton PM & Hay DL (2016). An allosteric role for receptor activity-modifying proteins in defining GPCR pharmacology. Cell Discov 2, 16012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Lyczek A, Berger BT, Rangwala AM, Paung Y, Tom J, Philipose H, Guo J, Albanese SK, Robers MB, Knapp S, Chodera JD & Seeliger MA (2021). Mutation in Abl kinase with altered drug-binding kinetics indicates a novel mechanism of imatinib resistance. Proc Natl Acad Sci U S A 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Xie T, Saleh T, Rossi P & Kalodimos CG (2020). Conformational states dynamically populated by a kinase determine its function. Science 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Saleh T, Rossi P & Kalodimos CG (2017). Atomic view of the energy landscape in the allosteric regulation of Abl kinase. Nat Struct Mol Biol 24, 893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Xie T, Saleh T, Rossi P, Miller D & Kalodimos CG (2021). Imatinib can act as an Allosteric Activator of Abl Kinase. J Mol Biol 434, 167349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Knight ZA, Lin H & Shokat KM (2010). Targeting the cancer kinome through polypharmacology. Nat Rev Cancer 10, 130–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Nussinov R, Tsai CJ & Jang H (2021). Anticancer drug resistance: An update and perspective. Drug Resist Updat, 100796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Luttman JH, Hoj JP, Lin KH, Lin J, Gu JJ, Rouse C, Nichols AG, MacIver NJ, Wood KC & Pendergast AM (2021). ABL allosteric inhibitors synergize with statins to enhance apoptosis of metastatic lung cancer cells. Cell Rep 37, 109880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Eck MJ & Manley PW (2009). The interplay of structural information and functional studies in kinase drug design: insights from BCR-Abl. Curr Opin Cell Biol 21, 288–95. [DOI] [PubMed] [Google Scholar]
- 225.Lu H & Tonge PJ (2010). Drug-target residence time: critical information for lead optimization. Curr Opin Chem Biol 14, 467–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Bradshaw JM, McFarland JM, Paavilainen VO, Bisconte A, Tam D, Phan VT, Romanov S, Finkle D, Shu J, Patel V, Ton T, Li X, Loughhead DG, Nunn PA, Karr DE, Gerritsen ME, Funk JO, Owens TD, Verner E, Brameld KA, Hill RJ, Goldstein DM & Taunton J (2015). Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat Chem Biol 11, 525–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Robers MB, Friedman-Ohana R, Huber KVM, Kilpatrick L, Vasta JD, Berger BT, Chaudhry C, Hill S, Muller S, Knapp S & Wood KV (2020). Quantifying Target Occupancy of Small Molecules Within Living Cells. Annu Rev Biochem 89, 557–581. [DOI] [PubMed] [Google Scholar]
- 228.Tiwary P, Mondal J & Berne BJ (2017). How and when does an anticancer drug leave its binding site? Sci Adv 3, e1700014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Jones JK & Thompson EM (2020). Allosteric Inhibition of ABL Kinases: Therapeutic Potential in Cancer. Mol Cancer Ther 19, 1763–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Burslem GM, Schultz AR, Bondeson DP, Eide CA, Savage Stevens SL, Druker BJ & Crews CM (2019). Targeting BCR-ABL1 in Chronic Myeloid Leukemia by PROTAC-Mediated Targeted Protein Degradation. Cancer Res 79, 4744–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Troup RI, Fallan C & Baud MGJ (2020). Current strategies for the design of PROTAC linkers: a critical review. Explor Target Antitumor Ther 1, 273–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Schreiber SL (2021). The Rise of Molecular Glues. Cell 184, 3–9. [DOI] [PubMed] [Google Scholar]
- 233.Mullard A (2021). Targeted protein degraders crowd into the clinic. Nat Rev Drug Discov 20, 247–250. [DOI] [PubMed] [Google Scholar]
- 234.Che Y, Gilbert AM, Shanmugasundaram V & Noe MC (2018). Inducing protein-protein interactions with molecular glues. Bioorg Med Chem Lett 28, 2585–2592. [DOI] [PubMed] [Google Scholar]
- 235.Bueno AB, Sun B, Willard FS, Feng D, Ho JD, Wainscott DB, Showalter AD, Vieth M, Chen Q, Stutsman C, Chau B, Ficorilli J, Agejas FJ, Cumming GR, Jimenez A, Rojo I, Kobilka TS, Kobilka BK & Sloop KW (2020). Structural insights into probe-dependent positive allosterism of the GLP-1 receptor. Nat Chem Biol 16, 1105–1110. [DOI] [PubMed] [Google Scholar]
- 236.Rudolph J, Settleman J & Malek S (2021). Emerging Trends in Cancer Drug Discovery-From Drugging the “Undruggable” to Overcoming Resistance. Cancer Discov 11, 815–821. [DOI] [PubMed] [Google Scholar]
- 237.Vavers E, Zvejniece L, Maurice T & Dambrova M (2019). Allosteric Modulators of Sigma-1 Receptor: A Review. Front Pharmacol 10, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.St-Cyr D, Ceccarelli DF, Orlicky S, van der Sloot AM, Tang X, Kelso S, Moore S, James C, Posternak G, Coulombe-Huntington J, Bertomeu T, Marinier A, Sicheri F & Tyers M (2021). Identification and optimization of molecular glue compounds that inhibit a noncovalent E2 enzyme-ubiquitin complex. Sci Adv 7, eabi5797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Lv L, Chen P, Cao L, Li Y, Zeng Z, Cui Y, Wu Q, Li J, Wang JH, Dong MQ, Qi X & Han T (2020). Discovery of a molecular glue promoting CDK12-DDB1 interaction to trigger cyclin K degradation. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Burslem GM & Crews CM (2017). Small-Molecule Modulation of Protein Homeostasis. Chem Rev 117, 11269–11301. [DOI] [PubMed] [Google Scholar]
- 241.Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM & Deshaies RJ (2001). Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A 98, 8554–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Bondeson DP, Mares A, Smith IE, Ko E, Campos S, Miah AH, Mulholland KE, Routly N, Buckley DL, Gustafson JL, Zinn N, Grandi P, Shimamura S, Bergamini G, Faelth-Savitski M, Bantscheff M, Cox C, Gordon DA, Willard RR, Flanagan JJ, Casillas LN, Votta BJ, den Besten W, Famm K, Kruidenier L, Carter PS, Harling JD, Churcher I & Crews CM (2015). Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol 11, 611–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Lowe D (2021). Hard Thinking About Protein Degradation and Bifunctional Molecules. In In the pipeline, Blogs, Science. American Association for the Advancement of Science. [Google Scholar]
- 244.Zaidman D, Prilusky J & London N (2020). PRosettaC: Rosetta Based Modeling of PROTAC Mediated Ternary Complexes. J Chem Inf Model 60, 4894–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Bai N, Miller SA, Andrianov GV, Yates M, Kirubakaran P & Karanicolas J (2021). Rationalizing PROTAC-Mediated Ternary Complex Formation Using Rosetta. J Chem Inf Model 61, 1368–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Liao J, Nie X, Unarta I, Ericksen S & Tang W (2021). In Silico Modeling and Scoring of PROTAC-Mediated Ternary Complex Poses. ChemRxiv, DOI: 10.26434/chemrxiv-2021-ldzzz-v2. [DOI] [PubMed] [Google Scholar]
- 247.Casement R, Bond A, Craigon C & Ciulli A (2021). Mechanistic and Structural Features of PROTAC Ternary Complexes. Methods Mol Biol 2365, 79–113. [DOI] [PubMed] [Google Scholar]
- 248.Zorba A, Nguyen C, Xu Y, Starr J, Borzilleri K, Smith J, Zhu H, Farley KA, Ding W, Schiemer J, Feng X, Chang JS, Uccello DP, Young JA, Garcia-Irrizary CN, Czabaniuk L, Schuff B, Oliver R, Montgomery J, Hayward MM, Coe J, Chen J, Niosi M, Luthra S, Shah JC, El-Kattan A, Qiu X, West GM, Noe MC, Shanmugasundaram V, Gilbert AM, Brown MF & Calabrese MF (2018). Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc Natl Acad Sci U S A 115, E7285–E7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.de Castro GV & Ciulli A (2021). Estimating the cooperativity of PROTAC-induced ternary complexes using (19)F NMR displacement assay. RSC Med Chem 12, 1765–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Weerakoon D, Carbajo RJ, De Maria L, Tyrchan C & Zhao H (2022). Impact of PROTAC Linker Plasticity on the Solution Conformations and Dissociation of the Ternary Complex. J Chem Inf Model 62, 340–349. [DOI] [PubMed] [Google Scholar]
- 251.Smith BE, Wang SL, Jaime-Figueroa S, Harbin A, Wang J, Hamman BD & Crews CM (2019). Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat Commun 10, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Cyrus K, Wehenkel M, Choi EY, Han HJ, Lee H, Swanson H & Kim KB (2011). Impact of linker length on the activity of PROTACs. Mol Biosyst 7, 359–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Donoghue C, Cubillos-Rojas M, Gutierrez-Prat N, Sanchez-Zarzalejo C, Verdaguer X, Riera A & Nebreda AR (2020). Optimal linker length for small molecule PROTACs that selectively target p38alpha and p38beta for degradation. Eur J Med Chem 201, 112451. [DOI] [PubMed] [Google Scholar]
- 254.Weng G, Shen C, Cao D, Gao J, Dong X, He Q, Yang B, Li D, Wu J & Hou T (2021). PROTAC-DB: an online database of PROTACs. Nucleic Acids Res 49, D1381–D1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Nagar B, Hantschel O, Young MA, Scheffzek K, Veach D, Bornmann W, Clarkson B, Superti-Furga G & Kuriyan J (2003). Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell 112, 859–71. [DOI] [PubMed] [Google Scholar]
- 256.Zhang J, Adrian FJ, Jahnke W, Cowan-Jacob SW, Li AG, Iacob RE, Sim T, Powers J, Dierks C, Sun F, Guo GR, Ding Q, Okram B, Choi Y, Wojciechowski A, Deng X, Liu G, Fendrich G, Strauss A, Vajpai N, Grzesiek S, Tuntland T, Liu Y, Bursulaya B, Azam M, Manley PW, Engen JR, Daley GQ, Warmuth M & Gray NS (2010). Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature 463, 501–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Wylie AA, Schoepfer J, Jahnke W, Cowan-Jacob SW, Loo A, Furet P, Marzinzik AL, Pelle X, Donovan J, Zhu W, Buonamici S, Hassan AQ, Lombardo F, Iyer V, Palmer M, Berellini G, Dodd S, Thohan S, Bitter H, Branford S, Ross DM, Hughes TP, Petruzzelli L, Vanasse KG, Warmuth M, Hofmann F, Keen NJ & Sellers WR (2017). The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature 543, 733–737. [DOI] [PubMed] [Google Scholar]
- 258.Ichim CV (2014). Kinase-independent mechanisms of resistance of leukemia stem cells to tyrosine kinase inhibitors. Stem Cells Transl Med 3, 405–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Rodriguez-Rivera FP & Levi SM (2021). Unifying Catalysis Framework to Dissect Proteasomal Degradation Paradigms. ACS Cent Sci 7, 1117–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Ling Y, Jing M & Wang XD (2015). Allosteric therapies for lung cancer. Cancer Metastasis Rev 34, 303–12. [DOI] [PubMed] [Google Scholar]
- 261.Meijer FA, Oerlemans GJM & Brunsveld L (2021). Orthosteric and Allosteric Dual Targeting of the Nuclear Receptor RORgammat with a Bitopic Ligand. ACS Chem Biol 16, 510–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Meijer FA, van den Oetelaar MCM, Doveston RG, Sampers ENR & Brunsveld L (2021). Covalent Occlusion of the RORgammat Ligand Binding Pocket Allows Unambiguous Targeting of an Allosteric Site. ACS Med Chem Lett 12, 631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Valant C, Robert Lane J, Sexton PM & Christopoulos A (2012). The best of both worlds? Bitopic orthosteric/allosteric ligands of g protein-coupled receptors. Annu Rev Pharmacol Toxicol 52, 153–78. [DOI] [PubMed] [Google Scholar]
- 264.Henzler-Wildman K & Kern D (2007). Dynamic personalities of proteins. Nature 450, 964–72. [DOI] [PubMed] [Google Scholar]
- 265.Tsai CJ & Nussinov R (2011). Gene-specific transcription activation via long-range allosteric shape-shifting. Biochem J 439, 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Kumar S, Ma B, Tsai CJ, Wolfson H & Nussinov R (1999). Folding funnels and conformational transitions via hinge-bending motions. Cell Biochem Biophys 31, 141–64. [DOI] [PubMed] [Google Scholar]
- 267.Nussinov R, Tsai CJ & Ma B (2013). The underappreciated role of allostery in the cellular network. Annu Rev Biophys 42, 169–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Tzeng SR & Kalodimos CG (2011). Protein dynamics and allostery: an NMR view. Curr Opin Struct Biol 21, 62–7. [DOI] [PubMed] [Google Scholar]
- 269.Rohban MH, Singh S, Wu X, Berthet JB, Bray MA, Shrestha Y, Varelas X, Boehm JS & Carpenter AE (2017). Systematic morphological profiling of human gene and allele function via Cell Painting. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Noe F & Fischer S (2008). Transition networks for modeling the kinetics of conformational change in macromolecules. Curr Opin Struct Biol 18, 154–62. [DOI] [PubMed] [Google Scholar]
- 271.Guo J & Zhou HX (2016). Protein Allostery and Conformational Dynamics. Chem Rev 116, 6503–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.O’Rourke KF, D’Amico RN, Sahu D & Boehr DD (2021). Distinct conformational dynamics and allosteric networks in alpha tryptophan synthase during active catalysis. Protein Sci 30, 543–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Kannan S, Venkatachalam G, Lim HH, Surana U & Verma C (2018). Conformational landscape of the epidermal growth factor receptor kinase reveals a mutant specific allosteric pocket. Chem Sci 9, 5212–5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Dixit A & Verkhivker GM (2011). The energy landscape analysis of cancer mutations in protein kinases. PLoS One 6, e26071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Verkhivker GM, Agajanian S, Hu G & Tao P (2020). Allosteric Regulation at the Crossroads of New Technologies: Multiscale Modeling, Networks, and Machine Learning. Front Mol Biosci 7, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Fantini M, Lisi S, De Los Rios P, Cattaneo A & Pastore A (2020). Protein Structural Information and Evolutionary Landscape by In Vitro Evolution. Mol Biol Evol 37, 1179–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Phillips R & Milo R (2009). A feeling for the numbers in biology. Proc Natl Acad Sci U S A 106, 21465–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Tasic B, Yao Z, Graybuck LT, Smith KA, Nguyen TN, Bertagnolli D, Goldy J, Garren E, Economo MN, Viswanathan S, Penn O, Bakken T, Menon V, Miller J, Fong O, Hirokawa KE, Lathia K, Rimorin C, Tieu M, Larsen R, Casper T, Barkan E, Kroll M, Parry S, Shapovalova NV, Hirschstein D, Pendergraft J, Sullivan HA, Kim TK, Szafer A, Dee N, Groblewski P, Wickersham I, Cetin A, Harris JA, Levi BP, Sunkin SM, Madisen L, Daigle TL, Looger L, Bernard A, Phillips J, Lein E, Hawrylycz M, Svoboda K, Jones AR, Koch C & Zeng H (2018). Shared and distinct transcriptomic cell types across neocortical areas. Nature 563, 72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Raghavan S, Winter PS, Navia AW, Williams HL, DenAdel A, Lowder KE, Galvez-Reyes J, Kalekar RL, Mulugeta N, Kapner KS, Raghavan MS, Borah AA, Liu N, Vayrynen SA, Costa AD, Ng RWS, Wang J, Hill EK, Ragon DY, Brais LK, Jaeger AM, Spurr LF, Li YY, Cherniack AD, Booker MA, Cohen EF, Tolstorukov MY, Wakiro I, Rotem A, Johnson BE, McFarland JM, Sicinska ET, Jacks TE, Sullivan RJ, Shapiro GI, Clancy TE, Perez K, Rubinson DA, Ng K, Cleary JM, Crawford L, Manalis SR, Nowak JA, Wolpin BM, Hahn WC, Aguirre AJ & Shalek AK (2021). Microenvironment drives cell state, plasticity, and drug response in pancreatic cancer. Cell 184, 6119–6137 e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Inoue M & Horimoto K (2017). Relationship between regulatory pattern of gene expression level and gene function. PLoS One 12, e0177430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Mitsis T, Efthimiadou A, Bacopoulou F, Vlachakis D, Chrousos GP & Eliopoulos E (2020). Transcription factors and evolution: An integral part of gene expression (Review). World Acad Sci J 2, 3–8. [Google Scholar]
- 282.Crow M, Lim N, Ballouz S, Pavlidis P & Gillis J (2019). Predictability of human differential gene expression. Proc Natl Acad Sci U S A 116, 6491–6500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Lee PR & Fields RD (2021). Activity-Dependent Gene Expression in Neurons. Neuroscientist 27, 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Nussinov R, Zhang M, Maloney R & Jang H (2021). Ras isoform-specific expression, chromatin accessibility, and signaling. Biophys Rev 13, 489–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Alvarez-Benayas J, Trasanidis N, Katsarou A, Ponnusamy K, Chaidos A, May PC, Xiao X, Bua M, Atta M, Roberts IAG, Auner HW, Hatjiharissi E, Papaioannou M, Caputo VS, Sudbery IM & Karadimitris A (2021). Chromatin-based, in cis and in trans regulatory rewiring underpins distinct oncogenic transcriptomes in multiple myeloma. Nat Commun 12, 5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Wang T, Song J, Qu M, Gao X, Zhang W, Wang Z, Zhao L, Wang Y, Li B, Li J & Yang J (2021). Integrative Epigenome Map of the Normal Human Prostate Provides Insights Into Prostate Cancer Predisposition. Front Cell Dev Biol 9, 723676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Gui Y, Grzyb K, Thomas MH, Ohnmacht J, Garcia P, Buttini M, Skupin A, Sauter T & Sinkkonen L (2021). Single-nuclei chromatin profiling of ventral midbrain reveals cell identity transcription factors and cell-type-specific gene regulatory variation. Epigenetics Chromatin 14, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Li R, Grimm SA & Wade PA (2021). Low-input ATAC&mRNA-seq protocol for simultaneous profiling of chromatin accessibility and gene expression. STAR Protoc 2, 100764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Xu B, Wang H, Wright S, Hyle J, Zhang Y, Shao Y, Niu M, Fan Y, Rosikiewicz W, Djekidel MN, Peng J, Lu R & Li C (2021). Acute depletion of CTCF rewires genome-wide chromatin accessibility. Genome Biol 22, 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Bum-Erdene K, Liu D, Gonzalez-Gutierrez G, Ghozayel MK, Xu D & Meroueh SO (2020). Small-molecule covalent bond formation at tyrosine creates a binding site and inhibits activation of Ral GTPases. Proc Natl Acad Sci U S A 117, 7131–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Monod J, Wyman J & Changeux JP (1965). On the Nature of Allosteric Transitions: A Plausible Model. J Mol Biol 12, 88–118. [DOI] [PubMed] [Google Scholar]
- 292.Tsai CJ, Kumar S, Ma B & Nussinov R (1999). Folding funnels, binding funnels, and protein function. Protein Sci 8, 1181–90. [DOI] [PMC free article] [PubMed] [Google Scholar]