

Abstract
Membrane transport proteins are involved in the absorption, disposition, efficacy, and/or toxicity of many drugs. Numerous mechanisms (e.g., nuclear receptors, epigenetic gene regulation, microRNAs, alternative splicing, post‐translational modifications, and trafficking) regulate transport protein levels, localization, and function. Various factors associated with disease, medications, and dietary constituents, for example, may alter the regulation and activity of transport proteins in the intestine, liver, kidneys, brain, lungs, placenta, and other important sites, such as tumor tissue. This white paper reviews key mechanisms and regulatory factors that alter the function of clinically relevant transport proteins involved in drug disposition. Current considerations with in vitro and in vivo models that are used to investigate transporter regulation are discussed, including strengths, limitations, and the inherent challenges in predicting the impact of changes due to regulation of one transporter on compensatory pathways and overall drug disposition. In addition, translation and scaling of in vitro observations to in vivo outcomes are considered. The importance of incorporating altered transporter regulation in modeling and simulation approaches to predict the clinical impact on drug disposition is also discussed. Regulation of transporters is highly complex and, therefore, identification of knowledge gaps will aid in directing future research to expand our understanding of clinically relevant molecular mechanisms of transporter regulation. This information is critical to the development of tools and approaches to improve therapeutic outcomes by predicting more accurately the impact of regulation‐mediated changes in transporter function on drug disposition and response.
Transport proteins of the solute carrier (SLC) and ATP‐binding cassette (ABC) superfamilies are widely recognized as key determinants of the absorption, distribution, and excretion of many endogenous compounds and xenobiotics, including drugs, bile acids, hormones, and nutrients, which may thereby directly or indirectly impact medication efficacy and/or safety. Not surprisingly, intersubject variability in drug response due to transporters has been attributed to altered transporter function in organs, such as the intestine, liver, and kidneys. 1 , 2 Mechanisms of alterations in drug transporter activity have focused primarily on (i) differences in gene expression and/or protein abundance due to genetic polymorphisms; and (ii) drug‐drug interactions (DDIs) predominantly involving direct inhibition of transporters. 1 These factors have been reviewed previously, as detailed in numerous International Transporter Consortium White Papers (https://www.itc‐transporter.org/publications.html). Whereas incorporation of these factors into drug disposition predictions has been successful to some extent, current knowledge of tissue protein levels and direct inhibitory DDI mechanisms do not always adequately explain clinically observed transporter‐mediated DDIs, provide accurate predictions of drug disposition in vivo, or explain the extent of intersubject variability in drug exposure and response. Possible reasons for this may be due to alterations in mechanisms beyond genetic polymorphisms that regulate transporter expression, localization, and/or function.
Transport proteins are synthesized in the endoplasmic reticulum, assembled in the Golgi apparatus, and translocated to the apical or basolateral plasma domain of cells where they may be localized directly in the plasma membrane or reside in intracellular storage vesicles. 3 Transporters stored in intracellular compartments may be recruited “on demand” to the membrane, and cycle on and off membranes by exocytic insertion and endocytic retrieval prior to lysosomal degradation. 4 Recently, more complex drug‐transporter interactions and disease‐related changes in transporter expression, localization, and function 5 have emphasized the need for a deeper understanding of the various factors that regulate the structural modifications and requisite localization for transport activity.
For example, reduced systemic exposure of numerous organic anion transporting polypeptide (OATP) 1B1 uptake transporter substrates (e.g., rosuvastatin, pravastatin, pitavastatin, and coproporphyrin I) has been reported following multiple dose administration of the pregnane X receptor (PXR) activators rifampin and/or carbamazepine (see detailed review by Zamek‐Gliszczynski et al.). 6 Although no consistent effect on SLCO1B1/OATP1B1 mRNA or protein could be demonstrated in vitro or in vivo, and PXR does not regulate OATP1B, 7 physiologically based pharmacokinetic (PBPK) modeling suggested that increased OATP1B1 activity could describe the clinical results. These data suggest that mechanisms other than PXR‐mediated transcriptional activation of SLCO1B1 may be involved. Unexpected clinical DDIs with some US Food and Drug Administration (FDA)‐approved tyrosine kinase inhibitors (TKIs) may be explained by the recent discovery that OATP1B1 function is regulated by tyrosine kinase‐mediated phosphorylation. 8 Another intriguing example involves unexpected discrepancies between gene expression and protein levels of intestinal MDR1 P‐glycoprotein (P‐gp), multidrug resistance‐associated protein (MRP) 2, and breast cancer resistance protein (BCRP) after chronic treatment with the PXR ligands rifampin and carbamazepine, which were associated with different expression patterns of regulatory microRNAs (miRNAs) correlating with differential effects on protein amounts of these transporters in the human intestine. 9 Thus, in addition to well‐established genetic and transcriptional regulation, epigenetic and post‐transcriptional factors also may contribute to variability and poor predictability of pharmacokinetics, efficacy, as well as DDIs. 10
Transporter regulation mechanisms also are important from a drug safety standpoint. For example, accurate assessment of drug‐induced liver injury liability associated with bile salt export pump (BSEP) inhibition is a widely recognized challenge in drug development. BSEP inhibition is typically assessed in drug discovery using in vitro screening tools that lack functional cellular regulatory machinery. 11 Increased hepatic bile acids due to direct inhibition of BSEP activate nuclear receptors (NRs), most notably farnesoid X receptor (FXR). FXR regulates compensatory mechanisms that control bile acid homeostasis, including bile acid synthesis, qualitative and quantitative composition, and excretion by other transporters (e.g., organic solute transporter α/β (OSTα/β), MRP3, and MRP4). The interplay of these factors may contribute to the low predictive accuracy for assessing drug‐induced liver injury liability when only measuring direct BSEP inhibition. 11 These clinically relevant examples emphasize the importance of considering the regulation of transport proteins by intrinsic and/or extrinsic factors in drug development.
The science of understanding relevant underlying mechanisms of transporter regulation is at an early stage and many of the published studies have used in vitro and animal models. Lack of in vitro‐to‐in vivo correlations and species differences in transporters and regulatory pathways may limit translation of data from these systems. 12 This white paper reviews important mechanisms that can influence the activity of transporters that are relevant in drug development with a focus on new information that may contribute to intersubject variability in transporter expression and/or function. It further highlights experimental approaches to investigate regulation mechanisms of drug transporters, including established tools and emerging techniques, as well as modeling and simulation approaches to predict the clinical impact of transporter regulation.
MECHANISMS OF TRANSPORTER REGULATION
Nuclear receptors
One of the most prominent mechanisms of transporter regulation involves NRs, which act as transcription factors. Table 1 provides a snapshot of the importance of NRs in transporter regulation. After binding endogenous or exogenous ligands (e.g., drugs and nutrients), NRs typically dimerize with another NR (usually retinoid X receptor) and subsequently bind to specific DNA sequences (receptor‐specific DNA‐binding domains) to initiate transcription of multiple detoxification genes (Figure 1a ). 13 Generally, NR activation results in markedly increased expression and function of drug transporters and metabolizing enzymes. Whereas this mechanism is highly desirable from an evolutionary standpoint to accelerate elimination of potentially harmful xenobiotics from the human body, it causes challenges for pharmacotherapy with certain combinations of drugs. Co‐administration of NR ligands (i.e., potent inducers of drug transporter expression) such as rifampin, hyperforin (the ingredient in St. John’s wort that activates PXR), and carbamazepine with drug transporter substrates can result in clinically relevant DDIs due to diminished systemic or tissue drug exposure, which may reduce drug efficacy. 14 With respect to transporter regulation, the NRs PXR, constitutive androstane receptor (CAR) and FXR are of high relevance. Although it is known that the activation of different NRs is associated with a receptor‐specific induction profile (e.g., PXR regulates P‐gp, MRP2, and cytochrome P‐450 (CYP) 3A4/2C enzymes), knowledge of the actual impact of NR agonists and antagonists on transporter function in humans remains incomplete because studies have been performed in vitro or via indirect inference in vivo. Direct regulation of human drug transporters (mRNA and/or protein abundance) was demonstrated in only a few studies for the human intestine and focused on the ABC transporters P‐gp and MRP2 after oral administration of PXR ligands. 7 , 15 However, the underlying mechanism(s) cannot always be derived directly from such rather descriptive studies. Although there is also functional evidence from many pharmacokinetic DDI studies with NR ligands, data interpretation is complex and does not allow discrimination between transporter regulation in the intestine, liver, kidneys, or other organs. 14
Table 1.
Snapshot of mechanisms of regulation for drug transporters a
| Transport proteins | Nuclear receptors/Transcription factors | Epigenetic | miRNA | Alternative splicing | Post‐translational | Trafficking |
|---|---|---|---|---|---|---|
| MATEs | ||||||
| MATE1 | Methylation (CL‐K) | |||||
| MATE2‐K | Methylation (CL‐K) | |||||
| OATs | ||||||
| OAT1 | HNF1 (NC‐K); HNF4 (CL‐K) | Methylation (PC‐K) |
Phosphorylation (CL‐K) Glycosylation (CL) Ubiquitination (CL‐K) |
Glycosylation (CL) Ubiquitination (CL‐K) | ||
| OAT3 | HNF1 (PC‐K); HNF4 (NC‐K) | Methylation (PC‐K) |
Phosphorylation (CL‐K) Glycosylation (CL) SUMOylation (CL‐K) |
Glycosylation (CL) SUMOylation (CL‐K) | ||
| OATPs | ||||||
| OATP1B1 |
FXR (PC‐H) LXRα (PC‐H) HNF4α (PC‐H) |
Methylation (C‐H) | CL‐H | C‐H |
Phosphorylation (CL‐H) |
CL‐H |
| OATP1B3 |
FXR (PC‐H) HNF1α (CL‐H) |
Methylation (C‐H) | CL‐H | Phosphorylation (PC‐H) | CL‐H | |
| OATP2B1 |
HNF4α (CL‐H) TNFα (C‐P) |
CL‐I CL‐B, NC‐B C‐H |
C‐H, CL‐H C‐I |
CL‐I | ||
| OCTs | ||||||
| OCT1 |
HNF1α (CL, C‐H) HNF4α (CL‐H) |
Methylation (C‐H) | Phosphorylation (CL‐H) | |||
| OCT2 | HNF4 (NC‐K) |
Methylation (PC‐K) Acetylation (PC‐K) |
Phosphorylation (CL‐K) Glycosylation (CL‐K) | Glycosylation (CL‐K) | ||
| BCRP |
AhR (CL‐I), PPARα/γ (CL‐I) PPARα (CL‐B) TNFα (CL‐B, C‐P) EGF (PC‐P) HNF2 (C‐P) CAR (PC‐H) PXR (PC, C‐H) AhR (PC‐H) NRF2 (PC‐H, CL‐LU) |
CL‐I C‐P |
Tyr‐Phosphorylation (C‐I) | CL‐I, CL‐LU | ||
| BSEP | FXR (PC‐H) | Ubiquitination CL‐H | ||||
| P‐gp |
PXR (C‐I) PXR (PC‐H) CAR (PC, C‐H) VDR (CL‐B) |
Methylation (CL‐B) |
CL‐I; C‐I CL‐H, C‐P, CL‐LU |
C‐B, CL‐B, CL‐K | Phosphorylation (CL‐K) | CL‐K, CL‐LU |
| MRPs | ||||||
| MRP1 | NRF2 (CL‐LU) | NC‐B, CL‐LU | CL‐LU | CL‐LU | ||
| MRP2 |
PXR (C‐I) PXR (PC‐H) CAR (PC‐H) NRF2 (PC‐H) |
CL‐I CL‐H |
CL‐H, CL‐LU | |||
Not included are single nucleotide polymorphisms, disease‐related, age, gender, race, or other intrinsic/extrinsic factors. Focus is on clinically relevant findings and transporters highlighted by the ITC. 2 Nonclinical data are only included if human‐relevant data are not available. For references, the reader is referred to Tables 2 , 3 , 4 ; Tables [Link] , [Link] , [Link] .
B, brain; C, clinical; CL, cell line; H, hepatic; I, intestine; K, kidney; NC, nonclinical in vivo organ; P, placenta; PC, primary cell; LU, lung.
AhR, aryl hydrocarbon receptor; BCRP, breast cancer resistance protein; BSEP, bile salt export pump; CAR, constitutive androstane receptor; EGF, epidermal growth factor; FXR, farnesoid X receptor; HNF, hepatocyte nuclear factor; LXR, liver X receptor; MATE, multidrug and toxin extrusion; miRNA, microRNA; MRP, multidrug resistance‐associated protein; NRF2, nuclear factor‐erythroid factor 2‐related factor 2; OAT, organic anion transporter; OATP, organic anion transporting polypeptide; OCT, organic cation transporter; P‐gp, MDR1 P‐glycoprotein; PPAR, peroxisome proliferator‐activated receptors; PXR, pregnane X receptor; TNFα, tumor necrosis factor alpha; Tyr, tyrosine; VDR, vitamin D receptor.
Figure 1.
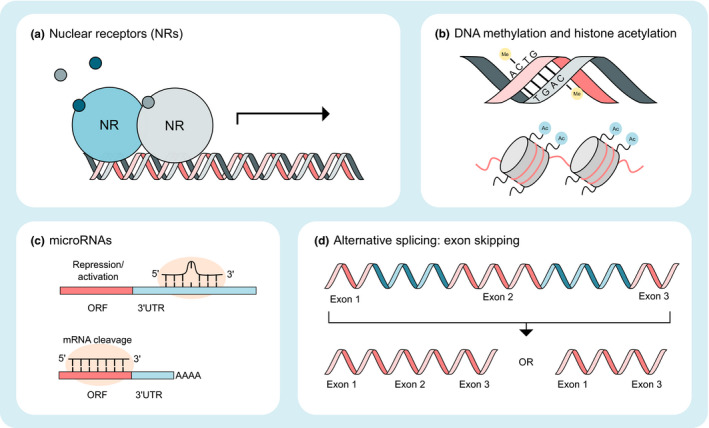
Mechanisms of transporter regulation. (a) Nuclear receptors (NRs) bind ligands and attach to specific DNA sequences, often dimerized with another NR, to initiate transcription. (b) DNA methylation can decrease gene expression by disturbing the binding of transcription factors or co‐activators. Histone acetylation can unfold chromatin leading to a decrease in the binding affinity between histones and DNA, thereby resulting in an increase in gene expression. (c) microRNAs (miRNAs) are small non‐coding RNAs that can suppress (or potentially activate) translation by binding to 3′‐UTR regions of mRNA or initiate mRNA degradation through perfect complementarity with the mRNA. (d) With alternative splicing, multiple mRNAs can be produced from one gene, which can then result in different proteins. In humans, exon skipping is the most common form of alternative splicing. ORF, open reading frame; UTR, untranslated region.
Moreover, very little is known about the expression pattern of NRs in different tissues, and the distinct tissue concentrations of NR ligands, which are indispensable prerequisites to conclude that transporter regulation occurs in vivo. Tissue‐specific NR expression profiles suggest profound differences in the induction potential of NR ligands in different organs depending on their binding affinity to the various NRs and the respective abundance of NRs and transporters. 16 Based on current knowledge, PXR seems to play an important role in the clinically relevant regulation of human intestinal drug transporters, whereas in the liver, CAR and to a lesser extent PXR appear to be more relevant for DDIs involving hepatic transporter regulation. 9 , 17 Hence, the route of drug administration (i.e., oral vs. intravenous) can have a profound impact on the extent of DDIs for drugs undergoing significant NR‐mediated regulation (e.g., CYP3A4 metabolism and/or P‐gp efflux). 15
In addition to DDIs caused by drug‐mediated activation of NRs, interindividual differences in the plasma concentration of endogenous NR ligands (e.g., hormones and bile acids) may contribute to clinical variability in transporter expression and function. For example, in cholestatic liver diseases, intrahepatic bile acid accumulation leads to FXR and PXR activation and to significant changes in hepatic transporter expression. 5 Likewise, lifestyle factors, including diet, alcohol consumption, and smoking, can affect transporter expression and function. 18 However, for several transport proteins, including BCRP, organic cation transporter (OCT) 1, OCT3, peptide transporter (PEPT) 1, and OATP2B1, although some evidence for transcriptional regulation exists based on results obtained in cell lines (Tables 1 , 2 , 3 , 4 ), the in vivo regulation by NRs remains uncertain and requires further research.
Table 2.
Mechanisms of regulation for transporters in the intestine a
| Transport proteins | Mechanism | Model system | Agonist/Causes | mRNA | Protein | Activity | Reference |
|---|---|---|---|---|---|---|---|
| BCRP | PXR | Duodenal biopsy (N = 12) | Rifampin (600 mg, 6 days) | ↔ | ↔ | ND | 9 |
| PXR/CAR | Duodenal biopsy (N = 7) | Carbamazepine (600 mg, 14–18 days) | ↑ | ↔ | ND | 9 | |
| AhR | Caco‐2 | Various AhR ligands | ↑↑ | ↑ |
Apical transport of benzo[a]pyrene‐sulfate ↑ |
57 | |
| DNA methylation, miRNA (indirect) | Colon carcinoma and adjacent “normal” tissue, colon (cancer) cell lines | DNA methyltransferase DNMT3b, siRNA miRNA‐203 (indirect) | ↑↑ | ND | ↑↑ | 119 | |
| miRNA and RNA binding protein | S1 and S1M1‐80, Caco‐2, HT‐29, and SW‐620 cells | miRNA‐519c | ↓ | ↓↓ | ↓↓ | 60 | |
| Localization | HT‐29 and Caco‐2 | Uric acid (6 or 8 mg/dL) | ↑ | ↑ | ↑ | 120 | |
| Post‐translational | Colon biopsy from patients with chronic low‐grade inflammation–associated obesity | Loss of tyrosine phosphorylation (Janus kinase 3) | ND | ↓↓ | ND | 121 | |
| MRP2 | PXR | Duodenal biopsy (N = 16) | Rifampin (600 mg, 9 days) | ↑ | ↑ | ND | 122 |
| PXR/CAR | Duodenal biopsy (N = 7) | Carbamazepine (600 mg, 14–18 days) | ↑↑ | ↔ | ND | 9 | |
| OATP2B1 | miRNA | Caco‐2 and HEK‐OATP2B1 | miRNA‐24 mimic |
Caco‐2: ↓↓ HEK‐OATP2B1: ↓ |
Caco‐2: ↓↓ HEK‐OATP2B1: ↓↓ |
Caco‐2: ↔ HEK‐OATP2B1: ↓ |
123 |
| Post‐translational, internalization |
MDCKII‐OATP2B1 Caco‐2 |
Phorbol 12‐myristate 13‐acetate induced PKC activation | ND | ↓ |
↓ |
51 | |
| Localization | HEK‐OATP2B1 Caco‐2 |
Amiodarone Rutin Insulin (via Rab1) |
ND |
Plasma membrane↑ Total ↔ |
↑ |
124 | |
| P‐gp | PXR | Duodenal biopsy | Rifampin (600 mg, 10 days) | ND | ↑↑ | ↑ | 125 |
| PXR | Duodenal biopsy | Rifampin (600 mg, 9 days) | ↑ | ↑↑ | AUC/Cmax of talinolol reduced by 35%/38%; ↑ | 126 | |
| PXR/CAR | Duodenal biopsy (N = 7) | Carbamazepine (600 mg, 14–18 days) | ↑↑ | ↔ | ND | 9 | |
| miRNA | Intestinal tissue, luciferase reporter assay (HepG2) | miRNA‐27a‐3p and miRNA‐409‐3p | ND | ↓ | ND | 22 |
↑, < 2‐fold increase; ↑↑, ≥ 2‐fold increase; ↓, < 2‐fold decrease; ↓↓, ≥ 2‐fold decrease; ↔, no change; ND, not determined.
AhR, aryl hydrocarbon receptor; AUC, area under the curve; BCRP, breast cancer resistance protein; Caco‐2, human colorectal adenocarcinoma cell; CAR, constitutive androstane receptor; Cmax, maximum concentration; HEK, human embryonic kidney; HT‐29, human colonic adenocarcinoma cell line; MDCK, Madin‐Darby canine kidney; miRNA, microRNA; MRP, multidrug resistance‐associated protein; OATP, organic anion transporting polypeptide; P‐gp, P‐glycoprotein; PKC, protein kinase C; PXR, pregnane X receptor; siRNA, small interfering RNA; S1, human colon cancer cell line S1; S1M1‐80, mitoxantrone resistant S1 cell line derivative; SW‐620, human colonic adenocarcinoma cell line.
Table 3.
Mechanisms of regulation for transporters in the liver a
| Transport proteins | Mechanism | Model system | Agonist/Causes | mRNA | Protein | Activity | Reference |
|---|---|---|---|---|---|---|---|
| OATP1B1 | HNF4α | HH | HNF4α siRNA | ↓↓ | ND | ND | 127 |
| LXRα |
Huh7 HH |
TO901317 GW3965 |
↑↑ |
Huh7, ND HH, Relative Increase |
Huh7 (TO901317, GW3965), ↑↑ HH (TO91317), ↑↑ |
68 | |
| PXR | HH | Rifampin | ↔ or ↑↑ | ND | ND | ||
| FXR |
Huh7 HH |
CDCA GW4064 Fexaramine |
↑↑ |
HH, CDCA, Relative Increase |
Huh7, ↑ HH, CDCA, ↑↑ |
68 | |
| miRNA |
Huh7 Human liver tissue Chang liver cells |
miRNA‐206 miRNA‐511 |
↓↓ | ↓ | ↓; ND | 71 , additional references in Table S2 | |
| Epigenetics (DNA methylation) | Human liver tissue | Hypomethylated regions in liver were identified around the transcriptional start site | ND | ND | ND | 74 | |
| LYN kinase‐mediated tyrosine phosphorylation | HEK‐OATP1B1 | Nilotinib | ND | ND | ↓↓ | 8 | |
| PKC | HEK‐OATP1B1 | Phorbol 12‐myristate 13‐acetate | ND |
Plasma membrane ↓ |
↓↓ | 128 | |
| Alternative splicing | Postmortem human livers | Alternative splicing of gene occurs frequently in children | ND | ND | ND | 32 | |
| OATP1B3 | FXR |
HH HG2 Huh7 |
CDCA | ↑↑ | ND | ND | 68 , additional references in Table S2 |
| HNF1α | HG2 | Overexpression HNF1α | Promoter ↑↑ | ND | ND | 129 | |
| PKC | HH | Phorbol 12‐myristate 13‐acetate | ↔ |
Phosphorylated protein ↑ |
↓↓ | 76 | |
| Epigenetics (methylation) | HG2 | 5‐aza‐2′‐deoxycytidine (DNA methylation inhibitor) | ↑↑ | ND | ND | 130 | |
| Alternative splicing | Tumor specific OATP1B3 variant | Lacking N‐terminal 28 amino acids | ND |
Abundance in plasma membrane ↓; predominantly cytoplasmic expression |
↓↓ | 73 | |
| OATP2B1 | HNF4α | Huh7 | Overexpression of HNF4α siRNA | Liver‐enriched OATP2B1 mRNA variant ↓ | ND | ND | 131 |
| Post‐translational internalization | MDCKII | Phorbol 12‐myristate 13‐acetate induced PKC activation | ND |
Plasma membrane ↓↓ |
ND | 51 | |
| miRNA |
Human liver HG2 HepaRG |
miRNA‐24 | Human liver: expression level of miRNA‐24 negatively correlated with OATP2B1 mRNAHG2: promoter activity ↓↓;HG2 and HepaRG: mRNA ↓↓ |
Human liver: expression level of miRNA‐24 negatively correlated with OATP2B1 protein HG2: ND HepaRG: ↓↓ |
ND | 72 | |
| OCT1 | HNF4α | HepaRG | HNF4α siRNA |
↓↓ |
ND | ND | 31 and Table S2 |
| HNF1α |
HG2 Huh7 Human liver samples (correlation study) |
In silico assay and expression correlation study | Promoter activity; correlation between HNF1 and OCT1 mRNA expression ↑ | ND | ND | 132 | |
| Epigenetic (methylation) | Human liver tissue biopsies | Hepatocellular carcinoma | Relative decrease | ↓↓ | ND | 133 | |
| YES1 kinase‐mediated tyrosine phosphorylation |
HEK Mouse liver |
Dasatinib | ND | ND | ↓↓ | 82 | |
| BCRP | CAR | HH | Phenobarbital | ↑ | ND | ND | 69 |
| PXR |
HH Human liver biopsy |
Rifampin Carbamazepine |
↑↑ | ND | ND | 7 | |
| NRF2 | HH | Oltipraz | In 5/7 donors ↑↑ | ND | ND | 134 | |
| AhR | HH | TCDD | ↑↑ | ND | ND | 69 | |
| BSEP | FXR |
HH HG2 |
CDCA OCA Oxysterol 22(R)‐Hydroxycholesterol GW4064 |
↑↑ ↑↑ ↑↑ ↑↑ |
Increase | ND | 135 |
| P‐gp | PXR |
HH Human liver slices |
Rifampin | ↑↑ | ↑ (HH only) | ND | 7 |
| CAR |
HH Human liver slices |
Phenobarbital | ↑↑ | ↑↑ (HH only) | ND | 7 | |
| NRF2 |
HG2 Liver (mice) |
Bajijiasu herb |
↑↑ ↑↑ |
↑↑ ↑↑ |
↑ ND |
136 | |
| NF‐κB and AKT dependent MAPK activation | HG2 | Deoxynivalenol | ↑↑ | ↑↑ | ND | 137 | |
| miR‐223 | HCC cell lines | miRNA | ↓↓ | Relative down regulation | miRNA‐223 over‐expression increases HCC cell sensitivity to doxorubicin and paclitaxel | 138 | |
| MRP2 | PXR | HH | Rifampin | ↑↑ | ND | ND | 134 |
| CAR | HH | Phenobarbital | 7/7 donors ↑↑ | ND | ND | 134 | |
| NRF2 | HH | Oltipraz | 5/7 donors ↑↑ | ND | ND | 134 | |
| miRNA and alternative polyadenylation | HG2 | miRNA‐379 | Longer 3'‐UTR variants ↓ | ND | ND | 59 |
↑, < 2‐fold increase; ↑↑, ≥ 2‐fold increase; ↓, < 2‐fold decrease; ↓↓, ≥ 2‐fold decrease; ↔, no change; ND, not determined.
AhR, aryl hydrocarbon receptor; BCRP, breast cancer resistance protein; BSEP, bile salt export pump; CAR, constitutive androstane receptor; CDCA, chenodeoxycholic acid; CHO, chinese hamster ovary cells; FXR, farnesoid X receptor; HCC, hepatocellular carcinoma; HEK, human embryonic kidney cells; HepaRG, hepatic progenitor cell line; HG2, human hepatoma (HepG2) cells; HH, human hepatocytes; HL, HeLa cells; HNF, hepatocyte nuclear factor; Huh7, human hepatocyte carcinoma derived cell line; LXR, liver X receptor; MAPK, mitogen‐activated protein kinase; miRNA, microRNA; MDCK, Madin‐Darby canine kidney cells; MRP, multidrug resistance‐associated protein; NRF2, nuclear factor‐erythroid factor 2‐related factor 2; NF‐κB, nuclear factor kappa B; OATP, organic anion transporting polypeptide; OCA, obeticholic acid; OCT, organic cation transporter; P‐gp, P‐glycoprotein; PKC, protein kinase C; PRL, prolactin; PXR, pregnane X receptor; siRNA, small interfering RNA; TCCD, 2,3,7,8 ‐Tetrachlorodibenzo‐p‐dioxin; UTR, untranslated region.
Table 4.
Mechanisms of regulation for transporters in the kidneys a
| Transport proteins | Mechanism | Model system | Agonist/Causes | mRNA | Protein | Activity | Reference |
|---|---|---|---|---|---|---|---|
| MATE1 | Promoter methylation | LS174T and HaCat cells |
Demethylation 5‐aza‐2deoxycytidine |
↑↑ | ND | ND | 139 |
| MATE2K | Histone methylation | 769‐P and 786‐O cells | H3K4me3 enrichment | ↓↓ | ND | ND | 140 |
| OAT1 | PKA activity | OK cells | Forskolin | ND | ND | ↑ | 141 |
| PKC activity | OK cells | Parathyroid hormone (0.1 µM) | ND | ND | ↓↓ | ||
| MAPK pathway activity | OK cells | PD98059 | ND | ND | ↑ | 141 | |
| Ubiquitination and internalization | COS‐7 cells | USP8 overexpression | ND | ↑ | ↑ | 89 | |
| Ubiquitination and degradation | Overexpressing HEK‐293 cells |
Bortezomib Carfilzomib |
ND | ↑ | ↑ | 142 | |
| OAT3 | Promoter methylation | HepG2, Caco‐2, and HEK293 cells | 5‐aza‐2deoxycytidine | ND | ↑↑ | ND | 141 |
| PKA activity; SUMOylation ↑ Ubiquitination ↓ | Overexpressing COS‐7 cells | Bt2‐cAMP | ND |
Plasma membrane abundance ↑ Total abundance ↔ |
↑ | ||
| OCT2 | Unknown | MDCK cells | Dexamethasone Hydrocortisone | ↑↑ | ND | ↑ | 83 |
| Promoter methylation | Patient tissue, Renal Cell Carcinoma Cells, HEK‐293 cells |
Demethylation Decitabine |
↑↑ | ↑↑ | ↑↑ | 85 | |
| Histone acetylation | Patient tissue, Renal Cell Carcinoma Cells, HEK‐293 cells | Vorinostat | ↑↑ | ↑↑ | ↑↑ | 21 | |
| PKA‐mediated phosphorylation | Overexpressing HEK‐293 cells | Forskolin | ND | ND | ↓ | 86 | |
| PI3K‐mediated phosphorylation | Overexpressing HEK‐293 cells | Wortmannin | ND | ND | ↑ | 86 | |
| Tyrosine phosphorylation (YES1‐mediated) | Overexpressing HEK‐293 cells, FVB mice |
Dasatinib siRNA Y362F mutant |
ND | ↔ | ↓↓ | 39 | |
| Glycosylation | Overexpressing CHO cells | N96Q | ND | ↔ | ↓↓ | 87 | |
| P‐gp | Post‐transcriptional/Alternative splicing | SA7K cells (pseudo‐immortalized primary RPTECs) | ADAR1 mouse knockout | ↓↓ | ↓ | ND | 30 |
| NRF2 activation | HK‐2 shKEAP1 (stable KEAP1 knockdown) | KEAP1 mouse knockout | ↑↑ | ↑↑ | ↑ | 144 | |
| Transcriptional (decreased Src signalling, JNK activation) | Caki‐1 | 5‐aza‐20‐deoxycytidine | ↓ | ND | ↓ | 145 | |
| Localization (lipid rafts) | MDCK‐MDR1 cells | Methyl‐β‐cyclodextrin | ND |
Plasma membrane abundance ↓ |
↓↓ | 146 | |
| PKC activation | LLC‐GA5 Col300 cell (LLC‐PK1‐MDR1) | Phorbol 12,13‐dibutyrate | ND | ND | ↑ | 147 |
↑, < 2‐fold increase; ↑↑, ≥ 2‐fold increase; ↓, < 2‐fold decrease; ↓↓, ≥ 2‐fold decrease; ↔, no change; ND, not determined.
aADAR, adenosine deaminase; Caki‐1, human kidney clear cell carcinoma cell line; COS‐2, cercopithecus aethiops kidney cell line; CHO, Chinese hamster ovaries cell line; COS‐7, CV‐1 in origin with SV‐40 genes cells; H3K4me3, trimethylation on histone H3; HaCat, immortalized human keratinocytes; HEPG2, human hepatoma cell line; HK‐2, human kidney proximal tubule cells; KEAP, Kelch‐like ECH protein; LLC‐PK1, Lilly Laboratories culture‐porcine kidney cells; MDCK, Madin‐Darby canine kidney cells; MAPK, mitogen‐activated protein kinase; MATE, multidrug and toxin extrusion; NRF2, nuclear factor‐erythroid factor 2‐related factor 2; OAT, organic anion transporter; OCT, organic cation transporter; OK, opossum kidney cells; P‐gp, MDR1 P‐glycoprotein; PI3K, phosphoinositide 3‐kinase; PK, protein kinase; RPTECs, renal proximal tubule epithelial cells; SA7K, human proximal tubule epithelial cell line; siRNA, small interfering RNA; USP, ubiquitin specific peptidase 8.
Epigenetic gene regulation
Epigenetics is defined as heritable variation of the expression of a gene that is not caused by changes in the DNA sequence. Epigenetic mechanisms, such as DNA methylation and histone acetylation, are also known to regulate transporters (Figure 1b ). The process of DNA methylation causes the addition of methyl groups to the DNA strands, and predominately occurs at the dinucleotide CpG sequences in the promoter region of a gene. As a result, transcription of the corresponding gene may be inhibited. 19 For example, the expression of the uptake transporter OCTN1 in acute myeloid leukemia cell lines and potentially in patients is dependent on a DNA methylation‐based mechanism that is sensitive to modulation by hypomethylating agents such as cytarabine (see reference 150 in Table S1 ). Histone acetylation and deacetylation primarily occur at lysine residues at the N‐terminus of histones that are enriched around the transcription start site and are associated with gene activation and inactivation, respectively. 20 Histone deacetylation, in addition to DNA hypermethylation, has been found to be relevant for repression of SLC22A2 (encoding OCT2) in renal cell carcinoma (RCC). 21 This may be particularly relevant in the case of treatment of patients with platinum compounds, such as the OCT2 substrate oxaliplatin. In RCC cell lines, the DNA methylation inhibitor decitabine and the histone deacetylase inhibitor vorinostat increased OCT2 abundance, which sensitized the cells to oxaliplatin. 21
Post‐transcriptional regulation by miRNAs
In addition to transcriptional regulation by NRs, post‐transcriptional regulation by miRNAs—a form of epigenetics—may be another source of interindividual variability in the expression and function of drug transporters (Table 1 ). The miRNAs are small non‐coding RNAs that bind to certain mRNA molecules, thereby initiating their degradation or blocking subsequent translation processes (Figure 1c ). 10 One example highlighting the potential relevance of miRNAs and drug transporters relates to the differences in the protein abundance of P‐gp, PEPT1, and MRP3 along the length of the human intestine, which correlated with the expression of regulatory miRNAs. 22 Therefore, environmental factors that affect the expression pattern of miRNAs, such as smoking, diet, and certain drugs, may result in modified transporter protein abundance. 10 Moreover, pathological conditions, such as cancer or inflammatory diseases, have been associated with changes in the expression pattern of transporter‐regulating miRNAs, and, in turn, with changes in the mRNA expression and/or protein abundance of the respective transporters. 23 , 24 Furthermore, the highly variable expression of NRs was attributed not only to genetic factors, 25 but also to miRNA regulation. 9 Vice versa, NR ligands also demonstrated significant induction of miRNA expression, indicating a complex coordinated network between the different regulatory factors. 26 This may also explain, in part, the observed differences in the induction properties between different NR ligands (e.g., rifampin vs. carbamazepine) that cause different induction patterns of regulatory miRNAs. 9
Alternative splicing
RNA splicing refers to the editing process of precursor mRNA transcripts to form mature mRNAs. Alternative splicing, the process where differential splicing can produce multiple mRNAs from one gene, results in proteins that potentially can have different functions. 27 In humans, the most prevalent form of alternative splicing is exon skipping (Figure 1d , Table 1 ). For ABCB11 (encoding BSEP), patients with the c.1445A>G variant develop PFIC‐2 due to the use of a cryptic splice site in exon 14 resulting in the insertion of 14 new amino acids and a premature stop codon. 28 Exon skipping or aberrant splicing in ABCC2 (encoding MRP2) has been reported to result in Dubin‐Johnson syndrome. 29 For ABCB1 (encoding P‐gp), alternative splicing in intron 27 resulted in significant 24‐hour oscillation in P‐gp protein levels in human renal proximal tubular epithelial cells and adenosine deaminase acting on RNA expression correlated with the alternative splicing event. 30 Whether this effect will translate to humans is not clear yet.
Alternative splicing in SLC transporters is common. In SLC22A1 (encoding OCT1), multiple spliced isoforms generated by exon skipping have been identified, several of which result in the loss of transporter function or poor outcomes in patients with cancer treated with imatinib or sorafenib. 31 A study investigating alternative splicing of the SLCO1B1 gene (encoding OATP1B1) as a function of age revealed that alternative splicing occurred commonly in pediatric liver tissue. Most of the splice variants, however, were predicted to code for truncated forms that may lack transporter activity or to translate into the same amino acid sequence as the reference isoform of OATP1B1 (Table 3 ). 32 An interesting example of alternative splicing resulting in a new transporter protein is OATP1B3‐1B7, which is the combination product of splicing of exons encoding SLCO1B3 and the pseudo gene SLCO1B7. 33 The result is a protein with 12 transmembrane domains mostly localized in the endoplasmic reticulum. Whether OATP1B3‐1B7 is of clinical relevance remains to be tested in pharmacogenetic studies. 34
Nuclear factor kappa B (NF‐kB) and inflammatory responses
Signaling pathways underlying stress‐induced regulation of transporters are complex and not well‐understood. For instance, in isolated rat blood‐brain‐barrier (BBB) capillaries, protein levels of efflux transporters were upregulated in response to chronic inflammation, oxidative stress, DNA damage, and seizures. 35 Each of these stimuli activates distinct multistep signaling cascades leading to activation of the transcription factor NF‐kB resulting in increased P‐gp protein levels. Conversely, inhibition of NF‐kB activation or nuclear translocation can block increased P‐gp activity following exposure to pro‐inflammatory cytokines, such as tumor necrosis factor alpha. Interestingly, the effect of NF‐kB‐mediated stimulation of P‐gp expression in the rat brain is different from the mouse liver, where inhibition of NF‐kB was associated with an attenuation of the endotoxin‐mediated decrease in Abc and Slco transporter expression. 36 Although acute inflammation is known to downregulate Pxr in mice, this does not seem to explain the endotoxin‐mediated downregulation of transporters. 36 Whether the effects of NF‐kB are due to a direct or indirect effect on transporter gene transcription is unclear. Of note, it is not known whether the mechanistic studies conducted in rodent models to understand the effect of stress and inflammation on transporters translate to humans.
Post‐translational modifications
The most common post‐translational modifications (PTMs) involved in transporter regulation include phosphorylation, glycosylation, ubiquitination, SUMOylation, acetylation, and palmitoylation. A comprehensive review on this topic was published recently. 37 PTMs can modulate transporter function, expression, efficiency, structure, and trafficking, as described below.
Phosphorylation without changes in membrane expression can result in an on/off switch as has been demonstrated for OCT2. 38 Clinically, this could be relevant in patients treated with cisplatin, because TKIs, such as dasatinib, reduced OCT2 function in both cell lines and in kidney tissue. 39 As discussed later in this review, TKIs also affect the function of other transporters.
N‐glycosylation of transporters occurs on extracellular asparagine residues. 37 Although the role of glycosylation is unclear in most cases, evidence suggests that it affects resistance against proteases and, therefore, protein stability. Interestingly, differential glycosylation of MRP4 affected substrate specificity. 40 In OATP1B1, disruption of all glycosylation sites resulted in lower protein stability with reduced total protein levels. Non‐glycosylated OATP1B1 was detected in the endoplasmic reticulum and not in the plasma membrane. 41 N‐glycosylation plays three roles in the functional expression of SLC26A2 and SLC26A3 proteins: to retain misfolded proteins in the endoplasmic reticulum, to stabilize the protein at the cell surface, and to maintain the transport protein in a functional state. 42
Ubiquitination is a common mechanism resulting in degradation of SLC and ABC transporters, and the balance between ubiquitination and de‐ubiquitination may regulate transporter activity in the plasma membrane. For instance, ubiquitination has been implied in P‐gp degradation in cancer cell lines and this may lead to reduced resistance to chemotherapeutic drugs. 43 Small ubiquitin‐like modifier (SUMO) is a small protein that, like ubiquitin, is conjugated to a lysine residue. However, SUMO modification does not cause degradation but rather internalization, and can even result in elevated protein levels as was shown for the glucose transporter (GLUT) 4. 44 Mrp2 can also undergo SUMO modification, which decreased expression, although it was not clear whether expression at the cell surface was altered. 45
Palmitoylation can occur at cysteine residues close to the protein‐lipid interface and is highly dynamic. By association with lipid rafts, it has been hypothesized that palmitoylation can result in rapid endocytosis or increased transporter efficiency by transporter clustering. 37 This mechanism has been described for SLC and ABC transporters involved in the translocation of cholesterol, but it is not clear whether this mechanism is relevant for drug transporters.
Trafficking
Many mechanisms regulate the movement of mature transport proteins from the site of synthesis to the plasma membrane, cycling on and off the membrane, and storage in intracellular compartments, in some cases for ready‐response when needed. 4 Misfolded proteins are retained in the endoplasmic reticulum prior to undergoing degradation. Properly folded proteins are transported from the endoplasmic reticulum to the Golgi apparatus for further processing and covalent modification, followed by packaging into secretory vesicles and transport to the plasma membrane. N‐linked glycosylation on asparagine residues in the large extracellular loops of some organic anion transporters (OATs) and OATPs, 46 as well as sodium taurocholate cotransporting polypeptide (NTCP) 37 control membrane targeting. However, N‐linked glycosylation of asparagine 596 was not essential for routing of BCRP to the plasma membrane. 47 In the case of P‐gp, N‐glycosylation did not directly affect drug transport capacity, but appeared to improve the routing or sorting efficiency of the protein en route to the plasma membrane, or to affect the stability of the protein, either by protection from proteolysis or by facilitating/stabilizing correct protein folding. 48 In interpreting effects of glycosylation, it is important to distinguish between effects of N‐glycosylation on transport protein stability vs. proper routing. Alterations in the Golgi complex by colchicine, a microtubule disruptor, or in the glycosylation status of transporters, can increase intracellular localization of these proteins leading to decreased transporter activity. For example, Western blot analysis revealed that protein amounts of non‐glycosylated OATP1B1, OATP1B3, OATP2B1, NTCP, and MRP2, representing nonfunctional proteins, were increased in liver tissue from patients with nonalcoholic steatohepatitis. 49 Phosphorylation of transporters (either directly or indirectly) catalyzed by kinases, and dephosphorylation by protein phosphatases, can rapidly regulate transporter function by causing internalization or reinsertion of protein into the membrane. 37 Protein kinase C (PKC) activation triggered OAT1 and OATP2B1 internalization from the surface of COS‐7 cells to intracellular compartments and reduced transporter activity. 50 , 51 PKC often initiates cellular signaling that involves other PTMs, as demonstrated for OAT1 where PKC‐mediated internalization occurred through ubiquitination. 37 , 50 Ubiquitination may also serve as a sorting signal to modulate movement to endosomal compartments, as shown for BSEP, and for lysosomal degradation of internalized MRP2. 52
Several molecular partners that interact directly or indirectly with transporters clearly play a role in trafficking of some transporters, which may impact function. For example, p38 mitogen‐activated protein kinase, protein kinase A (PKA), PKC, proto‐oncogene serine/threonine‐protein kinase, and phosphoinositide 3‐kinase (PI3K) are important for ABC transporters, and specific inhibitors of these kinases decreased protein levels of some ABC transporters in the canalicular membrane of hepatocytes. 53 In addition, small GTPases, class V myosins, and adaptor proteins regulate canalicular ABC transporter endocytosis and recycling. Rab11a and myosin Vb regulate recycling of BSEP from the subapical compartment to the bile canalicular membrane, and disruption of the actin cytoskeleton or microtubules can inhibit BSEP trafficking. 53 Ezrin, radixin, and moesin are scaffolding proteins that attach actin filaments to the plasma membrane and serve as anchors for transporters. Cytoskeletal proteins are important regulators of hepatic transporter function, as shown in radixin‐knockout mice that developed conjugated hyperbilirubinemia and liver injury due to loss of canalicular Mrp2. 54 Ezrin, radixin, and moesin proteins have PDZ binding domains that play a role in stabilization, proper membrane localization, and regulation of proteins, including MRP2 and MRP4. 54
Mechanisms regulating transporter trafficking are, by nature, complex and difficult to precisely evaluate in vivo. In part, this is due to lack of specific activators or inhibitors of signaling pathways and transporters. In addition, inhibition of these processes in vivo may result in toxicity. Direct inhibition of transporters by modulators, and the compensatory mechanisms that influence overall disposition of transporter substrates, often confound data interpretation. Recognizing potential alterations in transporter trafficking by drugs, disease, and other intrinsic or extrinsic factors that regulate these processes is imperative to understand the potential impact on drug disposition.
TRANSPORTER PROTEIN REGULATION IN ABSORPTION, DISTRIBUTION, METABOLISM, AND EXCRETION ORGANS
Intestine
P‐gp, MRP2, MRP3, BCRP, OATP2B1, PEPT1, and apical sodium bile acid transporter (ASBT) are functionally relevant in the human intestine. 2 Examples of intestinal transporter regulation are provided below with additional details summarized in Tables 1 and 2 ; Table S1 .
Transcriptional regulation
One of the best understood mechanisms in terms of intestinal transporter regulation from a clinical perspective involves NRs (Tables 1 and 2 ). Human small intestinal tissue shows high expression of PXR followed by FXR, but only traces of CAR. 17 Although transporters are expected to be inducible by respective ligands, PXR‐regulated transporters in the intestine (i.e., P‐gp and MRP2) may be most likely to cause clinically relevant DDIs after oral drug administration. 14 Similarly, bile acids regulate the expression of ASBT, OSTα/β, and MRP3 via FXR activation.
Several studies investigated transporter expression in intestinal biopsies taken from the upper small intestine of healthy volunteers after dosing with prototypical inducers such as rifampin (PXR ligand), carbamazepine (PXR and CAR ligand), or efavirenz (CAR ligand). Potent induction of mRNA and protein was demonstrated for intestinal P‐gp and MRP2 after treatment with rifampin for up to 9 days, resulting in a significant decrease in the oral bioavailability of P‐gp and MRP2 substrates (Table 2 ). 55 Interestingly, intestinal BCRP abundance was not affected by rifampin, carbamazepine, or efavirenz treatment, suggesting that it was not regulated by either intestinal PXR or CAR. 9 , 56 Evidence from expression and functional in vitro studies suggests that aryl hydrocarbon receptor (AhR), peroxisome proliferator‐activated receptor (PPAR) α and/or PPARγ may be involved in the regulation of BCRP, 57 , 58 but significance in humans has not been demonstrated.
Epigenetic/post‐transcriptional regulation
Many miRNAs known to regulate drug transporters are expressed in the intestine and thus could affect proteins involved in drug absorption. 24 However, there are limited in vivo and in vitro data characterizing miRNA‐mediated transporter regulation in intestine‐derived cells or tissue. Differences in miRNA levels in tissue samples from multiple intestinal regions have been linked to regional transporter protein levels. 22 For example, miR‐27a‐3p and miR‐409‐3p regulate ABCB1 and their elevated expression in the colon coincides with significantly lower P‐gp protein levels compared with the small intestine. Alternative polyadenylation, resulting in different lengths of 3′‐UTR regions, as detected for ABCC1, ABCC2, and ABCC3 in cell lines and tissues, could affect tissue‐dependent regulation: for example, ABCC2 with the shortest 3′‐UTR was not regulated by miR‐397, as shown in a luciferase gene reporter assay in HepG2 cells. 59 Similarly, in colorectal cancer, ABCG2 3′‐UTR length was shown to affect ABCG2 regulation mediated by the interplay of miR‐519c and RNA binding protein human antigen R. 60
The role of miRNAs in drug‐mediated transporter induction has been studied in human duodenal biopsy samples. 9 A striking disconnect between the induction of ABCB1 and ABCC2 mRNA but no significant increase in P‐gp or MRP2 protein abundance was reported after carbamazepine treatment. Levels of several miRNAs were increased by rifampin and carbamazepine treatment and inversely correlated with P‐gp, MRP2, and BCRP abundance or protein/mRNA ratios. It is hypothesized that post‐transcriptional regulation by miRNAs, which bind to ABCB1 and ABCC2 mRNA and block translation of the respective proteins, may help explain this discrepancy and suggests the involvement of NR‐independent pathways in transcriptional regulation by some NR ligands. Additional studies are needed to confirm these interesting hypotheses and observations. Furthermore, epigenetics may affect NR expression, in turn influencing NR‐mediated regulation of intestinal transporters. 9
Post‐translational regulation
Mutagenesis of asparagine residues increased murine PEPT1 activity, indicating that N‐glycosylation can affect the function of this intestinal transporter. 61 Mutagenesis of the cysteine residues in OATP2B1 also led to misprocessing and intracellular accumulation in CHO‐OATP2B1 cells. 62 Similarly, S‐acylated ASBT, the predominant form in ileal brush border membrane vesicles derived from human donor ileum, is important for activity, plasma membrane localization, and protection from degradation. 63 Localization of transporters like P‐gp and BCRP in plasma membrane “lipid rafts” impacts efflux activity 64 ; for example, depletion of cellular cholesterol content by methyl‐beta‐cyclodextrin caused a 40% decrease in BCRP activity in MDCKII‐BCRP cells. 64 The activation of PKC accelerated OATP2B1 internalization via the clathrin‐dependent pathway and subsequent lysosomal degradation in MDCKII‐OATP2B1 and Caco‐2 cell monolayers. 51 , 65
Taken together, transporter regulation via intestinal NR activation has resulted in clinically relevant DDIs and intersubject variability in the efficacy and safety of drugs. In contrast, whereas exemplar studies show the effects of PTMs and localization on intestinal drug transporter activity, these have been conducted mostly in vitro and, therefore, require further investigation to document in vivo relevance. Similarly, miRNA‐mediated transporter regulation has been demonstrated mostly in vitro, but the limited in vivo data may provide an explanation for the tissue specific expression of transporters.
Liver
Hepatic transport proteins of clinical relevance include OATP1B1, OATP1B3, BCRP, P‐gp, OCT1, OATP2B1, BSEP, and MRP2. 2 Examples of clinically relevant regulation of hepatic transporters are discussed below with detailed information summarized in Tables 1 and 3 ; Table S2 .
Transcriptional regulation
The mRNA of hepatic transporters can be induced by NRs, including liver X receptor (LXR) (SLCO1B1 and ABCC2), FXR (SLCO1B1, SLCO1B3, ABCC2, ABCB11, SLC10A1, and SLC51A/B), CAR (ABCB1, ABCG2, ABCC2, and ABCC4), and PXR (ABCG2, ABCB1, and ABCC2; Table 3 , Table S2 ). The induction of ABCB11 and SLC51A/B, and increased bile acid transport, have been reported in sandwich‐cultured human hepatocytes by the FXR agonist obeticholic acid, at clinically relevant concentrations, and the endogenous bile acid chenodeoxycholic acid. 66
Among examples of drug‐mediated regulation of hepatic transporters, induction of OATP1B by rifampin has received the most attention in vitro and in clinical studies using OATP1B probe substrates. As thoroughly reviewed, 6 , 7 several clinical studies using multiple‐dose rifampin as an inducer and OATP1B1 probe substrates as victim drugs support the potential induction of OATP1B1 activity, 6 but the induction of SLCO1B1 mRNA and OATP1B1 protein and transport function has not been demonstrated consistently in human hepatocytes (Table 3 ). One major challenge with mechanistic clinical studies on NR regulation is the fact that NRs modulate gene networks; hence, it may be difficult to separate the impact on OATP1B regulation from that of efflux transporters. To address this challenge, a PBPK model incorporating transporter induction and suppression was developed, 67 which has potential to dissect differential regulation and inhibition of various transporters. Induction of hepatic transporters including SLCO1B1 by LXR and FXR has been well‐characterized. 68 For drugs that are agonists of LXR or FXR (e.g., obeticholic acid) in hepatocytes, it will be important to understand whether they are inducers of transporter activity in humans. Induction of ABCC2, ABCC3, ABCC4, and ABCG2 by PXR, CAR, and aryl hydrocarbon receptor agonists was reported in cultured human hepatocytes, but based on more recent literature, clinical relevance likely is low. 69 , 70
Epigenetic/post‐transcriptional regulation
At the post‐transcriptional level, miRNAs were involved in downregulation of expression of SLCO1B1, 71 SLCO2B1, 72 and ABCC2. 59 Expression levels of miR‐206 and miR‐24 were associated with expression of SLCO1B1 71 and OATP2B1 72 in the human liver, respectively. In cancer cell lines and cancer tissues, an alternatively spliced variant of OATP1B3 lacking the N‐terminal 28 amino acids present in wild‐type OATP1B3 was identified. 73 The OATP1B3 variant was localized predominantly intracellularly and had negligible transport activity when overexpressed in vitro. 73 At the epigenetic level, there is evidence that DNA methylation plays a role in the liver specific expression of SLCO1B1 and SLCO1B3. 74 Tissue‐dependent hypomethylated regions in liver DNA relative to kidney DNA were identified around the transcriptional start site of SLCO1B1 and SLCO1B3.
Post‐translational regulation
Altered glycosylation of OATP1B1, OATP1B3, OATP2B1, NTCP, and MRP2 has been implicated in disease states, such as nonalcoholic steatohepatitis. 49 , 75 Phosphorylated OATP1B1, OATP1B3, and OATP2B1 has been detected. 51 , 76 , 77 The OATP1B1 variant c.521T>C (rs4149056; p.174Val>Ala) had reduced transport activity in vitro and this variant showed a modest increase in phosphorylation status compared with wild‐type OATP1B1. 78 The causal relationship between altered phosphorylation of the c.521T>C variant and reduced transport function has yet to be demonstrated.
Short‐term pre‐incubation with inhibitors may reduce transporter activity in vitro to a greater extent than simultaneous incubation with the substrate, even after washing the cells. This “pre‐incubation‐dependent inhibition” phenomenon has been reported for several SLC transporters, including OATP1B1 and OATP1B3, OATP2B1, and OCT1 in recombinant cell lines, 38 , 79 and in sandwich‐cultured primary human hepatocytes. 80 For several inhibitors, including cyclosporin A, 79 the half‐maximal inhibitory concentration values after inhibitor preincubation were close to the estimated in vivo inhibition constant values. Many of the reported inhibitors or modulators that show pre‐incubation‐dependent inhibition of transporters are phosphorylation modulators, including TKIs. 81 Transporter‐mediated DDIs involving TKIs have received considerable attention due to the chronic use of these drugs. Although multiple mechanisms have been proposed to explain pre‐incubation‐dependent inhibition, 79 inhibition of LYN and YES1 kinase‐mediated tyrosine phosphorylation was implicated in reduced transport function of OATP1B1 and OCT1, respectively, following treatment with TKIs. 8 , 82 The clinical implication of altered transporter phosphorylation, as shown for TKIs, warrants further investigation.
Kidneys
In the kidneys, transporters that are relevant to drug or endogenous compound disposition include multidrug and toxin extrusion (MATE) 1, MATE2K, OCT2, OAT1, OAT2, OAT3, OAT4, OATP4C1, PEPT1, PEPT2, P‐gp, MRP2, and MRP4. 2 Examples of regulation of clinically relevant kidney transporters are summarized below, and additional details are included in Tables 1 and 4 ; Table S1 .
Transcriptional regulation
Despite the established overall protein abundance profiles of kidney transporters, the regulatory factors contributing to modulation of transporter function in renal tissue are understudied and, thus far, a significant role for NRs in gene regulation has not been reported. However, various regulatory mechanisms have been characterized in preclinical models. For example, corticosteroids altered canine Slc22a2/Oct2 expression and activity 83 and murine Slc22a2 mRNA expression fluctuated based on circadian rhythm. 84 Collectively, this suggests that hormone and external signaling factors play a role in modulating OCT2 function, although, to date, no human data have been reported to confirm these observations.
Epigenetic/post‐transcriptional regulation
The impact of epigenetics on the regulation of OCT2 activity is the most established; promoter methylation and loss of histone acetylation in human tissue samples were associated with a renal‐specific reduction in SLC22A2/OCT2 mRNA expression and protein abundance in RCC. 21 , 85 Epigenetics also has been demonstrated as a mechanism that regulates activity of the urate transporter 1, OAT1 and OAT3 in vitro and in vivo (Figure 1 , Table 4 ).
Post‐translational regulation
Evidence continues to accumulate that OCT2 transport activity is dependent on various forms of PTMs: (i) phosphorylation‐dependent signaling has long been reported to modulate OCT2‐mediated transport. This was first shown with pharmacological activators or inhibitors of PKA, or phosphoinositide 3‐kinase, which revealed that these kinases reduce OCT2‐dependent uptake. 86 However, the thesis that PKA regulates OCT2 remains exclusively dependent on nonspecific multi‐kinase inhibitors or activators and has not been verified directly with genetic strategies. Thus, it remains unknown which kinases affect OCT2 function or whether such kinases act directly or indirectly on this transporter. (ii) In vitro genetic studies showed the functional importance of a YES1‐dependent tyrosine phosphorylation event at tyrosine 362. 39 This site is located at the substrate binding domain, according to predictive computational modeling, and the negative charge of tyrosine phosphorylation is expected to play a role in substrate binding. Support for this idea came from a mutagenesis approach and a YES1 TKI that abolished transport activity. 39 The dependence of OCT2 activity on tyrosine phosphorylation and the sensitivity of OCT2 activity to TKIs was confirmed in rodent models; and (iii) in addition to phosphorylation, glycosylation status also is reported to be involved in OCT2 activity. To date, data are limited to in vitro models, but loss of glycosylation at N96 reduced substrate affinity, and N112 glycosylation played a major role in OCT2 cell surface localization. 87 Protein kinase signaling has been investigated in OAT1 and OAT3 regulation, where PKA promotes phosphorylation of these transporters (along with SUMOylation of OAT3) and enhanced transport activity. 88 PKC can also influence OAT1 transport through promotion of ubiquitination, internalization, and degradation. 89 However, these mechanisms have yet to be confirmed in vivo or in human tissues. Furthermore, there is a plethora of data lacking for other renal transporters regarding regulatory factors and their contribution to in vivo transport function.
Collectively, whereas the information described above and in Table 4 demonstrate some progress in clarifying factors that can contribute to transport activity in the kidneys, it is evident that more detailed investigations are needed, particularly in translating findings from in vitro and animal models to the clinic to confirm the contribution of each regulatory mechanism to drug disposition and/or patient outcomes. Regarding use of animal models, the species‐specific expression profile of some transporters currently considered important in the active tubular transport of drugs must be emphasized. One example involves the expression of Slc22a1/SLC22A1, which is present in rodent kidneys, but not in monkey and human kidneys. 12 Nonetheless, data gathered particularly for transporters involved in tubular uric acid handling and summarized in the “urate transportosome” 90 support that novel methods are required to translate mechanistic studies into human phenotypes. Moreover, the studies on uric acid handling emphasize that tubular transporters are part of networks, where regulatory mechanisms coordinately change expression of transporters and work in concert to mediate tubular reabsorption and secretion.
Brain
Clinically relevant transporters for drugs and/or endogenous compounds in the brain are localized in the BBB, the blood‐cerebrospinal fluid‐barrier, in glial cells, and/or at other brain barriers. Transporters at the BBB, ranked based on currently available abundance data, include glucose transporter 1 with the highest levels, followed by BCRP, OAT2, P‐gp, OCT3, OCT1, OATP1A2, OAT7, OAT1, OATP1B3, OATP2B1, OAT3, OATP1C1, and MRP4. However, there is still some controversy regarding the location of brain transporters in polarized brain endothelial cells, especially for the OATPs. 91 Studies on underlying mechanisms in patients with brain diseases (e.g., Alzheimer’s, Parkinson’s, and epilepsy) have demonstrated that regulatory factors and pathways contribute to modulation of transporter function in the brain leading to changes in transporter expression and abundance 92 (Table S1 ). P‐gp is the brain transporter that has been characterized best at different regulatory levels (Table 1 , Table S1 ).
Lungs
The influence of transport proteins in the lungs (e.g., OCTs, P‐gp) on pulmonary drug disposition and toxicity is an emerging area of research. Human‐relevant data documenting transporter regulation in the lungs are limited and are primarily based on cells or lung tissue obtained from patients with pulmonary disease, or in vitro cell models (Table S3 ). The expression, subcellular localization, function, and regulation of OCT transporters in lung tissue and cell lines has been reviewed. 93 Pulmonary diseases (e.g., asthma) increased ABCC2 mRNA. 94 ABCB1 mRNA was significantly lower in bronchoalveolar lavage cells from smokers compared with nonsmokers. 95 Understanding how regulation of pulmonary transporters may impact transporter function is critical in the development of medications for respiratory diseases.
Placenta
The placenta plays an important role in nutrient and waste exchange between mother and fetus and several transporters are expressed in the apical and basolateral membranes of the syncytiotrophoblast. The most abundant and clinically relevant placental drug transporters include BCRP, P‐gp, OATP2B1, OCT3, and OAT4. 96 , 97 These transporters prevent entry or facilitate efflux of potentially harmful endogenous and exogenous substrates from the fetal compartment. While the placenta develops and grows throughout pregnancy, abundance of the transporters changes: BCRP and P‐gp levels decrease while OAT4 and OCT3 levels increase from the first or second trimester to term (Table S1 ). 97 Protein levels of both P‐gp and BCRP decline almost two‐fold near term, leaving the fetus more susceptible to potentially harmful drugs.
Transcriptional regulation
In the placenta, steroid hormones, such as progesterone and estrogen, which physiologically fluctuate during pregnancy or by treatment with glucocorticoids, regulate drug transporter expression. 98 , 99 Importantly, pathological conditions, such as preeclampsia, gestational diabetes, growth restriction, or infection, can lead to dysregulation of drug transporters. 96 Transporter expression changed along with alterations in transcript levels of cytokines and growth factors. 96 Differences in the extent or direction of mRNA vs. protein changes point to involvement of both transcriptional and translational regulatory pathways. In vitro studies in primary cultures of placental cells and tissues have demonstrated that growth factors, hypoxia, and pro‐inflammatory cytokines can alter transporter levels and/or activities. 100 , 101 However, the regulatory factors contributing to modulation of placental transporters in health and disease are relatively unknown. A better understanding of placental drug transporter regulation in normal and pathological pregnancies will aid in predicting fetal drug exposure and the development of more targeted medication for the mother.
EXPERIMENTAL APPROACHES TO INVESTIGATE REGULATION OF TRANSPORT PROTEINS
Knowledge of transporter protein regulation is relatively limited, and many questions still need to be addressed to better understand and accurately predict the impact of alterations in regulatory pathways on drug efficacy and toxicity. Considerable advances have been made in the methods used to elucidate the mechanisms and factors that regulate transporter activity. Techniques that can be used to study transcriptional regulation, epigenetic alterations, PTMs, and changes in trafficking, transporter expression, and/or abundance are listed in Table 5 . Immunohistochemistry/immunocytochemistry and immunofluorescence techniques applied to in vitro, nonclinical in vivo, or clinical samples can provide insight into tissue levels and, in some cases, more detailed information regarding transporter localization than cell fractionation or biotinylation methods. Protein quantification can be obtained, for instance, by global or targeted proteomics. 102 Global proteomics provides a relative picture of all the proteins in a given sample, but may lack precision relative to targeted proteomics, which measures protein abundance of single or selected proteins with higher reproducibility and sensitivity, particularly for low abundance proteins. Both methods may be able to detect PTMs. However, various forms of protein modifications potentially can be difficult to measure simultaneously. In addition to protein analysis, mRNA expression assessment is possible in various tissues or cells using current sequencing technology. In fact, this analysis is useful in combination with bisulfite sequencing or chromatin immunoprecipitation to measure epigenetic modifications or transcription factor binding. Each of the procedures outlined are suitable to measure various factors that regulate transporter activity. However, these approaches often cannot be used simultaneously due to sample preparation differences or limited quantities of sample. As such, experiments need to be carefully designed.
Table 5.
Experimental methods to study regulation of transporters
| Level | Methods | Strengths | Limitations | Application (+) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Nuclear receptors | Epigenetics | Post‐transcriptional | Alternative splicing | Post‐translational | Trafficking | Expression or abundance | Activity | ||||
| Preclinical | Cell lines |
|
|
+ | + | + | + | + | + | + | + |
| Stem cells |
|
|
+ | + | + | + | + | + | + | + | |
| Primary cells |
|
|
+ | + | + | + | + | + | + | + | |
| Animals |
|
|
+ | + | + | + | + | + | + | + | |
| Tissue | Bisulfite Sequencing |
|
|
+ | |||||||
| ChIP |
|
|
+ | + | |||||||
| RNA Seq |
|
|
+ | + | + | ||||||
| Western Blot |
|
|
+ | + | + | ||||||
| LC/MS global proteomics |
|
|
+ | + | + | ||||||
| Targeted LC/MS proteomics |
|
|
+ | + | + | ||||||
| IHC/IF |
|
|
+ | + | |||||||
| Surface biotinylation |
|
|
+ | + | + | ||||||
| Cell fractionation |
|
|
+ | + | + | ||||||
| Clinical (non‐invasive) | Endogenous biomarkers |
|
|
+ | |||||||
| Drug substrates (e.g., cocktails) |
|
|
+ | ||||||||
| Liquid biopsy |
|
|
+ | + | |||||||
ChIP, chromatin immuno‐precipitation; IHC/IF, immunohistochemistry/immunofluorescence; LC/MS, liquid chromatography/mass spectrometry; miRNA, microRNA; PTMs, post‐translational modifications.
Most studies to date have used in vitro models, such as cell lines, cells overexpressing transport proteins of interest, or primary cells, to assess transporter regulation. Key considerations in selection of in vitro models include: (i) the need for adequate transporter or regulatory protein expression (i.e., NRs); (ii) cell‐, organ‐, or species‐specific differences in transporter expression, membrane localization (i.e., apical vs. basolateral), and/or regulatory pathways; and (iii) laboratory‐ or culture‐specific conditions (e.g., cell source, passage number, media content). It is reasonable to expect that epigenetic‐ or transcription factor‐mediated control of gene promoter activity (if expressed at sufficient levels) would be consistent across in vitro, in vivo, or clinical conditions. However, the impact of disease or exposure to potential modifiers on these regulatory mechanisms in various human tissues remains to be investigated. This need is similarly essential for RNA and proteomic quantification of transporters in various tissues or disease states. Gene expression and protein abundance using RNA sequencing and proteomics is well‐established in cell lines and tissues involved in drug disposition in both animal models and humans. However, these techniques have not been applied robustly to assess changes or variability in mRNA or protein levels throughout development, disease states, gender, or race, which can vary markedly from observations in cell lines or animal models. Additionally, RNA sequencing and proteomics can be applied to clarify changes in splice variant expression, which is understudied, particularly in human tissues where expression may differ significantly from that observed in vitro. Likewise, the approaches used to assess changes in PTMs by intrinsic and extrinsic factors, and the impact on transporter localization and activity can be highly dependent on intracellular signaling. These signaling events can differ based on distinct protein abundance among in vitro cell models or primary tissues, and also could be changed following drug exposure. Such events, along with regulatory protein expression and activity may differ among species. Thus, care needs to be taken when translating data from animal models to humans, particularly with proteins that are not well‐conserved. Therefore, given the lack of characterization in humans, use of primary tissues or clinical samples should be the major focus of future investigations. Method standardization is required to validate cellular models as surrogates for in vivo studies, including consistent use of drug and modulator concentrations, exposure times, and culture conditions (e.g., media and additives) for valid comparisons across investigations.
Various clinical approaches may be used to investigate transporter regulation, including use of exogenous transporter substrate probes, endogenous biomarkers, and tissue and liquid biopsies. Exogenous transporter substrate probe cocktails, with multiple transporter and metabolic enzyme substrates, have been used successfully in numerous clinical studies to characterize DDIs or the effect of disease on transporter activity. 103 , 104 Although this approach is informative for identification of DDIs, experimental execution is complex and requires administration of multiple drugs. In contrast, endogenous transporter substrate biomarkers 105 overcome some of these challenges. More specifically, transporter biomarkers do not require additional compound administration, can easily assess baseline characteristics, can be measured over longer periods of time, and require a low volume of blood. However, use of biomarkers to assess transporter regulation has some limitations. For example, synthesis of these endogenous compounds needs to be stable during assessment, and the profile of transporters and/or metabolic enzymes that recognize these molecules must be well‐defined. 106 Similar to exogenous probes, changes in systemic biomarker concentrations do not provide mechanistic insight into regulatory alterations caused by DDIs. Liquid biopsies are an emerging technique that may overcome the lack of such mechanistic insights into transporter regulation. Liquid biopsies allow for the collection of circulating exosomes isolated from human plasma that originate from various cells. 107 Following isolation, protein levels can be measured. Theoretically, PTMs can be measured as well, although this strategy has not been reported to date. Current methods require large volumes of plasma to isolate sufficient exosomes, 107 and it remains unknown whether abundance or modifications reflect transport activity at the moment of extraction.
PHARMACOKINETIC MODELING FOR TRANSPORTER INDUCTION AND SUPPRESSION
With the growing understanding that drugs and other xenobiotics can cause clinical DDIs via transporter induction 6 , 108 , 109 and suppression, 110 it is imperative to incorporate such complexity into physiologically based mathematical models to accurately predict DDI risks (Table 6 ). To date, the dynamics of NR‐mediated transporter regulation has not been systematically studied, and a widely accepted predictive mathematical and/or biology‐based model evaluating the dynamics of transporter induction after NR activation and the effects of post‐transcriptional or post‐translational modifications is not available. Hence, it is challenging to build suitable mechanistic models for DDIs involving transporter regulation due to induction/suppression. Besides an empirical model that uses a fixed relative scalar, 111 , 112 , 113 the turnover model that was established for induction/suppression modeling of CYPs and UGTs, 114 can be applied to transporters, 67 because transporters are regulated through mechanisms likely similar to CYP enzymes (Figure 1 ). In vivo transporter levels are governed by the rates of de novo protein synthesis and degradation, defined as the protein turnover half‐life. The current lack of in vivo turnover half‐life values for human transporters places a significant limitation on the accurate prediction of changes in drug concentration‐time profiles associated with interactions involving transporter induction. Protein turnover is the only system data (i.e., population information) required for the turnover model, besides the baseline protein level (Figure 2 ), but only sparse data are available for transporter turnover. P‐gp is inducible and relevant for clinical DDIs, whereas the induction of OATP1B is more controversial as discussed above. The turnover number for P‐gp is ~ 0.054/hour based on a meta‐analysis of 13 independent published studies from seven laboratories, and the turnover number for OATP1B1 is ~ 0.031/hour based on two independent studies (author S.N., personal communication). However, robust turnover numbers are still lacking for most transporters. In addition, clinical data for validation of turnover numbers are sparse. In most clinical studies, the concentration of the perpetrator is not reported. For example, only a few concentration‐time profiles with low doses of rifampin are available to validate the maximum fold induction/suppression over vehicle control (IndC50). Although the turnover model is semimechanistic, it does not link the underlying induction process to the change in the protein level. This requires more detailed in vitro experimental design and mathematical models, such as quantitative systems pharmacology models. 115
Table 6.
Physiologically based mathematical models to assess transporter induction or suppression a
| Application (transporter/location) | Substrate | Inducer/suppressor | Advantages | Limitations | Reference |
|---|---|---|---|---|---|
| Empirical model | |||||
|
Intestinal P‐gp Hepatic OATP |
Dabigatran Pravastatin Rosuvastatin Midazolam Tolbutamide Caffeine |
Rifampin |
|
Each additional compound to the matrix brings it's own uncertainties, however, this limitation will change to an advantage the more compounds can be successfully estimated and therefore included in the matrix | 108 |
|
Intestinal P‐gp Hepatic OATP |
Sofosbuvir |
Rifabutin Carbamazepine |
|
Same as above | 109 |
| REF or RAF scaling within an IVIVE‐PBPK framework | |||||
| Intestinal (and hepatic) P‐gp | Digoxin | Rifampin |
|
|
111 |
| Intestinal P‐gp |
Dabigatran etexilate Digoxin Quinidine Talinolol |
Rifampin |
|
Same as above | 112 |
|
Intestinal P‐gp |
Abemaciclib Acalabrutinib Bosutinib Crizotinib Dabigatran etexilate Digoxin Naldemedine Naloxegol Olaparib Quinidine Talinolol Verapamil |
Rifampin |
|
Same as above The more compounds that can be correctly estimated with the same REF, the higher the confidence in this system parameter |
113 |
| Hepatic OATP1B1 | CP‐I | Rifampin; OATP1B1 521CC polymorphism |
|
|
148, 149 |
| Turnover model within an IVIVE‐PBPK model | |||||
| Hepatic OATP1B, MRP2 |
Glibenclamide Repaglinide CP‐I |
Rifampin |
|
Model not accessible to everyone due to program used ‐ low practicality | 67 |
| Hepatic OATP1B1 | Repaglinide | Rifampin | Using the transporter induction turnover model, DDIs could be recovered reasonably well when an OATP1B1 induction Indmax value (2.3) and a kdeg for OATP1B1 of 0.0311/h was included for rifampin in simulations across a range of dosing regimens |
Further investigations and data are required to assess the validity of the derived rifampin in vitro OATP1B1 Indmax value (i.e., using different rifampin doses and OATP1B1 substrates) and whether inclusion of an IndC50 value would reduce the overpredictions OATP1B1 is a polymorphic transporter and no genotype information was available from the clinical studies. Thus, understanding the effect of polymorphisms was not assessed in this investigation and may contribute to some of the variability observed due to the small sample size of the clinical studies |
(SN, Personal communication) |
| Intestinal P‐gp |
Digoxin Dabigatran etexilate |
Rifampin |
|
Although these preliminary results are encouraging, the model needs to be verified against other clinical studies with different rifampin doses to confirm the utility of the model | (SN, Personal communication) |
| QSP | |||||
| Hepatic OATP1B1 and P‐gp |
Diclofenac Celecoxib |
Rifampin | Changes in hepatic OATP1B1 and P‐gp can be included in the model | Transporter alterations have not been evaluated in the models for diclofenac and celecoxib | 150 |
Caco‐2, human colorectal adenocarcinoma cell; CL, clearance; CP‐1, coproporphyrin I; CYP, cytochrome P450; DDI, drug‐drug interaction; IndC50, test compound concentration that supports half‐maximal induction/suppression; Indmax, maximum fold induction/suppression over vehicle control; IVIVE‐PBPK, in vitro‐to‐in vivo extrapolation linked physiologically based pharmacokinetics; Jmax, maximum rate of transport; kdeg,, rate of degradation, defined by a first‐order rate constant; Km, Michaelis constant; MRP, multidrug resistance‐associated protein; OATP, organic anion transporting polypeptide; PBPK, physiologically based pharmacokinetics; P‐gp, MDR1 P‐glycoprotein; PXR, pregnane X receptor; QSP, quantitative systems pharmacology; RAF, relative activity factor; REF, relative expression factor.
Figure 2.
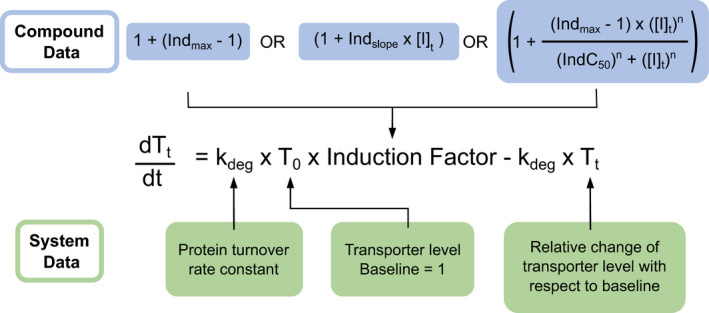
The turnover model as applied to drug‐drug interactions. The dynamics of transporter regulation via induction/suppression is represented by a turnover model 114 in which the amount of transporter at equilibrium reflects the balance between its rate of synthesis and its rate of degradation, defined by a first‐order rate constant (kdeg). The required perpetrator data are either: (1) maximum fold induction/suppression over vehicle control (Indmax; Indmax < 1 indicates suppression, Indmax > 1 indicates induction); (2) The slope of the fold induction/suppression versus transporter inhibitor concentration ([I]t) plot when induction/suppression is linear within the range of perpetrator concentrations (Indslope in µM−1); or (3) perpetrator concentration that supports half‐maximal induction/suppression (IndC50 in µM) together with Indslope. This option will link the perpetrator concentration over a range of concentrations directly to induction/suppression and hence will best translate to changes in protein levels. For the IndC50 determination, the fraction of unbound drug in the in vitro incubation (fu inc) also should be considered. Required system data (population information) are: baseline protein level (T0) and kdeg. Tt is the state variable describing the relative change in the transporter level with respect to baseline because of induction/suppression. Without induction/suppression Tt = 1.
CONCLUSIONS AND FUTURE PERSPECTIVE
The preclinical and clinical evidence presented in this white paper clearly demonstrates that regulation of transporters is highly orchestrated (Table 1 ), and that the net effect of various regulation mechanisms contributes to variability in drug response in healthy individuals and in patients with various stages of disease. In contrast to the well‐established role of NRs, the importance of regulation involving epigenetic, post‐transcriptional and post‐translational mechanisms, and effects on transporter expression, membrane localization, and function is still fragmented and requires more systematic research to establish to what extent these various mechanisms affect drug response in the clinic. In addition, the effect of food and the intestinal microbiome on transporter regulation is an area where further studies are needed. Currently, the number of drug transporters for which clinically significant regulatory mechanisms have been demonstrated is relatively small (Tables 2 , 3 , 4 ). Studying transporter regulation in human subjects in the various organs, tissues, and epithelial barriers where membrane transporters play a critical role (including organs not covered in this white paper such as heart, skeletal muscle, and testes) is challenging and, in most cases, based on current experimental methods with limitations, requires invasive procedures to obtain tissue. Thus, there is an urgent need for biorelevant in vitro models, such as primary cells and microphysiological systems that possess similar regulatory plasticity to that in vivo. Studying regulation in immortalized cell lines can be hypothesis generating, but results obtained in such cells should be interpreted with caution and confirmed in physiologically relevant models. In addition, continued efforts to understand the translatability of endogenous biomarkers and endpoints that can be quantified in liquid biopsies will be valuable. This is also true for analytical advances enabling the highly sensitive and tissue specific analysis of gene and miRNA expression as well as protein abundance of transporters and NRs from liquid biopsies. 116
Although the role of NRs in the induction of intestinal P‐gp and MRP2 is well‐established, there are still gaps in understanding which organs are most significantly impacted by transporter induction. Current data indicate that PXR‐regulated transporters (i.e., P‐gp and MRP2) are inducible by respective ligands and may cause clinically relevant DDIs after oral drug administration, 14 whereas ligands of CAR do not induce intestinal P‐gp or other CAR‐regulated enzymes or transporters. 9 , 56 To what extent this translates to other PXR activators and NRs is less clear. To increase knowledge in this area, the route of administration (intravenous vs. oral) of a perpetrator drug should be considered carefully to allow a mechanistic interpretation of DDI results.
The role of miRNAs in transporter regulation also is emerging. The correlation of miRNAs with downregulation of transport activity is implied based on data obtained in the intestine and liver. Currently, no systematic data are available to identify the functional significance of miRNAs, and methods to identify miRNA binding sites are indirect and time consuming. To address this gap, application of the chimeric‐eCLIP assay recently described to map the hepatocyte miRNA‐mRNA interactome 117 should be explored to identify miRNAs binding to transporter mRNAs in hepatocytes and cells in other organs. Another class of regulatory RNAs that is understudied are the lncRNAs. Evidence suggests that lncRNAs may be involved in transporter regulation in multidrug resistant tumor cells by either interacting with NRs or with mRNAs encoding ABC transporters, 118 but their role in healthy tissue is unclear.
The impact of PTMs and altered protein trafficking on transporter localization and function remains poorly understood und understudied. More focus on the use of human‐based in vitro and ex vivo models with intact regulatory machinery is needed. Proteomic methods used to quantify transporters must distinguish functional from inactive proteins, for example, due to PTMs that affect activity. Transporter localization in the relevant membrane, and appropriate corrections for loss during sample preparation, must be considered when using quantitative cell or tissue abundance data to predict in vivo drug disposition.
In summary, based on the evidence presented in this white paper, regulation of transport proteins is highly complex and can occur at multiple levels to impact transporter function and drug disposition. Preclinical and clinical research advances are needed to close knowledge gaps in this field and support the development of improved PBPK and quantitative systems pharmacology models to predict drug response and interpatient variability. As such, these investigations are anticipated among the next frontiers in transporter research.
FUNDING
K.L.R.B. is supported, in part, by the National Institute of General Medical Sciences of the National Institutes of Health (NIH) under Award Number R35 GM122576. S.H. is supported in part by NIH grant R01CA238946. J.A.S. is supported, in part, by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number 1R01GM139936‐01A1. W.Y. is supported, in part, by the University of Oklahoma College of Pharmacy Seed Grant.
CONFLICT OF INTEREST
K.L.R.B. is co‐inventor of the sandwich‐cultured hepatocyte technology for quantification of biliary excretion (B‐CLEAR) and related technologies, which have been licensed exclusively to Qualyst Transporter Solutions, acquired by BioIVT. R.E. is an employee of Johnson & Johnson and may own stock or stock options of Johnson & Johnson. C.Y.L. is an employee of Amgen and may be a stockholder of Amgen. S.N. is an employee of Certara UK Ltd. and stockholder of Certara. S.H.S. is an employee of AstraZeneca and holds AstraZeneca stock. All other authors declared no competing interests for this work.
Supporting information
Table S1
Table S2
Table S3
ACKNOWLEDGMENTS
The authors are grateful to Drs. Per Artursson, Richard Kim, Hong Shen, Daniel Tatosian, Mitch Taub, and Joe Ware for providing valuable and insightful comments on the manuscript. We apologize to many of the scientists who contributed to this area of research but whose work could not be cited due to reference limitations.
Contributor Information
Kim L.R. Brouwer, Email: kbrouwer@unc.edu.
Raymond Evers, Email: revers1@its.jnj.com.
- 1. Giacomini, K.M. et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 9, 215–236 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zamek‐Gliszczynski, M.J. et al. Transporters in drug development: 2018 ITC recommendations for transporters of emerging clinical importance. Clin. Pharmacol. Ther. 104, 890–899 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Keogh, J. , Hagenbuch, B. , Rynn, C. , Stieger, B. & Nicholls, G. Chapter 1. Membrane transporters: fundamentals, function and their role in ADME. In Drug Transporters: Role and Importance in ADME and Drug Development Vol. 1, 1–56 (The Royal Society of Chemistry, Cambridge, 2016). [Google Scholar]
- 4. Wakabayashi, Y. , Kipp, H. & Arias, I.M. Transporters on demand: intracellular reservoirs and cycling of bile canalicular ABC transporters. J. Biol. Chem. 281, 27669–27673 (2006). [DOI] [PubMed] [Google Scholar]
- 5. Evers, R. et al. Disease‐associated changes in drug transporters may impact the pharmacokinetics and/or toxicity of drugs: a white paper from the International Transporter Consortium. Clin. Pharmacol. Ther. 104, 900–915 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zamek‐Gliszczynski, M.J. et al. Intestinal P‐gp and putative hepatic OATP1B induction: International Transporter Consortium perspective on drug development implications. Clin. Pharmacol. Ther. 109, 55–64 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rodrigues, A.D. , Lai, Y. , Shen, H. , Varma, M.V.S. , Rowland, A. & Oswald, S. Induction of human intestinal and hepatic organic anion transporting polypeptides: where is the evidence for its relevance in drug‐drug interactions? Drug Metab. Dispos. 48, 205–216 (2020). [DOI] [PubMed] [Google Scholar]
- 8. Hayden, E.R. et al. Regulation of OATP1B1 function by tyrosine kinase‐mediated phosphorylation. Clin. Cancer Res. 27, 4301–4310 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brueck, S. et al. Transcriptional and post‐transcriptional regulation of duodenal P‐Glycoprotein and MRP2 in healthy human subjects after chronic treatment with rifampin and carbamazepine. Mol. Pharm. 16, 3823–3830 (2019). [DOI] [PubMed] [Google Scholar]
- 10. Hirota, T. , Tanaka, T. , Takesue, H. & Ieiri, I. Epigenetic regulation of drug transporter expression in human tissues. Expert Opin. Drug Metab. Toxicol. 13, 19–30 (2017). [DOI] [PubMed] [Google Scholar]
- 11. Kenna, J.G. et al. Can bile salt export pump inhibition testing in drug discovery and development reduce liver injury risk? An International Transporter Consortium perspective. Clin. Pharmacol. Ther. 104, 916–932 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chu, X. , Bleasby, K. & Evers, R. Species differences in drug transporters and implications for translating preclinical findings to humans. Expert Opin. Drug Metab. Toxicol. 9, 237–252 (2013). [DOI] [PubMed] [Google Scholar]
- 13. Evans, R.M. & Mangelsdorf, D.J. Nuclear receptors, RXR, and the big bang. Cell 157, 255–266 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gessner, A. , Konig, J. & Fromm, M.F. Clinical aspects of transporter‐mediated drug‐drug interactions. Clin. Pharmacol. Ther. 105, 1386–1394 (2019). [DOI] [PubMed] [Google Scholar]
- 15. Rodrigues, A.D. & Rowland, A. Profiling of drug‐metabolizing enzymes and transporters in human tissue biopsy samples: a review of the literature. J. Pharmacol. Exp. Ther. 372, 308–319 (2020). [DOI] [PubMed] [Google Scholar]
- 16. Nishimura, M. , Naito, S. & Yokoi, T. Tissue‐specific mRNA expression profiles of human nuclear receptor subfamilies. Drug Metab. Pharmacokinet. 19, 135–149 (2004). [DOI] [PubMed] [Google Scholar]
- 17. Fritz, A. , Busch, D. , Lapczuk, J. , Ostrowski, M. , Drozdzik, M. & Oswald, S. Expression of clinically relevant drug‐metabolizing enzymes along the human intestine and their correlation to drug transporters and nuclear receptors: an intra‐subject analysis. Basic Clin. Pharmacol. Toxicol. 124, 245–255 (2019). [DOI] [PubMed] [Google Scholar]
- 18. Nigam, S.K. What do drug transporters really do? Nat. Rev. Drug Discov. 14, 29–44 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sperling, S. Transcriptional regulation at a glance. BMC Bioinform. 8 Suppl 6 (S2), (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang, Y. et al. Overview of histone modification. Adv. Exp. Med. Biol. 1283, 1–16 (2021). [DOI] [PubMed] [Google Scholar]
- 21. Zhu, Q. et al. Regulation of OCT2 transcriptional repression by histone acetylation in renal cell carcinoma. Epigenetics 14, 791–803 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bruckmueller, H. et al. Clinically relevant multidrug transporters are regulated by microRNAs along the human intestine. Mol. Pharm. 14, 2245–2253 (2017). [DOI] [PubMed] [Google Scholar]
- 23. Erdmann, P. et al. Dysregulation of mucosal membrane transporters and drug‐metabolizing enzymes in ulcerative colitis. J. Pharm. Sci. 108, 1035–1046 (2019). [DOI] [PubMed] [Google Scholar]
- 24. Ikemura, K. , Iwamoto, T. & Okuda, M. MicroRNAs as regulators of drug transporters, drug‐metabolizing enzymes, and tight junctions: implication for intestinal barrier function. Pharmacol. Ther. 143, 217–224 (2014). [DOI] [PubMed] [Google Scholar]
- 25. Mbatchi, L.C. , Brouillet, J.P. & Evrard, A. Genetic variations of the xenoreceptors NR1I2 and NR1I3 and their effect on drug disposition and response variability. Pharmacogenomics 19, 61–77 (2018). [DOI] [PubMed] [Google Scholar]
- 26. Ramamoorthy, A. et al. Regulation of microRNA expression by rifampin in human hepatocytes. Drug Metab. Dispos. 41, 1763–1768 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Park, J.E. , Ryoo, G. & Lee, W. Alternative splicing: expanding diversity in major ABC and SLC drug transporters. AAPS J. 19, 1643–1655 (2017). [DOI] [PubMed] [Google Scholar]
- 28. Byrne, J.A. et al. Missense mutations and single nucleotide polymorphisms in ABCB11 impair bile salt export pump processing and function or disrupt pre‐messenger RNA splicing. Hepatology 49, 553–567 (2009). [DOI] [PubMed] [Google Scholar]
- 29. Nies, A.T. & Keppler, D. The apical conjugate efflux pump ABCC2 (MRP2). Pflugers Arch. 453, 643–659 (2007). [DOI] [PubMed] [Google Scholar]
- 30. Omata, Y. , Yamauchi, T. , Tsuruta, A. , Matsunaga, N. , Koyanagi, S. & Ohdo, S. RNA editing enzyme ADAR1 governs the circadian expression of P‐glycoprotein in human renal cells by regulating alternative splicing of the ABCB1 gene. J. Biol. Chem. 296, 100601 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hyrsova, L. , Smutny, T. , Trejtnar, F. & Pavek, P. Expression of organic cation transporter 1 (OCT1): unique patterns of indirect regulation by nuclear receptors and hepatospecific gene regulation. Drug Metab. Rev. 48, 139–158 (2016). [DOI] [PubMed] [Google Scholar]
- 32. van Groen, B.D. et al. Alternative splicing of the SLCO1B1 gene: an exploratory analysis of isoform diversity in pediatric liver. Clin. Transl. Sci. 13, 509–519 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Malagnino, V. , Hussner, J. , Seibert, I. , Stolzenburg, A. , Sager, C.P. , & Meyer Zu Schwabedissen, H.E. LST‐3TM12 is a member of the OATP1B family and a functional transporter. Biochem. Pharmacol. 148, 75–87 (2018). [DOI] [PubMed] [Google Scholar]
- 34. Meyer Zu Schwabedissen, H.E. et al. Genetic variants of SLCO1B7 are of relevance for the transport function of OATP1B3‐1B7. Pharmacol. Res. 161, 105155 (2020). [DOI] [PubMed] [Google Scholar]
- 35. Miller, D.S. & Cannon, R.E. Signaling pathways that regulate basal ABC transporter activity at the blood‐ brain barrier. Curr. Pharm. Des. 20, 1463–1471 (2014). [DOI] [PubMed] [Google Scholar]
- 36. Abualsunun, W.A. & Piquette‐Miller, M. Involvement of nuclear factor kappaB, not pregnane X receptor, in inflammation‐mediated regulation of hepatic transporters. Drug Metab. Dispos. 45, 1077–1083 (2017). [DOI] [PubMed] [Google Scholar]
- 37. Czuba, L.C. , Hillgren, K.M. & Swaan, P.W. Post‐translational modifications of transporters. Pharmacol. Ther. 192, 88–99 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Minematsu, T. & Giacomini, K.M. Interactions of tyrosine kinase inhibitors with organic cation transporters and multidrug and toxic compound extrusion proteins. Mol. Cancer Ther. 10, 531–539 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sprowl, J.A. et al. A phosphotyrosine switch regulates organic cation transporters. Nat. Commun. 7, 10880 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Miah, M.F. , Conseil, G. & Cole, S.P. N‐linked glycans do not affect plasma membrane localization of multidrug resistance protein 4 (MRP4) but selectively alter its prostaglandin E2 transport activity. Biochem. Biophys. Res. Commun. 469, 954–959 (2016). [DOI] [PubMed] [Google Scholar]
- 41. Yao, J. , Hong, W. , Huang, J. , Zhan, K. , Huang, H. & Hong, M. N‐Glycosylation dictates proper processing of organic anion transporting polypeptide 1B1. PLoS One 7, e52563 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rapp, C.L. , Li, J. , Badior, K.E. , Williams, D.B. , Casey, J.R. & Reithmeier, R.A.F. Role of N‐glycosylation in the expression of human SLC26A2 and A3 anion transport membrane glycoproteins (1). Biochem. Cell Biol. 97, 290–306 (2019). [DOI] [PubMed] [Google Scholar]
- 43. Katayama, K. , Noguchi, K. & Sugimoto, Y. FBXO15 regulates P‐glycoprotein/ABCB1 expression through the ubiquitin–proteasome pathway in cancer cells. Cancer Sci. 104, 694–702 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sadler, J.B. , Bryant, N.J. , Gould, G.W. & Welburn, C.R. Posttranslational modifications of GLUT4 affect its subcellular localization and translocation. Int. J. Mol. Sci. 14, 9963–9978 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Minami, S. et al. Posttranslational regulation of Abcc2 expression by SUMOylation system. Am. J. Physiol. Gastrointest. Liver Physiol. 296, G406–G413 (2009). [DOI] [PubMed] [Google Scholar]
- 46. Murray, M. & Zhou, F. Trafficking and other regulatory mechanisms for organic anion transporting polypeptides and organic anion transporters that modulate cellular drug and xenobiotic influx and that are dysregulated in disease. Br. J. Pharmacol. 174, 1908–1924 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mohrmann, K. , van Eijndhoven, M.A. , Schinkel, A.H. & Schellens, J.H. Absence of N‐linked glycosylation does not affect plasma membrane localization of breast cancer resistance protein (BCRP/ABCG2). Cancer Chemother. Pharmacol. 56, 344–350 (2005). [DOI] [PubMed] [Google Scholar]
- 48. Schinkel, A.H. , Kemp, S. , Dolle, M. , Rudenko, G. & Wagenaar, E. N‐glycosylation and deletion mutants of the human MDR1 P‐glycoprotein. J. Biol. Chem. 268, 7474–7481 (1993). [PubMed] [Google Scholar]
- 49. Clarke, J.D. , Novak, P. , Lake, A.D. , Hardwick, R.N. & Cherrington, N.J. Impaired N‐linked glycosylation of uptake and efflux transporters in human non‐alcoholic fatty liver disease. Liver Int. 37, 1074–1081 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yu, Z. , Liu, C. , Zhang, J. , Liang, Z. & You, G. Protein kinase C regulates organic anion transporter 1 through phosphorylating ubiquitin ligase Nedd4‐2. BMC Mol. Cell Biol. 22, 53 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Köck, K. et al. Rapid modulation of the organic anion transporting polypeptide 2B1 (OATP2B1, SLCO2B1) function by protein kinase C‐mediated internalization. J. Biol. Chem. 285, 11336–11347 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Aida, K. , Hayashi, H. , Inamura, K. , Mizuno, T. & Sugiyama, Y. Differential roles of ubiquitination in the degradation mechanism of cell surface‐resident bile salt export pump and multidrug resistance‐associated protein 2. Mol. Pharmacol. 85, 482–491 (2014). [DOI] [PubMed] [Google Scholar]
- 53. Ben Saad, A. et al. Molecular regulation of canalicular ABC transporters. Int. J. Mol. Sci. 22, 2113 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Crawford, R.R. , Potukuchi, P.K. , Schuetz, E.G. & Schuetz, J.D. Beyond competitive inhibition: regulation of ABC transporters by kinases and protein‐protein interactions as potential mechanisms of drug‐drug interactions. Drug Metab. Dispos. 46, 567–580 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Oswald, S. et al. Intestinal expression of P‐glycoprotein (ABCB1), multidrug resistance associated protein 2 (ABCC2), and uridine diphosphate‐glucuronosyltransferase 1A1 predicts the disposition and modulates the effects of the cholesterol absorption inhibitor ezetimibe in humans. Clin. Pharmacol. Ther. 79, 206–217 (2006). [DOI] [PubMed] [Google Scholar]
- 56. Oswald, S. et al. Impact of efavirenz on intestinal metabolism and transport: insights from an interaction study with ezetimibe in healthy volunteers. Clin. Pharmacol. Ther. 91, 506–513 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ebert, B. , Seidel, A. & Lampen, A. Identification of BCRP as transporter of benzo[a]pyrene conjugates metabolically formed in Caco‐2 cells and its induction by Ah‐receptor agonists. Carcinogenesis 26, 1754–1763 (2005). [DOI] [PubMed] [Google Scholar]
- 58. Xie, Q.S. et al. Short‐chain fatty acids exert opposite effects on the expression and function of p‐glycoprotein and breast cancer resistance protein in rat intestine. Acta Pharm. Sin. 42, 470–481 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bruhn, O. et al. Alternative polyadenylation of ABC transporters of the C‐family (ABCC1, ABCC2, ABCC3) and implications on posttranscriptional micro‐RNA regulation. Mol. Pharmacol. 97, 112–122 (2020). [DOI] [PubMed] [Google Scholar]
- 60. To, K.K. , Leung, W.W. & Ng, S.S. Exploiting a novel miR‐519c‐HuR‐ABCG2 regulatory pathway to overcome chemoresistance in colorectal cancer. Exp. Cell Res. 338, 222–231 (2015). [DOI] [PubMed] [Google Scholar]
- 61. Stelzl, T. , Geillinger‐Kästle, K.E. , Stolz, J. & Daniel, H. Glycans in the intestinal peptide transporter PEPT1 contribute to function and protect from proteolysis. Am. J. Physiol. Gastrointest. Liver Physiol. 312, G580–G591 (2017). [DOI] [PubMed] [Google Scholar]
- 62. Hanggi, E. , Grundschober, A.F. , Leuthold, S. , Meier, P.J. & St‐Pierre, M.V. Functional analysis of the extracellular cysteine residues in the human organic anion transporting polypeptide, OATP2B1. Mol. Pharmacol. 70, 806–817 (2006). [DOI] [PubMed] [Google Scholar]
- 63. Ticho, A.L. et al. S‐acylation modulates the function of the apical sodium‐dependent bile acid transporter in human cells. J. Biol. Chem. 295, 4488–4497 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Storch, C.H. , Ehehalt, R. , Haefeli, W.E. & Weiss, J. Localization of the human breast cancer resistance protein (BCRP/ABCG2) in lipid rafts/caveolae and modulation of its activity by cholesterol in vitro. J. Pharmacol. Exp. Ther. 323, 257–264 (2007). [DOI] [PubMed] [Google Scholar]
- 65. Mayati, A. et al. Protein kinases C‐mediated regulations of drug transporter activity, localization and expression. Int. J. Mol. Sci. 18, 764 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Guo, C. , LaCerte, C. , Edwards, J.E. , Brouwer, K.R. & Brouwer, K.L.R. Farnesoid X receptor agonists obeticholic acid and chenodeoxycholic acid increase bile acid efflux in sandwich‐cultured human hepatocytes: Functional evidence and mechanisms. J. Pharmacol. Exp. Ther. 365, 413–421 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Asaumi, R. et al. Expanded physiologically‐based pharmacokinetic model of rifampicin for predicting interactions with drugs and an endogenous biomarker via complex mechanisms including organic anion transporting polypeptide 1B induction. CPT Pharmacometrics Syst. Pharmacol. 8, 845–857 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Meyer Zu Schwabedissen, H.E. , Böttcher, K. , Chaudhry, A. , Kroemer, H.K. , Schuetz, E.G . & Kim, R.B . Liver X receptor alpha and farnesoid X receptor are major transcriptional regulators of OATP1B1. Hepatology 52, 1797–1807 (2010). [DOI] [PubMed] [Google Scholar]
- 69. Niu, C. , Smith, B. & Lai, Y. Transporter gene regulation in sandwich cultured human hepatocytes through the activation of constitutive androstane receptor (CAR) or aryl hydrocarbon receptor (AhR). Front. Pharmacol. 11, 620197 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Niu, C. et al. Organic anion‐transporting polypeptide genes are not induced by the pregnane X receptor activator rifampin: Studies in hepatocytes in vitro and in monkeys in vivo. Drug Metab. Dispos. 47, 1433–1442 (2019). [DOI] [PubMed] [Google Scholar]
- 71. El Saadany, T. et al. microRNA‐206 modulates the hepatic expression of the organic anion‐transporting polypeptide 1B1. Liver Int. 39, 2350–2359 (2019). [DOI] [PubMed] [Google Scholar]
- 72. Tajiri, A. , Hirota, T. , Kawano, S. , Yonamine, A. & Ieiri, I. Regulation of organic anion transporting polypeptide 2B1 expression by microRNA in the human liver. Mol. Pharm. 17, 2821–2830 (2020). [DOI] [PubMed] [Google Scholar]
- 73. Thakkar, N. et al. A cancer‐specific variant of the SLCO1B3 gene encodes a novel human organic anion transporting polypeptide 1B3 (OATP1B3) localized mainly in the cytoplasm of colon and pancreatic cancer cells. Mol. Pharm. 10, 406–416 (2013). [DOI] [PubMed] [Google Scholar]
- 74. Imai, S. , Kikuchi, R. , Kusuhara, H. & Sugiyama, Y. DNA methylation and histone modification profiles of mouse organic anion transporting polypeptides. Drug Metab. Dispos. 41, 72–78 (2013). [DOI] [PubMed] [Google Scholar]
- 75. Ali, I. et al. Transporter‐mediated alterations in patients with NASH increase systemic and hepatic exposure to an OATP and MRP2 substrate. Clin. Pharmacol. Ther. 104, 749–756 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Powell, J. et al. Novel mechanism of impaired function of organic anion‐transporting polypeptide 1B3 in human hepatocytes: post‐translational regulation of OATP1B3 by protein kinase C activation. Drug Metab. Dispos. 42, 1964–1970 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Farasyn, T. et al. Preincubation with everolimus and sirolimus reduces organic anion‐transporting polypeptide (OATP)1B1‐ and 1B3‐mediated transport independently of mTOR kinase inhibition: implication in assessing OATP1B1‐ and OATP1B3‐mediated drug‐drug interactions. J. Pharm. Sci. 108, 3443–3456 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Crowe, A. et al. Characterization of plasma membrane localization and phosphorylation status of organic anion transporting polypeptide (OATP) 1B1 c.521 T>C nonsynonymous single‐nucleotide polymorphism. Pharm. Res. 36, 101 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Shitara, Y. & Sugiyama, Y. Preincubation‐dependent and long‐lasting inhibition of organic anion transporting polypeptide (OATP) and its impact on drug‐drug interactions. Pharmacol. Ther. 177, 67–80 (2017). [DOI] [PubMed] [Google Scholar]
- 80. Farasyn, T. , Pahwa, S. , Xu, C. & Yue, W. Pre‐incubation with OATP1B1 and OATP1B3 inhibitors potentiates inhibitory effects in physiologically relevant sandwich‐cultured primary human hepatocytes. Eur. J. Pharm. Sci. 165, 105951 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Garrison, D.A. , Talebi, Z. , Eisenmann, E.D. , Sparreboom, A. & Baker, S.D. Role of OATP1B1 and OATP1B3 in drug‐drug interactions mediated by tyrosine kinase inhibitors. Pharmaceutics 12, 856 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Uddin, M.E. et al. Influence of YES1 kinase and tyrosine phosphorylation on the activity of OCT1. Front. Pharmacol 12, 644342 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Shu, Y. , Bello, C.L. , Mangravite, L.M. , Feng, B. & Giacomini, K.M. Functional characteristics and steroid hormone‐mediated regulation of an organic cation transporter in Madin‐Darby canine kidney cells. J. Pharmacol. Exp. Ther. 299, 392–398 (2001). [PubMed] [Google Scholar]
- 84. Oda, M. , Koyanagi, S. , Tsurudome, Y. , Kanemitsu, T. , Matsunaga, N. & Ohdo, S. Renal circadian clock regulates the dosing‐time dependency of cisplatin‐induced nephrotoxicity in mice. Mol. Pharmacol. 85, 715–722 (2014). [DOI] [PubMed] [Google Scholar]
- 85. Liu, Y. et al. Epigenetic activation of the drug transporter OCT2 sensitizes renal cell carcinoma to oxaliplatin. Sci. Transl. Med. 8, 348ra97 (2016). [DOI] [PubMed] [Google Scholar]
- 86. Çetinkaya, I. et al. Regulation of human organic cation transporter hOCT2 by PKA, PI3K, and calmodulin‐dependent kinases. Am. J. Physiol. Gastrointest. Liver Physiol. 284, F293–F302 (2003). [DOI] [PubMed] [Google Scholar]
- 87. Pelis, R.M. , Suhre, W.M. & Wright, S.H. Functional influence of N‐glycosylation in OCT2‐mediated tetraethylammonium transport. Am. J. Physiol. Gastrointest. Liver Physiol. 290, F1118–F1126 (2006). [DOI] [PubMed] [Google Scholar]
- 88. Zhang, J. , Yu, Z. & You, G. Insulin‐like growth factor 1 modulates the phosphorylation, expression, and activity of organic anion transporter 3 through protein kinase A signaling pathway. Acta Pharmacol. Sin. B 10, 186–194 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zhang, J. , Liu, C. & You, G. Ubiquitin‐specific peptidase 8 regulates the trafficking and stability of the human organic anion transporter 1. Biochim. Biophys. Acta Gen. Subj. 1864, 129701 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Anzai, N. , Kanai, Y. & Endou, H. New insights into renal transport of urate. Curr. Opin. Rheumatol. 19, 151–157 (2007). [DOI] [PubMed] [Google Scholar]
- 91. Schafer, A.M. , Meyer zu Schwabedissen, H.E . & Grube, M . Expression and function of organic anion transporting polypeptides in the human brain: physiological and pharmacological implications. Pharmaceutics 13, 834–849 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Qosa, H. , Miller, D.S. , Pasinelli, P. & Trotti, D. Regulation of ABC efflux transporters at blood‐brain barrier in health and neurological disorders. Brain Res. 1628, 298–316 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Selo, M.A. , Sake, J.A. , Ehrhardt, C. & Salomon, J.J. Organic cation transporters in the lung‐Current and emerging (patho)physiological and pharmacological concepts. Int. J. Mol. Sci. 21, 9168 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Aguiar, J.A. et al. The impact of cigarette smoke exposure, COPD, or asthma status on ABC transporter gene expression in human airway epithelial cells. Sci. Rep. 9, 153 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Berg, T. et al. Expression of MATE1, P‐gp, OCTN1 and OCTN2, in epithelial and immune cells in the lung of COPD and healthy individuals. Respir. Res. 19, 68 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Anger, G.J.C. , Dai, W. & Piquette‐Miller, M. Pharmacokinetics in pregnancy. In: Reproductive and Developmental Toxicology. 3rd edn., ( Gupta, R. ed.), pp. 33–46 (Elsevier Inc., San Diego, CA, 2022). [Google Scholar]
- 97. Anoshchenko, O. , Prasad, B. , Neradugomma, N.K. , Wang, J. , Mao, Q. & Unadkat, J.D. Gestational age‐dependent abundance of human placental transporters as determined by quantitative targeted proteomics. Drug Metab. Dispos. 48, 735–741 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Manceau, S. et al. ABC drug transporter and nuclear receptor expression in human cytotrophoblasts: influence of spontaneous syncytialization and induction by glucocorticoids. Placenta 33, 927–932 (2012). [DOI] [PubMed] [Google Scholar]
- 99. Lye, P. , Bloise, E. , Nadeem, L. , Gibb, W. , Lye, S.J. & Matthews, S.G. Glucocorticoids modulate multidrug resistance transporters in the first trimester human placenta. J. Cell. Mol. Med. 22, 3652–3660 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Evseenko, D.A. et al. Independent regulation of apical and basolateral drug transporter expression and function in placental trophoblasts by cytokines, steroids, and growth factors. Drug Metab. Dispos. 35, 595–601 (2007). [DOI] [PubMed] [Google Scholar]
- 101. Meyer zu Schwabedissen, H.E . et al. Epidermal growth factor‐mediated activation of the map kinase cascade results in altered expression and function of ABCG2 (BCRP). Drug Metab. Dispos. 34, 524–533 (2006). [DOI] [PubMed] [Google Scholar]
- 102. Prasad, B. et al. Toward a consensus on applying quantitative liquid chromatography‐tandem mass spectrometry proteomics in translational pharmacology research: a white paper. Clin. Pharmacol. Ther. 106, 525–543 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Tatosian, D.A. et al. A microdose cocktail to evaluate drug interactions in patients with renal impairment. Clin. Pharmacol. Ther. 109, 403–415 (2021). [DOI] [PubMed] [Google Scholar]
- 104. Nguyen, J.T. et al. Assessing transporter‐mediated natural product‐drug interactions via in vitro‐in vivo extrapolation: clinical evaluation with a probe cocktail. Clin. Pharmacol. Ther. 109, 1342–1352 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Li, Y. , Talebi, Z. , Chen, X. , Sparreboom, A. & Hu, S. Endogenous biomarkers for SLC transporter‐mediated drug‐drug interaction evaluation. Molecules 26, 5500 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Rodrigues, A.D. , Taskar, K.S. , Kusuhara, H. & Sugiyama, Y. Endogenous probes for drug transporters: balancing vision with reality. Clin. Pharmacol. Ther. 103, 434–448 (2018). [DOI] [PubMed] [Google Scholar]
- 107. Rodrigues, A.D. et al. Exploring the use of serum‐derived small extracellular vesicles as liquid biopsy to study the induction of hepatic cytochromes P450 and organic anion transporting polypeptides. Clin. Pharmacol. Ther. 110, 248–258 (2021). [DOI] [PubMed] [Google Scholar]
- 108. Lutz, J.D. et al. Cytochrome P450 3A induction predicts P‐glycoprotein induction; Part 1: establishing induction relationships using ascending dose rifampin. Clin. Pharmacol. Ther. 104, 1182–1190 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Lutz, J.D. et al. Cytochrome P450 3A induction predicts P‐glycoprotein induction; Part 2: prediction of decreased substrate exposure after rifabutin or carbamazepine. Clin. Pharmacol. Ther. 104, 1191–1198 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Takano, M. , Higa, S. , Furuichi, Y. , Naka, R. & Yumoto, R. Suppression of P‐glycoprotein by cigarette smoke extract in human lung‐derived A549/P‐gp cells. Drug Metab. Pharmacokinet. 35, 214–219 (2020). [DOI] [PubMed] [Google Scholar]
- 111. Neuhoff, S. , Yeo, K.R. , Barter, Z. , Jamei, M. , Turner, D.B. & Rostami‐Hodjegan, A. Application of permeability‐limited physiologically‐based pharmacokinetic models: part II – prediction of P‐glycoprotein mediated drug‐drug interactions with digoxin. J. Pharm. Sci. 102, 3161–3173 (2013). [DOI] [PubMed] [Google Scholar]
- 112. Yamazaki, S. , Costales, C. , Lazzaro, S. , Eatemadpour, S. , Kimoto, E. & Varma, M.V. Physiologically‐based pharmacokinetic modeling approach to predict rifampin‐mediated intestinal P‐glycoprotein induction. CPT Pharmacometrics Syst. Pharmacol. 8, 634–642 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Pan, X. , Yamazaki, S. , Neuhoff, S. , Zhang, M. & Pilla Reddy, V. Unraveling pleiotropic effects of rifampicin by using physiologically based pharmacokinetic modeling: assessing the induction magnitude of P‐glycoprotein‐cytochrome P450 3A4 dual substrates. CPT Pharmacometrics Syst. Pharmacol. 10, 1485–1496 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Almond, L.M. et al. Prediction of drug‐drug interactions arising from CYP3A induction using a physiologically based dynamic model. Drug Metab. Dispos. 44, 821–832 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Yamashita, F. et al. Modeling of rifampicin‐induced CYP3A4 activation dynamics for the prediction of clinical drug‐drug interactions from in vitro data. PLoS One 8, e70330 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Rodrigues, D. & Rowland, A. From endogenous compounds as biomarkers to plasma‐derived nanovesicles as liquid biopsy; Has the golden age of translational pharmacokinetics‐absorption, distribution, metabolism, excretion‐drug‐drug interaction science finally arrived? Clin. Pharmacol. Ther. 105, 1407–1420 (2019). [DOI] [PubMed] [Google Scholar]
- 117. Powell, N.R. , Zhao, H. , Ipe, J. , Liu, Y. & Skaar, T.C. Mapping the miRNA‐mRNA interactome in human hepatocytes and identification of functional mirSNPs in pharmacogenes. Clin. Pharmacol. Ther. 110, 1106–1118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Wang, Y. , Fang, Z. , Hong, M. , Yang, D. & Xie, W. Long‐noncoding RNAs (lncRNAs) in drug metabolism and disposition, implications in cancer chemo‐resistance. Acta Pharmacol. Sin. B 10, 105–112 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. To, K.K. , Leung, W.W. & Ng, S.S. A novel miR‐203‐DNMT3b‐ABCG2 regulatory pathway predisposing colorectal cancer development. Mol. Carcinog. 56, 464–477 (2017). [DOI] [PubMed] [Google Scholar]
- 120. Chen, M. et al. Soluble uric acid increases PDZK1 and ABCG2 expression in human intestinal cell lines via the TLR4‐NLRP3 inflammasome and PI3K/Akt signaling pathway. Arthritis Res. Ther. 20, 20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Mishra, J. , Simonsen, R. & Kumar, N. Intestinal breast cancer resistance protein (BCRP) requires Janus kinase 3 activity for drug efflux and barrier functions in obesity. J. Biol. Chem. 294, 18337–18348 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Fromm, M.F. et al. The effect of rifampin treatment on intestinal expression of human MRP transporters. Am. J. Clin. Pathol. 157, 1575–1580 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Liu, W. , Nakano, M. , Nakanishi, T. , Nakajima, M. & Tamai, I. Post‐transcriptional regulation of OATP2B1 transporter by a microRNA, miR‐24. Drug Metab. Pharmacokinet. 35, 515–521 (2020). [DOI] [PubMed] [Google Scholar]
- 124. Ogura, J. , Yamaguchi, H. & Mano, N. Stimulatory effect on the transport mediated by organic anion transporting polypeptide 2B1. Asian J. Pharm. Sci. 15, 181–191 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Greiner, B. et al. The role of intestinal P‐glycoprotein in the interaction of digoxin and rifampin. J. Clin. Invest. 104, 147–153 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Westphal, K. et al. Induction of P‐glycoprotein by rifampin increases intestinal secretion of talinolol in human beings: a new type of drug/drug interaction. Clin. Pharmacol. Ther. 68, 345–355 (2000). [DOI] [PubMed] [Google Scholar]
- 127. Kamiyama, Y. , Matsubara, T. , Yoshinari, K. , Nagata, K. , Kamimura, H. & Yamazoe, Y. Role of human hepatocyte nuclear factor 4alpha in the expression of drug‐metabolizing enzymes and transporters in human hepatocytes assessed by use of small interfering RNA. Drug Metab. Pharmacokinet. 22, 287–298 (2007). [DOI] [PubMed] [Google Scholar]
- 128. Hong, M. , Hong, W. , Ni, C. , Huang, J. & Zhou, C. Protein kinase C affects the internalization and recycling of organic anion transporting polypeptide 1B1. Biochim. Biophys. Acta 1848, 2022–2030 (2015). [DOI] [PubMed] [Google Scholar]
- 129. Jung, D. , Hagenbuch, B. , Gresh, L. , Pontoglio, M. , Meier, P.J. & Kullak‐Ublick, G.A. Characterization of the human OATP‐C (SLC21A6) gene promoter and regulation of liver‐specific OATP genes by hepatocyte nuclear factor 1 alpha. J. Biol. Chem. 276, 37206–37214 (2001). [DOI] [PubMed] [Google Scholar]
- 130. Ichihara, S. , Kikuchi, R. , Kusuhara, H. , Imai, S. , Maeda, K. & Sugiyama, Y. DNA methylation profiles of organic anion transporting polypeptide 1B3 in cancer cell lines. Pharm. Res. 27, 510–516 (2010). [DOI] [PubMed] [Google Scholar]
- 131. Knauer, M.J. , Girdwood, A.J. , Kim, R.B. & Tirona, R.G. Transport function and transcriptional regulation of a liver‐enriched human organic anion transporting polypeptide 2B1 transcriptional start site variant. Mol. Pharmacol. 83, 1218–1228 (2013). [DOI] [PubMed] [Google Scholar]
- 132. O'Brien, V.P. et al. Hepatocyte nuclear factor 1 regulates the expression of the organic cation transporter 1 via binding to an evolutionary conserved region in intron 1 of the OCT1 gene. J. Pharmacol. Exp. Ther. 347, 181–192 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Schaeffeler, E. et al. DNA methylation is associated with downregulation of the organic cation transporter OCT1 (SLC22A1) in human hepatocellular carcinoma. Genome Med. 3, 82 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Jigorel, E. , Le Vee, M. , Boursier‐Neyret, C. , Parmentier, Y. & Fardel, O. Differential regulation of sinusoidal and canalicular hepatic drug transporter expression by xenobiotics activating drug‐sensing receptors in primary human hepatocytes. Drug Metab. Dispos. 34, 1756–1763 (2006). [DOI] [PubMed] [Google Scholar]
- 135. Ren, T. , Pang, L. , Dai, W. , Wu, S. & Kong, J. Regulatory mechanisms of the bile salt export pump (BSEP/ABCB11) and its role in related diseases. Clin. Res. Hepatol. Gastroenterol. 45, 101641 (2021). [DOI] [PubMed] [Google Scholar]
- 136. Yang, X. et al. Regulation of P‐glycoprotein by Bajijiasu in vitro and in vivo by activating the Nrf2‐mediated signalling pathway. Pharm. Biol. 57, 184–192 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Li, X. , Mu, P. , Qiao, H. , Wen, J. & Deng, Y. JNK‐AKT‐NF‐kappaB controls P‐glycoprotein expression to attenuate the cytotoxicity of deoxynivalenol in mammalian cells. Biochem. Pharmacol. 156, 120–134 (2018). [DOI] [PubMed] [Google Scholar]
- 138. Yang, T. et al. MiR‐223 modulates multidrug resistance via downregulation of ABCB1 in hepatocellular carcinoma cells. Exp. Biol. Med. 238, 1024–1032 (2013). [DOI] [PubMed] [Google Scholar]
- 139. Tanaka, T. , Hirota, T. & Ieiri, I. Relationship between DNA methylation in the 5' CpG island of the SLC47A1 (multidrug and toxin extrusion protein MATE1) gene and interindividual variability in MATE1 expression in the human liver. Mol. Pharmacol. 93, 1–7 (2018). [DOI] [PubMed] [Google Scholar]
- 140. Yu, Q. et al. Histone H3 lysine 4 trimethylation, lysine 27 trimethylation, and lysine 27 acetylation contribute to the transcriptional repression of solute carrier family 47 member 2 in renal cell carcinoma. Drug Metab. Dispos. 45, 109–117 (2017). [DOI] [PubMed] [Google Scholar]
- 141. Saito, H. Pathophysiological regulation of renal SLC22A organic ion transporters in acute kidney injury: pharmacological and toxicological implications. Pharmacol. Ther. 125, 79–91 (2010). [DOI] [PubMed] [Google Scholar]
- 142. Fan, Y. & You, G. Proteasome inhibitors bortezomib and carfilzomib stimulate the transport activity of human organic anion transporter 1. Mol. Pharmacol. 97, 384–391 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Wang, H. , Zhang, J. & You, G. Activation of protein kinase A stimulates SUMOylation, expression, and transport activity of organic anion transporter 3. AAPS J. 21, 30 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Jeong, H.S. , Ryoo, I.G. & Kwak, M.K. Regulation of the expression of renal drug transporters in KEAP1‐knockdown human tubular cells. Toxicol. In Vitro 29, 884–892 (2015). [DOI] [PubMed] [Google Scholar]
- 145. Takano, Y. et al. Up‐regulation of connexin 32 gene by 5‐aza‐2'‐deoxycytidine enhances vinblastine‐induced cytotoxicity in human renal carcinoma cells via the activation of JNK signalling. Biochem. Pharmacol. 80, 463–470 (2010). [DOI] [PubMed] [Google Scholar]
- 146. Kamau, S.W. , Kramer, S.D. , Gunthert, M. & Wunderli‐Allenspach, H. Effect of the modulation of the membrane lipid composition on the localization and function of P‐glycoprotein in MDR1‐MDCK cells. In Vitro Cell. Dev. Biol. Anim. 41, 207–216 (2005). [DOI] [PubMed] [Google Scholar]
- 147. Sampaio‐Maia, B. , Gomes, R. & Soares‐da‐Silva, P. P‐glycoprotein phosphorylation/dephosphorylation and cellular accumulation of L‐DOPA in LLC‐GA5 Col300 cells. J. Auton. Pharmacol. 19, 173–179 (1999). [DOI] [PubMed] [Google Scholar]
- 148. Takita, H. et al. PBPK model of coproporphyrin I: evaluation of the impact of SLCO1B1 genotype, ethnicity, and sex on its inter‐individual variability. CPT Pharmacometrics Syst. Pharmacol. 10, 137–147 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Yoshikado, T. et al. PBPK modeling of coproporphyrin I as an endogenous biomarker for drug interactions involving inhibition of hepatic OATP1B1 and OATP1B3. CPT Pharmacometrics Syst. Pharmacol. 7, 739–747 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Thiel, C. et al. Using quantitative systems pharmacology to evaluate the drug efficacy of COX‐2 and 5‐LOX inhibitors in therapeutic situations. NPJ Syst. Biol. Appl. 4, 28 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Table S2
Table S3