

Summary:
X-chromosome inactivation (XCI) is a random, permanent, and developmentally early epigenetic event that occurs during mammalian embryogenesis. We harness these features to investigate characteristics of early lineage specification events during human development. We initially assess the consistency of X-inactivation and establish a robust set of XCI-escape genes. By analyzing variance in XCI ratios across tissues and individuals, we find that XCI is shared across all tissues, suggesting XCI is completed in the epiblast (in at least 6-16 cells) prior to specification of the germ layers. Additionally, we exploit tissue-specific variability to characterize the number of cells present during tissue lineage commitment, ranging from approximately 20 cells in liver and whole blood tissues to 80 cells in brain tissues. By investigating variability of XCI ratios using adult tissue, we characterize embryonic features of human XCI and lineage specification otherwise difficult to ascertain experimentally.
Keywords: X-chromosome Inactivation, allele-specific expression, developmental lineage, human development, embryonic stochasticity, escape from XCI
Graphical Abstract
eTOC:
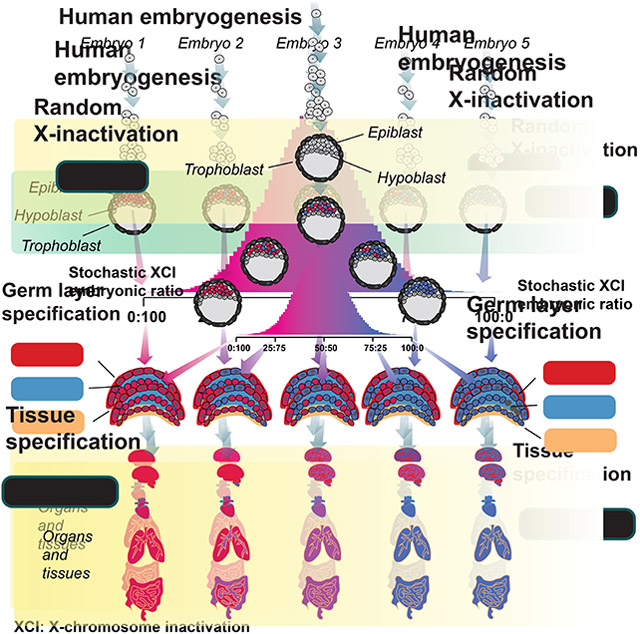
Werner et al. model variability in human XCI ratios across tissues and individuals using the GTEx dataset, determining XCI ratios are consistent across all tissue lineages. This suggests observed XCI variability in adult populations is explained by the statistics of a stochastic embryonic event occurring in a small cell pool.
Introduction:
Every cell within female mammalian embryos undergoes the process of X-chromosome inactivation (XCI), which silences expression from a single randomly chosen X-allele via epigenetic mechanisms (Dossin and Heard, 2021; Lyon, 1961; Migeon, 2013). The random choice of which allele to inactivate occurs early in development and is permanent thereafter with the inactivated allele propagated through each cell’s developmental lineage (Lyon, 1972). As a result, adult females exhibit mosaic X-linked allelic expression throughout every tissue within the body, an enduring phenotypic consequence of an early embryonic milestone. The random, permanent, and developmental early nature of XCI positions the whole-body mosaicism of X-linked allelic expression as a lineage marker reaching back to the earliest embryonic stages (Mclaren, 1972; Nesbitt, 1971). Careful analysis of X-linked allelic expression across individuals and tissues can thus reveal whole-body lineage relationships stemming from some of the first lineage decisions made during embryogenesis (Bittel et al., 2008; Fialkow, 1973; Monteiro et al., 1998; Nesbitt, 1971).
While the probability for inactivation is equal between the X-alleles in humans, variation in XCI allelic ratios across individuals is a salient feature of XCI. Deviation from the expected XCI allelic ratio of 0.5 can arise through various mechanisms (Brown and Robinson, 2000; Naumova et al., 1996; Schmidt and Sart, 1992; Wu et al., 2014) with the most basic being the inherent stochasticity of the initial choice of allelic inactivation (Shvetsova et al., 2019). The variability of the initial XCI ratio within the embryo is directly linked to the number of cells present during inactivation where smaller cell numbers result in increased variability of XCI ratios (Nesbitt, 1971). In fact, one can estimate the number of cells present at the time of inactivation by analyzing the variance of XCI ratios across a population. Several studies using this approach (Amos-Landgraf et al., 2006; Shvetsova et al., 2019) , as well as studies utilizing in vitro embryonic models (Moreira de Mello et al., 2017; Petropoulos et al., 2016; van den Berg et al., 2009), have estimated that XCI occurs in a small stem cell pool within the human embryo with estimates as little as 8 cells. The combination of the random nature and small pool of cells present during XCI imparts an ever-present basal-level of variability in XCI ratios within adult human populations.
The stability of XCI down lineages means that minor cell sampling variation can be used as a marker for any process involving selection of a set of cells, i.e., lineage specification (Fialkow, 1973; Nesbitt, 1971). While growing evidence indicates XCI is initiated early (Moreira de Mello et al., 2017; Petropoulos et al., 2016; van den Berg et al., 2009) , the exact timing of XCI as it relates to early lineage specification is unclear (Geens and Chuva De Sousa Lopes, 2017) and has important implications for the variance in XCI ratios across early lineages. Specifically, the extent of variability in XCI across adult tissues, those derived from the embryonic lineage during embryogenesis, is a long-standing question (Bittel et al., 2008; Hoon et al., 2015) and directly linked to the timing of XCI and early lineage events. Germ layer specification is the first lineage decision made for all future embryonic tissues and occurs during post-implantation embryonic development (Ghimire et al., 2021), a similar timeframe to XCI. If XCI is completed before germ layer specification each germ layer would be specified from the same pool of cells with a set XCI ratio (Fig. 1A). The germ layer-specific XCI ratio would be dependent on the initial XCI ratio resulting in shared XCI ratios across germ layers (Fig. 1A) and the subsequently derived adult tissues. In contrast, if XCI is completed after germ layer specification, germ layer-specific XCI ratios are set independently and are not expected to be shared across the different germ layers (Fig. 1B), producing variance in XCI ratios across adult tissues. Consequently, comparing XCI ratios for tissues within either the same or different germ layer lineages can reveal the temporal ordering of XCI and germ layer specification.
Figure 1: Timing of XCI determines lineage-specific XCI ratio probability.

A, Schematic representing completed XCI before germ layer specification. Each germ layer inherits the same randomly determined XCI ratio set prior to germ layer lineage specification. The probability distribution of XCI is determined by the number of cells present during inactivation. B, Schematic representing completed XCI after germ layer specification. The XCI ratio for each germ layer is set independent of one another, together along with variation in cell numbers fated for each germ layer results in variable XCI ratios across the germ layer lineages.
An additional early lineage event that may overlap with XCI is extraembryonic/embryonic lineage specification (Moreira de Mello et al., 2017; Petropoulos et al., 2016), which precedes germ layer lineage specification. If XCI occurs before or during extraembryonic/embryonic lineage specification, variance in XCI ratios across adult tissues will be influenced by the initial stochasticity of XCI and the subsequent cell selection for the embryonic lineage. In other words, variance in XCI ratios across the germ layer lineages is tied to their last developmental common denominator: the specification of the embryonic epiblast. Since extraembryonic tissues do not contribute to adult tissues, the timing of XCI and extraembryonic/embryonic lineage specification provides the developmental context that variance in adult tissues is potentially tied to the specification of the embryonic epiblast.
In this study, we develop an approach to determine the tissue XCI ratio from unphased bulk RNA-sequencing data, allowing us to assess XCI ratios from any publicly available RNA-sequencing dataset. Utilizing the tissue sampling scheme of the Genotype-Tissue Expression (GTEx v8) project (Lonsdale et al., 2013), we analyze XCI ratios for 49 tissues both within and across individuals for 311 female donors (Fig. S1). We establish that XCI ratios are shared for tissues both within and across germ layers demonstrating that XCI is completed before any significant lineage decisions are made for embryonic tissues. Additionally, we extend population-level modeling of variance in XCI ratios to all well-powered tissues, deriving estimates for the number of cells present at the time of embryonic epiblast and tissue-specific lineage commitment. By providing cell counts, temporal ordering of lineage events, and lineage relationships across tissues, capturing the statistical commonalities that underlie the inherently stochastic nature of XCI is a powerful approach for resolving questions of early developmental lineage specification.
Results:
The folded-normal model accurately estimates XCI ratios from unphased data
A practical consequence of bulk RNA-sequencing is that the XCI ratio of a tissue can be estimated from the direction and magnitude of X-linked allele-specific expression. For a tissue with 75% of cells carrying an active maternal X-allele, approximately 75% of RNA-sequencing reads for heterozygous loci are expected to align to the maternal X-allele (Fig. 2A). However, allelic expression for any given gene is affected by a variety of factors both biological (e.g., eQTLs) and technical (e.g., read sampling). To derive robust estimates, we aggregate allelic expression ratios across well-powered intra-genic heterozygous SNPs for a given tissue, providing a chromosome-wide estimate of the tissue XCI ratio (Fig, 2A).
Figure 2: The folded-normal model accurately estimates XCI ratios from unphased bulk RNA-sequencing data.

A, Schematic demonstrating how allelic expression of heterozygous SNPs reflect the XCI ratio of bulk tissue samples. Aligning expression data to a reference genome scrambles the parental haplotypes. Folding the reference allelic expression ratios captures the magnitude of the tissue XCI ratio. B, Distributions of reference allelic expression ratios for identified heterozygous SNPs across tissue samples exhibiting a range of bulk XCI ratios. Both the unfolded (top row) and folded distributions with the fitted folded normal model (bottom row) are shown. C, For the EN-TEx tissue samples, the phased median gene XCI ratio is plotted against the unphased XCI ratio estimate from the folded normal model. The folded normal model produces near identical XCI ratio estimates for samples with XCI ratios greater than or equal to 0.60. D, Deviation of the folded normal model from the phased median gene XCI ratio when excluding or including known escape genes. E, Aggregated folded reference allelic expression distributions for known escape and inactive genes in EN-TEx tissues with XCI ratios >= 0.70. F, Root mean squared error distributions for GTEx tissue samples binned by their original estimated XCI ratio as read depth per SNP is gradually reduced. See also Figure S1.
When aligned to a reference genome, reference alleles will be composed of both maternal and paternal alleles for a given sample. It follows that reference allelic expression ratios represent the expected expression ratios from both the maternal and paternal alleles given the XCI ratio of the tissue (Fig. 2A). To account for this, folding the reference allelic expression ratios about 0.5 aggregates the imbalanced allelic expression within the tissue across the two alleles. This enables the magnitude of the XCI ratio to be estimated from unphased expression data by fitting a folded distribution (Gart, 1970; Urbakh, 1967) (see methods, Fig. 2A-B).
To assess the accuracy of the folded-normal model in estimating XCI ratios, we test our approach with phased bulk RNA-sequencing data from the EN-TEx (Rozowsky et al., 2021) consortium, a total of 49 tissue samples from 2 female donors spanning 26 different tissues. Comparing the unphased estimates derived with the folded-normal model to the phased median allelic expression per sample, we find nearly perfect XCI ratio estimate correspondence for ratios greater than 0.6 (Fig. 2C). For samples skewed closer to the folding point of 0.5, model misspecification of the underlying distribution makes the estimate overconservative.
Our approach for estimating XCI ratios aggregates allelic expression across numerous heterozygous loci, averaging away mechanisms outside of XCI that may impact X-linked allelic expression. A widespread mechanism that may still impact our XCI ratio estimates is escape from inactivation, where a gene is biallelically expressed from the active and inactive X-alleles (Tukiainen et al., 2017). Between 15-30% of genes on the X-chromosome have documented evidence for escape (Carrel and Willard, 2005; Tukiainen et al., 2017). While we exclude known escape genes (Tukiainen et al., 2017) from our folded-normal XCI ratio estimates, it is very likely unannotated escape genes are present within the data. To identify the impact of escape on our XCI ratio estimates, we compare folded-normal XCI ratio estimates derived with either excluding or including known escape genes to the phased XCI ratio of tissues excluding the known escape genes (Fig. 2D). Including known escape genes biases the folded-normal XCI ratio estimates towards 0.5 (Fig. 2D). By comparing allelic ratios of known escape genes to all other genes in EN-TEx tissues with XCI ratios >= 0.7, we clearly see escape genes trend towards balanced biallelic expression contributing to the underestimated XCI ratios when including escape genes (Fig. 2E).
To assess variance in XCI and escape more broadly, we capitalize on the tissue sampling structure of the Genotype-Tissue Expression (GTEx v8) dataset (Fig. S1). From an average of 56 +− 23.5 (SD) well-powered heterozygous SNPs (genes, see methods) per sample (Fig. S1), we derive robust XCI ratio estimates for 4658 GTEx tissue samples spanning 49 different tissues (Fig. S1).
In addition to biological sources of variation (escape), read depth is a critical source of technical variation to assess when analyzing allelic expression. Sampled allelic expression is the result of a binomial sampling event dependent on the number of reads sampled and the probability of allelic expression. While we employ stringent read count requirements (see methods), we additionally explore how robust our tissue-level XCI ratio estimates are in the face of global decreases in read depths across genes (Fig. 2F). As read depths per gene are decreased (10%, 20%, 30%, etc.), the vast majority of increased error in the XCI ratio estimates is constrained to the estimates below 0.6 (Fig. 2F), whereas the most skewed tissue samples (XCI ratio estimates above 0.9) display nearly zero additional error even up to an 80% reduction in read depth (Fig. 2F). These results are in line with our phased vs unphased comparisons demonstrating XCI ratio estimates above 0.6 (Fig. 2C) are highly accurate. Additionally, these results appear to be independent of the number of genes used to estimate the tissue XCI ratio (Fig. S1), where we use a minimum of 10 genes per sample. This suggests that aggregating allelic expression over even a modest number of genes is powered to accurately estimate tissue XCI ratios above 0.6 from bulk RNA-sequencing data.
Escape genes exhibit consistent cross-tissue biallelic expression
Our method to quantitatively determine the tissue XCI ratio via aggregating signal across genes is especially well-suited to explore escape from XCI within the GTEx dataset (Fig. 2E). Our basic strategy for detecting escape genes is to calculate each gene’s consistency with the aggregate chromosomal inactivation ratio. Assessing all X-linked genes utilized in our GTEx XCI ratio estimates (Fig. 3A) and previously annotated constitutively escape genes (Tukiainen et al., 2017) results in a wide range of correlations between gene and tissue XCI ratios, exemplified by the genes SHROOM4 and TCEAL3 (Fig. 3B). As expected, the transcripts associated with XCI, namely, XIST and TSIX, show some of the highest correlations to the tissue XCI ratio (i.e., top 8.7%, Fig. 3B). Similarly, known escape genes exhibit some of the smallest correlations (Fig. 3B). Interestingly, several genes previously annotated as escape do exhibit rather strong correlations to the XCI ratio of tissues. We find that increased gene expression is linked to increased correlation to the tissue XCI ratio (Fig. 3C) suggesting that some gene variation with respect to the tissue XCI ratio is technical, reflecting read sampling at low expression. At matched expression levels, previously annotated escape genes have smaller tissue-gene XCI ratio correlations compared to all other genes (Fig. 3C), demonstrating that known escape genes are less correlated to the tissue XCI ratio as expected by expression levels alone.
Figure 3: Genes that escape XCI exhibit balanced biallelic expression across XCI skewed tissues.

A, The genomic location and number of GTEx samples each gene is detected for the 542 genes that pass our quality control filters. B, All 542 genes and 45 known escape genes ranked by the Pearson correlation coefficient for each gene’s allelic expression and the XCI ratio of the tissue for samples that detect that gene. C, Distributions of gene-tissue XCI ratio correlations for all 542 genes and 45 escape genes, binned by average expression. The range of average expression is binned into 4 equally spaced bins. We label the top 50% of ‘all other genes’ in each expression bin as ‘inactive genes’ and the bottom 50% as ‘unknown’ genes, as they are potentially a mix of inactive and unannotated escape genes. D, An example for how the empirical p-values are calculated for a given test gene across tissue samples. For a given tissue sample, we calculate each gene’s allelic expression ratio deviation from 0.5, where the black histogram represents the deviations from the inactive genes in the sample and the blue dotted line represents the deviation of the given test gene in the sample, ARHGAP4 in this example. We apply Fisher’s method to aggregate each test gene’s distribution of empirical p-values to calculate a meta-analytic p-value to determine significance (ARHGAP4 meta-analytic p-value: 4.44e−21, SLC6A8 meta analytic p-value: 0.997). E, The aggregated empirical p-value distributions for inactive, known escape, and the unknown genes now classified as confident inactive and novel escape are plotted. The unknown genes are classified as either confident inactive or novel escape by using a significance threshold of meta-analytic p-value < .001. F, The percent of genes previously annotated for escape per sample is plotted against the difference between the sample’s XCI ratio estimates derived when either including or excluding the previously annotated escape genes. The inset plot compares the XCI ratio estimates derived without the known escape genes (x-axis) or including the known escape genes (y-axis). See also Figure S2 and Table S1.
From our analysis in the EN-TEx dataset, escape from inactivation trends toward balanced biallelic expression rather than achieving completely equal allelic expression (Fig. 2E), explaining how some escape genes retain significant correlations to tissue XCI ratios in the GTEx dataset. To comprehensively test the degree to which escape produces balanced allelic expression, we construct a one-sided test to detect whether a gene consistently trends towards balanced biallelic expression regardless of the XCI ratio of the tissue (see methods, Fig. S2). Against a null distribution of inactivated genes, we are able to identify genes with consistent biallelic expression in opposition to the aggregate imbalanced tissue XCI ratio, indicating escape from XCI (Fig. 3D).
Testing the known escape genes using this approach results in significant escape signal (Fig. 3E). Similarly, we are able to identify 19 genes previously unannotated for constitutive escape to have significant escape signal (p-value < .001): ARHGAP4, BTK, CASK, CHRDL1, CLIC2, COX7B, CTPS2, CXorf36, F8, ITM2A, MECP2, MPP1, NLGN4X, PGK1, RPL36A, SASH3, SEPT6, STARD8, VSIG4 (Fig. 3E, Fig. S4). Revisiting these genes within the literature, several have prior evidence for escape, though typically limited in the tissues assessed: BTK (Hagen et al., 2020; Zito et al., 2021), CASK (Zito et al., 2021), CHRDL1 (Zito et al., 2021), CLIC2 (Tukiainen et al., 2017; Zito et al., 2021), COX7B (Larsson et al., 2019), CTPS2 (Balaton et al., 2021), CXorf36 (Winham et al., 2019), MPP1 (Zito et al., 2021), NLGN4X (Tukiainen et al., 2017; Zito et al., 2021), SASH3 (Zito et al., 2021), SEPT6 (Zhang et al., 2013), VSIG4 (Berletch et al., 2015). Our results suggest these genes escape inactivation more broadly than previously reported. In addition, our analysis provides supporting evidence of escape for 34 previously annotated escape genes and supporting evidence of inactivation for 143 genes (Table S1). While in this analysis we are powered to identify more constitutively escape genes, variability in escape across tissues and individuals is well documented. As such, our escape annotations are robust to the GTEx data we sample over and will benefit greatly from future experimental follow up.
To test the impact of including escape genes on our GTEx tissue XCI ratio estimates, we compare our original tissue XCI ratio estimates to estimates calculated while including the known escape genes (Fig. 3F). The inclusion of escape genes results in slightly underestimated XCI ratios (Fig. 3F), though the impact is minimal with an average absolute deviation of 0.0088 (± 0.010 SD) between XCI ratio estimates including/excluding the known escape genes. This demonstrates our folded aggregation of allelic expression across genes to estimate XCI ratios is robust to noise generated by escape from inactivation.
XCI is completed prior to germ layer specification
Having developed a robust approach to measure XCI ratios from unphased data, we turn to assessing the degree XCI ratios are shared across tissues within individuals. As an initial visualization of XCI ratios across tissues, we order all female GTEx donors by their average XCI ratio and plot the ratio for all tissues grouped by germ layer (Fig. 4A). XCI ratios qualitatively appear consistent across all tissues and the three germ layers (Fig. 4A). We then ask how well do individual tissues predict all other tissues’ XCI ratios, which we quantify with the AUROC (area under receiver operating characteristic curve) metric (Fig. S3). For a given tissue, we take the average XCI ratio of all other tissues for each donor and use this average to classify the donors as low/high XCI ratio donors. If the given tissue’s XCI ratio can recapitulate the same low/high classifications of the donors, this indicates that tissue’s XCI ratio is in concordance with the average of all other tissues and would result in an AUROC close to 1. Across various thresholds for defining low/high donors, we see that performance is high and consistent across all tissues, suggesting XCI ratios are generally shared across all tissues for an individual (Fig. S3).
Figure 4: XCI ratios are shared across germ layer lineages.

A, Heatmap of all estimated XCI ratios for the tissues of each donor, with donors ordered by their mean XCI ratio across tissues and tissues grouped by germ layer lineage. Black indicates no tissue donation for that donor-tissue pair. B, Examples of within and across germ layer lineage comparisons of XCI ratios. Each data point represents the estimated XCI ratios of the two indicated tissues for a single donor. C, All significant (FDR corrected p-value <= 0.5, permutation test n = 10000) Pearson correlation coefficients for within and across germ layer lineage comparisons. D, Stacked bar plots for the germ layer percentage composition for each sample in the Lung, Esophagus Mucosa, and Skin Lower Leg GTEx tissues. The deconvolved cell type percentages and their germ layer annotations are provided in Fig. S2. E-G, the folded allelic expression ratios for germ layer markers and all other genes (Not markers) are plotted for several example donors per tissue, E: Lung, F: Skin Lower Leg, G: Esophagus Mucosa. The adjacent scatter plots compare the median folded allelic expression between germ layer markers for all donors. E: Lung mesodermal and endodermal markers, Pearson correlation of 0.626 (p-value < .001), F: Skin Lower Leg mesodermal and ectodermal markers, Pearson correlation of 0.621 (p-value < .001), G: Esophagus Mucosa endodermal and ectodermal markers, Pearson correlation 0.603 (p-value < .001), mesodermal and ectodermal markers, Pearson correlation 0.360, (p-value < .001), mesodermal and endodermal markers Pearson correlation 0.537 (p-value < .001). See also Figure S3-4 and Table S2.
Stratifying tissue comparisons of XCI ratios by germ layer lineage relationships should resolve the temporal ordering of XCI and germ layer specification within the human embryo. If XCI occurs before germ layer specification, tissue XCI ratios are expected to positively covary across tissues from different germ layer lineages (Fig.1A). In contrast, if XCI occurs after germ layer specification, the XCI ratio of each germ layer is set independently and there is little expected covariance in XCI ratios for tissues from different germ layers (Fig.1B). We compute correlations of the XCI ratio for combinations of tissues derived from either the same or different germ layers, exemplified in Figure 4 panel B. Tissues sharing the same germ layer lineage produce strictly positive significant correlation values ranging from 0.25 to 0.90 (Fig. 4C), demonstrating XCI ratios are shared within individual germ layer lineages. Strikingly, significant positive ratio correlations for tissues derived from different germ layers are on the same order as the within germ layer comparisons, ranging from 0.24 to 0.87 (Fig. 4C, Fig. S3). The fact tissues derived from different germ layers covary for their XCI ratio strongly suggests XCI is completed prior to germ layer specification and the initial embryonic XCI ratio is propagated through all germ layer lineages.
While we annotate individual tissues to belong to a single primary germ layer, tissues are compositions of cell types derived from different germ layers. This may impact the observed variance in XCI ratios across tissues if there is a strong germ layer-specific effect in XCI ratio variance. We take advantage of the recently released single-nucleus RNA-sequencing (Eraslan et al., 2022) GTEx data to deconvolve (Newman et al., 2019) several of the bulk tissues into their germ layer components, allowing us to explore variance in XCI ratios across germ layers within single tissues. Figure 4D provides examples of the deconvolved germ layer proportions of three tissues with the remaining 6 tissues provided in Figure S4, demonstrating there is variation in germ layer composition within tissues. We extract germ layer-specific markers for the lung, skin, and esophagus mucosa tissues (Table S2, see methods) to explore variance in XCI ratios across germ layers within single tissues. The XCI ratios of germ layer-specific markers positively covary in each tissue (Fig. 4E-G, Pearson correlations: lung mesoderm and endoderm 0.626, skin mesoderm and ectoderm 0.621, esophagus endoderm and ectoderm 0.603, esophagus mesoderm and ectoderm 0.360, esophagus mesoderm and endoderm 0.537), recapitulating the result of shared XCI ratios across germ layers we demonstrate with the non-deconvolved tissues.
Specific tissue lineages have increased probability for switching the parental direction of XCI
In addition to demonstrating that XCI ratios are broadly shared across all tissues, our cross-tissue analysis reveals there is a degree of variability in XCI ratios across tissues within individuals. Comparing distributions of gene-level allelic expression across tissues for individual donors reveals there are often individual tissues that exhibit divergence in XCI ratios in opposition to the general trend of shared XCI ratios (Fig. 5A-B). This is evidenced by the divergent distributions of gene-level allelic-expression for the Whole Blood, Vagina, and Skin tissues in donor 11P81 (Fig. 5A), and the Esophagus – Mucosa, Vagina, and Skin tissues in donor 1J1OQ (Fig. 5B). The presence of individual tissues exhibiting divergent XCI ratios within an individual suggests there may be lineage-specific effects contributing to variance in XCI ratios across tissues.
Figure 5: Individual tissue lineages exhibit increased variance in XCI ratios.

A, Folded allele-specific expression distributions for individual tissues from the 11P81 donor with the aggregated germ layer distributions in the top panel. B, Folded allele-specific expression distributions for individual tissues from the 1J1OQ donor with the aggregated germ layer distributions in the top panel. C, Pearson correlation distributions calculated from all pairwise comparisons of shared heterozygous SNPs between two tissues for all of donor 11P81 ‘s tissues. Positive correlations indicate the same parental direction of XCI, negative correlations indicate opposite parental directions of XCI. D, Similar to C, displaying results for donor 1J1OQ’s tissues. E, Box plots of the per donor proportion of tissues that switched parental XCI directions with donors binned by their mean XCI ratio across tissues. F, Bar plot indicating the proportion of donors where the specified tissue switched directions compared to other tissues. Asterisks indicate significance from Fisher’s Exact test (FDR corrected p-value <= .05), identifying tissues enriched for switching XCI directions.
To further investigate the degree of variation in XCI ratios across tissues, we take advantage of the cross-tissue sampling of individual donors to determine the parental direction of XCI. If an expressed heterozygous SNP is captured for two different tissues of an individual, the reference allele is on the same haplotype and maintains directional allelic information. Thus, calculating the correlation of reference SNP allelic ratios for shared SNPs between two tissues can reveal whether those tissues share the same XCI direction (Fig. 5C-D, see methods). When examining a donor with generally high XCI ratios across all tissues (Fig. 5C Donor 11P81), we find that all tissues share the same parental direction in allelic inactivation. Whereas a less skewed donor (Fig. 5D Donor 1J10Q, Ovary and Vagina tissues) exhibits a subset of tissues with opposite parental inactivation compared to the majority of tissues for that donor. Across all donors, as the average XCI ratio of their tissues increases, the proportion of their tissues exhibiting switched parental XCI decreases (Fig. 5E), with the most skewed donors exhibiting zero tissues with switched parental XCI (Fig. 5E). Interestingly, switching parental direction of XCI is in fact concentrated in a subset of tissues, with 12 out of 49 tissues being significantly enriched for instances of switched XCI (Fig. 5F, fisher’s exact test, p-value <= 0.5). The existence of individual tissues with increased probability for switching parental directions of XCI is indicative of increased variance in XCI ratios for those particular tissue lineages. We explore this model further in the Results section ‘Cell population estimates at the time of tissue-specific lineage commitment’.
Cell population estimate at the time of embryonic epiblast lineage specification
The fact XCI ratios are broadly shared across tissues suggests the initial embryonic XCI ratio determined at the time of inactivation is propagated through development. This is strongly evidenced by the consistency of XCI ratios across the developmental distant germ layer lineages (Fig. 4, Fig. 5A-B). Population level variance in adult XCI ratios thus, in part, reflects the sample distribution during XCI, which depends on the number of cells present during inactivation. We derive estimates for the number of cells present at the time of inactivation by modeling XCI ratio variance from tissue-specific ratio distributions across donors (Fig. 6A, Fig. S8). Using a maximum likelihood approach, we fit estimated models to the tails of the empirical XCI ratio distributions to account for the uncertain unfolded XCI ratio estimates between 0.4 and 0.6 (Fig. 6A, see methods). The cell number estimates derived from all well-powered tissues range from 6 to 16 cells (Fig. 6B), i.e., approximately within a single cell division, demonstrating a striking degree of similarity in population level XCI ratio variance across the assessed tissues. We model variance in XCI ratios as a random binomial sampling event that is then propagated through development. The consistency in XCI ratios across developmentally distant tissues supports this model, though there are likely additional contributors to the observed variance in XCI ratios, such as genetic variation which might drive allelic selection (Brown and Robinson, 2000; Schmidt and Sart, 1992) as well as stochastic deviations during development (Sun et al., 2021). In the simplest case, observed variance in XCI ratios is derived from the initial stochasticity of XCI, positing our cell number estimates as lower bounds for the number of cells that must be involved in XCI.
Figure 6: XCI and tissue lineage specification can be timed to a pool of cells by exploiting observed variability.

A, Example tissue demonstrating the model for estimating cell numbers at the time of XCI using the population-level variance in XCI ratios. We fit normal distributions, as a continuous approximation of the underlying binomial distribution of XCI ratios, to the tails of tissue-specific XCI ratio distributions (shaded in blue), which accounts for the uncertain 0.40-0.60 unfolded XCI ratio estimates (shaded in grey). B, The resulting estimated cell numbers present during XCI derived from the XCI ratio variance of all tissues with at least 10 donors. Error bars are 95% confidence intervals and tissues are grouped by germ layer lineage. C, Schematic for our model of tissue lineage specification and the implications for tissue-specific XCI ratios. The XCI ratio of a tissue is dependent on the prior XCI ratio of the embryo and the number of cells selected for that tissue lineage. These two features define the binomial distribution for that tissue’s XCI ratio. D, Estimated number of cells selected for individual tissue lineage specification of 46 different tissues. Error bars represent 95% confidence intervals. The top bar graph plots the variance in the distribution of tissue XCI ratio deviation from the average XCI ratio of each donor for that tissue. The inset plot compares the estimated number of cells present at the time of tissue specification to the proportion of that tissue’s samples that switched parental XCI directions, Pearson correlation −0.663 (p-value < .001).
Notably, we sample variance in XCI of tissues derived from the embryonic lineage. If XCI occurs before extraembryonic/embryonic lineage specification, the variance we observe in adult tissues is a combination of the initial variance at the time of XCI and additional sampling variance linked to the lineage specification of the embryonic epiblast. This contextualizes our 6-16 cell number estimate as a potential lower bound for the number of cells present during embryonic epiblast lineage specification in the human embryo.
Cell population estimates at the time of tissue-specific lineage commitment
Tissue-specific lineage commitment can be modeled as a random sampling event from a pool of unspecified progenitor cells. In the context of XCI, the XCI ratio of the newly specified tissue is dependent on the prior XCI ratio of the progenitor pool and the number of cells fated for that tissue and can be modeled as a binomial sampling event (Fig. 6C). As such, the GTEx dataset offers a unique opportunity to capture this tissue-specific XCI variance and model the lower bound for the number of cells present at the time of tissue-specific lineage commitment across a broad range of human tissues.
To capture the tissue-specific variance in XCI as it relates to the prior embryonic XCI ratio, we model the deviation of tissue-specific XCI ratios from the average donor XCI ratios for all donors of a given tissue (see methods, Fig. 6D, 46 well-powered tissues). Our model follows the logic that tissues with large variation in their deviation from average donor XCI ratios are derived from a smaller pool of cells, a consequence of increased variability due to small sample size effects. On the low end of the estimated cell numbers, we have liver, whole blood, and adrenal tissues with ~20 estimated cells compared to the brain tissues which occupy most of the higher estimated cell numbers, ranging from ~40-140 estimated cells. In line with our model that tissues derived from smaller stem cell pools are subject to increased variability in XCI ratios, we find a strong negative relationship between our estimated tissue lineage-specific cell numbers and the probability of a tissue switching the direction of parental XCI (Fig. 6D inset, Pearson correlation: −0.663, p-value < .001). A tissue derived from a small number of cells is more likely to result in a sample of oppositely skewed cells compared to the parental XCI ratio of the unspecified progenitor pool simply through increased sampling variance. Our estimated lineage-specific cell numbers and lineage-specific probability for switching parental XCI are internally consistent with a model of lineage-specific variance in XCI ratios being driven by cell sampling variation at the time of lineage specification.
Discussion:
In this work, we exploited the random, permanent, and developmentally early nature of XCI to investigate characteristics of early lineage specification events during human development. By analyzing variance in XCI ratios across tissues and individuals, we showed human XCI is completed before tissue specification and the stochastically determined XCI ratio set during embryogenesis is a shared feature across all tissue lineages. We estimate a lower bound of 6-16 cells are fated for the embryonic epiblast lineage based on population-level variance in XCI ratios. Additionally, we provide lower bound estimates of the number of cells present during tissue-specific lineage specification for 46 different tissues. To conduct this analysis, we developed a method to estimate the ratio of XCI using unphased allele-specific expression, a highly scalable approach applicable to any bulk RNA-sequencing sample.
This work provides insight into the observed variance of XCI ratios in normal female populations, an area of ongoing debate (Brown and Robinson, 2000; Clerc and Avner, 2006; Migeon, 1998; Peeters et al., 2016). Our results indicate that the initial embryonic XCI ratio is propagated through development and is a shared feature across all tissue lineages. This demonstrates the stochasticity of the initial choice for inactivation within the embryo has a measurable impact on XCI ratios in adult females. Importantly, GTEx donors presumably represent a phenotypically normal population; as such, our analysis captures XCI variance in the absence of potential drivers (X-linked diseases) of allelic-selection, representing the null distribution of XCI variation in adult females.
Additional contributors to the observed variance in XCI ratios across tissues may be genetic variation that can drive allelic selection over development (Brown and Robinson, 2000; Schmidt and Sart, 1992) or stochastic deviations in XCI ratios caused by developmental proliferation (Sun et al., 2021). In contrast to these models, we report strikingly consistent XCI ratios across tissues for individual donors, and, importantly, across tissues derived from different germ layers. If allelic-selection or stochastic deviations from proliferation were strong contributors to variance in XCI, we would not expect consistent XCI ratios across developmentally distant adult tissues. Nevertheless, it is unlikely that the initial embryonic XCI ratio is propagated through development with perfect fidelity, which contextualizes our cell number estimates as lower bound estimates for the number of cells that must have been involved in XCI or lineage specification events. In general, our results suggest XCI ratios are broadly shared across tissues with lineage-specific stochasticity due to cell sampling effects during lineage-specification.
For the timing of XCI, there is a wealth of complimentary research on the exact molecular mechanisms (Dossin and Heard, 2021; Vallot et al., 2017) that define the highly complex biological process of XCI. XCI is a continuous molecular process and recent studies from human embryos suggest the timing of XCI may overlap the lineage specification of the extraembryonic and embryonic tissues (Moreira de Mello et al., 2017; Petropoulos et al., 2016), which precedes germ layer specification. In this study, we aimed to interrogate timing of XCI as it relates to germ layer specification within the embryonic lineage. Any overlap in timing for the molecular process of XCI and extraembryonic/embryonic lineage specification will have no impact on our results and conclusions of shared variance in XCI within the embryonic lineage. The consideration of extraembryonic tissues provides the developmental context that XCI ratio variance within the germ layer lineages may be a combination of XCI stochasticity and cell sampling during embryonic epiblast specification.
One alternative model consistent with our results is the potential for rapid allelic changes in the time between XCI and germ layer specification, allowing for selection or drift to occur, with the XCI ratio then stabilized after germ layer specification. While possible, we find this improbable due to the small number of cell divisions estimated to occur between XCI and germ layer specification, as well as the lack of evidence for any continued effects after germ layer specification.
Our work is part of a broader history of using X-linked mosaicism as a useful tool for studying lineage relationships, with studies ranging from investigations of early lineage events in mice (Nesbitt, 1971) to ascertaining tumor clonality (Linder and Gartler, 1965). Typically, these approaches will capitalize on a single locus of the X-chromosome to determine XCI status (Boudewijns et al., 2007). One of our methodological contributions is demonstrating the allelic expression imbalance generated via XCI can be aggregated across multiple loci to provide near-perfect estimates of XCI ratios, even in the absence of phased information.
While GTEx represents the premier dataset for human cross-tissue functional genomics, more data is always helpful. As our approach for estimating XCI ratios is applicable to any bulk RNA-sequencing data, we envision this work providing an informative control for any future functional genomic investigations involving the X-chromosome.
Limitations of study
While the GTEx dataset aims to sample non-diseased tissues, we cannot rule out potential disease-states, genetic or otherwise, for all tissue samples, where disease may impact allelic selection and contribute to variance inn XCI ratios. When assessing escape from XCI, we focus on genes with constitutive rather than facultative signal and cannot make conclusions on likely tissue- or donor-specific escape. Our tissue-specific cell count estimations depend on the sample size of the given tissue and the number of tissues sampled for individual donors, both of which vary considerably across tissues and individuals. As such, these estimates are likely rough approximations that can be improved with additional tissue and donor sampling.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Requests for further information should be directed to and will be fulfilled by the lead contact, Jesse Gillis (jesse.gillis@utoronto.ca).
Materials availability
This study did not generate new unique reagents
Data and code availability
This paper analyzes existing, publicly available data. Links to access these datasets are listed in the key resources table. The generated allele-specific expression information per sample (variant information removed) and the CIBERSORTx deconvolution results are made available at the FTP site: http://labshare.cshl.edu/shares/gillislab/people/werner/werner_et_al_Dev_Cell_2022/data. Descriptions of the data are available at github.com/JonathanMWerner/human_cross_tissue_XCI
All original code has been deposited at figshare (DOI: 10.6084/m9.figshare.20216816) and at Github (github.com/JonathanMWerner/human_cross_tissue_XCI) and is publicly available as of the date of publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Detailed explanation of donor enrollment, sample collection, and ethical details of the GTEx dataset are provided in Lonsdale et al., 2013.
METHOD DETAILS
GTEx and EN-TEx data
Fastq files for all female donors from the GTEx project v7 release (Lonsdale et al., 2013) were obtained from dbGaP accession number phs000424.vN.pN. BAM files for additional female samples from the v8 release were obtained from the associated AnVIL repository (gtexportal.org/home/protectedDataAcccess). All GTEx v7 data files can also be accessed in the GTEx v8 AnVIL repository. Phased expression data from the EN-TEx project (Rozowsky et al., 2021) were obtained in collaboration with the ENCODE consortium. EN-TEx data is available on the online portal. Expression data and annotations for the GTEx single nucleus RNA-sequencing data were obtained from the GTEx data portal.
RNA-seq alignment and SNP identification
For aligning RNA-sequencing data, the GRCh38.p7 human reference genome using GENCODE v.25 (Frankish et al., 2021) annotations was generated with STAR v2.4.2a (Dobin et al., 2013) and data was aligned with STAR v2.4.2a or STAR v2.5.2b. STAR was run using default parameters with per sample 2-pass mapping. BAM files for the additional GTEx v8 samples (originally aligned to GRCh38.p10 with GENCODE v.26 annotations) were sorted using samtools v1.9 (Li et al., 2009) and converted to fastq files using bedtools v.2.26.0 (Quinlan and Hall, 2010). For each sample, alignment to the X-chromosome was extracted using samtools and passed to GATK (McKenna et al., 2010) for SNP identification. Using GATK v.4.1.3.0 and following the best practices workflow for RNAseq short variant discovery (GATK best practices), we utilized the following pipeline of GATK tools using default parameters unless otherwise stated: AddorReplaceReadGroups -> MarkDuplicates -> SplitNCigarReads -> HaplotypeCaller (-stand-call-conf 0.0) -> SelectVariants (-select-type SNP) -> VariantFiltration. The following filters were used in VariantFiltration to set flags for downstream filtering: QD < 2.0, QUAL < 30.0, SOR > 3.0, FS > 60.0, MQ < 40.0, MQRankSum < −12.5, and ReadPosRankSum < −8.0. These filters were determined from GATK recommendations and empirical evaluation of the identified SNPs’ metrics.
SNP quality control
SNPs identified through GATK were further filtered on various metrics to increase confidence in SNPs identified from RNA-sequencing data and ensure well-powered SNPs for allele-specific expression analysis. The resulting .vcf files from GATK were filtered to only contain SNPs present within dbSNP (Sherry et al., 2001). The remaining SNPs were filtered to be heterozygous with 2 identified alleles and at least 10 reads mapped to each allele for a minimum threshold of 20 reads per SNP. Additionally, SNPs were required to pass the SOR, FS, and ReadPosRankSum filters set in the GATK pipeline. Only SNPs located within annotated genes (excluding the PAR regions of the X-chromosome) were considered and in the case of multiple identified SNPs in the same gene for a sample, the SNP with the highest total read count was taken as the max-powered representative for that gene. SNPs with a total read count above 3000 were excluded as they demonstrated a uniform distribution of allelic expression.
Gene filtering (reference bias and XCI escape)
From the observation of a heavy tail towards allelic expression in the reference direction across all called SNPs in the GTEx dataset, we compiled gene specific distributions of allelic expression to determine if a select few genes/SNPs were at fault. The majority of genes demonstrated distributions of relative allelic expression centered around 0.5 with several considerable exceptions, some genes exhibited bimodal or extremely biased distributions. We excluded genes that failed the dip test for unimodality as well as the top and bottom 5% of genes ranked by the deviation of their mean reference expression ratio from 0.5. Additionally, we excluded genes previously annotated to constitutively escape XCI (Tukiainen et al., 2017). In total, we end up with well-powered SNPs from 542 genes along the X-chromosome for modeling XCI ratios.
Folded normal model for estimating XCI ratios
We aggregate the allelic expression imbalance of the X-chromosome over both alleles by folding the reference allelic expression ratios about 0.5 (Fig 2A-B). To obtain our XCI ratio estimates we fit a folded normal distribution to the folded reference allelic expression ratios of each sample, using the maximum log likelihood estimate as the estimated XCI ratio. Theoretically, the captured bulk allelic expression for a heterozygous X-linked SNP follows a binomial distribution characterized by the read depth of the SNP and the XCI ratio of the sample. Without phasing information, the allelic expression of heterozygous X-linked SNPs can be characterized by the folded-binomial model (Gart, 1970; Urbakh, 1967). Since SNPs vary in read depth and various biological factors (e.g. eQTLs) are not accounted for in the binomial model, we take the folded normal model as a continuous approximation. We require samples to have XCI ratio estimates derived from at least 10 filtered SNPs for downstream analysis, resulting in 4659 samples with a mean of 56 well-powered SNPs per sample (Fig. S1). Additionally, we calculate 95% confidence intervals (CI) for each XCI ratio estimate via a nonparametric bootstrap percentile approach (n = 200), excluding XCI ratio estimates with a CI width >= .15 from downstream analysis. For donors with multiple samples for the same tissue, we average the XCI ratio estimates together, duplicated tissue samples have minor differences in estimated XCI ratios (mean difference in XCI ratios for duplicate tissue samples: 0.018 +− 0.023 SD).
Modeling read sampling error when estimating XCI ratios
The sampled allelic reads for any expressed heterozygous loci will follow a binomial distribution defined by the total number of reads sampled (n) and the probability for allelic expression (p). For a given GTEx sample, we define SNP-specific binomial distributions as Binomial(n = total number of reads, p = sampled reference allelic expression ratio). For each individual GTEx tissue sample, we randomly sample a single instance from each SNP-specific binomial distribution to simulate SNP expression ratios with noise from allelic read sampling. We estimate the XCI ratio using the folded normal model on the simulated SNP expression ratios and repeat the simulation 50 times to generate a distribution of estimated tissue XCI ratios. We compute the root mean squared error of the simulated tissue XCI ratios about the original estimated tissue XCI ratio. We repeat the entire analysis with a percent reduction in each SNP’s total read count (10%, 20%, 30%, etc.) to model variance in our estimated XCI ratios as read depth decreases.
Gene-tissue XCI ratio correlations
To test individual gene’s propensity to follow the aggregate chromosomal XCI ratio, we calculate Pearson correlations between a gene’s reference allelic expression ratio and the estimated XCI ratio leaving out that gene for all samples the gene is detected. We calculate these correlations for each of the 542 filtered genes described above and for 45 previously annotated constitutively escape genes detected in our dataset. We only consider genes detected in at least 30 samples and with an FDR corrected (Benjamini-Hochberg) correlation p-value <= .05 determined by a permutation test (n = 10000) for further investigation of escape status, resulting in 380 putative inactive genes and the 45 previously annotated escape genes.
Testing for escape from XCI
To detect escape genes, it is necessary to compare against genes that undergo complete inactivation and do not escape. After stratifying by mean expression, we reason the genes most likely to undergo complete inactivation are genes with high gene-tissue XCI ratio correlations within each expression bin (Fig. 3C). Accordingly, we take the top 50% of putative inactive genes within each bin to define the null distribution of allelic expression under the hypothesis of complete inactivation (191 genes). The remaining 189 putative inactive genes and the 45 known escape genes comprise our test set. We reason a gene that escapes XCI will be biased for balanced biallelic expression regardless of the XCI ratio of the tissue. Using only tissues with an estimated XCI ratio >= 0.70, we compute the deviation from 0.5 (balanced allelic expression) for all inactive genes and the test gene. We rank the gene deviations and calculate the empirical p-value as the rank of the test gene divided by the total number of ranks i.e. the number of null inactive genes + 1 (Fig. S2). We only consider empirical p-values derived from samples with at least 20 null inactive genes detected. Additionally, we only consider test genes with at least 50 empirical p-values. For each remaining test gene, we aggregate the distribution of empirical p-values using Fisher’s method and apply an FDR correction (Benjamini-Hochberg) to the resulting meta-analytic p-values. We use a threshold of meta-analytic p-value < .001 to call significance for escape. For Fisher’s method, under the null hypothesis, the log sum of all p-values follows a chi-squared distribution with 2k degrees of freedom, where k is the number of independent tests being combined. We use R’s pchisq function to compute the meta-analytic p-value for the following test statistic:
Tissue XCI ratio predicting donor XCI ratio
For the donors that contribute to a given tissue, we calculate the mean XCI ratio across all other tissues for each donor and use that mean as an approximation for the true XCI ratio for each donor. We classify donors as low/high XCI ratio donors if they have a mean XCI ratio greater than or equal to various thresholds (0.65, 0.7, 0.75). We calculate the AUROC of a given tissue’s XCI ratio predicting the low/high donors via the Mann-Whitney U test statistic where
Cross-tissue XCI ratio correlations
For all pairwise combinations of the 49 tissues present within the GTEx dataset, we take the subset of donors that contribute both tissues for a given comparison and calculate the Pearson correlation for the folded XCI ratio of the tissues. Figure 4c1-c2 depicts only the correlation values derived with a sample size of at least 20 donors and an FDR corrected (Benjamini-Hochberg) p-value <= .05 derived from a permutation test (n = 10000). Supplemental Figure 3 depicts all computed correlations regardless of sample size or p-value.
CIBERSORTx deconvolution and germ layer-specific marker identification
CIBERSORTx (https://cibersortx.stanford.edu, (Newman et al., 2019)) was run using the recommended settings following the “Build a signature matrix file from single-cell RNA sequencing data” and “Impute cell fractions” tutorials, batch correction was enabled when imputing cell fractions. Briefly, the annotated single-cell RNA sequencing data from GTEx is used to build a signature matrix that identifies genes that define the annotated cell types. This signature matrix is used to impute the cell type composition of bulk RNA sequencing samples. We extract germ layer-specific marker genes from the signature matrices identified from CIBERSORTx, classifying a gene as a germ layer marker if it is a gene that identifies cell types exclusively from a single germ layer. Our annotated germ layer markers, the cell types they define, and the tissue they are derived from are available in Supplementary Table 2. The signature matrices and imputed cell types per tissue with associated statistics from CIBERSORTx are made available on the FTP site http://labshare.cshl.edu/shares/gillislab/people/werner/werner_et_al_Dev_Cell_2022/data.
Inference on direction of XCI ratios
To infer the direction of XCI ratios from unphased data, we look at allelic expression of heterozygous SNPs captured in multiple tissues for an individual donor. The reference allele of a heterozygous SNP captured in two different tissues of a single donor represents the same parental X-allele in both tissues. If the direction of XCI is the same for both tissues, the heterozygous SNP is expected to exhibit the same degree of reference allelic expression across the two tissues (positive correlation). If the direction of XCI is different, reference allelic expression will be inverted for one of the tissues resulting in a negative correlation. For each donor, for all pairwise combinations of their donated tissues with XCI ratios >= 0.6, we calculate Pearson correlations for unfolded reference allelic expression ratios using only SNPs detected in both tissues (Fig. 5). We only use SNPs that are within the previously filtered 542 genes described above and only consider correlations derived from tissue comparisons with at least 30 shared SNPs. Using positive or negative correlations as a readout for switched XCI direction between tissues, we perform Fisher’s exact test with a Benjamini-Hochberg correction to identify any tissue significantly enriched for switching XCI directions. We use the hypergeometric distribution to calculate raw p-values for Fisher’s Exact Test. For a given tissue, we input the number of times that tissue switched XCI directions minus 1, the total number of switched XCI cases across all tissues, the total number of non-switched XCI cases across all tissues, and the sample size for the given tissue.
Evaluating XCI cell number estimates
XCI is a binomial sampling event defined by the number of cells present during inactivation and the equal probability of inactivation between the alleles Binomial(N = # of cells, p = 0.5). As such, the variance in XCI ratios within a population is directly linked to the number of cells present during XCI. We derive estimates for the number of cells present during XCI by fitting a normal model to tissue-specific XCI skew distributions as a smoothened estimate for the underlying binomial distribution. We take the theoretical variance from the binomial model as the variance for the normal approximation.
, where p,q = probability of allelic inactivation.
For a range of cell numbers (N = 2:50), we select the normal model with minimum error between its CDF and the empirical XCI ratio CDF of a given tissue for the tails of the distribution (XCI ratio <= 0.4 and XCI ratio >= 0.6). This accounts for the uncertain folded 0.5 – 0.6 XCI ratios estimates in the unfolded space. We calculate 95% CIs for each estimated cell number via a nonparametric bootstrap percentile approach (n = 2000). We only consider cell number estimates from tissues with at least 10 donors.
Evaluating tissue-specific lineage cell number estimates
We model tissue-specific lineage specification as a cell sampling event from a large pool of cells. As such, the XCI ratio of a tissue will follow a binomial model defined by the number of cells fated for that tissue and the XCI ratio of the embryo (Fig. 6c).
For a given tissue, across donors with variable XCI ratios (XCIembryo) the variation in the tissue XCI ratio is defined by the constant Ntissue, the number of cells fated for that tissue. To estimate this constant, we calculate z-scores for each tissue-donor pair of a given tissue using the mean XCI ratio of all other tissues for each donor as an approximation for the XCIembryo.
As the standard deviation of a distribution of z-scores is 1, we solve for Ntissue:
, where m = number of donors for a given tissue
We calculate 95% CIs for each Ntissue via a nonparametric bootstrap percentile approach (n = 2000) using the ttissue distribution. We require a tissue to have at least 10 donors in order to calculate Ntissue.
Data analysis and visualization
All analysis was conducted in R version 4.0.5 (R Core Team, 2021). Graphs were generated using the ggplot2 (Wickham, 2016), ComplexHeatmap (Gu et al., 2016), karyoploteR (Gel and Serra, 2017), and base R packages.
QUANTIFICATION AND STATISTICAL ANALYSIS
When correcting p-values, we use the Benjamini-Hochberg procedure implemented by R's p.adjust function with “method = BH” parameter. Significance is determined with p-value <= 0.05 unless otherwise stated. We use the R dip.test function from the diptest package to perform Hartigan’s dip test of unimodality. For Fisher’s method of aggregating p-values, we use the R function pchisq with ‘lower.tail = FALSE’ parameter to compute the meta-analytic p-value from the calculated chi-square test statistic. All confidence intervals are computed using a nonparametric bootstrap percentile approach, where the underlying data is sampled with replacement to generate a bootstrapped distribution of the variable in question (tissue XCI ratio estimates, cell number estimates). The 95% confidence interval is defined by the 2.5th and 97.5th percentile of the bootstrapped distribution. We determine if tissues are enriched for switching parental XCI directions using the hypergeometric implementation of Fisher’s Exact Test, using R’s phyper function. When fitting normal distributions to tissue XCI ratio distributions, we use the R quantile function with parameter “type = 1” to compute the empirical CDF and the R qnorm function to compute the theoretical normal CDF. For any given correlation calculated, we permute the underlying data to get a null distribution of correlations under the hypothesis of independence, using R’s cor function with “method = pearson” parameter. We derive a raw p-value for the original correlation value from the empirical null distribution of correlations (permutation test). In the analyses where we generate many correlations, we apply a Benjamini-Hochberg FDR correction to the associated distribution of raw p-values to call significance, using a threshold of p-value <= 0.05.
Supplementary Material
Table S1. Escape annotations. Related to Figure 3.
Assigned labels of inactive, known escape, confident inactive, or novel escape for the 189 genes powered enough to investigate cross-tissue XCI escape.
Table S2. Germ layer-specific marker genes. Related to Figure 4.
Extracted germ layer-specific markers from the CIBERSORT deconvolution. Germ layer-specific markers are defined as genes that are identified through CIBERSORT as gene signatures exclusively for cell types of a single germ layer.
Highlights:
Tissue XCI ratios can be determined using reference-aligned bulk RNA-seq data
XCI ratios are shared across all human tissues
XCI variance in adult populations is explained by the inherent stochasticity of XCI
Human XCI occurs when the embryonic epiblast is composed of at least 6-16 cells
Acknowledgements
JMW was supported by NSF Award No. DGE-1938105. JG, SB and JH were supported by NIH Grants R01MH113005 and R01LM012736.
This material is based upon work supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. DGE-1938105. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
We thank all members of the Gillis lab and particularly Manthan Shah for assisting in some of the initial downloading.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests
The authors declare no competing interests.
References
- Amos-Landgraf JM, Cottle A, Plenge RM, Friez M, Schwartz CE, Longshore J, Willard HF, 2006. X Chromosome–Inactivation Patterns of 1,005 Phenotypically Unaffected Females. Am J Hum Genet 79, 493–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaton BP, Fornes O, Wasserman WW, Brown CJ, 2021. Cross-species examination of X-chromosome inactivation highlights domains of escape from silencing. Epigenetics Chromatin 14, 12. 10.1186/s13072-021-00386-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berletch JB, Ma W, Yang F, Shendure J, Noble WS, Disteche CM, Deng X, 2015. Escape from X Inactivation Varies in Mouse Tissues. PLoS Genet 11, e1005079. 10.1371/journal.pgen.1005079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittel DC, Theodoro MF, Kibiryeva N, Fischer W, Talebizadeh Z, Butler MG, 2008. Comparison of X-chromosome inactivation patterns in multiple tissues from human females. J Med Genet 45, 309–313. 10.1136/jmg.2007.055244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudewijns M, van Dongen JJM, Langerak AW, 2007. The Human Androgen Receptor X-Chromosome Inactivation Assay for Clonality Diagnostics of Natural Killer Cell Proliferations. J Mol Diagn 9, 337–344. 10.2353/jmoldx.2007.060155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown C, Robinson W, 2000. The causes and consequences of random and non-random X chromosome inactivation in humans: X chromosome inactivation in humans. Clinical Genetics 58, 353–363. 10.1034/j.1399-0004.2000.580504.x [DOI] [PubMed] [Google Scholar]
- Carrel L, Willard HF, 2005. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 434, 400–404. 10.1038/nature03479 [DOI] [PubMed] [Google Scholar]
- Clerc P, Avner P, 2006. Random X-chromosome inactivation: skewing lessons for mice and men. Current Opinion in Genetics & Development, Genetics of disease 16, 246–253. 10.1016/j.gde.2006.04.001 [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dossin F, Heard E, 2021. The Molecular and Nuclear Dynamics of X-Chromosome Inactivation. Cold Spring Harb Perspect Biol a040196. 10.1101/cshperspect.a040196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eraslan G, Drokhlyansky E, Anand S, Fiskin E, Subramanian A, Slyper M, Wang J, Van Wittenberghe N, Rouhana JM, Waldman J, Ashenberg O, Lek M, Dionne D, Win TS, Cuoco MS, Kuksenko O, Tsankov AM, Branton PA, Marshall JL, Greka A, Getz G, Segrè AV, Aguet F, Rozenblatt-Rosen O, Ardlie KG, Regev A, 2022. Single-nucleus cross-tissue molecular reference maps toward understanding disease gene function. Science 376, eab14290. 10.1126/science.abl4290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fialkow PJ, 1973. Primordial cell pool size and lineage relationships of five human cell types*. Annals of Human Genetics 37, 39–48. 10.1111/j.1469-1809.1973.tb01813.x [DOI] [PubMed] [Google Scholar]
- Frankish A, Diekhans M, Jungreis I, Lagarde J, Loveland JE, Mudge JM, Sisu C, Wright JC, Armstrong J, Barnes I, Berry A, Bignell A, Boix C, Carbonell Sala S, Cunningham F, Di Domenico T, Donaldson S, Fiddes IT, García Girón C, Gonzalez JM, Grego T, Hardy M, Hourlier T, Howe KL, Hunt T, Izuogu OG, Johnson R, Martin FJ, Martínez L, Mohanan S, Muir P, Navarro FCP, Parker A, Pei B, Pozo F, Riera FC, Ruffier M, Schmitt BM, Stapleton E, Suner M-M, Sycheva I, Uszczynska-Ratajczak B, Wolf MY, Xu J, Yang YT, Yates A, Zerbino D, Zhang Y, Choudhary JS, Gerstein M, Guigó R, Hubbard TJP, Kellis M, Paten B, Tress ML, Flicek P, 2021. GENCODE 2021. Nucleic Acids Research 49, D916–D923. 10.1093/nar/gkaa1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gart JJ, 1970. A Locally Most Powerful Test for the Symmetric Folded Binomial Distribution. Biometrics 26, 129–138. 10.2307/2529049 [DOI] [PubMed] [Google Scholar]
- Geens M, Chuva De Sousa Lopes SM, 2017. X chromosome inactivation in human pluripotent stem cells as a model for human development: back to the drawing board? Hum Reprod Update 23, 520–532. 10.1093/humupd/dmx015 [DOI] [PubMed] [Google Scholar]
- Gel B, Serra E, 2017. karyoploteR : an R / Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics 33, 3088–3090. 10.1093/bioinformatics/btx346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghimire S, Mantziou V, Moris N, Martinez Arias A, 2021. Human gastrulation: The embryo and its models. Developmental Biology, Synthetic Embryology 474, 100–108. 10.1016/j.ydbio.2021.01.006 [DOI] [PubMed] [Google Scholar]
- Gu Z, Eils R, Schlesner M, 2016. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. [DOI] [PubMed] [Google Scholar]
- Hagen SH, Henseling F, Hennesen J, Savel H, Delahaye S, Richert L, Ziegler SM, Altfeld M, 2020. Heterogeneous Escape from X Chromosome Inactivation Results in Sex Differences in Type I IFN Responses at the Single Human pDC Level. Cell Rep 33, 108485. 10.1016/j.celrep.2020.108485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoon B. de, Monkhorst K, Riegman P, Laven JSE, Gribnau J, 2015. Buccal swab as a reliable predictor for X inactivation ratio in inaccessible tissues. Journal of Medical Genetics 52, 784–790. 10.1136/jmedgenet-2015-103194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson AJM, Coucoravas C, Sandberg R, Reinius B, 2019. X-chromosome upregulation is driven by increased burst frequency. Nat Struct Mol Biol 26, 963–969. 10.1038/s41594-019-0306-y [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup, 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder D, Gartler SM, 1965. Glucose-6-Phosphate Dehydrogenase Mosaicism: Utilization as a Cell Marker in the Study of Leiomyomas. Science 150, 67–69. 10.1126/science.150.3692.67 [DOI] [PubMed] [Google Scholar]
- Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, Hasz R, Walters G, Garcia F, Young N, Foster B, Moser M, Karasik E, Gillard B, Ramsey K, Sullivan S, Bridge J, Magazine H, Syron J, Fleming J, Siminoff L, Traino H, Mosavel M, Barker L, Jewell S, Rohrer D, Maxim D, Filkins D, Harbach P, Cortadillo E, Berghuis B, Turner L, Hudson E, Feenstra K, Sobin L, Robb J, Branton P, Korzeniewski G, Shive C, Tabor D, Qi L, Groch K, Nampally S, Buia S, Zimmerman A, Smith A, Burges R, Robinson K, Valentino K, Bradbury D, Cosentino M, Diaz-Mayoral N, Kennedy M, Engel T, Williams P, Erickson K, Ardlie K, Winckler W, Getz G, DeLuca D, MacArthur D, Kellis M, Thomson A, Young T, Gelfand E, Donovan M, Meng Y, Grant G, Mash D, Marcus Y, Basile M, Liu J, Zhu J, Tu Z, Cox NJ, Nicolae DL, Gamazon ER, Im HK, Konkashbaev A, Pritchard J, Stevens M, Flutre T, Wen X, Dermitzakis ET, Lappalainen T, Guigo R, Monlong J, Sammeth M, Koller D, Battle A, Mostafavi S, McCarthy M, Rivas M, Maller J, Rusyn I, Nobel A, Wright F, Shabalin A, Feolo M, Sharopova N, Sturcke A, Paschal J, Anderson JM, Wilder EL, Derr LK, Green ED, Struewing JP, Temple G, Volpi S, Boyer JT, Thomson EJ, Guyer MS, Ng C, Abdallah A, Colantuoni D, Insel TR, Koester SE, Little AR, Bender PK, Lehner T, Yao Y, Compton CC, Vaught JB, Sawyer S, Lockhart NC, Demchok J, Moore HF, 2013. The Genotype-Tissue Expression (GTEx) project. Nat Genet 45, 580–585. 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon MF, 1972. X-CHROMOSOME INACTIVATION AND DEVELOPMENTAL PATTERNS IN MAMMALS. Biological Reviews 47, 1–35. 10.1111/j.1469-185X.1972.tb00969.x [DOI] [PubMed] [Google Scholar]
- Lyon MF, 1961. Gene Action in the X -chromosome of the Mouse ( Mus musculus L.). Nature 190, 372–373. 10.1038/190372a0 [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA, 2010. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mclaren A, 1972. Biological Sciences: Numerology of Development. Nature 239, 274–276. 10.1038/239274a0 [DOI] [PubMed] [Google Scholar]
- Migeon B, 2013. Females Are Mosaics: X Inactivation and Sex Differences in Disease, Females Are Mosaics. Oxford University Press. [DOI] [PubMed] [Google Scholar]
- Migeon BR, 1998. Non-random X chromosome inactivation in mammalian cells. Cytogenet Cell Genet 80, 142–148. 10.1159/000014971 [DOI] [PubMed] [Google Scholar]
- Monteiro J, Derom C, Vlietinck R, Kohn N, Lesser M, Gregersen PK, 1998. Commitment to X Inactivation Precedes the Twinning Event in Monochorionic MZ Twins. The American Journal of Human Genetics 63, 339–346. 10.1086/301978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira de Mello JC, Fernandes GR, Vibranovski MD, Pereira LV, 2017. Early X chromosome inactivation during human preimplantation development revealed by single-cell RNA-sequencing. Sci Rep 7, 10794. 10.1038/s41598-017-11044-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumova AK, Plenge RM, Bird LM, Leppert M, Morgan K, Willard HF, Sapienza C, 1996. Heritability of X chromosome--inactivation phenotype in a large family. Am J Hum Genet 58, 1111–1119. [PMC free article] [PubMed] [Google Scholar]
- Nesbitt MN, 1971. X chromosome inactivation mosaicism in the mouse. Developmental Biology 26, 252–263. 10.1016/0012-1606(71)90125-4 [DOI] [PubMed] [Google Scholar]
- Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, Khodadoust MS, Esfahani MS, Luca BA, Steiner D, Diehn M, Alizadeh AA, 2019. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 37, 773–782. 10.1038/s41587-019-0114-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeters SB, Yang C, Brown CJ, 2016. Have humans lost control: The elusive X-controlling element. Seminars in Cell & Developmental Biology, X chromosome inactivation 56, 71–77. 10.1016/j.semcdb.2016.01.044 [DOI] [PubMed] [Google Scholar]
- Petropoulos S, Edsgärd D, Reinius B, Deng Q, Panula SP, Codeluppi S, Plaza Reyes A, Linnarsson S, Sandberg R, Lanner F, 2016. Single-Cell RNA-Seq Reveals Lineage and X Chromosome Dynamics in Human Preimplantation Embryos. Cell 165, 1012–1026. 10.1016/j.cell.2016.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM, 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team, 2021. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- Rozowsky J, Drenkow J, Yang YT, Gursoy G, Galeev T, Borsari B, Epstein CB, Xiong K, Xu J, Gao J, Yu K, Berthel A, Chen Z, Navarro F, Liu J, Sun MS, Wright J, Chang J, Cameron CJ, Shoresh N, Gaskell E, Adrian J, Aganezov S, Balderrama-Gutierrez G, Banskota S, Corona GB, Chee S, Chhetri SB, Martins GCC, Danyko C, Davis CA, Farid D, Farrell NP, Gabdank I, Gofin Y, Gorkin DU, Gu M, Hecht V, Hitz BC, Issner R, Kirsche M, Kong X, Lam BR, Li S, Li B, Li T, Li X, Lin KZ, Luo R, Mackiewicz M, Moore JE, Mudge J, Nelson N, Nusbaum C, Popov I, Pratt HE, Qiu Y, Ramakrishnan S, Raymond J, Salichos L, Scavelli A, Schreiber JM, Sedlazeck FJ, See LH, Sherman RM, Shi X, Shi M, Sloan CA, Strattan JS, Tan Z, Tanaka FY, Vlasova A, Wang J, Werner J, Williams B, Xu M, Yan C, Yu L, Zaleski C, Zhang J, Cherry JM, Mendenhall EM, Noble WS, Weng Z, Levine ME, Dobin A, Wold B, Mortazavi A, Ren B, Gillis J, Myers RM, Snyder MP, Choudhary J, Milosavljevic A, Schatz MC, Guigó R, Bernstein BE, Gingeras TR, Gerstein M, 2021. Multi-tissue integrative analysis of personal epigenomes. bioRxiv 2021.04.26.441442. 10.1101/2021.04.26.441442 [DOI] [Google Scholar]
- Schmidt M, Sart DD, 1992. Functional disomies of the X chromosome influence the cell selection and hence the X inactivation pattern in females with balanced X-autosome translocations: A review of 122 cases. American Journal of Medical Genetics 42, 161–169. 10.1002/ajmg.1320420205 [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward M-H, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K, 2001. dbSNP: the NCBI database of genetic variation. Nucleic Acids Research 29, 308–311. 10.1093/nar/29.1.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shvetsova E, Sofronova A, Monajemi R, Gagalova K, Draisma HHM, White SJ, Santen GWE, Chuva de Sousa Lopes SM, Heijmans BT, van Meurs J, Jansen R, Franke L, KieŁbasa SM, den Dunnen JT, ‘t Hoen PAC, 2019. Skewed X-inactivation is common in the general female population. Eur J Hum Genet 27, 455–465. 10.1038/s41431-018-0291-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun KY, Oreper D, Schoenrock SA, McMullan R, Giusti-Rodríguez P, Zhabotynsky V, Miller DR, Tarantino LM, Pardo-Manuel de Villena F, Valdar W, 2021. Bayesian modeling of skewed X inactivation in genetically diverse mice identifies a novel Xce allele associated with copy number changes. Genetics 218, iyab034. 10.1093/genetics/iyab034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukiainen T, Villani A-C, Yen A, Rivas MA, Marshall JL, Satija R, Aguirre M, Gauthier L, Fleharty M, Kirby A, Cummings BB, Castel SE, Karczewski KJ, Aguet F, Byrnes A, Lappalainen T, Regev Aviv, Ardlie KG, Hacohen N, MacArthur DG, 2017. Landscape of X chromosome inactivation across human tissues. Nature 550, 244–248. 10.1038/nature24265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbakh V.Yu., 1967. Statistical Testing of Differences in Causal Behaviour of Two Morphologically Indistinguishable Objects. Biometrics 23, 137–143. 10.2307/2528286 [DOI] [PubMed] [Google Scholar]
- Vallot C, Patrat C, Collier AJ, Huret C, Casanova M, Liyakat Ali TM, Tosolini M, Frydman N, Heard E, Rugg-Gunn PJ, Rougeulle C, 2017. XACT Noncoding RNA Competes with XIST in the Control of X Chromosome Activity during Human Early Development. Cell Stem Cell 20, 102–111. 10.1016/j.stem.2016.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg IM, Laven JSE, Stevens M, Jonkers I, Galjaard R-J, Gribnau J, Hikke van Doorninck J, 2009. X Chromosome Inactivation Is Initiated in Human Preimplantation Embryos. Am J Hum Genet 84, 771–779. 10.1016/j.ajhg.2009.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H, 2016. ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag; New York. [Google Scholar]
- Winham SJ, Larson NB, Armasu SM, Fogarty ZC, Larson MC, McCauley BM, Wang C, Lawrenson K, Gayther S, Cunningham JM, Fridley BL, Goode EL, 2019. Molecular signatures of X chromosome inactivation and associations with clinical outcomes in epithelial ovarian cancer. Hum Mol Genet 28, 1331–1342. 10.1093/hmg/ddy444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Luo J, Yu H, Rattner A, Mo A, Wang Y, Smallwood PM, Erlanger B, Wheelan SJ, Nathans J, 2014. Cellular resolution maps of X-chromosome inactivation: implications for neural development, function, and disease. Neuron 81, 103–119. 10.1016/j.neuron.2013.10.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Castillo-Morales A, Jiang M, Zhu Y, Hu L, Urrutia AO, Kong X, Hurst LD, 2013. Genes That Escape X-Inactivation in Humans Have High Intraspecific Variability in Expression, Are Associated with Mental Impairment but Are Not Slow Evolving. Mol Biol Evol 30, 2588–2601. 10.1093/molbev/mst148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zito A, Roberts AL, Visconti A, Rossi N, Andres-Ejarque R, Nardone S, Moustafa JES, Falchi M, Small KS, 2021. Escape from X-inactivation in twins exhibits intra- and inter-individual variability across tissues and is heritable. 10.1101/2021.10.15.463586 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Escape annotations. Related to Figure 3.
Assigned labels of inactive, known escape, confident inactive, or novel escape for the 189 genes powered enough to investigate cross-tissue XCI escape.
Table S2. Germ layer-specific marker genes. Related to Figure 4.
Extracted germ layer-specific markers from the CIBERSORT deconvolution. Germ layer-specific markers are defined as genes that are identified through CIBERSORT as gene signatures exclusively for cell types of a single germ layer.
Data Availability Statement
This paper analyzes existing, publicly available data. Links to access these datasets are listed in the key resources table. The generated allele-specific expression information per sample (variant information removed) and the CIBERSORTx deconvolution results are made available at the FTP site: http://labshare.cshl.edu/shares/gillislab/people/werner/werner_et_al_Dev_Cell_2022/data. Descriptions of the data are available at github.com/JonathanMWerner/human_cross_tissue_XCI
All original code has been deposited at figshare (DOI: 10.6084/m9.figshare.20216816) and at Github (github.com/JonathanMWerner/human_cross_tissue_XCI) and is publicly available as of the date of publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table