

Summary
Unlike eukaryotes and archaea, which have multiple replication origins on their chromosomes, bacterial chromosomes usually contain a single replication origin 1. Here, we discovered a dicentric bacterial chromosome with two replication origins, which has resulted from the fusion of the circular and linear chromosomes in Agrobacterium tumefaciens. The fused chromosome is well tolerated, stably maintained, and retains similar subcellular organization and genome-wide DNA interactions found for the bipartite chromosomes. Strikingly, the two replication origins and their partitioning systems are both functional and necessary for cell survival. Finally, we discovered that the site-specific recombinases XerC and XerD 2 are essential in cells harboring the fused chromosome but not in cells with bipartite chromosomes. Analysis of actively dividing cells suggests a model in which XerC/D are required to recombine the sister fusion chromosomes when the two centromeres on the same chromosome are segregated to opposite cell poles. Thus, faithful segregation of dicentric chromosomes in bacteria can occur because of site-specific recombination between the sister chromatids during chromosome partitioning. Our study provides a natural comparative platform to examine a bacterial chromosome with multiple origins and a possible explanation for the fundamental difference in bacterial genome architecture relative to eukaryotes and archaea 1.
Keywords: chromosome fusion, chromosome segregation, multipartite genome, Agrobacterium tumefaciens, C58, XerC, XerD
In Brief:
Bacterial chromosomes usually contain a single replication origin. Liao et al. identify a dicentric, linear bacterial chromosome that contains two active replication origins and their independent partitioning systems. This study reveals that XerC/D site-specific recombinases are required to enable this dramatic shift in genomic architecture.
Grapphical Abstract
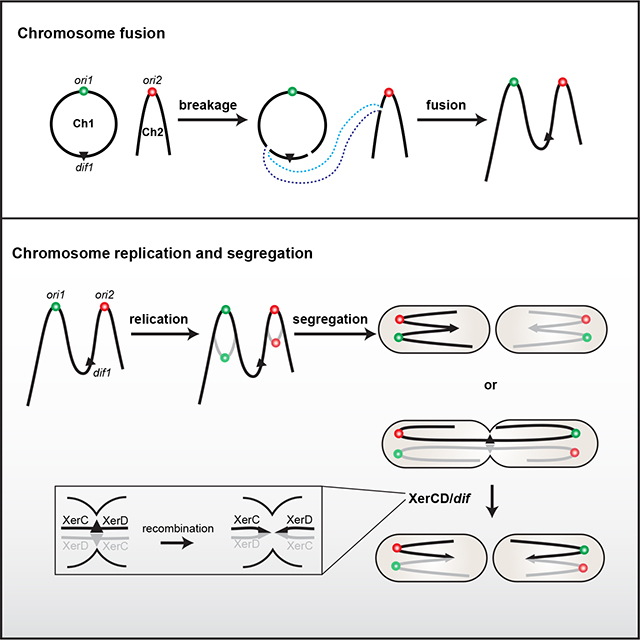
Results and Discussion
A. tumefaciens C58 contains a circular chromosome (Ch1), a linear chromosome (Ch2) and two large plasmids (pAtC58 and pTiC58), and has been used as a model to study multipartite genome organization 3–7. Using genome-wide chromosome conformation capture (Hi-C) assays and fluorescence microscopy, recent studies showed that in the C58 wild-type strain, the origins of all the four replicons are clustered at the cell pole, and each chromosome has the two replication arms juxtaposed from origin to terminus 6,7 (Figure 1A). However, when we performed Hi-C on a different laboratory lineage of C58, the Hi-C interaction map showed discontinuities (Figure 1B), indicating a genome rearrangement in this particular genetic isolate.
Figure 1. A commonly used wild-type strain of A. tumefaciens has the two chromosomes fused into one large linear chromosome.

(A-C) Top: Schematic diagrams of the replicons in A. tumefaciens C58 original strain (A) and the fusion strain (B and C). The original strain contains a circular chromosome (Ch1), a linear chromosome (Ch2) and two plasmids (pAt and pTi). The fusion strain harbors a linear chromosome resulting from the fusion of Ch1 and Ch2, pAt with a deletion, and pTi. Middle: normalized Hi-C contact maps of exponentially growing A. tumefaciens C58 original strain (A) 6 and fusion strain (B and C). Hi-C map displays contact frequencies for pairs of 10-kb bins across the genome. In A and B, Hi-C data were mapped to the original C58 reference genome, in which ori1 was placed at the center of Ch1. In C, Hi-C data from B were mapped to the rearranged reference genome in which deletions were removed. x-axis and y-axis indicate genomic coordinates. The ends of each replicon are indicated by dark red dotted lines. Bottom: Marker frequency analysis (MFA) of exponentially growing original strain (A) 6 and fusion strain (B and C). y-axis indicates relative copy number normalized to the copy number of ter1 (A) or ter2L (B and C).
(D) Schematic diagrams depicting regions on Ch1 (green) and Ch2 (red) in the original strain. Single nucleotide polymorphisms are shown as green or red bars. F1/R1 and F2/R2 indicate primer pairs to amplify these regions.
(E) Schematic diagrams depicting fusion junctions on the fused chromosome. Primer pairs of F2/R1 and F1/R2 were used to amplify junction1 (top) and juction2 (bottom), respectively.
(F) Gel electrophoresis analysis of fusion junctions of multiple “wild-type” A. tumefaciens C58 strains from different sources.
See also Figure S1.
The sequence information garnered in Hi-C prompted us to literally dissect the map and rearrange the pieces to obtain a confluent pattern (Figure S1A). Indeed, we achieved a map with a connected primary diagonal and a continuous secondary diagonal, revealing a putative 4.9 Mb linear chromosome containing both Ch1 and Ch2. We analyzed the Hi-C map and identified the breakpoints to be ~1790 kb on Ch1 and ~710 kb on Ch2 on NCBI Reference Sequence GCA_000092025.1 (Figure 1B Top panel). We hypothesized that Ch1 and Ch2 were recombined at these two breakpoints, generating a fused linear chromosome. For simplicity, we refer to this strain as the “fusion strain” as opposed to the wild-type “original strain” with bipartite chromosomes. We note that the fusion strain also contains a previously described 194-kb deletion from the plasmid pAt (from 206.308 kb to 400.732 kb) 8 and a ~10-kb deletion on Ch1 (from 1175.9 kb to 1186.2 kb) which appears to contain cryptic phage genes with no obvious relevance to chromosome topology.
To test the hypothesis of a fused chromosome, we used three approaches. First, we rearranged the reference sequence and remapped our Hi-C reads. This regenerated the map seen in the “solved puzzle” (Figure 1C, S1A). Secondly, we performed marker frequency analysis (MFA) of the fusion strain by whole genome sequencing. When mapped to the original reference genome, the fusion strain showed a disconnected profile with interruptions in slopes (Figure 1B bottom). When mapped to the rearranged reference genome, MFA plot showed a continuous and smooth curve (Figure 1C bottom). Finally, we checked the continuity of the DNA molecules using PCR. We designed two pairs of PCR primers flanking the breakpoints (Figure 1D). When amplified from the original strain, 3.1 kb and 3.2 kb PCR products were obtained using F1+R1 and F2+R2, respectively, but no product using F1+R2 or F2+R1. However, the fusion strain gave a reverse pattern (Figure 1F). Altogether, our results show that the fusion strain contains a single large linear chromosome with two replication origins.
To narrow down the fusion site, we compared DNA sequences at the two breakpoints and found two highly similar 1.5 kb regions at 1793 kb on Ch1 (Atu1813-Atu1814) and 713 kb on Ch2 (Atu3663-ephA) (Figure 1D) that only differ by 11 single nucleotide polymorphisms. The high degree of similarity perhaps explains why this rearrangement was not detected in previous whole-genome-sequencing experiments 8. Comparison of the Sanger sequencing results of the PCR products indicated a strand exchange between Atu1814 and ephA within the first ~300 bp (Figure 1EF). It is likely that a double-stranded break happened at ephA, and the highly similar sequence on Atu1814 provided the repair template for homologous recombination; this single crossover event may have led to the fusion of the two chromosomes.
To test whether the fusion strain is commonly used in the Agrobacterium research community, we analyzed over 20 of C58 WT strains or direct derivatives from 16 labs using PCR and Hi-C. To our surprise, WT stocks from 7 labs contain the fused chromosome 5,8–11 (Figure 1F, Table S1). It is likely that this fusion strain is even more widely dispersed than revealed through this limited analysis. By tracing the historical distribution of these strains, we suspect that this fusion event could have happened while the strain was propagated by the laboratory of Stephen K. Farrand. Then the fusion strain was distributed to other labs.
Despite that the two chromosomes were fused into a single molecule, the replication “arms” on either side of each origin interacted along their length and the origins of all the replicons remained clustered (Figure 1AC). Fluorescence images and detailed analysis showed that the localization of the two origins was very similar between the original and the fusion strains (Figure 2A). In shaken liquid cultures, these two strains had near identical doubling times (~204 min in defined ATGN medium and ~90 min in nutrient rich LB medium) and very similar growth curves, albeit the fusion strain showed a marginal but reproducible growth advantage (Figure 2B), which is consistent with growth competition assays published previously 8. Thus, the fused chromosome was well tolerated by the cell, behaved similarly to the bipartite chromosomes in terms of localization and genome-wide organization, and did not pose an obvious growth difference, although subtle growth differences dependent on environmental conditions are possible. We note that fusion strain is fully virulent 12,13, even though the level of virulence was not directly compared with the original strain.
Figure 2. The fusion and original strains exhibit similar characteristics for growth, chromosome localization, replication, and partition.

(A) Localization of ori1 and ori2 in the original strain (AtWX263) and the fusion strain (AtWX366). The replication origins were visualized by expressing GFP-ParB1 (green, ori1) and RFP-RepBCh2 (red, ori2) from a pSRKKm-based plasmid. Top panel, fluorescence images. Scale bar represents 2 μm. The doubling time of these cells growing in ATGN medium were indicated. Bottom panel: plots showing relative position of foci. Black lines indicate positions of two cell poles.
(B) Growth curves of the WT original and WT fusion strains growing in LB broth (left panel) or ATGN medium (right panel) in a microplate at 30°C on a plate reader with shaking. Optical density at 600 nm was measured at an interval of 30 minutes. The error bars indicate the standard deviation of six biological replicates combined from two independent experiments.
(C) 10-fold serial dilutions of indicated strains were spotted on LB plates. ParB1 depletion strains (AtWX192 and AtWX194) have the endogenous parB1 gene deleted and an ectopic copy of parB1 expressed from PtraI-riboswitch at the tetRA locus 6. 1 μM AHL and 2 mM theophylline were added into ParB1+ plates. Although the cells were extremely sick, repBCh2 was successfully deleted from the original strain (ΔrepBCh2, AtWX089) but not from the fusion strain. To deplete RepBCh2 from the fusion strain (AtWX025), the endogenous repBch2 was deleted and an ectopic copy of repBch2 was expressed from Plac at the T7 attachment site. 0.5 mM IPTG was added into RepBCh2 + plates.
(D) Tn-seq plots showing transposon intolerance in the two replication origins and two centromeres in WT original strain (top) and WT fusion strain (bottom). y-axis indicates the number of sequencing reads at each insertion site. x-axis indicates gene locus. Black dotted rectangles highlight regions of interest. This screen was carried out in LB medium. Similar results were observed when cells were grown in minimal ATGN medium (Figure S1B).
See also Figure S1.
Bacterial chromosomes typically contain a single replication origin and one centromere. However, our MFA plots indicated that both origins on the fused chromosome were functional and fired at levels similar to the original strain 6; the pair of replication forks tracking from the same origin progressed at the similar speed as indicated by the slope on the curve (Figure 1A, 1C). We next questioned whether both origins are necessary for cell survival. We performed transposon mutagenesis coupled to sequencing (Tn-seq) to identify all essential genes in the original and fusion strains. Mutant libraries were constructed by introducing a mariner transposon into both strains. Approximately 1×106 mutants derived from each strain were harvested and processed for next-generation sequencing. In both strains, no transposon insertion was detected at the replication origins (ori1 and ori2), the replication initiators (dnaA for ori1 and repC for ori2), or the centromeric or partitioning factors (parAB for ori1 and repAB for ori2), regardless of growth media (Figure 2D, S1B). Consistent with the Tn-seq results, we were unable to delete the genes encoding the centromeric proteins, ParB1 or RepBCh2, from the fused chromosome. Instead, the endogenous deletions were successful only in depletion strains in which we expressed an ectopic copy: Figure 2C shows that in the absence of ParB1 or RepBCh2 the fusion strain was as debilitated as the original strain. These results indicate that replication origins and centromeres of both ori1 and ori2 are required for the viability of the fusion strain. This surprising finding is being investigated further in a separate study.
For the original strain, our Tn-Seq experiments identified all the essential genes reported previously 14. We next investigated whether genes that were not essential in the original strain had become essential in the fusion strain. In Tn-seq, xerC and xerD 2 were the only two genes that had dramatically decreased insertions in the fusion strain compared with the original strain. Similar results were obtained using either LB or ATGN medium (Figure 3A, S2A). To validate the Tn-seq results, we sought to delete xerC and xerD. As expected, we successfully deleted these genes in the original strain (Fig S2E) but not in the fusion strain. Therefore, we generated xerC and xerD depletion strains in which the endogenous promotors of these two genes were disrupted by inserting an IPTG-inducible promoter combined with a theophylline-inducible riboswitch (Plac-riboswitch) 15,16. We found that the fusion strain was not able to form colonies when xerC or xerD was depleted, while the original strains grew similarly with or without inducers (Figure 3B, S2B). Thus, xerC and xerD are required for the survival of the fusion strain but not of the original strain. Given that the fusion strain has a 10-kb deletion on Ch1 and a 194-kb deletion on pAt, we queried whether xerC/D has a synthetic lethal relationship with any of missing genes. We performed Tn-seq on a ΔxerD original strain and found that the genes that were missing in the fusion strain tolerated transposon insertions (Figure S3). Thus, xerC/D’s essentiality in the fusion strain is not due to these missing genes.
Figure 3. XerC and XerD are required for the survival of the fusion strain.

(A) Tn-seq plots showing transposon insertions at xerC/xerD loci and flanking regions in the WT original (top) and WT fusion (bottom) strains. xerC and xerD genes tolerated insertions in the original strain but not in the fusion strain. This screen was performed in LB. Similar results were observed for ATGN medium (Figure S2A).
(B) Validation of essentiality of xerD and xerC using depletion constructs (AtWX323, AtWX327, AtWX331 and AtWX332). 10-fold serial dilutions of indicated strains were spotted on LB plates. The depletion constructs had the promotor of xerD or xerC replaced by Plac-riboswitch. 2 mM theophylline and 0.5 mM IPTG were added as inducers. Similar results were observed for ATGN medium (Figure S2B).
(C) Tn-seq performed in ΔrecA original strain (AtWX398, top) and ΔrecA fusion strain (AtWX387, bottom).
(D) Tn-seq performed in Δsmc original strain (AtWX108, top) and Δsmc fusion strain (AtWX035, bottom).
(E) 10-fold serial dilutions of indicated strains were spotted on LB plates. XerC/D depletion strains contain cumate-inducible plasmids expressing A. tumefaciens Topo IV (AtWX502 and AtWX503) or E. coli Topo IV (AtWX540 and AtWX541). 300 μg/ml of kanamycin were applied on all plates for plasmid maintenance. The following inducers were added: 0.5 mM IPTG (left), none (middle), 2 mM cumate (right).
See also Figures S2 and S3.
The xerC and xerD gene products are broadly conserved site-specific recombinases, which act on a 28-bp dif site at the terminus region of circular bacterial chromosomes to resolve chromosome dimers 2,17–23. This recombination is activated by the very C-terminal (gamma) domain of FtsK, FtsKγ, which is directed by the FtsK-orienting polar sequences (KOPS) to translocate to the dif site 24–27. Although the N-terminal domain of FtsK is essential, it is common that xerC, xerD, and ftsKγ are non-essential 24,25. To understand why xerC and xerD became essential in the fusion strain, we first experimentally determined the binding sites of XerC and XerD in A. tumefaciens. We tagged endogenous copies of XerC and XerD with GFP and performed chromatin immunoprecipitation sequencing (ChIP-seq) using anti-GFP antibodies. ChIP-seq analysis indicated that in both the fusion and original strains, XerC-GFP and XerD-GFP had two enrichment peaks, one on Ch1 and one on pAt (Figure S2C). Using the MEME suite (http://meme-suite.org/) for motif search, we identified dif1 on Ch1 and difpAt on pAt (Figure S2D). To test the essentiality of these dif sites, we successfully deleted both dif sites from the original strain and difpAt from the fusion strain (Figure S2E), but we were not able to delete dif1 from the fusion strain. Consistent with this, our Tn-seq results showed that only dif1 in the fusion strain did not tolerate transposon insertions (Figure S2F). Finally, although ftsK was indicated as essential for both strains in Tn-seq, a closer examination revealed that ftsKγ tolerated transposon insertions in the original strain but not in the fusion strain (Figure S2G). Altogether, these results indicate that XerC/D recombination at dif1 activated by FtsKγ is essential for the survival of the fusion strain. We note that dif1 is still at the convergence of KOPS in the fusion strain, indicating proper function of this system (Figure S2H).
Based on the established functions of XerC/D recombinases in resolving chromosome dimers generated by homologous recombination in RecA+ strains 28, unloading of structural maintenance of chromosome (SMC) complexes from the replication terminus 29, and decatenate the intertwined sister chromosomes in a stepwise manner 30–32, we tested whether the essentiality of XerC/D for the fused chromosome could be suppressed by ΔrecA, Δsmc, or increasing decatenation activity by overexpressing Topoisomerase IV (Topo IV). We performed Tn-seq in ΔrecA or Δsmc in the backgrounds of both original and fusion strains and found that xerC and xerD were still essential in the mutated fusion strains (Figure 3C, 3D). Furthermore, overexpression of A. tumefaciens Topo IV or E. coli Topo IV was unable to rescue the growth defect of XerC/D depletion strains (Figure 3E). Thus, the essentiality of XerC/D is not related to RecA-dependent recombination, the presence of SMC, or insufficient decatenation of the fused chromosome.
Thus, we hypothesized that the essentiality of XerC/D is due to the topology of the fused chromosome: if the segregation of the origins is random, in every cell generation, in 50% of cells, the two origins on the same chromosome will migrate to opposite poles, resulting in segregation problems, breakage of DNA, and potentially cell death unless resolved by the XerC/D/dif1 system (Figure 4A). To directly observe the effect of XerC and XerD depletion on dif1 segregation in the fusion strain, we visualized dif1 locus and tracked the progress of its segregation in time-lapse microscopy. In the WT fusion strain, cells were born with a single dif1 focus localized and remained at the new cell pole as the cell grew. The dif1 focus then moved to the midcell and split into two foci before cell division (Figure 4B). When XerC was depleted, the majority of cells started with a single dif1 focus remaining at the deeply constricted septum. Then the cells did not seem to completely divide; the daughter cells were still connected to each other, forming a cluster of branched cells with randomly distributed dif1 foci (Figure 4C). For comparison, we simultaneously labeled the end of the linear chromosome by inserting an mcherry-parBP1-parSP1 visualization cassette at 39.8 kb from right terminus of the fused chromosome (ter2R). When XerC was depleted, unlike dif1, ter2R was replicated and segregated into two daughter cells normally (Figure S4A). Thus, XerC depletion impaired cell division and dif1 segregation but not ter2R segregation.
Figure 4. XerC and XerD are required to segregate the fused chromosome.

(A) Schematic model. After the initiation of replication, two copies of ori1 and ori2 independently move into opposite daughter cells. In the original strain (top), either combination leads to viable cells. In the fusion strain (bottom), the cells will survive when ori1 and ori2 on the same DNA molecule are distributed in the same cell half. However, when they are positioned in opposite cell halves, cell division is blocked until the sister fusion chromosomes are recombined by XerC/D at dif1.
(B and C) Time-lapse progression of dif1 dynamics during the cell cycle in WT fusion (AtWX455) (B) and XerC-depleted fusion strain (AtWX467) (C) growing on LB agarose pad. dif1 was visualized by inserting the visualization cassette, ygfp-parBpMT1-parSpMT1, at the position ~20 kb away from dif1. For (C), XerC was pre-depleted in liquid LB medium without inducers for 6 hr before the start of time-lapse microscopy. The time interval was 10 minutes. Scale bar depicts 2 μm.
(D) Localization of dif1 in time-course of XerC depletion (AtWX467, top) and XerD depletion (AtWX471, bottom) in the fusion strain. Cells were grown in LB medium with the addition of 0.5 mM IPTG and 2 mM theophylline, collected and resuspended in medium without inducers to start depletion. The quantitative analysis can be found in Figure S4B.
See also Figure S4.
For quantitative measurement, we analyzed a large number of cells in a time course experiment during XerC or XerD depletion (Figure S4B). In the WT fusion strain during exponential growth, 67.7% of cells contained a single dif1 focus at the cell pole or the midcell, 4.8% of cells had two foci before constriction, and 20.5% of cells contained two foci and a constricting septum. In XerC or XerD depletion strain, prior to depletion, the dif1 locus behaved similarly to that in WT fusion strain. However, within the 6 hr depletion of XerC or XerD, we observed a roughly two-fold increase in the number of deeply constricted cells with one or two dif1 foci. After 12 hr of depletion, the percentage of deeply constricted cells remained while the number of cells without foci increased over three fold compared with the 6 hr time point. After 18 hr of depletion, the majority of cells did not contain a dif1 focus and cells exhibited branchy and other abnormalities in cell shape. Our time-lapse and snapshot imaging experiments are consistent with the idea that XerC/D are required to resolve the dif1 region of the fused chromosome. The depletion of XerC/D led to the delayed segregation of dif1 site, blocked cell division, frequent guillotining of DNA at the septum, and cell death.
Here, we discovered a linear dicentric chromosome that has resulted from the fusion of a circular and a linear chromosome in the multipartite genome of A. tumefaciens. We showed that this fusion event did not confer to the cell dramatic growth advantages or disadvantages and the dicentric chromosome exhibited cellular localization and the features of genome-wide organization resembling those observed for the original strain. Moreover, both replication origins on this fused chromosome retained independent replication and segregation programs. We found that this fusion strain is stably maintained and has been commonly used in the research community (Table S1). Although non-engineered chromosome fusions have been reported in several bacterial species 33–38, they frequently lead to slower growth, are unstable, and can be reversed. It is thus surprising that the fusion strain discussed here can cope so effectively with its complex genome structure. Our genetic screen revealed that the fusion strain requires the XerC/D/dif1 system for proper chromosome segregation and cell division, which is likely a consequence of the independent origin segregation.
Roughly 10% of sequenced bacteria, most of which are pathogens or symbionts, have multipartite genomes 39. It has been proposed that the secondary chromosomes have evolved from plasmids acquiring essential genes 40. Multipartite genomes allow more rapid genome duplication which is advantageous for those species. However, compared with a single chromosome, a split genome is more complicated to maintain and requires coordination between replicons during replication and segregation. Why would bacteria not evolve multiple replication origins on a single chromosome to confer a faster genome duplication rate and simplify the management of multiple replicons, analogous to eukaryotes and many archaea? Our discovery here suggests that the concurrent DNA replication and segregation in bacteria can be incompatible with multiple replication origins or centromeres on the same chromosome, if it is not co-evolved with a XerC/D/dif-like system between each of the multiple origins, and/or properly oriented KOPS for FtsK to pump the DNA into the correct daughter cell.
The segregation of a bacterial chromosome is driven by partitioning proteins which bind to the centromere region of each chromosomal copy and drag them to opposite cell poles 41. In a multipartite genome, faithful segregation of each replicon relies on their own partitioning systems 6,42. The dicentric chromosome we discovered here generates a segregation problem that a multipartite genome avoids: independent segregation of the origins that translocate different parts of the same chromosome into opposite cell poles (Figure 4A). In contrast, eukaryotes and many archaea have multiple replication origins but a single centromere for each chromosome. These species employ temporal separation of DNA replication and segregation to ensure genome integrity. The dicentric bacterial chromosome we discovered here has survived and flourished, but this might be a serendipitous situation in which the XerC/D/dif system happens to be at the appropriate location to correct the opposite localization of each origin. The stable maintenance of this remarkable dicentric chromosome captures a snapshot of genome and species evolution through the survival of an accident and has revealed a possible explanation for the fundamental difference in genome architecture between bacteria and eukaryotes/archaea.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Xindan Wang (xindan@indiana.edu).
Materials availability
Plasmids and strains generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
Unprocessed microscopy images are available at Mendeley data: https://doi.org/10.17632/crv96srkp4.1. Hi-C, ChIP-seq and WGS data were deposited to the NCBI Sequence Read Archive (accession no. PRJNA824072). This paper does not report original code. Any additional information required to analyze the data reported in this paper is available from the Lead Contact upon request without restriction.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial strains and growth
A. tumefaciens cells were grown in defined ATGN minimal medium 46 or LB broth as specified at 30°C with aeration. In liquid media, when appropriate, antibiotics or supplements were added at the following concentrations: kanamycin (IBI, IB02120) 150 μg/ml, carbenicillin (GoldBio, C-103-5) 25 μg/ml, gentamicin (ACROS Organics, AC613980010) 150 μg/ml, IPTG (Dot Scientific, DS102125) 0.25 or 0.5 mM, theophylline (Sigma, T1633-100G) 2 mM and cumate (VWR, 103852-040) 2 mM. Antibiotic concentrations were doubled when applied on solid media. Lists of Next-Generation-Sequencing samples, bacterial strains and plasmids, and oligonucleotides used in this study can be found in Tables S2, S3 and S4.
METHODS DETAILS
Hi-C
The Hi-C procedure was carried out as previously described 6,47. Specifically, cells grown at the desired condition were crosslinked with 3% formaldehyde at room temperature for 30 min then quenched with 125 mM glycine. Cells were lysed using Ready-Lyse Lysozyme (Epicentre, R1802M) and treated by 0.5% SDS. Solubilized chromatin was digested with HindIII for 2 hours at 37°C. The digested ends were filled in with Klenow and Biotin-14-dATP, dGTP, dCTP, dTTP. The products were ligated with T4 DNA ligase at 16°C for about 20 hr. Crosslinks were reversed at 65°C for about 20 hr in the presence of EDTA, proteinase K and 0.5% SDS. The DNA was then extracted twice with phenol/chloroform/isoamylalcohol (25:24:1) (PCI), precipitated with ethanol, and resuspended in 20 μl of 0.1XTE buffer. Biotin from non-ligated ends was removed using T4 polymerase (4 hr at 20°C) followed by extraction with PCI. The DNA was then sheared by sonication for 12 min with 20% amplitude using a Qsonica Q800R2 water bath sonicator. The sheared DNA was used for library preparation with the NEBNext UltraII kit (E7645). Biotinylated DNA fragments were purified using 10 μl streptavidin beads. DNA-bound beads were used for PCR in a 50 μl reaction for 14 cycles. PCR products were purified using Ampure beads (Beckman, A63881) and sequenced at the Indiana University Center for Genomics and Bioinformatics using NextSeq500. Paired-end sequencing reads were mapped to the combined genome files of A. tumefaciens C58 (NCBI Reference Sequence GCA_000092025.1). The genome was divided into 10-kb bins. Subsequent analysis and visualization were done using R. To put ori1 at the center of Ch1, the reference genome of Ch1 starts at 1400 kb. The rearranged genome of the fusion strain was pieced together in the following order: Ch2 0.001 kb - 712.671 kb, Ch1 1793.369 kb - 2841.580 kb, Ch1 0.001 kb - 1793.368 kb, Ch2 712.672 kb - 2075.577 kb, pAt 0.001 kb - 542.868 kb, pTi 0.001 kb - 214.233 kb. Note that the genome of the fusion strain had deletions of Ch1 1175.9 kb - 1186.195 kb and pAt 206.308 kb - 400.732 kb.
ChIP-seq
Chromatin immunoprecipitation (ChIP) for A. tumefaciens was performed as described previously 47. Briefly, cells were crosslinked using 3% formaldehyde for 30 min at room temperature and then quenched using 125 mM glycine, washed using PBS, and lysed using lysozyme. Crosslinked chromatin was sheared to an average size of 250 bp by sonication using Qsonica Q800R2 water bath sonicator. The lysate was precleared using ProteinA magnetic beads (GE Healthcare/cytiva 28951378) and was then incubated with anti-GFP antibodies 43 overnight at 4°C. Next day, the lysate was incubated with ProteinA magnetic beads for 1h at 4°C. After washes and elution, the immunoprecipitate was incubated at 65°C overnight to reverse the crosslinks. The DNA was further treated with RNaseA, Proteinase K, extracted with PCI, resuspended in 100 μl EB and used for library preparation with the NEBNext UltraII kit (E7645). The library was sequenced using Illumina NextSeq500 at IU Center for Genomics and Bioinformatics. The sequencing reads were mapped to the combined A. tumefaciens C58 genome (NCBI GCA_000092025.1) using CLC Genomics Workbench (CLC Bio, QIAGEN). Sequencing reads from each ChIP and input sample were normalized by the total number of reads. The ChIP enrichment (ChIP/Input) was plotted and analyzed using R.
Identification of dif sites
In ChIP plots of XerC-GFP and XerD-GFP, three peaks with greater than 15-fold ChIP/Input enrichment were identified in all ChIP samples. By checking accurate locations of each peak using CLC Genomics Workbench (CLC Bio, QIAGEN), we excluded the one on pAt plasmid which was observed in every ChIP-seq experiment we have done on A. tumefaciens using anti-GFP antibodies regardless of strains used (Figure 2SC, asterisk marked). The other two sites on Ch1 and pAt were seen in all XerC-GFP and XerD-GFP samples (Figure 2SC). We used these two strong sites to search for the binding motif by MEME (http://meme-suite.org/) and identified a 26 bp consensus sequence which contains 23 consensus nucleotide positions and 3 degenerate positions (Figure S2D).
Whole Genome Sequencing (WGS)
Approximately 5×108 exponentially growing cells were collected for each WGS. Genomic DNA was extracted using Qiagen DNeasy Kit (69504), sonicated using a Qsonica Q800R2 water bath sonicator, prepared using the NEBNext UltraII kit (E7645), and sequenced using Illumina NextSeq500. The reads were mapped to the A. tumefaciens C58 genome (NCBI GCA_000092025.1) using CLC Genomics Workbench (CLC Bio, QIAGEN). The mapped reads were normalized by the total number of reads. Relative copy numbers were calculated by dividing normalized reads with the averaged total number of reads at the terminus of Ch1 or at the left terminus of the fused chromosome. Plotting and analysis were performed using R.
Transposon insertion sequencing (Tn-seq)
To prepare transposon libraries, Mariner transposon-based plasmid (pTND2823, gift from Triana Dalia and Ankur Dalia at Indiana University) was transformed into an auxotrophic donor strain, MFDpir E. coli 48, and then conjugated into A. tumefaciens. To generate a deep library, 1×106 kanamycin-resistant conjugants were evenly plated on 10 large plates (150 mm diameter, VWR 25384-326) containing LB or ATGN with 300 μg/ml Kanamycin. The plates were incubated at 30°C for two days for LB or three days for ATGN. Colonies of the 10 plates from the same condition were scraped and combined into a single pool. 5 OD600 units of cells from the pool were used for genomic DNA (gDNA) isolation using QIAGEN DNeasy blood & tissue kit (69504). 3 μg of gDNA was digested with MmeI (NEB R0637S) for 90 min, and quick CIP (NEB M0525L) for 60 min at 37°C. The DNA was extracted using Phenol-Chloroform, precipitated using ethanol and resuspended in 15 μl ddH2O. The digested end was ligated to an annealed adapter 49 using T4 DNA ligase and incubated at 16°C for about 16 hr. Adapter-ligated DNA was amplified with the primers complementary to the adapter and transposon inverted repeat sequence. The PCR product was gel purified using Monarch DNA Gel Extraction Kit (NEB T1020S) and sequenced at the IU Center for Genomics and Bioinformatics using NextSeq500. Sequencing reads were mapped to the combined genome files of A. tumefaciens C58 (NCBI Reference Sequence GCA_000092025.1) and analyzed using a procedure that was described before 49,50. The results were visualized using Artemis (https://www.sanger.ac.uk/tool/artemis/).
Growth curve measurement
In Figure 2B, WT original strain (AtWX063) and WT fusion strain (AtWX001) were grown in LB or ATGN liquid medium overnight at 30°C with aeration. Overnight cultures were back-diluted to OD600 of 0.02 for LB cultures and 0.05 for ATGN cultures. 200 μl of each diluted culture was transferred into 96-well microplate with lid (Corning 3603). To monitor cell growth, the OD600 was recorded every 30 min for a total of 24 hr using a Synergy H1 multimode microplate reader at 30°C with shaking. For each medium per strain, three biological replicates were set up by inoculating cells in three independent culture tubes. This experiment was performed twice on two different days and the results were combined. Plotting and analysis were performed using GraphPad Prism 8.
Fluorescence microscopy analysis
Fluorescence microscopy was performed on a Nikon Ti2E microscope equipped with Plan Apo 100x/1.4NA phase contrast oil objective and an sCMOS camera. Cells were immobilized using 2% agarose pads containing growth media. To determine cellular localization of origins, image analyses were performed using the MathWorks MATLAB-based program Oufti 45. Cell outline and localizations of fluorescent foci were detected and plotted as described previously 6.
In XerC/D depletion time-course experiments, cells were grown in LB medium containing inducers (0.5 mM IPTG and 2 mM theophylline) overnight. For XerC(+) and XerD(+) cells, subcultures were set up in 25 mL of LB medium containing inducers. Exponentially growing cells were collected for imaging. For XerC(−) and XerD(−) cells, inducers were washed off using LB medium for 3 times, and cells were sub-cultured in LB medium without inducers. At 3 hr, 6 hr, 12 hr, 18 hr post-subculturing, cells were collected for snapshot imaging. To prevent cells from entering stationary phase, when the OD600 reached 0.6, cultures were diluted to prewarmed fresh medium.
In time-lapse imaging, for WT fusion strain having dif1 labeled (AtWX455), cells were grown in LB medium overnight and then were sub-cultured in 25 ml of LB medium at initial OD600 of 0.04. For XerC(−) strains (AtWX467 and AtWX514), cells were grown in LB medium containing inducers (0.5 mM IPTG and 2 mM theophylline) overnight. Then inducers were washed off using LB medium for 3 times and then cells were sub-cultured in 25 ml of LB medium at initial OD600 of 0.04. After 6-hour depletion, cells were collected for time-lapse imaging. A glass bottom dish (Willco dish HBSt-5040; Willco Wells) was used as a coverslip 51. Cells were concentrated and spotted onto the glass bottom dish. A 2% LB agarose strip was then laid on top of the cells. The agarose strip was fully exposed to adequate oxygen for cell growth. The dish was placed in a temperature-controlled incubator 51. Cells were imaged every 10 min. Images were processed using MetaMorph software (Molecular Devices).
Plasmid construction
pWX811 [pNPTS138 ΔrepBCh2 (Atu3923/ATU_RS18280) (kan)] was constructed by an isothermal assembly reaction containing three gel-purified fragments: 1) pNPTS138 digested by EcoRV; 2) repBCh2 upstream region amplified using oWX2021 and oWX2022 from C58 gDNA; 3) repBCh2 downstream region amplified using oWX2023 and oWX2024 from C58 gDNA. The construct was sequenced using oWX1854 and oWX1855.
pWX813 [pMiniTn7 pLac repBCh2 (Atu3923/ATU_RS18280) kan (gen)] was constructed by a ligation reaction containing two DNA fragments: 1) pUC18-mini-Tn7T-GM-Plac-HA 9 digested by NdeI and XhoI; 2) repBCh2 amplified using oWX2027 and oWX2028 from C58 gDNA. The construct was sequenced using oWX2031 and oWX2042.
pWX855 [pNPTS138 ΔxerD (Atu3629/ATU_RS16850)::amp (kan)] was constructed by an isothermal assembly reaction containing four gel-purified fragments: 1) EcoRV-digested pNPTS138 52; 2) At xerD (Atu3629/ATU_RS16850) upstream region amplified using oWX2144 and oWX2208 from C58 genomic DNA; 3) At xerD (Atu3629/ATU_RS16850 downstream region amplified using oWX2146 and oWX2209 from C58 genomic DNA; 4) amp amplified using oWX2210 and oWX2211 from plasmid pHP45omega 53. The construct was sequenced using oWX1854 and oWX1855. pWX923 [pACYC terminator Ppen cfp-parBP1-parSP1 terminators amp] was constructed by an isothermal assembly reaction containing two gel-purified fragments: 1) SaII/Eagl-digested pWX916 7; 2) terminators amplified using oWX2403 and oWX2404 from genomic DNA of BWX925 54. The construct was sequenced using oWX2395.
pWX926 [pNPTS138 Ppen ygfp-parBMT1-parSMT1 at Atu1460/ATU_RS07195 (kan)] was constructed by an isothermal assembly reaction containing four gel-purified fragments: 1) EcoRV-digested pNPTS138 52; 2) a part of Atu1460/ATU_ RS07195 amplified using oWX2409 and oWX2410 from C58 gDNA; 3) ygfp-parBMT1-parSMT1 amplified using oWX2407 and oWX2408 from pWX924 6; 4) a part of Atu1461/ATU_ RS07195 amplified using oWX2411 and oWX2412 from C58 gDNA. The construct was sequenced using oWX2379, oWX2418, oWX2419 and oWX2426.
pWX943 [pNPTS138 terminator gen lacI terminators Plac-riboswitch (kan)] was constructed by an isothermal assembly reaction containing three gel-purified fragments: 1) EcoRV-digested pNPTS138 52; 2) terminator gen lacI terminators Plac was generated using oWX2482 and oWX2450 on pUC18-mini-Tn7T-GM-Plac-HA 9; 3) Riboswitch was amplified using oWX2451 and oWX2452 from pJZ274 15. The construct was sequenced oWX1854, oWX1855, oWX2491, oWX2492 and oWX2493.
pWX944 [pNPTS138 terminator gen lacI terminators Plac-riboswitch inserted before xerC (Atu2628/ATU_RS12790) (kan)] was constructed by an isothermal assembly reaction containing four gel-purified fragments: 1) EcoRV-digested pNPT138 52; 2) At xerC (Atu2628/ATU_RS12790) upstream region amplified using oWX2453 and oWX2483 from C58 genomic DNA; 3) gen lacI-Plac-riboswitch amplified using oWX2484 and oWX2456 from plasmid pWX943; 4) A part of At xerC (Atu2628/ATU_RS12790) amplified using oWX2457 and oWX2458 from C58 genomic DNA. The construct was sequenced using oWX1854 and oWX1855.
pWX945 [pNPTS138 terminator gen lacI terminators Plac-riboswitch inserted before xerD (Atu3629/ATU_RS16850) (kan)] was constructed by an isothermal assembly reaction containing four gel-purified fragments: 1) EcoRV-digested pNPT138 52; 2) At xerD (Atu3629/ATU_RS16850) upstream region amplified using oWX2459 and oWX2485 from C58 genomic DNA; 3) gen lacI-Plac-riboswitch amplified using oWX2486 and oWX2462 from plasmid pWX943; 4) A part of At xerD (Atu3629/ATU_RS16850) amplified using oWX2463 and oWX2464 from C58 genomic DNA. The construct was sequenced using oWX1854 and oWX1855.
pWX965 [pNPTS138 PT7strong ygfp-parBMT1-parSMT1 at Atu1460/ATU_RS07195] was constructed by an isothermal assembly reaction containing one gel-purified fragments: pWX926 backbone amplified using oWX2431 and oWX2432. The construct was sequenced using oWX2379, oWX2418, oWX2419 and oWX2426.
pWX968 [pNPTS138 PT7strong cfp-parBP1-parSP1 at Atu4854 (kan)] was constructed by an isothermal assembly reaction containing four gel-purified fragments: 1) EcoRV-digested pNPTS138 52; 2) PT7strong cfp-parBP1-parSP1 amplified using oWX2407 and oWX2408 from pWX936 7; 3) a part of Atu4854 amplified using oWX2514 and oWX2515 from C58 gDNA; 4) a part of Atu4855/ATU_22800 amplified using oWX2516 and oWX2517 from C58 gDNA. The construct was sequenced using oWX2377, oWX2426, oWX2518 and oWX2519.
pWX998 [pNPTS138 PT7strong mcherry-parBP1-parSP1 at Atu4854 (kan)] was constructed by an isothermal assembly reaction containing two gel-purified fragments: 1) pWX968 backbone amplified using oWX2589 and oWX2590 on pWX968; 2) mcherry amplified using oWX2584 and oWX2585 from gDNA of BWX2208 55. The construct was sequenced using oWX2377, oWX2426, oWX2518 and oWX2519.
pWX1006 [pNPTS138 ΔxerC (Atu2628/ATU_RS12790)::amp (kan)] was constructed by an isothermal assembly reaction containing four gel-purified fragments: 1) EcoRV-digested pNPTS138 52; 2) At xerC (Atu2628/ATU_RS12790) upstream region amplified using oWX2601 and oWX2602 from C58 genomic DNA; 3) At xerC (Atu2628/ATU_RS12790) downstream region amplified using oWX2603 and oWX2604 from C58 genomic DNA; 4) amp amplified using oWX2210 and oWX2211 from plasmid pHP45omega 53. The construct was sequenced using oWX2210, oWX2624 and oWX2625.
pWX1007 [pNPTS138 ΔrecA (Atu1873/ATU_RS09160)::amp (kan)] was constructed by an isothermal assembly reaction containing four gel-purified fragments: 1) EcoRV-digested pNPTS138 52; 2) At recA (Atu1873/ATU_RS09160) upstream region amplified using oWX2605 and oWX2606 from C58 genomic DNA; 3) At recA (Atu1873/ATU_RS09160) downstream region amplified using oWX2607 and oWX2608 from C58 genomic DNA; 4) amp amplified using oWX2210 and oWX2211 from plasmid pHP45omega 53. The construct was sequenced using oWX2210, oWX2624 and oWX2625.
pWX1008 [pNPTS138 xerC-gfpmut3 (Atu2628/ATU_RS12790) (kan)] was constructed by an isothermal assembly reaction containing four gel-purified fragments: 1) EcoRV-digested pNPTS138 52; 2) a part of C terminal region of At xerC (Atu2628/ATU_RS12790) amplified using oWX2609 and oWX2610 from C58 genomic DNA; 3) At xerC (Atu2628/ATU_RS12790) downstream region amplified using oWX2613 and oWX2614 from C58 genomic DNA; 4) gfpmut3 amplified using oWX2611 and oWX2612 on BWX2030 47. The construct was sequenced using oWX2497, oWX2626, oWX2627 and oWX2628.
pWX1009 [pNPTS138 xerD-gfpmut3 (Atu3629/ATU_RS16850) (kan)] was constructed by an isothermal assembly reaction containing four gel-purified fragments: 1) EcoRV-digested pNPTS138 52; 2) a part of C terminal region of At xerD (Atu3629/ATU_RS16850) amplified using oWX2615 and oWX2616 from C58 genomic DNA; 3) At xerD (Atu3629/ATU_RS16850) downstream region amplified using oWX2617 and oWX2618 from C58 genomic DNA; 4) gfpmut3 amplified using oWX2611 and oWX2612 on BWX2030 47. The construct was sequenced using oWX2497, oWX2626, oWX2627 and oWX2628.
pWX1039 [pNPTS138 Δdif1::amp (kan)] was constructed by an isothermal assembly reaction containing four gel-purified fragments: 1) EcoRV-digested pNPTS138 52; 2) dif1 upstream region amplified using oWX2796 and oWX2797 from C58 genomic DNA; 3) dif1 downstream region amplified using oWX2800 and oWX2801 from C58 genomic DNA; 4) amp amplified using oWX2798 and oWX2799 from plasmid pHP45omega 53. The construct was sequenced using oWX2210, oWX2624 and oWX2625.
pWX1040 [pNPTS138 ΔdifpAt::amp (kan)] was constructed by an isothermal assembly reaction containing four gel-purified fragments: 1) EcoRV-digested pNPTS138 52; 2) difpAt upstream region amplified using oWX2802 and oWX2803 from C58 genomic DNA; 3) difpAt downstream region amplified using oWX2806 and oWX2807 from C58 genomic DNA; 4) amp amplified using oWX2804 and oWX2805 from plasmid pHP45omega 53. The construct was sequenced using oWX2210, oWX2624 and oWX2625.
pWX1076 [pSRKKm cymR cuO Plac cuO At parC (Atu1158/ATU_RS05720) At parE (Atu1622/ATU_RS07965) (kan)] was constructed by an isothermal assembly reaction containing three gel-purified fragments: 1) NdeI/NheI-digested plasmid pSRKKm cuO Plac cuO msfgfp (gift from Pam Brown); 2) At parC (Atu1158/ATU_RS05720) amplified using oWX2912 and oWX2913 from C58 genomic DNA; 3) optimized ribosomal binding site plus At parE (Atu1622/ATU_RS07965) amplified using oWX2914 and oWX2915 from C58 genomic DNA. The construct was sequenced using oWX2720, oWX2920, oWX2922, oWX2923, oWX2924, oWX2925, oWX2926 and oWX2927. Based on protein sequence alignment and gene synteny analysis, we note that At parE (Atu1622/ATU_RS07965) was mis-annotated as gyrB in NCBI Reference Sequence GCA_000092025.1; the bona fide gyrB is (Atu0012/ATU_RS00060).
pWX1080 [pSRKKm cymR cuO Plac cuO Ec parE-parC (kan)] was constructed by an isothermal assembly reaction containing two gel-purified fragments: 1) NdeI/NheI-digested plasmid pSRKKm cuO Plac cuO msfgfp (gift from Pam Brown); 2) E.coli parE-parC amplified using oWX2936 and oWX2939 on pLEXRparEC3 56. The construct was sequenced using oWX2720, oWX2922, oWX2940, oWX2941, oWX2942, oWX2943, oWX2944 and oWX2945.
Strain construction
In general, in-frame deletions of C58 A. tumefaciens strains were constructed using a previously described allelic replacement method 46. Briefly, regions flanking the gene to be deleted were PCR amplified using Q5 polymerase (NEB M0491) and cloned into pNPTS138 52, a ColE1 suicide plasmid that confers kanamycin resistance and sucrose sensitivity, by isothermal assembly reactions. See Plasmid construction section for details. The pNPTS138 deletion plasmids were then introduced into A. tumefaciens C58 via mating with E. coli S17-1/λpir 57 carrying the appropriate construct. Screening for plasmid integration and target gene deletion was performed as previously described 46,58. The deletion mutants were confirmed by colony PCR. Specific information about each strain can be found below.
Fusion strain, ori1 and ori2 visualized by EGFP-ParB1 and RFP-RepBCh2 (AtWX366) was generated by directly electroporating pWX970 into WT fusion strain (AtWX001).
Fusion strain, tetRA::a-attTn7 pLac repBch2 gen, ΔrepBCh2 (AtWX025) was constructed in two steps: 1) pLac repBch2 gen was inserted at the engineered a-attTn7 site using pWX813 in AtWX003 9 as pervious described 9; 2) pWX811 was used to delete the endogenous repBCh2 in presence of 0.5 mM IPTG. The deletion was confirmed oWX2021 and oWX2024.
Fusion strain, terminator gen lacI terminators Plac-riboswitch-xerC (AtWX323) was generated using pWX944 to introduce terminator gen LacI terminators Plac-riboswitch before start codon of xerC (Atu2628/ATU_RS12790) in the fusion strain (AtWX001) and confirmed using oWX487 and oWX2029.
Fusion strain, terminator gen lacI terminators Plac-riboswitch-xerD (AtWX327) was generated using pWX945 to introduce terminator gen LacI terminators Plac-riboswitch before start codon of xerD (Atu3629/ATU_RS16850) in the fusion strain (AtWX001) and confirmed using oWX487 and oWX2029.
Original strain, terminator gen lacI terminators Plac-riboswitch-xerC (AtWX331) was generated using pWX944 to introduce terminator gen LacI terminators Plac-riboswitch before start codon of xerC (Atu2628/ATU_RS12790) in the original strain (AtWX063) and confirmed using oWX487 and oWX2029.
Original strain, terminator gen lacI terminators Plac-riboswitch-xerD (AtWX332) was generated using pWX945 to introduce terminator gen LacI terminators Plac-riboswitch before start codon of xerD (Atu3629/ATU_RS16850) in the original strain (AtWX063) and confirmed using oWX487 and oWX2029.
ΔrecA fusion strain (AtWX387) was generated by conjugating pWX1007 into the fusion strain (AtWX001) and confirmed using oWX2210, 2624, 2625.
ΔrecA original strain (AtWX398) was generated by conjugating pWX1007 into the original strain (AtWX063) and confirmed using oWX2210, 2624, 2625.
Δsmc fusion strain (AtWX035) was generated by conjugating pWX832 into the fusion strain (AtWX001) and confirmed using oWX2085 and oWX2086.
Fusion strain, yGFP-parBMT1-parSMT1 inserted between Atu1460/ATU_RS07195 and Atu1461/ ATU_RS07200 20 kb from dif1 (AtWX455) was generated by conjugating pWX965 into WT fusion strain (AtWX001) and confirmed using oWX2418 and oWX2419.
Fusion strain with XerC depletion, yGFP-parBMT1-parSMT1 inserted between Atu1460/ATU_RS07195 and Atu1461/ ATU_RS07200 20 kb from dif1 (AtWX467) was generated by conjugating pWX965 into XerC depletion strain (AtWX323) and confirmed using oWX2418 and oWX2419.
Fusion strain with XerD depletion, yGFP-parBMT1-parSMT1 inserted between Atu1460/ATU_RS07195 and Atu1461/ ATU_RS07200 20 kb from dif1 (AtWX471) was generated by conjugating pWX965 into XerD depletion strain (AtWX327) and confirmed using oWX2418 and oWX2419.
Original strain, xerC-gfpmut3 (AtWX377) was generated by conjugating pWX1008 into the original strain (AtWX063) and confirmed using oWX2497 and oWX2627.
Original strain, xerD-gfpmut3 (AtWX370) was generated by conjugating pWX1009 into the original strain (AtWX063) and confirmed using oWX2497 and oWX2627.
Fusion strain, xerC-gfpmut3 (AtWX367) was generated by conjugating pWX1008 into the fusion strain (AtWX001) and confirmed using oWX2497 and oWX2627.
Fusion strain, xerD-gfpmut3 (AtWX379) was generated by conjugating pWX1009 into the fusion strain (AtWX001) and confirmed using oWX2497 and oWX2627.
ΔxerD original strain (AtWX092) was generated by conjugating pWX855 into the original strain (AtWX063) and confirmed using oWX2148 and oWX2149.
ΔxerC original strain (AtWX375) was generated by conjugating pWX1006 into the original strain (AtWX063) and confirmed using oWX2210, 2624, 2625.
Δdif1 original strain (AtWX439) was generated by conjugating pWX1039 into the original strain (AtWX063) and confirmed using oWX2847 and oWX2848.
ΔdifpAt fusion strain (AtWX440) was generated by conjugating pWX1040 into the fusion strain (AtWX001) and confirmed using oWX2849 and oWX2850.
ΔdifpAt original strain (AtWX441) was generated by conjugating pWX1040 into the original strain (AtWX063) and confirmed using oWX2849 and oWX2850.
Fusion strain with XerC depletion, yGFP-parBMT1-parSMT1 inserted between Atu1460/ATU_RS07195 and Atu1461/ ATU_RS07200 20 kb from dif1, mcherry-parBP1-parSP1 inserted between Atu4854 and Atu4855, 39.8 kb from right ter (AtWX514) was generated by conjugating pWX998 into AtWX467 and confirmed using oWX2518 and oWX2519.
For strains containing plasmids for overexpressing A. tumefaciens Topo IV or E. coli Topo IV, pWX1076 or pWX1080 was directly electroporated into fusion strains with XerC depletion (AtWX323) or XerD depletion (AtWX327) as previously described 46 to generate AtWX502, AtWX503, AtWX540 AtWX541.
QUANTIFICATION AND STATISTICAL ANALYSIS
Localization of ori1 and ori2
To quantify distribution and colocalization of fluorescence foci, we analyzed images using the MathWorks MATLAB-based program Oufti 45 as described previously 6. Briefly, cell outlines were detected using the cellDetection module. Localizations of fluorescent foci were identified using the spotDetection module. Subsequently, manual inspection was employed to remove the cell meshes with wrongly detected cell outline or spots. The data were further analyzed and plotted in MATLAB. In our snapshot images, the average diameter of a fluorescence focus was 6 pixels. As such, the spatial relationship of foci was defined as colocalizing when red and green foci had inter-focal distance of less than 6 pixels.
Measurements of doubling time and growth curve
Measurements were performed in biological triplicates. Two independent growth curve measurements were carried out. Plotting and analysis were performed using GraphPad Prism 8. Data were presented as the Means ± Standard Deviations (SDs).
Quantitative analysis of dif1 localization patterns
To quantitatively analyze dif1 localization pattern, we analyzed snapshot images from time-course experiments using NIS-Elements software. Cells were classified in terms of cell cycle and foci number. 700~1200 cells were analyzed for each sample and counted manually using counter mode in NIS-Elements AR.
Supplementary Material
KEY RESOURCE TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-GFP polyclonal rabbit antibody, affinity purified | 43 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Biotin-14-dATP | ThermoFisher | Cat # 19524016 |
| Carbenicillin | GoldBio | Cat # C-103-5 |
| Chloroform | VWR | Cat # MK444004 |
| CIP | NEB | Cat # M0525L |
| Cumate | VWR | Cat # 103852-040 |
| dCTP, dGTP, dTTP | Fisher | Cat # R0182 |
| EDTA | VWR | Cat # EM-4050 |
| Formaldehyde 37% | Sigma | Cat # F8775 |
| Gentamicin | ACROS Organics | Cat # AC613980010 |
| Glycine | VWR | Cat # JT4059-6 |
| HindIII | NEB | Cat # R0104M |
| Isoamyl alcohol | VWR | Cat # JT9054-1 |
| IPTG | Dot Scientific | Cat # DS102125 |
| Kanamycin | IBI | Cat # IB02120 |
| Klenow | NEB | Cat # M0210L |
| MmeI | NEB | Cat # R0637S |
| Proteinase K | NEB | Cat # P8107S |
| Phenol | VWR | Cat # 97064-822 |
| Ready-Lyse Lysozyme | Epicenter | Cat # R1802M |
| RNase A | Promega | Cat # R7973 |
| SDS | Calbiochem | Cat # 7910-500GM |
| T4 DNA Ligase | NEB | Cat # M0202M |
| T4 DNA Polymerase | NEB | Cat # M0203L |
| Theophylline | Sigma | Cat # T1633-100G |
| Critical commercial assays | ||
| NEBNext Ultra II DNA Library Prep Kit | NEB | Cat # E7645 |
| QIAGEN DNeasy blood & tissue kit | QIAGEN | Cat # 69504 |
| Monarch DNA Gel Extraction Kit | NEB | Cat # T1020S |
| Deposited data | ||
| Hi-C, ChIP-Seq, WGS data | This study | NCBI SRA: PRJNA824072); See Table S2 |
| Unprocessed Microscopy Images | Mendeley Data | https://doi.org/10.17632/crv96srkp4.1 |
| Experimental models: organism and strains | ||
| Agrobacterium tumefaciens strains, see Table S3 | N/A | N/A |
| Oligonucleotides | ||
| See Table S4 | N/A | N/A |
| Recombinant DNA | ||
| See Table S3 | N/A | N/A |
| Software and algorithms | ||
| Artemis | Sanger | https://www.sanger.ac.uk/tool/artemis/ |
| CLC Genomics Workbench | QIAGEN | https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-clc-genomics-workbench/ |
| GraphPad Prism 8 | GraphPad Software, Inc | https://www.graphpad.com/ |
| Hiclib | 44 | https://github.com/mirnylab/hiclib-legacy |
| MATLAB | Mathworks | https://www.mathworks.com/products.html?s_tid=gn_ps |
| MEME | N/A | http://meme-suite.org/) |
| NIS-Elements AR | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements/nis-elements-advanced-research |
| MetaMorph | Molecular Devices | https://www.moleculardevices.com/ |
| Oufti | 45 | http://www.oufti.org/ |
| R | N/A | https://www.r-project.org/ |
| Other | ||
| 96-well microplate | Corning | Cat #3603 |
| Ampure Beads | Beckman | Cat # A63881 |
| Glass-bottom dish | Willco Wells | Cat # HBSt-5040 |
| ProteinA Magnetic Beads | GE Healthcare/cytiva | Cat # 28951378 |
| Streptavidin Beads MyOne | Invitrogen | Cat# 65-001 |
Highlight:
A commonly used A. tumefaciens C58 strain has a linear dicentric chromosome
The fused chromosome resulted from integration of the circular Ch1 into linear Ch2
The two replication origins are active and their partitioning systems are essential
Recombinases XerC/D are essential to resolve this dicentric chromosome
Acknowledgements
We thank the Indiana University Center for Genomics and Bioinformatics for assistance with high throughput sequencing. We thank Ankur Dalia for the transposon plasmid (pTND2823); Julia van Kessel for plate reader; Pam Brown, Patricia Zambryski, John Zupan for strains and plasmids; Dan Kearns and David Rudner for reading the manuscript. We thank the entire Agrobacterium community for sharing their C58 strains for characterization. Support for this work comes from National Institutes of Health R01GM141242 (X.W.) and R01GM120337 (C.F.).
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Robinson NP, and Bell SD (2005). Origins of DNA replication in the three domains of life. FEBS J 272, 3757–3766. 10.1111/j.1742-4658.2005.04768.x. [DOI] [PubMed] [Google Scholar]
- 2.Blakely G, May G, McCulloch R, Arciszewska LK, Burke M, Lovett ST, and Sherratt DJ (1993). Two related recombinases are required for site-specific recombination at dif and cer in E. coli K12. Cell 75, 351–361. 10.1016/0092-8674(93)80076-q. [DOI] [PubMed] [Google Scholar]
- 3.Kahng LS, and Shapiro L (2003). Polar localization of replicon origins in the multipartite genomes of Agrobacterium tumefaciens and Sinorhizobium meliloti. J Bacteriol 185, 3384–3391. 10.1128/JB.185.11.3384-3391.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Slater SC, Goldman BS, Goodner B, Setubal JC, Farrand SK, Nester EW, Burr TJ, Banta L, Dickerman AW, Paulsen I, et al. (2009). Genome sequences of three agrobacterium biovars help elucidate the evolution of multichromosome genomes in bacteria. J Bacteriol 191, 2501–2511. 10.1128/JB.01779-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robalino-Espinosa JS, Zupan JR, Chavez-Arroyo A, and Zambryski P (2020). Segregation of four Agrobacterium tumefaciens replicons during polar growth: PopZ and PodJ control segregation of essential replicons. Proc Natl Acad Sci U S A 117, 26366–26373. 10.1073/pnas.2014371117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ren Z, Liao Q, Karaboja X, Barton IS, Schantz EG, Mejia-Santana A, Fuqua C, and Wang X (2022). Conformation and dynamic interactions of the multipartite genome in Agrobacterium tumefaciens. Proc Natl Acad Sci U S A 119. 10.1073/pnas.2115854119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ren Z, Liao Q, Barton IS, Wiesler EE, Fuqua C, and Wang X (2022). Centromere Interactions Promote the Maintenance of the Multipartite Genome in Agrobacterium tumefaciens. mBio, e0050822. 10.1128/mbio.00508-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morton ER, Merritt PM, Bever JD, and Fuqua C (2013). Large deletions in the pAtC58 megaplasmid of Agrobacterium tumefaciens can confer reduced carriage cost and increased expression of virulence genes. Genome Biol Evol 5, 1353–1364. 10.1093/gbe/evt095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Figueroa-Cuilan W, Daniel JJ, Howell M, Sulaiman A, and Brown PJ (2016). Mini-Tn7 Insertion in an Artificial attTn7 Site Enables Depletion of the Essential Master Regulator CtrA in the Phytopathogen Agrobacterium tumefaciens. Appl Environ Microbiol 82, 5015–5025. 10.1128/AEM.01392-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuqua C, Burbea M, and Winans SC (1995). Activity of the Agrobacterium Ti plasmid conjugal transfer regulator TraR is inhibited by the product of the traM gene. J Bacteriol 177, 1367–1373. 10.1128/jb.177.5.1367-1373.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farrand SK, and Dessaux Y (1986). Proline biosynthesis encoded by the noc and occ loci of Agrobacterium Ti plasmids. J Bacteriol 167, 732–734. 10.1128/jb.167.2.732-734.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heindl JE, Hibbing ME, Xu J, Natarajan R, Buechlein AM, and Fuqua C (2015). Discrete Responses to Limitation for Iron and Manganese in Agrobacterium tumefaciens: Influence on Attachment and Biofilm Formation. J Bacteriol 198, 816–829. 10.1128/JB.00668-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hibbing ME, and Fuqua C (2011). Antiparallel and interlinked control of cellular iron levels by the Irr and RirA regulators of Agrobacterium tumefaciens. J Bacteriol 193, 3461–3472. 10.1128/JB.00317-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Curtis PD, and Brun YV (2014). Identification of essential alphaproteobacterial genes reveals operational variability in conserved developmental and cell cycle systems. Mol Microbiol 93, 713–735. 10.1111/mmi.12686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zupan JR, Grangeon R, Robalino-Espinosa JS, Garnica N, and Zambryski P (2019). GROWTH POLE RING protein forms a 200-nm-diameter ring structure essential for polar growth and rod shape in Agrobacterium tumefaciens. Proc Natl Acad Sci U S A 116, 10962–10967. 10.1073/pnas.1905900116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Topp S, Reynoso CM, Seeliger JC, Goldlust IS, Desai SK, Murat D, Shen A, Puri AW, Komeili A, Bertozzi CR, et al. (2010). Synthetic riboswitches that induce gene expression in diverse bacterial species. Appl Environ Microbiol 76, 7881–7884. 10.1128/AEM.01537-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carnoy C, and Roten CA (2009). The dif/Xer recombination systems in proteobacteria. PLoS One 4, e6531. 10.1371/journal.pone.0006531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Recchia GD, and Sherratt DJ (1999). Conservation of xer site-specific recombination genes in bacteria. Mol Microbiol 34, 1146–1148. 10.1046/j.1365-2958.1999.01668.x. [DOI] [PubMed] [Google Scholar]
- 19.Yen MR, Lin NT, Hung CH, Choy KT, Weng SF, and Tseng YH (2002). oriC region and replication termination site, dif, of the Xanthomonas campestris pv. campestris 17 chromosome. Appl Environ Microbiol 68, 2924–2933. 10.1128/AEM.68.6.2924-2933.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Val ME, Kennedy SP, El Karoui M, Bonne L, Chevalier F, and Barre FX (2008). FtsK-dependent dimer resolution on multiple chromosomes in the pathogen Vibrio cholerae. PLoS Genet 4, e1000201. 10.1371/journal.pgen.1000201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sciochetti SA, Piggot PJ, and Blakely GW (2001). Identification and characterization of the dif Site from Bacillus subtilis. J Bacteriol 183, 1058–1068. 10.1128/JB.183.3.1058-1068.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neilson L, Blakely G, and Sherratt DJ (1999). Site-specific recombination at dif by Haemophilus influenzae XerC. Mol Microbiol 31, 915–926. 10.1046/j.1365-2958.1999.01231.x. [DOI] [PubMed] [Google Scholar]
- 23.Nunes-Duby SE, Kwon HJ, Tirumalai RS, Ellenberger T, and Landy A (1998). Similarities and differences among 105 members of the Int family of site-specific recombinases. Nucleic Acids Res 26, 391–406. 10.1093/nar/26.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crozat E, and Grainge I (2010). FtsK DNA translocase: the fast motor that knows where it’s going. Chembiochem 11, 2232–2243. 10.1002/cbic.201000347. [DOI] [PubMed] [Google Scholar]
- 25.Grainge I (2013). Simple topology: FtsK-directed recombination at the dif site. Biochem Soc Trans 41, 595–600. 10.1042/BST20120299. [DOI] [PubMed] [Google Scholar]
- 26.Sivanathan V, Allen MD, de Bekker C, Baker R, Arciszewska LK, Freund SM, Bycroft M, Lowe J, and Sherratt DJ (2006). The FtsK gamma domain directs oriented DNA translocation by interacting with KOPS. Nat Struct Mol Biol 13, 965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bigot S, Saleh OA, Cornet F, Allemand JF, and Barre FX (2006). Oriented loading of FtsK on KOPS. Nat Struct Mol Biol 13, 1026–1028. [DOI] [PubMed] [Google Scholar]
- 28.Blakely G, Colloms S, May G, Burke M, and Sherratt D (1991). Escherichia coli XerC recombinase is required for chromosomal segregation at cell division. New Biol 3, 789–798. [PubMed] [Google Scholar]
- 29.Karaboja X, Ren Z, Brandao HB, Paul P, Rudner DZ, and Wang X (2021). XerD unloads bacterial SMC complexes at the replication terminus. Mol Cell 81, 756–766 e758. 10.1016/j.molcel.2020.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ip SC, Bregu M, Barre FX, and Sherratt DJ (2003). Decatenation of DNA circles by FtsK-dependent Xer site-specific recombination. EMBO J 22, 6399–6407. 10.1093/emboj/cdg589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grainge I, Bregu M, Vazquez M, Sivanathan V, Ip SC, and Sherratt DJ (2007). Unlinking chromosome catenanes in vivo by site-specific recombination. EMBO J 26, 4228–4238. 10.1038/sj.emboj.7601849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shimokawa K, Ishihara K, Grainge I, Sherratt DJ, and Vazquez M (2013). FtsK-dependent XerCD-dif recombination unlinks replication catenanes in a stepwise manner. Proc Natl Acad Sci U S A 110, 20906–20911. 10.1073/pnas.1308450110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo X, Flores M, Mavingui P, Fuentes SI, Hernandez G, Davila G, and Palacios R (2003). Natural genomic design in Sinorhizobium meliloti: novel genomic architectures. Genome Res 13, 1810–1817. 10.1101/gr.1260903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xie G, Johnson SL, Davenport KW, Rajavel M, Waldminghaus T, Detter JC, Chain PS, and Sozhamannan S (2017). Exception to the Rule: Genomic Characterization of Naturally Occurring Unusual Vibrio cholerae Strains with a Single Chromosome. Int J Genomics 2017, 8724304. 10.1155/2017/8724304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Val ME, Kennedy SP, Soler-Bistue AJ, Barbe V, Bouchier C, Ducos-Galand M, Skovgaard O, and Mazel D (2014). Fuse or die: how to survive the loss of Dam in Vibrio cholerae. Mol Microbiol 91, 665–678. 10.1111/mmi.12483. [DOI] [PubMed] [Google Scholar]
- 36.Bruhn M, Schindler D, Kemter FS, Wiley MR, Chase K, Koroleva GI, Palacios G, Sozhamannan S, and Waldminghaus T (2018). Functionality of Two Origins of Replication in Vibrio cholerae Strains With a Single Chromosome. Front Microbiol 9, 2932. 10.3389/fmicb.2018.02932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mori JF, and Kanaly RA (2022). Natural Chromosome-Chromid Fusion across rRNA Operons in a Burkholderiaceae Bacterium. Microbiol Spectr 10, e0222521. 10.1128/spectrum.02225-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mavingui P, Flores M, Guo X, Davila G, Perret X, Broughton WJ, and Palacios R (2002). Dynamics of genome architecture in Rhizobium sp. strain NGR234. J Bacteriol 184, 171–176. 10.1128/JB.184.1.171-176.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.diCenzo GC, and Finan TM (2017). The Divided Bacterial Genome: Structure, Function, and Evolution. Microbiol Mol Biol Rev 81. 10.1128/MMBR.00019-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harrison PW, Lower RP, Kim NK, and Young JP (2010). Introducing the bacterial ‘chromid’: not a chromosome, not a plasmid. Trends Microbiol 18, 141–148. 10.1016/j.tim.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 41.Wang X, Montero Llopis P, and Rudner DZ (2013). Organization and segregation of bacterial chromosomes. Nat Rev Genet 14, 191–203. 10.1038/nrg3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deghelt M, Mullier C, Sternon JF, Francis N, Laloux G, Dotreppe D, Van der Henst C, Jacobs-Wagner C, Letesson JJ, and De Bolle X (2014). G1-arrested newborn cells are the predominant infectious form of the pathogen Brucella abortus. Nat Commun 5, 4366. 10.1038/ncomms5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rudner DZ, Fawcett P, and Losick R (1999). A family of membrane-embedded metalloproteases involved in regulated proteolysis of membrane-associated transcription factors. Proc Natl Acad Sci U S A 96, 14765–14770. 10.1073/pnas.96.26.14765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Imakaev M, Fudenberg G, McCord RP, Naumova N, Goloborodko A, Lajoie BR, Dekker J, and Mirny LA (2012). Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat Methods 9, 999–1003. 10.1038/nmeth.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paintdakhi A, Parry B, Campos M, Irnov I, Elf J, Surovtsev I, and Jacobs-Wagner C (2016). Oufti: an integrated software package for high-accuracy, high-throughput quantitative microscopy analysis. Mol Microbiol 99, 767–777. 10.1111/mmi.13264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morton ER, and Fuqua C (2012). Laboratory maintenance of Agrobacterium. Curr Protoc Microbiol Chapter 1, Unit3D 1. 10.1002/9780471729259.mc03d01s24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X, Le TB, Lajoie BR, Dekker J, Laub MT, and Rudner DZ (2015). Condensin promotes the juxtaposition of DNA flanking its loading site in Bacillus subtilis. Genes Dev 29, 1661–1675. 10.1101/gad.265876.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ferrieres L, Hemery G, Nham T, Guerout AM, Mazel D, Beloin C, and Ghigo JM (2010). Silent mischief: bacteriophage Mu insertions contaminate products of Escherichia coli random mutagenesis performed using suicidal transposon delivery plasmids mobilized by broad-host-range RP4 conjugative machinery. J Bacteriol 192, 6418–6427. 10.1128/JB.00621-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Opijnen T, Lazinski DW, and Camilli A (2014). Genome-Wide Fitness and Genetic Interactions Determined by Tn-seq, a High-Throughput Massively Parallel Sequencing Method for Microorganisms. Curr Protoc Mol Biol 106, 7 16 11–24. 10.1002/0471142727.mb0716s106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meeske AJ, Sham LT, Kimsey H, Koo BM, Gross CA, Bernhardt TG, and Rudner DZ (2015). MurJ and a novel lipid II flippase are required for cell wall biogenesis in Bacillus subtilis. Proc Natl Acad Sci U S A 112, 6437–6442. 10.1073/pnas.1504967112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang X, and Montero Llopis P (2016). Visualizing Bacillus subtilis During Vegetative Growth and Spore Formation. Methods Mol Biol 1431, 275–287. 10.1007/978-1-4939-3631-1_19. [DOI] [PubMed] [Google Scholar]
- 52.Hinz AJ, Larson DE, Smith CS, and Brun YV (2003). The Caulobacter crescentus polar organelle development protein PodJ is differentially localized and is required for polar targeting of the PleC development regulator. Mol Microbiol 47, 929–941. 10.1046/j.1365-2958.2003.03349.x. [DOI] [PubMed] [Google Scholar]
- 53.Fellay R, Frey J, and Krisch H (1987). Interposon mutagenesis of soil and water bacteria: a family of DNA fragments designed for in vitro insertional mutagenesis of gram-negative bacteria. Gene 52, 147–154. 10.1016/0378-1119(87)90041-2. [DOI] [PubMed] [Google Scholar]
- 54.Wang X, Montero Llopis P, and Rudner DZ (2014). Bacillus subtilis chromosome organization oscillates between two distinct patterns. Proc Natl Acad Sci U S A 111, 12877–12882. 10.1073/pnas.1407461111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang X, Tang OW, Riley EP, and Rudner DZ (2014). The SMC condensin complex is required for origin segregation in Bacillus subtilis. Curr Biol 24, 287–292. 10.1016/j.cub.2013.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Madabhushi R, and Marians KJ (2009). Actin homolog MreB affects chromosome segregation by regulating topoisomerase IV in Escherichia coli. Mol Cell 33, 171–180. 10.1016/j.molcel.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Simon RP, U; Pühler, Alfred (1983). A Broad Host Range Mobilization System for In Vivo Genetic Engineering: Transposon Mutagenesis in Gram Negative Bacteria. Nature Biotechnology 1, pages 784–791. 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 58.Barton IS, Platt TG, Rusch DB, and Fuqua C (2019). Destabilization of the Tumor-Inducing Plasmid from an Octopine-Type Agrobacterium tumefaciens Lineage Drives a Large Deletion in the Co-resident At Megaplasmid. G3 (Bethesda) 9, 3489–3500. 10.1534/g3.119.400554. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Unprocessed microscopy images are available at Mendeley data: https://doi.org/10.17632/crv96srkp4.1. Hi-C, ChIP-seq and WGS data were deposited to the NCBI Sequence Read Archive (accession no. PRJNA824072). This paper does not report original code. Any additional information required to analyze the data reported in this paper is available from the Lead Contact upon request without restriction.