

Abstract
The success of chimeric antigen receptor T cell (CAR-T) therapy in the treatment of hematologic malignancies has prompted the development of numerous CAR-T technologies, including switchable CAR-T (sCAR-T) systems that combine a universal CAR-T with bispecific adapter proteins. Owing to their controllability and versatility, sCAR-Ts have received considerable attention. To explore the therapeutic utility of sCAR-Ts targeting the receptor tyrosine kinase ROR1, which is expressed in hematologic and solid malignancies, and to identify bispecific adaptor proteins that efficiently mediate universal CAR-T engagement, a panel of switches based on ROR1-targeting Fabs with different epitopes and affinities was compared in in vitro and in vivo models of ROR1-expressing cancers. For switches targeting overlapping or identical epitopes, potency correlated with affinity. Surprisingly, however, we identified a switch targeting a unique epitope with low affinity but mediating potent and selective antitumor activity in vitro and in vivo. Converted to a conventional CAR-T, the same anti-ROR1 mAb (324) outperformed a clinically investigated conventional CAR-T that is based on an anti-ROR1 mAb (R12) with ~200-fold higher affinity. Thus, demonstrating therapeutic utility on their own, sCAR-Ts also facilitate higher throughput screening for the identification of conventional CAR-T candidates for preclinical and clinical studies.
Introduction
Chimeric antigen receptor T (CAR-T) cell therapy has emerged as a promising strategy for cancer therapy [1,2,3,4,5]. In addition to five Food and Drug Administration (FDA)-approved CAR-Ts on the U.S. market targeting CD19 and BCMA for the treatment of hematologic malignancies, approximately 600 clinical trials are currently underway that target over 25 different cell surface glycoproteins and proteoglycans in all major cancer indications [6, 7]. Unlike most of these targets, the receptor tyrosine kinase ROR1 is frequently and restrictively expressed in both hematologic and solid malignancies, making it a highly attractive target for CAR-T cell therapy [8, 9]. A phase I clinical trial (ClinicalTrials.gov Identifier: NCT02706392) is ongoing to assess the safety and potency of ROR1-targeting CAR-Ts, which are based on rabbit monoclonal antibody (mAb) R12 [10–13], in leukemia, lymphoma, and carcinoma.
Accumulating evidence has demonstrated that ROR1 plays important roles in cancer development by activating cell signaling events, particularly the non-canonical WNT signaling pathway, to promote cell proliferation, survival and migration [14]. Studies have shown that high expression of ROR1 on tumor cells correlated well with poor overall survival of cancer patients [15]. As an oncofetal antigen, ROR1 is expressed mainly in embryonic tissues and essential for embryogenesis. Undetectable in adult vital organs and most healthy tissues, ROR1 generally has been considered a safe target for cancer therapeutics [16]. Using conventional CAR-Ts based on R12, which recognizes human and macaque ROR1, both preclinical activity and safety of ROR1-targeting CAR-Ts was shown in rodents and non-human primates, despite low expression levels in some healthy tissues [8, 11–13, 17]. However, conventional CAR-Ts based on rabbit mAb R11 targeting the membrane-proximal kringle domain of ROR1 which is fully conserved between human and mouse were found able to induce lethal bone marrow failure in mice. This was due to significant upregulation of mouse ROR1 expression on stromal cells in bone marrow and spleen upon preconditioning with sublethal radiation or chemotherapy, raising needs to develop novel strategies to control the activity of CAR-Ts targeting ROR1 [18, 19]. Although SynNotch CAR-Ts targeting EpCAM and ROR1 or B7-H3 and ROR1 in an AND gate fashion have been developed and demonstrated the ability to rescue mice when tumor cells were segregated from bone marrow, these Boolean logic-gated CAR-Ts failed to prevent cytotoxicity if tumor cells were co-localized with normal cells in bone marrow. Thus, the treatment of ROR1-positive hematologic and solid malignancies may require systematically and precisely controllable CAR-Ts to provide a safe and effective therapy.
Strategies to improve the safety of CAR-Ts [20, 21] have included the use of, for example, (i) inducible caspase-9 to trigger apoptosis of CAR-Ts in the presence of small molecules [22]; (ii) small molecules to activate CAR-Ts upon dimerization of split CARs [23]; or (iii) SUPRA zipCAR to control the activity of CAR-Ts [24]. However, these approaches are limited by either the irreversible elimination of therapeutic CAR-Ts, fixed epitope/antigen specificity, immunogenicity of the CARs, or a less confined biodistribution and shorter circulatory half-life of the small molecules. Addressing these shortcomings, a platform that provides precise control of the activation of switchable CAR-Ts (sCAR-Ts) with more versatile epitope/antigen specificity and limited immunogenicity was developed and shown to be effective for targeting CD19, HER2, and FOLR1 [25–30]. In this system, sCAR-Ts express mAb 52SR4 in scFv format as universal CAR. Mouse mAb 52SR4 was evolved by ribosome display for picomolar affinity to a 14-amino acid (aa) peptide of yeast transcription factor GCN4 [31]. Recombinantly fused to Fabs that target tumor cells, the GCN4 peptide-tagged Fabs constitute bispecific adapter proteins that link the universal CAR on T cells to a specific antigen on tumor cells. As such, they serve as a switch to control sCAR-Ts’ activity and specificity. In the current study, we applied the sCAR-T platform to target ROR1 in hematologic and solid malignancies. ROR1 is a type I single-pass transmembrane protein with an extracellular domain (ECD) that consists of ~375 aa folding into immunoglobulin (Ig), frizzled (Fz), and kringle (Kr) domains and displaying numerous epitopes. Considering the importance of the affinity and the location of the epitope to the formation of cytolytic synapses between T cells and tumor cells [11, 32–36], we included a panel of rabbit anti-human ROR1 mAbs previously generated from immune and naïve rabbit antibody repertoires with different affinities and epitopes on the ROR1 ECD [10, 37], aiming to identify the best candidate for potent and controllable sCAR-Ts.
Results
Design, generation, and validation of ROR1-targeting sCAR-Ts
Our sCAR-T system was built on an established platform that uses a Fab with a 14-aa tag (NYHLENEVARLKKL, derived from yeast transcription factor GCN4) to control a universal CAR-T [25, 26, 29, 30]. Unlike conventional CAR-Ts which recognize tumor antigens directly, the generic and inert CAR of this sCAR-T system is based on mAb 52SR4 in scFv format that binds specifically to the GCN4 tag instead. Equipping a Fab targeting a tumor antigen with the GCN4 tag affords a bispecific adaptor protein that bridges effector and target cells and as such serves as a switch to turn on-tumor cell killing by the sCAR-T (Fig. 1A and B). In this study, a panel of anti-human ROR1 Fabs with different affinities (Kd ranging from 0.56 nM to ~110 nM), different epitopes (distal and proximal to the cell membrane), and with or without cross-reactivity to mouse ROR1 were chosen to build a diverse assortment of switches (Fig. 1C). All Fabs were derived from naïve and immune rabbit antibody libraries by phage display and described previously [10, 37]. Considering the dominant contribution of most heavy chains to antigen binding, we elected to recombinantly fuse the GCN4 tag to the light chain, either at the N- or C-terminus (Fab-N or Fab-C). After transient co-transfection of light and heavy chain plasmids into Expi293F cells, 20 switches including two negative controls (10 Fab-N and 10 Fab-C) were purified by using CaptureSelect CH1-XL Affinity Matrix beads. High yields ranging from 20-100 mg/L were obtained. SDS-PAGE under reducing and nonreducing conditions revealed the expected bands (Suppl. Fig. S1A), except for both Fab-N and Fab-C formats of clone R11 which revealed a ~5-kDa shift to higher molecular weight of either light or heavy chain. Analysis of the aa sequence uncovered two putative N-glycosylation sites, one (NLT) in framework 1 of VH (HFR1) and the other (NVS) in the third complementarity-determining region (LCDR3) of VL. Mutation experiments confirmed the N-glycosylation site in HFR1 but not LCDR3 (data not shown). The N-glycosylated R11 Fab was found to have retained its ability to bind both human and mouse ROR1 in an ELISA that confirmed the binding of all 18 switches but not the 2 negative controls (Fig. 1D).
Fig. 1. A ROR1-targeting sCAR-T system.
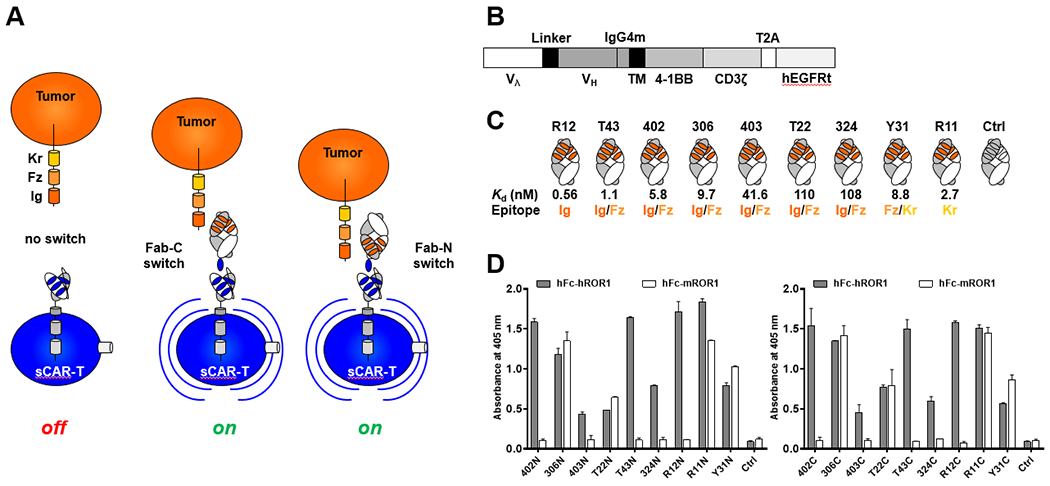
(A) Depiction of tumor cell (orange) expressing ROR1. The ectodomain of ROR1 consists of immunoglobulin (Ig), frizzled (Fz), and kringle (Kr) domains. The universal CAR of sCAR-Ts (blue) binds with high affinity to a GCN4 peptide (NYHLENEVARLKKL, also shown in blue) appended to the C-terminus (Fab-C) or N-terminus (Fab-N) of the light chain of a ROR1-targeting Fab that serves as switch for tumor cell engagement. In the presence of the switch, sCAR-Ts are turned on to kill tumor cells. (B) Lentiviral universal CAR cassette encompassing GCN4 peptide-binding scFv 52SR4 fused to a mutant IgG4 hinge domain and CD28 transmembrane domain, followed by 4-1BB and CD3ζ cytoplasmic signaling domains. Truncated human EGFR (hEGFRt) enables the detection of CAR expression on T cells and purification of CAR-positive T cells by MACS. (C) Nine switch candidates of ROR1-targeting Fabs with various epitopes and affinities. (Note, the epitopes of Fabs R12, T43, 402, 306, 403, and T22 overlap; Fabs 324, Y31, and R11 have unique epitopes that do not overlap with epitopes of other Fabs). (D) ROR1 binding of Fabs equipped with a GCN4 peptide at the N- (left) or C-terminus (right) was detected by ELISA.
The encoding sequence of the universal CAR controlled by a human EF-1α promoter in lentivirus vector epHIV7 [38], shown in (Fig. 1B), contained an expression cassette of scFv 52SR4 in Vλ-(G4S)3-VH format and a mutated human IgG4 hinge sequence (12 aa with S108P substitution), followed by the CD28 transmembrane segment and the cytoplasmic signaling domains of 4-1BB and CD3ζ [11]. A truncated human epidermal growth factor receptor (hEGFRt) encoding sequence linked by a T2A ribosomal skip element was introduced to facilitate measuring transduction rate and enrichment of CAR-expressing T cells by magnetic activated cell sorting (MACS) [39]. After spinofection and MACS-purification, >90% of human primary T cells were CAR-positive and recognized the GCN4 tag on all switches as determined by flow cytometry. (Fig. 2A and C). In addition, we used flow cytometry to analyze the binding of the switches to cell surface ROR1. This study included a stable K562 cell line ectopically expressing ROR1 (K562/ROR1) along with mantle cell lymphoma cell line JeKo-1 and triple negative breast cancer (TNBC) cell line MDA-MB-231, both expressing ROR1 endogenously (Fig. 2B). With one exception (clone Y31), all ROR1-targeting switches bound to K562/ROR1 but not K562 cells, and we observed an approximate correlation of mean fluorescence intensity (MFI) and affinity. However, both Fab-N and Fab-C switches of clone R11 showed much weaker binding to cell surface ROR1 by flow cytometry (Fig. 2C) compared to purified ROR1 by ELISA (Fig. 1C). This is consistent with previous data [10] we attributed to diminished accessibility of the R11 epitope due to its membrane-proximal location in the kringle domain of ROR1 [40]. Similar distinctive binding of the switches to ROR1-positive cell lines JeKo-1 and MDA-MB-231 but not ROR1-negative cell line Raji was observed (Suppl. Fig. S1B).
Fig. 2. Orthogonal binding of anti-ROR1 Fab switches to effector cells and target cells.
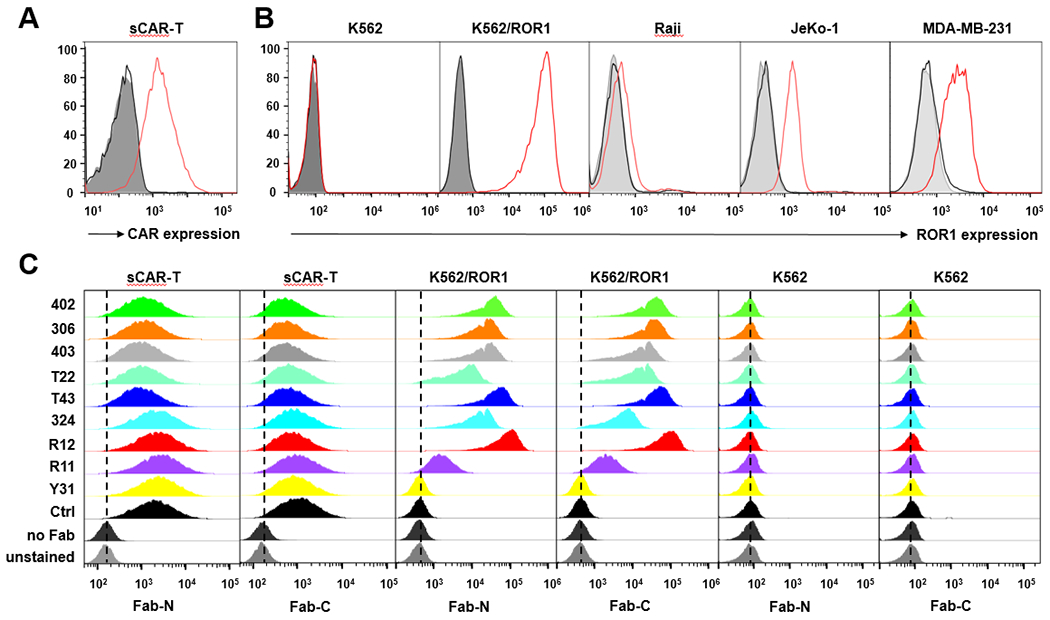
(A) Lentiviral CAR transduction on MACS-purified human primary T cells was determined by flow cytometry using cetuximab (5 μg/mL) conjugated to Alexa Fluor 647; (B) The expression level of ROR1 on different target cells was compared by flow cytometry using R12 Fab-N (10 μg/mL) followed by R-Phycoerythrin-conjugated goat F(ab’)2 anti-human IgG, F(ab’)2 fragment pAbs (K562 and K562/ROR1) or Alexa Fluor 647-conjugated 52SR4 scFv-Fc (Raji, JeKo-1, and MDA-MB-231). (C) The binding of anti-ROR1 Fab switches (5 μg/mL) to sCAR-T cells and target cells was determined by flow cytometry using R-Phycoerythrin-conjugated goat F(ab’)2 anti-human IgG, F(ab’)2 fragment pAbs (y axis: normalized count; x axis: fluorescence).
Switch-mediated in vitro activation of sCAR-Ts against ROR1-expressing hematologic malignancies
We next tested the capability of the 20 switches for mediating sCAR-T activation in vitro. Specific lysis of ROR1-expressing hematologic cancer cells after 20-h incubation with concentrations ranging from 10 fM to 10 nM for K562/ROR1 and from 64 fM to 5 nM for JeKo-1 cells at an effector-to-target ratio of 10:1 was assessed. K562/ROR1 and JeKo-1 cells, but not K562 cells or Raji cells, were killed in the presence of anti-ROR1 but not negative control Fab switches, indicating that target cell lysis was strictly dependent on ROR1 (Fig. 3A–D). Consistent with the finding that the anti-ROR1 Fab switches based on clone Y31 did not bind to cell surface ROR1, they were not able to mediate sCAR-T activation (Fig. 3A and C). Interestingly, all Fab-N switches revealed higher activity than the corresponding Fab-C switches in terms of both EC50 values and maximum killing capacity. This was more obvious against JeKo-1 cells, which express lower levels of ROR1 compared to K562/ROR1 cells. The top 3 switches in both Fab-N and Fab-C format that emerged from these experiments were clones R12 ≥ T43 > 402. We previously showed that R12 has an epitope in Ig domain that overlaps with the epitopes of T43 and 402 in Ig/Fz domain, which all bind ROR1 with the highest affinities (Fig. 1B). By contrast, clones 306, 403, and T22, which also have overlapping epitopes with T43 and 402 in the Ig/Fz domain but bind ROR1 with lower affinities, were significantly less potent. To further investigate the contribution of the switches’ affinity to the potency of sCAR-Ts, we generated a pair of affinity variants of clone R12 that bind ROR1 with either higher or lower affinity than parental clone R12 (Suppl. Fig. S2). The EC50 values of R12_20, R12_WT, R12_16 were 1.9 pM, 5.4 pM, 104.4 pM against K562/ROR1 cells, respectively, and 6.0 pM, 24.4 pM, 367.3 pM against JeKo-1 cells, respectively, correlating well with their affinities of 0.46 nM, 1.21 nM, and 80.8 nM. We previously showed that clone 324 has a unique epitope in the Ig/Fz domain that does not overlap with any of the other epitopes. Notably, despite its low affinity (Kd = 108 nM), which is comparable to 403 and T22, both 324 switches were significantly and consistently more potent than the corresponding 403 and T22 switches, demonstrating that the epitope of the switch was also critical for controlling the activity of sCAR-Ts. Although R11 switches showed much weaker binding to target cells, they still demonstrated strong activation of sCAR-Ts, especially R11N. We hypothesize that this was caused by the cell membrane proximal location of the epitope of R11 which may enable tighter cytolytic synapse formation between sCAR-T and target cell. Recently, Nerreter et. al. [41] revealed that as low as ~100 copies of a cell surface antigen can mediate strong cytotoxicity of CAR-Ts. Considering this, R11N, despite the reduced accessibility of its epitope, might engage enough copies of ROR1 to interact with the sCAR on T cells and trigger potent target cell killing. Given these unique features of switches 324N and R11N, we tried their systematic combination with each other and with other switches to assess additive or synergistic effects. However, none of the combinations showed an EC50 improvement compared to its most potent switch alone (Suppl. Fig. S3).
Fig. 3. Switch-mediated in vitro cytotoxicity of sCAR-Ts.
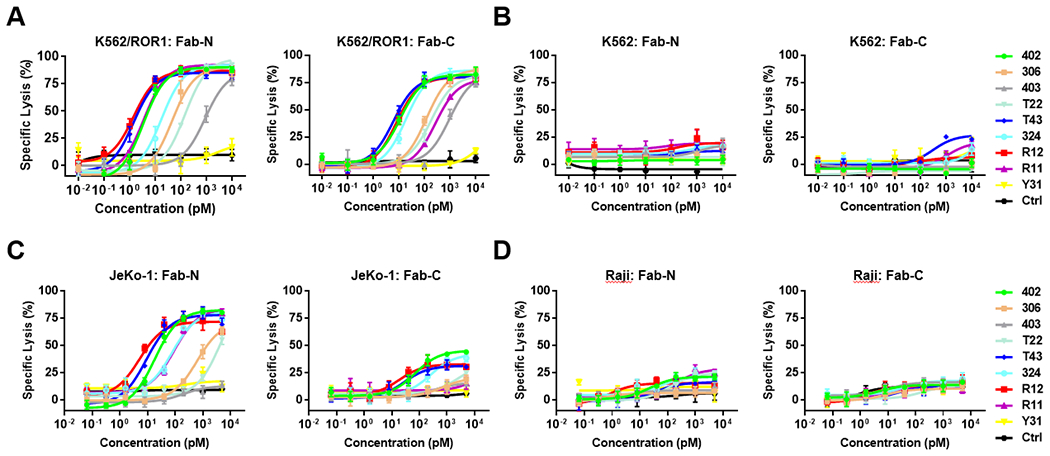
Using a concentration range of 10 fM to 10 nM of anti-ROR1 Fab-N and Fab-C switches, the cytotoxicity of sCAR-Ts against ROR1-positive K562/ROR1 (A) and JeKo-1 (C) cells, and ROR1-negative K562 (B) and Raji (D) cells was determined at an E:T ratio of 10:1 and measured after 20-h incubation.
Next, we examined the switches for inducing sCAR-T activation by determining the expression of T-cell activation marker CD25 and the release of type 1 cytokines, including TNFα, IL2, and IFNγ (Fig. 4). Analysis by flow cytometry showed that CD25 was upregulated after 20-h incubation with 100 pM switches in the presence of K562/ROR1 but not K562 cells (Fig. 4A and C). Consistent with the cytotoxicity of the switches, R12 ≥ T43 > 402 were the top 3 ranked switches, followed by 324 and R11, in terms of the ability to induce CD25 expression on sCAR-T. As analyzed by ELISA, type 1 cytokine release was also strictly dependent on the presence of switch and target (Fig. 4B and D). Interestingly, whereas switches R12, T43, and 402 along with 324 induced high secretion of IL2 and IFNγ, R11N induced significantly higher secretion of TNFα (Fig. 4B).
Fig. 4. Switch-mediated in vitro activation of sCAR-Ts.
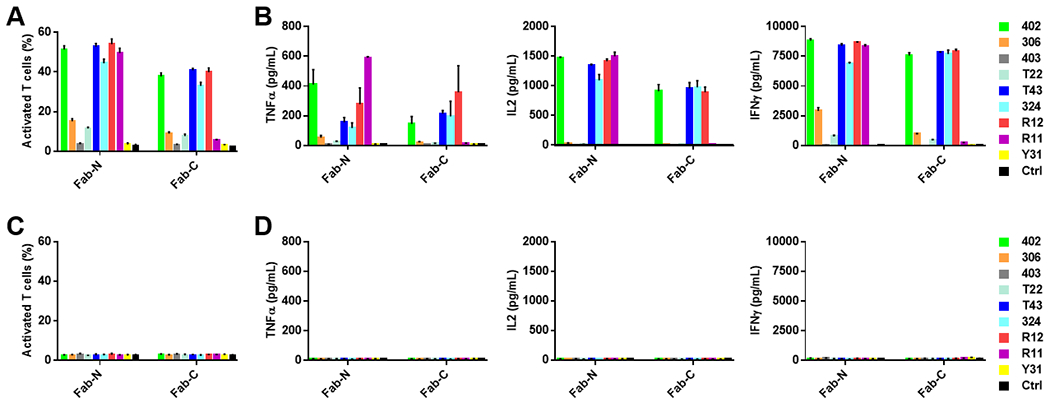
Using 100 pM of anti-ROR1 Fab-N and Fab-C switches, the activation of sCAR-Ts in the presence of K562/ROR1 (A and B) and K562 (C and D) cells was determined at an E:T ratio of 10:1 and measured after 20-h incubation. In (A) and (C), the percentage of activated sCAR-Ts (CD3-FITC+ plus CD25-APC+) was detected by flow cytometry. In (B) and (D), cytokines TNFα, IL2, and IFNγ released from sCAR-Ts were measured by ELISA. All data are represented as mean ± SD.
Switch-mediated in vitro activation of sCAR-Ts against ROR1-expressing solid malignancies
In addition to subsets of B-cell leukemia and lymphoma, ROR1 is also expressed in certain carcinomas including TNBC. Thus, we tested the broader therapeutic utility of the switches to activate sCAR-Ts using TNBC cell line MDA-MB-231 as ROR1-expressing target cells. As before, we found that Fab-N switches were consistently more potent than Fab-C switches in a dose-dependent manner in the cytotoxicity assay (Fig. 5A). The high affinity switches R12, T43, and 402 were the most potent for activating sCAR-Ts and killing MDA-MB-231 cells. Interestingly, switch 324C outperformed the other Fab-C switches in terms of mediating maximum killing of MDA-MB-231 cells although with a significantly higher EC50 value than switches R12, T43, and 402 (Table 1) (Fig. 5A). The compilation of all EC50 values in Table 1 revealed that the switches fell into similar orders of potency in terms of turning on sCAR-Ts to kill different target cells. sCAR-T activation in the presence of MDA-MB-231 cells and switches was determined as before by CD25 expression and type 1 cytokine secretion. Again, Fab-N induced significantly more expression of CD25 on sCAR-Ts compared to Fab-C, with switches R12, T43, 402, and 324 being the most potent (Fig. 5B). Cytokine secretion was highly dependent on the presence of the ROR1 switches (Fig. 5C). While Fab-N switches induced elevated levels of cytokines, Fab-C switches were much weaker and control switches did not trigger any cytokine secretion from sCAR-Ts. Consistently, R12, T43, 402, and 324 were the most potent for switching on sCAR-Ts.
Fig. 5. Switch-mediated in vitro cytotoxicity and activation of sCAR-Ts against TNBC cells.
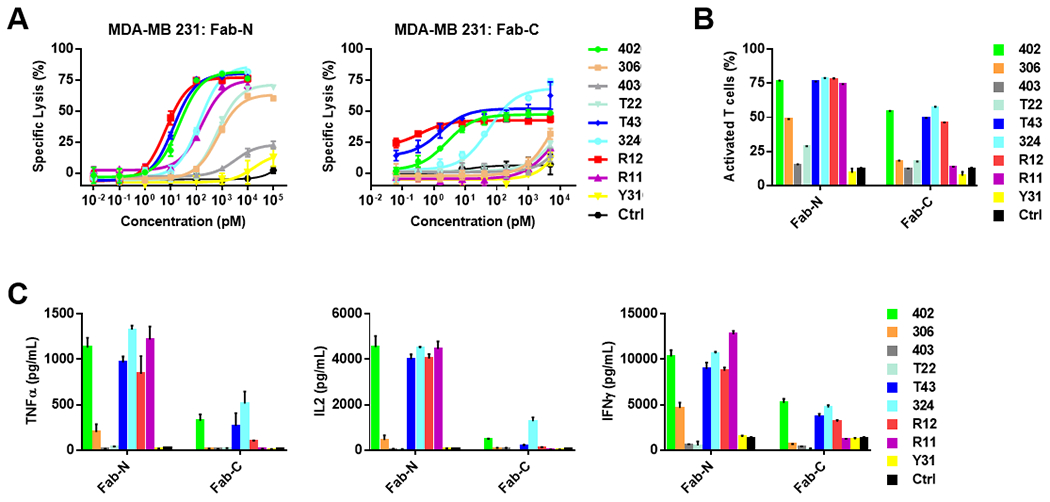
(A) Using a concentration range of 10 fM to 10 nM of anti-ROR1 Fab-N and Fab-C switches, the cytotoxicity of sCAR-Ts against ROR1-positive TNBC cell line MDA-MB-231 was determined at an E:T ratio of 10:1 and measured after 20-h incubation. Using 1 nM of anti-ROR1 Fab-N and Fab-C switches, the activation of sCAR-Ts in the presence of MDA-MB-231 cells was determined at an E:T ratio of 10:1 and measured after 20-h incubation. The percentage of activated sCAR-Ts (CD3-FITC+ plus CD25-APC+) was detected by flow cytometry (B) and cytokines TNFα, IL2, and IFNγ released from sCAR-Ts were measured by ELISA (C). All data are represented as mean ± SD.
Table 1.
Summary of EC50 values determined in switch-mediated in vitro cytotoxicity assays with sCAR-Ts.
| EC50 (pM) | R12 | T43 | 402 | 306 | 403 | T22 | 324 | Y31 | R11 | Ctrl | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| K562/ROR1 | Fab-N | 1.4 | 1.4 | 3.5 | 46 | 890 | 150 | 18 | - | 3.7 | - |
| Fab-C | 8.1 | 6.1 | 11 | 120 | 950 | 190 | 19 | - | 260 | - | |
| JeKo-1 | Fab-N | 5.2 | 9.2 | 18 | 620 | - | 4700 | 77 | - | 91 | - |
| Fab-C | 18 | 28 | 71 | - | - | - | 260 | - | - | - | |
| MDA-MB-231 | Fab-N | 6.5 | 13 | 21 | 700 | - | 660 | 130 | - | 160 | - |
| Fab-C | 0.38 | 1.5 | 2.6 | - | - | - | 56 | - | - | - |
Switch 324N-mediated in vitro activation of sCAR-Ts in presence of human PBMC
Modeling a more natural environment that includes the presence of human lymphoid and myeloid cells, we conducted additional in vitro experiments to test the potency and specificity of switch 324N in the presence and absence of human peripheral blood mononuclear cells (PBMC) from three different healthy donors. Using K562/ROR1 target cells, 324N revealed higher potency in the presence of human PBMC (Suppl. Fig. S4B), at least some of which can be attributed to sCAR-T-independent background killing of K562/ROR1 cells, likely mediated by NK cells (Suppl. Fig. S4C). Cytokine release was strictly dependent on the presence of the switch and was not significantly different in the presence or absence of human PBMC with respect to TNFα, IL2, and IFNγ (Suppl. Fig. S4A). However, significantly higher levels of IL6 were detected in the presence of human PBMC (Suppl. Fig. S4A), presumably due to the presence of monocytes. Background killing of K562/ROR1 cells by human PBMC, likely mediated by NK cells, Notably, 324N did not significantly change the percentage of healthy B cells, T cells, NK cells, and monocytes, regardless of whether ROR1+ target cells were present (Suppl. Fig. S4D) or not (Suppl. Fig. S4E). The gating strategy for the lymphoid and myeloid compartments of human PBMC is shown in Suppl. Fig. S5. These data demonstrate the high specificity of ROR1-targeting sCAR-Ts for ROR1-positive cancer cells in the presence of ROR1-negative lymphoid and myeloid cells.
Switch-mediated in vivo activity of sCAR-Ts in a xenograft mouse model of a ROR1-expressing hematologic malignancy
To investigate whether the in vitro sCAR-T activation controlled by ROR1-targeting Fab-N switches would translate into in vivo activity, we evaluated tumor growth of JeKo-1 cells stably transfected with firefly luciferase (JeKo-1_ffluc) and xenografted systemically in immunodeficient NOD scid gamma (NSG) mice (Fig. 6A). For this, 0.5 × 106 JeKo-1_ffluc cells in 100 μL PBS were injected intravenously (i.v.; tail vein) into six cohorts of five NSG mice per cohort. On day 6 when the tumor was disseminated, 5 × 106 MACS-purified and expanded sCAR-Ts in 100 μL PBS were injected i.v. (tail vein) into mice. Six hours later, vehicle (PBS) alone (cohort 2), or 1 μg of switches 324N (cohort 3), 402N (cohort 4), and R12N (cohort 5) in 100 μL PBS were administered intraperitoneally (i.p.). The dosage was then increased gradually to 3 μg and 10 μg on days 7 and 8, respectively, and kept at 10 μg thereafter on days 10, 12, and 14 to avoid cytokine release syndrome-like toxicity [42]. Starting on day 16, another round of treatment was given. In cohort 6, 5 × 106 R12 CAR-Ts, a conventional ROR1-targeting CAR-T therapy in a phase I clinical trial [11–13], were injected i.v. (tail vein) as positive control. To assess the response to treatment, in vivo bioluminescence imaging was performed before the first dose and then every 4-6 days (Fig. 6B) until day 27 when the signals were saturated in the negative control cohorts. Control cohorts 1 (no sCAR-Ts, no switches) and 2 (no switches) revealed aggressive tumor growth (Fig. 6C). Positive control R12 CAR-Ts in cohort 6 efficiently eradicated tumor cells. In cohorts 3, 4, and 5, which received switches 324N, 402N, and R12N, respectively, we observed significant tumor growth retardation on day 13 after one dose of sCAR-Ts and five doses of switches but relapse on day 17 when the second round of treatment just started, indicating a shortage of effector cells and the requirement to supply more sCAR-Ts. This was confirmed when we saw a second tumor growth retardation on day 21. Note that similar studies used substantially more sCAR-Ts (up to 5 × 107) in single dose treatment [25, 26, 29], which was not acceptable for JeKo-1 cells as the allogeneic T cells on their own were found to kill JeKo-1 cells (cohort 2 vs cohort 1, Fig. 6B and C), and high titers of sCAR-Ts were enough to clear the tumor cells in vivo (data not shown). Interestingly, switch 324N was more potent in vivo compared to 402N and R12N (Fig. 6C and D), which demonstrated higher efficiency to activate sCAR-Ts in vitro. Pharmacokinetic (PK) studies revealed that all three switches had similar circulatory half-lives, with t1/2 values around 9 h (324N, 8.89 ± 0.32 h; 402, 9.22 ± 0.54 h; and R12N, 9.40 ± 0.77 h) (Suppl. Fig. S6). Adding the switches to the sCAR-Ts did not induce obvious toxicity in the mice, with no significant changes in body weights during treatment (Fig. 6E).
Fig. 6. Switch-mediated in vivo cytotoxicity of sCAR-Ts in an NSG/JeKo-1_ffluc xenograft mouse model.
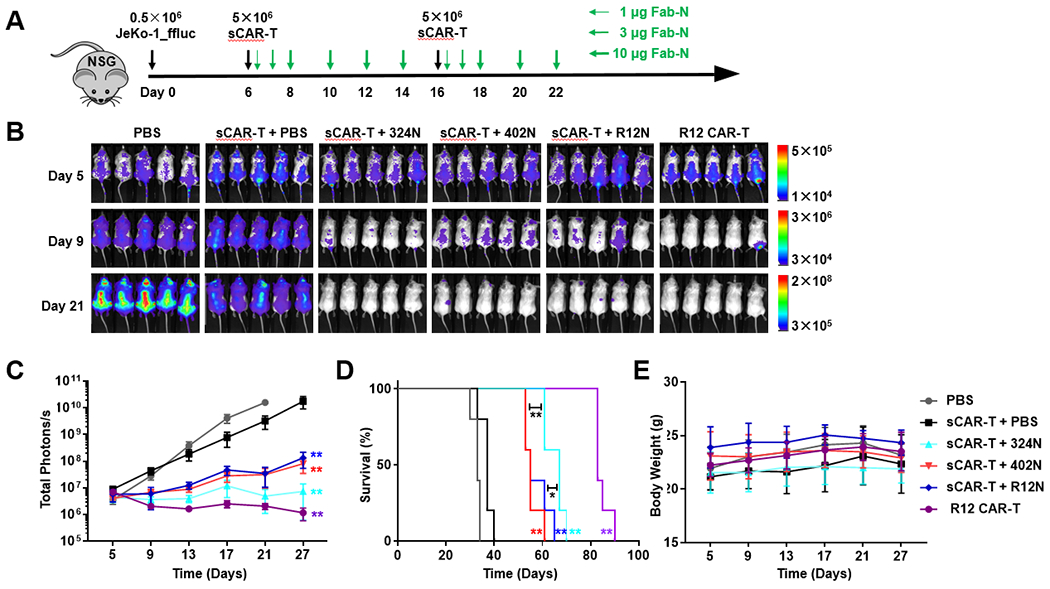
(A) Six cohorts of NSG mice (n = 5) were inoculated with 0.5 × 106 JeKo-1_ffluc cells via i.v. (tail vein) injection and subsequently treated with or without i.v. (tail vein) administered 5 × 106 sCAR-Ts and with or without i.p. administered 1, 3, or 10 μg Fab-N switches at the indicated time points. A conventional R12 CAR-T was included as positive control. (B) Representative bioluminescence images of all 30 mice taken on day 5 (before treatment) and days 9 and 21 (after treatment). (C) The radiance of bioluminescence was recorded on days 5, 9, 13, 17, 21, and 27 and plotted as mean ± SD. Significant differences between cohorts treated with sCAR-T plus Fab-N switches and sCAR-T only were calculated using an unpaired two-tailed t-test (**, p < 0.01). (D) Corresponding Kaplan-Meier survival curves with p-values using a log-rank (Mantel-Cox) test (*, p < 0.05; **, p < 0.01). (E) The body weight of all 30 mice was recorded on the indicated days as mean ± SD.
An additional in vivo experiment was conducted to demonstrate the retained potency and safety of sCAR-Ts in the presence of healthy human PBMC. In this humanized mouse model of mantle cell lymphoma (NSG/JeKo-1 + human PBMC; Suppl. Fig. S7A), the sCAR-T with switch 324N was as potent as the conventional 324-based CAR-T (Suppl. Fig. S7B and S7D) with similar IFNγ levels measured in the serum of treated mice at three different time points (Suppl. Fig. S7C). No weight loss (Suppl. Fig. S7E) or other observable adverse events were seen.
Switch-mediated in vivo activity of sCAR-Ts in a xenograft mouse model of a ROR1-expressing solid malignancy
We next investigated the potential of ROR1-targeting Fab-N switches to activate sCAR-Ts to kill solid tumor cells in vivo. Considering the high expression of PD-L1 on MDA-MB-231 cells (Suppl. Fig. S8A) which is likely to mitigate the activity of sCAR-Ts in vivo, we elected to use HT29, a colon cancer cell line with high ROR1 (Fig. 7A) but low PD-L1 expression (Suppl. Fig. S8B). Before carrying out in vivo studies, we first evaluated the ability of R12N, 402N, and 324N to switch on sCAR-Ts to kill HT29 cells in vitro. We observed comparable results as before, with strong activation of sCAR-Ts by all three Fab-N switches and dose-dependent killing of HT29 cells with picomolar EC50 values (Fig. 7B). For comparison, we also tested conventional R12 and 324 CAR-Ts and found that both had very similar cytotoxicity against HT29 cells (Fig. 7C). For the in vivo study, we first introduced ffluc into HT29 cells. To establish an NSG/HT-29_ffluc xenograft mouse model, 0.5 × 106 HT29_ffluc cells in 100 μL PBS were injected i.p. into six cohorts of five NSG mice per cohort. On day 3, 1 × 107 MACS-purified and expanded sCAR-Ts in 100 μL PBS were injected i.p. into mice. Six hours later, vehicle (PBS) alone (cohort 2), or 1 μg of switches 324N (cohort 3) and R12N (cohort 4) in 100 μL PBS were administered i.v. (tail vein). The dosage was then increased gradually to 3 μg and 10 μg on days 4 and 5, respectively, and kept at 10 μg thereafter on days 7, 9, and 11. Only one round of treatment was given. For cohorts 5 and 6, 2 × 106 conventional 324 and R12 CAR-Ts, respectively, were injected i.p. as positive controls. Bioluminescence imaging revealed aggressive tumor growth in control cohorts 1 (no sCAR-Ts, no switches) and 2 (no switches) (Suppl. Fig. S9 and Fig. 7D). In cohort 4, which received R12N, we did not observe significant inhibition of tumor growth compared to cohort 2. In contrast, tumor growth was significantly impeded with 324N in cohort 3. Interestingly, among the two conventional CAR-Ts, 324 also outperformed R12 in terms of impeding tumor growth and prolonging survival (Fig. 7D and E). Again, no significant changes in body weights were detected across all treatments (Fig. 7F).
Fig. 7. In vitro and in vivo activity of sCAR-Ts targeting colon cancer cells.
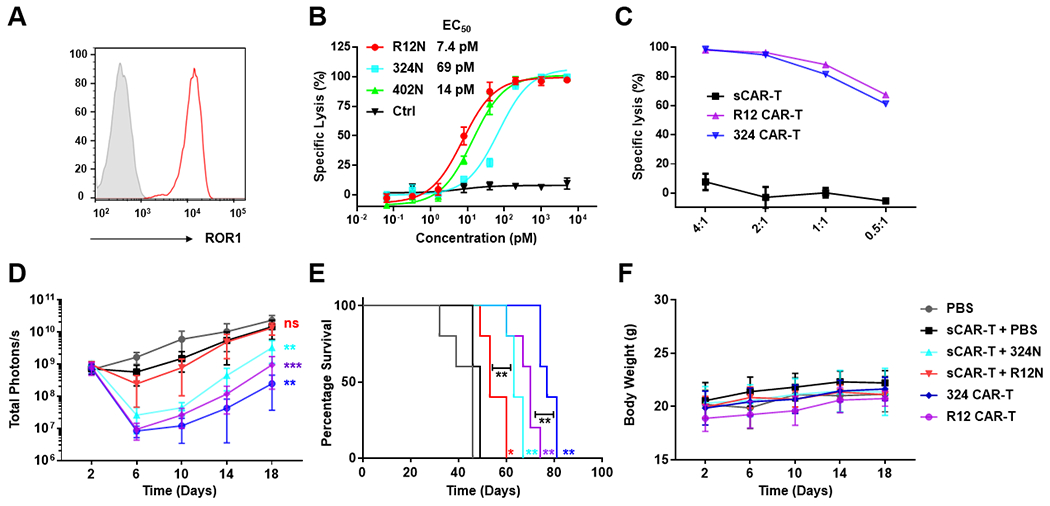
(A) Expression of ROR1 on colon cancer cell line HT29 detected by 402 IgG1 conjugated to Alexa Fluor 647 (y axis: normalized count; x axis: fluorescence). (B) Using a concentration range of the indicated anti-ROR1 Fab-N switches, the cytotoxicity of sCAR-Ts against HT29 cells was determined at an E:T ratio of 10:1 and measured after 16-h incubation. (C) Using the indicated E:T ratios, the cytotoxicity of conventional CAR-Ts against HT29 cells was determined after 16-h incubation; sCAR-Ts in the absence of switches were used as negative control. (D) Six cohorts of NSG mice (n = 5) were inoculated with 0.5 × 106 HT29_ffluc cells via i.p. injection and subsequently treated with or without i.p. administered 1 × 107 sCAR-Ts or 2 × 106 conventional 324 and R12 CAR-Ts on day 3. For the sCAR-T-treated mice this was followed by i.v. (tail vein) injection of 1 μg (day 3), 3 μg (day 4), and 10 μg (days 5, 7, 9, and 11) of 324N or R12N. Injection of vehicle (PBS) alone was included as negative control. The radiance of bioluminescence was recorded on the indicated days and plotted as mean ± SD. Significant differences between cohorts treated with sCAR-T plus Fab-N switches and sCAR-T only were calculated using an unpaired two-tailed t-test (**, p < 0.01; ***, p < 0.001; ns, not significant). (E) Corresponding Kaplan-Meier survival curves with p-values using a log-rank (Mantel-Cox) test (*, p < 0.05; **, p < 0.01). (F) The body weight of all 30 mice was recorded on the indicated days (mean ± SD).
Discussion
Currently, the five CAR-Ts approved by the FDA to treat leukemia, lymphoma, and myeloma are targeting only two different antigens, CD19 and BCMA, which are mainly expressed on B cells and plasma cells, respectively, that can be depleted without fatal side effects. The extremely high potency of CAR-Ts strictly limits the candidate antigens that can be targeted. To control the activity and ensure the safety, and also to expand the target range of CAR-Ts, multiple switchable CAR-T platforms have been developed, including Unum’s ACTR (antibody-coupled T cell receptor), the leucine zipper-mediated dimerizing strategy of SUPRA CAR [24], the covalent attachment of targeting ligands in the Spy-Catcher system [43], the IMPACT system in which persistent CD19 CAR-Ts in patients can be reutilized and reactivated by a bifunctional protein such as a fusion of the extracellular domain of CD19 to an anti-CD20 scFv [44], and universal CARs recognizing tagged antibodies [45, 46]. Among the latter, bispecific adapter proteins that fuse a GCN4 peptide, which binds a universal CAR with picomolar affinity, to a tumor-targeting Fab were developed and clinically translated by Calibr at The Scripps Research Institute. Targeting CD19, phase 1 clinical trials (NCT04450069 and NCT04488354) for the treatment of patients with relapsed or refractory (r/r) B-cell malignancies are ongoing. In the current study, we applied this system to targeting ROR1 in both hematologic and solid malignancies. We explored the potential of a diverse panel of GCN4 peptide-tagged Fab switches to control the activity of sCAR-Ts both in vitro and in vivo and found unexpectedly that a switch based on rabbit mAb 324, which binds to a unique epitope in the Ig/Fz domain of ROR1, was highly active in vitro and significantly more potent in two in vivo models compared to other switches binding to different epitopes with higher or similar affinities.
A critical step of T cell-mediated cytotoxicity of tumor cells is the formation of cytolytic synapses between effector cells and target cells, which could be affected by multiple factors, including the antigen density and mobility on tumor cells, the CAR expression level on T cells, the affinity/avidity between antigen and CAR, the concentration of the switch, and the bridged distance between effector cells and target cells [32, 34–36, 47]. There is evidence that increasing affinity above a ceiling or threshold can adversely affect T cell behavior [48, 49], and a low-affinity anti-CD19 CAR (KD = 14 nM), compared with the commonly used FMC63-based anti-CD19 CAR (KD = 0.33 nM) of all four FDA-approved CD19-targeting CAR-Ts enhanced CAR T cell expansion and prolonged persistence in patients with ALL [35]. It is also believed that an optimal space between effector cells and target cells exists [50–53]. Previously, we have shown that conventional ROR1-targeting CARs based on rabbit mAbs R12 and 402 targeting a membrane-distal epitope (Ig/Fz domain) were more potent with a shorter spacer between scFv and transmembrane segment, whereas a longer spacer exerted higher activity of an R11-based CAR targeting a membrane-proximal epitope (Kr domain) [11, 37]. To identify suitable anti-ROR1 mAb candidates for the GCN4 peptide-tagged Fab switches of sCAR-Ts allowing the formation of appropriate cytolytic synapses, we screened a panel of mAbs with diverse epitopes and affinities [10, 37]. In addition, we also fused the GCN4 peptide tag on either N or C terminus of the light chains of the Fabs to further tailor the space between T cells and tumor cells. Interestingly, we found that Fabs with an N-terminal tag were generally more potent compared to those with a C-terminal tag. Among the Fab-N switches with overlapping epitopes in the Ig/Fz domain, T43N, R12N, 402N with the highest affinities mediated the strongest sCAR-T activation in vitro. Fab-N switches such as T22N that engage the same dominant epitope cluster with low affinity were significantly less potent. Notably, 324N, which binds a unique non-overlapping epitope in the Ig/Fz domain, had high potency (EC50 = 18 pM, 77 pM, and 130 pM for K562/ROR1, JeKo-1, and MDA-MB-231 cells, respectively) despite its low affinity (Kd = 108 nM). In contrast, T22N with essentially the same affinity (Kd = 110 nM) was substantially less potent (EC50 = 150 pM, 4700 pM, and 660 pM for K562/ROR1, JeKo-1, and MDA-MB-231 cells, respectively). Variants of 324N with different affinities would be interesting to test. Moreover, 324N showed even stronger potential to control tumor growth in vivo compared with R12N and 402N. As the three switches had similar circulating half-life in mice, the unique epitope of 324N might facilitate the formation and duration of cytolytic synapse between effector cells and target cells. Given its low affinity, optimizing the frequency and dosage of 324N administration will be critical for achieving maximum therapeutic efficacy. Solving the structure of Fab 324 in complex with the Ig/Fz domain by X-ray crystallography or cryo-electron microscopy may help elucidate its unique activity. Interestingly, after conversion to conventional CAR-Ts, we found that the 324 CAR-T also outperformed the R12 CAR-T, indicating that the sCAR-T platform could facilitate higher throughput screening for the identification of conventional CAR-T candidates for preclinical and clinical studies, which could be hampered by variables related to T-cell product composition, virus production, gene transfer rate, and CAR expression level.
Unexpectedly, R11-based Fab switches that target a membrane-proximal epitope in the Kr domain and thereby shorten the space between effector cells and target cells [40], were inferior in terms of potency compared to the high-affinity Fab switches targeting the Ig/Fz domain. This may be due to steric hindrance, as evidenced by their weaker binding of cell surface ROR1 observed by flow cytometry. We had previously noted that R11 in scFv-Fc format reveals stronger staining of cell surface ROR1 compared to R11 in IgG1 format [10]. Furthermore, ROR1 × CD3 bispecific antibodies based on the R11 scFv-Fc format were the most potent T-cell recruiting and activating bispecific antibodies in in vitro and in vivo models of ROR1-expressing cancers [40]. Thus, replacing the 50-kDa Fab with a 25-kDa scFv switch could allow better access to the R11 epitope and as such perform better as sCAR-T switch. In addition to Fab and scFv, other antibody formats or antibody-like scaffolds for ROR1-targeting sCAR-Ts could serve as switches. In general, bispecific adapter proteins have different circulatory half-lives, ranging from less than an hour for small molecules to approximately 200 h for IgG in mice. Smaller sized switches may require frequent dosing or even continuous administration. In contrast, larger sized switches do not need to be administrated frequently but may be more challenged with efficiently and homogeneously penetrating dense tumor tissue. As sCAR-T switches, Fab has an attractive intermediate size with a circulatory half-life of approximately 8 h in mice, permitting once a day (q.d.) dosing that can be tailored in terms of increasing or decreasing dosage and frequency while closely monitoring potency and safety. As such, administration of Fab with flexible dosing regimen could not only control the activity of sCAR-Ts, but also create a “rest” period between dosing cycles to induce memory and expansion of sCAR T cells, thus enhancing memory formation and improving the efficacy and persistence of sCAR-Ts [27].
Due to its highly restricted expression in a wide range of hematologic and solid malignancies, ROR1 has emerged as an attractive target for antibody-based protein and cell therapy [16, 54–56]. Several ROR1-targeting antibody-based therapeutics are at different stages in preclinical investigations and early clinical trials, including naked mAb cirmtuzumab (NCT03088878 and NCT02776917), antibody-drug conjugates NBE-002 (NCT04441099) and zilovertamab vedotin (NCT02222688), T-cell recruiting and activating bispecific antibody NVG-111 (NCT04763083), and a CAR-T based on R12 (NCT02706392). Cirmtuzumab, zilovertamab vedotin, and the conventional R12-based CAR-T were well tolerated in cancer patients [13, 57, 58]. However, on-target toxicity has been observed in mouse models for CAR-Ts cross-reactive for human and mouse ROR1 [19, 59]. A conventional CAR-T based on R11 targeting a membrane-proximal epitope demonstrated lethal bone marrow failure in mice due to induced expression of ROR1 on stromal cells in response to pre-conditioning with radiation, raising concerns of the utilization of conventional CAR-Ts targeting ROR1 [13]. In the same study, it was shown that AND-gate CAR-Ts programmed to express the R11-based CAR only after induction by a synthetic Notch receptor binding to a second antigen expressed on tumor but not stromal cells, could dodge this toxicity. As an alternative to CAR-Ts with Boolean logic gates, sCAR-Ts are a safer option compared to conventional CAR-Ts. Adjusting frequency and dosage of switch administration and having the option to alternate switches of different specificity and affinity could address off-tumor-on-target and off-tumor-off-target toxicity immediately without compromising activity. With ROR1-targeting antibody-based therapeutics expected to enter pivotal clinical trials over the next few years, it is possible that evading mutations in the ROR1 ECD that lead to cancer resistance emerge. The ability to administer different anti-ROR1 Fab switches simultaneously or sequentially could be used to prevent epitope escape. As such, sCAR-Ts constitute a polyclonal assault that mimics the natural adaptive immune system more closely than monoclonal treatments including conventional CAR-Ts.
Materials and methods
Cell lines
K562, JeKo-1, and Raji cell lines were obtained from ATCC and cultured in RPMI-1640 supplemented with L-glutamine, 100 U/mL penicillin/streptomycin, and 10% (v/v) fetal bovine serum (FBS; BioFluid Technology). K562/ROR1 and JeKo-1_ffluc cell lines were provided by Drs. Stanley R. Riddell (The Fred Hutchinson Cancer Research Center, Seattle, WA) and Michael Hudecek (University of Würzburg, Würzburg, Germany), respectively. MDA-MB-231 (ATCC), and HT29 cells (an NCI-60 panel cell line obtained from UF Scripps Biomedical Research’s Cell-Based HighThroughput Screening Core) were grown in DMEM supplemented with L-glutamine, 100 U/mL penicillin/streptomycin, and 10% (v/v) FBS. Raji_ffluc, MDA-MB-231_ffluc, and HT29_ffluc cells were generated by transduction with lentivirus expressing firefly luciferase and selection with puromycin. The lentivector used here was made by cloning ffluc gene fused with T2A and puroR into pepHIV7 (pepHIV7_ffluc-T2A-puroR) and the lentivirus was produced as described below for making CAR-Ts. Expi293 cells were grown in Expi293 Expression Medium supplemented with 100 U/mL penicillin/streptomycin (all from Thermo Fisher Scientific). Cell lines were regularly authenticated by DNA fingerprinting using custom short tandem repeat (STR) profiling (Iddex or ATCC).
Cloning, expression, and purification of recombinant proteins
hFc-hROR1 and hFc-mROR1 were produced as described [37]. Gene fragments encoding the variable domains of the Fab switches were PCR-amplified from previously described phagemids [10, 37] and fused to the constant domains of Fabs by overlap extension PCR. The GCN4 14-aa peptide (NYHLENEVARLKKL) encoding sequence was introduced to the N- or C-terminus of light chains by PCR using oligonucleotides (Integrated DNA Technologies). The resulting light and heavy chain encoding sequences were cloned into mammalian cell expression vector pCEP4 and transiently co-transfected into Expi293 cells using the ExpiFectamine 293 Transfection Kit (Thermo Fisher Scientific). Five days later, supernatants were collected and loaded on a 1-mL CaptureSelect IgG-CH1 Column (Thermo Fisher Scientific) in conjunction with an ÄKTA FPLC instrument (Cytiva). The purity of the Fabs was confirmed by SDS-PAGE followed by Coomassie Blue staining and their concentration was determined by Pierce BCA Protein Assay Kit according to the instruction (Thermo Fisher Scientific). The anti-GCN4 peptide mAb 52SR4 in scFv-Fc format was generated as described [30].
Generation of affinity variants of clone R12
A 5 amino acid sequence of the R12 VH CDR3 or a 4 amino acid sequence of the R12 VL CDR3 were randomly mutated by NNK saturation mutagenesis, minimizing both codon redundancy and thus the screening effort but also the frequency of premature stop codons [60, 61]. The randomly mutated R12 Fab were cloned into the phage display vector pC3C and the resulting libraries were enriched against immobilized human ROR1 protein [62], followed by determination of the Fab sequences via deep sequencing. Sequence reads of the two phage display libraries were organized by grouping nucleotide sequence variants encoding the same amino acid sequence and ranking them by frequency. Being very well optimized, the original R12 was found to be the most frequent aa sequence in both libraries. The two R12 variants used in this work arose from an extensive testing campaign and represent a less frequent variant from the VL CDR3 library (R12_16) and a frequent variant from the VH CDR3 library (R12_20).
Surface plasmon resonance
Surface plasmon resonance (SPR) for the measurement of kinetic and thermodynamic parameters of the binding of purified R12 affinity variant switches to human ROR1 was performed on a Biacore X100 instrument using Biacore reagents and software (Cytiva). A mouse anti-human IgG CH2 mAb was immobilized on a CM5 sensor chip using reagents and instructions supplied with the Human Antibody Capture Kit (Cytiva). hFc-hROR1 was captured at a density not exceeding 1,000 RU. Each sensor chip included an empty flow cell for instantaneous background depletion. All binding assays used 1x HBS-EP+ running buffer (10 mM HEPES, 150 mM NaCl, 3 mM EDTA (pH 7.4), and 0.05% (v/v) Surfactant P20) and a flow rate of 30 μL/min. For affinity measurements, all switches were injected at five different concentrations, one of which was tested in duplicate. The sensor chips were regenerated with 3 M MgCl2 from the Human Antibody Capture Kit without any loss of binding capacity. Calculation of association (kon) and dissociation (koff) rate constants was based on a 1:1 Langmuir binding model. The equilibrium dissociation constant (Kd) was calculated from koff /kon.
ELISA
Switch binding assay to captured Fc fusion proteins.
Each well of a 96-well Costar 3690 plate (Corning) was coated with 100 ng hFc-hROR1 or hFc-mROR1 in 25 μL coating buffer (0.1 M Na2CO3, 0.1 M NaHCO3, pH 9.6) for 1 h at 37oC. After blocking with 150 μL 3% (w/v) BSA/TBS for 1 h at 37oC, 100 ng Fab switches in 50 μL 1% (w/v) BSA/TBS were captured by incubation for 1 h at 37oC. The wells were washed three times with 150 μL TBS. Next, 50 μL of a 1:1,000 dilution of HRP-conjugated goat F(ab’)2 anti-human IgG, F(ab’)2 fragment (Jackson ImmunoResearch) in 1% (w/v) BSA/TBS was added and incubated for 1 h at 37oC. The wells were washed four times and colorimetric detection was performed using 2,2’-azino-bis(3-ethylbenzothiazoline)-6-sulfonic acid (Roche) as substrate according to the manufacturer’s directions. The absorbance was measured at 405 nm using a SpectraMax M5 microplate reader (Molecular Devices) and SoftMax Pro software (Molecular Devices).
Cytokine release.
TNFα, IL2, IFNγ, and IL6 released in the culture medium from CAR-Ts or human PBMC after 20-h coincubation with target cells at E:T ratios of 10:1 in presence or absence of switches were quantified using the corresponding ELISA MAX Deluxe Sets (BioLegend) and following the manufacturer’s instructions.
Flow cytometry
Cells were stained using standard flow cytometry methodology. The expression level of ROR1 on target cells was determined with R12 Fab-N (10 μg/mL) followed by R-Phycoerythrin-conjugated goat F(ab’)2 anti-human IgG, F(ab’)2 fragment-specific pAbs (Jackson ImmunoResearch) for K562 and K562/ROR1 cells, or 52SR4 scFv-Fc conjugated to Alexa Fluor 647 for Raji, JeKo-1, MDA-MB-231, and HT29 cells. The binding of switches (5 μg/mL) to sCAR-Ts and target cells was detected by R-Phycoerythrin-conjugated goat F(ab’)2 anti-human IgG, F(ab’)2 fragment-specific pAbs (Jackson ImmunoResearch). To detect CAR-T activation, mouse anti-human CD3 mAb OKT3-FITC and mouse anti-human CD25 mAb BC96-APC (both from BioLegend) were used. T-cell transduction efficiency was detected indirectly by staining EGFRt using 5 μg/mL cetuximab (CTX; a chimeric mouse/human IgG1) conjugated to Alexa Fluor 647. To determine the percentage of different cell subtypes in human PBMC (human CD45+), spectral flow cytometry was performed on a Cytek Aurora flow cytometry instrument to allow accurate gating, compensation, and analysis with multicolor fluorescence staining for B cells (CD19+/CD3-), T cells (CD3+/CD56-), NK cells (CD56+/CD3-), and monocytes (CD3-/CD19-/CD14+/HLA-DR+). eFluor 780 (Thermo Fisher Scientific, 1:5,000 dilution) was included to exclude dead cells. Human CD45-PE-Cy7 (clone 2D1, 1:400), CD3-FITC (clone OKT3, 1:200), CD19-BV421 (clone HIB19, 1:200), and CD56-PE (clone 5.1H11, 1:200) were purchased from BioLegend. Human CD14-BUV395 (clone MφP9, 1:400) and HLA-DR-BUV 805 (clone G46-6, 1:400) were purchased from BD Biosciences. Data were analyzed with FlowJo software v10.
Generation of CAR-Ts
A lentiviral vector encoding the universal CAR cassette based on scFv 52SR4 was generated as described [30]. Conventional 324 and R12 CARs were generated by replacing scFv 52SR4 of the universal CAR cassette with scFv 324 and scFv R12 using overlap extension PCR and then cloned into lentiviral vector pepHIV7. CAR lentivirus was produced in Lenti-X 293T (Takara Bio) using the packaging vectors pCHGP-2, pCMV-Rev2, and pCMV-G. Pan-T cells were isolated from fresh PBMC of healthy donors (Allcells) by negative selection (Pan T Cell Isolation Kit; Miltenyi Biotec), activated using Dynabeads Human T-Activator CD3/CD28 (Thermo Fisher Scientific), and transduced on day 2 by spinfection (800 × g for 1 h at 32℃) with concentrated lentivirus (MOI = 3:1) and 1 μg/mL polybrene (Millipore) as described [30]. CAR-Ts were expanded in RPMI 1640 supplemented with L-glutamine, 100 U/mL penicillin/streptomycin (all from Thermo Fisher Scientific), 10% (v/v) FBS (Thermo Fisher Scientific) and 100 U/mL of IL2 (Cell Sciences). On day 8, 65-85% of the Pan-T cells expressing the CAR were purified by MACS using biotinylated cetuximab and LS Columns (Miltenyi Biotec). CAR-Ts were maintained at a concentration of 0.5-2 × 106/mL during expansion and used within 21 days.
In vitro cytotoxicity assay
Cytotoxicity mediated by conventional CAR-Ts.
CAR-Ts (4 / 2 / 1 / 0.5 × 104/well) in triplicate were added into a 96-well tissue culture plate seeded with 1 × 104/well HT29_ffluc cells at E:T ratios of 4:1, 2:1, 1:1, and 0.5:1. Specific lysis was measured with 0.15 mg/mL D-luciferin (Biosynth) following 16-h of co-culture as described [37].
Cytotoxicity mediated by sCAR-Ts.
CAR-Ts (10 × 104/well) in triplicate were incubated with target cells expressing firefly luciferase at E:T ratios of 10:1 for 20 h in the presence or absence of a concentration range of 10 fM - 100 nM of Fab-N and Fab-C switches. Specific lysis was then measured with the luciferase-based cytotoxicity assay as above.
Xenograft mouse models
NSG/JeKo-1 xenograft.
Thirty 6-8 weeks old NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ NSG mice (The Jackson Laboratory #005557) were each given 0.5 × 106 JeKo-1_ffluc via i.v. (tail vein) injection on day 0. On day 5, the animals were i.p. injected with 150 mg/kg D-luciferin (Biosynth) and divided into 6 groups of 5 animals each by average bioluminescence. On day 6, each mouse was i.v. injected with 5 × 106 CAR-Ts, and 6 h later, with i.p. injection of 1 μg switches, which was increased gradually to 3 μg and 10 μg on days 7 and 8, respectively, and kept at 10 μg thereafter on days 10, 12, and 14. Starting on day 16, another round of treatment was given. In cohort 6, 5 × 106 R12-based conventional CAR-Ts were injected i.v. (tail vein) as positive control.
Humanized NSG/JeKo-1 xenograft.
Twenty 6-8 weeks old NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ NSG mice (The Jackson Laboratory #005557) were each given 0.5 × 106 JeKo-1_ffluc via i.v. (tail vein) injection on day 0. On day 5, the animals were i.p. injected with 150 mg/kg D-luciferin (Biosynth) and divided into 5 groups of 4 animals each by average bioluminescence. On day 6, each mouse was i.v. injected with 3 × 106 human PBMC (isolated from fresh buffy coats provided by OneBlood, Orlando, FL) and 5 × 106 CAR-Ts, and 6 h later, with i.p. injection of 1 μg switch 324N, which was increased gradually to 3 μg and 10 μg on days 7 and 8, respectively, and kept at 10 μg thereafter on days 10, 12, 14, 16, and 18. In cohort 5, 5 × 106 324-based conventional CAR-Ts were injected i.v. (tail vein) as positive control. To monitor IFNγ levels in mouse blood, 100 μL blood was collected by retro-orbital bleeding on day 9, 14, and 20 and diluted 5-fold for quantitative ELISA using the ELISA MAX Deluxe Set Human IFN-γ (BioLegend).
NSG/HT-29 xenograft.
Similarly, 30 6-8 weeks old NSG mice were each i.p. injected with 0.5 × 106 HT29_ffluc on day 0. On day 2, the animals were i.p. injected with 150 mg/kg D-luciferin (Biosynth) and divided into 6 groups of 5 animals each by average bioluminescence. On day 3, each mouse was i.p. injected with 10 × 106 CAR-Ts, and 6 h later, with i.v. injection of 1 μg switches, which was increased gradually to 3 μg and 10 μg on days 4 and 5, respectively, and kept at 10 μg thereafter on days 7, 9, and 11. Only one round of treatment was given. In cohort 5 and 6, 2 × 106 324 and R12-based, respectively, conventional CAR-Ts were injected i.p. as positive control. To monitor tumor growth, bioluminescence images of all 30 mice were taken by LagoX (Spectrum Instruments Imaging) before and after treatment and their radiance was recorded (mean ± SD) every 3-5 days. The body weight of the mice was measured and euthanasia was performed when the mice gained more than 25% body weight due to tumor burden. All procedures were approved by the Institutional Animal Care and Use Committee of UF Scripps Biomedical Research and were performed in compliance with the NIH Guide for the Care and Use of Laboratory Animals.
PK study
Three female CD-1 mice (~25 g; Charles River) were injected i.p. with Fab-N at 8 mg/kg. Blood was collected in heparinized capillary tubes at 5 min, 30 min, 1 h, 3 h, 6 h, 12 h, 24 h, 48 h, and 72 h after injection. Plasma was obtained by centrifuging the samples at 2,000 × g for 5 min in a microcentrifuge and stored at −80°C until analysis. The concentrations of Fabs in the plasma samples were measured by ELISA. For this, each well of a 96-well Costar 3690 plate (Corning) was incubated with 100 ng 52SR4 scFv-Fc in 25 μL carbonate/bicarbonate buffer (pH 9.6) at 37°C for 1 h. After blocking with 150 μL 3% (w/v) BSA/PBS solution for 1 h at 37°C, the prepared plasma samples were added. Peroxidase AffiniPure F(ab’)2 fragment goat anti-human IgG F(ab’)2 fragment-specific pAbs (Jackson ImmunoResearch) were used for detection as described above. The concentration of the Fabs in the plasma samples was extrapolated from a four-variable fit of the standard curve. PK parameters were analyzed by using Phoenix WinNonlin PK/PD Modeling and Analysis software (Pharsight). All procedures were approved by the Institutional Animal Care and Use Committee of UF Scripps Biomedical Research and were performed in compliance with the NIH Guide for the Care and Use of Laboratory Animals.
Statistical analyses
Statistical analyses were performed using GraphPad Prism 7. For in vivo studies, 4-5 mice were randomized and treated per experimental group based on the resource equation method [66]. Treatment and analysis were unblinded. No treated mice were excluded from the analysis. Variance within experimental groups was similar between experimental groups that were statistically compared. Bioluminescence data shown in Fig. 6C, Fig. 7D, and Suppl. Fig. S7D were subjected to an unpaired two-tailed t-test. Statistical analysis of mouse survival (Fig. 6D and Fig. 7E) was done by log-rank (Mantel-Cox) testing. Results with a p-value of p < 0.05 were considered significant.
Supplementary Material
Acknowledgements:
CR acknowledges support by National Institutes of Health (NIH) grants R01 CA174844, R01 CA181258, R01 CA204484, R21 CA229961, and R21 CA263240, and by the Klorfine Foundation.
Footnotes
Conflicts of interest: CR and HP are named inventors on a licensed patent family (assignee, University of Florida and Boehringer Ingelheim) that claims a set of anti-ROR1 mAbs used in this study including 324 (United States Patent 10,618,959). CR is named inventor on a licensed patent family (assignee, United States of America) that claims another set of anti-ROR1 mAbs used in this study including R12 (United States Patent 9,758,586).
References
- 1.June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science 2018; 359: 1361–1365. [DOI] [PubMed] [Google Scholar]
- 2.Feins S, Kong W, Williams EF, Milone MC, Fraietta JA. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am J Hematol 2019. [DOI] [PubMed] [Google Scholar]
- 3.Dana H, Chalbatani GM, Jalali SA, Mirzaei HR, Grupp SA, Suarez ER et al. CAR-T cells: early successes in blood cancer and challenges in solid tumors. Acta Pharm Sin B 2021; 11: 1129–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hong M, Clubb JD, Chen YY. Engineering CAR-T cells for next-generation cancer therapy. Cancer Cell 2020; 38: 473–488. [DOI] [PubMed] [Google Scholar]
- 5.Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov 2020; 19: 185–199. [DOI] [PubMed] [Google Scholar]
- 6.Townsend MH, Shrestha G, Robison RA, O’Neill KL. The expansion of targetable biomarkers for CAR T cell therapy. J Exp Clin Cancer Res 2018; 37: 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moreno-Cortes E, Forero-Forero JV, Lengerke-Diaz PA, Castro JE. Chimeric antigen receptor T cell therapy in oncology - Pipeline at a glance: analysis of the ClinicalTrials.gov database. Crit Rev Oncol Hematol 2021; 159: 103239. [DOI] [PubMed] [Google Scholar]
- 8.Balakrishnan A, Goodpaster T, Randolph-Habecker J, Hoffstrom BG, Jalikis FG, Koch LK et al. Analysis of ROR1 protein expression in human cancer and normal tissues. Clin Cancer Res 2017; 23: 3061–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baskar S, Kwong KY, Hofer T, Levy JM, Kennedy MG, Lee E et al. Unique cell surface expression of receptor tyrosine kinase ROR1 in human B-cell chronic lymphocytic leukemia. Clin Cancer Res 2008; 14: 396–404. [DOI] [PubMed] [Google Scholar]
- 10.Yang J, Baskar S, Kwong KY, Kennedy MG, Wiestner A, Rader C. Therapeutic potential and challenges of targeting receptor tyrosine kinase ROR1 with monoclonal antibodies in B-cell malignancies. PLoS One 2011; 6: e21018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin Cancer Res 2013; 19: 3153–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berger C, Sommermeyer D, Hudecek M, Berger M, Balakrishnan A, Paszkiewicz PJ et al. Safety of targeting ROR1 in primates with chimeric antigen receptor-modified T cells. Cancer Immunol Res 2015; 3: 206–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Srivastava S, Furlan SN, Jaeger-Ruckstuhl CA, Sarvothama M, Berger C, Smythe KS et al. Immunogenic chemotherapy enhances recruitment of CAR-T cells to lung tumors and improves antitumor efficacy when combined with checkpoint blockade. Cancer Cell 2021; 39: 193–208 e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao Y, Zhang D, Guo Y, Lu B, Zhao ZJ, Xu X et al. Tyrosine kinase ROR1 as a target for anti-cancer therapies. Front Oncol 2021; 11: 680834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Menck K, Heinrichs S, Baden C, Bleckmann A. The WNT/ROR pathway in cancer: from signaling to therapeutic intervention. Cells 2021; 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hojjat-Farsangi M, Moshfegh A, Daneshmanesh AH, Khan AS, Mikaelsson E, Osterborg A et al. The receptor tyrosine kinase ROR1--an oncofetal antigen for targeted cancer therapy. Semin Cancer Biol 2014; 29: 21–31. [DOI] [PubMed] [Google Scholar]
- 17.Hudecek M, Schmitt TM, Baskar S, Lupo-Stanghellini MT, Nishida T, Yamamoto TN et al. The B-cell tumor-associated antigen ROR1 can be targeted with T cells modified to express a ROR1-specific chimeric antigen receptor. Blood 2010; 116: 4532–4541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dave H, Anver MR, Butcher DO, Brown P, Khan J, Wayne AS et al. Restricted cell surface expression of receptor tyrosine kinase ROR1 in pediatric B-lineage acute lymphoblastic leukemia suggests targetability with therapeutic monoclonal antibodies. PLoS One 2012; 7: e52655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Srivastava S, Salter AI, Liggitt D, Yechan-Gunja S, Sarvothama M, Cooper K et al. Logic-gated ROR1 chimeric antigen receptor expression rescues T cell-mediated toxicity to normal tissues and enables selective tumor targeting. Cancer Cell 2019; 35: 489–503 e488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jensen MC, Riddell SR. Designing chimeric antigen receptors to effectively and safely target tumors. Curr Opin Immunol 2015; 33: 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sahillioglu AC, Schumacher TN. Safety switches for adoptive cell therapy. Curr Opin Immunol 2021. [DOI] [PubMed] [Google Scholar]
- 22.Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia 2010; 24: 1160–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 2015; 350: aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cho JH, Collins JJ, Wong WW. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell 2018; 173: 1426–1438 e1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodgers DT, Mazagova M, Hampton EN, Cao Y, Ramadoss NS, Hardy IR et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc Natl Acad Sci U S A 2016; 113: E459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao Y, Rodgers DT, Du J, Ahmad I, Hampton EN, Ma JS et al. Design of switchable chimeric antigen receptor T cells targeting breast cancer. Angew Chem Int Ed Engl 2016; 55: 7520–7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Viaud S, Ma JSY, Hardy IR, Hampton EN, Benish B, Sherwood L et al. Switchable control over in vivo CAR T expansion, B cell depletion, and induction of memory. Proc Natl Acad Sci U S A 2018; 115: E10898–E10906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Darowski D, Kobold S, Jost C, Klein C. Combining the best of two worlds: highly flexible chimeric antigen receptor adaptor molecules (CAR-adaptors) for the recruitment of chimeric antigen receptor T cells. mAbs 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raj D, Yang MH, Rodgers D, Hampton EN, Begum J, Mustafa A et al. Switchable CAR-T cells mediate remission in metastatic pancreatic ductal adenocarcinoma. Gut 2019; 68: 1052–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qi J, Tsuji K, Hymel D, Burke TR Jr., Hudecek M, Rader C et al. Chemically programmable and switchable CAR-T therapy. Angew Chem Int Ed Engl 2020; 59: 12178–12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zahnd C, Spinelli S, Luginbuhl B, Amstutz P, Cambillau C, Pluckthun A. Directed in vitro evolution and crystallographic analysis of a peptide-binding single chain antibody fragment (scFv) with low picomolar affinity. J Biol Chem 2004; 279: 18870–18877. [DOI] [PubMed] [Google Scholar]
- 32.Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res 2015; 75: 3596–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Benmebarek MR, Karches CH, Cadilha BL, Lesch S, Endres S, Kobold S. Killing mechanisms of chimeric antigen receptor (CAR) T cells. Int J Mol Sci 2019; 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Larson RC, Maus MV. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat Rev Cancer 2021; 21: 145–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med 2019; 25: 1408–1414. [DOI] [PubMed] [Google Scholar]
- 36.Greenman R, Pizem Y, Haus-Cohen M, Goor A, Horev G, Denkberg G et al. Shaping functional avidity of CAR T cells: affinity, avidity, and antigen density that regulate response. Mol Cancer Ther 2021; 20: 872–884. [DOI] [PubMed] [Google Scholar]
- 37.Peng H, Nerreter T, Chang J, Qi J, Li X, Karunadharma P et al. Mining naive rabbit antibody repertoires by phage display for monoclonal antibodies of therapeutic utility. J Mol Biol 2017; 429: 2954–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yam PY, Li S, Wu J, Hu J, Zaia JA, Yee JK. Design of HIV vectors for efficient gene delivery into human hematopoietic cells. Mol Ther 2002; 5: 479–484. [DOI] [PubMed] [Google Scholar]
- 39.Wang X, Chang WC, Wong CW, Colcher D, Sherman M, Ostberg JR et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 2011; 118: 1255–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qi J, Li X, Peng H, Cook EM, Dadashian EL, Wiestner A et al. Potent and selective antitumor activity of a T cell-engaging bispecific antibody targeting a membrane-proximal epitope of ROR1. Proc Natl Acad Sci U S A 2018; 115: E5467–E5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nerreter T, Letschert S, Gotz R, Doose S, Danhof S, Einsele H et al. Super-resolution microscopy reveals ultra-low CD19 expression on myeloma cells that triggers elimination by CD19 CAR-T. Nat Commun 2019; 10: 3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee YG, Chu H, Lu Y, Leamon CP, Srinivasarao M, Putt KS et al. Regulation of CAR T cell-mediated cytokine release syndrome-like toxicity using low molecular weight adapters. Nat Commun 2019; 10: 2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Minutolo NG, Sharma P, Poussin M, Shaw LC, Brown DP, Hollander EE et al. Quantitative control of gene-engineered T-cell activity through the covalent attachment of targeting ligands to a universal immune receptor. J Am Chem Soc 2020; 142: 6554–6568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rennert P, Su L, Dufort F, Birt A, Sanford T, Wu L. et al. A novel CD19-anti-CD20 bridging protein prevents and reverses CD19-negative relapse from CAR19 T cell treatment in vivo. Blood 2019; 134 Suppl 1: 252.31118164 [Google Scholar]
- 45.Arndt C, Fasslrinner F, Loureiro LR, Koristka S, Feldmann A, Bachmann M. Adaptor CAR platforms - next generation of T cell-based cancer immunotherapy. Cancers (Basel) 2020; 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Minutolo NG, Hollander EE, Powell DJ Jr. The emergence of universal immune receptor T cell therapy for cancer. Front Oncol 2019; 9: 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hombach AA, Schildgen V, Heuser C, Finnern R, Gilham DE, Abken H. T cell activation by antibody-like immunoreceptors: the position of the binding epitope within the target molecule determines the efficiency of activation of redirected T cells. J Immunol 2007; 178: 4650–4657. [DOI] [PubMed] [Google Scholar]
- 48.Chmielewski M, Hombach A, Heuser C, Adams GP, Abken H. T cell activation by antibody-like immunoreceptors: increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J Immunol 2004; 173: 7647–7653. [DOI] [PubMed] [Google Scholar]
- 49.Schmid DA, Irving MB, Posevitz V, Hebeisen M, Posevitz-Fejfar A, Sarria JC et al. Evidence for a TCR affinity threshold delimiting maximal CD8 T cell function. J Immunol 2010; 184: 4936–4946. [DOI] [PubMed] [Google Scholar]
- 50.Lindner SE, Johnson SM, Brown CE, Wang LD. Chimeric antigen receptor signaling: functional consequences and design implications. Sci Adv 2020; 6: eaaz3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Picanco-Castro V, Swiech K, Malmegrim KCR, Covas DT. CAR-T cells for cancer treatment: current design and next frontiers. Methods Mol Biol 2020; 2086: 1–10. [DOI] [PubMed] [Google Scholar]
- 52.Jayaraman J, Mellody MP, Hou AJ, Desai RP, Fung AW, Pham AHT et al. CAR-T design: elements and their synergistic function. EBioMedicine 2020; 58: 102931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res 2015; 3: 125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shabani M, Naseri J, Shokri F. Receptor tyrosine kinase-like orphan receptor 1: a novel target for cancer immunotherapy. Expert Opin Ther Targets 2015; 19: 941–955. [DOI] [PubMed] [Google Scholar]
- 55.Kamrani A, Mehdizadeh A, Ahmadi M, Aghebati-Maleki L, Yousefi M. Therapeutic approaches for targeting receptor tyrosine kinase like orphan receptor-1 in cancer cells. Expert Opin Ther Targets 2019; 23: 447–456. [DOI] [PubMed] [Google Scholar]
- 56.Peng H Perspectives on the development of antibody-drug conjugates targeting ROR1 for hematological and solid cancers. Antib Ther 2021; 4: 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Choi MY, Widhopf GF 2nd, Ghia EM, Kidwell RL, Hasan MK, Yu J et al. Phase I trial: cirmtuzumab inhibits ROR1 signaling and stemness signatures in patients with chronic lymphocytic leukemia. Cell Stem Cell 2018; 22: 951–959 e953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang ML, Barrientos JC, Furman RR, Mei M, Barr PM et al. Zilovertamab vedotin targeting of ROR1 as therapy for lymphoid cancers. N Engl J Med Evid 2022; 1:1–11. [DOI] [PubMed] [Google Scholar]
- 59.Labanieh L, Majzner RG, Klysz D, Sotillo E, Fisher CJ et al. Enhanced safety and efficacy of protease-regulated CAR-T cell receptors. Cell 2022; 185: 1745–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Acevedo-Rocha CG, Reetz MT, Nov Y. Economical analysis of saturation mutagenesis experiments. Sci Rep 2015; 5: 10654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rydzek J, Nerreter T, Peng H, Jutz S, Leitner J, Steinberger P et al. Chimeric antigen receptor library screening using a novel NF-kappaB/NFAT reporter cell platform. Mol Ther 2019; 27: 287–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goydel RS, Weber J, Peng H, Qi J, Soden J, Freeth J et al. Affinity maturation, humanization, and co-crystallization of a rabbit anti-human ROR2 monoclonal antibody for therapeutic applications. J Biol Chem 2020; 295: 5995–6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Charan J, Kantharia ND. How to calculate sample size in animal studies? J Pharmacol Pharmacother 2013; 4: 303–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.