

Abstract
Generalized force fields (FFs) act as extensions to biomolecular FFs to provide a wide coverage of organic molecules. However, their precise application to an arbitrary molecule presents a separate challenge. We show that MATCH assigns different atom types and bonded and nonbonded parameters than CGenFF, and the AM1-BCC charge model, commonly used with GAFF/GAFF2, does not exactly reproduce the performance of the RESP charge model. The results indicate the need for caution when employing FFs to ensure their integrity with respect to their implementation and validation.
Graphical Abstract
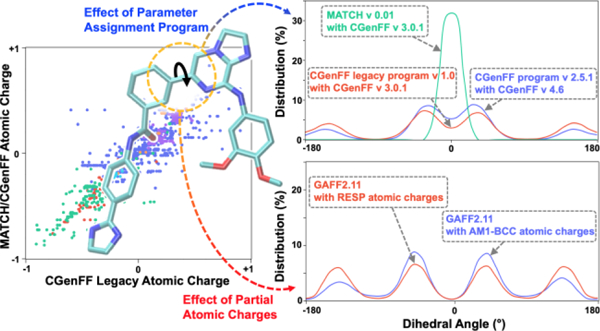
Empirical force fields (FFs) represent the foundation of molecular modeling and simulation. Currently, a range of FFs are available, dominated by additive, pairwise all-atom models optimized primarily for macromolecular simulations, including CHARMM1–10 and AMBER,11–16 among others,17, 18 that are implemented in different simulation programs.19–22 In the context of drug design as well as studies of a wide range of diverse systems such as nanomaterials and ionic liquids, additive FFs have been extended to small, organic compounds. These include the CHARMM General FF (CGenFF)23–26 in the context of CHARMM1–10 and General Amber FF (GAFF)27–31 and GAFF232 in the context of AMBER.11–16
While the small-molecule empirical FFs have seen wide use, users must be aware of the fidelity of their implementations from different sources and simulation programs. For example, a simple challenge is the use of different constants for Coulombs law, leading to differences in electrostatic energies and forces. Other examples include using the correct units, joules (International System of Units) versus calories (centimeter-gram-seconds unit system), or Rmin versus σ values for the Lennard–Jones term. Such problems are typically solved through careful implementation of an FF in a simulation program, an ideally one-time task. However, a more insidious problem is the actual implementation of the FF itself, given the requirement for assigning atom types and parameters to a wide range of diverse chemical structures.
Recently, a study was published applying a high-throughput workflow to leverage the capabilities of high-performance supercomputers for free-energy perturbation calculations.33 While the computational methods and workflows in the study were well described and validated, the definition and implementation of the FFs used raised important concerns that went beyond that specific study.33 Motivated by this33 and other studies,34–36 three issues are addressed in this study. First is the integrity of CGenFF,23–26 where the program MATCH37 rather than the CGenFF program38, 39 was used for atom typing and parameter assignment.33 As will be shown, the fidelity of MATCH37 in producing CGenFF23–26 atom types and parameters is quite low, such that the “CGenFF” referenced in that study did not represent CGenFF23–26 of the MacKerell Laboratory and collaborating laboratories. What is presented as representing a widely used FF associated with a specific parameter assignment program is actually a very different FF than that implied based on the nomenclature used in the publication.33 Second is the use of Austin Model 1-bond charge correction (AM1-BCC) model40, 41 to assign atomic charges to assess the quality of GAFF2. GAFF2 parameters were developed using HF/6–31G* restrained electrostatic potential (RESP) charges;42 RESP charges have been shown to out-perform AM1-BCC charges in hydration free-energy calculations with GAFF2.32 Thus, the reported GAFF2 is assessed based on a charge model that may not be able to achieve the original performance of GAFF2. Third, is the opposite issue, where two FFs, OpenFF 1.2.043 and GAFF,27–31 are presented as independent models, while detailed analysis of the nonbonded parameters show them to be nearly identical. This may reflect the fact that SMIRNOFF99Frost,44 the predecessor of OpenFF 1.2.0,43 originated from parent FFs AMBER parm9945 and parm@Frosst46 and has been reported to be related to, “but not identical” to, GAFF.44, 43 Accordingly, the goal of the present study is to provide clarification of the integrity of small-molecule FFs and to raise user awareness of the importance of such clarification when applying such FFs.
Coverage of Parameter Assignment Programs.
Nine parameter sets were investigated (Table 1) on the 264 ligands reported in Gapsys et al.33 The parameter set (first column, Table 1) was used in conjunction with different parameter assignment programs (second column, Table 1) to assign atom types and parameters, yielding the corresponding generalized FF (third column, Table 1), to the investigated set of ligands. All parameter assignment programs, except for MATCH,37 successfully generated parameters for all 264 ligands. Gapsys/CGenFF, Gapsys/GAFF2, and OpenFF parameters for all 264 ligands were already available in the database extracted from the Github repository provided by Gapsys et al.33 MATCH successfully assigned parameters for 243 ligands and failed to assign parameters for 21 ligands. Thus, all reported data regarding MATCH/CGenFF exclude these 21 ligands.
Table 1.
Overview of the Parameter Sets Investigated.
| Parameter Set | Parameter Assignment Program | Force Field |
|---|---|---|
| CGenFF | CGenFF v 2.5.1 | CGenFF v4.6 |
| CGenFF Legacy | CGenFF legacy v 1.0 | CGenFF v3.0.1 |
| MATCH/CGenFF | MATCH v 0.01 | CGenFF v3.0.1 |
| Gapsys/CGenFFa | MATCH v 0.01b | CGenFF v3.0.1 |
| GAFF2/RESP | Psi4, Antechamber, AmberTools21 | GAFF2.11 |
| GAFF2/AM1-BCC | Antechamber, AmberTools21 | GAFF2.11 |
| Gapsys/GAFF2a | Antechamber, ACPYPE | GAFF2.11 |
| GAFF/AM1-BCC | Antechamber, AmberTools21 | GAFF1.81 |
| OpenFF* | OpenFF Toolkit | OpenFF 1.2.0 |
Parameters were extracted directly from Gapsys et al.33
As shown in Figures S1 and S2, while the parameters were indicated to be generated using MATCH, there are differences between the MATCH charges calculated in the present study and those reported in the Gapsys et al.33 study.
Discrepancies in Assigned CGenFF Atom Types and Parameters.
None of the studied ligands were assigned the same set of CGenFF or CGenFF Legacy atom types and parameters in MATCH/CGenFF or Gapsys/CGenFF; all studied ligands were assigned at least one parameter that differed across the three programs. Despite Gapsys/CGenFF being based on MATCH and CGenFF v3.0.1,33 the Gapsys/CGenFF parameters include lone pairs (LPs). Neither MATCH nor CGenFF v3.0.1 has LP functionality; thus, it appears that the parameters of ligands containing σ-holes were modified for Gapsys/CGenFF based on the procedure described by CGenFF developers applied to CGenFF v3.0.1 FF26 and released in subsequent versions of CGenFF, including CGenFF v4.6 used in the present study. In addition, over 80% of the ligands contain atoms that are typed differently in MATCH/CGenFF and/or Gapsys/CGenFF (Table S1). Furthermore, nearly half of the studied ligands contain atoms that are typed differently between MATCH/CGenFF and Gapsys/CGenFF (Table S1).
Unsurprisingly, given the discrepancies in atom typing, discrepancies were observed in the bonded parameters (Table S2). Despite the bonded parameters of Gapsys/CGenFF being extracted from CGenFF v3.0.1 independently of MATCH,33 discrepancies are also present between Gapsys/CGenFF and CGenFF Legacy as well as CGenFF (Table S2). All ligands contain at least one differently assigned bonded parameter in Gapsys/CGenFF and MATCH/CGenFF. Notably, MATCH does not assign Urey-Bradley terms to angle parameters where appropriate. Thus, all angle parameters containing Urey-Bradley terms are described differently by MATCH/CGenFF regardless of atom type assignment.
Comparing the assigned partial atomic charges also revealed significant differences among the parameters (Figures 1A, S1, S2). Compared to CGenFF and CGenFF Legacy, over half of the atoms have different charges in MATCH/CGenFF and Gapsys/CGenFF, affecting all the studied ligands (Table S1). The differences in charges are often significant, with several charges differing by more than 0.5 e and including changes in the sign (Figure S2).
Figure 1.
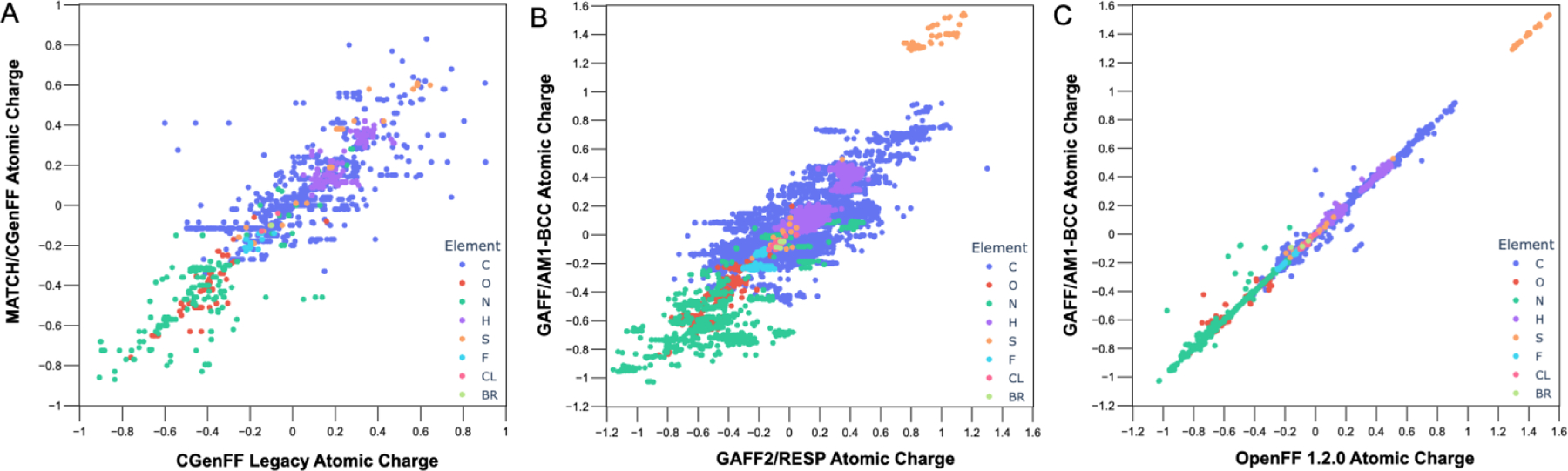
Correlation plots of atomic charges for (A) CGenFF Legacy with MATCH/CGenFF, (B) GAFF2/RESP with GAFF2/AM1-BCC, and (C) OpenFF with GAFF/AM1-BCC. Each datapoint corresponds to an atom.
Differences in Assigned GAFF2 Atom Types and Parameters.
Similarly to Gapsys/CGenFF, Gapsys/GAFF2 also includes LPs.33 Currently, GAFF2 does not have LP functionality, thus it appears that the authors followed the published rules for off-site-charge generation for GAFF family of FFs,47, 48 although details of the charge assignment approach were not presented.33 Beyond the presence of added LPs, nearly 20% of the ligands contain discrepancies in atom typing by Gapsys/GAFF2 compared to GAFF2/AM1-BCC and GAFF2/RESP (Table S3). These discrepancies also affect the assigned bonded parameters (Table S4).
Beyond atom typing and bonded parameters, a comparison of the atomic charges assigned in GAFF2/RESP, GAFF2/AM1-BCC, and Gapsys/GAFF2 shows that the AM1-BCC and RESP charge assignments are not equivalent (Figures 1B and S3A,B). While AM1-BCC provides an efficient method to generate high-quality atomic charges for small molecules, GAFF and GAFF2 were originally developed with HF/6–31G* RESP charges.27–32 Additionally, the use of GAFF2/AM1-BCC has been shown to result in systemic errors greater than 1.0 kcal/mol in solvation free-energy calculations for several specific functional groups,32 which may significantly limit the accuracy of computationally predicted protein–ligand binding free energies. A new AM1-BCC model (ABCG2)32 developed specifically for GAFF2 may be used to circumvent quantum mechanics calculations needed to derive RESP charges. This charge model paves the road for the development of the next generation of GAFF-GAFF3. How well ABCG2 performs in protein–ligand binding calculations is under investigation. Aside from the fact that AM1-BCC atomic charges with GAFF2 may introduce systemic errors in free-energy calculations,32 the atomic charges of Gapsys/GAFF2 are not equivalent to those ofGAFF2/AM1-BCC for several atoms (Figure S3C).
Similarities in GAFF and OpenFF 1.2.0 Parameters.
Comparisons of GAFF and OpenFF showed striking similarities between the two FFs; this may be expected as OpenFF has been reported to share “common ancestry” with (yet be distinct from) GAFF.44, 43 Unsurprisingly, due to the use of the AM1-BCC model to derive atomic charges in both GAFF/AM1-BCC and OpenFF, the assigned atomic charges are nearly identical (Figure 1C). Interestingly, the assigned Lennard–Jones parameters σ and ε of GAFF/AM1-BCC and OpenFF are also similar (Figure S4A,B). Despite OpenFF assigning unique atom types to each individual atom, only a handful of σ and ε values are used, similar to GAFF/AM1-BCC (Figure S4 A,B). There are larger apparent differences between the assigned σ and ε values of GAFF/AM1-BCC with GAFF2/AM1-BCC compared with OpenFF (Figure S4C,D).
In addition, the assigned bonded parameters by GAFF/AM1-BCC and OpenFF are often similar (Table S5). Approximately a sixth of the bond, angle, and dihedral force constants of OpenFF are within 10% difference of GAFF/AM1-BCC (Table S5), while all equilibrium bond parameters and the majority of equilibrium angle, dihedral periodicity, and dihedral phase parameters are within 10% difference. Moreover, nearly all the bond and angle parameters and over half of the dihedral parameters of OpenFF are within 35% difference of GAFF/AM1-BCC (Table S6). None of the improper parameters of OpenFF are similar to GAFF/AM1-BCC as the two FFs treat improper torsions differently.
Structural and Energetic Consequences of Assigned Parameters.
Molecular dynamics (MD) simulations of two representative ligands (lig_CHEMBL3265002 and lig_8e from Gapsys et al.33), followed by dihedral angle distribution calculations, root mean square deviation (RMSD)-based clustering, and interaction energy calculations show that the differences (and similarities) in assigned parameters directly affect the structural and energetic properties of the ligands.
CGenFF and CGenFF Legacy allow more conformational flexibility in the simulated ligands compared to MATCH/CGenFF and Gapsys/CGenFF (Figures 2, S6, S7); MATCH/CGenFF and Gapsys/CGenFF frequently yield significantly different dihedral angle distributions from the simulations, with those distributions being more narrow (Figure 2, S6, S7). The atoms associated with these dihedral angles are assigned alternate atom types, have large discrepancies in charge, and/or are involved in the assignment of different bonded parameters (Figure S5, Tables S7, S8). The rigidity imposed by MATCH/CGenFF and Gapsys/CGenFF disallows the ligands from adopting conformations allowed by CGenFF and CGenFF Legacy (Figure S8). Furthermore, the average interaction energies of the ligands with the surrounding solvent are more favorable using CGenFF and CGenFF Legacy compared to MATCH/CGenFF or Gapsys/CGenFF (Figure S9).
Figure 2.
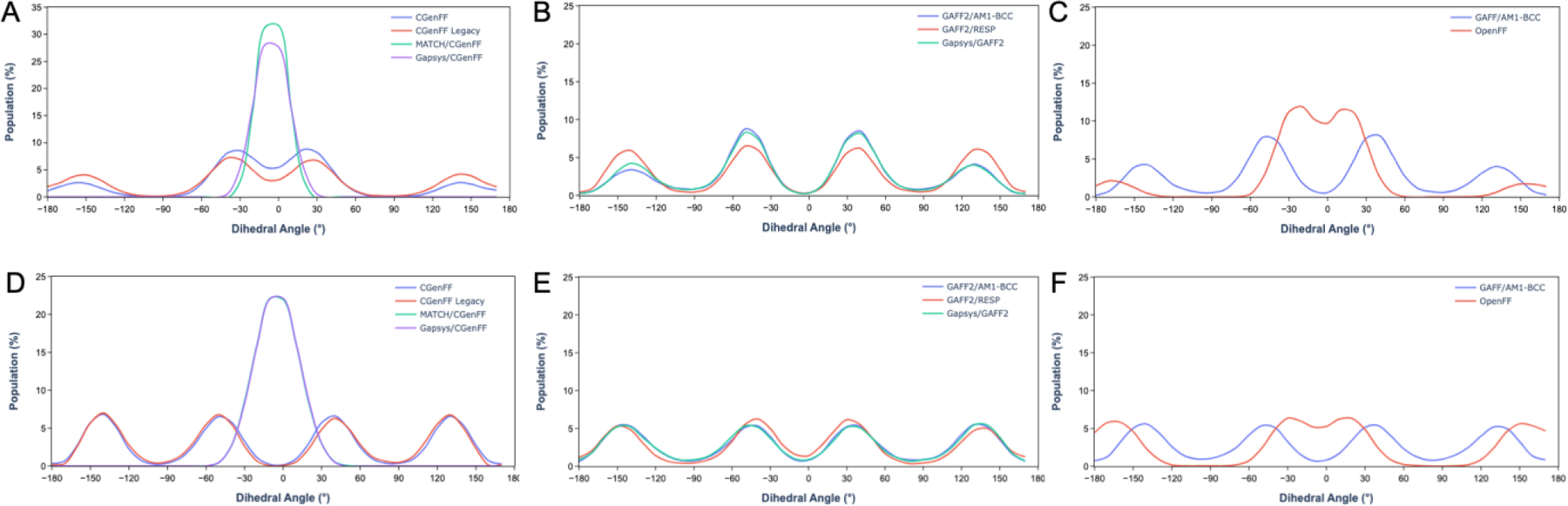
Dihedral angle distributions for the dihedral of atoms (A–C) 11–10-17–18 of lig_CHEMBL3265002 from the syk protein system of Gapsys et al.33 and (D–F) 5–11-12–17 of lig_8e from the tnks2 protein system of Gapsys et al.33
Differences in the conformational behavior of the ligands assigned using GAFF2/RESP and GAFF2/AM1-BCC or Gapsys/GAFF2 were also observed. The dihedral angle distribution of the ligands are slightly different with GAFF2/RESP compared to GAFF2/AM1-BCC and Gapsys/GAFF2 despite the use of the same bonded parameters due to the differences in assigned atomic charges (Figure 2 S6, S7, Table S9). The difference in the dihedral angle distribution with GAFF2/RESP is also reflected in the slightly larger number of distinct conformations adopted by the ligands with GAFF2/RESP (Figure S8). Additionally, despite the use of the same parameters, the assigned charges of lig_CHEMBL3265002 affect its water interaction energy, making it less favorable with GAFF2/RESP compared to GAFF2/AM1-BCC and Gapsys/GAFF2 (Figure S9). Variations in the conformations and interaction energies for lig_8e are relatively small, likely due to the small number of rotatable bonds (two rotatable bonds in lig_8e compared to nine in lig_CHEMBL3265002, Figure S5).
The overall conformational behavior of both ligands is similar with GAFF/AM1-BCC and OpenFF, despite the differences in their dihedral angle distributions. The dihedral angle distributions of OpenFF appear to loosely mimic those of GAFF/AM1-BCC, with a wider spread for several dihedral angles (Figure 2, S6, S7). Additionally, the ligands adopt nearly the same distinct conformations with OpenFF as with GAFF/AM1-BCC; only a few structures fall into mutually exclusive clusters (Figure S8). The average interaction energy of the ligands with OpenFF is similar to GAFF/AM1-BCC (Figure S9). Overall, it appears that OpenFF1.2.0 generally reproduces the behavior of the original GAFF through similar nonbonded and bonded parameters, despite being derived from refitting SMIRNOFF99Frost44 parameters against several sets of high-level quantum mechanical data.43
Best Practices for Nomenclature of Small-Molecule FFs.
The present results show that the specific parameter assignment programs and charge models used have a notable effect on the generalized atom types and parameters assigned. This in turn affects the properties of the assigned generalized FF in molecular simulations. Thus, it is important to apply a descriptive naming scheme in the scientific literature to avoid misrepresenting FFs and misleading readers as to what atom types and parameters were used.
We propose the nomenclature presented in Table 2. It requires that the FF version, parameter assignment program version, and/or charge model used be explicitly stated prior to using any abbreviation for the generalized FF being used. Further, researchers should reserve the use of the abbreviations “CGenFF” and “GAFF2” for the most current (at the time of their studies) versions of the FFs and their respective parameter assignment programs. Proper specification of the version number is particularly important as with any machine-learning based models, and the assigned parameters of newer versions of the CGenFF program may differ as the training sets and associated FF are updated. For CGenFF, the version of the CGenFF program and FF used is available in the header of the stream file output of the CGenFF program (Figure S10). For GAFF and GAFF2, the charge model is explicitly entered by the user in the Antechamber command line (Figure S11A), and the versions of GAFF and GAFF2 are available in the “gaff.dat” and “gaff2.dat” files, respectively, provided by AmberTools (Figure S11B). This information should be included in the description of the FF as shown in Table 2.
Table 2.
Proposed Nomenclature for Generalized FFs, CGenFF and GAFF/GAFF2, Reported in Scientific Literature.
| Parameter Assignment Program | Force Field | Charge Model | Ideal Nomenclature | Abbreviated Nomenclature |
|---|---|---|---|---|
| CGenFF v 2.5.1 | CGenFFv4.6 | N/A | CGenFF program v2.5.1/CGenFF v4.6 | CGenFFa |
| CGenFF legacy v 1.0 | CGenFFv3.0.1 | N/A | CGenFF legacy program v1.0/CGenFFv 3.0.1 | CGenFF Legacy |
| MATCH v 0.01 | CGenFFv3.0.1 | N/A | MATCH/CGenFF v3.0.1 | MATCH/CGenFF |
| Antechamber, AmberTools21 | GAFF2.11 | RESP | GAFF2.11/RESP | GAFF2/RESP |
| Antechamber, AmberTools21 | GAFF2.11 | AM1-BCC | GAFF2.11/AM1-BCC | GAFF2/AM1-BCC |
| Antechamber, AmberTools21 | GAFF1.81 | RESP | GAFF1.81/RESP | GAFF/RESP |
| Antechamber, AmberTools21 | GAFF1.81 | AM1-BCC | GAFF1.81/AM1-BCC | GAFF/AM1-BCC |
The abbreviation “CGenFF” should be reserved only for the latest versions of CGenFF program and FF at the time of the study.
Conclusions.
Parameter assignment programs allow the application of generalized small-molecule FFs to diverse chemical structures. Here, we showed that (1) MATCH frequently assigns atom types and parameters that differ from CGenFF, affecting all ligands investigated in this study, (2) AM1-BCC charges are distinct from RESP charges, and thus GAFF2 performs differently based on the charge model used, and (3) OpenFF and GAFF, particularly their nonbonded parameters, are similar. These differences and similarities directly affect the conformational and energetic properties of ligands in MD simulations, as presented for two example compounds. Accordingly, researchers must carefully implement generalized FFs using the appropriate atom typing and parameter assignment programs when considering their research aims. For simulation studies using the CHARMM36 biomolecular FF in which the standard CGenFF parameters are desired, the CGenFF program with the latest CGenFF parameters (currently v4.6) should be used, while MATCH represents a viable option to assign CGenFF parameters, though the resulting FF differs significantly. For AMBER studies in which high accuracy is required, we recommend GAFF2/RESP over GAFF2/AM1-BCC, although AMB1-BCC is a reasonable substitute in studies on a large number of molecules. Users should also inspect the format of their input ligand structure (e.g., correctly assigned bond orders in MOL2 files) to ensure the peak performance of the parameter assignment programs. The CGenFF program robustly assigns parameters with the use of MOL2 files converted from PDB by Open Babel49 (as in this study), Molecular Operating Environment (MOE),50 Schrödinger Maestro,51 and Marvin Sketch,52 resulting in the same topologies and parameters. However, variation with MATCH when using MOE generated MOL2 files were observed as described in the Supporting Information (Impact of method for PDB to MOL2 conversion, Figure S12). Nonetheless, the presented results remain largely the same regardless of the source of the initial ligand files, which was further tested through use of SDF files, directly from Schindler et al.,53 as input rather than PDB files, directly from Gapsys et al.33 (Tables S12, S13). In addition, users should be aware of differences in the standard54 and CHARMM specific versions55, 10 of the TIP3P water model and use the model recommended for the specific FF being used. Importantly, we encourage future publications to follow standard nomenclature, explicitly stating the versions of the FFs, parameter assignment program, and/or charge models used. Overall, the presented results highlight the importance of the selected parameter assignment program as well as the small-molecule FF to maintain the integrity of the FF used.
Supplementary Material
ACKNOWLEDGMENT
The authors acknowledge financial support from NIH GM131710.
Footnotes
The authors declare the following conflict of interest: A.D.M. Jr. is co-founder and CSO of SilcsBio LLC.
ASSOCIATED CONTENT
Supporting Information. The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.2c00615.
Details of methods, correlation plots of non-bonded parameters, distribution of atomic charge differences, atom numbers of simulated ligands, dihedral angle distributions of simulated ligands, population of RMSD-based clusters, average interaction energies of the simulated ligands with the surrounding solvent molecules, identification of CGenFF program and FF version through CGenFF program output, identification of GAFF version and charge model, tabulated data of discrepancies in atom types and partial atomic charges along with the number of ligands affected by discrepancies, tabulated data of percent bonded parameters with discrepancies and ligands affected by the discrepancies, tabulated data of similarities in assigned bonded parameters, and assigned atom types and atomic charges for simulated ligands (PDF)
DATA AND SOFTWARE AVAILABILITY
All methodological details are provided in the Supporting Information. All simulation data are available upon request. The generated ligand topology and parameter files are available at https://github.com/mackerell-lab/small_mol_FF_toppar_JCIM
REFERENCES
- (1).Mackerell AD Jr.; Feig M; Brooks CL 3rd, Extending the Treatment of Backbone Energetics in Protein Force Fields: Limitations of Gas-Phase Quantum Mechanics in Reproducing Protein Conformational Distributions in Molecular Dynamics Simulations. J Comput Chem 2004, 25 (11), 1400–15. [DOI] [PubMed] [Google Scholar]
- (2).Guvench O; Hatcher E; Venable RM; Pastor RW; MacKerell AD, Charmm Additive All-Atom Force Field for Glycosidic Linkages between Hexopyranoses. J Chem Theory Comput 2009, 5 (9), 2353–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Klauda JB; Venable RM; Freites JA; O’Connor JW; Tobias DJ; Mondragon-Ramirez C; Vorobyov I; MacKerell AD; Pastor RW, Update of the Charmm All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J Phys Chem B 2010, 114 (23), 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Raman EP; Guvench O; MacKerell AD, Charmm Additive All-Atom Force Field for Glycosidic Linkages in Carbohydrates Involving Furanoses. J Phys Chem B 2010, 114 (40), 12981–12994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Denning EJ; Priyakumar UD; Nilsson L; Mackerell AD Jr., Impact of 2’-Hydroxyl Sampling on the Conformational Properties of Rna: Update of the Charmm All-Atom Additive Force Field for Rna. J Comput Chem 2011, 32 (9), 1929–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Guvench O; Mallajosyula SS; Raman EP; Hatcher E; Vanommeslaeghe K; Foster TJ; Jamison FW; MacKerell AD, Charmm Additive All-Atom Force Field for Carbohydrate Derivatives and Its Utility in Polysaccharide and Carbohydrate–Protein Modeling. J Chem Theory Comput 2011, 7 (10), 3162–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Best RB; Zhu X; Shim J; Lopes PEM; Mittal J; Feig M; MacKerell AD, Optimization of the Additive Charmm All-Atom Protein Force Field Targeting Improved Sampling of the Backbone Φ, Ψ and Side-Chain Χ1 and Χ2 Dihedral Angles. J Chem Theory Comput 2012, 8 (9), 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hart K; Foloppe N; Baker CM; Denning EJ; Nilsson L; MacKerell AD, Optimization of the Charmm Additive Force Field for DNA: Improved Treatment of the Bi/Bii Conformational Equilibrium. J Chem Theory Comput 2012, 8 (1), 348–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Mallajosyula SS; Guvench O; Hatcher E; MacKerell AD, Charmm Additive All-Atom Force Field for Phosphate and Sulfate Linked to Carbohydrates. J Chem Theory Comput 2012, 8 (2), 759–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Huang J; Rauscher S; Nawrocki G; Ran T; Feig M; de Groot BL; Grubmüller H; MacKerell AD, Charmm36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat Methods 2017, 14 (1), 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bergonzo C; Cheatham TE, Improved Force Field Parameters Lead to a Better Description of Rna Structure. J Chem Theory Comput 2015, 11 (9), 3969–3972. [DOI] [PubMed] [Google Scholar]
- (12).Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C, Ff14sb: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99sb. J Chem Theory Comput 2015, 11 (8), 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Zgarbová M; Šponer J; Otyepka M; Cheatham TE; Galindo-Murillo R; Jurečka P, Refinement of the Sugar–Phosphate Backbone Torsion Beta for Amber Force Fields Improves the Description of Z- and B-DNA. J Chem Theory Comput 2015, 11 (12), 5723–5736. [DOI] [PubMed] [Google Scholar]
- (14).Galindo-Murillo R; Robertson JC; Zgarbová M; Šponer J; Otyepka M; Jurečka P; Cheatham TE, Assessing the Current State of Amber Force Field Modifications for DNA. J Chem Theory Comput 2016, 12 (8), 4114–4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Tian C; Kasavajhala K; Belfon KAA; Raguette L; Huang H; Migues AN; Bickel J; Wang Y; Pincay J; Wu Q; Simmerling C, Ff19sb: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J Chem Theory Comput 2020, 16 (1), 528–552. [DOI] [PubMed] [Google Scholar]
- (16).Dickson CJ; Walker RC; Gould IR, Lipid21: Complex Lipid Membrane Simulations with Amber. J Chem Theory Comput 2022, 18 (3), 1726–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Oostenbrink C; Villa A; Mark AE; van Gunsteren WF, A Biomolecular Force Field Based on the Free Enthalpy of Hydration and Solvation: The Gromos Force-Field Parameter Sets 53a5 and 53a6. J Comput Chem 2004, 25 (13), 1656–76. [DOI] [PubMed] [Google Scholar]
- (18).Harder E; Damm W; Maple J; Wu C; Reboul M; Xiang JY; Wang L; Lupyan D; Dahlgren MK; Knight JL; Kaus JW; Cerutti DS; Krilov G; Jorgensen WL; Abel R; Friesner RA, Opls3: A Force Field Providing Broad Coverage of Drug-Like Small Molecules and Proteins. J Chem Theory Comput 2016, 12 (1), 281–296. [DOI] [PubMed] [Google Scholar]
- (19).Brooks BR; Bruccoleri RE; Olafson BD; States DJ; Swaminathan S. a.; Karplus M, Charmm: A Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J Comput Chem 1983, 4 (2), 187–217. [Google Scholar]
- (20).Case DA; Cheatham TE 3rd; Darden T; Gohlke H; Luo R; Merz KM Jr.; Onufriev A; Simmerling C; Wang B; Woods RJ, The Amber Biomolecular Simulation Programs. J Comput Chem 2005, 26 (16), 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Abraham MJ; Murtola T; Schulz R; Páll S; Smith JC; Hess B; Lindahl E, Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar]
- (22).Phillips JC; Hardy DJ; Maia JDC; Stone JE; Ribeiro JV; Bernardi RC; Buch R; Fiorin G; Hénin J; Jiang W; McGreevy R; Melo MCR; Radak BK; Skeel RD; Singharoy A; Wang Y; Roux B; Aksimentiev A; Luthey-Schulten Z; Kalé LV; Schulten K; Chipot C; Tajkhorshid E, Scalable Molecular Dynamics on Cpu and Gpu Architectures with Namd. J Chem Phys 2020, 153 (4), 044130–044130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Vanommeslaeghe K; Hatcher E; Acharya C; Kundu S; Zhong S; Shim J; Darian E; Guvench O; Lopes P; Vorobyov I; Mackerell AD Jr., Charmm General Force Field: A Force Field for Drug-Like Molecules Compatible with the Charmm All-Atom Additive Biological Force Fields. J Comput Chem 2010, 31 (4), 671–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Yu W; He X; Vanommeslaeghe K; MacKerell AD Jr., Extension of the Charmm General Force Field to Sulfonyl-Containing Compounds and Its Utility in Biomolecular Simulations. J Comput Chem 2012, 33 (31), 2451–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Hegazy L; Richards NG, Optimized Cgenff Force-Field Parameters for Acylphosphate and N-Phosphonosulfonimidoyl Functional Groups. J Mol Model 2013, 19 (11), 5075–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Soteras Gutierrez I; Lin FY; Vanommeslaeghe K; Lemkul JA; Armacost KA; Brooks CL 3rd; MacKerell AD Jr., Parametrization of Halogen Bonds in the Charmm General Force Field: Improved Treatment of Ligand-Protein Interactions. Bioorg Med Chem 2016, 24 (20), 4812–4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA, Development and Testing of a General Amber Force Field. J Comput Chem 2004, 25 (9), 1157–74. [DOI] [PubMed] [Google Scholar]
- (28).Ozpinar GA; Peukert W; Clark T, An Improved Generalized Amber Force Field (Gaff) for Urea. J Mol Model 2010, 16 (9), 1427–40. [DOI] [PubMed] [Google Scholar]
- (29).Jambeck JP; Lyubartsev AP, Update to the General Amber Force Field for Small Solutes with an Emphasis on Free Energies of Hydration. J Phys Chem B 2014, 118 (14), 3793–804. [DOI] [PubMed] [Google Scholar]
- (30).Boyd NJ; Wilson MR, Optimization of the Gaff Force Field to Describe Liquid Crystal Molecules: The Path to a Dramatic Improvement in Transition Temperature Predictions. Phys Chem Chem Phys 2015, 17 (38), 24851–65. [DOI] [PubMed] [Google Scholar]
- (31).Torsello M; Pimenta AC; Wolters LP; Moreira IS; Orian L; Polimeno A, General Amber Force Field Parameters for Diphenyl Diselenides and Diphenyl Ditellurides. J Phys Chem A 2016, 120 (25), 4389–400. [DOI] [PubMed] [Google Scholar]
- (32).He X; Man VH; Yang W; Lee TS; Wang J, A Fast and High-Quality Charge Model for the Next Generation General Amber Force Field. J Chem Phys 2020, 153 (11), 114502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Gapsys V; Hahn DF; Tresadern G; Mobley DL; Rampp M; de Groot BL, Pre-Exascale Computing of Protein-Ligand Binding Free Energies with Open Source Software for Drug Design. J Chem Inf Model 2022, 62 (5), 1172–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Lundborg M; Lindahl E, Automatic Gromacs Topology Generation and Comparisons of Force Fields for Solvation Free Energy Calculations. J Phys Chem B 2015, 119 (3), 810–23. [DOI] [PubMed] [Google Scholar]
- (35).Elisee E; Gapsys V; Mele N; Chaput L; Selwa E; de Groot BL; Iorga BI, Performance Evaluation of Molecular Docking and Free Energy Calculations Protocols Using the D3r Grand Challenge 4 Dataset. J Comput Aided Mol Des 2019, 33 (12), 1031–1043. [DOI] [PubMed] [Google Scholar]
- (36).Gapsys V; Pérez-Benito L; Aldeghi M; Seeliger D; van Vlijmen H; Tresadern G; de Groot BL, Large Scale Relative Protein Ligand Binding Affinities Using Non-Equilibrium Alchemy. Chem Sci 2020, 11 (4), 1140–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Yesselman JD; Price DJ; Knight JL; Brooks CL 3rd, Match: An Atom-Typing Toolset for Molecular Mechanics Force Fields. J Comput Chem 2012, 33 (2), 189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Vanommeslaeghe K; MacKerell AD Jr., Automation of the Charmm General Force Field (Cgenff) I: Bond Perception and Atom Typing. J Chem Inf Model 2012, 52 (12), 3144–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Vanommeslaeghe K; Raman EP; MacKerell AD Jr., Automation of the Charmm General Force Field (Cgenff) Ii: Assignment of Bonded Parameters and Partial Atomic Charges. J Chem Inf Model 2012, 52 (12), 3155–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Jakalian A; Bush BL; Jack DB; Bayly CI, Fast, Efficient Generation of High-Quality Atomic Charges. Am1-Bcc Model: I. Method. J Comput Chem 2000. [DOI] [PubMed] [Google Scholar]
- (41).Jakalian A; Jack DB; Bayly CI, Fast, Efficient Generation of High-Quality Atomic Charges. Am1-Bcc Model: Ii. Parameterization and Validation. J Comput Chem 2002, 23 (16), 1623–41. [DOI] [PubMed] [Google Scholar]
- (42).Bayly CI; Cieplak P; Cornell W; Kollman PA, A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The Resp Model. J Phys Chem 1993, 97 (40), 10269–10280. [Google Scholar]
- (43).Qiu Y; Smith DGA; Boothroyd S; Jang H; Hahn DF; Wagner J; Bannan CC; Gokey T; Lim VT; Stern CD; Rizzi A; Tjanaka B; Tresadern G; Lucas X; Shirts MR; Gilson MK; Chodera JD; Bayly CI; Mobley DL; Wang L-P, Development and Benchmarking of Open Force Field V1.0.0—the Parsley Small-Molecule Force Field. J Chem Theory Comput 2021, 17 (10), 6262–6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Mobley DL; Bannan CC; Rizzi A; Bayly CI; Chodera JD; Lim VT; Lim NM; Beauchamp KA; Slochower DR; Shirts MR; Gilson MK; Eastman PK, Escaping Atom Types in Force Fields Using Direct Chemical Perception. J Chem Theory Comput 2018, 14 (11), 6076–6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Wang J; Cieplak P; Kollman PA, How Well Does a Restrained Electrostatic Potential (Resp) Model Perform in Calculating Conformational Energies of Organic and Biological Molecules? J Comput Chem 2000, 21 (12), 1049–1074. [Google Scholar]
- (46).Bayly C; McKay D An Informal Amber Small Molecule Force Field: Parm@Frosst 2010. http://www.ccl.net/cca/data/parm_at_Frosst/.
- (47).Ibrahim MA, Molecular Mechanical Study of Halogen Bonding in Drug Discovery. J Comput Chem 2011, 32 (12), 2564–74. [DOI] [PubMed] [Google Scholar]
- (48).Kolář M; Hobza P, On Extension of the Current Biomolecular Empirical Force Field for the Description of Halogen Bonds. J Chem Theory Comput 2012, 8 (4), 1325–1333. [DOI] [PubMed] [Google Scholar]
- (49).O’Boyle NM; Banck M; James CA; Morley C; Vandermeersch T; Hutchison GR, Open Babel: An Open Chemical Toolbox. Journal of Cheminformatics 2011, 3 (1), 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).ULC, C. C. G. Molecular Operating Environment (Moe), 2020.09, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2022. [Google Scholar]
- (51).Schrödinger L Maestro. Schrödinger Release 2022–2, New York, NY, 2021. [Google Scholar]
- (52).Chemaxon Marvinsketch Version 17.1.30.0, 2017. [Google Scholar]
- (53).Schindler CEM; Baumann H; Blum A; Böse D; Buchstaller H-P; Burgdorf L; Cappel D; Chekler E; Czodrowski P; Dorsch D; Eguida MKI; Follows B; Fuchß T; Grädler U; Gunera J; Johnson T; Jorand Lebrun C; Karra S; Klein M; Knehans T; Koetzner L; Krier M; Leiendecker M; Leuthner B; Li L; Mochalkin I; Musil D; Neagu C; Rippmann F; Schiemann K; Schulz R; Steinbrecher T; Tanzer E-M; Unzue Lopez A; Viacava Follis A; Wegener A; Kuhn D, Large-Scale Assessment of Binding Free Energy Calculations in Active Drug Discovery Projects. J Chem Inf Model 2020, 60 (11), 5457–5474. [DOI] [PubMed] [Google Scholar]
- (54).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML, Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys 1983, 79 (2), 926–935. [Google Scholar]
- (55).Durell SR; Brooks BR; Ben-Naim A, Solvent-Induced Forces between Two Hydrophilic Groups. J Phys Chem 1994, 98 (8), 2198–2202. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All methodological details are provided in the Supporting Information. All simulation data are available upon request. The generated ligand topology and parameter files are available at https://github.com/mackerell-lab/small_mol_FF_toppar_JCIM