

Abstract
While vaccines remain at the forefront of global healthcare responses, pioneering therapeutics against SARS-CoV-2 are expected to fill the gaps for waning immunity. Rapid development and approval of orally available direct-acting antivirals targeting crucial SARS-CoV-2 proteins marked the beginning of the era of small-molecule drugs for COVID-19. In that regard, the papain-like protease (PLpro) can be considered a major SARS-CoV-2 therapeutic target due to its dual biological role in suppressing host innate immune responses and in ensuring viral replication. Here, we summarize the challenges of targeting PLpro and innovative early-stage PLpro-specific small molecules. We propose that state-of-the-art computer-aided drug design (CADD) methodologies will play a critical role in the discovery of PLpro compounds as a novel class of COVID-19 drugs.
Keywords: papain-like protease, artificial intelligence, small molecules, computer-aided drug design, COVID-19, SARS-CoV-2
Waning immunity opens the door for COVID-19 drugs
In the early stages of the coronavirus disease 2019 (COVID-19) pandemic, many scientific groups around the world deployed a large variety of drug discovery pipelines to identify chemical entities acting against its etiologic agent, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Though SARS-CoV-2 was classified as a novel pathogen, it shared many features with previously known coronavirus species including SARS-CoV-1 and Middle East respiratory syndrome (MERS) [1]. From the initial studies of SARS-CoV-2, it became evident that several proteins produced by the virus, including the main protease [Mpro, also known as 3-chymotrypsin-like protease (3Clpro)], the RNA-dependent RNA polymerase (RdRp), and the PLpro, had significant potential as therapeutic targets [2., 3., 4.]. Accordingly, Pfizer developed nirmatrelvir, an anti-COVID-19 orally bioavailable small-molecule agent targeting Mpro, which received FDA authorization for emergency use in combination with the CYP3A4 inhibitor, ritonavir [5]. Simultaneously, Merck developed molnupiravir, a ribonucleoside analog directly targeting the RdRp, which also received FDA approval for emergency use as an oral antiviral for adults [6]. Despite these groundbreaking COVID-19 therapeutic options, subsequent analysis has shown that these drugs exhibited subpar pharmacological properties: Paxlovid, the drug cocktail of nirmatrelvir and ritonavir, has been associated with several concerning drug–drug interactions, while molnupiravir has demonstrated lower-than-expected efficacy, requiring cautious management [7,8]. Nevertheless, the two drugs are pioneering the postvaccine COVID-19 era, where powerful oral antiviral drugs could fill the gaps for the immunocompromised, the elderly, and children while vaccines rightly remain at the forefront of global healthcare responses [9,10].
Even though Mpro and RdRp are well under investigation, it is important to study other SARS-CoV-2 constituents, such as the aforementioned PLpro enzyme, as therapeutic targets in the event that Mpro or RdRp develops resistant mutations under therapeutic pressure [11,12]. It is expected that combination therapies of PLpro inhibitors with other SARS-CoV-2-specific drugs will represent viable options to deal with potential drug resistance in COVID-19 [13]. Additionally, both Mpro and PLpro should be considered for synergistic targeting with antiviral drug cocktails as these were shown to be very effective against numerous serious viral infections, including HIV [14]. Consequently, interest in SARS-CoV-2 PLpro as the next major therapeutic target against COVID-19 grew significantly since approval of Paxlovid and molnupiravir.
A number of high-quality reviews covering SARS-CoV-2 PLpro’s biological functions, structural features, and drug re-repurposing opportunities were already reported with great details [15., 16., 17., 18.]. These reviews also provide excellent and extensive summaries of currently known PLpro-specific inhibitors and their binding mechanisms. Here, we review SARS-CoV-2 PLpro enzyme as a potential COVID-19 therapeutic target and consider the challenges associated with its drug development efforts. We discuss the transformative role of computer-aided drug design (CADD) (see Glossary) methodologies, and we propose the deployment of artificial intelligence (AI)-enhanced CADD methodologies as possible guides to identify and design novel SARS-CoV-2 PLpro small-molecule inhibitors. We anticipate that CADD will play an important role in overcoming the hurdles faced by PLpro drug development. Thus, it is both timely and important to review the emergence of state-of-the-art CADD tools implementing AI to support the development of innovative PLpro-directed drugs.
Targeting the dual biological role of PLpro as therapeutic opportunity
SARS-CoV-2 PLpro plays a dual biological role in (i) suppressing host innate immune responses which leads to increased viral proliferation and (ii) processing the viral polyproteins which ensures viral RNA replication (Figure 1A) [19]. Specifically, PLpro enables SARS-CoV-2 to evade host antiviral responses as the protease is characterized by deISGylating and deubiquitinating activities [20]. ISGylation and ubiquitination post-translational modifications normally initiate the release of necessary interferons (IFNs) to upregulate the production of antiviral cytokines and instigate immune responses [21]. However, it was demonstrated that PLpro interferes with the host's capabilities to respond to infections by removing these post-translational modifications from selected proteins [22,23]. PLpro recognizes both interferon-stimulated gene 15 (ISG15) and ubiquitin (Ub) to cleave them from proteins, thus diminishing the host's abilities to respond adequately to SARS-CoV-2 infections. PLpro also cleaves the viral polyproteins to generate nonstructural proteins (nsp) 1–3 that are required in the replication and transcription of the viral RNA [24]. Mpro is responsible for generating the other necessary nsp ensuring adequate replication of SARS-CoV-2. Therefore, inhibition of PLpro should restore antiviral signaling and natural defenses of the host against SARS-CoV-2 as well as blocking replication of the virus.
Figure 1.

The dual role of SARS-CoV-2 PLpro.
(A) Inhibition of PLpro's dual activities is a viable venue for the development of therapeutics for COVID-19. Briefly, following SARS-CoV-2 infection, ISG15 interacts with interferon responsive factor 3 (IRF3) to initiate release of interferons (IFNs) and nuclear factor kappa B (NF-κB), leading to increase host responses against viral infections. However, PLpro is involved in deISGylation activity and interferes with the host’s ability to respond to viral infections. Specifically, SARS-CoV-2 PLpro cleaves ISG15 from IRF3 to attenuate immune responses. PLpro is also involved in proteolytic activity by cleaving the viral polyproteins at three specific positions to generate three nonstructural proteins (nsp 1–3). The nsp are critical to facilitate the replication/transcription of the virus. Successful inhibition of PLpro should suppress antiviral infections, as SARS-CoV-2 will not be able to establish a functional replication/transcription process, and it should enhance antiviral immune responses, as ISG15 will remain intact to carry out its objectives. (B) To perform its dual biological role, PLpro relies on its capacity to bind to host and viral proteins via its active site. Development of new small-molecule therapeutics that target and block the active site should effectively inhibit PLpro’s capacities. The active site of PLpro is centered on the catalytic triad of Cys111-His272-Asp286 (shown as magenta sticks). PLpro can be divided into different sections as colored. Orange: Ubl domain; pink: thumb subdomain; yellow: Zn-binding domain (fingers subdomain); teal: active site (palm subdomain). PDB ID: 7D7K. This figure was created with BioRender.com. Abbreviation: PLpro, papain-like protease.
SARS-CoV-2 PLpro, like other human proteases, adopts a ‘thumb-palm-fingers’ structure, where three distinct domains can be distinguished. Those include a cysteine cleavage site (also referred as the active site) that contains the catalytic triad of Cys111, His272, and Asp286 (Figure 1B), the zinc-binding domain, and an N-terminal ubiquitin-like domain (Ubl). One of the roles of the zinc-binding domain is to maintain PLpro's structural integrity [25], while Ubl domain has been suggested to play a role in blocking the innate immune responses, but its specific functions in SARS-CoV-2 are not yet well understood [26]. The active site of PLpro, the most studied of the three domains, is responsible for generating nsp 1–3 from the viral polyproteins, as well as processing ISG15 and Ub. Herein, we broadly define the active site to include both the catalytic site centered around the catalytic triad, and the substrate binding cleft centered on the interface of PLpro and ISG15/Ub’s C-terminus. This active site recognizes a consensus residue sequence of ‘LXGG’ along the viral polyproteins and cleaves them after the second Gly. SARS-CoV-2 PLpro identifies the same consensus sequence on ISG15 and Ub conjugates – with a preference toward ISG15 due to reduced ability to process Ub, to cleave them from targeted proteins. This consensus motif delimits the active site into four distinct subsites with each named per their relative position to the catalytic triad: S4 being the furthest away from the catalytic triad, followed by S3, S2, and S1, closest to it. The residues of the substrates recognized in each subsite are named following the same nomenclature, from P4 to P1 – with P4 corresponding to Leu in S4, P3 to X in S3, P2 to Gly in S2, and P1 to Gly in S1 (Figure 2A) [26]. The interaction mechanisms between the substrates and PLpro map out the residues to be occupied by ligands for successful inhibition. P4(Leu) forms H-bonds with the side chain of Asp164 while P3(Arg) forms H-bonds with Glu161 and Leu162, and an additional H-bond with Tyr268. Hydrophobic interactions are still the major contributors for binding interactions in the S3 and S4, mainly with Leu162, Tyr264, Tyr268, and Tyr273. In S2 and S1, the P2(Glu) forms two backbone H-bonds with Gly163. Thus, the disposition of PLpro's active site is an important structural blueprint for rational drug design.
Figure 2.

The active site of SARS-CoV-2 PLpro.
(A) PLpro's active site recognizes a ‘LXGG’ consensus sequence on viral polyproteins, ISG15, and ubiquitin. The PLpro is characterized by a Gly-Gly tunnel in the S1 and S2 subsites, and more accommodating S3 and S4 subsites. The active site is delimited into four subsites, each occupied by a residue of the substrate sequence. Residues of ISG15 and ubiquitin substrates are colored per the occupied position in the active site: green: P4 Leu; teal: P3 Arg; red: P2 Gly; yellow: P1 Gly. The surface of the subsites is colored per the substrate residues, in order from S4 (blue), S3 (orange), S2 (magenta), and S1 (gray). Important PLpro active site residues are shown in thin gray sticks and labeled appropriately. PDB used: 7RBR for ubiquitin and 7RBS for ISG15. Pink ribbons: PLpro with ISG15; gray ribbons: PLpro with ubiquitin. (B) The BL2 loop, spanning residues 267-271 in the S3 and S4 subsites, is highly flexible. The BL2 loop undergoes induced-fitted effects and enables the active site to accommodate different binding partners by transitioning between open and closed states. S4 subsite shown in the blue oval and S3 subsite shown in the green oval. The ‘BL2 groove’ is adjacent to the S4 and is formed when the BL2 loop is in its closed conformation. Orange: apo (PDB ID: 6W9C); magenta: Ub (PDB ID: 6XAA); teal: VIR250 (PDB ID: 6WUU); white: ISG15 (PDB ID: 6XA9); green: GRL-0617 (PDB: 7CMD).
To summarize, for PLpro to perform its dual biological role, the protease relies on its capacity to bind to host and viral proteins via its active site. Development of new small-molecule therapeutics that target and block the active site should effectively inhibit PLpro in a ‘two birds, one stone’ scenario [16]. Firstly, SARS-CoV-2 would not be able to establish functional replication/transcription, suppressing antiviral infection, and secondly, ISG15/Ub modifications would remain intact and initiate antiviral immune responses [27]. Accordingly, it is crucial to understand the structural features and characteristics of the active site for successful drug designs and optimization campaigns.
Structural challenges of targeting the SARS-CoV-2 PLpro
Rational drug design against SARS-CoV-2 PLpro remains challenging due to its unique structural features. The SARS-CoV-2 PLpro active site is characterized by (i) a highly dynamic and flexible blocking loop 2 (BL2 loop) located near the S3 and S4 subsites, (ii) amino acid specificity in the S4 subsite, and (iii) tunnel-like rigidity in the S1 and S2 subsites.
Several residues in PLpro’s active site have dynamic side/main chains to accommodate different substrates. Notably, the BL2 loop, spanning residues 267–271 in the S3 and S4 subsites, is highly flexible (Figure 2B). The two main residues at the center of the BL2 loop, Tyr268, and Gln269 are heavily involved in the induced-fit effects observed in PLpro’s active site and its ability to adopt a ‘closed’ or ‘open’ configuration. The two residues were shown to have a critical role in the active site’s ability to interact with a wider range of binding partners [24]. Hence, the interaction network in PLpro's S3 and S4 subsites is partly dictated by the dynamic BL2 loop. Furthermore, as a cysteine protease, PLpro can also modulate its structural plasticity via protonation and deprotonation of its Cys and His residues. Molecular dynamics (MD) simulations revealed that modifying Cys270's charge affected the structure of the BL2 loop [28]. Likewise, other simulation techniques revealed that the fluctuations in the BL2 loop could disrupt inhibitors from binding to the active site [29]. Therefore, it should be viable to design small molecules that can disable the BL2 loop's movements and lock it in place. Expanding an inhibitor's interaction network specifically with the BL2 loop could help achieve this objective, instead of only optimizing interactions with the S3 and S4 subsites.
The S4 subsite in PLpro's active site is highly conserved between SARS-CoV-1 and SARS-CoV-2 species. This subsite dictates the specificity of the consensus ‘LXGG’ motif by recognizing the Leu side chain in its pocket [30]. Comparatively, the S3 subsite lacks residue preference as it recognizes the backbone of the P3 residue. In addition, due to the obligatory Gly-Gly recognition, the S1 and S2 subsites adopt a narrow tunnel-shaped conformation [24]. Inevitably, most small-molecule designs have focused on the open-ended S3 and S4 subsites in the active site, as they present more space to accommodate ligands.
Early-stage small molecules targeting SARS-CoV-2 PLpro
One of the most studied inhibitors of SARS-CoV-2 PLpro is GRL-0617 (Table 1 ). GRL-0617 is a noncovalent binder developed in 2008 for SARS-CoV-1 PLpro, occupying the S3 and S4 subsites to inhibit the protease's proteolytic activity [31]. As both PLpro enzymes from SARS-CoV-1 and SARS-CoV-2 share many structural and biological features, GRL-0617 unsurprisingly maintained its activity against SARS-CoV-2 PLpro with an IC50 of 1.5 ± 0.08 μM in enzymatic assays [20]. This value is in line with that observed for SARS-CoV-1 (IC50: 0.66 ± 0.09 μM) [31]. Cocrystal structures of SARS-CoV-2 PLpro with GRL-0617 reveal that residues Tyr268 and Gln269 on the BL2 loop are shifted toward the compound to interact strongly with it. Gln269 is oriented toward the inhibitor to form optimal H-bonds, anchoring the flexible BL2 loop to GRL-0617 and fixing the loop in a stable and closed conformation. Additional hydrophobic interactions with Asp164, Pro247, Pro248, Tyr264, and Tyr268 also maintain the compound tightly in the S3 and S4 of PLpro's active site [27]. These promising results encouraged several studies to seek out new PLpro hits with a similar mode of action, where a compound occupies the S3 and S4 subsites and interacts with the BL2 loop to block access to the catalytic triad, thus stopping PLpro from carrying out its proteolytic functions.
Table 1.
Early-stage noncovalent inhibitors against SARS-CoV-2 PLproa
| Inhibitor | Name in study | Potency in biochemical assays (IC50) | Potency in cellular assays (EC50) | Discovery method | Targeted subsites | Refs |
|---|---|---|---|---|---|---|
GRL-0617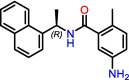
|
GRL-0617 | 1.5 μM | 21 μM (Vero E6) | HTS | S3, S4 | [20] |
Tanshinone IIA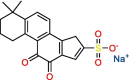
|
Tanshinone IIA sulfonate sodium | 1.65 μM | Not tested | HTS | S3, S4 | [34] |
C1
|
DY2-97 | ~100 μM | Not tested | Derivative of GRL-0617 | S3, S4 | [37] |
C2
|
ZN-3-61 | >>10 μM | Not tested | Derivative of GRL-0617 | S3, S4 | [37] |
C3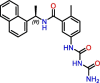
|
Compound 2 | 5.1 μM | No inhibition (Vero E6) | Derivative of GRL-0617 | S3, S4 | [26] |
C4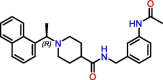
|
Compound 12 | 2.69 μM | Not tested | Derivative of GRL-0617 | S3, S4 | [33] |
C5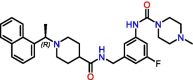
|
Compound 19 | 0.44 μM | 0.18 μM (HeLa-hACE2) | Derivative of GRL-0617 | S3, S4 | [33] |
C6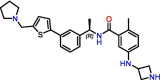
|
XR8-24 | 0.56 μM | 1.2 μM (Vero E6) | Derivative of GRL-0617 | S3, S4, BL2 groove | [37] |
C7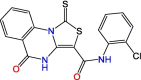
|
F0213 | 7.4 μM | 4.5 μM (Vero E6) | HTS | S3, S4 | [46] |
C8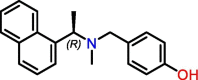
|
Jun9-72-2 | 0.67 μM | 6.62 μM (Vero E6); 7.90 μM (Caco2-hACE2) | HTS | S3, S4 | [47] |
C9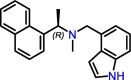
|
Jun9-75-4 | 0.62 μM | 7.88 μM (Vero E6); 12.48 μM (Caco2-hACE2) | HTS | S3, S4 | [47] |
C10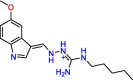
|
Compound 56 | 1.42 μM | Not tested | VS | S3, S4 | [51] |
C11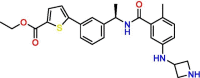
|
Compound 2 | 0.6 μM | Not tested | VS | S3, S4 | [52] |
C12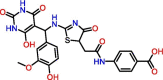
|
Compound 7 | 0.085 μM | Not tested | VS | S3, S4 | [53,54] |
C13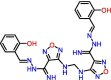
|
Compound 13 | 0.063 μM | Not tested | VS | S3, S4 | [53,54] |
Abbreviations: BL2, blocking loop 2; hACE2, human angiotensin-converting enzyme 2.
Significant efforts were made to experimentally screen medium- to large-sized chemical libraries against SARS-CoV-2 PLpro's active site [23,32., 33., 34., 35.]. However, experimentally identifying diverse PLpro-specific chemical scaffolds proved more challenging than expected. Even though repurposed drugs such as tanshinone IIA (Table 1) showed potency in high-throughput screening (HTS) and offered valuable insight into the topology of PLpro's active site, their scaffolds represent challenging starting points for optimization due to their limited design space and lack of extended chemical structure (Table 1) [34]. Hence, most of the published compounds are still at the early discovery stage. Large-scale HTS of commercial libraries reported low hit rates (0.017% to 0.1%), with few or no reports of subsequent optimization of hits [36]. We hypothesize that a driving factor for the limited success of HTS campaigns against PLpro is the specific binding requirements of the active site. For example, a polar region located in the S4 subsite of PLpro was revealed to be occupied by multiple water molecules [27], and GRL-0617 contains a chiral methyl group that points toward this specific region. Shen et al. attempted to occupy this region by substituting the methyl group for larger groups to enhance interaction with PLpro, as shown by their compounds, C1 and C2 (Table 1) [37]. These molecules lost their activity, indicating that the chiral methyl is required for inhibitory activity. This also suggests that this polar region is a stable water-binding site with a specific role, or that a very specific interaction from a ligand would be required with the correct combination of flexibility and H-bonding opportunities. We posit that a systematic exploration of this water site would substantially improve the understanding of PLpro dynamics and potentially lead to increasing compound potency.
Due to the active site's topology requiring more investigation and the lack of novel high-quality small-molecule leads, PLpro drug discovery efforts focused on optimizing GRL-0617. Studies adopted two general design strategies. Firstly, design analogs to improve the interaction network in the S3 subsite, as demonstrated by compounds C3, C4, and C5 (Table 1). Secondly, design analogs like compound C6 (Table 1) that expand outside of the active site and along PLpro's surface, toward the ‘BL2 groove’ adjacent to the S4 subsite (Figure 2B). This BL2 groove is composed primarily of prolines and is formed only when the BL2 loop is in its closed conformation. Most of these GRL-0617 analogs showed improvement over the parental compound, and all of them engaged the BL2 loop to block physical access to the catalytic triad. In particular, C6 demonstrated that prolonged interactions with Glu167 could potentially lead to reinforced interactions with Asp164 as the main contact required for strong binding with PLpro [37]. C6 also appeared to maintain strong interactions with Pro247, Pro248, and Pro299 near the BL2 groove through its bi-aryl group. New contacts with Gly266 in the BL2 groove itself are interesting novel interactions requiring further analysis to determine their full potential. Nonetheless, these compounds should still be considered as early stage, and likely could be improved further for efficacy and drug likeness. We speculate that a current limitation in PLpro drug discovery could be attributed to the lack of consequential analysis and optimization toward drug-like properties. This possibly stems from a combination of PLpro being the less attractive protease compared with Mpro, and Mpro receiving the most drug development efforts. Thus, there exist very few reports on the pharmacokinetic and pharmacodynamic properties of the literature compounds. However, with the recent development of nirmatrelvir, there is more interest in designing novel oral small-molecule drugs against PLpro. The work of Shen et al. in designing the XR8 series represents a substantial leap forward in identifying successful medicinal chemistry design strategies leading to potent in vitro compounds [37]. However, we do have concerns over the drop in ligand efficiency and an increase in the number of rotatable bonds, H-bond donors, and amine functional groups. These properties may lead to difficulty in optimizing in vivo stability, availability, and formulation toward an oral direct-acting PLpro drug.
One major argument against targeting the SARS-CoV-2 PLpro's active site is a significant structural similarity with catalytic sites of other human proteases. However, there are few or no reports on PLpro inhibitors with significant off-target effects. When testing compounds C4 and C5 in human lung epithelial A549 cells overexpressing human ACE2 receptor (a preclinical model relevant to SARS-CoV-2), no cytotoxicity was observed at concentrations lower than 30 μM. A counter-screen of compounds C4 and C5 against a wide range of human deubiquitinating enzymes (DUB) or DUB-like proteases did not show any significant inhibition. Finally, another counter-screen of C6 analogs against the catalytic domain of human USP7 – the closest human structural homolog of PLpro – revealed no off-target inhibition even at a high concentration of 30 μM. The aforementioned compounds are potent and selective SARS-CoV-2 PLpro inhibitors, and therefore, the active site of SARS-CoV-2 PLpro can be considered as sufficiently unique and suitable for selective targeting [23].
To summarize, empirical HTS campaigns had limited success in identifying high-quality small molecules with preferable bioactive properties. This has biased PLpro drug discovery efforts toward the scaffold of GRL-0617, skewing subsequent analog designs [38]. Hence, the diversity of starting cores is critical to expand our portfolio of PLpro-specific scaffolds and to develop the next-generation PLpro small-molecule therapeutics. This plays to the strength of CADD methodologies that can facilitate the identification of these novel chemotypes.
CADD-guided hit discovery in SARS-CoV-2
With the emergence of COVID-19, structure-based virtual screening (SBVS) studies initially investigated opportunities to repurpose existing drugs or natural products against SARS-CoV-2 PLpro. Although the characterization and optimization of natural compounds is challenging [39], they can act as a structural reference for the development of novel therapeutic agents. The libraries that were virtually screened against PLpro primarily covered public databases including late-phase clinical trial drugs, along with databases of antiviral, anticancer, and antifungal compounds [40., 41., 42., 43., 44.]. But, empirical studies on in silico hits produced by different studies demonstrated that the majority of available drugs and natural products were not potent enough to be relevant against PLpro [45]. Instead, identification of potent hits against SARS-CoV-2 PLpro should come from screening libraries of novel chemical entities. This was demonstrated by Yuan et al. when they identified compound C7 (Table 1) from a collection of 50 080 diverse small-molecule compounds with drug-like properties [46]. The broad-spectrum anti-coronaviral candidate demonstrated promising potency, but poor binding affinity limited its clinical value. Significant optimization will be necessary to enhance its binding and pharmacokinetic profile. Similarly, Ma et al. identified two potent hit compounds, C8 and C9 (Table 1), in a HTS of Enamine's library of 50 000 top-quality and diverse lead-like compounds [47]. The C8 and C9 compounds shared similar features to GRL-0617, highlighting the need to screen even larger libraries for novel hit discovery. However, traditional HTS of ultralarge libraries (>1 000 000 compounds) is not feasible for most scientific groups [48], and large-scale SBVS offers a cost-efficient and faster alternative.
Computational methodologies are able to rapidly identify diverse and unique small-molecule inhibitor candidates for SARS-CoV-2 Mpro [49,50]. Two studies performed smaller scale SBVS followed by biological validation to identify noncovalent small-molecule inhibitors, C10 and C11 [51,52]. As their scaffolds are different from GRL-0617, further binding validation will be required on C10 to fully ascertain its binding profile as it lacks the characteristics often observed in other noncovalent PLpro-specific inhibitors. Notably, C11 which was identified using a pharmacophore (ph4) model and CADD filters is characterized by a scaffold similar to the one developed through extensive medicinal chemistry efforts by Shen et al. Therefore, it could be anticipated that SBVS of ultralarge libraries of novel chemical entities could also yield promising SARS-CoV-2 PLpro hits. Correspondingly, Elseginy et al. virtually screened large commercial libraries of 500 000 molecules [53,54] and identified two hits, C12 and C13 (Table 1), that could efficiently inhibit the growth of SARS-CoV-2-infected Vero E6 cells. However, these two compounds are marred by a high number of H-bond donors, likely leading to poor gut permeability and limiting their clinical value; thus, additional optimization will be required [55].
The aforementioned modern CADD pipeline successfully identified novel small-molecule therapeutics potentially suitable for hit-to-lead optimization while experimental methods struggled. These preliminary studies highlighted the transitioning role of CADD in SARS-CoV-2 drug discovery. Notably, most of the relevant compounds initially identified in SARS-CoV-1 PLpro drug discovery came from costly experimental HTS campaigns, while computationally driven methodologies did not play a significant role in the discovery stage [56,57]. In fact, throughout the SARS and MERS pandemics, in silico methodologies, such as molecular modeling, were used to understand binding interactions and to help optimize compounds, rather than to discover new hits. During COVID-19, there was an urgent need to identify new antiviral therapeutics, and rapid discovery of small molecular hits became the primary research focus in lieu of hit optimization. Thus, in response to SARS-CoV-2, CADD projects could produce novel and potent chemotypes, which reinforces the applicability of computational pipelines against SARS-CoV-2 PLpro [50].
Notably, this highlights a methodological shift, moving from small-scale experimental screening of compounds followed by computational validation of binding hypotheses to ultralarge-scale computational screening of chemical libraries followed by experimental validation of on-target effects [58]. The recent emergence of powerful graphics processing units (GPUs) facilitated such transformation. This resulted in a massive increase in available computational power, and the deep learning (DL) revolution, allowing for the integration of advanced machine learning (ML) algorithms in modern CADD methodologies. Recent reviews by our group and others have extensively covered the emergence of GPUs and ML in CADD and its impact on democratizing drug discovery [59., 60., 61.]. Importantly, they highlight the possibility to deploy state-of-the-art ML-enhanced methodologies to expand our searches for new hits against SARS-CoV-2 PLpro with appropriate lead-likeness at a fraction of the traditional computational cost [62].
Although the usage of CADD methodology can guide and aid in the development of SARS-CoV-2 small-molecule inhibitors, there remain significant challenges for structure-based drug discovery against PLpro. Indeed, the active site of SARS-CoV-2 PLpro includes a highly flexible BL2 loop, while the corresponding induced-fit effects are rarely taken into consideration in SBVS campaigns due to the significant associated computational cost [63]. The concept of induced-fit effect proposes that the shape of the binding site is influenced by the interacting ligand; hence, it is critical to consider the flexibility and intrinsic mobility of both the ligand and the receptor in SBVS [64]. Developing methods that can accurately incorporate the global flexibility of PLpro (and notably the backbone flexibility) could provide important gains for SBVS against PLpro. In summary, balancing computational speed, precision, and accuracy of CADD approaches to ensure good hit rates and diversity of identified scaffolds represents an important goal for future CADD campaigns against the protease.
Deploying state-of-the-art ML approaches for PLpro
ML-enhanced virtual screening methods that can account for flexible protein targets could contribute to tackling the challenging features of PLpro, such as the dynamic BL2 loop. Useful computational methods have been recently developed that can automatically detect the conformations of proteins in ligand-bound states. For example, Degiacomi demonstrated that a generative autoencoder (a DL-based algorithm) – when trained on protein structures produced by MD simulations – could correctly capture the plausible conformations of a target protein (Figure 3A) [65]. In doing so, the autoencoder reduced thousands of possible protein structures to a handful of high-quality conformations that could describe the flexibility observed upon ligand binding. These predicted conformations can then be used to enhance ensemble docking pipelines as they can account for the lack of accurate structural information [66]. Due to their static nature, crystal structures may not represent the protein conformation that is present when a different ligand binds to the target. Being able to incorporate different conformations will enable SBVS to capture virtual hits that would otherwise be missed due to not having the correct crystal structure. This could potentially increase the overall accuracy of in silico hit discovery in general and for the case of PLpro.
Figure 3.

Machine-learning-assisted PLpro drug discovery.
(A) Physics-based neural networks can learn the protein conformational space. The method returns new, plausible near ligand-bound protein structures from the protein conformational space, complementing the existing ones. Specifically, molecular dynamics (MD) simulations generate many protein structures, encompassing all the possible conformations that a protein can adopt, from unbound to bound. A sample of the structures obtained from the MD simulations are used to train a generative autoencoder that correctly reconstructs protein conformations to account for broad flexibility upon ligand binding. High-quality structures (red dots on the 3D latent space) outputted from the autoencoder are selected for SBVS, such as ensemble docking. Being able to incorporate different conformations will enable SBVS to capture virtual hits that would otherwise be missed due to not having the correct crystal structure. (B) Pipeline of a modern deep reinforcement learning (DRL)-based methodology for de novo compound generation used to enhance new drug design. A generative model takes in a seed atom to start the process and continually predicts subsequent atoms of a compound (in SMILES format) until the model produces a valid chemical structure. The ‘drug property prediction model’ checks the biological, chemical, and physical properties of the newly generated molecules, and the model calculates a reward based on the selected features. Depending on the reward threshold, the generative model either further modifies the molecule or stops the generative process. The exploration of this space is important to generate compounds designed for the target of interest. Abbreviation: SBVS, structure-based virtual screening.
Newly developed DL generative models could also be very practical for the design of novel PLpro inhibitors. These methods involve taking reported hits and automatically generating a database of analogs specifically designed for the target of interest to enhance their binding features (Figure 3B) [67,68]. In a proof-of-concept study, Merk et al. deployed a DL model to design new drug-like compounds against two oncogenic agents, retinoid X receptor and peroxisome proliferator-activated receptor [69]. They trained their generative model to efficiently capture the chemical and biological properties of known bioactive compounds. Their model generated fine-tuned inhibitors against their two targets of interest. Following testing in a cell-based assay, top-ranked de novo compounds were revealed to be highly potent with EC50 values ranging in nanomolar to low-micromolar potency against the two receptors. These positive results suggest that the design of PLpro-specific hits could follow a similar optimized approach. Notably, the interplay between the SBVS and AI-enhanced CADD tools such as generative models will represent an intriguing venue for PLpro drug discovery. Due to the protease's unique structural features, SBVS can be used to identify a wide variety of diverse compounds to build an essential interaction profile for PLpro inhibitors. This interaction profile can then be exploited in CADD-led de novo designs, optimized specifically for SARS-CoV-2 PLpro. This would enable the exploration of different and unknown medicinal chemistry optimization design paths.
Finally, there is a booming interest in utilizing computational methods to search exhaustively the large chemical space in response to the need to discover new biologically relevant small-molecule therapeutics against SARS-CoV-2 PLpro. Conventional molecular docking methods and computational resources are unsuitable to undertake these large VS campaigns [70]. For this, it will be critical to deploy modern DL-based methods like our Deep Docking platform and other methodologies exploiting GPU and cloud computing such as OpenEye’s Orioni, or the virtual synthon-based approach (V-SYNTHES) [71,72]. These timely innovations should enable virtual screening of ultralarge libraries containing billions of compounds, such as the Enamine REAL Space (22.7B molecules), to identify unique and potent scaffolds. This expanding chemical space is ripe for the development of new therapeutics, and these new DL-based methods – only recently feasible – will enable ultralarge-scale SBVS to initiate drug discovery campaigns against SARS-CoV-2 PLpro.
Concluding remarks and future perspectives
The SARS-CoV-2 PLpro is a very promising and interesting therapeutic target against COVID-19 due to its dual biological roles, but there are considerations to address before initiating large-scale SBVS campaigns against it (see Outstanding questions). Currently, PLpro's active site is the most studied thanks to its importance, but the active site itself has shown to be quite a challenging target for rational drug design due to its structural features. Ergo, it would be practical to explore other potentially targetable sites on PLpro, such as the Zn-binding functionality, or allosteric sites in the Ubl domain, and to determine if inhibition of these sites would also bring beneficial therapeutic responses. Additionally, adequate standardization of appropriate cell lines will be important as the variety of different cells used to investigate SARS-CoV-2 is a challenge to correctly interpreting discrepancies from different studies.
Outstanding questions.
As demonstrated in treatment plans against other viral infections such as HIV, can new SARS-CoV-2 PLpro therapeutics be used as part of antiviral cocktails with existing SARS-CoV-2 Mpro or RdRp drugs to treat COVID-19 patients?
If using PLpro’s catalytic site as a target, which of the four subsites is the most critical one to occupy? What is the key protein–substrate interaction in each subsite, and how can it be exploited for rational drug designs?
As SARS-CoV-2 Mpro drugs revealed that covalent inhibitors had the most potential in inhibiting Mpro's activity, are covalent modifications the direction to adopt as well for PLpro drug design?
As they remain largely unexplored sites on SARS-CoV-2 PLpro, is it plausible to target the Zn-binding site to disrupt the protease's stability or the allosteric Ubl domain to inhibit the protease's enzymatic activity?
Will the integration of state-of-the-art CADD tools significantly improve drug discovery efforts against SARS-CoV-2 PLpro? Considering the protease's difficult features such as flexibility in the active site, amino acid specificity, and tunnel-like rigidity that limit the efficacy of traditional CADD tools, will ML-enhanced methods, such as DL-assisted ensemble docking or ultralarge-scale virtual screening that can take into consideration some of these difficult features, be able to overcome the hurdles limiting PLpro drug discovery?
On the same idea, will ML-enhanced tools and methodologies be able to surpass expert's knowledge and experience to become the better solution for hit-to-lead optimization for PLpro candidate drugs? Will it be possible to automate the final stages of drug design to ensure minimal human biases in drug discovery?
Alt-text: Outstanding questions
Although select groups have designed potent PLpro compounds, these molecules are all derived from the same starting template and lack diversity. Additionally, medicinal chemist virtuosos with decades of experience designed these compounds. Therefore, there is a need for better computational tools to guide medicinal chemistry of PLpro compounds. It would be significantly advantageous to complement expert-guided designs with advanced CADD methods, such as automated fragment-based optimization, and generative models for de novo drug design [73., 74., 75.]. This will subsequently increase the efficiency of the optimization process. Covalent conversion of noncovalent hits represents another interesting and prospective option. Thus, Sanders et al. created covalent analogs of GRL-0617 by attaching a reactive warhead to the inhibitor's scaffold, and their compounds showed excellent potency and selectivity against SARS-CoV-2 PLpro, without exhibiting any cytotoxicity or off-target effects [76]. Their promising study highlighted the possibility of designing covalent inhibitors for the PLpro active site. Finally, we propose that AI-enhanced CADD methods be placed at the forefront of antiviral drug discovery and should often be incorporated as early as possible. They will play a critical role in democratizing the development of new PLpro-directed drugs with clinical value against COVID-19 and future pandemics.
Acknowledgments
Acknowledgments
The authors acknowledge contribution from Ms Mariia Radaeva and Ms Olivia Garland for their help with literature review of reported SARS-CoV-2 PLpro inhibitors. This work is supported by the Canadian Institutes of Health Research (CIHR) (AWD-013505, AWD-015079, AWD-017671, and AWD-018223) and donations from the Tai Hung Fai Charitable Foundation.
Declaration of interest
No interests are declared.
Glossary
- Chemical scaffolds
concept used to represent the common core structure of a series of molecules with bioactive properties.
- Chemical space
concept defined as the set of all possible chemical compounds. The bioactive small-molecule chemical space is estimated at 1063 compounds, and current empirical methods are still not able to efficiently sample it (106 compounds).
- Computer-aided drug design (CADD)
computational technique aimed at discovering, designing, and developing therapeutic chemical agents. Used to predict properties of known and unknown molecular species and is extensively involved in rational drug design.
- Deep learning (DL)
subset of machine learning based on artificial neural networks that detect features from large sets of data.
- Drug-like
set of pharmacokinetic and physicochemical properties used quantitatively to assess the probability of a compound to be an oral drug, with respect to bioavailability. Embodied by the Lipinski's rule-of-five guidelines.
- Ensemble docking
molecular docking of ligands to a set of the same receptor structure in different conformations to capture different possible states.
- ISGylation
ISGylation is a process involved in the innate immune responses induced by type I interferon. ISG15 exerts strong antiviral effects by binding to host and viral target proteins to inhibit release of viral particles and hinder viral replication.
- Lead-likeness
a lead compound is one deemed of sufficient pharmacological properties that it could be developed into a drug compound. Lead-like properties are physicochemical properties used to select starting points for chemical optimization to obtain ‘drug-like’ candidates.
- Machine learning (ML)
algorithms that imitate human learning behavior using observed data.
- Molecular docking (docking)
computational method used to model the atomic interactions between a ligand and the target to characterize their behavior. Docking usually involves a two-step sampling and scoring method: prediction of the ligand conformation, position, and orientation within the receptor site (pose) and assessment of their complementarity and binding affinity (docking score).
- Molecular dynamics (MD)
computer simulation method used to study and understand the dynamic protein–ligand interplay in the presence of a solvent and under realistic physiological conditions.
- Pharmacophore (ph4) model
ensemble of steric and electronic features necessary to ensure optimal supramolecular interactions with a specific biological target structure and to trigger (or to block) its biological response (IUPAC recommendations, 1998). Ph4 models can be used during VS by comparing how well compounds align with the ph4 in 2D or 3D space.
- Structure-based virtual screening (SBVS)
set of computational methods used for identification of hits by analyzing large databases of compounds and the structure of a target. Considered as the computational equivalent of high-throughput screening, but quicker and more cost effective.
Resources
iwww.eyesopen.com/webinars/giga-docking-structure-based-virtual-screeningReferences
- 1.Naqvi A.A.T., et al. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: structural genomics approach. Biochim. Biophys. Acta Mol. basis Dis. 2020;1866 doi: 10.1016/j.bbadis.2020.165878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aftab S.O., et al. Analysis of SARS-CoV-2 RNA-dependent RNA polymerase as a potential therapeutic drug target using a computational approach. J. Transl. Med. 2020;18:275. doi: 10.1186/s12967-020-02439-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jin Z., et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 4.Dai W., et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parums D.V. Editorial: Current status of oral antiviral drug treatments for SARS-CoV-2 infection in non-hospitalized patients. Med. Sci. Monit. 2022;28 doi: 10.12659/MSM.935952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burki T.K. The role of antiviral treatment in the COVID-19 pandemic. Lancet Respir. Med. 2022;10 doi: 10.1016/S2213-2600(22)00011-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kozlov M. Merck’s COVID pill loses its lustre: what that means for the pandemic. Nature. 2021 doi: 10.1038/d41586-021-03667-0. Published online December 13, 2021 . [DOI] [PubMed] [Google Scholar]
- 8.Heskin J., et al. Caution required with use of ritonavir-boosted PF-07321332 in COVID-19 management. Lancet. 2022;399:21–22. doi: 10.1016/S0140-6736(21)02657-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levin E.G., et al. Waning immune humoral response to BNT162b2 Covid-19 vaccine over 6 months. N. Engl. J. Med. 2021;385 doi: 10.1056/NEJMoa2114583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collier D.A., et al. Age-related immune response heterogeneity to SARS-CoV-2 vaccine BNT162b2. Nature. 2021;596:417–422. doi: 10.1038/s41586-021-03739-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sacco M.D., et al. The P132H mutation in the main protease of Omicron SARS-CoV-2 decreases thermal stability without compromising catalysis or small-molecule drug inhibition. Cell Res. 2022;32:498–500. doi: 10.1038/s41422-022-00640-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mótyán J.A., et al. Potential resistance of SARS-CoV-2 main protease (Mpro) against protease inhibitors: lessons learned from HIV-1 protease. IJMS. 2022;23:3507. doi: 10.3390/ijms23073507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ledford H. COVID antiviral pills: what scientists still want to know. Nature. 2021;599:358–359. doi: 10.1038/d41586-021-03074-5. [DOI] [PubMed] [Google Scholar]
- 14.Narayanan A., et al. Identification of SARS-CoV-2 inhibitors targeting Mpro and PLpro using in-cell-protease assay. Commun. Biol. 2022;5:169. doi: 10.1038/s42003-022-03090-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petushkova A.I., Zamyatnin A.A. Papain-like proteases as coronaviral drug targets: current inhibitors, opportunities, and limitations. Pharmaceuticals. 2020;13:277. doi: 10.3390/ph13100277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang H., et al. Potential inhibitors targeting papain-like protease of SARS-CoV-2: two birds with one stone. Front. Chem. 2022;10 doi: 10.3389/fchem.2022.822785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan H., et al. Progress and challenges in targeting the SARS-CoV-2 papain-like protease. J. Med. Chem. 2022;65:7561–7580. doi: 10.1021/acs.jmedchem.2c00303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calleja D.J., et al. Inhibitors of SARS-CoV-2 PLpro. Front. Chem. 2022;10 doi: 10.3389/fchem.2022.876212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McClain C.B., Vabret N. SARS-CoV-2: the many pros of targeting PLpro. Signal. Transduct. Target Ther. 2020;5:223. doi: 10.1038/s41392-020-00335-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shin D., et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. 2020;587:657–662. doi: 10.1038/s41586-020-2601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li W., et al. SARS-CoV-2 Nsp5 activates NF-κB pathway by upregulating SUMOylation of MAVS. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.750969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Freitas B.T., et al. Characterization and noncovalent inhibition of the deubiquitinase and deISGylase activity of SARS-CoV-2 papain-like protease. ACS Infect. Dis. 2020;6:2099–2109. doi: 10.1021/acsinfecdis.0c00168. [DOI] [PubMed] [Google Scholar]
- 23.Klemm T., et al. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 2020;39 doi: 10.15252/embj.2020106275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao X., et al. Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm. Sin. B. 2021;11:237–245. doi: 10.1016/j.apsb.2020.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maiti B.K. Can papain-like protease inhibitors halt SARS-CoV-2 replication? ACS Pharmacol. Transl. Sci. 2020;3:1017–1019. doi: 10.1021/acsptsci.0c00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Osipiuk J., et al. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 2021;12:743. doi: 10.1038/s41467-021-21060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fu Z., et al. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat. Commun. 2021;12:488. doi: 10.1038/s41467-020-20718-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Henderson J.A., et al. Assessment of proton-coupled conformational dynamics of SARS and MERS coronavirus papain-like proteases: implication for designing broad-spectrum antiviral inhibitors. J. Chem. Phys. 2020;153 doi: 10.1063/5.0020458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sohraby F., Aryapour H. Unraveling the unbinding pathways of SARS-CoV-2 Papain-like proteinase known inhibitors by supervised molecular dynamics simulation. PLoS One. 2021;16 doi: 10.1371/journal.pone.0251910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rut W., et al. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: a framework for anti–COVID-19 drug design. Sci. Adv. 2020;6:eabd4596. doi: 10.1126/sciadv.abd4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ratia K., et al. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. U. S. A. 2008;105:16119–16124. doi: 10.1073/pnas.0805240105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith E., et al. High-throughput screening for drugs that inhibit papain-like protease in SARS-CoV-2. SLAS Discov. 2020;25:1152–1161. doi: 10.1177/2472555220963667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shan H., et al. Development of potent and selective inhibitors targeting the papain-like protease of SARS-CoV-2. Cell Chem. Biol. 2021;28:855–865.e9. doi: 10.1016/j.chembiol.2021.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu Y., et al. Repurposing clinically approved drugs for COVID-19 treatment targeting SARS-CoV-2 papain-like protease. Int. J. Biol. Macromol. 2021;188:137–146. doi: 10.1016/j.ijbiomac.2021.07.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Napolitano V., et al. Acriflavine, a clinically approved drug, inhibits SARS-CoV-2 and other betacoronaviruses. Cell Chem. Biol. 2022;29:774–784.e8. doi: 10.1016/j.chembiol.2021.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weglarz-Tomczak E., et al. Identification of ebselen and its analogues as potent covalent inhibitors of papain-like protease from SARS-CoV-2. Sci. Rep. 2021;11:3640. doi: 10.1038/s41598-021-83229-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen Z., et al. Design of SARS-CoV-2 PLpro inhibitors for COVID-19 antiviral therapy leveraging binding cooperativity. J. Med. Chem. 2021;65:2940–2955. doi: 10.1021/acs.jmedchem.1c01307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jamalan M., et al. Structure-based screening to discover new inhibitors for papain-like proteinase of SARS-CoV-2: an in silico study. J. Proteome Res. 2021;20:1015–1026. doi: 10.1021/acs.jproteome.0c00836. [DOI] [PubMed] [Google Scholar]
- 39.Atanasov A.G., et al. Natural products in drug discovery: advances and opportunities. Nat. Rev. Drug Discov. 2021;20:200–216. doi: 10.1038/s41573-020-00114-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ibrahim T.M., et al. Supporting SARS-CoV-2 papain-like protease drug discovery: in silico methods and benchmarking. Front. Chem. 2020;8 doi: 10.3389/fchem.2020.592289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Delre P., et al. Repurposing known drugs as covalent and non-covalent inhibitors of the SARS-CoV-2 papain-like protease. Front. Chem. 2020;8 doi: 10.3389/fchem.2020.594009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li D., et al. Molecular docking of potential SARS-CoV-2 papain-like protease inhibitors. Biochem. Biophys. Res. Commun. 2021;538:72–79. doi: 10.1016/j.bbrc.2020.11.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elekofehinti O.O., et al. Molecular docking studies, molecular dynamics and ADME/tox reveal therapeutic potentials of STOCK1N-69160 against papain-like protease of SARS-CoV-2. Mol. Divers. 2021;25:1761–1773. doi: 10.1007/s11030-020-10151-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang D.-H., et al. In silico screening of Chinese herbal medicines with the potential to directly inhibit 2019 novel coronavirus. J. Integr. Med. 2020;18:152–158. doi: 10.1016/j.joim.2020.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ma C., Wang J. Validation and invalidation of SARS-CoV-2 papain-like protease inhibitors. ACS Pharmacol. Transl. Sci. 2022;5:102–109. doi: 10.1021/acsptsci.1c00240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yuan S., et al. Targeting papain-like protease for broad-spectrum coronavirus inhibition. Protein Cell. 2022;13:940–953. doi: 10.1007/s13238-022-00909-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma C., et al. Discovery of SARS-CoV-2 papain-like protease inhibitors through a combination of high-throughput screening and a FlipGFP-based reporter assay. ACS Cent. Sci. 2021;7:1245–1260. doi: 10.1021/acscentsci.1c00519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szymański P., et al. Adaptation of high-throughput screening in drug discovery-toxicological screening tests. Int. J. Mol. Sci. 2012;13:427–452. doi: 10.3390/ijms13010427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Z., et al. Identify potent SARS-CoV-2 main protease inhibitors via accelerated free energy perturbation-based virtual screening of existing drugs. Proc. Natl. Acad. Sci. U. S. A. 2020;117:27381–27387. doi: 10.1073/pnas.2010470117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gentile F., et al. Automated discovery of noncovalent inhibitors of SARS-CoV-2 main protease by consensus Deep Docking of 40 billion small molecules. Chem. Sci. 2021;12:15960–15974. doi: 10.1039/d1sc05579h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao G., et al. Hydrogen bonding penalty used for virtual screening to discover potent inhibitors for Papain-Like cysteine proteases of SARS-CoV-2. Chem. Biol. Drug Des. 2022 doi: 10.1111/cbdd.14115. Published online July 6, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tian X., et al. Discovery of novel and highly potent inhibitors of SARS CoV-2 papain-like protease through structure-based pharmacophore modeling, virtual screening, molecular docking, molecular dynamics simulations, and biological evaluation. Front. Pharmacol. 2022;13 doi: 10.3389/fphar.2022.817715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Elseginy S.A., et al. Promising anti-SARS-CoV-2 drugs by effective dual targeting against the viral and host proteases. Bioorg. Med. Chem. Lett. 2021;43 doi: 10.1016/j.bmcl.2021.128099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Elseginy S.A., Anwar M.M. In silico analysis of SARS-CoV-2 papain-like protease potential inhibitors. RSC Adv. 2021;11:38616–38631. doi: 10.1039/d1ra07845c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dahlgren D., Lennernäs H. Intestinal permeability and drug absorption: predictive experimental, computational and in vivo approaches. Pharmaceutics. 2019;11:411. doi: 10.3390/pharmaceutics11080411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen X., et al. Thiopurine analogue inhibitors of severe acute respiratory syndrome-coronavirus papain-like protease, a deubiquitinating and deISGylating enzyme. Antivir. Chem. Chemother. 2009;19:151–156. doi: 10.1177/095632020901900402. [DOI] [PubMed] [Google Scholar]
- 57.Chou C.-Y., et al. Thiopurine analogues inhibit papain-like protease of severe acute respiratory syndrome coronavirus. Biochem. Pharmacol. 2008;75:1601–1609. doi: 10.1016/j.bcp.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lyu J., et al. Ultra-large library docking for discovering new chemotypes. Nature. 2019;566:224–229. doi: 10.1038/s41586-019-0917-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pandey M., et al. The transformational role of GPU computing and deep learning in drug discovery. Nat. Mach. Intell. 2022;4:211–221. [Google Scholar]
- 60.Paul D., et al. Artificial intelligence in drug discovery and development. Drug Discov. Today. 2021;26:80–93. doi: 10.1016/j.drudis.2020.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vamathevan J., et al. Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 2019;18:463–477. doi: 10.1038/s41573-019-0024-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hoffmann T., Gastreich M. The next level in chemical space navigation: going far beyond enumerable compound libraries. Drug Discov. Today. 2019;24:1148–1156. doi: 10.1016/j.drudis.2019.02.013. [DOI] [PubMed] [Google Scholar]
- 63.Baumgartner M.P., Evans D.A. Lessons learned in induced fit docking and metadynamics in the Drug Design Data Resource Grand Challenge 2. J. Comput. Aided Mol. Des. 2018;32:45–58. doi: 10.1007/s10822-017-0081-y. [DOI] [PubMed] [Google Scholar]
- 64.Gimeno A., et al. The light and dark sides of virtual screening: what is there to know? Int. J. Mol. Sci. 2019;20:1375. doi: 10.3390/ijms20061375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Degiacomi M.T. Coupling molecular dynamics and deep learning to mine protein conformational space. Structure. 2019;27:1034–1040.e3. doi: 10.1016/j.str.2019.03.018. [DOI] [PubMed] [Google Scholar]
- 66.Amaro R.E., et al. Ensemble docking in drug discovery. Biophys. J. 2018;114:2271–2278. doi: 10.1016/j.bpj.2018.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Meyers J., et al. De novo molecular design and generative models. Drug Discov. Today. 2021;26:2707–2715. doi: 10.1016/j.drudis.2021.05.019. [DOI] [PubMed] [Google Scholar]
- 68.Popova M., et al. Deep reinforcement learning for de novo drug design. Sci. Adv. 2018;4:eaap7885. doi: 10.1126/sciadv.aap7885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Merk D., et al. De novo design of bioactive small molecules by artificial intelligence. Mol. Inf. 2018;37:1700153. doi: 10.1002/minf.201700153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Irwin J.J., et al. ZINC20—a free ultralarge-scale chemical database for ligand discovery. J. Chem. Inf. Model. 2020;60:6065–6073. doi: 10.1021/acs.jcim.0c00675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gentile F., et al. Artificial intelligence–enabled virtual screening of ultra-large chemical libraries with deep docking. Nat. Protoc. 2022;17:672–697. doi: 10.1038/s41596-021-00659-2. [DOI] [PubMed] [Google Scholar]
- 72.Sadybekov A.A., et al. Synthon-based ligand discovery in virtual libraries of over 11 billion compounds. Nature. 2022;601:452–459. doi: 10.1038/s41586-021-04220-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bilsland A.E., et al. Automated generation of novel fragments using screening data, a Dual SMILES autoencoder, transfer learning and syntax correction. J. Chem. Inf. Model. 2021;61:2547–2559. doi: 10.1021/acs.jcim.0c01226. [DOI] [PubMed] [Google Scholar]
- 74.de Souza Neto L.R., et al. In silico strategies to support fragment-to-lead optimization in drug discovery. Front. Chem. 2020;8:93. doi: 10.3389/fchem.2020.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tong X., et al. Generative models for de novo drug design. J. Med. Chem. 2021;64:14011–14027. doi: 10.1021/acs.jmedchem.1c00927. [DOI] [PubMed] [Google Scholar]
- 76.Sanders B., et al. Potent and selective covalent inhibitors of the papain-like protease from SARS-CoV-2. Res. Sq. 2021 doi: 10.21203/rs.3.rs-906621/v1. Published online October 8, 2021. [DOI] [Google Scholar]