

Abstract
Natural products have been included in our dietary supplements and have been shown to have numerous therapeutic properties. With the looming danger of many zoonotic agents and novel emerging pathogens mainly of viral origin, many researchers are launching various clinical trials, testing these compounds for their antiviral activity. The present work deals with some of the available natural compounds from the literature that have demonstrated activity in counteracting pathogen infections. Accordingly, we screened, using in silico methods, this subset of natural compounds for searching potential drug candidates able to interfere in the recognition of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike protein and its target human angiotensin-converting enzyme 2 (hACE2) receptor, leading to the viral entry. Disrupting that recognition is crucial for slowing down the entrance of viral particles into host cells. The selected group of natural products was examined, and their interaction profiles against the host cell target protein ACE2 were studied at the atomic level. Based on different computer-based procedures including molecular docking, physicochemical property evaluation, and molecular dynamics, butein was identified as a potential hit molecule able to bind the hACE2 receptor. The results indicate that herbal compounds can be effective for providing possible therapeutics for treating and managing coronavirus disease 2019 (COVID-19) infection.
Graphical abstract
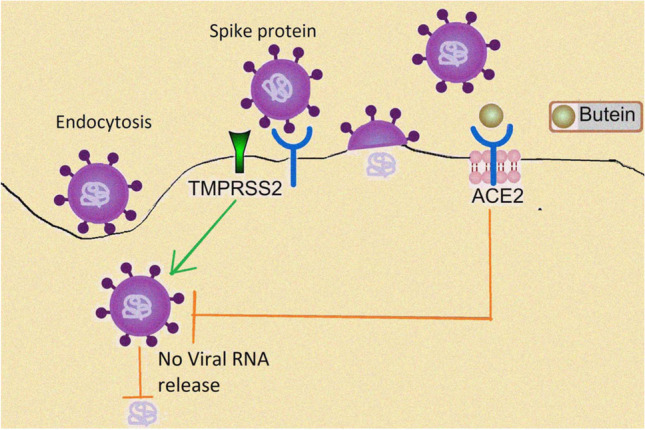
Keywords: SARS-CoV-2, hACE-2, Phytoconstituents, Natural products, Butein, Molecular modeling
Introduction
The still unfolding coronavirus disease 2019 (COVID-19) pandemic exacted a heavy toll worldwide during the second wave. It led to more deaths than the first wave, and as on 3rd March 2022, confirmed COVID-19 cases around the world stood at 522 million and more than 6.2 million deaths (https://covid19.who.int/). COVID-19 caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infects humans as well as different animals and is continuously mutating to new variants such as the B.1.617.2 (Delta) variant, which had a devastating outcome owing to its ability to evade our immune system and recently the B.1.1.529 (Omicron) variant that has almost engulfed the globe due to its stronger transmission potential. Apart from the popularity of vaccines, a series of non-pharmacological interventions (NPIs), like more frequent hand washing, wearing face masks, stay-at-home orders, terminating travel, and social distancing, are advocated to resist further spread. Although many studies have aimed at preventing and treating COVID-19, its overall frequency has not changed [1]. This issue requires new approaches to overcome this global health issue and considering some natural compounds in combating the viral spread may show some light at the end of the tunnel. Providentially, the genome of SARS-CoV-2 is almost 82% similar to its predecessor SARS-CoV, and 50% similar to MERS-CoV, which caused severe pandemics at the turn of the century [2–4], indicating a common mechanism of infection. As previously mentioned, during these years, several attempts to develop therapeutic agents and diagnostic tools have been described [5–7]. Although these efforts led to medicines able to contrast the effects of pandemic health emergency, the need of novel therapeutic options is still urgent due to the SARS-CoV-2 variants that can arise from the virus circulation since coronavirus species unceasingly evolve by changing the genetic code (genetic mutations) throughout the replication phase of the genome (https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-classifications.html) [8]. A possible strategy for developing therapeutic agents against SARS-CoV-2 is to investigate natural products and nutraceutical components to provide novel molecules as potential drug candidates that could interfere with the viral life cycle [9].
To this end, ancient India has been a land of Ayurveda, Siddha, and Unani medicinal practices. Overall, 21,000 plants are enlisted to possess therapeutic potential around the world; India alone has 2500 varieties of medicinal plants that are widely used commercially. [10]. Some of the natural compounds listed in Siddha medicine namely Cucurbitacin E, Cucurbitacin B, Isocucurbitacin B, bis-andrographolide, Piperine, Orientin, Vitexin, Berberine, Bryonolic acid, and Magnoflorine could represent possible lead compounds possessing the ability to interfere with the viral replication of SARS-CoV-2 [11]. Our earlier work indicated that Cucurbitacin G 2-glucoside and Cucurbitacin H showed favorable pharmacological profiles against SARS-CoV [12]. Furthermore, natural phenolic compounds like Quercetin, Luteolin, Resveratrol, and Amentoflavone have been shown to have immunomodulatory and antiviral activities against SARS-CoV and MERS-CoV [13].
Rather than the complex structure of some bioactive compounds, more structurally simpler molecules can be proposed to modulate immune response and inflammation using the phytoconstituents as prototypes. Moreover, targeting specific viral proteins with such molecules may pave the way to the identification of targeted anti-viral drugs. The binding of coronavirus glycosylated spike protein with the host human angiotensin-converting enzyme-2 (hACE2) is a crucial element to initiate the viral infection. Assuming renewed interest toward hACE2 as a possible drug target, many natural products and marketed drugs could be potentially repositioned as off-target for hACE2-modulatory effects [14]. Many such natural small molecules bind to the hACE2 receptor and alter the binding of spike protein with the hACE2 receptor. The contagion of SARS-CoV-2 mainly depends on the affinity and specificity of the recognition between the spike protein and hACE2 receptor. As a result, structural modifications in hACE2 caused by one or more of these natural small molecules disrupt the proper recognition of hACE2 and the viral spike protein, preventing viral entry. Some popular flavonoids exhibit varied structural complexities that have been shown to induce different effects on hACE2, which may be associated with their ability to change the hACE2 structure conformation [15]. In this work, molecular docking, and molecular dynamics (MD) experiments were employed to analyze a targeted database of natural products. From that investigation Butein, an important dietary polyphenol, has been selected as a potential hACE2 receptor ligand deserving further investigation in this field. Based on this hypothesis, this work aims to unravel the relationship between Butein and hACE2 receptor, so that it can potentially be harnessed as a relatively low-cost treatment for SARS-CoV-2.
Materials and Methods
Molecular modeling
Selection and preparation of target proteins and ligands
On the basis of the literature, SARS-CoV-2 chimeric receptor-binding domain in complex with its receptor hACE2 (PDB ID 6VW1; Resolution = 2.68 Å, Chain A, B with 597 amino acid length) [16] was downloaded from the RCSB Protein Data Bank (PDB) (http://www.rcsb.org/) [17]. The designated receptor was prepared employing a protein preparation script supplied with Schrödinger/Maestro suites (Maestro, release 2018, Schrödinger, LLC, New York, NY, 2018). The protein was stripped of their associated ligands and water molecules. The missing sidechains of the crystallized protein were added using a specific module accessible in Prime (Prime, release 2018, Schrödinger, LLC, New York, NY, 2018). The complex was submitted to the energy minimization using OPLS-2005 force field. Ligand structures were optimized employing LigPrep application (LigPrep, release 2018, Schrödinger, LLC, New York, NY, 2018) to determine the correct Lewis structure, tautomeric and ionization state at pH 7.0 ± 2.0.
Grid generation and screening
For appropriate generation of the receptor grid for docking simulation studies, we analyzed the ACE2 protein structure by Sitemap tool (Sitemap, release 2018, Schrödinger, LLC, New York, NY, 2018) available in Maestro for mapping suitable binding sites. The most suitable identified binding site (Dscore > 1) was used as the entry selection in receptor grid generation. Furthermore, the prepared ligands were screened with the respective hACE2 receptor using “virtual screening workflow” available in Glide software.
Protein–protein docking
Protein–protein docking, using the RDB of viral spike protein as the ligand and hACE2 as the receptor, was conducted employing the HDOCK webserver (http://hdock.phys.hust.edu.cn/ accessed on May 12, 2022) [18, 19]. The server performed protein–protein and protein-DNA/RNA docking automatically and can predict interactions by employing a hybrid algorithm of template-based and template-free docking. For this study, during the input step, we specified the interacting residues of the viral spike protein and hACE2, namely, hotspots [16, 20, 21]. Further protein–protein docking studies were conducted by HADDOCK webserver (High Ambiguity Driven protein–protein DOCKing) (https://wenmr.science.uu.nl/haddock2.4/ accessed on May 16, 2022), This server employs ab-initio docking methods to encode information from identified or predicted protein interfaces in ambiguous interaction restraints (AIRs) to drive the docking process [22]. Also, in this case, the interacting residues of the viral spike protein and hACE2 were specified during the input step.
Molecular dynamics
Desmond 5.6 academic version via Maestro was used to perform MD simulation studies (Desmond Molecular Dynamics System, version 5.6, D. E. Shaw Research, New York, NY, 2018. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2018). MD simulations were executed on two NVIDIA GPUs, employing the Compute Unified Device Architecture (CUDA) API [23]. The complex resulting from molecular docking studies was inserted into an orthorhombic box together with water molecules, simulated by the solvent model TIP3P, using Desmond system builder available in Maestro [24, 25]. MD simulation was performed adopting OPLS as the force field [26]. Na+ and Cl− ions (0.15 M) were added for mimicking the physiological concentration of the monovalent ions. The ensemble class NPT (constant number of particles, pressure, and temperature) were used adopting a constant temperature of 310 K and a pressure of 1.01325 bar. For integrating the equations of motion, RESPA integrator [27] was used. Nose–Hoover thermostats [28] and the Martyna-Tobias-Klein method [29] were employed to keep constant temperature and pressure of the simulation, respectively. Particle-mesh Ewald technique (PME) was employed for calculating long-range electrostatic interactions [30]. 9.0 Å was chosen as the threshold for the van der Waals and short-range electrostatic interactions. The system was equilibrated using the default procedure, which consists of a series of restrained minimization and MD simulations to gradually relax the system. As a result, a single 500 ns trajectory was determined. MD simulation studies were independently repeated three times for providing a more reliable output. Simulation Event Analysis tools, included in the software package, were used to examine the trajectory files. All charts relating to MD simulation presented in this article were created using the same tools. Therefore, the root mean square deviation (RMSD) was evaluated using the following equation:
RMSDx: calculation for a frame x; N: number of selected atoms; tref: reference time (normally the first frame is utilized as the reference at time t = 0); r′: position of chosen atoms in frame x at time tx, after the superimposition with the reference frame. Every frame in the simulation trajectory is subjected to the same technique. The following formula was used to calculate the root mean square fluctuation (RMSF):
RMSFi: generic residue; T: trajectory time considered for the calculation of RMSF; tref: reference time, ri: position of residue i; r′: position of atoms in residue i after superposition on the reference. The square distance is averaged over the atoms in the residue, as indicated by the angle brackets.
Physicochemical properties and Lipinski’s Rule
The next step followed was the identification of those natural components among the selected natural/herbal compounds, based on their potential antioxidant, antiviral, and immunomodulatory properties. The structures of natural products were downloaded from the NCBI PubChem database [31] and evaluated for their drug-likeness through the free online web-server tool, namely, Swiss ADME [32]. The pharmacokinetics parameters, lipophilicity, and toxicity of all 32 natural products were examined and the best molecules were selected. Butein was finally selected for further studies based on its binding score and ADMET analysis.
Results
Mapping possible binding site on hACE2 receptor
The sitemap analysis of all 32 potent inhibitors provided hints about the crucial contacts governing their binding to the binding cavity of the hACE2 receptor, interacting with residues that are involved in catalysis and substrate specificity. hACE2, a type I integral membrane protein, is composed by 805 residues comprising a zinc binding consensus sequence (HEXXH + E). The native and inhibitor-bound crystal structures of the hACE2 extracellular domains show considerable inhibitor-dependent hinge-bending movement (~ 16°) of one catalytic subdomain in comparison to the other, bringing significant residues into the site for catalysis [33]. The catalytic domain, also known as the zinc metallopeptidase domain, is comprised between residues 19–611, and it is also subdivided into two subdomains (I and II). The subdomain I (painted in blue in Fig. 1) is composed by residues 19–102, 290–397, and 417–430), while subdomain II (painted in magenta in Fig. 1) is composed by residues 103–289, 398–416, and 431–615. These two subdomains are separated by a long α-helix composed by 17 residues (511–531) (painted in yellow, Fig. 1). We hereby, provide the detailed sitemap of the interactions used for the location of the receptor binding site for Butein (Dscore more than 1). The potential receptor binding domain (shown in sky-blue mesh along with coordinating residues in Fig. 1) of Butein is found sandwiched between the two catalytic subdomains.
Fig. 1.
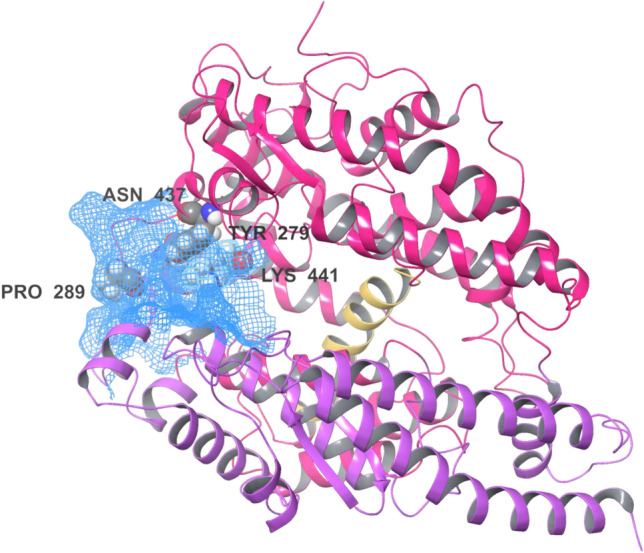
Pictorial representation of receptor binding site through sitemap analysis
Docking of natural products against the hACE2 receptor
The focused natural product chemical library was screened to identify plausible drug candidates considering their binding affinities and structure-based prospects. All compounds were arranged in congeneric series as per their Dscore of binding affinity, which engages protein–ligand complex geometries, free energy calculation or MM-GBSA and the peptide linkages. Table 1 enlists the docking score of all the compounds. After a careful analysis of the docking score and visual inspection of the binding poses, we selected Arctiin, Wedelolactone, Butein, Emodin, and Curcumin as best performing molecules based on their potential binding affinity for hACE2.
Table 1.
Docking of naturally occurring biomolecules within hACE2

Furthermore, out of the above most promising natural products selected by molecular docking against the receptor binding site of hACE2, Butein was identified as one of the best hit molecules (Fig. 2). The binding profile of Butein was found interesting due to the relevant interactions observed within the selected hACE2 binding site. The residues N437, Y279, P289, and D367 showed strong H-bonding interaction with the hydroxyl group of Butein, while the delocalized electrons of benzene moiety strongly interacted with the residue K441. This binding mode showed a docking score of − 7.28 kcal/mol, indicating a satisfactory affinity of Butein for the selected binding site. Remarkably, Butein is considered a suitable molecule for further investigation due to its simple structure and the straightforward synthetic eco-friendly and cost-effective route.
Fig. 2.
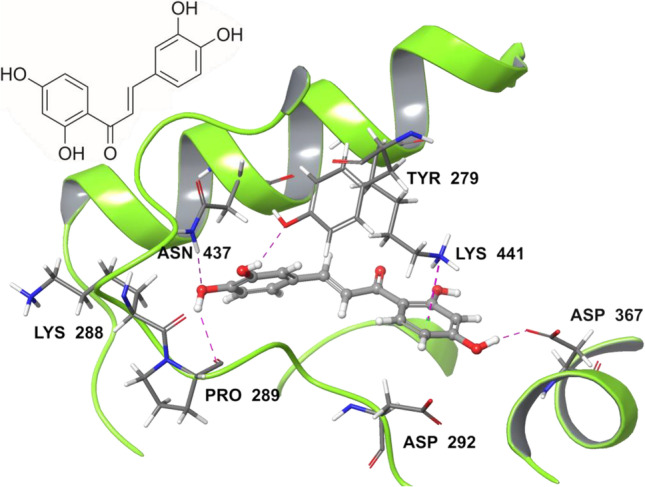
Chemical structure of Butein and its putative binding mode (gray sticks) within hACE2 binding site (PDB ID 6VW1)
Notably, the potential binding site of Butein is a well-established allosteric binding site of hACE2 that if targeted can influence the normal function of the enzyme modifying the conformational dynamics [34–36]. This site, composed by the following amino acids F428, P289, R288, N290, E430, L418, P415, I291, T434, E435, N437, K541, T414, M366, F438, L439, K441, Y279, A413, H540, C542, A412, L539, Y587, Q442, L410, L370, and Q526 (cavity volume of 448.4 Å3). According to the AlloFinder software, this allosteric site showed an AlloScore of 8.18, indicating that it can be considered druggable [34]. Remarkably, small molecules able to bind an allosteric site on hACE2 can disrupt the recognition between the viral spike protein and hACE2, limiting the capability of SARS-CoV-2 to infect human cells, maintaining the function of the human enzyme. In a recent work Wang and collaborators, based on the fact that dexamethasone, chloroquine, and telmisartan have been demonstrated in vitro to bind hACE2, investigated their mechanism of action, demonstrated that the compounds interacted with an allosteric site of hACE2 comprising the mentioned region, causing a conformational change of the hACE2, precluding the recognition with the viral spike protein [35]. Very recently, Hochuli and co-workers, using a virtual screening approach, identified five small molecules able to allosterically bind hACE2, with a slight reduction in its enzymatic activity, inhibiting the viral replication in human cells [36]. Accordingly, the allosteric modulation of hACE2 can be a valuable approach for identifying effective antivirals.
Analysis of ADMET data based on Lipinski’s rule of five
Based on the docking analysis, the selected compounds were screened using computer-based applications to evaluate the ADMET (absorption, distribution, metabolism, excretion, Toxicity) profile of the considered molecules. The identification of a drug-like molecule is based on many factors, namely, the physicochemical properties, water solubility, lipophilicity, and the pharmacokinetics of their molecular structures. Through analyzing the inhibitory effects using the Lipinski’s rule of 5, Emodin, Curcumin, and Butein showed promise as potential “small molecule” modulators with a good druggability quotient (Table 2). In fact, Emodin showed a significant drug-like profile, but Butein was chosen in this particular study for both its high binding score and ADMET analysis.
Table 2.
Drug profile and ADME/toxicity analysis of the best performing natural products selected in this study
| Physicochemical parameters | Arctiin | Wedelolactone | Butein | Emodin | Curcumin | Range |
|---|---|---|---|---|---|---|
| mol_MW (g/mol) | 534.55 | 314.25 | 272.25 | 270.24 | 368.38 | 150–500 g/mol |
| #Rotatable bonds | 10 | 1 | 3 | 0 | 7 | < 9 |
| #H-bond acceptors | 11 | 7 | 5 | 5 | 6 | < 10 |
| #H-bond donors | 4 | 3 | 4 | 3 | 3 | < 5 |
| XLOGP3 | 1.78 | 2.38 | 2.82 | 2.72 | 3.98 | between − 0.7 and + 5.0 |
| TPSA | 153.37 | 113.27 | 97.99 | 94.83 | 96.22 | 20–130 Å2 |
| Log S | − 4.62 | − 4.4 | − 4.54 | − 4.37 | − 5.7 | < 6 |
Lipophilicity: XLOGP3 between − 0.7 and + 5.0, size: MW between 150 and 500 g/mol, polarity: TPSA between 20 and 130Å2, solubility: log S not higher than 6, saturation: fraction of carbons in the sp3 hybridization not less than 0.25, and flexibility: no more than 9 rotatable bonds
Molecular dynamics
In order to validate the docking output and to assess the influence of the binding of Butein on the conformational dynamics of hACE2, evaluating the stability of the selected complex and related time-line behavior, we conducted 500 ns MD simulation study on the biological system hACE2/Butein. The resulting trajectory was comprehensively examined employing diverse standard simulation parameters, including RMSD assessment for each backbone atom and ligand, RMSF of each protein residue (Fig. 3). The analysis revealed that the system is quite stable until 200 ns, after that we observed a significant variation in RMSD with a reduction of the structure stability for the rest of the simulation, ascribable to the Butein binding (Fig. 3A).
Fig. 3.
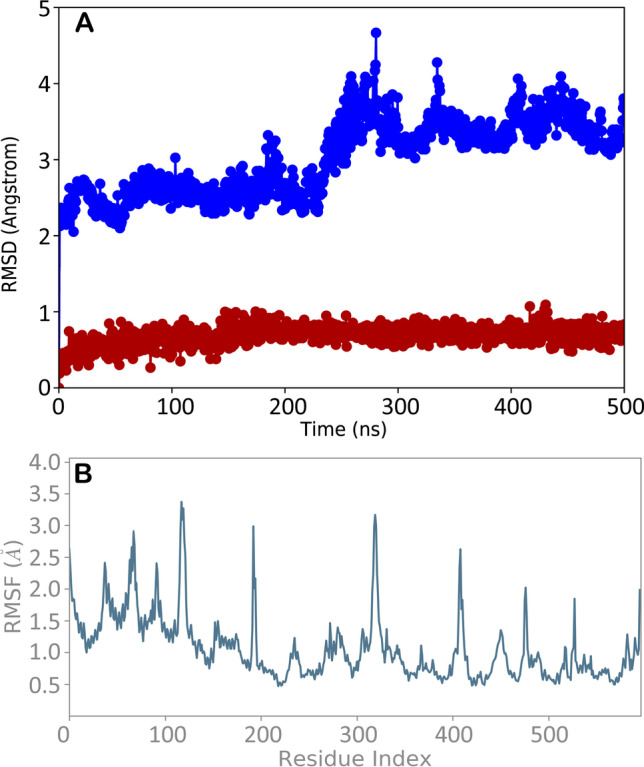
A RMSD regarding the complex hACE2 (blue line) and Butein (red line); B RMSF of all residues of hACE2. Pictures were created by Simulation Event Analysis available in Desmond
Interestingly, Butein maintained the main interactions found by docking studies, with no relevant movement within the binding site and maintaining the interacting conformation (Fig. 3A, red line) with no relevant conformational changes, also highlighted by superposing the complexes obtained at the time 0 of the MD simulation and at the end of the MD run (Fig. 4). However, the binding determined some changes in the protein arrangement as highlighted by the RMSD and RMSF. The RMSF represents the discrepancy between the protein atomic Cα coordinates and its average position throughout the MD run. This kind of estimation is significantly advantageous for determining the flexibility of specific protein backbone amino acids. In fact, although the considered complex did not exhibit dramatic fluctuation events, we observed some groups of hACE2 residues that showed a high fluctuation ratio (Fig. 3B). In addition, the conformational alterations of key amino acids in the hACE2 binding cavity (lowest RMSF value) proved the capability of the molecule to establish stable contacts with hACE2 enzyme.
Fig. 4.
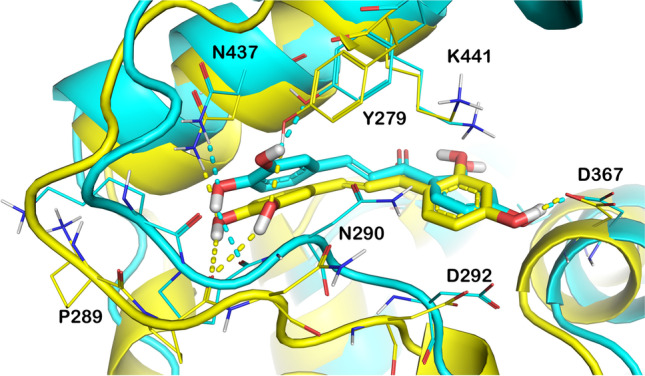
Superposition between initial docked structure (cyan) and final structure (yellow) after 500 ns of MD simulation of Butein bound to hACE2 binding cleft
As previously mentioned, the analysis of the MD run revealed some conformational changes in the protein structure and destabilization/fluctuation of precise cluster residues within the protein structure. Accordingly, we investigated which residues were interested from this perturbation induced by the binding of Butein. We analyzed whether the well-established hotspots on hACE2 [16, 20, 21], crucial for the binding of viral spike protein, were involved in these conformational changes of the protein structure. In Fig. 5, we reported the main hotspots responsible for the recognition between hACE2 and viral spike protein, named hotspots A-D (Fig. 5A). Considering the residues that are perturbated by the binding of Butein during the MD run, showing high fluctuation, we noted that amino acids involved in the fluctuation phenomena mainly belong to the hotspots A and D, while the residues from hotspots B and C are more stable with lower fluctuation especially for the hotspot B (Fig. 5B). In fact, as detailed in Fig. 5B, by superposing the structure at the time 0 and the structure after 500 ns is clearly observable that residues from hotspots A and D, completely lack their original conformation, precluding the correct recognition between the RDB of the spike protein and hACE2. This event could be relevant in reducing the affinity of the viral protein to its human target and possibly slowing down the capacity of the virus to enter the host cell.
Fig. 5.

A Hotspots of hACE2 crucial for the binding with SARS-CoV-2 spike protein. B Main conformational changes in the hotspots after 500 ns of MD in the presence of butein
Finally, to determine whether there is a difference in the binding affinity of the two mentioned conformations of hACE2, at time 0 and after 500 ns, with the viral spike protein interacting domain, we performed a calculation using the HDOCK webserver. Regarding the original structure, we observed that the viral spike protein interacted with the hACE2 with a binding affinity of − 326.16 kcal/mol, while the structure obtained after 500 ns in the presence of Butein was bound by the viral spike protein with a binding affinity of − 175.08 kcal/mol. These results highlighted a dramatic reduction of the binding affinity of the viral spike protein toward hACE2 after the inclusion of Butein, indicating its possible role in influencing the ability of the virus to enter the host cells. To confirm the obtained results, we employed another method for protein–protein docking implemented in the HADDOCK webserver. Using the same input, we obtained a similar result. In fact, considering the original structure, we observed that the viral spike protein interacted with the hACE2 with an HADDOCK score of − 142.7 a.u., while the resulting conformation of hACE2 after 500 ns in the presence of Butein was bound by the viral spike protein with an HADDOCK score of − 126.9 a.u., indicating that after the conformational changes of hACE2, the affinity of the viral spike protein for the mentioned target exhibited a dramatic decrease since the HADDOCK score is a weighted sum of electrostatics, van der Waals, restraint energies, buried surface area and an empirical desolvation term [22, 37].
Discussion
The purpose of this computational investigation was to explore, using a computational approach, the possibility of targeting hACE2 with small-molecule ligands and possibly to cause conformational alterations inside the hACE2 receptor, thus changing the dynamics with which it can bind to viral spike protein [14, 34]. hACE2 receptor that is ubiquitously present in the heart, kidney and the epithelia of the lung and small intestine, is a multifaceted proteolytic enzyme of the renin-angiotensin system (RAS). A synthetic small-molecule XNT has been proposed as a cardiovascular drug by activating hACE2 receptor [38]. Surprisingly, this molecule obliterates the binding between hACE2 and anti-ACE2 IgG autoantibodies, indicating a conformational change within hACE2 receptor, thus affecting the binding potential of ACE2-binding proteins [39]. According to the Protein Data Bank of Transmembrane Proteins database (PDBTM), hACE2 receptor has three domains, a cytoplasmic domain, an extracellular signal sequence and a transmembrane helical region [40]. It is now proven that hACE2 is an allosteric protein containing two binding sites, one for the modulator and the other for the ligand [41]. The first binding site is the functional site where the protein’s physiological function is carried out; while the second binding site is the regulatory unit, which modulates the shape and thus the functional activity of the hACE2 receptor. The spike protein of SARS-CoV-2 binds as a ligand to the functional site of the hACE2 receptor and participates in the biological process of the viral entry into the host cells. The presence of small molecules as modulators allosterically regulates the binding capacity of the spike proteins. Thus, anti-viral activity could be achieved by destabilizing the binding of the host hACE2 receptor with the viral spike protein and certain natural compounds were shown to bind as non-competitive molecules and act as modulators. Here, 32 natural phytochemicals were considered potential ligands for protein–ligand molecular docking study and were screened for their ability to bind the functional site of the hACE2 receptor.
Since Butein exhibited one of the lowest binding energies and a satisfactory drug-like profile, this phytochemical was identified as the most promising compound for targeting the selected biomolecule. The natural compound Butein (3,4,2′,4′-tetrahydroxychalcone), which is also a crucial dietary polyphenol that shows the ability to suppress the signaling pathways adapted by protein tyrosine kinases [42]. This simple chalcone is widely present in various plants belonging to the families of Anacardiaceae, Adoxaceae, Asparagaceae, Asteraceae, Fabaceae, Pinaceae, Rubiaceae, Schisandraceae, and Sonanaceae. This molecule can be found in the leaves, heartwood, stem, rhizome, roots, flowers, seeds, fruits, tubers and even in whole plants [43]. The hydroxyl groups present in the structure (Fig. 2) help Butein effortlessly mitigate hepatic, renal and a neurological condition as well it is effective against diabetes and hypertension [44]. Its high nutraceutical value makes it a potent antioxidant and anti-angiogenic agent [45]. With its ability to inhibit phosphorylation and many enzymatic pathways, it imparts protection against many cancers and inflammatory diseases [46]. Notwithstanding evidence from literature suggests lack of harnessing pure Butein for human clinical trials. Also, it is plausible its potential role as anti-pathogenic agent, possessing antimicrobial or antiviral properties that are not considered yet.
Butein showed a docking score of − 7.38 kcal/mol, which indicates a possible affinity against hACE2 receptor. Thus, Butein can be considered a significant candidate for targeting the viral entry. Furthermore, Butein did not contravene the Lipinski’s rule of five and showed a satisfactory pharmacokinetic profile as indicated by the computational investigation. To gain further insight into the behavior of Butein into hACE2 receptor active site, we performed a comprehensive assessment of the MD simulation, examining the main interactions formed by Butein within the binding cavity. The result of the evaluation regarding the hACE2 in complex with Butein is illustrated in Fig. 6. In general, observing the trajectory of MD simulation, Butein maintained the interactions found by molecular docking calculation, targeting Y279, P289, I291, D367, N437, and K441. Furthermore, during the MD simulation, we observed further additional contacts with the backbone of G286 and the sidechain of K288, which can be relevant in stabilizing the discussed binding mode. Notably, the computational outcome undeniably validated the crucial contacts of Butein found by docking studies, showing acceptable thermodynamic stability within hACE2 binding site, indicating that the compound can behave as a possible hACE2 binder. This binding provoked a destabilization of the conformational dynamics of hACE2 influencing its binding with SARS-CoV-2 spike protein, as demonstrated by analyzing the MD run in which we observed relevant conformational changes in the interacting residues (hotspots) relevant for the recognition of SARS-CoV-2 spike protein and hACE2 and by performing docking calculations between the viral spike protein and hACE2 considering the starting conformation and the resulting conformation of hACE2 after 500 ns of MD run in the presence of Butein. The results highlighted that after the inclusion of Butein that influenced the conformational dynamics, the affinity of the viral spike protein for hACE2 was dramatically reduced, possibly precluding the correct recognition between the mentioned proteins, slowing down the viral entry.
Fig. 6.

Butein monitored in the course of the MD run. The interactions can be grouped into four types: H-bonds (green), hydrophobic (gray), ionic (magenta), and water bridges (blue). The subsequent diagram of the figure illustrates a timeline description of the main interactions. A darker hue of orange indicates that some residues make many distinct contacts with the ligand
Butein belongs to the chalcone family of flavonoids, which possesses numerous biological properties as previously discussed. However, the extraction of the Butein from plant suffers low yield. Therefore, total synthesis of Butein was conducted via Claisen-Schmidt condensation between 3,4-dihydroxybenzaldehyde and 2,4-dihydroxyacetophenone using a basic catalyst [47]. It was also synthesized via aldol condensation catalyzed by thionyl chloride (SOCl2) following the method reported by Hu and coworkers [48].
Conclusion
A total of 32 phytoconstituents were screened, out of which 4 phytochemicals (Arctiin, Wedelolactone, Butein, and Emodin) showed potential affinity for hACE2 binding site along with satisfactory drug-like profile. The other best performing compounds (Garcinol, Brassinin, Arctiin, and Butein) also showed potential as possible hACE2 ligands. Thus, Arctiin and Butein are the common polyphenols that show tremendous promise as potential binders of the hACE2 receptor. In this study, Butein was finally chosen due to its satisfactory computational scores regarding its behavior as a binder of hACE2, its simple structure, and its synthetic accessibility. The binding to hACE2 is assumed to be crucial for interfering with the recognition step between hACE2 and SARS-CoV-2 spike protein. Accordingly, Butein as a possible hACE2 receptor ligand could be potentially able to induce conformational changes in the native structure of the hACE2 receptor, precluding the correct recognition of spike viral protein with the human receptor, slowing down the viral entry. Furthermore, Butein showed a satisfactory ADMET profile, paving the way for developing innovative anti-SARS-CoV-2 therapeutic agents.
Author contribution
Conceptualization, N.K. and S.B.; methodology, N.K., S.M.G., P.K.K., R.B., and S.B.; investigation, N.K., S.M.G., P.K.K., R.B., and S.B.; data curation, N.K., S.M.G., and S.B.; validation, N.K. and S.B.; writing—original draft preparation, N.K. and S.B.; writing—review and editing, N.K., S.M.G., and S.B.; supervision, N.K. and S.B. All authors have read and agreed to the published version of the manuscript.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Code availability
No software code is associated with this article.
Declarations
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Neha Kapoor, Email: nehakapoor@hindu.du.ac.in.
Simone Brogi, Email: simone.brogi@unipi.it.
References
- 1.Hu B, Guo H, Zhou P, Shi ZL. Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol. 2021;19(3):141–154. doi: 10.1038/s41579-020-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Naqvi AAT, Fatima K, Mohammad T, Fatima U, Singh IK, Singh A, Atif SM, Hariprasad G, Hasan GM. Hassan MI (2020) Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: structural genomics approach. Biochim Biophys Acta Mol Basis Dis. 1866;10:165878. doi: 10.1016/j.bbadis.2020.165878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khan MI, Khan ZA, Baig MH, Ahmad I, Farouk AE, Song YG, Dong JJ. Comparative genome analysis of novel coronavirus (SARS-CoV-2) from different geographical locations and the effect of mutations on major target proteins: an in silico insight. PLoS ONE. 2020;15(9):e0238344. doi: 10.1371/journal.pone.0238344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang YY, Li BR, Ning BT. The comparative immunological characteristics of SARS-CoV, MERS-CoV, and SARS-CoV-2 coronavirus infections. Front Immunol. 2020;11:2033. doi: 10.3389/fimmu.2020.02033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Han F, Liu Y, Mo M, Chen J, Wang C, Yang Y, Wu J (2021) Current treatment strategies for COVID19 (Review). Mol Med Rep 24(6). 10.3892/mmr.2021.12498 [DOI] [PMC free article] [PubMed]
- 6.Safarchi A, Fatima S, Ayati Z, Vafaee F. An update on novel approaches for diagnosis and treatment of SARS-CoV-2 infection. Cell Biosci. 2021;11(1):164. doi: 10.1186/s13578-021-00674-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marcianò G, Roberti R, Palleria C, Mirra D, Rania V, Casarella A, De Sarro G, Gallelli L. SARS-CoV-2 treatment: current therapeutic options and the pursuit of tailored therapy. Appl Sci. 2021;11(16):7457. doi: 10.3390/app11167457. [DOI] [Google Scholar]
- 8.Aleem A, Akbar Samad AB, Slenker AK (2022) Emerging variants of SARS-CoV-2 and novel therapeutics against coronavirus (COVID-19). In: StatPearls. Treasure Island (FL), [PubMed]
- 9.Kim CH. Anti-SARS-CoV-2 natural products as potentially therapeutic agents. Front Pharmacol. 2021;12:590509. doi: 10.3389/fphar.2021.590509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anand U, Jacobo-Herrera N, Altemimi A, Lakhssassi N (2019) A comprehensive review on medicinal plants as antimicrobial therapeutics: potential avenues of biocompatible drug discovery. Metabolites 9(11). 10.3390/metabo9110258 [DOI] [PMC free article] [PubMed]
- 11.Alagu Lakshmi S, Shafreen RMB, Priya A, Shunmugiah KP. Ethnomedicines of Indian origin for combating COVID-19 infection by hampering the viral replication: using structure-based drug discovery approach. J Biomol Struct Dyn. 2021;39(13):4594–4609. doi: 10.1080/07391102.2020.1778537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kapoor N, Ghorai SM, Kushwaha PK, Shukla R, Aggarwal C, Bandichhor R. Plausible mechanisms explaining the role of cucurbitacins as potential therapeutic drugs against coronavirus 2019. Inform Med Unlocked. 2020;21:100484. doi: 10.1016/j.imu.2020.100484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dejani NN, Elshabrawy HA, Bezerra Filho C, de Sousa DP (2021) Anticoronavirus and immunomodulatory phenolic compounds: opportunities and pharmacotherapeutic perspectives. Biomolecules 11(8). 10.3390/biom11081254 [DOI] [PMC free article] [PubMed]
- 14.Brogi S, Calderone V. Off-target ACE2 ligands: possible therapeutic option for CoVid-19? Br J Clin Pharmacol. 2020;86(6):1178–1179. doi: 10.1111/bcp.14343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, Graham BS, McLellan JS. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H, Geng Q, Auerbach A, Li F. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;581(7807):221–224. doi: 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan Y, Zhang D, Zhou P, Li B, Huang SY. HDOCK: a web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017;45(W1):W365–W373. doi: 10.1093/nar/gkx407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mondal SK, Mukhoty S, Kundu H, Ghosh S, Sen MK, Das S, Brogi S. In silico analysis of RNA-dependent RNA polymerase of the SARS-CoV-2 and therapeutic potential of existing antiviral drugs. Comput Biol Med. 2021;135:104591. doi: 10.1016/j.compbiomed.2021.104591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ali A, Vijayan R. Dynamics of the ACE2-SARS-CoV-2/SARS-CoV spike protein interface reveal unique mechanisms. Sci Rep. 2020;10(1):14214. doi: 10.1038/s41598-020-71188-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jawad B, Adhikari P, Podgornik R, Ching WY. Key interacting residues between RBD of SARS-CoV-2 and ACE2 receptor: combination of molecular dynamics simulation and density functional calculation. J Chem Inf Model. 2021;61(9):4425–4441. doi: 10.1021/acs.jcim.1c00560. [DOI] [PubMed] [Google Scholar]
- 22.van Zundert GCP, Rodrigues J, Trellet M, Schmitz C, Kastritis PL, Karaca E, Melquiond ASJ, van Dijk M, de Vries SJ, Bonvin A (2016) The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J Mol Biol 428(4):720–725. 10.1016/j.jmb.2015.09.014 [DOI] [PubMed]
- 23.Nickolls J, Buck I, Garland M, Skadron K. Scalable parallel programming with CUDA. Queue. 2008;6(2):40–53. doi: 10.1145/1365490.1365500. [DOI] [Google Scholar]
- 24.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79(2):926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 25.Sirous H, Chemi G, Gemma S, Butini S, Debyser Z, Christ F, Saghaie L, Brogi S, Fassihi A, Campiani G, Brindisi M. Identification of novel 3-hydroxy-pyran-4-one derivatives as potent HIV-1 integrase inhibitors using in silico structure-based combinatorial library design approach. Front Chem. 2019;7:574. doi: 10.3389/fchem.2019.00574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118(45):11225–11236. doi: 10.1021/ja9621760. [DOI] [Google Scholar]
- 27.Humphreys DD, Friesner RA, Berne BJ. A multiple-time-step molecular dynamics algorithm for macromolecules. J Phys Chem. 2002;98(27):6885–6892. doi: 10.1021/j100078a035. [DOI] [Google Scholar]
- 28.Hoover WG. Canonical dynamics: equilibrium phase-space distributions. Phys Rev A Gen Phys. 1985;31(3):1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 29.Martyna GJ, Tobias DJ, Klein ML. Constant pressure molecular dynamics algorithms. J Chem Phys. 1994;101(5):4177–4189. doi: 10.1063/1.467468. [DOI] [Google Scholar]
- 30.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103(19):8577–8593. doi: 10.1063/1.470117. [DOI] [Google Scholar]
- 31.Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, Li Q, Shoemaker BA, Thiessen PA, Yu B, Zaslavsky L, Zhang J, Bolton EE. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 2021;49(D1):D1388–D1395. doi: 10.1093/nar/gkaa971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Towler P, Staker B, Prasad SG, Menon S, Tang J, Parsons T, Ryan D, Fisher M, Williams D, Dales NA, Patane MA, Pantoliano MW. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J Biol Chem. 2004;279(17):17996–18007. doi: 10.1074/jbc.M311191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dutta K. Allosteric site of ACE-2 as a drug target for COVID-19. ACS Pharmacol Transl Sci. 2022;5(3):179–182. doi: 10.1021/acsptsci.2c00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang D-S, Hayatshahi HS, Jayasinghe-Arachchige VM, Liu J (2021) Allosteric modulation of small molecule drugs on ACE2 conformational change upon binding to SARS-CoV-2 spike protein. 2587–2594. 10.1109/bibm52615.2021.9669438
- 36.Hochuli JE, Jain S, Melo-Filho C, Sessions ZL, Bobrowski T, Choe J, Zheng J, Eastman R, Talley DC, Rai G, Simeonov A, Tropsha A, Muratov EN, Baljinnyam B, Zakharov AV (2022) Allosteric binders of ACE2 are promising anti-SARS-CoV-2 agents. bioRxiv. 10.1101/2022.03.15.484484 [DOI] [PMC free article] [PubMed]
- 37.Karaca E, Bonvin AM. Advances in integrative modeling of biomolecular complexes. Methods. 2013;59(3):372–381. doi: 10.1016/j.ymeth.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 38.Raizada MK, Ferreira AJ. ACE2: a new target for cardiovascular disease therapeutics. J Cardiovasc Pharmacol. 2007;50(2):112–119. doi: 10.1097/FJC.0b013e3180986219. [DOI] [PubMed] [Google Scholar]
- 39.Haga S, Takahashi Y, Ishizaka Y, Mimori A (2012) The small molecule activator to ACE2 prevents the inhibition of ACE2 activity by autoantibodies. In Arthritis and Rheumatism 64(No. 10):S284-S285. 111 River St, Hoboken 07030–5774, NJ USA: Wiley-Blackwell
- 40.Kozma D, Simon I, Tusnady GE (2013) PDBTM: Protein Data Bank of transmembrane proteins after 8 years. Nucleic Acids Res 41(Database issue):D524–529. 10.1093/nar/gks1169 [DOI] [PMC free article] [PubMed]
- 41.Basu A, Sarkar A, Maulik U. Molecular docking study of potential phytochemicals and their effects on the complex of SARS-CoV2 spike protein and human ACE2. Sci Rep. 2020;10(1):17699. doi: 10.1038/s41598-020-74715-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang EB, Zhang K, Cheng LY, Mack P. Butein, a specific protein tyrosine kinase inhibitor. Biochem Biophys Res Commun. 1998;245(2):435–438. doi: 10.1006/bbrc.1998.8452. [DOI] [PubMed] [Google Scholar]
- 43.Semwal RB, Semwal DK, Combrinck S, Viljoen A. Butein: from ancient traditional remedy to modern nutraceutical. Phytochem Lett. 2015;11:188–201. doi: 10.1016/j.phytol.2014.12.014. [DOI] [Google Scholar]
- 44.Nerya O, Musa R, Khatib S, Tamir S, Vaya J. Chalcones as potent tyrosinase inhibitors: the effect of hydroxyl positions and numbers. Phytochemistry. 2004;65(10):1389–1395. doi: 10.1016/j.phytochem.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 45.Padmavathi G, Roy NK, Bordoloi D, Arfuso F, Mishra S, Sethi G, Bishayee A, Kunnumakkara AB. Butein in health and disease: a comprehensive review. Phytomedicine. 2017;25:118–127. doi: 10.1016/j.phymed.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 46.Jayasooriya R, Molagoda IMN, Park C, Jeong JW, Choi YH, Moon DO, Kim MO, Kim GY. Molecular chemotherapeutic potential of butein: a concise review. Food Chem Toxicol. 2018;112:1–10. doi: 10.1016/j.fct.2017.12.028. [DOI] [PubMed] [Google Scholar]
- 47.Ma L, Yang Z, Li C, Zhu Z, Shen X, Hu L. Design, synthesis and SAR study of hydroxychalcone inhibitors of human beta-secretase (BACE1) J Enzyme Inhib Med Chem. 2011;26(5):643–648. doi: 10.3109/14756366.2010.543420. [DOI] [PubMed] [Google Scholar]
- 48.Hu Z, Liu J, Dong Z, Guo L, Wang D. Zeng P (2019) Synthesis of chalcones catalysed by SOCl2/EtOH. J Chem Res. 2004;2:158–159. doi: 10.3184/030823404323000594. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
No software code is associated with this article.