

Abstract
Introduction:
Telomere biology disorders (TBDs) encompass a group of illnesses caused by germline mutations in genes regulating telomere maintenance, resulting in very short telomeres. Possible TBD manifestations range from complex multisystem disorders with onset in childhood such as dyskeratosis congenita (DC), Hoyeraal-Hreidarsson syndrome, Revesz syndrome and Coats plus to adults presenting with one or two DC-related features.
Areas covered:
The discovery of multiple genetic causes and inheritance patterns has led to the recognition of a spectrum of clinical features affecting multiple organ systems. Patients with DC and associated TBDs are at high risk of bone marrow failure, cancer, liver and pulmonary disease. Recently, vascular diseases, including pulmonary arteriovenous malformations and gastrointestinal telangiectasias, have been recognized as additional manifestations. Diagnostics include detection of very short leukocyte telomeres and germline genetic testing. Hematopoietic cell transplantation and lung transplantation are the only current therapeutic modalities but are complicated by numerous comorbidities. This review summarizes the pathophysiology underlying TBDs, associated clinical features, management recommendations and therapeutic options.
Expert opinion:
Understanding TBDs as complex, multisystem disorders with a heterogenous genetic background and diverse phenotypes, highlights the importance of clinical surveillance and the urgent need to develop new therapeutic strategies to improve health outcomes.
Keywords: Telomere biology disorder, dyskeratosis congenita, inherited bone marrow failure syndrome, Hoyeraal-Hreidarsson syndrome, Coats plus, Revesz syndrome, pulmonary fibrosis, telomere
1. INTRODUCTION
Dyskeratosis congenita (DC) was first described by Zinsser in 1906 in a case report of two brothers presenting with leukoplakia, abnormal skin pigmentation, nail dystrophy [1]. Subsequent detailed case reports by Engman in 1926 [2] and Cole in 1930 [3] led to its designation Zinsser-Engman-Cole syndrome. The X-linked gene, dyskerin (DKC1), was discovered in affected males in 1998 [4] and mutations were shown to result in decreased telomerase activity and short telomeres, establishing the connection between telomere biology and human disease [5].
Over the last two decades, advances in genomics and an increased understanding of telomeres in disease have led to the discovery of pathogenic germline genetic variants in at least fourteen different telomere biology genes [6–9] associated with DC and related disorders. This has led to a growing appreciation of variable genetic penetrance and expressivity as well as recognition of a broad DC-related phenotypic spectrum (Table 1). The classical phenotype consists of the mucocutaneous triad of dysplastic finger and toenails, oral leukoplakia, and lacy, reticular skin pigmentation (Figure 1). Some patients present in early childhood with complex multisystem illnesses (e.g., Hoyeraal-Hreidarsson syndrome, Revesz syndrome, or Coats plus) whereas others present later in life with fewer medical problems. This variability occurs even within the same family. Patients with DC are at very high risk of bone marrow failure, cancer, pulmonary fibrosis, liver disease, and other medical problems. The exact prevalence of DC in the general population is unknown but it is generally estimated at about one in a million[10] with approximately 900–1000 cases published, to date [11–19].
Table 1:
Germline genetics of telomere biology disorders
| Gene (protein product names) | Protein complex/function | Functional consequence of disease- associated mutation | TBD subtype | Mode of inheritance1 |
|---|---|---|---|---|
| DKC1 (DKC1, dyskerin) | Telomerase enzyme complex/telomerase assembly, TERC stability | Reduced TERC stability and telomerase activity | DC, HH, PF Female carriers may have subtle findings | XLR |
| TERC (hTR, human telomerase RNA component) | Telomerase enzyme complex/telomere elongation | Reduction of telomerase activity | DC, AA, PF, LD, MDS, AML, HH | AD |
| TERT (TERT, telomerase reverse transcriptase) | Telomerase enzyme complex/telomere elongation/telomerase recruitment | Reduction telomerase recruitment, processivity and/or activity | DC, AA, PF, LD, MDS, AML | AD |
| HH | AR | |||
| NOP 10 (NOP10, NOLA3, NOLA nuclear protein family A, member 3) | Telomerase enzyme complex/telomerase assembly, TERC stability | Reduced TERC stability and telomerase activity | DC | AR |
| NHP2 (NHP2, NOLA2, nucleolar protein family A, member 2) | Telomerase enzyme complex/telomerase assembly, TERC stability | Reduced TERC stability and telomerase activity | DC | AR |
| NAF1 (NAF1, nuclear assembly factor 1 ribonucleoprotein) | Telomerase enzyme complex/telomerase assembly, TERC stability | Reduced TERC stability and telomerase activity | PF, LD, MDS | AD |
| PARN (PARN, poly(A)-specific ribonuclease) | Associated with telomerase complex/TERC RNA maturation and stabilization | Reduced TERC stability and telomerase activity | PF | AD |
| DC, HH | AR | |||
| WRAP53 (TCAB1, telomere Cajal body associated protein 1) | Associated with telomerase complex/telomerase trafficking through Cajal bodies and recruitment | Impaired telomerase trafficking though Cajal body and recruitment to telomeres | DC, HH | AR |
| ACD (TPP1, telomere protection protein 1) | Shelterin enzyme complex: Telomerase recruitment, activity and processivity | Impaired telomerase recruitment | AA | AD |
| HH | AR | |||
| STN1 (STN1, CST complex subunit) | CST-complex/C-strand fill in, telomere replication | Impaired telomere replication | CP | AR |
| CTC1 (CTC1, conserved telomere maintenance component 1) | CST-complex/C-strand fill in, telomere replication | Impaired telomere replication, fragile telomeres | DC, CP | AR |
| RTEL1 (RTEL1, regulator of telomere elongation helicase 1) | Telomeric DNA replication/repair, t-loop stability and unwinding, prevention of telomere loss during cell division | Impaired telomere replication/stability | PF, AA, LD, DC | AD |
| DC, HH | AR | |||
| TINF2 (TIN2, TERF1 [TRF1]-interacting nuclear factor 2) | Shelterin enzyme complex/telomerase regulation, sister telomere cohesion, telomere protection from DDR, telomere recruitment | Multifactorial disruption of telomere maintenance | DC, HH, RS, PF | AD |
| POT1 (POT1, protection of telomeres 1) | Shelterin enzyme complex, interaction with CST complex, negative telomerase regulation, telomere protection from DDR | Defective telomerase regulation, dysfunctional telomere replication | CP | AR |
All mentioned gene alterations can occur de novo. Most frequently it is reported in TINF2 [84,85], and has also been reported for DKC1 [175]
Abbreviations: DC, dyskeratosis congenita; HH, Hoyeraal-Hreidarsson syndrome; RS, Revesz syndrome, CP, Coats plus; PF, pulmonary fibrosis; LD, liver disease; AA, aplastic anemia; XLR, X-linked recessive; AD, autosomal dominant; AR, autosomal recessive; DDR, DNA damage response
Figure 1. Examples of clinical manifestations of telomere biology disorders.
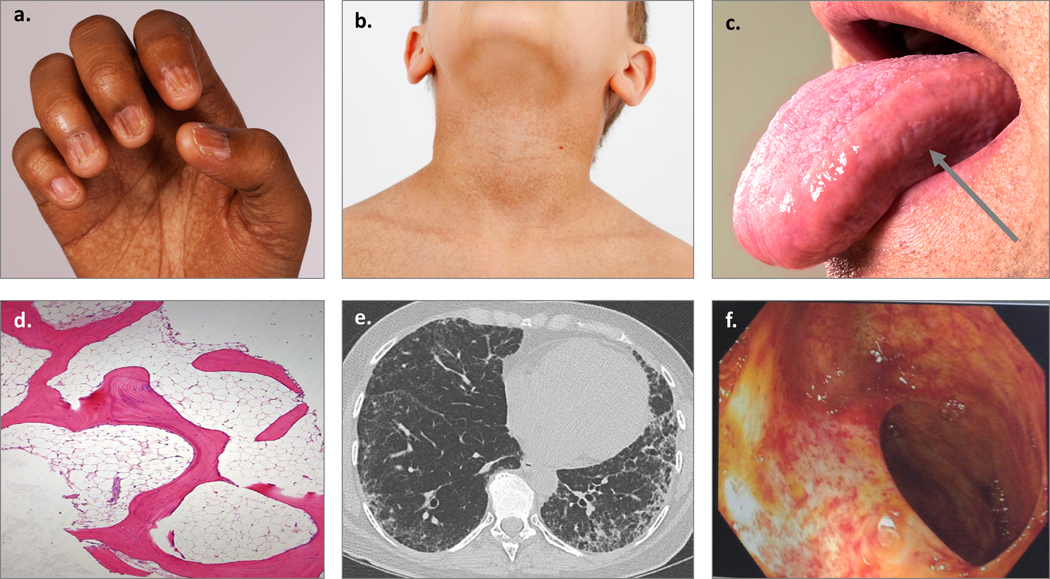
a) Nail dysplasia and palmar pigmentation changes; b) Hyperpigmentation of the neck and upper chest; c) Oral leukoplakia on the tongue, designated by arrow; d) Hypocellular bone marrow biopsy; e) Peripheral interstitial and ground-glass opacities with honeycombing consistent with usual interstitial pneumonia pattern of pulmonary fibrosis; f) Telangiectasias of the small bowel lumen.
This group of DC-related illnesses has been termed in the literature as telomeropathies, short telomere syndromes, impaired telomere maintenance spectrum disorders, and telomere biology disorders (TBDs). We favor the TBD designation because it reflects the underlying biology that unites these disorders [6,20–22].
2. TELOMERES AND TELOMERE BIOLOGY DISORDERS
2.1. TELOMERE BIOLOGY
Telomeres are specialized nucleoprotein structures at the ends of eukaryotic chromosomes that protect chromosome ends and are essential for maintaining genome stability. They consist of double-stranded TTAGGG nucleotide repeats and a six-protein complex called shelterin [23]. Human telomere lengths range from two to 14 kilobases [24] and vary depending on cell type, age, inheritance, ancestry, and measurement method [24–27]. The addition of nucleotide repeats to telomeric ends is accomplished by the telomerase holoenzyme complex consisting of telomerase reverse transcriptase (encoded by TERT), its RNA component TERC (also known as hTR, encoded by TERC), dyskerin (encoded by DKC1), NOP10, NHP2, NAF1 and GAR1[28].
Telomere structure poses two main challenges related to DNA repair and replication: 1) they may be recognized by DNA damage repair machinery as double-stranded DNA breaks, and 2) the “end replication problem” that results in ongoing telomere nucleotide repeat loss due to semiconservative replication of DNA ends during DNA replication.
The DNA damage response (DDR) could lead to recognition of telomere ends as double-strand DNA breaks, resulting in checkpoint activation, degradation and initiation of inadequate repair. Telomeres are protected from DDR, in part, by their T-loop structure. Telomeres contain both a C-rich lagging strand and a G-rich leading strand, which contains a 3’overhang consisting of single-stranded nucleotide repeats. The G-strand overhang forms a lariat-like structure through invasion of the terminal single stranded 3’ overhang into the proximal double stranded telomeric region, forming the so-called T-loop and displacing the G-rich strand, generating a displacement loop (D-loop)[29]. Additionally, telomeres are shielded by a specialized group of six proteins called the shelterin complex (Figure 2) [23]. Shelterin consists of homodimers telomeric repeat binding factor 1 (TRF1, encoded by TERF1) and TRF2 (encoded by TERF2) that bind to duplex telomeric sequences, protection of telomeres 1 (POT1, encoded by POT1) localizing to the single 3’ telomeric overhang, and linked by three additional proteins, TRF1-interacting nuclear factor 2 (TIN2, encoded by TINF2), telomere protection protein 1 (TPP1, encoded by ACD) and RAP1 (encoded by TERF2IP), all of which are recruited to the telomere through their interaction with TRF1 and TRF2. Shelterin subunits and the complex as a whole are involved in generating and stabilizing the T-loop structure (TRF2), preventing DDR (TRF2, POT1, TIN2), recruiting telomerase (TPP1, POT1, TIN2) and regulating telomere elongation (TRF1, POT1) [8,30–35]. Shelterin components also interact with the telomerase complex. For example, TPP1 interacts directly with TERT, is essential for its recruitment to telomeres and also binds POT1 [35,36].
Figure 2: Schematic depiction of the telomere and key components of telomere maintenance.

Protein names are shown. DKC1: dyskerin (encoding gene DKC1); TERC: hTR, human telomerase RNA component (TERC); TERT: human telomerase reverse transcriptase (TERT); NOP10: nuclear protein family A, member 3 (NOP10); NHP2: NOLA2 nucleolar protein family A, member 2 (NHP2); NAF1: nuclear assembly factor 1 ribonucleoprotein (NAF1); GAR1: nucleolar protein family A, member 1 (GAR1); PARN: poly (A)-specific ribonuclease (PARN); TCAB1: telomere Cajal body associated protein 1 (WRAP53); TPP1: telomere protection protein 1 (ACD); STN1: CST complex subunit (STN1); CTC1: conserved telomere maintenance component 1 (CTC1); RTEL1: Regulator of telomere elongation helicase 1 (RTEL1); TIN2: TERF1 (TRF1)-interacting nuclear factor 2 (TINF2); TRF1: telomeric repeat binding factor 1 (TERF1); TRF2: telomeric repeat binding factor 2 (TERF2); RAP1: TERF2 interacting protein (RAP1).
Components are grouped based on their function. Colored shapes indicate proteins with known telomere biology disorders associated mutations and inheritance pattern is implicated by type coloring. Blue: autosomal recessive inheritance; Yellow: autosomal dominant inheritance; Green: autosomal recessive and autosomal dominant inheritance; Red: X-linked recessive inheritance. Proteins shown but not yet reported associated with TBD are colored in gray.
The end replication problem occurs due to the inability of DNA polymerases to completely replicate the telomeric C-rich lagging strand [37,38]. When the last RNA primer at the 3′ end of DNA is removed, the newly synthesized strand is a few nucleotides shorter and causes gradual telomere shortening. Hayflick and Moorhead were the first to report the replication limit of cells in culture, which is now known to occur due to progressive telomere shortening with each cell division [39]. When telomeres reach a critically short length, a non-replicative state known as cellular senescence is triggered and some cells may then undergo apoptosis [40]. Notably the shortest telomere on a single chromosome, not average telomere length, triggers the onset of cell senescence [41]. TERT expression is silenced in most human cells after the first few weeks of embryogenesis [42,43] and therefore, telomeres shorten during life and are a cellular marker of aging. Some stem cells such as germ cells, hematopoietic stem cells, expanding lymphocytes, skin cells and the intestinal lining continue to express telomerase and maintain telomeres at a constant length, evading cellular senescence [44–47].
In addition to shelterin and the telomerase complex, the CST complex (CTC1, STN1, and TEN1) and other proteins (RTEL1, TCAB1, PARN), which directly or indirectly interact with any of the before mentioned proteins, play essential roles in telomere maintenance (see Figure 2 and Table 1). The CST complex is involved in telomere capping, interacts with shelterin proteins, and can also modulate telomerase access to the telomere [48]. RTEL1 is a DNA helicase with various functions in the telomeric context such as t-loop unwinding, duplex telomeric DNA replication and prevention of catastrophic telomere loss during cell division [49]. In interaction with TPP1 and TERT, TCAB1 functions in the recruitment of the assembled telomerase at the Cajal bodies and its trafficking to the telomere [50]. PARN is a deadenylase that processes mRNAs and non-coding RNA, including TERC [51–53].
2. 2. MOLECULAR MECHANISMS OF TELOMERE BIOLOGY DISORDERS
The central component in the etiology of DC is defective telomere biology, resulting in critically short telomeres limiting cellular replicative capacity. To date, pathogenic germline variants (i.e., mutations) in 14 genes encoding for telomere biology proteins have been described to cause TBDs. These mutations are can be inherited in X-linked recessive (XLR), autosomal dominant (AD), or autosomal recessive (AR) patterns or de novo. Genetic anticipation in which disease severity increases with successive generations has been reported seen in AD DC in association with TERC, TERT, and TINF2 mutations [54–57]. Given the complexity of telomere maintenance and the multiple genes known to be associated with telomere biology, it has been proposed to divide the mutations in these genes into five categories based on the biological effects [6]. The details are shown in Table 1.
Due to the direct association of dyskerin with active telomerase, pathogenic XLR DKC1 variants lead to destabilized TERC levels and reduced telomerase activity, resulting in extremely short telomeres. Most males with DC due to DKC1 manifest with the mucocutaneous triad and early onset bone marrow failure [58]. However, some patients may not be diagnosed until later in life and males in their fifth decade with apparently isolated idiopathic pulmonary fibrosis and liver cirrhosis have been reported [59]. Female carriers of DKC1 pathogenic variants occasionally present with subtle DC-associated features, such as isolated mucocutaneous symptoms, due to skewed X chromosome inactivation and possibly additional mechanisms, such as germline mosaicism or epigenetics [59–61].
The first pathogenic variants implicated in autosomal inheritance were detected in TERC [62] followed by the discovery of TERT mutations [63], which both impair telomerase catalytic activity. TERT mutations are predominantly heterozygous missense variants leading to haploinsufficiency. Rarely bi-allelic variants are found, leading to dramatically shortened telomeres and the HH phenotype [64]. TERC mutations may include deletion of RNA segments or affect central components, such as its template region [6]. AD pathogenic variants in TERC frequently associate with adult-onset of DC-associated manifestations, but early onset, severe disease has also been reported [65]. AR recessive DC can also be the result of bi-allelic mutations in the telomerase protein complex cofactors NOP10 and NHP2 [66,67], both affecting telomerase assembly and stability. Recently, AD NAF1 frameshift mutations, causing low telomerase RNA levels, were reported in pulmonary fibrosis-emphysema patients [9]. Heterozygous PARN mutations were first reported in familial pulmonary fibrosis probands, but later bilallelic pathogenic variants in PARN have been also been found in patients with HH [52,68,69]. Fitting its role in TERC maturation, PARN mutations are assumed to destabilize TERC levels, resulting in reduced telomerase activity [51,70]. However further investigations are warranted to elucidate whether this is the only effect of PARN alterations in defective telomere biology.
Compound heterozygote missense mutations in WRAP53, encoding for the telomerase trafficking protein TCAB1, have been shown to result in AR DC [71]. TCAB1 depletion prevents TERC from associating with Cajal bodies and therefore disrupts telomerase-telomere association [50], resulting in impaired telomere elongation [71]. Another process required for telomerase recruitment is the interaction of TERT with a specific set of amino acids (TEL patch) on the surface of TPP1. Mutations in the TPP1 encoded by ACD, affecting the TPP1 TEL patch, have been found to cause AD DC and AR HH [72,73].
Pathogenic changes in genes encoding for the components of the telomere capping CST complex, CTC1 and STN1, lead to impairment in duplex telomere replication and C-strand fill in[6]. CTC1 and STN1 alterations primarily cause Coats plus disease, which was added to the TBD spectrum after the discovery that CTC1 mutations in AR Coats plus resulted in short telomeres and also in DC phenotypes[74–79]. Biallelic mutations in STN1 were recently identified as cause of Coats plus disease in two patients [7].
Due to its role in telomere replication and the prevention of telomere loss during cell division, changes in RTEL1 significantly disrupt telomere stability. Homozygous or compound heterozygous RTEL1 mutations are associated with very short telomeres and result in HH [80–83] while heterozygous RTEL1 mutations were identified in pulmonary fibrosis patients [68].
TINF2 was the first subunit of the shelterin complex found to cause AD DC [84]. TINF2 mutations are heterozygous and often de novo, causing severe telomere shortening and are associated with HH and RS [85–87]. Rarely, TINF2 variants may also cause adult onset pulmonary fibrosis [88–90], implicating that some mutations might have a less severe impact on TIN2 function or that somatic mosaicism might play a role [91]. Biallelic POT1 mutations have been recently described in siblings with Coats plus and were proposed to lead to defective C-strand maintenance, and possibly to impaired interaction with the CST complex [8].
3. CLINICAL MANIFESTATIONS OF TELOMERE BIOLOGY DISORDERS
The discovery of germline mutations in telomere biology genes resulting in very short telomeres has unified a set of complicated disorders. Tables 1 and 2 summarize the key features of each of these disorders and their genetic etiologies. The underlying pathophysiology described above leads to disease manifestations across all organ systems. Many patients lack all mucocutaneous triad features whereas others develop it over time [92]. The spectrum of clinical complications includes neurological, ophthalmic, dental, skeletal, pulmonary, gastrointestinal, liver, hematological and immunologic abnormalities (Table 2). BMF, pulmonary disease, and malignancy represent the main causes of mortality in DC/TBD affected individuals [12]. Due to improved medical care, better diagnostics, and identifying affected individuals of older age, the overall survival in a recent analysis has improved compared to previous analyses, however the median lies still at only 51 years of age [12]. The following section details the spectrum of medical problems by TBD subtype and also by organ system (Table 2).
Table 2:
Clinical features of telomere biology disorders.
| Organ system | Features |
|---|---|
| Mucocutaneous | Classic triad: Nail dysplasia, abnormal skin pigmentation (hyper/hypopigmentation), oral leukoplakia Additional features [92]: Premature greying, scalp or eyelash hair loss, adermatoglyphia, palmoplantar hyperkeratosis, hyperhidrosis, epiphora, blepharitis |
| Hematologic | Cytopenias, bone marrow failure, isolated aplastic anemia |
| Immunologic | Immunodeficiency (lymphopenia, decreased, T, B and NK cell count, hypogammaglobulinemia)[100,176,177] |
| Central Nervous System | Brain structure: Microcephaly, cerebellar hypoplasia/atrophy, intracranial calcifications, intracranial cysts Neurological: Learning difficulty, developmental delay (psychomotor and mental), ataxia Psychiatric: mood disorders [178], schizophrenia [179] |
| Ophthalmologic | Lacrimal duct stenosis, epiphora, blepharitis, entropion, trichiasis, keratoconjunctivitis, cataracts, ulcers [131] Retinal abnormalities: retinal detachment [131], pigmentary changes [131], exudative retinopathy, proliferative retinopathy Corneal limbal insufficiency [180] |
| Oropharynx | Reduced hearing/deafness [110,181] |
| Dental | Caries, perodontitis, decreased crown/root ratio, taurodontism [110,182] |
| Cardiac | Atrial septal defect, ventricular septal defect, dilated cardiomyopathy, reported but not common [112,183] |
| Pulmonary | Pulmonary fibrosis, hepatopulmonary syndrome, pulmonary arteriovenous malformations, interstitial pneumonitis, hypersensitivity pneumonitis[117], pleuroparenchymal fibroelastosis [117], pulmonary emphysema [9,13], combined pulmonary fibrosis and emphysema [9,184] |
| Gastrointestinal and liver | Esophageal narrowing/stricture/webs, dysphagia, failure to thrive, enteropathy/enterocolitis [185] Noninfectious/nonalcoholic liver fibrosis/cirrhosis, nodular regenerative hyperplasia/non-cirrhotic portal hypertension |
| Vascular | Gastrointestinal telangiectatic anomalies and GI bleeding, pulmonary arteriovenous malformations, retinal vessel abnormalities |
| Genitouritary | Urethral stenosis/strictures/phimosis [110], undecended testes (rare)[110], males: hypospadias, penile leukoplakia, females: urethral stricture, vaginal atrophy and leukoplakia [112] |
| Reproductive | Infertility reported in one case study [89], normal fertility in women with DC/TBD analyzed for pregnancy related complications [186]. |
| Endocrine | Short stature [110], hypogonadism (male) [110], reported by not common |
| Skeletal | Osteopenia, osteoporosis [110,187], avascular necrosis of hips and shoulders [55] |
| Other | Intrauterine growth retardation, low birthweight [110] |
| Reported malignancies | AML, MDS, HNSCC (especially tongue), NHL, anal SCC, Skin: BCC, SCC Reported but rare: esophagus, rectal adenocarcinomas, cervix, thyroid, Hodgkin, PTLD (all reported in [12]), lung [117], stomach [188], pancreas, colon, hepatic adenoma [112], hepatic angiosarcoma [189,190], |
| Most common causes of death | Bone marrow failure, malignancies, pulmonary fibrosis |
References are noted for clinical features not specifically mentioned or explained in the text.
Abbreviations: AML, acute myeloid leukemia; MDS, myelodysplastic syndrome; HNSCC, head and neck squamous cell carcinoma; SCC, squamous cell carcinoma; NHL, non-Hodgkin lymphoma; PTLD, post-transplant lymphoproliferative disease; BCC, basal cell carcinoma; GI, gastrointestinal
3.1. Hoyeraal-Hreidarsson Syndrome
The original description of two brothers with cerebellar hypoplasia and pancytopenia was published by Hoyeraal in 1970, followed by a case description of a boy with progressive pancytopenia, microcephaly, cerebellar hypoplasia, and growth retardation by Hreidarsson in 1988 [93] [94]. The name Hoyeraal-Hreidarsson syndrome (HH) was subsequently proposed by the following fourth case report in 1995 [95] and the identification of DKC1 mutations [96] as well as short telomeres in HH patients connected it to the TBD spectrum. To date germline pathogenic variants causing HH have been identified in DKC1[97], TERT [64], TERC [65], TINF2 [85,98], WRAP53 [71], RTEL1 [83], PARN [99], and ACD [73]. Most HH is due to XLR or AR inheritance, except for all patients with heterozygous TINF2 and one reported patient with heterozygous TERC [65]. HH typically presents in infancy with numerous complications including cerebellar hypoplasia, microcephaly, developmental delay, immunodeficiency, intrauterine growth retardation (IUGR), as well as progressive bone marrow failure. HH-related immunodeficiency may be non-specific and challenging to diagnose [80,97,100]. Due to the young age at onset, the DC-associated mucocutaneous triad might not be present at diagnosis of HH but often develops over time[69,101]. Other HH-associated clinical features may include nonspecific enteropathy and intracranial calcifications [102]. Cerebellar hypoplasia is considered a requirement to establish the diagnosis of HH in the setting of DC-related features.
3.2. Revesz Syndrome
The defining of Revesz syndrome is bilateral exudative retinopathy, which was initially described in 1992 by Revesz et al in a 6 month old patient who subsequently developed severe BMF [103]. Notably, it should be distinguished from proliferative retinopathy, which can also occur in TBDs [104]. Additional clinical features of Revesz syndrome include IUGR, fine and sparse hair, intracerebral calcifications, cerebellar hypoplasia and psychomotor retardation, as well as other DC overlapping features. The discovery of very short telomeres and germline mutations in TINF2 have verified Revesz syndrome as a severe form of a TBD [84,87].
3.3. Coats Plus
Coats plus is characterized by bilateral exudative retinopathy, retinal telangiectasias, IUGR, intracranial calcifications, osteopenia with tendency to fracture with poor bone healing, and gastrointestinal vascular ectasias [74,105–107]. Due to the vascular ectasias, Coats plus affected individuals are at a high risk of life-threatening gastrointestinal bleeding [107,108]. Other clinical findings may include DC-related mucocutaneous changes, such as dystrophic nails and sparse or graying hair [105,107]. The clinical manifestations overlap with Revesz syndrome [103], though in Coats plus intracranial calcification may be associated with leukencephalopathy and intracranial cysts [74,106,107]. The majority of Coats plus is caused by AR pathogenic variants in components of the CST telomere capping complex including CTC1 [74,106] and rarely in STN1 [7]. The connection to TBDs was further established with the discovery of pathogenic CTC1 variants in patients with DC [75,76]. Pathogenic variants in POT1 have also been identified in patients with Coats plus [8]. In addition, many Coats plus syndrome patients feature telomeres at or below the first percentile for age, while heterozygous CTC1 carriers have telomere lengths below average but still within normal limits [74].
3.4. Bone Marrow Failure
It is important to distinguish immune-mediated acquired aplastic anemia from an inherited bone marrow failure syndrome (IBMFS), such as a TBD, because patients with IBMFS do not respond to immunosuppressive therapy [109]. BMF is a common manifestation of a TBD with a probability of about 80% for patients with classic DC to develop at least a single lineage cytopenia by the age of 30 years [110,111]. Initially, only one cell lineage may be involved which then progresses into severe pancytopenia and may later evolve into MDS. In a prospective study, clinically significant BMF was seen in 50% and manifest MDS in 20% of patients by the age of 50 years [12]. Some patients may present with aplastic anemia in the absence of DC-associated features.
3.5. Pulmonary Manifestations
Pulmonary complications have been reported in up to 20% of patients with DC and related TBDs, but may be more frequent than appreciated [12,110,112], [Giri et al, under review]. A connection between telomere biology and PF was established in 2007 with the discovery of germline mutations in telomere biology genes as the cause of familial PF [113,114]. Heterozygous pathogenic variants in TERT and TERC were the first reported [113,115]. Additionally, mutations in DKC1, PARN, RTEL1, NAF1 and rarely in TINF2 have been found in patients with familial PF [9,57,59,68,89,116–118]. Taken together, these mutations account for approximately 20–25% of familial cases, but also up to 10% sporadic PF cases are associated with pathogenic variants in telomerase biology genes (TERT, TERC, RTEL1, PARN) [113,119]. Sporadic and familial PF with underlying heterozygous pathogenic variants in TERT, TERC, RTEL1, PARN, or NAF1 predominantly manifest in late adulthood, with a median age of onset between 50 and 60 years of age [13,117]. The interstitial lung changes in PF seen on computerized tomography (CT) scans often show a pattern consistent with usual interstitial pneumonia, but atypical patterns have been found as well [120].
The most common pulmonary manifestation in TBDs is PF, a multifactorial disease leading to progressive lung scarring and fibrotic changes (Figure 1). Importantly, PF can present as isolated disease without BMF or other overt TBD-related symptoms. Children with complex phenotypes including classic DC and HH, have been reported to develop PF as teenagers [121]. PF is also a frequent complication in patients with DC after bone marrow transplantation [56,121,122] [Giri et al., under review]. It is a rapidly progressive disease with a mean survival of 2 to 5 years in clinically symptomatic patients [117,123], with a possibly more aggressive course in the TBD context [115] and the only curative therapy to date is lung transplantation.
Pulmonary function in TBDs can also be affected as part of a hepatopulmonary syndrome and might be the first presentation of portal hypertension [15]. Pulmonary arteriovenous malformation (PAVM) is increasingly recognized as being part of the TBD-related phenotypic spectrum. PAVMs have been reported in patients with and without prior hematopoietic cell transplantation (HCT) and may occur alone or in the setting of hepatopulmonary syndrome [15,124].
We recently investigated baseline pulmonary function test and outcomes in 43 patients with TBDs [Giri et al., under review]. Nearly half of the patients had asymptomatic pulmonary function abnormalities at baseline testing (42% of 43 tested subjects). These findings were associated with progression to PF at a young age in patients with XLR, AR and TINF2 DC as well as after HCT (Giri et al, under review).
3.6. Liver Disease
There is a growing body of literature of complex liver disease in TBDs, which includes non-alcoholic, non-infectious liver cirrhosis, nodular regenerative hyperplasia, non-cirrhotic portal hypertension, and hepatopulmonary syndrome (Table 2). The reported prevalence of liver disease in DC/TBD appears to range between 5–10%, but this number is highly dependent on co-morbid conditions, follow-up time, and ascertainment [15,110]. DC/TBD patients may present with lung disease caused by hepatopulmonary syndrome, due to an underlying liver disease as noted above [15]. Notably, patients presenting with hepatopulmonary syndrome have shown a progressive course of disease with median time of dyspnea symptom onset to death or liver transplantation of 6 years [15], and life-threatening gastrointestinal bleeding in the context of liver dysfunction has been reported [125].
Liver fibrosis has been reported as the initial or sole manifestation of an underlying TBD and may also be present in patients with apparently isolated PF [126]. However, most studies of liver disease in TBDs, to date, have consisted of small case series reported in the context of other manifestations. Patients with isolated liver fibrosis with or without PF typically have heterozygous germline mutations in TERT or, less often, in TERC [126–128]. A 2019 study of liver disease in 40 patients with TBDs reported the frequent occurrence of liver enzyme elevations and ultrasound abnormalities[128]. Heterozygous pathogenic variants in RTEL1, TERT, TINF2, or NHP2 were recently reported in 18 of 86 (20%) patients with end stage liver disease due to heterogenous causes [129].
3.7. Malignancies in Telomere Biology Disorders
Telomeres play a significant role in chromosomal stability and telomere dysfunction is implicated in cancer biology[130]. Patients with DC have a significantly increased lifetime risk in developing cancer [12,110]. The 2017 update on cancer incidence in 197 patients with DC registered in the NCI IMBFS longitudinal cohort study reported an approximately four-fold higher incidence of cancer in DC when compared with the general population. Patients who had undergone HCT had an approximately 30-times higher risk of cancer than healthy individuals [12]. The major cancer types in this analysis included head and neck squamous cell carcinoma (HNSCC), MDS, acute myeloid leukemia (AML) and non-Hodgkin lymphoma (NHL): MDS and AML appeared at a 578- and 24-fold greater incidence, respectively, than in the general population. The observed/expected (O/E) ratio for any HNSCC was 74, the predominant subtype being tongue HNSCC with an O/E ratio of 216, and for NHL 11-fold. The median age for developing any solid tumor was 38 years (range 18–61). The excess in tongue HNSCC highlights leukoplakia as precancerous lesion and the importance of regular surveillance as well as early diagnostic. Additional malignancies reported in DC/TBD patients are listed in Table 2. Nonmelanoma skin cancer has also been observed [12,112].
3.8. Vascular Diseases
Life-threatening gastrointestinal (GI) bleeding, similar to that seen in Coats plus, has recently been reported as a significant cause of morbidity in DC-associated TBDs [125]. At a 2017 workshop on vascular abnormalities in TBDs, significant GI bleeding, mostly due to teleangiectatic lesions (Figure 1), but in some cases without identified origin, was reported in 16 patients [125]. Additional vascular abnormalities reported include retinal vascular disease and PAVMs [124,131]. PAVMs were reported to have occurred with and without underlying hepatopulmonary syndrome [15,132–135]. In a recent retrospective case study, the presence of PAVMs independent of liver disease, including hepatopulmonary syndrome, was established [124], leading to the conclusion that PAVM can be an independent pulmonary phenotype of TBDs. Several pathogenic mechanisms are currently being discussed, such as a possible connection between short telomeres, vascular dysfunction, and impaired wound healing [125,136,137], or a link between abnormal Wnt/beta-catenin signaling, vasculopathy and telomere dysfunction [138,139].
3.9. Central Nervous System
Central nervous system involvement (CNS) has been reported in 10–25% of patients with DC/TBDs [110,112]. Structural brain abnormalities, microcephaly and developmental delay have been described in HH, RS, and Coats plus. In 2012, a report on 14 patients with DC or DC-like found primary psychiatric disorders in 64% and neurocognitive disorders in 36% [140]. More recently, we systematically evaluated 44 TBD patients with brain MRI, neurology and psychiatry evaluations and found that about half of the patients had at least one structural brain abnormality or variant, most commonly cerebellar hypoplasia (39%). Twenty-one patients (48%) had a neurologic deficit such as developmental delay or psychomotor abnormality. Twelve had psychiatric illnesses, including depression and/or anxiety (Bhala et al, under review).
3.10. Genotype-Phenotype Correlations
Evaluating the correlation between genotype and phenotype in TBDs is difficult due to small patient numbers, variable penetrance and expressivity of telomere biology defects, numerous genetic causes, and the occurrence of genetic anticipation in some families. Nevertheless, there is some evidence for the associations of certain genes and the complexities of their clinical manifestations. Heterozygous mutations in TERT, TERC, RTEL1, and PARN appear to be more likely to occur in adults with isolated disease (e.g., PF or BMF alone). More complications, including HH and RS phenotypes are more likely in XLR disease (DKC1), AR, and heterozygous TINF2 disease (Table 1). Coats plus has been primarily associated with aberrations in CTC1, STN1, or POT1. Individuals with sporadic or familial IPF with a known TERT, TERC, RTEL1, or PARN mutation, may not have telomeres as short as patients experiencing an early onset of DC/TBD symptoms [68,114,141,142]
A 2012 genotype–phenotype study found that patients with the shortest lymphocyte telomeres, measured by flow FISH, had the most severe disease, youngest age of onset, earliest disease-related mortality [58]. In this study, the shortest telomeres were associated with DKC1, AD TINF2, or no known causative mutation. In contrast, the UK DC Registry did not find a relationship between telomere length, measured by quantitative PCR, and clinical severity in their DC-cohort [65]. Notably, relatives with very short telomeres but only mild clinical symptoms have been identified in an HH family [73].
A recent study on the association of genotype and severity of mucocutaneous manifestation, found that patients with higher numbers of triad and total mucocutaneous features were more likely to present with a severe phenotype and to have AR DC or AD TINF2 mutations, while all patients lacking triad features had AD DC[92]. Additionally, a report on neurological findings in DC patients found that shorter telomeres were associated with an increased number of cerebral MRI findings and neurodevelopmental abnormalities [Bhala et al under review].
4. DIAGNOSING TELOMERE BIOLOGY DISORDERS
The diagnosis of DC and related TBDs can be complicated due to the variable, complex, and time-dependent nature of medical problems in this spectrum of illnesses (Tables 1 and 2, Figure 1). The mucocutaneous triad is often subtle, but also progressive with age [92]. Apparently isolated bone marrow failure (BMF), liver disease or pulmonary fibrosis may be the initial symptom and may first manifest in adulthood. [89] [68,143].
Classic DC should be considered in individuals with 1) all three mucocutaneous triad features (nail dysplasia, lacy skin pigmentation and oral leukoplakia); 2) any one feature of the triad in combination with BMF and two other physical findings consistent with DC; 3) BMF, myelodysplastic syndrome (MDS), or pulmonary fibrosis (PF) associated with a previously described pathogenic germline variant in a TBD-associated gene; or 4) two or more features seen in DC associated with telomere length below the first percentile for age [10].
All patients with new-onset BMF should be assessed for Fanconi anemia by chromosome breakage analysis and if that test is normal, clinical telomere length testing is recommended.
Adult-onset TBDs may not be readily diagnosed due to the heterogenous germline genetics, variable penetrance and expressivity of the phenotype, including lack of or minimally affected mucocutaneous features. Early recognition of adults with TBDs is important because the diagnosis has direct implications for surveillance and treatment decisions [144]. For example, IST is not an effective treatment for BMF due to a TBD [109]. Evaluation for a TBD is recommended for adult patients presenting with a family history of PF, aplastic anemia, early-onset HNSCC, and/or unexplained liver disease (or combinations thereof), as well as those with early-onset sporadic PF.
Flow cytometry with fluorescent in situ hybridization (flow FISH) in leukocyte subsets is the only clinically validated test, to date, proven to be reliable in DC/TBD diagnostics (Figure 3) [26,58,145–147]. Lymphocyte telomeres measured by flow FISH less than the first percentile for age are more than 95% sensitive and highly specific for differentiating patients with DC from their unaffected relatives or patients with other inherited bone marrow failure syndromes [58]. Terminal restriction fragment (TRF) measurement by Southern blot, quantitative PCR (qPCR), and single telomere length assays are useful in the research setting, but not yet validated for clinical diagnostics [26,145,148].
Figure 3. Example results of lymphocyte telomere lengths measured by flow cytometry with in situ hybridization.

Circle color codes, sex, and causative gene: red, male with heterozygous TINF2; green, male with DKC1; gray, male with DKC1; orange, female with autosomal recessive RTEL1; blue, male with heterozygous TERT; female with heterozygous TERC
Genetic testing is an important complement to clinical evaluations and telomere length measurement. Genetic education and counseling are essential for all individuals undergoing a DC/TBD evaluation because the results may have far reaching implications related to clinical prognostication and family planning. Prior to testing, individuals being tested should be given information on the clinical spectrum of the TBDs, on the modes of inheritance, and the implications of genetic testing for the entire family. Unpredicted results may occur due to variable modes of inheritance, incomplete penetrance, variable expressivity, and genetic anticipation in successive generations. Individuals undergoing testing need to understand that future medical complications cannot be predicted, but through monitoring and early detection, outcomes can be improved. Currently, genetic testing for IBMFS or other cancer predisposition syndromes contain most, but not consistently all, of the known DC/TBD-associated genes (DKC1, TERC, TERT, NOP10, NHP2, ACD, TINF2, POT1, CTC1, STN1, WRAP53, RTEL1, PARN, NAF1). Genetic testing may be inconclusive because about 20% to 30% of patients with classic DC do not have an identifiable genetic cause of their disease [10,12].
5. CLINICAL MANAGEMENT
As described above, individuals with TBDs are at high risk of suffering from severe, life-threatening complications. In 2015, the first diagnosis and management guidelines for DC and related TBDs was published and is available online (https://teamtelomere.org/resources/#research). In 2016, the American Association for Cancer Research’s Childhood Cancer Predisposition Workshop published further recommendations for cancer surveillance [149]. Based on these resources, an overview of the current surveillance and management recommendations for DC/TBD affected patients is given in Table 3. Resources for patients and families are available at Team Telomere, Inc (www.teamtelomere.org) and/or DC Action (United Kingdom, http://dcaction.org/).
Table 3:
Surveillance Recommendations for Patients with Telomere Biology Disorders
| Specialty | Recommendation |
|---|---|
| Hematology | Baseline CBC, bone marrow aspiration and biopsy with careful morphologic examination and cytogenetic studies (G-banding and FISH). If CBCs are normal and stable, annual CBC to identify trends and early manifestations. Annual bone marrow evaluation based on clinical features. Individuals with abnormalities may need more frequent monitoring due to often progressive disease. If patient has stable blood counts, less frequent monitoring may be necessary. CBCs and bone marrow evaluation should be obtained more frequently if − cytopenias are present/falling blood counts occur − or emergence of clonal cytogenetic abnormality (such as 5q-, 7q-/monosomy 7, trisomy 8, 20q-, 11q23 translocation, 3q abnormalities) is seen − if on androgen therapy: prior to therapy, repeat every 4–6 weeks, when counts are stable every 2–3 months Early referral to hematopoietic cell transplant center |
| Immunology | Consider a complete immunological evaluation including lymphocyte subsets, lymphocyte proliferation response, serum IgG, IgM, IgA levels, tetanus/diphteria/poliomyelitis/pneumococcal antibodies, based on patient’s presentation. Consider IgM isohemagglutinin titers measurement. Follow-up according to findings and immunologist recommendation Childhood vaccines, including human papilloma virus and influenza, if not contraindicated due to immunodeficiency or HCT |
| Dermatology | Regular skin self-examination and annual full body skin exam by dermatologist Regular use of sunscreen, advise to avoid excessive sun exposure |
| Neurology | MRI assessment for cerebellar hypoplasia at diagnosis in children or individuals with developmental delay or learning problems Regular evaluation for developmental delay and early intervention if needed |
| Ophthalmology | Annual examination to detect/correct vision problems, abnormally growing eyelashes, lacrimal duct stenosis, retinal changes, bleeding, cataracts and glaucoma |
| Otolaryngology | Annual cancer screening by a dentist and an otolaryngologist beginning in adolescence. Patient should be taught how to perform a monthly self-examination for oral, head and neck cancer Follow oral leukoplakia carefully and biopsy any changes or suspicious sites Baseline hearing evaluation |
| Dental | Dental hygiene and screening every 6 months Maintain good oral hygiene Inform the primary dentist of the patient’s increased risk of oral, head and neck squamous cell cancers |
| Cardiology | Baseline evaluation for arteriovenous malformations and cardiac malformations. Bubble echocardiogram for pulmonary symptoms in the absence of pulmonary fibrosis |
| Pulmonary | Baseline PFTs at diagnosis and annually, beginning at an age when the patient can properly perform the test. Early referral to specialist for shortness of breath or unexplained cough Counsel patients to avoid exposure to cigarette smoke |
| Gastroenterology/nutrition | Evaluate for clinical history suspicious for esophageal stenosis and/or enteropathy and refer as needed. Liver function tests at least annually If on androgen therapy: Check liver function tests prior to starting, then every 6–12 weeks Check lipid profile prior to starting and every 6–12 months. Perform liver ultrasound examination prior to initiation of androgens and semiannually to evaluate for adenomas, carcinomas or fibrosis. |
| Genitourinary | Baseline assessment for genitourinary anomalies, including symptoms of urethral stenosis |
| Endocrinology | Follow growth carefully If on androgen therapy: Evaluation prior to therapy, on treatment regular (annually) evaluation for side effects |
| Orthopedics | Baseline bone density scan to evaluate for osteopenia at approximately 14 years of age. Follow-up bone density scans yearly or as recommended by physician. Evaluation of hip and shoulder avascular necrosis based on symptoms. Vitamin D and calcium as needed to optimize bone health If on androgen therapy: In growing child baseline prior to treatment, then every 6–12 months |
Recommendations based on the guidelines published on www.teamtelomere.org and the results of the 2016 American Association for Cancer Research Childhood Cancer Predisposition Workshop [149].
Abbreviations: CBC, complete blood count; HCT, hematopoietic cell transplantation; PFTs, pulmonary function tests
Classification of bone marrow failure
Mild: ANC 1000-<1500/mm3, Platelets 50.000-<150.000/mm3, Hb ≥8g/dl-less than normal for age
Moderate: ANC 500-<1000/mm3, Platelets 20.000-< 50.0000/mm3, Hb ≥8g/dl-less than normal for age
Severe: ANC < 500/mm3, Platelets < 20.000/mm3, Hb < 8.0 g/dl
5.1. Bone Marrow Failure
BMF is a major clinical complication in DC and related TBDs, may be the first clinical manifestation, and remains the most common cause of mortality [12,110]. Regular monitoring of asymptomatic individuals with mild or moderate cytopenias is recommended while those with persistent severe BMF should receive therapy (Table 3). Unlike in acquired aplastic anemia, BMF in the TBDs does not respond to immunosuppressive therapy (IST) [109].
Oral androgens, such as oxymethalone or halotestin, have successfully been used for over 50 years in the treatment for aplastic anemia including for BMF in Fanconi anemia and DC [112,150] [151], Androgen related side effects include virilization, dyslipidemia, liver function abnormalities and elevated risk of liver adenomas. Individuals with TBDs may be more susceptible to these therapy-limiting side effects because of TBD-related liver pathology. Patients taking androgens should not simultaneously use hematopoietic growth factors because splenic peliosis and splenic rupture have been reported with concurrent use[152]. The synthetic androgen derivative danazol is currently the preferred androgen for TBD-related BMF because it has fewer virilizing side effects.
Retrospective analyses on the use of danazol in patients with DC have reported response, defined as independence from red blood cell or platelet transfusions, rates of up to 70% [153,154]. One prospective trial using danazol over 24 months in adults with TBDs reported a hematologic response rate of approximately 80% [155]. However, several patients on that study discontinued treatment early and 41% showed increased liver enzyme levels. The authors of this trial reported telomere elongation in peripheral blood DNA, measured by qPCR, over the treatment duration. Based on a previous preclinical study [156], the authors hypothesized that danazol may lead to activation of an estrogen-responsive element in the reporter region of the telomerase gene, resulting in upregulation of TERT expression and subsequently in telomere elongation. However, danazol and its derivates are not capable of being aromatized to estrogens [157]. A subsequent retrospective study using flow FISH to measure telomere length did not find a significant difference in telomere attrition between androgen-treated and untreated patients with DC, but that study had only a few danazol-treated patients. [158]. Overall, androgens such as danazol, are a feasible therapy option in patients with a hematologic treatment indication who are not candidates for HCT, either due to medical reasons, lacking donor, or personal choice.
Eltombopag, a thrombopoietin receptor (c-Mpl) agonist, is now used in front-line therapy for severe aplastic anemia (SAA) in combination with IST. There has been no systematic evaluation of Eltrombopag in patients with TBDs. There is one report, to date, of two patients with DC patients treated with Eltrombopag for BMF in whom there was no therapeutic response [159].
HCT is the only curative therapeutic approach for BMF in DC/TBDs and when possible a matched, related donor HCT is the treatment of choice [122,160]. Related donors must be proven not to be affected by DC/TBD by genetic and/or telomere length testing because of the phenotypic variability in the TBDs and poor outcomes associated with using affected donors [143] . HCT from an unrelated donor can be considered for those lacking a matched, related donor. HCT has been performed for DC-related BMF since the 1980s [161], but outcomes for patients undergoing conventional HCT were dismal with a high frequency of fatal lung complications [162]. With the increased use of reduced intensity conditioning regimen, HCT outcomes have improved after 2000 [160,162] and a retrospective study of 34 DC patients transplanted since 1981 documented an increase of 5-year post HCT survival from 46% to 65% [162]. However, the 10-year post transplant survival still ranges only between 20 – 30 % [160,162]. A recent, and to date largest, retrospective analysis of HCT data of 94 DC patients showed better outcomes in patients of younger age (3-year overall survival 72% in patients <20 years of age vs 43% in patients ≥20 years of age) and in patients with no pre-existing organ damage[122]. Prior organ impairment was associated with chronic GvHD and nearly all patients showed lung damage post-transplant, which was irreversible in most cases [122].
A prospective multi-institutional clinical trial HCT TBD-associated BMF (ClinicalTrials.gov Identifier: NCT01659606) is currently ongoing. This study tests whether a regimen that avoids DNA alkylators and radiation can permit successful HCT without compromising survival in patients with DC.
The comorbidities in DC/TBD such as PF, liver disease and vascular abnormalities and risk of secondary malignancies make careful post-HCT clinical management challenging and especially important [12,163]. Patients are at high risk of avascular necrosis of hips and shoulders and fractures which could be exacerbated by corticosteroid use. Medications known to be associated with lung or liver toxicity should be avoided, if possible. Further studies are warranted to optimize HCT strategies for this unique patient group and reduce therapy-related toxicity.
5.2. Pulmonary and Liver Disease
Management of pulmonary and liver disease in patients with TBDs is complicated due to lack of proven therapies and the frequent presence of significant co-morbidities. Anti-fibrotic medications, such as pirfenidone and nintedanib, used in PF have not been prospectively studied in TBD-associated PF. One retrospective analysis suggested that PF patients with TERT/TERC associated TBD may not be as likely to respond to pirfenidone as non-carriers [164]. Also, there are no data to suggest IST for PF would be effective [165]. The question has been raised if androgen treatment, specifically danazol, could slow down the progression of pulmonary fibrosis [155,166]. One case report showed amelioration of pulmonary fibrosis symptoms during Danazol treatment in a TINF2 patient after HCT, however there was no long-term follow-up since the patients in that report succumbed to infection [166]. The prospective trial on the efficacy of danazol in patients with DC mentioned above stated that there was no further decrease in lung function as measured by diffusing capacity of the lungs for carbon monoxide (DLCO) in seven patients with TBDs [155]. To date there is no systematic data on possible positive or negative androgen effects on PF in TBDs. Lung transplantation as curative approach has been successfully performed in patients with DC/TBD but there are limited long term outcome data [121,167–169]. The three published studies on lung transplantation in TBD patients showed a high rate of renal complications and a high incidence of hematological abnormalities after lung transplantation [167–169]. Thrombocytopenia after lung transplant was common and progressive BMF has also been reported [168]. Pre-lung transplant screening by bone marrow biopsy in TBD patients is advisable to evaluate whether the hematological stress of immunosuppressive medication can be tolerated.
The literature on liver transplantation in patients with TBDs is scarce. There is one case report of liver transplantation after HCT in a patient with DC who had cirrhosis, hepatopulmonary syndrome, and numerous co-morbid conditions [170] and in a population of patients with liver cirrhosis awaiting liver transplantation possibly pathogenic TBD gene variants were identified [129]. These reports suggest that liver transplantation in TBDs is a growing need and additional study is warranted to develop the optimal approaches.
5.4. Multi-organ Transplantation
The multisystemic manifestations of TBDs can lead to the co-occurrence of complex co-morbidities. For example, severe pulmonary disease and bone marrow failure [167,171] or pulmonary fibrosis and liver cirrhosis [172] have been reported. In selected cases this might lead to the clinical dilemma on the sequence of multi-organ transplantation, for which to date no general recommendation exists. Successful combined lung and liver transplantation in TBD patients has been reported, but there are no long-term outcome data available [172]. There is one trial underway for lung transplant in tandem with bone marrow transplant for patients (ClinicalTrials.gov Identifier: NCT03500731), but results need to be awaited to determine if this would be a therapy option in the context of DC/TBD.
5.5. Vascular Diseases
PAVMs and GI telangiectasias are especially challenging to manage because of limited therapeutic options[125]. PAVMs may occur in the setting of PF and/or hepatopulmonary syndrome and thus have to be considered in this context. GI bleeding due to telangiectasis can be life threatening and difficult to control. Platelet and/or red blood cell transfusions may be required. Upper and lower endoscopy may identify specific lesions for intervention. Propranolol, estrogens, and anti-angiogenic agents have been postulated to potentially be effective. However, these data are limited to case reports and/or anecdotes [125].
5.6. Cancer
Early cancer diagnosis through surveillance is important to reduce cancer-related morbidity and mortality in patients with TBDs. Screening can identify HNSCC when it is still amenable to surgical resection only. TBD patients with hematopoietic malignancies may have more therapy-related complications than non-TBD patients and need to be monitored carefully for side effects [163].
5.7. An Urgent Need for New Therapeutics
The complex medical problems in the TBDs illustrate an urgent need for new therapies. Improvements in supportive care and HCT have increased survival in patients with DC, HH, Revesz syndrome, and BMF. Consequently, patients are living longer and developing additional complications that may or may not have been previously recognized. Lung and liver transplantation remain viable options, even after HCT, but ideally, future therapies would not involve transplantation of any organ(s).
Preclinical studies are underway based on the increased understanding of telomere biology underlying the TBDs. For example, a recently recognized connection between the WNT signaling pathway and telomere maintenance proteins has been described, suggesting a possible role of this pathway in DC/TBD pathology [173]. Targeting the WNT pathway might therefore offer a novel therapeutic approach and further research exploring this option is currently ongoing. Similarly, therapies targeting specific mutations may eventually prove useful. A short peptide derived from dyskerin showed some utility in cell culture[174].
6. EXPERT OPINION
The direct connection between pathogenic germline variants, telomere biology and human disease was established 20 years ago [5,175]. Aberrations in at least 14 genes are now associated with TBDs and the gene list is likely to grow as more and more individuals and their families are studied. More patients are now recognized to have TBDs due to, in part, telomere length testing by flow FISH of lymphocyte, and the increasing use of clinical sequencing via large gene panels or exome sequencing. However, we are now faced with the challenge of a growing clinical spectrum of disorders related to germline mutations in telomere biology genes and associated with all modes of inheritance. This also comes with a growing degree of uncertainty regarding genetic test results in patients undergoing panel or exome sequencing in the absence of a clear TBD phenotype. How does one counsel the parents of a child with a neurologic disorder whose exome sequencing identified a likely pathogenic variant in TERT?
Basic science studies of TBD-associated mutations are yielding important insights into the specific functions of each gene and variants within each gene. This has led to a growing appreciation of gene-gene interactions and the multiple functions each one may possess, even within the same pathway. TBDs are no longer “just” associated with alterations related to telomerase activity, but also to telomere protection, capping, RNA acetylation and helicase activity. We must constantly be on the look-out for non-telomeric functions of these genes and consider how they could contribute to clinical manifestations.
Short germline telomeres and the biological connections have led us to recognize that TBDs, like other disorders, occur along a phenotypic spectrum with some patients presenting in infancy and others not until middle-age or older. This is important clinically as we monitor patients and develop new therapies for complications. For example, could a drug effective in elderly-onset TBD PF be of use in a child with TBD BMF and liver disease?
In all rare diseases, international collaboration between clinicians of all specialties, basic scientists, and patient support and advocacy groups are absolutely essential for improving our understanding of TBD etiology, the development of new therapeutics, and providing optimal care of all TBD families. The Clinical Care Consortium of Telomere-associated ailments (CCCTAA) is an international group of clinicians and scientists who are working together with Team Telomere, Inc to develop clinical trials aimed to improve the lives of patients with TBDs [124,125].
It is our hope that in the next five years, novel therapeutic agents will be in clinical trials targeting key manifestations of the TBDs, such as PF, PAVM, GI bleeding, and/or liver disease. The current multi-center HCT trial will have generated critical data to optimize therapy for patients with BMF requiring HCT. Additional genetic causes will be identified, and genotype-phenotype correlations will be established to help in tailoring individual patient therapy. Disease modifiers may also be uncovered as multi-modal approaches are taken to further understand TBD etiology. Members of the CCCTAA will play a central role in these efforts through database creation, data sharing, and collaborative clinical trials.
ARTICLE HIGHLIGHTS.
Dyskeratosis congenita (DC) and related telomere biology disorders (TBDs) are caused by germline mutations in telomere biology genes resulting in very short telomeres.
The phenotypic spectrum affects all organ systems ranging from classic DC, Hoyeraal-Hreidarsson syndrome, Revesz syndrome and Coats plus to apparently isolated aplastic anemia, pulmonary fibrosis or liver disease.
Better understanding of the genetic etiology has led to the recognition of additional TBD-related manifestations such as vascular disease and structural brain abnormalities.
Patients with TBDs are at high risk of bone marrow failure, pulmonary disease, liver disease, and cancer.
To date, there are no curative therapeutic approaches other than hematopoietic cell transplantation for TBD-related bone marrow failure.
The complex medical problems in patients with TBDs require close clinical surveillance and highlight the need for new therapeutic approaches.
ACKNOWLEDGEMENTS
We thank the patients and their families for inspiring us every day. This work was supported by the intramural research program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute, and fellowship support for M.R.N. from the Mildred-Scheel-Postdoctoral Fellowship Program by the German Cancer Aid.
Financial & competing interest disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Footnotes
No writing assistance was utilized in the production of this manuscript.
REFERENCES
- 1.Zinsser F. Atrophia cutis reticularis cum pigmentatione dystrophia unguium et leukoplakia oris; poikiloderma atrophicans vascularis Jacobi. Inkonographia Dermatologia. 1906 1906;5:219–223. [Google Scholar]
- 2.Engman MF. A unique case of reticular pigmentation of the skin with atrophy. Arch Bleg Dermatol Syphiligr. 1926 1926;13:685–687. [Google Scholar]
- 3.COLE HN, RAUSCHKOLB JE, TOOMEY J. DYSKERATOSIS CONGENITA WITH PIGMENTATION, DYSTROPHIA UNGUIS AND LEUKOKERATOSIS ORIS. JAMA Dermatology. 1930;21(1):71–95. [DOI] [PubMed] [Google Scholar]
- 4.Heiss NS, Knight SW, Vulliamy TJ, et al. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nature Genetics. 1998 1998/May/01;19(1):32–38. [DOI] [PubMed] [Google Scholar]
- 5.Mitchell JR, Wood E, Collins K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature. 1999. Dec 2;402(6761):551–5. [DOI] [PubMed] [Google Scholar]
- 6.Bertuch AA. The molecular genetics of the telomere biology disorders. RNA Biol. 2016. Aug 2;13(8):696–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simon AJ, Lev A, Zhang Y, et al. Mutations in STN1 cause Coats plus syndrome and are associated with genomic and telomere defects. J Exp Med. 2016. Jul 25;213(8):1429–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takai H, Jenkinson E, Kabir S, et al. A POT1 mutation implicates defective telomere end fill-in and telomere truncations in Coats plus. Genes & Development. 2016. April 1, 2016;30(7):812–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stanley SE, Gable DL, Wagner CL, et al. Loss-of-function mutations in the RNA biogenesis factor NAF1 predispose to pulmonary fibrosis–emphysema. Science Translational Medicine. 2016;8(351):351ra107–351ra107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dokal I, Vulliamy T, Mason P, et al. Clinical utility gene card for: Dyskeratosis congenita - update 2015. Eur J Hum Genet. 2015. Apr;23(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vulliamy TJ, Marrone A, Knight SW, et al. Mutations in dyskeratosis congenita: their impact on telomere length and the diversity of clinical presentation. Blood. 2006;107(7):2680–2685. [DOI] [PubMed] [Google Scholar]
- 12.Alter BP, Giri N, Savage SA, et al. Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica. 2018. Jan;103(1):30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diaz de Leon A, Cronkhite JT, Katzenstein A-LA, et al. Telomere Lengths, Pulmonary Fibrosis and Telomerase (TERT) Mutations. PLOS ONE. 2010;5(5):e10680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gutierrez-Rodrigues F, Donaires FS, Pinto A, et al. Pathogenic TERT promoter variants in telomere diseases. Genet Med. 2018. Dec 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gorgy AI, Jonassaint NL, Stanley SE, et al. Hepatopulmonary syndrome is a frequent cause of dyspnea in the short telomere disorders. Chest. 2015. Oct;148(4):1019–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghemlas I, Li H, Zlateska B, et al. Improving diagnostic precision, care and syndrome definitions using comprehensive next-generation sequencing for the inherited bone marrow failure syndromes. Journal of Medical Genetics. 2015;52(9):575–584. [DOI] [PubMed] [Google Scholar]
- 17.Kirschner M, Maurer A, Wlodarski MW, et al. Recurrent somatic mutations are rare in patients with cryptic dyskeratosis congenita. Leukemia. 2018. Aug;32(8):1762–1767. [DOI] [PubMed] [Google Scholar]
- 18.Yamaguchi H, Sakaguchi H, Yoshida K, et al. Clinical and genetic features of dyskeratosis congenita, cryptic dyskeratosis congenita, and Hoyeraal-Hreidarsson syndrome in Japan. Int J Hematol. 2015. Nov;102(5):544–52. [DOI] [PubMed] [Google Scholar]
- 19.Alter BP, Giri N, Savage SA, et al. Cancer in dyskeratosis congenita. Blood. 2009. Jun 25;113(26):6549–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Savage SA, Dokal I, Armanios M, et al. Dyskeratosis congenita: the first NIH clinical research workshop. Pediatr Blood Cancer. 2009. Sep;53(3):520–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ballew BJ, Savage SA. Updates on the biology and management of dyskeratosis congenita and related telomere biology disorders. Expert Rev Hematol. 2013. Jun;6(3):327–37. [DOI] [PubMed] [Google Scholar]
- 22.Savage SA. Beginning at the ends: telomeres and human disease. F1000Res. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005. Sep 15;19(18):2100–10. [DOI] [PubMed] [Google Scholar]
- 24.de Lange T, Shiue L, Myers RM, et al. Structure and variability of human chromosome ends. Molecular and Cellular Biology. 1990;10(2):518–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hansen ME, Hunt SC, Stone RC, et al. Shorter telomere length in Europeans than in Africans due to polygenetic adaptation. Hum Mol Genet. 2016. Jun 1;25(11):2324–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gutierrez-Rodrigues F, Santana-Lemos BA, Scheucher PS, et al. Direct comparison of flow-FISH and qPCR as diagnostic tests for telomere length measurement in humans. PloS one. 2014;9(11):e113747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Factor-Litvak P, Susser E, Kezios K, et al. Leukocyte Telomere Length in Newborns: Implications for the Role of Telomeres in Human Disease. Pediatrics. 2016. Apr;137(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmidt JC, Cech TR. Human telomerase: biogenesis, trafficking, recruitment, and activation. Genes Dev. 2015. Jun 1;29(11):1095–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Griffith JD, Comeau L, Rosenfield S, et al. Mammalian telomeres end in a large duplex loop. Cell. 1999. May 14;97(4):503–14. [DOI] [PubMed] [Google Scholar]
- 30.Doksani Y, Wu JY, de Lange T, et al. Super-resolution fluorescence imaging of telomeres reveals TRF2-dependent T-loop formation. Cell. 2013. Oct 10;155(2):345–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Denchi EL, de Lange T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature. 2007. Aug 30;448(7157):1068–71. [DOI] [PubMed] [Google Scholar]
- 32.Sfeir A, Kosiyatrakul ST, Hockemeyer D, et al. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 2009. Jul 10;138(1):90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zimmermann M, Kibe T, Kabir S, et al. TRF1 negotiates TTAGGG repeat-associated replication problems by recruiting the BLM helicase and the TPP1/POT1 repressor of ATR signaling. Genes Dev. 2014. Nov 15;28(22):2477–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim SH, Beausejour C, Davalos AR, et al. TIN2 mediates functions of TRF2 at human telomeres. J Biol Chem. 2004. Oct 15;279(42):43799–804. [DOI] [PubMed] [Google Scholar]
- 35.Nandakumar J, Bell CF, Weidenfeld I, et al. The TEL patch of telomere protein TPP1 mediates telomerase recruitment and processivity. Nature. 2012. Dec 13;492(7428):285–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rice C, Shastrula PK, Kossenkov AV, et al. Structural and functional analysis of the human POT1-TPP1 telomeric complex. Nat Commun. 2017. Apr 10;8:14928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olovnikov AM. A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J Theor Biol. 1973. Sep 14;41(1):181–90. [DOI] [PubMed] [Google Scholar]
- 38.Watson JD. Origin of concatemeric T7 DNA. Nat New Biol. 1972. Oct 18;239(94):197–201. [DOI] [PubMed] [Google Scholar]
- 39.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961. Dec;25:585–621. [DOI] [PubMed] [Google Scholar]
- 40.Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990. May 31;345(6274):458–60. [DOI] [PubMed] [Google Scholar]
- 41.Hemann MT, Strong MA, Hao LY, et al. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell. 2001. Oct 5;107(1):67–77. [DOI] [PubMed] [Google Scholar]
- 42.Ulaner GA, Hu JF, Vu TH, et al. Telomerase activity in human development is regulated by human telomerase reverse transcriptase (hTERT) transcription and by alternate splicing of hTERT transcripts. Cancer Res. 1998. Sep 15;58(18):4168–72. [PubMed] [Google Scholar]
- 43.Kim NW, Piatyszek MA, Prowse KR, et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994. Dec 23;266(5193):2011–5. [DOI] [PubMed] [Google Scholar]
- 44.Härle-Bachor C, Boukamp P. Telomerase activity in the regenerative basal layer of the epidermis inhuman skin and in immortal and carcinoma-derived skin keratinocytes. Proc Natl Acad Sci U S A. 1996;93(13):6476–6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hiyama E, Tatsumoto N, Kodama T, et al. Telomerase activity in human intestine. Int J Oncol. 1996. Sep;9(3):453–8. [DOI] [PubMed] [Google Scholar]
- 46.Hiyama K, Hirai Y, Kyoizumi S, et al. Activation of telomerase in human lymphocytes and hematopoietic progenitor cells. J Immunol. 1995. Oct 15;155(8):3711–5. [PubMed] [Google Scholar]
- 47.Kim W, Shay JW. Long-range telomere regulation of gene expression: Telomere looping and telomere position effect over long distances (TPE-OLD). Differentiation. 2018. Jan-Feb;99:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rice C, Skordalakes E. Structure and function of the telomeric CST complex. Comput Struct Biotechnol J. 2016;14:161–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vannier JB, Sarek G, Boulton SJ. RTEL1: functions of a disease-associated helicase. Trends Cell Biol. 2014. Jul;24(7):416–25. [DOI] [PubMed] [Google Scholar]
- 50.Venteicher AS, Abreu EB, Meng Z, et al. A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science. 2009. Jan 30;323(5914):644–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moon DH, Segal M, Boyraz B, et al. Poly(A)-specific ribonuclease (PARN) mediates 3’-end maturation of the telomerase RNA component. Nat Genet. 2015. Dec;47(12):1482–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tummala H, Walne A, Collopy L, et al. Poly(A)-specific ribonuclease deficiency impacts telomere biology and causes dyskeratosis congenita. J Clin Invest. 2015. May;125(5):2151–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boyraz B, Moon DH, Segal M, et al. Posttranscriptional manipulation of TERC reverses molecular hallmarks of telomere disease. J Clin Invest. 2016. Sep 1;126(9):3377–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vulliamy T, Marrone A, Szydlo R, et al. Disease anticipation is associated with progressive telomere shortening in families with dyskeratosis congenita due to mutations in TERC. Nat Genet. 2004. May;36(5):447–9. [DOI] [PubMed] [Google Scholar]
- 55.Savage SA. Chapter Two - Human Telomeres and Telomere Biology Disorders. In: Calado RT, editor. Progress in Molecular Biology and Translational Science. Vol. 125: Academic Press; 2014. p. 41–66. [DOI] [PubMed] [Google Scholar]
- 56.Parry EM, Alder JK, Qi X, et al. Syndrome complex of bone marrow failure and pulmonary fibrosis predicts germline defects in telomerase. Blood. 2011;117(21):5607–5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Du H, Guo Y, Ma D, et al. A case report of heterozygous TINF2 gene mutation associated with pulmonary fibrosis in a patient with dyskeratosis congenita. Medicine (Baltimore). 2018. May;97(19):e0724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alter BP, Rosenberg PS, Giri N, et al. Telomere length is associated with disease severity and declines with age in dyskeratosis congenita. Haematologica. 2012. Mar;97(3):353–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alder JK, Parry EM, Yegnasubramanian S, et al. Telomere Phenotypes in Females with Heterozygous Mutations in the Dyskeratosis Congenita 1 (DKC1) Gene. Human Mutation. 2013;34(11):1481–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu J, Khincha PP, Giri N, et al. Investigation of chromosome X inactivation and clinical phenotypes in female carriers of DKC1 mutations. American Journal of Hematology. 2016;91(12):1215–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vulliamy TJ, Knight SW, Dokal I, et al. Skewed X-inactivation in carriers of X-linked dyskeratosis congenita. Blood. 1997. Sep 15;90(6):2213–6. [PubMed] [Google Scholar]
- 62.Vulliamy T, Marrone A, Goldman F, et al. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature. 2001. Sep 27;413(6854):432–5. [DOI] [PubMed] [Google Scholar]
- 63.Vulliamy TJ, Walne A, Baskaradas A, et al. Mutations in the reverse transcriptase component of telomerase (TERT) in patients with bone marrow failure. Blood Cells Mol Dis. 2005. May-Jun;34(3):257–63. [DOI] [PubMed] [Google Scholar]
- 64.Marrone A, Walne A, Tamary H, et al. Telomerase reverse-transcriptase homozygous mutations in autosomal recessive dyskeratosis congenita and Hoyeraal-Hreidarsson syndrome. Blood. 2007. Dec 15;110(13):4198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vulliamy TJ, Kirwan MJ, Beswick R, et al. Differences in disease severity but similar telomere lengths in genetic subgroups of patients with telomerase and shelterin mutations. PLoS One. 2011;6(9):e24383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Walne AJ, Vulliamy T, Marrone A, et al. Genetic heterogeneity in autosomal recessive dyskeratosis congenita with one subtype due to mutations in the telomerase-associated protein NOP10. Hum Mol Genet. 2007. Jul 1;16(13):1619–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vulliamy T, Beswick R, Kirwan M, et al. Mutations in the telomerase component NHP2 cause the premature ageing syndrome dyskeratosis congenita. Proceedings of the National Academy of Sciences. 2008;105(23):8073–8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stuart BD, Choi J, Zaidi S, et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nature Genetics. 2015. April/13/online;47:512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Burris AM, Ballew BJ, Kentosh JB, et al. Hoyeraal-Hreidarsson Syndrome due to PARN Mutations: Fourteen Years of Follow-Up. Pediatric Neurology. 2016 2016/March/01/;56:62–68.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roake CM, Chen L, Chakravarthy AL, et al. Disruption of Telomerase RNA Maturation Kinetics Precipitates Disease. Mol Cell. 2019. May 16;74(4):688–700 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhong F, Savage SA, Shkreli M, et al. Disruption of telomerase trafficking by TCAB1 mutation causes dyskeratosis congenita. Genes Dev. 2011. Jan 1;25(1):11–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guo Y, Kartawinata M, Li J, et al. Inherited bone marrow failure associated with germline mutation of ACD, the gene encoding telomere protein TPP1. Blood. 2014;124(18):2767–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kocak H, Ballew BJ, Bisht K, et al. Hoyeraal-Hreidarsson syndrome caused by a germline mutation in the TEL patch of the telomere protein TPP1. Genes Dev. 2014. Oct 1;28(19):2090–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Anderson BH, Kasher PR, Mayer J, et al. Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus. Nature Genetics. 2012. January/22/online;44:338. [DOI] [PubMed] [Google Scholar]
- 75.Keller RB, Gagne KE, Usmani GN, et al. CTC1 Mutations in a patient with dyskeratosis congenita. Pediatr Blood Cancer. 2012. Aug;59(2):311–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Walne AJ, Bhagat T, Kirwan M, et al. Mutations in the telomere capping complex in bone marrow failure and related syndromes. Haematologica. 2013. Mar;98(3):334–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Savage SA. Connecting complex disorders through biology. Nat Genet. 2012. Feb 27;44(3):238–40. [DOI] [PubMed] [Google Scholar]
- 78.Chen LY, Majerska J, Lingner J. Molecular basis of telomere syndrome caused by CTC1 mutations. Genes Dev. 2013. Oct 1;27(19):2099–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gu P, Chang S. Functional characterization of human CTC1 mutations reveals novel mechanisms responsible for the pathogenesis of the telomere disease Coats plus. Aging Cell. 2013. Dec;12(6):1100–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ballew BJ, Joseph V, De S, et al. A recessive founder mutation in regulator of telomere elongation helicase 1, RTEL1, underlies severe immunodeficiency and features of Hoyeraal Hreidarsson syndrome. PLoS Genet. 2013. Aug;9(8):e1003695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Deng Z, Glousker G, Molczan A, et al. Inherited mutations in the helicase RTEL1 cause telomere dysfunction and Hoyeraal-Hreidarsson syndrome. Proc Natl Acad Sci U S A. 2013. Sep 3;110(36):E3408–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Le Guen T, Jullien L, Touzot F, et al. Human RTEL1 deficiency causes Hoyeraal-Hreidarsson syndrome with short telomeres and genome instability. Hum Mol Genet. 2013. Aug 15;22(16):3239–49. [DOI] [PubMed] [Google Scholar]
- 83.Walne AJ, Vulliamy T, Kirwan M, et al. Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. Am J Hum Genet. 2013. Mar 7;92(3):448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Savage SA, Giri N, Baerlocher GM, et al. TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am J Hum Genet. 2008. Feb;82(2):501–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Walne AJ, Vulliamy T, Beswick R, et al. TINF2 mutations result in very short telomeres: analysis of a large cohort of patients with dyskeratosis congenita and related bone marrow failure syndromes. Blood. 2008. Nov 01;112(9):3594–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sarper N, Zengin E, Kilic SC. A child with severe form of dyskeratosis congenita and TINF2 mutation of shelterin complex. Pediatr Blood Cancer. 2010. Dec 01;55(6):1185–6. [DOI] [PubMed] [Google Scholar]
- 87.Sasa GS, Ribes-Zamora A, Nelson ND, et al. Three novel truncating TINF2 mutations causing severe dyskeratosis congenita in early childhood. Clin Genet. 2012. May;81(5):470–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kannengiesser C, Borie R, Ménard C, et al. Heterozygous RTEL1 mutations are associated with familial pulmonary fibrosis. European Respiratory Journal. 2015;46(2):474–485. [DOI] [PubMed] [Google Scholar]
- 89.Alder JK, Stanley SE, Wagner CL, et al. Exome Sequencing Identifies Mutant TINF2 in a Family With Pulmonary Fibrosis. Chest. 2015 2015/May/01/;147(5):1361–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fukuhara A, Tanino Y, Ishii T, et al. Pulmonary fibrosis in dyskeratosis congenita with TINF2 gene mutation. Eur Respir J. 2013. Dec;42(6):1757–9. [DOI] [PubMed] [Google Scholar]
- 91.Kannengiesser C, Borie R, Revy P. Pulmonary fibrosis associated with TINF2 gene mutation: is somatic reversion required? Eur Respir J. 2014. Jul;44(1):269–70. [DOI] [PubMed] [Google Scholar]
- 92.Ward SC, Savage SA, Giri N, et al. Beyond the triad: Inheritance, mucocutaneous phenotype, and mortality in a cohort of patients with dyskeratosis congenita. J Am Acad Dermatol. 2018. Apr;78(4):804–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hoyeraal HM, Lamvik J, Moe PJ. Congenital hypoplastic thrombocytopenia and cerebral malformations in two brothers. Acta Paediatr Scand. 1970. Mar;59(2):185–91. [DOI] [PubMed] [Google Scholar]
- 94.Hreidarsson S, Kristjansson K, Johannesson G, et al. A syndrome of progressive pancytopenia with microcephaly, cerebellar hypoplasia and growth failure. Acta Paediatr Scand. 1988. Sep;77(5):773–5. [DOI] [PubMed] [Google Scholar]
- 95.Aalfs CM, van den Berg H, Barth PG, et al. The Hoyeraal-Hreidarsson syndrome: the fourth case of a separate entity with prenatal growth retardation, progressive pancytopenia and cerebellar hypoplasia. Eur J Pediatr. 1995. Apr;154(4):304–8. [DOI] [PubMed] [Google Scholar]
- 96.Knight SW, Heiss NS, Vulliamy TJ, et al. Unexplained aplastic anaemia, immunodeficiency, and cerebellar hypoplasia (Hoyeraal-Hreidarsson syndrome) due to mutations in the dyskeratosis congenita gene, DKC1. Br J Haematol. 1999. Nov;107(2):335–9. [DOI] [PubMed] [Google Scholar]
- 97.Cossu F, Vulliamy TJ, Marrone A, et al. A novel DKC1 mutation, severe combined immunodeficiency (T+B-NK- SCID) and bone marrow transplantation in an infant with Hoyeraal-Hreidarsson syndrome. Br J Haematol. 2002. Dec;119(3):765–8. [DOI] [PubMed] [Google Scholar]
- 98.Vulliamy T, Beswick R, Kirwan MJ, et al. Telomere length measurement can distinguish pathogenic from non-pathogenic variants in the shelterin component, TIN2. Clin Genet. 2012. Jan;81(1):76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dhanraj S, Gunja SM, Deveau AP, et al. Bone marrow failure and developmental delay caused by mutations in poly(A)-specific ribonuclease (PARN). J Med Genet. 2015. Nov;52(11):738–48. [DOI] [PubMed] [Google Scholar]
- 100.Jyonouchi S, Forbes L, Ruchelli E, et al. Dyskeratosis congenita: a combined immunodeficiency with broad clinical spectrum--a single-center pediatric experience. Pediatr Allergy Immunol. 2011. May;22(3):313–9. [DOI] [PubMed] [Google Scholar]
- 101.Ward SC, Savage SA, Giri N, et al. Progressive reticulate skin pigmentation and anonychia in a patient with bone marrow failure. J Am Acad Dermatol. 2017. Dec;77(6):1194–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Glousker G, Touzot F, Revy P, et al. Unraveling the pathogenesis of Hoyeraal-Hreidarsson syndrome, a complex telomere biology disorder. Br J Haematol. 2015. Aug;170(4):457–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Revesz T, Fletcher S, al-Gazali LI, et al. Bilateral retinopathy, aplastic anaemia, and central nervous system abnormalities: a new syndrome? J Med Genet. 1992. Sep;29(9):673–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mason JO 3rd, Yunker JJ, Nixon PA, et al. Proliferative retinopathy as a complication of dyskeratosis congenita. Retin Cases Brief Rep. 2009. Summer;3(3):259–62. [DOI] [PubMed] [Google Scholar]
- 105.Tolmie JL, Browne BH, McGettrick PM, et al. A familial syndrome with coats’ reaction retinal angiomas, hair and nail defects and intracranial calcification. Eye. 1988 1988/May/01;2(3):297–303. [DOI] [PubMed] [Google Scholar]
- 106.Polvi A, Linnankivi T, Kivela T, et al. Mutations in CTC1, encoding the CTS telomere maintenance complex component 1, cause cerebroretinal microangiopathy with calcifications and cysts. Am J Hum Genet. 2012. Mar 9;90(3):540–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Linnankivi T, Valanne L, Paetau A, et al. Cerebroretinal microangiopathy with calcifications and cysts. Neurology. 2006;67(8):1437–1443. [DOI] [PubMed] [Google Scholar]
- 108.Briggs TA, Abdel-Salam GM, Balicki M, et al. Cerebroretinal microangiopathy with calcifications and cysts (CRMCC). Am J Med Genet A. 2008. Jan 15;146A(2):182–90. [DOI] [PubMed] [Google Scholar]
- 109.Al-Rahawan MM, Giri N, Alter BP. Intensive immunosuppression therapy for aplastic anemia associated with dyskeratosis congenita. Int J Hematol. 2006. Apr;83(3):275–6. [DOI] [PubMed] [Google Scholar]
- 110.Dokal I. Dyskeratosis congenita in all its forms. British Journal of Haematology. 2000;110(4):768–779. [DOI] [PubMed] [Google Scholar]
- 111.Dokal I. Dyskeratosis congenita. Hematology Am Soc Hematol Educ Program. 2011;2011:480–6. [DOI] [PubMed] [Google Scholar]
- 112.Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Reviews. 2010 2010/May/01/;24(3):101–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tsakiri KD, Cronkhite JT, Kuan PJ, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci U S A. 2007. May 1;104(18):7552–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Armanios MY, Chen JJ, Cogan JD, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007. Mar 29;356(13):1317–26. [DOI] [PubMed] [Google Scholar]
- 115.Borie R, Tabèze L, Thabut G, et al. Prevalence and characteristics of TERT and TERC mutations in suspected genetic pulmonary fibrosis. European Respiratory Journal. 2016;48(6):1721–1731. [DOI] [PubMed] [Google Scholar]
- 116.Hoffman TW, van der Vis JJ, van Oosterhout MFM, et al. TINF2 Gene Mutation in a Patient with Pulmonary Fibrosis. Case Reports in Pulmonology. 2016;2016:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Newton CA, Batra K, Torrealba J, et al. Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive. European Respiratory Journal. 2016;48(6):1710–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cogan JD, Kropski JA, Zhao M, et al. Rare variants in RTEL1 are associated with familial interstitial pneumonia. Am J Respir Crit Care Med. 2015. Mar 15;191(6):646–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Petrovski S, Todd JL, Durheim MT, et al. An Exome Sequencing Study to Assess the Role of Rare Genetic Variation in Pulmonary Fibrosis. Am J Respir Crit Care Med. 2017. Jul 1;196(1):82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lederer DJ, Martinez FJ. Idiopathic Pulmonary Fibrosis. N Engl J Med. 2018. May 10;378(19):1811–1823. [DOI] [PubMed] [Google Scholar]
- 121.Giri N, Lee R, Faro A, et al. Lung transplantation for pulmonary fibrosis in dyskeratosis congenita: Case Report and systematic literature review. BMC Blood Disord. 2011. Jun 15;11:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fioredda F, Iacobelli S, Korthof ET, et al. Outcome of haematopoietic stem cell transplantation in dyskeratosis congenita. Br J Haematol. 2018. Oct;183(1):110–118. [DOI] [PubMed] [Google Scholar]
- 123.Karimi-Shah BA, Chowdhury BA. Forced vital capacity in idiopathic pulmonary fibrosis--FDA review of pirfenidone and nintedanib. N Engl J Med. 2015. Mar 26;372(13):1189–91. [DOI] [PubMed] [Google Scholar]
- 124.Khincha PP, Bertuch AA, Agarwal S, et al. Pulmonary arteriovenous malformations: an uncharacterised phenotype of dyskeratosis congenita and related telomere biology disorders. Eur Respir J. 2017. Jan;49(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Higgs C, Crow YJ, Adams DM, et al. Understanding the evolving phenotype of vascular complications in telomere biology disorders. Angiogenesis. 2019. Feb;22(1):95–102. [DOI] [PubMed] [Google Scholar]
- 126.Calado RT, Regal JA, Kleiner DE, et al. A Spectrum of Severe Familial Liver Disorders Associate with Telomerase Mutations. PLOS ONE. 2009;4(11):e7926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Calado RT, Brudno J, Mehta P, et al. Constitutional telomerase mutations are genetic risk factors for cirrhosis. Hepatology. 2011. May;53(5):1600–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kapuria D, Ben-Yakov G, Ortolano R, et al. The Spectrum of Hepatic Involvement in Patients With Telomere Disease. Hepatology. 2019. Feb 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chiu V, Hogen R, Sher L, et al. Telomerase Variants in Patients with Cirrhosis Awaiting Liver Transplantation. Hepatology. 2019. Jun;69(6):2652–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Aviv A, Anderson JJ, Shay JW. Mutations, Cancer and the Telomere Length Paradox. Trends Cancer. 2017. Apr;3(4):253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tsilou ET, Giri N, Weinstein S, et al. Ocular and orbital manifestations of the inherited bone marrow failure syndromes: Fanconi anemia and dyskeratosis congenita. Ophthalmology. 2010. Mar;117(3):615–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Samuel BP, Duffner UA, Abdel-Mageed AS, et al. Pulmonary Arteriovenous Malformations in Dyskeratosis Congenita. Pediatric Dermatology. 2015;32(4):e165–e166. [DOI] [PubMed] [Google Scholar]
- 133.Gordijn SJ, Brand PLP. A boy with breathlessness, digital clubbing and central cyanosis. European Journal of Pediatrics. 2004 2004/February/01;163(2):129–130. [DOI] [PubMed] [Google Scholar]
- 134.Sands A, Dalzell E, Craig B, et al. Multiple Intrapulmonary Arteriovenous Fistulas in Childhood. Pediatric Cardiology. 2000 2000/September/01;21(5):493–496. [DOI] [PubMed] [Google Scholar]
- 135.Hirota T, Yamagami T, Nakamura T, et al. Small pulmonary arteriovenous fistulae revealed by scintigraphy during selective injection of 99Tcm-macroaggregated albumin. The British Journal of Radiology. 2004;77(917):445–448. [DOI] [PubMed] [Google Scholar]
- 136.Patel PL, Suram A, Mirani N, et al. Derepression of hTERT gene expression promotes escape from oncogene-induced cellular senescence. Proceedings of the National Academy of Sciences. 2016;113(34):E5024–E5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Liu Y, Bloom SI, Donato AJ. The role of senescence, telomere dysfunction and shelterin in vascular aging. Microcirculation. 2019;26(2):e12487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yang T- LB, Chen Q, Deng JT, et al. Mutual reinforcement between telomere capping and canonical Wnt signalling in the intestinal stem cell niche [Article]. Nature Communications. 2017. March/17/online;8:14766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Woo D-H, Chen Q, Yang T-Lin B, et al. Enhancing a Wnt-Telomere Feedback Loop Restores Intestinal Stem Cell Function in a Human Organotypic Model of Dyskeratosis Congenita. Cell Stem Cell. 2016 2016/September/01/;19(3):397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rackley S, Pao M, Seratti GF, et al. Neuropsychiatric Conditions Among Patients with Dyskeratosis Congenita: A Link with Telomere Biology? Psychosomatics. 2012 2012/May/01/;53(3):230–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Alder JK, Chen JJ-L, Lancaster L, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proceedings of the National Academy of Sciences. 2008;105(35):13051–13056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cronkhite JT, Xing C, Raghu G, et al. Telomere shortening in familial and sporadic pulmonary fibrosis. Am J Respir Crit Care Med. 2008. Oct 1;178(7):729–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Fogarty PF, Yamaguchi H, Wiestner A, et al. Late presentation of dyskeratosis congenita as apparently acquired aplastic anaemia due to mutations in telomerase RNA. The Lancet. 2003 2003/November/15/;362(9396):1628–1630. [DOI] [PubMed] [Google Scholar]
- 144.Alder JK, Hanumanthu VS, Strong MA, et al. Diagnostic utility of telomere length testing in a hospital-based setting. Proceedings of the National Academy of Sciences. 2018;115(10):E2358–E2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Khincha PP, Dagnall CL, Hicks B, et al. Correlation of Leukocyte Telomere Length Measurement Methods in Patients with Dyskeratosis Congenita and in Their Unaffected Relatives. Int J Mol Sci. 2017. Aug 13;18(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Alter BP, Baerlocher GM, Savage SA, et al. Very short telomere length by flow fluorescence in situ hybridization identifies patients with dyskeratosis congenita. Blood. 2007. Sep 1;110(5):1439–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gadalla SM, Khincha PP, Katki HA, et al. The limitations of qPCR telomere length measurement in diagnosing dyskeratosis congenita. Mol Genet Genomic Med. 2016. Jul;4(4):475–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Aubert G, Hills M, Lansdorp PM. Telomere length measurement—Caveats and a critical assessment of the available technologies and tools. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2012;730(1):59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Walsh MF, Chang VY, Kohlmann WK, et al. Recommendations for Childhood Cancer Screening and Surveillance in DNA Repair Disorders. Clinical Cancer Research. 2017;23(11):e23–e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Rose SR, Kim MO, Korbee L, et al. Oxandrolone for the treatment of bone marrow failure in Fanconi anemia. Pediatr Blood Cancer. 2014. Jan;61(1):11–9. [DOI] [PubMed] [Google Scholar]
- 151.Shahidi NT, Diamond LK. Testosterone-Induced Remission in Aplastic Anemia of Both Acquired and Congenital Types. New England Journal of Medicine. 1961;264(19):953–967. [DOI] [PubMed] [Google Scholar]
- 152.Giri N, Pitel PA, Green D, et al. Splenic peliosis and rupture in patients with dyskeratosis congenita on androgens and granulocyte colony-stimulating factor. Br J Haematol. 2007. Sep;138(6):815–7. [DOI] [PubMed] [Google Scholar]
- 153.Islam A, Rafiq S, Kirwan M, et al. Haematological recovery in dyskeratosis congenita patients treated with danazol. Br J Haematol. 2013. Sep;162(6):854–6. [DOI] [PubMed] [Google Scholar]
- 154.Khincha PP, Wentzensen IM, Giri N, et al. Response to androgen therapy in patients with dyskeratosis congenita. Br J Haematol. 2014. May;165(3):349–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Townsley DM, Dumitriu B, Liu D, et al. Danazol Treatment for Telomere Diseases. N Engl J Med. 2016. May 19;374(20):1922–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Calado RT, Yewdell WT, Wilkerson KL, et al. Sex hormones, acting on the TERT gene, increase telomerase activity in human primary hematopoietic cells. Blood. 2009. Sep 10;114(11):2236–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Grossmann M. Danazol Treatment for Telomere Diseases. N Engl J Med. 2016. Sep 15;375(11):1095. [DOI] [PubMed] [Google Scholar]
- 158.Khincha PP, Bertuch AA, Gadalla SM, et al. Similar telomere attrition rates in androgen-treated and untreated patients with dyskeratosis congenita. Blood Adv. 2018. Jun 12;2(11):1243–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Trautmann K, Jakob C, von Grunhagen U, et al. Eltrombopag fails to improve severe thrombocytopenia in late-stage dyskeratosis congenita and diamond-blackfan-anaemia. Thromb Haemost. 2012. Aug;108(2):397–8. [DOI] [PubMed] [Google Scholar]
- 160.Barbaro P, Vedi A. Survival after Hematopoietic Stem Cell Transplant in Patients with Dyskeratosis Congenita: Systematic Review of the Literature. Biol Blood Marrow Transplant. 2016. Jul;22(7):1152–1158. [DOI] [PubMed] [Google Scholar]
- 161.Agarwal S. Evaluation and Management of Hematopoietic Failure in Dyskeratosis Congenita. Hematol Oncol Clin North Am. 2018. Aug;32(4):669–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Gadalla SM, Sales-Bonfim C, Carreras J, et al. Outcomes of allogeneic hematopoietic cell transplantation in patients with dyskeratosis congenita. Biol Blood Marrow Transplant. 2013. Aug;19(8):1238–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Dietz AC, Savage SA, Vlachos A, et al. Late Effects Screening Guidelines after Hematopoietic Cell Transplantation for Inherited Bone Marrow Failure Syndromes: Consensus Statement From the Second Pediatric Blood and Marrow Transplant Consortium International Conference on Late Effects After Pediatric HCT. Biology of Blood and Marrow Transplantation. 2017 2017/September/01/;23(9):1422–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Justet A, Thabut G, Manali E, et al. Safety and efficacy of pirfenidone in patients carrying telomerase complex mutation. Eur Respir J. 2018. Mar;51(3). [DOI] [PubMed] [Google Scholar]
- 165.Rochwerg B, Neupane B, Zhang Y, et al. Treatment of idiopathic pulmonary fibrosis: a network meta-analysis. BMC Med. 2016. Feb 3;14:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zlateska B, Ciccolini A, Dror Y. Treatment of dyskeratosis congenita-associated pulmonary fibrosis with danazol. Pediatric Pulmonology. 2015;50(12):E48–E51. [DOI] [PubMed] [Google Scholar]
- 167.Silhan LL, Shah PD, Chambers DC, et al. Lung transplantation in telomerase mutation carriers with pulmonary fibrosis. Eur Respir J. 2014. Jul;44(1):178–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Borie R, Kannengiesser C, Hirschi S, et al. Severe hematologic complications after lung transplantation in patients with telomerase complex mutations. J Heart Lung Transplant. 2015. Apr;34(4):538–46. [DOI] [PubMed] [Google Scholar]
- 169.Tokman S, Singer JP, Devine MS, et al. Clinical outcomes of lung transplant recipients with telomerase mutations. The Journal of heart and lung transplantation : the official publication of the International Society for Heart Transplantation. 2015;34(10):1318–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mahansaria SS, Kumar S, Bharathy KG, et al. Liver Transplantation After Bone Marrow Transplantation for End Stage Liver Disease with Severe Hepatopulmonary Syndrome in Dyskeratosis Congenita: A Literature First. J Clin Exp Hepatol. 2015. Dec;5(4):344–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.George G, Rosas IO, Cui Y, et al. Short Telomeres, Telomeropathy, and Subclinical Extrapulmonary Organ Damage in Patients With Interstitial Lung Disease. CHEST. 2015;147(6):1549–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lebeer M, Wuyts WA, Cassiman D, et al. Multiple Solid Organ Transplantation in Telomeropathy: Case Series and Literature Review. Transplantation. 2018;102(10):1747–1755. [DOI] [PubMed] [Google Scholar]
- 173.Fernandez RJ 3rd, Johnson FB. A regulatory loop connecting WNT signaling and telomere capping: possible therapeutic implications for dyskeratosis congenita. Ann N Y Acad Sci. 2018. Apr;1418(1):56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Iarriccio L, Manguan-Garcia C, Pintado-Berninches L, et al. GSE4, a Small Dyskerin- and GSE24.2-Related Peptide, Induces Telomerase Activity, Cell Proliferation and Reduces DNA Damage, Oxidative Stress and Cell Senescence in Dyskerin Mutant Cells. PLoS One. 2015;10(11):e0142980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Knight SW, Heiss NS, Vulliamy TJ, et al. X-linked dyskeratosis congenita is predominantly caused by missense mutations in the DKC1 gene. Am J Hum Genet. 1999. Jul;65(1):50–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Giri N, Alter BP, Penrose K, et al. Immune status of patients with inherited bone marrow failure syndromes. American journal of hematology. 2015;90(8):702–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wagner CL, Hanumanthu VS, Talbot CC, Jr., et al. Short telomere syndromes cause a primary T cell immunodeficiency. J Clin Invest. 2018. Dec 3;128(12):5222–5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ungar RA, Giri N, Pao M, et al. Complex phenotype of dyskeratosis congenita and mood dysregulation with novel homozygous RTEL1 and TPH1 variants. American Journal of Medical Genetics Part A. 2018;176(6):1432–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Mahiques L, Febrer I, Vilata JJ, et al. A case of dyskeratosis congenita associated with schizophrenia and two malignancies. J Eur Acad Dermatol Venereol. 2006. Oct;20(9):1159–61. [DOI] [PubMed] [Google Scholar]
- 180.Aslan D, Akata RF, Holme H, et al. Limbal stem cell deficiency in patients with inherited stem cell disorder of dyskeratosis congenita. Int Ophthalmol. 2012. Dec;32(6):615–22. [DOI] [PubMed] [Google Scholar]
- 181.Kalejaiye A, Giri N, Brewer CC, et al. Otologic manifestations of Fanconi anemia and other inherited bone marrow failure syndromes. Pediatr Blood Cancer. 2016. Dec;63(12):2139–2145. [DOI] [PubMed] [Google Scholar]
- 182.Atkinson JC, Harvey KE, Domingo DL, et al. Oral and dental phenotype of dyskeratosis congenita. Oral Dis. 2008. Jul;14(5):419–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Li F, Li W, Qiao X, et al. Clinical features of dyskeratosis congenita in mainland China: case reports and literature review. Int J Hematol. 2019. Mar;109(3):328–335. [DOI] [PubMed] [Google Scholar]
- 184.Nunes H, Monnet I, Kannengiesser C, et al. Is Telomeropathy the Explanation for Combined Pulmonary Fibrosis and Emphysema Syndrome?: Report of a Family with TERT Mutation. American Journal of Respiratory and Critical Care Medicine. 2014 2014/March/15;189(6):753–754. [DOI] [PubMed] [Google Scholar]
- 185.Jonassaint NL, Guo N, Califano JA, et al. The gastrointestinal manifestations of telomere-mediated disease. Aging Cell. 2013. Apr;12(2):319–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Giri N, Stratton P, Savage SA, et al. Pregnancies in patients with inherited bone marrow failure syndromes in the NCI cohort. Blood. 2017. Oct 5;130(14):1674–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Shankar RK, Giri N, Lodish MB, et al. Bone mineral density in patients with inherited bone marrow failure syndromes. Pediatr Res. 2017. Sep;82(3):458–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Sahoo N, Padhi S, Patra S, et al. Dyskeratosis congenita, bone marrow failure, and gastric adenocarcinoma: an insight into telomere biology. Turk J Gastroenterol. 2017. Jul;28(4):319–321. [DOI] [PubMed] [Google Scholar]
- 189.Horiguchi N, Kakizaki S, Iizuka K, et al. Hepatic Angiosarcoma with Dyskeratosis Congenita. Internal Medicine. 2015;54(22):2867–2872. [DOI] [PubMed] [Google Scholar]
- 190.Olson TS, Chan ES, Paessler ME, et al. Liver failure due to hepatic angiosarcoma in an adolescent with dyskeratosis congenita. J Pediatr Hematol Oncol. 2014. May;36(4):312–5. [DOI] [PMC free article] [PubMed] [Google Scholar]